| CATEGORII DOCUMENTE | ||
|
||
| Astronomie | Biofizica | Biologie | Botanica | Carti | Chimie | Copii |
| Educatie civica | Fabule ghicitori | Fizica | Gramatica | Joc | Literatura romana | Logica |
| Matematica | Poezii | Psihologie psihiatrie | Sociologie |
Ustensile de laborator. Masurarea volumelor.
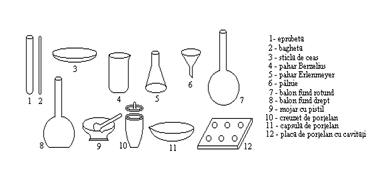
In laboratoarele chimice se utilizeaza vase si aparate confectionate din materiale rezistente la actiunea chimica si la variatiile bruste de temperatura (sticla, portelan, metal etc.).
Vase simple de sticla
Eprubeta are forma unui tub de sticla inchis la un capat si care se pastreaza in stativ de lemn sau metal. Eprubetele pot fi cilindrice, folosite pentru efectuarea reactiilor si conice, pentru separarea precipitatelor la centrifuga. Eprubetele se umplu cel mult pana la jumatate, iar agitarea continutului se face prin scuturare. La incalzire, eprubeta se inclina, pentru a asigura fierberea lichidului din partea superioara si se agita usor pentru a evita degajarea brusca a vaporilor.
Bagheta serveste la amestecarea si transvazarea lichidelor prin curgerea lor de-a lungul baghetei inclinate.
Sticla de ceas se foloseste pentru efectuarea reactiilor in picaturi sau pentru cantarire.
Paharele Berzelius si paharele conice Erlenmeyer se folosesc pentru prepararea solutiilor, pentru efectuarea de precipitari, filtrari sau transvazari de lichide.
Baloanele cu fund plat se folosesc pentru incalzirea lichidelor si distilari.
Palnia de sticla serveste la transvazarea lichidelor sau ca suport pentru materialul filtrant la efectuarea de filtrari simple.
Vase de portelan si cuart
Vasele se confectioneaza din portelan sau cuart atunci cand se cere o rezistenta chimica, termica sau mecanica mai ridicata.
Mojarul cu pistil serveste la sfaramarea si pulverizarea substantelor.
Capsula se foloseste pentru obtinerea de topituri.
Creuzetul de portelan se foloseste pentru calcinari si obtinerea de topituri.
Placa de portelan cu cavitati se utilizeaza la efectuarea reactiilor in picaturi.
Ustensile din metal
Cele mai intrebuintate ustensile din metal sunt: clestele, foarfeca, lingura, spatula, trepiedul, tringhiul de samota, stativul, inelul, clema, mufa.
Ustensile din lemn
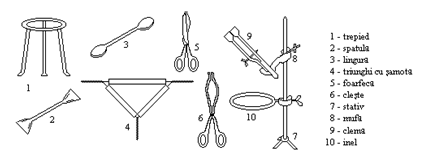
Cele mai utilizate sunt:
clestele pentru eprubete si stativul pentru eprubete.
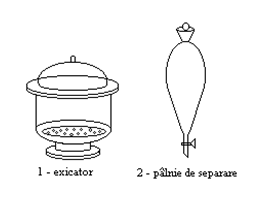
Vase si aparate speciale
Exicatorul se foloseste pentru uscarea lenta si pastrarea substantelor care absorb cu usurinta umiditatea din aer. In partea tronconica a exicatorului se introduce o substanta care absoarbe umiditatea (clorura de calciu anhidra, acid sulfuric concentrat, anhidrida fosforica). Deasupra absorbantului se aseaza o placa de portelan perforata ce permite circulatia aerului in exicator, iar pe aceasta placa se aseaza produsul destinat uscarii, intr-o sticla de ceas, fiola sau creuzet. Marginile slefuite ale exicatorului se ung cu vaselina pentru ca la inchiderea acestuia cu capacul sa se formeze intre cele doua parti un film subtire de vaselina care sa ermetizeze interiorul exicatorului. Deschiderea exicatorului se face prin culisare.
Palnia de separare serveste la spalarea si separarea lichidelor nemiscibile cu densitati diferite.
Masurarea volumelor
In analiza volumetrica, alaturi de cantarire, masurarea volumelor este operatia cea mai importanta. Unitatea de masura este litrul (l), definit ca volumul ocupat de un decimetru cub. Ca submultiplu se foloseste in mod curent mililitrul (ml) sau centimetrul cub (cm3)
Vasele de masurat volume se pot imparti in doua categorii:
- vase gradate pentru umplere, al caror semn indica volumul pe care-l masoara;
- vase gradate pentru golire, cu doua semne: semnul superior indica volumul de umplere, iar la golire se masoara volumul indicat de semnul inferior. Aceasta diferentiere trebuie facuta deoarece, la golire, o parte din lichidul din vas adera la pereti si astfel, volumul la golire este mai mic decat volumul la umplere.
Gradarea vaselor se face la 40C si la 200C. Pe fiecare vas este gravata capacitatea precum si temperatura la care este facuta etalonarea.
Citirea volumelor se face astfel incat raza vizuala sa fie perpendiculara pe diviziunea corespunzatoare volumului de masurat, iar limita inferioara a meniscului solutiei sa fie tangenta la aceasta diviziune, pentru lichidele incolore; pentru cele colorate, diviziunea va fi tangenta la limita superioara a meniscului.
Baloanele cotate sunt vase pentru umplere, cu fund plat si au pe gat un semn circular care arata pana unde se vor umple. Se folosesc pentru prepararea solutiilor titrate, plecand de la substante titrimetrice (etalon). Substanta cantarita se trece cantitativ in balon, se adauga apa distilata si se dizolva, aducandu-se apoi la semn cu apa distilata. Se omogenizeaza continutul prin agitare.
Cilindrii gradati sunt vase pentru umplere, mult mai putin precise decat baloanele cotate, de aceea se folosesc numai pentru masuratori aproximative.
Pipetele sunt vase pentru golire si sunt de doua categorii: pipete cotate (fara gradatii intermediare), cu ajutorul carora se masoara numai volumul inscris pe pipeta, si pipete gradate cu care se pot masura si volume mai mici decat cel inscris.
Pentru a masura cu pipeta un anumit volum, se introduce varful pipetei in lichid si se aspira pana cand lichidul depaseste gradatia dorita. Se astupa pipeta cu degetul aratator, se scoate din lichid si se lasa sa curga lichidul pana cand limita inferioara a meniscului lichidului atinge gradatia. La golire, lichidul se lasa sa curga liber, atingand varful pipetei de peretele vasului in care se goleste lichidul. Nu se sufla in pipeta pentru a goli si lichidul care ramane in varful pipetei.
Biuretele sunt vase pentru golire
alcatuite dintr-un tub gradat si care la partea inferioara au un
dispozitiv cu ajutorul caruia se poate inchide sau regla curgerea
lichidului (o clema sau un robinet). In timpul lucrului, biuretele se fixeaza
vertical pe un stativ cu ajutorul unei cleme. Umplerea lor se face cu ajutorul
unei palnii mici, iar prin deschiderea robinetului sau a clemei se lasa
lichidul sa se scurga pana cand ajunge la semn, dupa
regulile prezentate anterior (vezi 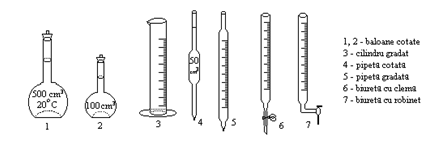
umplerea pipetelor).
Cantarirea la balanta tehnica si analitica
Determinarea masei unei substante se face prin cantarire la balanta tehnica sau analitica, in functie de gradul de precizie necesar.
Balanta tehnica
Este folosita pentru cantariri aproximative, putandu-se determina cel mult doua zecimale, cu ajutorul pieselor metalice etalon, de masa determinata.
Exista balante tehnice cu diferite sarcini maxime, de obicei folosindu-se in laborator balanta cu doua talere, pentru mase mici. Pentru cantarirea obiectelor ce depasesc 500 g se apeleaza la balanta tehnica cu un taler , de constructie mai solida.
Cutia cu mase etalonate
O serie completa de mase etalonate mari este alcatuita dintr-o masa etalonata de 100 g, una de 50 g, doua de 20 g, una de 10 g, una de 5 g, doua de 2 g si una de 1 g. Fractiunile de gram sunt confectionate din aluminiu si sunt de: 500, 200, 100, 100, 50, 20, 10, 10, 10 mg..
Cantarirea la balanta tehnica
Initial se verifica daca balanta este bine echilibrata prin deschiderea ei si verificarea punctului zero (acul balantei trebuie sa se aseze exact peste diviziunea zero a scalei gradate. Se inchide la loc balanta.
 Se
aseaza obiectul de cantarit pe talerul stang al balantei
(sau pe taler la cea masiva) iar pe talerul drept se aseaza cu
aproximatie mase etalonate pentru a contrabalansa cele doua talere.
Se deschide cu grija balanta, observand sensul de variatie al
acului. Se scad sau se adauga mase etalonate pe talerul drept pana
cand cele doua talere vor sustine aceeasi masa.
Se
aseaza obiectul de cantarit pe talerul stang al balantei
(sau pe taler la cea masiva) iar pe talerul drept se aseaza cu
aproximatie mase etalonate pentru a contrabalansa cele doua talere.
Se deschide cu grija balanta, observand sensul de variatie al
acului. Se scad sau se adauga mase etalonate pe talerul drept pana
cand cele doua talere vor sustine aceeasi masa.
Nici o operatie (adaugare sau retragere de mase etalonate sau substante) nu se va efectua cu balanta deschisa.
Pentru a determina masa obiectului cantarit se face suma maselor etalonate adaugate pe talerul din dreapta.
La celalalt tip de balanta tehnica, masa obiectului cantarit se citeste adunand la masa de plecare, stabilita dintr-un dispozitiv cu rotita(o, 100 g, 200g, 500g, 1000g etc.), valoarea citita pe ecranul cu scala gradata, la care se opreste acul balantei, dupa oprirea oscilatiilor.
Balanta analitica
Este folosita pentru cantariri exacte, putandu-se determina patru zecimale. Acest aparat are ca element principal o parghie cu doua brate egale si scurte. In cazul balantelor cu amortizor, dupa un numar mic de oscilatii, acul balantei se opreste.
Parghia se sprijina pe coloana centrala a balantei cu ajutorul unui cutit (o prisma de agat) prin intermediul unei placute tot din agat. La capetele parghiei sunt sprijinite cu ajutorul altor doua cutite doua furci in care sunt suspendate cele doua platane (talere). In centrul parghiei este fixat un ac indicator ce permite observarea celor mai mici devieri ale parghiei la cantarire. In acest scop este fixata in spatele acului o scala gradata. Balanta este prevazuta cu un dispozitiv de oprire ce permite ridicarea parghiei balantei, dispozitiv pus in functiune cu ajutorul unui buton asezat sub placa de suport a balantei.
Un sistem de proiectie optica mareste imaginea scalei pe un ecran de sticla mata. In interiorul cutiei balantei exista bare metalice care sustin mase etalonate de dimensiuni foarte mici, ce pot fi adaugate cand este necesar prin rotirea unor suruburi amplasate lateral.
Balanta analitica este amplasata intr-o cutie cu geamuri prevazuta cu usi ce o protejeaza de praf, curenti de aer si variatii de temperatura. Cand balanta este pusa in functiune, usile sunt inchise. De asemenea, balanta trebuie asezata in pozitie perfect orizontala, cu ajutorul picioarelor reglabile, de preferat pe o consola bine fixata in perete sau o masa foarte stabila.
Balanta analitica electronica
Este un aparat modern, mai usor de utilizat, care dispune de afisaj electronic si de posibilitatea de a efectua automat tara unui obiect pentru a cantari doar o alta substanta care se adauga ulterior (ex. - se cantareste o sticla de ceas, se face tara si apoi se adauga pe sticla de ceas o anumita cantitate de substanta; masa afisata va corespunde doar substantei adaugate). De asemenea, reglarea la zero se face automat, apasand pe tasta tara atunci cand talerul balantei este descarcat.
 La
efectuarea unei cantariri la balanta analitica trebuie sa
se respecte anumite etape si reguli:
La
efectuarea unei cantariri la balanta analitica trebuie sa
se respecte anumite etape si reguli:
Etape: - se conecteaza balanta la sursa decurent electric; - se deschide balanta; - se verifica punctul zero si eventual se regleaza, daca este necesar; - se inchide balanta (doar cea cu amortizor); - se aseaza obiectul de cantarit pe taler (pe cel din stanga, la balanta cu amortizor); - pentru balanta electronica, se face citirea si se inchide balanta; pentru balanta cu amortizor, se adauga pe talerul din dreapta mase etalonate, treptat, pana cand cele doua talere sunt echilibrate si acul balantei variaza in domeniul pozitiv al scalei gradate (pentru fiecare adaugare sau retragere de mase etalonate, mai mari sau mici de 1 g, se inchide balanta) ; - se citeste valoarea astfel: gramele se calculeaza facand suma maselor etalonate de pe taler, primele doua zecimale se citesc cu ajutorul celor doua suruburi situate lateral, de pe care s-au adaugat diverse subunitati ale gramului, iar a treia si a patra zecimala se citesc pe scala gradata a balantei; - se inchide balanta, se ia obiectul de cantarit de pe taler si se descarca balanta in ordinea descrescatoare a maselor adaugate. La sfarsitul cantaririlor se verifica din nou punctul zero.
Reguli : - nu se incarca niciodata balanta peste sarcina maxima;
- nu se aseaza pe taler obiecte calde sau foarte reci;
- substantele solide ce pot ataca metalul talerului se cantaresc pe sticle de ceas sau in alte vase, niciodata direct pe taler;
- in timpul cantarii, usile balantei se inchid;
- masele etalonate se manevreaza numai cu penseta.
Prepararea solutiilor de diferite concentratii
Exprimarea concentratiilor solutiilor
Concentratia unei solutii reprezinta cantitatea de substanta dizolvata intr-un anumit volum de solutie sau de solvent. Ea reprezinta raportul dintre solut (dizolvat) si solvent
Dupa concentratie, se deosebesc solutii concentrate, ce contin cantitati mari de substanta dizolvata si solutii diluate, cu cantitati mici de substanta dizolvata.
1. Concentratia procentuala (% ; C%)
Poate fi exprimata in unitati de masa sau in unitati de volum Concentratia procentuala exprimata in unitati de masa se defineste astfel:
Concentratia procentuala reprezinta gramele de substanta dizolvate in 100 grame de solutie.
Exemple: O solutie de concentratie 15 % va contine 15 grame de substanta dizolvata, indiferent de natura substantei.
Probleme:
1) Cate grame de NaOH se gasesc dizolvate in 450 grame de solutie de concentratie 25 % ?
Rezolvare:
100 g solutie NaOH25 g NaOH
450 g solutie ..x
x = 450 25 / 100 = 112,5 g NaOH
2) Calculati gramele de KI necesare prepararii a 700 g solutie 2 % .
Rezolvare:
100 g solutie KI .2 g KI
750 g solutie x
x = 750 2 / 100 = 15 g KI
3) Ce concentratie procentuala are solutia preparata prin dizolvarea a 60 g NaCl in 250 g solutie?
Rezolvare:
250 g solutie NaCl .60 g NaCl
100 g solutie .C %
C % = 100 60 / 250 = 24 %
2. Concentratia molara - molaritatea (m;M)
Concentratia molara reprezinta numarul de moli dizolvati in 1000 cm3 solutie (un litru solutie).
Molul reprezinta masa molara exprimata in grame. Aceasta se calculeaza prin insumarea maselor atomice ale elementelor componente, adunand, cand este cazul, masa moleculelor de apa de cristalizare.
Exemplu:
MNaCl = ANa + ACl = 23 + 35,5 = 58,5
MCuSO4 5 H2O = ACu + AS + 4 AO + 5 (2 AH + AO) = 64 + 32 + 4 !6 + 5 (2 1 + 16) = 250
Numarul de moli dintr-o substanta se calculeaza raportand masa in grame a substantei (cantarita la balanta) la masa ei molara.
nr.
moli = ![]()
Probleme:
1) Calculati cate grame de acid clorhidric se afla dizolvate in 500 ml solutie de concentratie 2 m , stiind ca masa molara a acidului MHCl = 36,5 .
Rezolvare:
1000 ml solutie HCl 2 moli HCl (236,5 g HCl)
500 ml solutie x
x = 500 2 36,5 / 1000 = 36,5 g HCl
2) Calculati molaritatea (concentratia molara) a unei solutii care contine dizolvat 1 g NaOH in 250 ml solutie, stiind ca MNaOH = 40 .
Rezolvare:
nr. moli NaOH = 1 / 40 = 0,025 moli NaOH
250 ml solutie 0,025 moli NaOH
1000 ml solutie.C molara
concentratia molara = 1000 0,025 / 250 = 0,1 m
O concentratie mai putin utilizata in practica analitica este concentratia molala, definita ca numarul de moli de substanta dizolvati in 1000 ml solvent.
3. Concentratia normala - normalitatea (n,N)
Concentratia normala reprezinta numarul de echivalenti gram de substanta dizolvati in 1000 ml solutie.
Numarul de echivalenti gram se calculeaza raportand masa in grame de substanta la echivalentul chimic al substantei respective.
nr. echivalenti = ![]()
Echivalentul chimic (E) reprezinta numarul de grame de substanta ce reactioneaza cu un atom gram de hidrogen (1 gram) sau cu un atom gram de oxigen (8 grame).
Echivalentul chimic notat E se calculeaza in functie de reactia chimica la care participa substanta.
- pentru elemente : E = masa atomica / valenta;
- pentru acizi : E = masa molara / numarul de protoni cedati in reactie;
- pentru baze : E = masa molara / numarul de protoni acceptati in reactie (de obicei, numarul de grupari -OH din molecula);
- pentru saruri : E = masa molara / valenta comuna a ionilor sarii;
- pentru saruri ce participa in reactii redox : E = masa molara / numarul de electroni cedati sau acceptati.
Probleme:
1) Ce cantitate de permanganat de potasiu este necesara prepararii a 300 ml solutie de concentratie 0,05 n , stiind ca masa molara a permanganatului de potasiu este 158 ?
Rezolvare: EKMnO4 = 158 / 5 = 31,6
1000 ml solutie ..0,05 e (0,0531,6 g) KMnO4
300 ml ..x
x = 300 0,05 31,6 / 1000 g KMnO4
2) Calculati normalitatea unei solutii de acid sulfuric ce contine 0,98 g H2SO4 dizolvati in 500 ml solutie, stiind ca masa molara a H2SO4este 98.
Rezolvare:
EH2SO4 = 98 / 2 = 49
nr. e H2SO4 = 0,98 / 49 = 0,02 e
500 ml solutie .0,02 e H2SO4
1000 ml .Cnormala
Cnormala = 1000 0,02 / 500 = 0,04 n
4. Titrul
Titrul unei solutii reprezinta gramele de substanta dizolvata in 1000 ml solutie sau intr-un mililitru solutie.
Titrul poate fi teoretic (Tt), atunci cand se calculeaza din date teoretice, sau real (Tr), cand se obtine prin calcul intr-o situatie data, in urma unei reactii chimice.
Relatia de calcul a titrului este: T = E N
Factorul de corectie volumetrica (F) reprezinta un numar care arata abaterea concentratiei reale a unei solutii de la concentratia dorita.
Factorul F se calculeaza ca raportul dintre titrul real si cel teoretic.
F = Treal / Tteoretic
Metode de purificare a substantelor:
dizolvarea, filtrarea, cristalizarea
Cristalizarea este una dintre tehnicile cele mai folosite in purificarea substantelor solide. In principiu, metoda se bazeaza pe proprietatea celor mai multe substante de a fi mai solubile intr-un solvent la cald decat la rece.
Prin solubilitate (grad de solubilitate) se intelege cantitatea maxima de substanta care la o anumita temperatura se poate dizolva intr-o anumita cantitate de solvent.
Solubilitatea substantelor variaza cu temperatura, aceasta variatie putand fi reprezentata grafic prin curbe de solubilitate.
Produsul de purificat se dizolva la cald intr-o cantitate cat mai mica de solvent. Dupa indepartarea impuritatilor insolubile prin filtrare, solutia se lasa sa se raceasca. O mare parte din produs se separa sub forma de cristale mai mult sau mai putin pure.
La recristalizarea unei substante se parcurg urmatoarele etape:
1 - alegerea solventului potrivit;
2 - dizolvarea substantei solide in solvent la o temperatura cat mai apropiata de punctul de fierbere al acestuia;
3 - filtrarea solutiei la cald pentru indepartarea impuritatilor insolubile;
4 - cristalizarea din solutie a substantelor pure;
5 - filtrarea si spalarea cristalelor;
6 - uscarea.
La efectuarea recristalizarii, un factor important este selectarea solventului. Un bun solvent de recristalizare indeplineste urmatoarele conditii:
- nu reactioneaza cu substanta dizolvata;
- dizolva o cantitate mai mare de substanta la cald decat la rece (diferenta se depune sub forma de cristale la racirea solutiei);
- nu dizolva la cald impuritatile din substanta de purificat;
- are un punct de fierbere mai coborat decat punctul de topire al substantei;
- are punctul defierbere suficient de coborat astfel incat sa poata fi usor indepartat la uscarea cristalelor;
- este accesibil si prezinta riscuri minime in ceeace priveste toxicitatea si inflamabilitatea.
Apa intruneste cele mai multe din conditiile de mai sus, de aceea este folosita ori de cate ori este posibil.
Dizolvarea. Substanta de purificat pulverizata se introduce intr-un pahar Berzelius. Se toarna peste substanta cantitatea necesara de solvent care la temperatura de fierbere sa asigure trecerea in solutie a substantei, cu exceptia impuritatilor insolubile. Este important sa nu se toarne dintr-o data prea mult solvent, deoarece la o dilutie excesiva substanta nu mai cristalizeaza.
Nici solutiile saturate la cald nu sunt de preferat, din cauza tendintei prea rapide de cristalizare, care are loc chiar in timpul filtrarii.
Filtrarea la cald. Pentru indepartarea impuritatilor insolubile, solutia se filtreaza pe o palnie conica prevazuta cu filtru obisnuit sau cutat. Solutia de filtrat se toarna fierbinte si in mod continuu pe filtru, pentru a evita racirea. Anterior, filtrul va fi umezit cu o cantitate mica din acelasi solvent. Filtratul se culege intr-un cristalizor sau pahar Erlenmeyer.
Cristalizarea. Filtratul fierbinte este lasat sa se raceasca la temperatura camerei, scaderea temperaturii micsorand solubilitatea substantei. Solutia devine suprasaturata si incep sa apara cristalele.
O racire rapida (cu gheata sau jet de apa rece) nu este indicata deoarece favorizeaza formarea de cristale fine (numar crescut de centre de cristalizare) care pot adsorbi pe suprafata impuritati din solutie; acelasi efect il are si agitarea.
La racirea lenta, numarul centrelor de cristalizare este mic si cristalele pot creste la o dimensiune convenabila. Daca dupa racire cristalizarea nu are loc, se insamanteaza solutia cu un cristal din substanta de purificat.
Filtrarea cristalelor. Amestecul de cristale si solutie se filtreaza la presiune redusa (la vid) folosind o palnie Bchner atasata la un flacon de vid.
Hartia de filtru se decupeaza la dimensiunea fundului palniei, se umezeste cu solvent apoi se transfera cristalele cu ajutorul unei baghete. Solutia filtrata se foloseste la trecerea completa a cristalelor pe filtru, apoi la spalarea acestora. Pentru operatia de spalare se intrerupe vidul.
Cristalele filtrate se preseaza bine cu o spatula sau cu un dop de sticla iar filtrarea are loc pana la aspirarea completa a lichidului despalare.
Uscarea cristalelor. Operatia are ca scop eliminarea urmelor de solvent. Cristalele se trec cu ajutorul baghetei pe o sticla de ceas sau pe o hartie de filtru. Uscarea se face fie in aer la temperatura camerei, fie intr-o etuva la o temperatura cu cel putin 200C mai mica decat punctul de topire al substantei purificate.
Determinarea constantelor fizice ale unor lichide : densitatea
Substantele pure contin o singura specie moleculara si poseda anumite proprietati fizice care le caracterizeaza: punct de topire, punct de fierbere, densitate, indice de refractie etc.
Substanta pura prezinta aceleasi proprietati fizice si chimice in toata masa sa si este caracterizata prin compozitie chimica bine determinata.
Densitatea
Reprezinta masa unitatii de volum si se exprima ca raportul dintre masa unei solutii si volumul ocupat de aceasta la o temperatura data.
ρ = m / V
Unitatea de masura a densitatii este de obicei g/cm3 dar se pot folosi si alte unitati de masura in functie de caz: kg/m3, g/l etc.
Determinarea densitatii unei solutii sau a unui lichid oarecare se poate face cu ajutorul unui dispozitiv numit densimetru. Acesta este format dintr-un tub de sticlacu o forma speciala care la partea inferioara are o dilatatie umpluta cu greutati (bile de plumb, de exemplu) care asigura imersarea dispozitivului in lichidul de cercetat si totodata si stabilitatea acestuia. La partea superioara ingustata prezinta in interior o scala gradata pe care se poate citi direct densitatea lichidului in g/cm3 (la densimetrele obisnuite).
Mod de lucru
Lichidul a carui densitate trebuie masurata se introduce intr-un tub de sticla vertical (cilindru gradat) apoi se coboara usor densimetrul in lichid si se asteapta ca acesta sa se stabilizeze. Se citeste gradatia la care ajunge nivelul superior al lichidului, apoi se scoate densimetru din lichid, se spala si se sterge cu grija.
O alta modalitate de a determina densitatea unei solutii sau a unui lichid oarecare implica folosirea picnometrului, un vas de sticla cu volum exact determinat.
Mod de lucru
Se cantareste picnometrul perfect curat si uscat la balanta analitica si se noteaza masa m1. Se umple picnometrul cu lichid de masurat iar prin fixarea capacului cu capilar se ajusteaza volumul la valoarea exact cunoscuta. Se sterge cu atentie picnometru plin pana cand este uscat la exterior si se cantareste din nou la balanta analitica, notandu-se masa m2. Prin diferenta intre cele doua valori dse determina masa lichidului din picnometru. Facand raportul dintre valoarea obtinuta (m2 - m1) si volumul cunoscut al picnometrului se obtine valoarea densitatii lichidului de cercetat.
Cromatografia
Identificarea cationilor Cu2+, Fe3+ si Co3+ si separarea lor prin cromatografie pe hartie
Cromatografia pe hartie ese o metoda cromatografica de repartitie,faza stationara fiind hartia cromatografica. Faza mobila (eluent) antreneaza componentele amestecului de analizat cu viteze diferite in functie de solubilitatea lor. Componentele se vor gasi in zone separate pe cromatograma.
In acest proces este important fenomenul de capilaritate al hartiei cromatografice, care permite inaintarea solventului (eluentului).
Etapele cromatografiei pe hartie
1. Prepararea hartiei
In centrul discului de hartie se efectueaza un orificiu prin care se introduce un fitil de hartie cromatografica. Cu ajutorul unei pipete fine se depune solutia de analizat in forma de cerc aproape de mijlocul discului de hartie, apoi se lasa la uscat.
2. Eluarea componentilor
Se utilizeaza o camera cromatografica in care se introduce un vas circular cu solvent. Se aseaza discul de hartie pe vas astfel ca fitilul sa patrunda in solvent. Se aseaza capacul cutiei deasupra discului in asa fel incat atmosfera din interior sa fie saturata in vaporii solventului.
3. Revelarea cromatogramei
Cromatograma uscata nu poate fi studiata direct deoarece substantele implicate sunt incolore. Pentru a localiza compusii se apeleaza la revelarea cu reactivi convenabili, care dau cu componentii derivati colorati sau fluorescenti in lumina ultravioleta.
Identificarea ionilor Cu2+, Fe3+ si Co3+
Mod de lucru
Se prepara solutii apoase de azotati ai metalelor de mai sus (concentratie 0,5 m).
Eluarea se face cu un amestec de apa:acid clorhidric:acetona in raport molar de 5:8:87, pana cand frontul de elutie este aproape de marginea discului.
Dupa uscare, se pulverizeaza cromatograma cu o solutie de acid rubeanic 1% in solvent de elutie, apoi se expune, dupa inca o uscare, la vapori de amoniac.
Se obtin complecsi colorati caracteristici: Cu2+ - verde masliniu, Fe3+ - verde brun si Co3+ - roscat.
Acidul rubeanic sufera o tautomerizare, trecand in forma imino, dupa care reactioneaza cu ionii metalici, formand combinatii chelatice polimere cu urmatoarea compozitie:
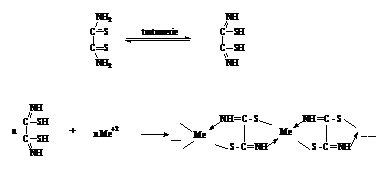
Reactiile specifice pentru
identificarea cationilor din proba de analizat sunt urmatoarele:
Fe3+
Tiocianatul de potasiu (sulfocianura de potasiu) produce o culoare rosu intens, datorita formarii tiocianatului feric:
(Fe3++3Cl-)+3(K+ + CN-) = (Fe3++3SCN-)+3(K++Cl-)
Co2+
Azotitul de potasiu in stae solida si in mediu de acidacetic precipita hexanitrocobaltiatul de potasiu, galben cristalin:
(Co2++2NO3-)+7(K++NO2-)+2(H++CH3COO-)=
K3[Co(NO2)6]+2(K++ CH3COO-)+2(K++NO3-)+NO+H2O
Cu2+
Hexacianoferatul tetrapotasic (ferocianura de potasiu) formeaza un precipitat gelatinos rosu-brun de ferocianura cuprica:
2 (Cu2++SO42-) + (4K+ + [Fe(CN)6]4-) =
Cu2[Fe(CN)6] + 2 (2K+ + SO42-)
pH - metria
Notiunea de pH a fost introdusa in anul 1909 de Srensen sub numele de exponent de hidrogen. El este definit ca logaritmul cu semn negativ al concentratiei ionilor de hidroniu: pH = - lg [H3O+] ; valoarea sa poate fi calculata daca se cunosc constantele de echilibru ale reactiilor chimice la care participa acest ion si compozitia analitica a solutiei.
Determinarea pH-ului se face prin metode colorimetrice, potentiometrice si spectrofotometrice.
Metode potentiometrice de determinare a pH-ului
Metodele potentiometrice de analiza se bazeaza pe variatia potentialului unui electrod indicator in functie de activitatea (respectiv concentratia) ionilor din solutie. Deoarece nu este posibila masurarea directa a potentialului unui singur electrod, practic se masoara tensiunea (forta) electromotoare a pilei formate din electrodul de cercetat si un electrod de comparatie. Ca electrod indicator se foloseste electrodul de hidrogen dar, datorita unor inconveniente de ordin experimental, acesta este inlocuit cu alti electrozi indicatori ai activitatii ionilor de hidroniu: electrodul de sticla (cel mai utilizat), electrodul de chinhidrona, electrodul de stibiu etc.
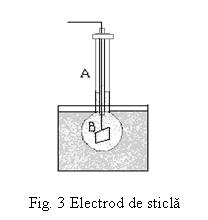 Electrodul de sticla face parte din clasa electrozilor cu membrana iar utilizarea sa
se bazeaza pe faptul ca potentialul care apare la interfata
solutie - membrana de sticla este functie de activitatea
ionilor de hidroniu din solutie.
Electrodul de sticla face parte din clasa electrozilor cu membrana iar utilizarea sa
se bazeaza pe faptul ca potentialul care apare la interfata
solutie - membrana de sticla este functie de activitatea
ionilor de hidroniu din solutie.
 Este alcatuit dintr-o membrana
de sticla speciala, de obicei de forma sferica. In
interiorul sferei care contine aceasta membrana se introduce o
solutie tampon cu pH constant si un electrod de referinta
intern. Sticla din care sunt confectionati electrozii are anumite
proprietati: sensibilitate la variatia pH-ului, durabilitate in
timp, higroscopicitate ridicata si rezistenta
electrica.
Este alcatuit dintr-o membrana
de sticla speciala, de obicei de forma sferica. In
interiorul sferei care contine aceasta membrana se introduce o
solutie tampon cu pH constant si un electrod de referinta
intern. Sticla din care sunt confectionati electrozii are anumite
proprietati: sensibilitate la variatia pH-ului, durabilitate in
timp, higroscopicitate ridicata si rezistenta
electrica.
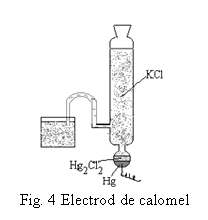
In determinarile de pH, potentialul electrodului indicator se masoara in raport cu potentialul constant al unui electrod de referinta. Drept electrod de referinta se foloseste electrodul de calomel format din mercur si calomel (clorura mercuroasa) in solutie de clorura de potasiu, cu urmatoarea schema:
Hg (l) | Hg2Cl2 (s) | KCl
El este format dintr-un vas de sticla in care se introduce amestecul format din mercur metalic si calomel uscat, deasupra unui strat de mercur metalic ce se gaseste in vasul de sticla. Contactul electric intre mercurul metalic si borna de conectare a electrodului se face printr-un fir de platina.
Aparatele folosite pentru determinarea tensiunii electromotoare se numesc pH-metre. Modelele noi folosesc un electrod combinat pentru a usura masurarea valorilor pH-ului, mai ales in cazul masuratorilor repetate, la probe diferite.
Etapele determinarii pH-ului la un astfel de aparat sunt:
- se conecteaza pH-metrul la sursa de energie electrica (sau se folosesc bateriile);
- se calibreaza aparatul cu ajutorul solutiilor tampon etalonate cu valori ale pH-ului 4,01; 7,0 si respectiv 10,0;
- se citesc valorile pH ale solutiilor de cercetat;
- electrodul se spala la schimbarea fiecarei solutii cu apa distilata si se sterge cu atentie cu hartie de filtru;
- se inchide aparatul, deconectand apoi electrodul.
Se vor efectua masuratori pentru determinarea pH-ului la diferite solutii si extracte, notand valorile obtinute.
O alta metoda uzuala pentru determinarea aproximativa a valorii pH-ului este folosirea hartiilor indicatoare. Exista mai multe tipurii de hartii indicatoare pentru domenii mai largi sau mai restranse de pH, pentru imprimarea carora se folosesc diferite solutii de indicatori acido-bazici.
Metode polarimetrice de analiza
Aceste metode se bazeaza pe masurarea unghiului de deviere a planului luminii polarizate.
Radiatia luminoasa ale carei vibratii au loc doar intr-un singur plan se numeste raza polarizata iar planul de vibratie, planul luminii polarizate.
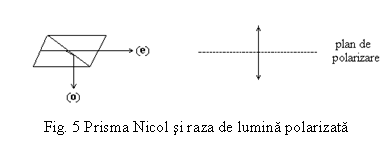 La
trecerea unei raze de lumina naturala printr-o prisma
speciala (nicol confectionat din spat de Islanda), ea se descompune
intr-o raza ordinara (o)
care este absorbita si o raza extraordinara (e) care vibreaza intr-un singur
plan.
La
trecerea unei raze de lumina naturala printr-o prisma
speciala (nicol confectionat din spat de Islanda), ea se descompune
intr-o raza ordinara (o)
care este absorbita si o raza extraordinara (e) care vibreaza intr-un singur
plan.
Substantele capabile sa modifice (sa roteasca) planul de polarizare al luminii se numesc optic active.
Activitatea optica este o marime caracteristica pentru o substanta data si se exprima prin unghiul de rotatie specifica [a]lt. Acesta se defineste ca fiind unghiul de rotatie al planului luminii polarizate la trecerea printr-un strat de solutie de lungime l dm, avand concentratia de 1 g substanta pura activa la 1 ml solutie si densitatea d la temperatura t.
[a]lt
= ![]()
Rotatia specifica depinde de lungimea de unda a radiatiei cu care se lucreaza si de temperatura, fapt care impune raportarea ei la o anumita lungime de unda (l) si la o anumita temperatura (de obicei lungimea de unda a liniei galbene a sodiului la temperatura de 200C).
[a]D20
= ![]()
Metodele polarimetrice sunt metode optice de analiza pe baza carora se poate determina concentratia unei solutii care prezinta activitate optica.
Deoarece rotatia specifica este o marime aditiva, determinarea concentratiei prin metode polarimetrice se poate face numai in cazul solutiilor care contin o singura componenta optic activa. Pentru o solutie care contine C grame substanta optic activa la 100 ml solutie, relatia devine:
[a]D20
= ![]() ,
,
de unde rezulta expresia concentratiei:
C = ![]()
Masurarea unghiului de deviere a planului luminii polarizate se face cu ajutorul aparatului numit polarimetru. Partile componente ale acestui aparat sunt:
- sursa de radiatie S;
- polarizor (dispozitiv pentru producerea radiatiei polarizate) P, P1 si P2;
- analizor (dispozitiv pentru stabilirea pozitiei planului de polarizare A), solidar cu un reper ce se deplaseaza in dreptul unei scale gradate;
- tub polarimetric T;
-
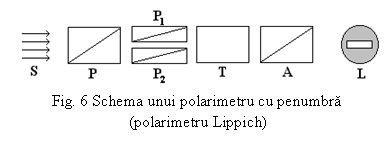 luneta
de observare L.
luneta
de observare L.
Modul de functionare
Radiatia luminoasa emisa de sursa S (lampa de sodiu) trece printr-o fanta, o lentila si o prisma, apoi strabate dispozitivul de polarizare compus din nicolul P si nicolii P1 si P2, paraleli intre ei si cu planul de polarizare al polarizorului P. Aceasta determina trei zone distincte in campul optic al lunetei L.
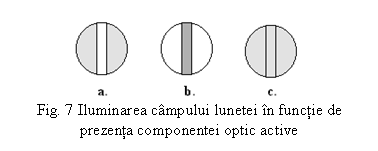
In orice pozitie a analizorului A fata de polarizorul P, cele doua zone laterale ale campului optic apar egal luminate (fig. 7a).
 Analizorul
este mobil in jurul axului sau si este in legatura cu o
scala gradata pe care se masoara unghiul de rotire.
Dupa ce a strabatut analizorul, radiatia trece printr-o
lentila si ajunge in campul optic al lunetei. Punctul "0" de pe scala
gradata corespunde si unei iluminari de intensitate minima,
uniforma, a campului optic.
Analizorul
este mobil in jurul axului sau si este in legatura cu o
scala gradata pe care se masoara unghiul de rotire.
Dupa ce a strabatut analizorul, radiatia trece printr-o
lentila si ajunge in campul optic al lunetei. Punctul "0" de pe scala
gradata corespunde si unei iluminari de intensitate minima,
uniforma, a campului optic.
Solutia ce contine substanta optic activa se introduce in tubul aparatului, se misca analizorul pana cand cele doua zone sunt egal luminate. Se face citirea unghiului pe scala vernierului aparatului.
Datorita activitatii optice a componentei analizate, planul de vibratie al radiatiei polarizate va fi deviat cu un anumit unghi. Campul optic al lunetei apare iluminat neuniform, impartit in trei zone (fig.7b). Se roteste analizorul pana cand campul optic apare din nou uniform intunecat (spectrul inregistrat pentru punctul "0") (fig. 7c).
Valoarea corespunzatoare rotatiei analizorului se citeste pe scala gradata si reprezinta unghiul cu care a fost deviat planul luminii (radiatiei) polarizate de catre solutia de analizat.
Determinarea fotocolorimetrica a ionului fosfat
Principiul metodei
Metoda se bazeaza pe reactia de culoare ce are loc in mediu acid intre ionul fosforic si molibdatul de amoniu, in prezenta unui amestec reducator format din clorura stanoasa si acid ascorbic.
Rezulta un complex de culoare albastra (MoO2∙4MoO3)2∙H3PO4∙4H2O iar intensitatea coloratiei variaza direct proportional cu concentratia solutiei. Se determina colorimetric aceasta concentratie la lungimea de unda de 650 nm.
Reactivi
1. Amestec reducator (clorura stanoasa - acid ascorbic)
a) 0,075 g SnCl2∙2H2O se dizolva in 0,5 ml HCl concentrat (d=1,19 g/ml) intr-un pahar de 25 ml. Se incalzeste usor pana la dizolvare apoi se adauga putina apa.
b) 0,75 g acid ascorbic se dizolva in 50 ml apa distilata intr-un balon de 100 ml, apoi se adauga solutia obtinuta la punctul a) si se aduce la semn continutul balonului cu apa distilata.
Solutia se foloseste dupa 15 minute si se prepara pentru fiecare determinare.
2. Solutie molibdenica in acid sulfuric diluat
Se cantaresc 16,67 g molibdat de amoniu (NH4)2MoO4∙4H2O apoi se trec intr-un pahar si se adauga 200 ml apa distilata cu temperatura de 500C. Intr-un balon cotat de 1000 ml se masoara 500 ml apa distilata peste care se adauga cu ajutorul unui cilindru gradat 187,6 ml acid sulfuric concentrat (d=1,84 g/ml). Dupa racirea amestecului se adauga solutia de molibdat de amoniu si se aduce la semn cu apa distilata. Aceasta solutie se pastreaza la intuneric si la rece, fiind stabila in aceste conditii.
3. Solutie stoc de fosfat (1 mg P/ml)
Se dizolva 0,4394 g KH2PO4 p.a. in apa distilata intr-un balon cotat de 100 ml. Pentru conservare se adauga cateva picaturi de cloroform apoi se aduce la semn cu apa distilata.
4. Solutie etalon de fosfat - 0,01 mg (10γ)P/ml
1 ml din solutia stoc de fosfat se aduce la un volum de 100 ml cu apa distilata intr-un balon cotat.
Prepararea extractului vegetal
In plante, fosforul se gaseste in cea mai mare parte sub forma de compusi organici. De aceea, pentru a analiza continutul in fosfor al materiilor vegetale se urmaresc trei etape:
- prelevarea probei de analizat si uscarea in etuva la 1050C pana la masa constanta;
- mineralizarea pe cale umeda in baloane Kjeldahl cu acid azotic concentrat si acid sulfuric concentrat;
- determinarea cantitativa a ionului fosfat (dozarea).
Se cantaresc la balanta analitica intre 1 - 2 g material vegetal fin maruntit si omogenizat, intr-o fiola de cantarire si se usuca la 1050C intr-o etuva pana la masa constanta. Substanta uscata se introduce cantitativ intr-un balon Kjeldahl apoi se adauga 10-20 ml acid azotic concentrat cu ajutorul unui cilindru gradat. Mineralizarea incepe printr-o incalzire usoara, sub nisa, urmata de o incalzire puternica, pe baie de nisip, pana la incetarea degajarii vaporilor bruni de oxizi de azot. In acest moment, solutia din balon este limpede, iar volumul a scazut la aproximativ 3 ml. Se lasa sa se raceasca balonul si se adauga cu ajutorul unui cilindru gradat 1 ml acid sulfuric concentrat. Se incalzeste balonul pana la disparitia vaporilor bruni de oxizi de azot si aparitia vaporilor albi de SO3. Mineralizarea se considera terminata atunci cand lichidul din balon devine perfect transparent si incolor.
Solutia din balon se trece cantitativ intr-un balon cotat de 100 ml, se aduce la semn cu apa distilata, se lasa sa se decanteze, iar din lichidul limpede se determina colorimetric fosforul.
Mod de lucru
Intr-un balon cotat de 50 ml se masoara cu pipeta 5 ml solutie de analizat, 2 ml reactiv sulfomolibdenic, 2 ml amestec reducator si se aduce la semn cu apa distilata. Se omogenizeaza, se lasa la intuneric 30 minute apoi se colorimetreaza la 650 nm.
Pregatirea curbei etalon
In sase baloane cotate de 50 ml se masoara cu pipeta volumele de solutie etalon de fosfat, amestec reducator si reactiv molibdenic conform tabelului, apoi se aduc baloanele la semn cu apa distilata. Probele se colorimetreaza dupa 30 de minute la 650 nm fata de o proba martor preparata din reactivi fara solutia care contine fosfat. Se construieste curba etalon avand in abscisa concentratia in γ P/ml iar in ordonata, valorile corespunzatoare ale extinctiilor. Se folosesc cuve de 1 cm, iar continutul in fosfor se exprima in functie de proba de analizat: mgP/100 g produs, ppm P, mg P2O5/1000 ml etc.
|
Nr. etalon Reactiv |
M |
1 |
2 |
3 |
4 |
5 |
|
Solutie etalon PO43- |
- |
1 |
3 |
5 |
7 |
9 |
|
Solutie sulfomolibdenica |
2 |
2 |
2 |
2 |
2 |
2 |
|
Amestec reducator |
2 |
2 |
2 |
2 |
2 |
2 |
|
Apa distilata |
46 |
45 |
43 |
41 |
39 |
37 |
|
Concentratie(γP/ml) |
0 |
0,2 |
0,6 |
1 |
1,4 |
1,8 |
|
Extinctie λ=650nm |
|
|
|
|
|
|
Reactiile ionului ortofosforic (fosforic) - PO43-
Pentru efectuarea reactiilor se foloseste o solutie de fosfat disodic, Na2HPO4.
1. Azotatul de argint, AgNO3, precipita in solutii neutre fosfatul de argint, Ag3PO4 galben.
(2Na+ + HPO42-) + 3(Ag+ + NO3-) Ag3PO4 + 2(Na+ + NO3-) + (H+ + NO3-)
Fosfatul de argint este solubil in acid azotic si hidroxid de amoniu.
Ag3PO4 + 6(NH4+ + OH-) = + 6H2O
2. Azotatul de bariu, Ba(NO3)2, formeaza un precipitat alb amorf de fosfat acid de bariu.
(2Na+ + HPO42-) + (Ba2+ + 2NO3-) BaHPO4 + 2(Na+ + NO3-)
In prezenta hidroxidului de amoniu se formeaza fosfatul neutru de bariu. Ambii fosfati sunt solubili in acid azotic.
2(2Na+ + HPO42-) + 3(Ba2+ + 2NO3-) + 2(NH4+ + OH-) = Ba3(PO4)2 + 4(Na++NO3-)+ 2(NH4+ + NO3-) + 2H2O
3 Acetatul de plumb, Pb(CH3COO)2, precipita fosfatul de plumb Pb3(PO4)2 alb, solubil in acid azotic si insolubil in acid acetic.
2 (2Na+ + HPO42-) + 3 (Pb2+ + 2CH3COO-) =
Pb3(PO4)2 + 4 (Na+ + CH3COO-) + 2 CH3COOH
4. Mixtura magneziana, (MgCl2 + NH4OH + NH4Cl), precipita fosfatul dublu de amoniu si magneziu, MgNH4PO4, alb cristalin.
(2Na+ + HPO42-) + (Mg2+ + 2Cl-) + (NH4+ + OH-) =
MgNH4PO4 + 2(Na+ + Cl-) + H2O
Precipitatul este solubil in acizi minerali si acid acetic. Clorura de amoniu se adauga pentru a nu se forma hidroxidul de magneziu.
5. Molibdatul de amoniu, (NH4)2MoO4, in mediu de acid azotic concentrat, precipita fosfomolibdatul de amoniu galben, cristalin, (NH4)3H4[P(Mo2O7)6].
(2Na+ + HPO42-) + 12(2NH4+ + MoO4-) + 23(H+ + NO3-) = (NH4)3H4[P(Mo2O7)6] + 2(Na++NO3-)+ 21(NH4+ + NO3-) + 10H2O
Este o reactie specifica de control.
Determinarea constantei de viteza pentru reactia de esterificare a acidului acetic cu alcool etilic
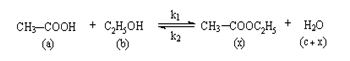
Reactia de esterificare a
acidului acetic cu etanol este o reactie de echilibru, cu formare de
acetat de etil si apa.
a = concentratia initiala a acidului acetic;
b = concentratia initiala a etanolului;
c = concentratia initiala a apei din amestec;
x= concentratia esterului;
(c+x) = cantitatea totala de apa din amestec.
Toate concentratiile sunt exprimate in moli/l.
Esterificarea se poate realiza fie in cataliza acida, omogena, folosind acidul sulfuric drept catalizator, in raport molar de 0,1 - 0,2 fata de acidul organic, fie in cataliza eterogena, folosind o rasina schimbatoare de ioni cu grupari active acide drept catalizator. Se efectueaza de obicei la temperatura de reflux a amestecului de reactie, sub agitare continua in cazul catalizei eterogene. Pentru a asigura un randament optim al procesului si a deplasa echilibrul reactiei spre formarea de ester, se va lucra cu un exces de alcool in raport molar de 15:1 fata de acidul acetic.
Pentru determinarea concentratiilor celor doi reactanti la echilibru, se va efectua esterificarea in cataliza eterogena cu catalizator Dowex 50 - R sau Amberlite IR - 150 (ambele cu grupari active sulfonice -SO3H) si se va determina concentratia acidului acetic prin titrare cu o solutie de hidroxid de sodiu de factor cunoscut.
Reactivi:
- acid acetic glacial;
- metanol 95 %;
- Dowex 50 - R sau Amberlite IR - 150;
- NaOH 0,1 n , F cunoscut;
- fenolftaleina 1 %, solutie alcoolica.
Mod de lucru
Initial se va efectua o proba martor din 1 ml metanol si 10 ml apa distilata, care se va titra cu solutia de hidroxid in prezenta de fenolftaleina.. Volumul de hidroxid consumat la aceasta titrare va fi scazut din toate titrarile ulterioare.
Se amesteca 0.1 moli acid acetic glacial (99%, d = 1,05 g/ml) = 5,75 ml cu 1,5 moli metanol (95% volum, d = 0.79 g/ml) = 63,95 ml. Volumul initial al amestecului de reactie este de 69,7 ml. Raportate la acest volum se calculeaza concentratiile initiale in acid acetic si metanol. Se va titra 1 ml amestec+10 ml apa distilata cu hidroxid de sodiu si se va nota volumul obtinut la titrare. Se incalzeste amestecul din balon pe plita, sub agitare, iar cand se atinge temperatura de reflux se adauga 2 g catalizator solid. Din acest moment, urmatoarea proba se va recolta peste 15 minute, apoi alta proba peste inca 15minute, efectuand titrarea in aceleasi conditii.
Calcul
1. Calculul cantitatii de NaOH din volumul consumat la titrare:
1000 ml 40 0,1 F g NaOH
V1 ml x1
x1 = 40 V1 0,1 F / 1000 g NaOH
2. Calculul gramelor de acid acetic dintr-un ml:
40 g NaOH. 60 g CH3COOH
40 V10,1F/1000 g NaOH ..x2
x2 = 60 V1 0,1 F / 1000 g CH3COOH
3. Calculul titrului in acid acetic:
1 ml 60 V1 0,1 F / 1000 g CH3COOH
1000 ml ..x3
X3 = 60 V1 0,1 F g CH3COOH/l
Concentratia molara a acidului acetic in amestecul de reactie va fi:
Cm = 60 V1 0,1 F / Macid = V1 0,1 F moli/l
Concentratia esterului obtinut se calculeaza facand diferenta dintre concentratia initiala a acidului acetic si ceaobtinuta la proba curenta. Concentratia in alcool scade proportional cu cu concentratia acidului (se va scadea din concentratia initiala a alcoolului concentratia esterului la momentul respectiv).
Se va completa tabelul de mai jos si se va trasa graficul variatiei concentratiilor in acid si ester in functie de timp.
|
Nr. proba |
Timp (ore) |
Cmolara acid acetic |
Cmolara ester |
Cmolara metanol |
|
1 |
0 |
|
|
|
|
2 |
0,25 |
|
|
|
|
3 |
0,5 |
|
|
|
|
4 |
0,75 |
|
|
|
|
5 |
1 |
|
|
|
|
6 |
1,25 |
|
|
|
|
7 |
1,5 |
|
|
|
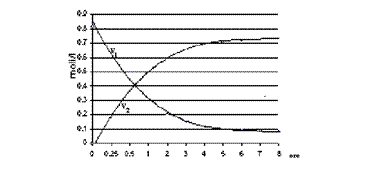
Graficul obtinut va avea
forma:
Sistemele disperse eterogene prezinta suprafete de separare intre componentele lor. Proprietatile acestor sisteme variaza in diferite puncte, interactiunile avand loc la limita de separare a fazelor.
Dintre fenomenele ce au loc la suprafata de separare dintre componente, foarte importante sunt cele care insotesc modificarile de concentratie ale componentelor. Fenomenele de sorbtie se petrec la interfata dintre doua componente. Atunci cand patrunderea moleculelor, ionilor, macromoleculelor are loc in intreaga masa a unei componente a sistemului, procesul se numeste absorbtie. Daca o componenta se concentreaza numai la suprafata de separare, procesul poarta numele de adsorbtie.
Acumularea la suprafata unei substante solide sau lichide a unei alte substante se numeste adsorbtie. Substanta pe a carei suprafata se produce fenomenul se numeste adsorbant iar substanta care se acumuleaza se numeste adsorbat.
Corpurile solide, prin suprafata lor specifica mare, au proprietatea de a retine diferite gaze sau lichide datorita fortelor necompensate de la nivelul interfazic. Exprimarea cantitativa a procesului se face prin relatia lui Freundlich:
X/m = k C1/n , in care:
X = cantitatea de adsorbat; m = cantitatea de adsorbant; C = concentratia de echilibru a adsorbatului lichid; k, n = constante.
Acesta relatie poarta numele de izoterma lui Freundlich. Ecuatia poate fi reprezentata grafic printr-o parabola.
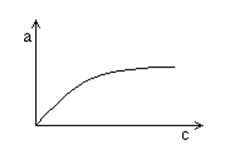 Izoterma de adsorbtie a lui Freundlich
Izoterma de adsorbtie a lui Freundlich
c = concentratia de echilibru in mediu fluid a substantei care se adsoarbe;
a = concentratia aceleiasi substante pe adsorbant.
Concentratia a exprima intensitatea adsorbtiei (coeficientul de adsorbtie).
Mod de lucru
Se pregatesc sase solutii de acid acetic de urmatoarele concentratii: 0,8n; 0.6n; 0,4 n ; 0,2 n ; 0,1 n ; 0,05 n. Titrul exact al acestor solutii se determina prin titrare cu o solutie de NaOH 0,1 n cu factor cunoscut in prezenta fenolftaleinei ca indicator.
Pentru titrare se masoara exact (cu ajutorul unor biurete sau pipete) cate 5 ml din solutiile de concentratie 0,05 n ; 0,1 n ; 0,2 n si cate 2 ml din solutiile de concentratie 0,4 n ; 0.6 n si 0,8 n, care se transvazeaza in cate un pahar Erlenmeyer, se adauga 1 - 2 picaturi de solutie alcoolica de fenolftaleina si se titreaza cu hidroxid de sodiu din biureta pana la virarea culorii in roz pal.
Se noteaza volumul consumat la titrare si se calculeaza gramele de acid acetic existente intr-un litru din acea solutie, dupa modelul:
1000 ml .0,1 ∙ 40 ∙ f g NaOH
V ml x g NaOH
x = V ∙ 0,1 ∙ 40 ∙ f / 1000 g NaOH
40 g NaOH .. 60 g acid acetic
x g NaOH . y g acid acetic
y = x ∙ 60 / 40 g acid acetic
5 ml (sau 2 ml) * y g acid acetic
1000 ml . C
C = 1000 ∙ y / 5 (sau 2)
______________________________________________________________________
* la ultima etapa a calculului se considera volumul probei de acid acetic luata in lucru initial.
Se calculeaza cantitatea de acid adsorbit de 1 g carbune, cu relatia:
a = (C - C') / 0,3 g acid/l g carbune
Se traseaza un grafic asezand pe abscisa concentratiile in acid acetic iar pe ordonata valorile coeficientului de adsorbtie a.
Pentru o buna reprezentare grafica a variatiei coeficientului de adsorbtie cu concentratia normala a acidului acetic, se recomanda trasarea punctelor si a curbei pe hartie milimetrica.
Datele obtinute se trec in tabelul urmator:
|
Nr. proba |
Cnormala CH3COOH |
Nr. ml NaOH (I) |
C (g/l) CH3COOH |
Nr. ml NaOH (II) |
C ' (g/l) CH3COOH |
a |
|
1 |
0,05 |
|
|
|
|
|
|
2 |
0,1 |
|
|
|
|
|
|
3 |
0,2 |
|
|
|
|
|
|
4 |
0,4 |
|
|
|
|
|
|
5 |
0,6 |
|
|
|
|
|
|
6 |
0,8 |
|
|
|
|
|
Metode de preparare a sistemelor disperse ultramicroeterogene (solurile)
Sistemele coloidale sunt sisteme intermediare intre cele grosiere, eterogene, cu grad de dispersie foarte mic si cele moleculare sau omogene.
Solurile pot fi obtinute prin doua metode:
a) prin dispersarea particulelor mari ale sistemelor eterogene in particule mai mici, de dimensiuni coloidale, proces ce duce la cresterea gradului de dispersie;
b) prin condensarea moleculelor sau ionilor dintr-un sistem omogen pana la formarea particulelor coloidale, prin scaderea gradului de dispersie al acelui sistem.
Formarea solurilor prin condensare poate avea loc prin procedee fizice sau chimice.
1. Procedee fizice: metoda inlocuirii solventului
Prepararea solului de parafina
Se prepara o solutie de aproximativ 2 % de parafina in alcool etilic; 10 ml din aceasta se toarna, picatura cu picatura si sub agitare energica, in 300 ml apa distilata. Se obtine un sol puternic opalescent de parafina in apa (hidrosol de parafina).
Pentru a elimina particulele macrodisperse, solul se filtreaza. Daca se doreste pastrarea solului un timp mai indelungat, este necesara indepartarea alcoolului prin dializa, deoarece acesta micsoreaza stabilitatea sistemului nou format.
Analog se pot prepara si solurile de colofoniu si mastic, folosind aceleasi volume de solutie de mai sus.
Se prepara o solutie saturata de sulf in alcool etilic, prin faramitarea sulfului in alcool la temperatura camerei. Sulful ramas nedizolvat se separa prin filtrare.
Se toarna apoi, picatura cu picatura, sub agitare, 5 ml din filtrat in 20 ml de apa distilata. Hidrosolul de sulf obtinut este putin solubil si puternic opalescent.
Prepararea solurilor halogenurilor de argint
Obtinerea acestor soluri se face printr-o reactie de dublu schimb. Halogenurile de argint (cu exceptia fluorurii) sunt foarte greu solubile in apa, solubilitatile lor fiind de : AgI - 9,7 10-9 moli/l; AgBr - 6,6 10-7 moli/l; AgCl - 1,25 10-5 moli/l.
Daca se amesteca solutii diluate de azotat de argint si halogenura alcalina, luandu-se unul dintre reactivi in exces, nu se obtine un precipitat, ci hidrosolul halogenurii de argint date. Stabilitatea solurilor obtinute depinde de solubilitatea halogenurii ce formeaza faza dispersa, stabilitatea fiind cu atat mai mare cu cat solubilitatea este mai mica.
Reactivul luat in exces constituie stabilizatorul.; ionii acestuia se adsorb pe suprafata particulelor coloidale, formand un dublu strat electric.
Pentru a prepara soluri de AgI de diferite stabilitati se toarna in trei pahare Berzelius cate 5 ml de solutie KI de concentratie 0,01 n. Se adauga apoi, picatura cu picatura, dintr-o biureta, urmatoarele volume de solutie de AgNO3 de concentratie 0,01 n: 3 ml in primul pahar, 4 ml in cel de-al doilea si 5 ml in al treilea pahar.
Se observa ca in ultimul pahar se obtine un preipitat, iar in primele doua, soluri relativ stabile.
In mod asemanator se pot prepara solurile de AgBr si AgCl, dar in special solul de AgCl este putin stabil.
Acest sol se obtine printr-o reactie de hidroliza a sarii unui metal trivalent.
Intr-un balon Erlenmeyer se incalzesc pana la fierbere 85 ml apa distilata. Se indeparteaza flacara si se toarna picatura cu picatura 15 ml solutie de clorura ferica de concentratie 2 %. Dupa cateva minute, in urma reactiei de hidroliza:
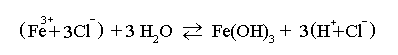
apare un sol rosu - brun
de hidroxid feric.
Deoarece prin scaderea temperaturii, reactia se petrece de la dreapta la stanga, pentru obtinerea unui sol de Fe(OH)3 suficient de stabil, se recomanda efectuarea unei dialize pentru indepartarea excesului de HCl.
Prepararea solului de sulf pe baza reactiei redox dintre Na2S2O3 si H2SO4 concentrat
La tratarea unei solutii de tiosulfat de sodiu 0,01 n cu acid sulfuric concentrat, in picaturi, are loc o reactie de oxido-reducere in urma careia se obtin, in solutie, sulf si SO2. Se formeaza, sub agitare continua, un sol de sulf de culoare alb-galbui, puternic opalescent.
Na2S2O3 + H2SO4 S + Na2SO4 + SO2 + H2O
Difuzia si osmoza joaca un rol foarte important in natura. Peretii celulelor vegetale si animale sunt constituite din membrane semipermeabile si functioneaza ca adevarate osmometre.
Introducand celula intr-o solutie a carei presiune osmotica este mai mare decat cea a sucului celular, adica intr-o solutie hipertonica, se observa cum celula se contracta si apoi se zbarceste. Acest fenomen numit plasmoliza, este datorat difuziei apei din interiorul celulei prin membrana semipermeabila.
Daca o celula este insa introdusa intr-o solutie mai diluata decat sucul celular, adica intr-o solutie hipotonica, sau chiar in apa, celula se umfla si poate crapa, datorita apei care patrunde din afara, prin membrana semipermeabila in celula. Acest fenomen numit turgescenta, este folosit in tehnica, de exemplu la extragerea zaharului (zaharozei) din sfecla. Sfecla taiata marunt este introdusa in vase cu apa (difuzoare). Datorita presiunii osmotice, celulele de sfecla crapa si zaharoza difuzeaza in apa, de unde se extrage.
Se ia o proba medie de radacini de sfecla si se marunteste cu ajutorul unei razatori. Din acest material se cantaresc 10 g si se trec cantitativ intr-un zaharimetru de 100 ml. Se adauga 50 ml de apa distilata, se amesteca si apoi se adauga sub agitare 15 ml solutie de acetat de plumb 10%, pentru defecare.
Zaharimetrul se introduce intr-o baie de apa la 800C, timp de 30 minute. Se scoate balonul, se completeaza la semn cu apa distilata fierbinte apoi se reintroduce in baie pentru inca 10-15 minute. Se raceste balonul, se controleaza cota si se filtreaza prin hartie de filtru uscata, aruncand primele portiuni.
Solutia zaharata finala trebuie sa fie limpede pentru a fi posibila masurarea unghiului de rotatie specifica a solutiei cu ajutorul unui polarimetru.
Se utilizeaza un tub polarimetric cu lungimea masurata in dm, iar cantitatea de zaharoza continuta in produsul cantarit, exprimata in grame, se calculeaza cu formula:
![]() in
care:
in
care:
- p = grade polarimetrice citite;
- l = lungimea tubului polarimetric, in decimetri;
- ad20 = deviatia polarimetrica specifica zaharozei, care in cazul solutiilor obtinute in modul de mai sus are valoarea medie de 66,5;
- V = volumul zaharimetrului (100 ml);
- G = cantitatea de material vegetal cantarita (10 g).
1. Procedee fizice
Prepararea gelului de amidon: se cantaresc la balanta tehnica aproximativ 10 g de amidon care se amesteca in 50 ml apa distilata rece.
Intr-un pahar Berzelius de 150 ml se aduc la fierbere 50 ml de apa distilata, pe o sita de azbest. Se adauga, sub agitare continua, amidonul amestecat cu apa rece.
Se lasa sa ajunga din nou la fierbere si se mentine timp de cateva minute la flacara cu intensitate redusa. Prin racirea solutiei coloidale se va obtine gelul de amidon.
2. Procedee chimice
In unele reactii chimice se obtin ca produsi de reactie precipitate gelatinoase.
a) Reactia ionului Fe3+ cu hidroxizi alcalini (NaOH, KOH)
Se formeaza un precipitat gelatinos rosu-brun de hidroxid feric, insolubil in exces de reactiv si solubil in acizi.
(Fe3++3 Cl-) + 3(Na++OH-) = Fe(OH)3 + 3(Na++Cl-)
b) Reactia ionului Mg2+ cu hidroxizi alcalini (NaOH, KOH)
Se formeaza un precipitat alb gelatinos de hidroxid de magneziu, solubil in acizi diluati, apa cu CO2, saruri de amoniu si in exces de reactiv.
(Mg2++SO42-) + 2(Na++OH-) = Mg(OH)2 +(2Na++SO42-)
c) Reactia ionului Cu2+ cu hidroxizi alcalini (NaOH, KOH)
Precipita hidroxidul cupric, albastru, gelatinos, solubil in acizi si in hidroxid de amoniu.
(Cu2++SO42-) + 2(Na++OH-) = Cu(OH)2 + (2Na++SO42-)
d) Reactia ionului Cu2+ cu hexacianoferatul tetrapotasic (ferocianura de potasiu)
Se formeaza un precipitat rosu-brun de ferocianura cuprica.
2 (Cu2++SO42-) + (4K++[Fe(CN)6]4-) = Cu2[Fe(CN)6] +
2(2K++SO42-)
e) Reactia ionului silicat (SiO32-) cu acidul sulfuric diluat
Prin actiunea acidului sulfuric (sau in general a acizilor) se formeaza in solutiile concentrate de silicati alcalini un precipitat alb gelatinos de acid silicic, care este de fapt un gel bogat in apa, cu formula [x SiO2 . y H2O]
(2Na++SiO32-) + (2H++SO42-) = H2SiO3 + (2Na++SO42-)
Precipitarea acidului silicic se explica prin hidroliza silicatilor alcalini in solutie apoasa, reactie catalizata de acizi.
(2Na+ + SiO32-) + 2 HOH = H2SiO3 + 2 (Na+ + OH-)
f) Reactia ionului silicat (SiO32-) cu clorura de amoniu
Sarurile de amoniu (in cantitati mari) formeaza in solutiile concentrate de silicati un precipitat gelatinos de acid silicic.
(2Na+ + SiO32-) + 2H2 O + 2(NH4+ + Cl-) = H2SiO3 +
2NH4OH + 2 (Na+ + Cl-)
g) Reactia ionului silicat (SiO32-) cu molibdatul de amoniu
La solutia de silicat alcalin se adauga o solutie proaspat preparata de molibdat de amoniu si se aciduleaza usor, formandu-se silicomolibdatul de amoniu, de culoare galbena, impregnat in gelul de acid silicic.
H2SiO3 + 12(2NH4+ + MoO42-) + 20 (H+ + NO3-) = (NH4)4H4[Si(Mo2O7)6] + 20(NH4+ + NO3-) + 9H2O
Prin peptizare se intelege trecerea unui gel sau a unui precipitat greu solubil in stare de sol, prin adaugarea unui agent chimic (de obicei un electrolit) numit peptizator. Dispersarea precipitatului sau a gelului se face prin spalarea repetata a acestuia cu apa sau prin adaugarea unor cantitati mici de solutii diluate de electroliti (HCl, NH4OH etc.) la precipitatele bine spalate.
In sase eprubete de centrifuga alese doua cate doua cu aceeasi masa, perfect curate si uscate se masoara cate 1 ml dintr-o solutie de FeCl3 0,25 m (0,014 g Fe/ml). Se adauga apoi in fiecare eprubeta cate 1 ml solutie de amoniac 25 % pentru precipitarea completa a Fe(OH)3.
Pentru separarea precipitatului se supun eprubetele centrifugarii la 3000 rotatii/minut, avand grija ca centrifuga sa fie echilibrata (se aseaza fata in fata eprubete cu aceeasi masa, selectate anterior).
Dupa separare se indeparteaza cu grija supernatantul (lichidul clar de deasupra precipitatului) si se spala precipitatele din eprubete cu cate 5 ml apa distilata, pana la reactia neutra a apelor de spalare (pH 7). De fiecare data se separa precipitatele prin centrifugare.
La sfarsitul acestor operatii, deasupra precipitatelor, in fiecare eprubeta, trebuie sa ramana 5 ml lichid.
Se adauga apoi in eprubete cate 1 ml de solutie de HCl, de urmatoarele concentratii: 0.02 n ; 0.04 n ; 0.06 n ; 0.08 n ; 0.16 n ; 0.32 n . Acidul clorhidric joaca rol de peptizator.
Se aseaza eprubetele intr-un termostat la aproximativ 500C timp de doua ore. Din 10 in 10 minute se agita continutul eprubetelor cu o bagheta mica de sticla. La sfarsit, eprubetele se supun din nou centrifugarii.
Deasupra precipitatelor apare un lichid rosietic, care este solul de hidroxid feric. Acesta se trece printr-o hartie de filtru pentru a indeparta orice urma de precipitat. Micela de hidroxid feric are urmatoarea formula
[m FeO(OH) ∙ n FeO+ ∙ (n-x)Cl‑ ]x+ ∙ x Cl‑
FeO(OH) + HCl FeOCl + H2O
FeOCl FeO+ + Cl‑ , sau
[m FeO(OH) ∙ p FeOCl , n FeO+ ∙ (n-x)Cl‑ ]x+ ∙ x Cl‑
FeCl3 + H2O FeOCl + 2 HCl
FeOCl + H2O FeO(OH) + HCl
In filtrat se determina apoi fierul prin metoda colorimetrica.
 Fierul
da cu acidul sulfosalicilic complecsi de culoare galbena in
mediu alcalin (cu un maxim de absorbtie la 424 nm) si rosie in
mediu acid (cu maxim de absorbtie la 505 nm).
Fierul
da cu acidul sulfosalicilic complecsi de culoare galbena in
mediu alcalin (cu un maxim de absorbtie la 424 nm) si rosie in
mediu acid (cu maxim de absorbtie la 505 nm).
Pentru prepararea solutiilor etalon se cantaresc la balanta analitica 0,025 g alaun feriamoniacal, se dizolva cu 100 ml apa distilata intr-un balon cotat de 250 ml, se adauga 0,5 ml acid sulfuric concentrat si se aduce la semn cu apa distilata. Solutia rezultata trebuie sa fie clara.
In sase baloane cotate de 50 ml se masoara, din solutia preparata anterior, urmatoarele volume: 25, 30, 35, 40, 45 si 50 ml si se adauga in primele cinci apa pana la semn. Concentratiile procentuale ale solutiilor obtinute vor fi : 0.5; 0.6; 0.7; 0.8; 0.9 si 1.0 . In aceste solutii se mai adauga cate 1 ml solutie NH4Cl de concentratie 2n si 1 ml acid sulfosalicilic solutie 20%.
Solutiile rosii se colorimetreaza la 505 nm si din datele experimentale se traseaza curba E (extinctie) - C (concentratie in Fe3+). Dupa trasarea curbei etalon se masoara extinctiile celor sase probe supuse peptizarii si prin interpolare se determina concentratiile in Fe3+ in solii obtinuti dupa peptizare. Se completeaza tabelul:
|
Nr. crt. |
Concentratie HCl |
Concentratie Fe3+ |
|
1. |
0.025 n |
|
|
2. |
0.05 n |
|
|
3. |
0.1 n |
|
|
4. |
0.2 n |
|
|
5. |
0.4 n |
|
|
6. |
0.6 n |
|
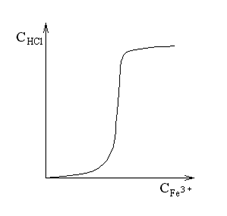 Se
reprezinta apoi grafic concentratia Fe3+
(corespunzatoare solu-bilitatii precipitatului) in functie
de con-centratia peptizatorului (HCl). Se obtine o curba in
forma de S. La concentratii mici se produce doar adsorbtia peptizatorului,
iar peptizarea este neglijabila.
Se
reprezinta apoi grafic concentratia Fe3+
(corespunzatoare solu-bilitatii precipitatului) in functie
de con-centratia peptizatorului (HCl). Se obtine o curba in
forma de S. La concentratii mici se produce doar adsorbtia peptizatorului,
iar peptizarea este neglijabila.
Peptizarea creste brusc intr-un anumit domeniu de concentratii apoi ramane relativ constanta, cand tot precipitatul a peptizat. Se precizeaza ca peptizatorul este de fapt FeCl3 ce se formeaza in urma dizolvarii partiale a Fe(OH)3 in HCl.
Imbibarea gelurilor
Gelurile, in special cele formate din substante macromoleculare, cum sunt gelatina. agar-agarul etc., au proprietatea de a-si mari volumul prin absorbtie de lichid. Fenomenul se numeste imbibare si se datoreaza vitezei mari cu care difuzeaza moleculele lichidului fata de moleculele din gel. Ca urmare, lichidul patrunde intre macromoleculele flexibile, indepartandu-le una de alta si gelul se umfla.
Concomitent cu imbibarea se produce si o dizolvare a gelului cu formarea unei solutii propriu-zise; dizolvarea poate fi partiala sau totala.
Imbibarea decurge ca o reactie de ordinul I. Daca se noteaza cu x - cantitatea de lichid absorbit in timpul t de 1 g de gel uscat si cu A - cantitatea maxima de lichid ce poate fi absorbita, viteza de imbibare este data de ecuatia:
![]() = k ( A - x )
= k ( A - x )
unde k este constanta de viteza. Marimea A se mai numeste si limita de imbibare.
Dispozitivul experimental este compus din :
P - o palnie din sticla, acoperita cu un capac prevazut cu un orificiu; C - cusca din sarma suspendata de capacul palniei; R - reper; T - tub de cauciuc ce leaga palnia de o micropipeta M.
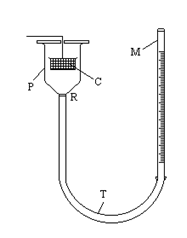 Mod de lucru
Mod de lucru
Se taie o fasie de agar-agar astfel incat sa incapa in cusca de sarma, apoi se cantareste la balanta analitica. Se noteaza cu m masa sa.
Se umple micropipeta si tubul de cauciuc (pana la un nivel superior reperului R) cu apa distilata, avand grija sa nu ramana bule de aer in tub. Se aseaza agar-agarul in cusca si se potriveste capacul la palnie.
Se ridica micropipeta pana cand apa acopera complet cusca, apoi se coboara imediat. Deoarece o cantitate oarecare de apa va ramane fixata prin aderenta de sarma, operatia are ca scop inlaturarea acestei surse de erori.
Se potriveste apoi nivelul apei in dreptul reperului R si se citeste pozitia initiala a meniscului in micropipeta.
Se ridica micropipeta pana cand apa acopera complet agarul si se mentine doua minute in aceasta pozitie. Imediat se coboara micropipeta pana cand apa ajunge din nou in dreptul reperului R si se citeste din nou pozitia meniscului. Diferenta dintre cele doua citiri reprezinta volumul de apa (in ml), absorbit de agar.
Se repeta operatiile de mai sus, lasand agarul in apa timp de inca 3, 5, 5, 5, 10, 10, 10 si 10 minute. Se noteaza de fiecare data pozitia meniscului in micropipeta fata de citirea initiala. Operatia de scoatere a agarului din apa si citirea pozitiei meniscului trebuie facuta cu rapiditate si atentie, pentru a evita "uscarea " gelului.
Volumele de apa masurate se raporteaza la 1 g de agar-agar, astfel:
Vapa = Vmasurat / m
Datele obtinute se trec in tabel:
|
Timp t (min) |
2 |
5 |
10 |
15 |
20 |
30 |
40 |
50 |
60 |
|
Apa absorbita x (ml) |
|
|
|
|
|
|
|
|
|
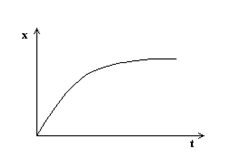 Pe
baza tabelului se reprezinta grafic cantitatile x de apa absorbita, in
functie de timpul t ,
obtinandu-se o curba.
Pe
baza tabelului se reprezinta grafic cantitatile x de apa absorbita, in
functie de timpul t ,
obtinandu-se o curba.
Trasand tangente la aceasta curba se pot calcula diferite valori dx/dt, ce reprezinta vitezele de imbibare pentru valorile respective ale lui x.
Se completeaza tabelul :
|
timp (min) |
ml H2O |
x / g |
dx / dt (tg) |
|
2 |
|
|
_________ = 2 |
|
5 |
|
|
_________ = 3 |
|
10 |
|
|
_________ = 5 |
|
15 |
|
|
_________ = 5 |
|
20 |
|
|
_________ = 5 |
|
30 |
|
|
_________ = 10 |
|
40 |
|
|
_________ = 10 |
|
50 |
|
|
_________ = 10 |
|
60 |
|
|
_________ = 10 |
Ecuatia initiala se poate scrie si sub forma:
![]()
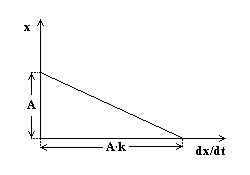 Daca
se reprezinta grafic x
functie de dx/dt se obtine
o dreapta. Panta acestei drepte reprezinta constanta de viteza k iar ordonata la origine - limita de
imbibare A.
Daca
se reprezinta grafic x
functie de dx/dt se obtine
o dreapta. Panta acestei drepte reprezinta constanta de viteza k iar ordonata la origine - limita de
imbibare A.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 39849
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved