| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
| електронен | изкуство култура | икономика | история | книга | компютри | медицина | психология |
| различни | социология | техника | управление | финанси | химия |
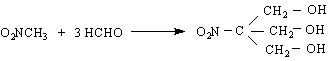
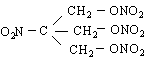
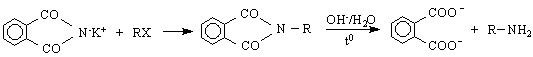
СЪЕДИНЕНИЯ – АЛДЕХИДИ И КЕТОНИ
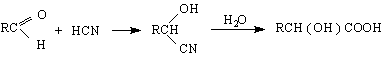
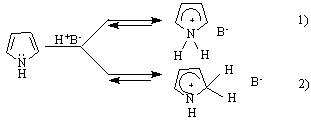
Едни от най-реактивоспособните съединения в органичната химия са алдехидите и кетоните. Алдехидите са съединения с обща формула RCHO, а кетоните – с обща формула RCOR′. Заместителите R и R′ могат да бъдат както алкилови, така и арилови. Наличието на карбонилната група С=О обединява двата вида съединения под общото название карбонилни.
![]() ни
показва
наличие на σ- и
π-връзки, sp2-хибридно
състояние на въглеродния
атом и две
свободни
електронни
двойки при
кислородния
атом. Според
съвременно
схващане при
алдехидите и
кетоните
кислородът
също е в sp2-хибридно
състояние. В
подкрепа на
това твърдение
се привежда ъгълът
на връзките
–С-О, който е ~1200,
дължината
на връзката
С=О, която е
равна на 1.22 Е и енергията
на тази
връзка, която
е 748 кДж/мол.
Обяснението
на факта за
по-голямата
здравина на
връзката в
карбонилната
група в
сравнение с
С=С връзката /610
кДж/мол /
трябва да се
търси в
по-голямото
отдалечаване
на
свободните
електронни
двойки.
ни
показва
наличие на σ- и
π-връзки, sp2-хибридно
състояние на въглеродния
атом и две
свободни
електронни
двойки при
кислородния
атом. Според
съвременно
схващане при
алдехидите и
кетоните
кислородът
също е в sp2-хибридно
състояние. В
подкрепа на
това твърдение
се привежда ъгълът
на връзките
–С-О, който е ~1200,
дължината
на връзката
С=О, която е
равна на 1.22 Е и енергията
на тази
връзка, която
е 748 кДж/мол.
Обяснението
на факта за
по-голямата
здравина на
връзката в
карбонилната
група в
сравнение с
С=С връзката /610
кДж/мол /
трябва да се
търси в
по-голямото
отдалечаване
на
свободните
електронни
двойки.
За алдехидите и кетоните се използват два вида наименования – тривиални /или рационални/ и по номенклатурата на IUPAC.
по IUPAC
- от наименованието на съответния
с въглеводород със същия брой С- ат
киселини + думата “алдехид” и окончанието -ал.
- от наименованието на съответния
въглеводородни остатъка + въглеводород със същия брой С-ат.
думата “кетон” и окончанието -он / с означение за
място /.
Алдехиди
І. Моноалдехиди
/ с една –СНО група/
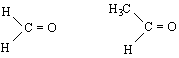
метанал етанал
/ацеталдехид/
![]()
2-бутенал 3-фенил-2-пропенал
/кротонов алдехид / / канелен алдехид /
![]()
3-бутинал
![]()

1-нафталенкарбалдехид
І. Монокетони
/с една -СО – група/
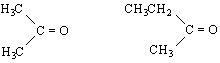
бутан-2-он
/метилетилкетон/
Алкенони
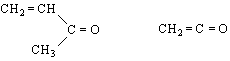
кетен
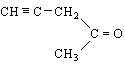
1-пентин-4-он
![]()
метилциклохексилкетон
![]()
дифенилкетон
/бензофенон /
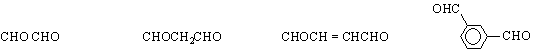
пропандиал 2-бутендиал 1,3-бензендикарбалдехид
![]()
пентан-2,4-дион 1,2-дифенилетандион 1,4-бензохинон
/ ацетилацетон/ / дибензил , бензил /

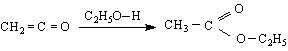
това е основната реакция от която произлиза и името “алдехид” – alcohol dehydrogenates
![]()
Когато се използва паладий като катализатор се отделя PdH
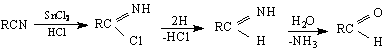
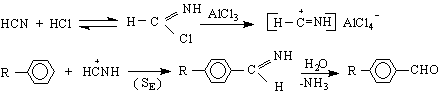
хлоримин алдимин
Като катализатор се използва безводен калаен хлорид и сух газообразен HCl.
![]()
Ако се използва катализатор на Линдлар ( Pd + BaSO
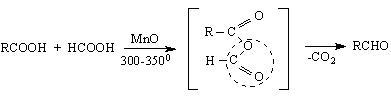
д/ чрез декарбоксилиране на карбоксилови киселини /по Sabatier
![]()
– това е технически метод, широко използван през Втората световна война в Германия. Работи се с Фишер-Тропшов катализатор /90% Co O ThO MgO
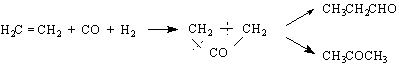
![]()
Най-често като катализатор се използва като окислител MnO + H SO
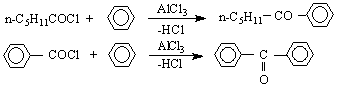
б/ реакция на Гатерман-Кох - частен случай на Фридел-Крафтсовите синтези. Реакцията се провежда като на ароматния въглеводород се действа със смес от сух СО и хлороводород /HCl/ в присъствие на AlCl CuCl Предполага се, че от двата газа се образува формилхлорид, който в присъствието на AlCl
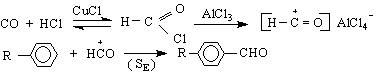
-
![]()
LiAlH O tret C CH
д/ редукция на киселинни амиди
![]()
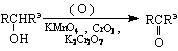
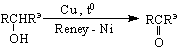
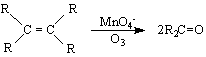
Ако окислението се извърши с KmnO
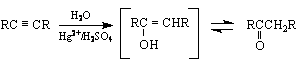
пиролиз на калциеви соли на съответни ароматни киселини ; пропускане на парите на съответните киселини върху нагрят ториев оксид или манганов оксид /методът е приложим, когато трябва да се получи СО-група в страничната верига/ ; окисление на вторични ароматни алкохоли.
при нагряване етери на фенола с AlCl
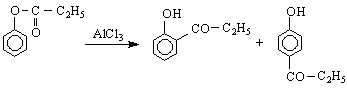
![]()
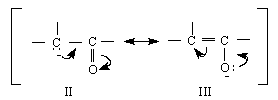
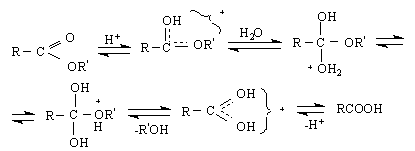
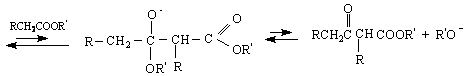
Нека се опитаме да направим аналогия с реакциите на двойната връзка при алкените, т.е.да сравним реакциите на С = О – групата с реакциите на С = С-групата. Нека си припомним, че присъединителните реакции към алкени са една атака на π-системата /π- връзката / от електрофил. Присъединяването към карбонилната група е основано на атака към карбонилния С-атом от нуклеофил. Карбонилната група много леко се атакува от нуклеофили главно по две причини : 1/ Тази група може да съществува в две резонансни структури, едната от които носи /+/ - заряд при С-атом и 2/ Електроотрицателността на О-атом обуславя и налага една постоянна поляризация на карбонилната група, при което С-атом се оказва зареден положително.
![]()
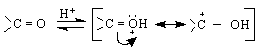
тях реакции на присъединяване. / За реакцията присъединяване към връзката С = О е характерна голяма обратимост, в резултат на което се регенерира /възстановява/ карбонилната група. Много от тези реакции могат да се разглеждат като истински равновесия. Напр.:
![]()
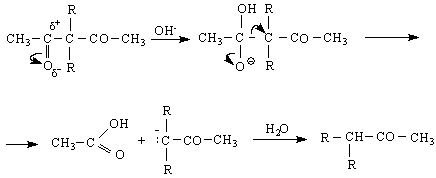
Да разгледаме сега от какво зависи присъединителната нуклеофилна реакция ( AN при карбонилните съединения ?
I
![]()
N
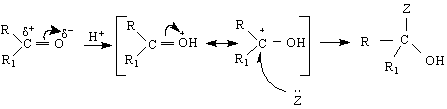
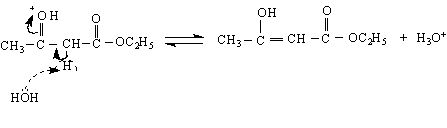
Вижда се, че нуклеофилната атака се улеснява от присъствието на електрофил, който засилва стойността на /+/ товар при карбонилния С-ат. Предварителното протониране снижава Еа за нуклеофилната атака, тъй като то позволява на кислорода да придобие чифта π-електрони без при това да се натоварва отрицателно.
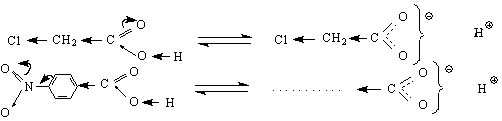
N-реакции, отколкото кетоните. Това се обяснява с факта, че кетона съдържа втора алкилова /или арилова/ група, а алдехида само водороден атом. Втората алкилова /арилова / група е по-обемиста от Н-атом и в по-голяма степен ще препятства образуването на междинно съединение в преходното състояние. Освен това алдехидната група подава електрони и по този начин дестабилизира преходното състояние за сметка на засилване на / - / товар при О-атом.
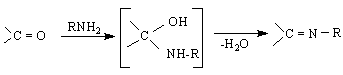
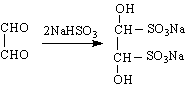
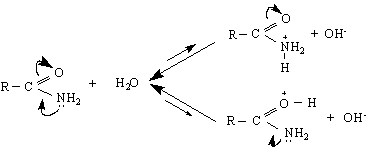
I - /бази/ или Н-съдържащи съединения, образуващи бази /аниони/ след дисоциация. Това са : NH3, RNH NH NH RNHNH C H NHNH NH OH / N – нуклеофили / ; H2O ROH O NaHSO RSH H S S
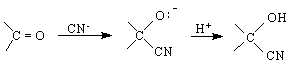
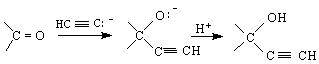
II - псевдокиселини /потенциални нуклеофили/. Това са СН-киселини, които се превръщат в основи /карбаниони/ след отцепване на Н+ под действие на протоноакцептор : HCN, C H CH CHO CH COR
III криптооснови. Това са съединения, в които анионната част е в “скрит” вид. Образува се при атаката върху карбонилния С-ат. или съществува в здрава контактна йонна двойка. Напр.органометални съединения R Li RMgX
От първата група нуклеофили N-съдържащите имат сравнително активен нуклеофилен център, а О- и S-съдържащите – по-слабо активен. Затова и АN N
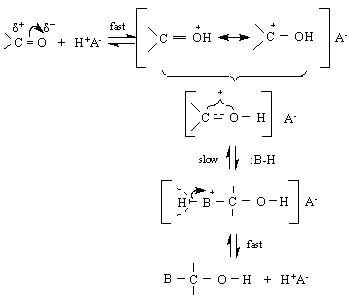
По-силно основните N-съдържащи нуклеофили реагират с карбонилните съединения при рН~7, докато по-слабо основните кислород и сяра-съдържащи нуклеофили – при рН < 7, т.е.в кисела среда.Отделните видове АN
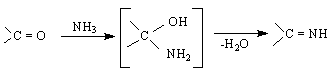
алдимин /или кетимин /
азометин /шифова база /
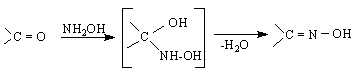
/алдоксим и кетоксим /
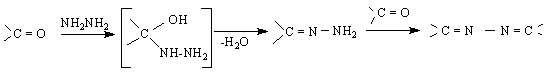
хидразон азин /алдазин и кетазин
![]()
/фенилхидразон /
N реакциите с О- и S-съдържащи нуклеофили са дадени на схемата :
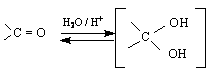
/нетрайни съединения ; бързо се разлагат /
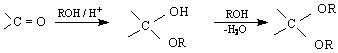
ацетал /или кетал /
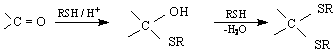
полутиоацетал тиоацетал

N СН-киселини /реакции от II CN N реакции в случая С = О групата предварително не се активира. Ролята на катализатора е да “активира” СН-киселината, т.е.да я превърне в карбанион /активен нуклеофил /, способен да атакаува карбонилния С-атом. Механизмът на тази катализирана от основи реакция /ANB
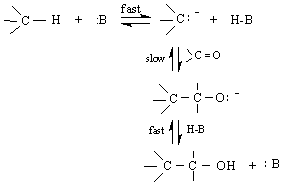
Вторият /бавния / стадий може да се ускорява чрез киселинен катализ и затова взаимодействието в този етап протича в прототен разтворител /вода, алкохол и др.
 /реакция на
Фаворски /
/реакция на
Фаворски /
алдол ненаситено
карбонилно с-ие
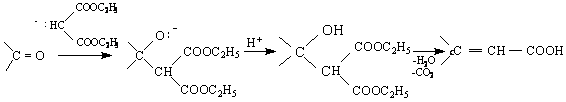
/реакция на
Кньовенагел 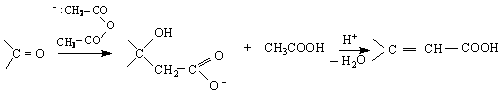
ненаситена киселина
/ Перкинова синтеза /
N
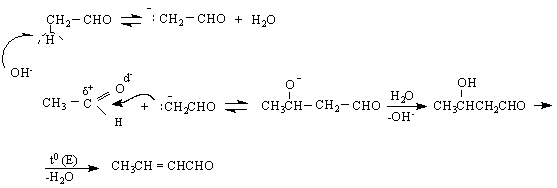
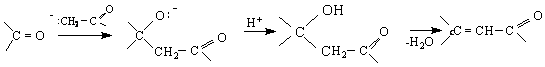
Характерното при алдолните реакции е, че за да се получи метиленовия компонент /карбаниона /, то съединението трябва да притежава α-Н-атоми, т.е.водородни атоми при съседния на карбонилната група въглероден атом. Йонизацията на α-водородния атом довежда до появата на карбаниона I, който е резонансен хибрид от двете структури /II и III/. Такъв резонанс е възможен само при участие на карбонилната група :
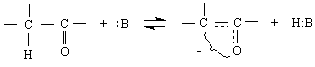
карбанион енолатен анион
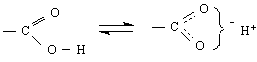
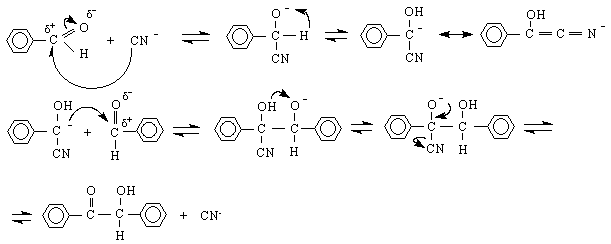
Присъединителни реакции протичат и между нуклеофили /криптооснови от III група/ и карбонилни съединения. Реакцията протича по хелатен механизъм. Нарича се още диспропорциониране или дисмутация. Има окислително-редукционен характер.
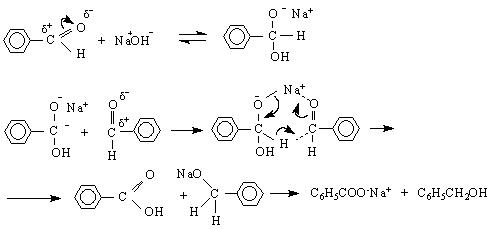
Такава реакция е т.нар.Каницарова реакция. Протича при взаимодействието между две молекули алдехид, който не съдържа α-водороден атом /обикновено бензалдехид/ в присъствие на конц.алкална основа. Механизмът на реакцията е следният :
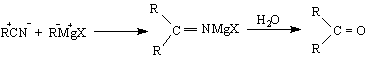
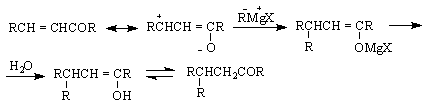
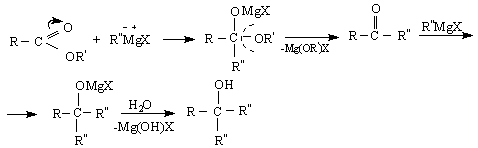
Друга важна реакция, която може да се отнесе към типа III /с криптооснови/, е взаимодействието на карбонилни съединения с Гринярови реактиви /алкилмагнезиеви халогениди RMgX / :
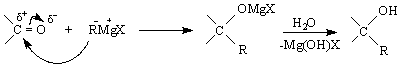
Реакцията може да се разглежда като редукционна, тъй като С = О-групата се превръща в С- ОН група.
Доказано е, че в реакцията участва димерна форма на Гриняровия реактив, който се отнася като криптооснова. Атаката се извършва от :R
1/ С каталитично активиран Н2 /в присъствие на катализатори Ni, Pt, Pd / алдехидите се редуцират до първични алкохоли, а кетоните - до вторични :
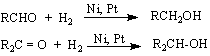
![]()
3/ Хидриране по Клеменсен - с аомалгамиран цинк в солнокисела среда. В този случай редукцията отива до парафини :
![]()
4/ Редукция по Кижнер-Волф /при кетоните / - косвена редукция ; отначало кетона се превръща в кетохидразон, който при нагряване до 2000 се разпада до парафин и азот :
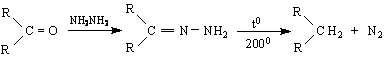
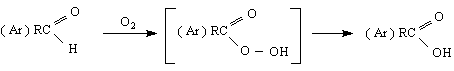
Кетоните се окисляват по-трудно - те са устойчиви на слаби окислители.Под действието на по-силни окислители се разкъсват С – С връзки във въглеводородните остатъци на кетоните и се получават смеси от различни окислителни продукти - киселини и по-леки кетони :
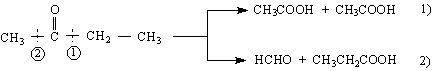
- амонячен разтвор на сребърен нитрат.Реакцията е известна като “реакция на сребърното огледало”, защото се отделя елементарно сребро, полепващо по стените на епруветката :
![]()
смес от слабокисел разтвор на CuSO с алкален разтвор на калиево-натриев тартарат /сол на винената киселина /. При нагряване с алдехид пада червена утайка от Cu O :
![]()
Реактив на Бенедикт -
използва се разтвор на Bi (NO
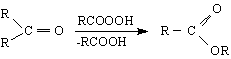
Кислородният атом в карбонилната група може да бъде заместен с хлорни атоми под действието напр.на PCl
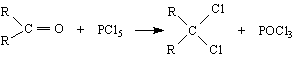
Една характерна реакция, с която също може да се замести кислородния атом в карбонилната група, е реакцията на Витиг :
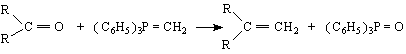
трифенилфосфинов
фосфоран / илиди / оксид
![]()
![]()
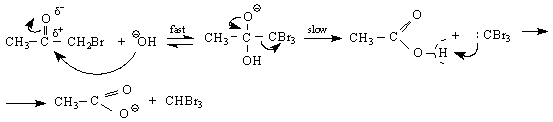
Реакцията носи името халоформна реакция и служи за качествена проба за наличие на метилкетони /терминални кетони /; особено в случая с йодоформа, тъй като той представлява малко разтворима яркожълта твърда утайка.
![]()
параформалдехид
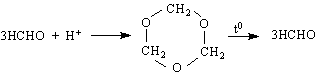
1,3,5-триоксан
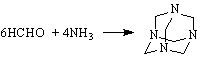
Уротропинът е сходен по структура с адамантана. Намира приложение като диуретик и за лечение на ревматизъм. Уротропинът при обработка с конц.HNO
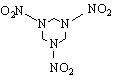
![]() етанол
с участието
на NADH и
декарбоксилаза
/.
етанол
с участието
на NADH и
декарбоксилаза
/.
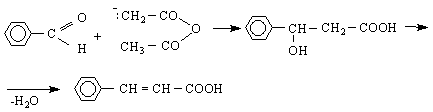
При смесването на ацеталдехид с няколко капки конц.H SO

![]()
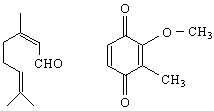
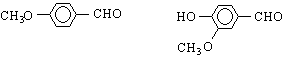
анисалдехид ванилин
Производни на 1,4-бензохиноните под името убихинони влизат в състава на коензим Q ( Q 10 ), който участва в реакции на пренос на електрони във веригите на процеса дишане.
Най-простият представител е глиоксалът /етандиал / ОНС – СНО. Получава се чрез окисление на ацеталдехид със SeO
![]()
Технически глиоксалът се получава от гликол при окисление в газова фаза в присъствие на катализатор Ag или Cu :
![]()
![]()
![]()
Образува и дифенилхидразони с фенилхидразина /C H NHNH
![]()
![]()
малоновия диалдехид ОНС-СН2-СНО, янтарния алдехид ОНС-(СН2)2-СНО. Общия метод за получаването им се състои в хидриране с Pd на съответните ди-киселинни хлориди, т.е.производни на съответните киселини.
Според положението на кетогрупата една спрямо друга различаваме : 1,2- или α- дикетони ; 1,3- или β-дикетони ; 1,4- или γ-дикетони и т.н. Тези групи дикетони имат доста различен химичен характер и затова ще се разглеждат поотделно /с по един техен представител /.
, диметилглиоксал, бутан-2,3-дион , СН3СОСОСН3. Получава се от метилетилкетон чрез окисление със SeO
![]()
![]()
![]()
![]()
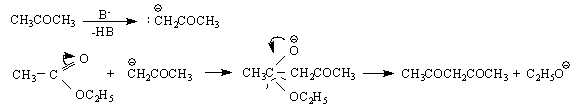
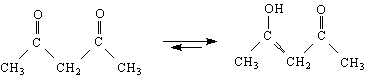
ацетилацетон енол
(15%) (85%)
При получаването на енолати /с алкален метал / процесът се изтегля значително надясно. Изместят ли се двата водородни атома с алкилови радикали / -С (R
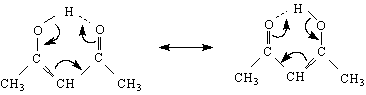
Характерно за 1,3-дикетоните е, че могат да образуват стабилни и слабополярни енолати с хелатообразуващи агенти /соли на тежки метали /. Солта на тривалентното желязо има червен цвят. Трябва да се знае, че FeCl3 е един реактив за β-дикетони, но по-скоро за еноли. Медните соли образуват вътрешнокомплексни соли :
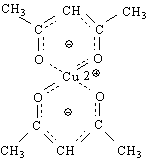
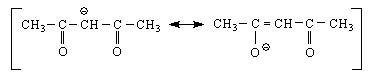
С тази структура се обяснява получаването на О- и С-производни. Реактивоспособността на 1,3-дикетоните и по-точно на тяхната метиленова группа се изразява в много реакции. На студено те реагират с Br2 /енолната форма /. Нитрират се също много лесно.
N
По-важни са α, β-етиленовите алдехиди. Те се получават като се действа на етиленови алкохоли с окислители, които не засягат двойната връзка, напр.MnO
![]()
Al
![]()
![]()
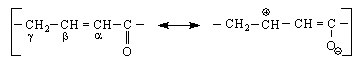
Все пак те реагират с Br HCl брома отива на β-място /обратно на Марковников, пак поради спрягане /. Карбонилната група реагира нормално с органомагнезиеви съединения RMgX, с хидроксиламин NH OH HCN LiAlH
Нуклеофилните присъединявания /вода, алкохол, амини, карбанион / става при С = С връзката, но всъщност след спрягане :
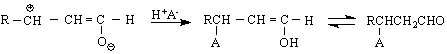
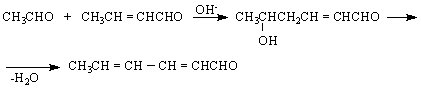
СН2 = СНСНО/. Той се получава при обезводняване на глицерол с KHSO
![]()
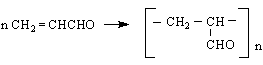 полиакрил
полиакрил
- чрез кротонизация /след алдолна кондензация / между кетони или между кетон и алдехид :
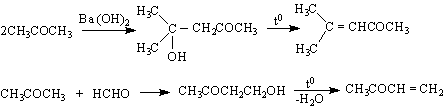
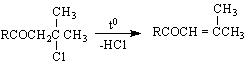
![]()
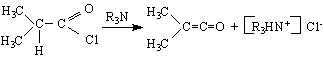
Най-простият представител е кетенът или етенон СН2 = С = О. Всички кетени се разглеждат като производни на първия член. По-висшите му аналози се наричат алдокетени R-CH = C = O или кетокетени (R) C C O R или арил. Напр. : СН3СН = С = О е метил-кетен, който е един алдокетен, а (CH C C O
т α-халогенирани киселинни халогениди като се действа с Zn в отсъствие на въздух :
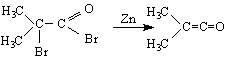
3/ От ацетонови пари при пиролиза /7500/ над Cr-Ni-спирала :
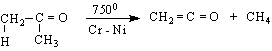
4/ Технически метод е пиролизата /8000/ на оцетна киселина при използване на катализатор триетилфосфат или AlPO4 :
![]()
N
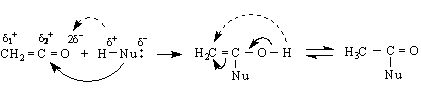
/ δ1+ < енол кетон
![]()
![]()
![]()
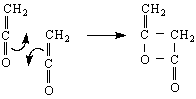
изисква наличието на катализатор. Тази роля изпълнява зеленото багрило хлорофил. Процесът се извърщва при осветяване /източник на енергия / :
![]()
При хранене на животните, а и на човека с нишесте, а в някои случаи и с целулоза, те се разрушават давайки отново изходната глюкоза. Последната с кръвта се пренася черния дроб и там се преврлъща в гликоген или животинско нишесте. В случай на необходимост гликогена може отново да бъде разрушен до гликоза. След това гликозата се пренася в ток от кръв до тъканите, където се окислява до СО2 и Н2О с отделяне на енергия, получена първоначално от слънчевата светлина. Известно количество глюкоза се превръща в мазнини, а друго количество реагира с N-съдържащи съединения, образувайки аминокиселини, които съединявайки се една с друга дават белтъци, основа на всички известни форми на живот.
Глюкозата, целулозата, нишестето и гликогена принадлежат към един и същи клас органични съединения, наречени въглехидрати. В крайна сметка въглехидратите са основен източник за нашата храна. Ние употребяваме зърно, съдържащо нишесте или храним с него животните, в организма на които то се превръща в месо и мазнини, които ние употребяваме. Нашето облекло е почти изцяло от целулоза /памук, лен, вискоза, ац;етатните влакна и др./. Мебели, къщи, хартия – всичко това е от дърво, а то е също целулоза. По такъв начин въглехидратите в буквалния смисъл обезпечават нашия живот .
Химията на въглехидратите е една от най-интересните области в органичната химия. Тя обхваща широк кръг въпроси от сложни проблеми при изучаване на процеса фотосинтеза до разплитането на доста сложните стадии във ферментативните превръщания на глюкозата до СО2 и Н2О. Та между тези два сложни биохимични проблема лежат и нашите задачи : да определим структурата и свойствата на някои по-прости въглехидрати и да изучим някои техни превръщания в други органични съединения.
въглехидрати е възникнало заради това, че съставът на голяма част от тези съединения може да се изрази с общата формула Cm H O n
. Тя се изгражда въз основата на състава и структурата на молекулите на тези съединения. И тривиалната номенклатура и тази на IUPAC са утвърдили окончанието –оза в наименованията на по-голяма част от въглехидратите. Поради сладкия вкус на мнага от въглехидратите те се наричат още и захари или захариди.
Въглехидрати
/захариди/
олигозахариди полизахариди
( монози, 3-8 С ат.) (2-10 монози ) (от 10 до няколко хиляди монози )
кетози дизахариди тризахариди хомополи- хетерополи-
захариди захариди
Това са бифункционални съединения. Молекулата им съдържа една алдехидна /-СНО / или кетонна / С = О / групи и повече от две ОН-групи. Те са полихидроксиалдехиди или полихидроксикетони.
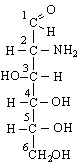
. Класификацията на монозахаридите се основава на съчетаването на два признака – вида на функционалната карбонилна група и броя на С-атоми в молекулата. Според първия признак монозахаридите се делят на алдози и кетози, а според втория – триози, тетрози, пентози, хексози и т.н. Трябва да се знае, че в молекулата на монозахаридите въглеродните атоми са свързани в права /нормална / верига и при един С-атом може да има само една –ОН-група.
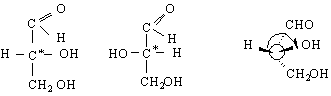
D R L(-) или S D
Конфигурацията на енантиомерите може да бъде определена по R, S-системата на Кан-Инголд-Прелог. А преди нея за означаване на монозахаридите и други хирални съединения е била създадена и използвана т.нар. D L За нейното изграждане като основа са послужили именно конфигурациите на енантиомерите на глицериналдехида. Условно е било прието с D (+) да се означава този негов енантиомер, в проекционната формула на който хидроксилната група е отдясно, когато СНО-групата е отгоре. Другият енантиомер е означен като L (-). Конфигурацията на D-енантиомера съответства на R-конфигурация, а тази на L-енантиомера – на S-конфигурацията.
D L-системата се основава на следната постановка : монозахариди, които могат да се разглеждат като произлезли от D чрез удължаване на въглеродната верига откъм СНО-групата, се обединяват в т.нар.D-стеричен ред енантиомери /стереоизомери/. По аналогичен начин се регламентира формирането и на L-стеричния ред. По-висшите от глицериналдехида алдози притежават нарастващ брой структурно различни асиметрични С-атоми и следователно имат 2n хирални стереоизомера. Те се систематизират съответно в D, L-стерични редове според конфигурацията на най-отдалечения от алдехидната група асиметричен С-атом, който се смята че е “наследен” от съответния енантиомер на глицериналдехида. Енантиомерите на дадената алдоза имат еднакви наименования /окончаващи на –оза /, чрез които се означава конфигурацията на всеки от тях : D или R , съотв.L или S.
От алдохексозите напр.са познати 16 стереоизомера /D и L/ : глюкоза, маноза, идоза, галактоза, алтроза, алоза, гулоза и талоза.
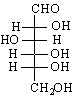
D (+) – глюкоза
.Те също биват кетотетрози, кетопентози, кетохексози и т.н. Разбира се, че тук карбонилния С-атом не може да бъде в края. Най-често това е втория С-атом. При кетозите също съществува енантиомерия /стереоизомерия/. Както при алдозите и тук стереоизомерите се причисляват към D, L-стерични редове. Особен интерес представляват кетохексозите и техния представител фруктозата. Фруктозата има три асиметрични С-атома и съществува като D- и L-форма, което се определя от най-отдалечения от кетогрупата асиметричен С-атом. Проекционната формула на фруктозата е следната :
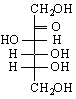
D (+)-фруктоза
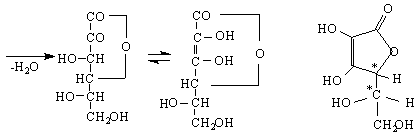
Това означаване на формулата е по Фишер – т.нар.Фишерови проекционни формули /оксоформули /. УВ и ИЧ-спектрите обаче не показват наличие на ивици на поглъщане за С = О група. Освен това разтвор на D (+)-глюкозата след известно време променя специфичното си въртене. Това явление за промяна на оптичното въртене на монозахаридите след разтварянето им се нарича мутаротация. Липсата на С = О група показва че те нямат оксоструктура и следва да се приеме, че те имат циклична полуацетална структура. Да си припомним, че полуацетали се образуват при взаимодействието на на молекула алдехид /или кетон/ с молекула алкохол. Така че при монозахаридите се образува един вътрешен полуацетал. Между молекулите на даден монозахарид с тези два вида структури /отворена и циклична / се осъществява оксоциклотавтомерно равновесие в разтвор, което обяснява и явлението мутаротация.
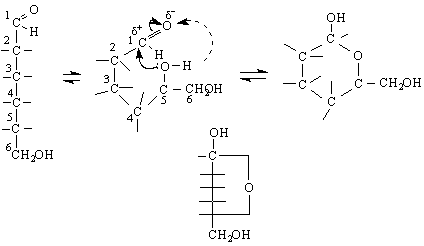
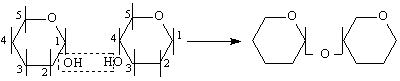
Когато в по-горната схема новополучената хидроксилна група при карбонилния въглероден атом, наречен гликозиден, се намира от страната на пръстена, получения изомер се означава като α-аномер. Когато хидроксилната група се намира от противната страна, тогава имаме β-аномер /от гр.ано – на върха /. Изразяването на тези два изомера може да стане или чрез цикличните форми, показани по-долу :
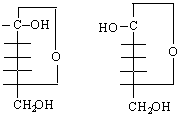
α – аномер β – аномер
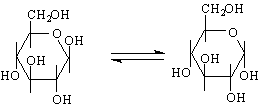
α – аномер
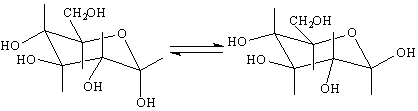
α – аномер β – аномер
По-стабилна е молекулата на β-изомера, тъй като всички хидроксилни групи се намират в по-изгодното екваториално положение. Това ще си припомним, когато разглеждаме огромната молекула на целулозата, съставена от β-D-глюкозни остатъци.Ето защо целулозата е най-разпространения въглехидрат в природата.
. Алдозите и най-вече хексозите са силни редуктори. Те лесно се поддават на окисление. При умерено окисление с разредена HNO или J
![]()
От по-силни, по-енергични окислители (конц.HNO ) алдозите преминават в хидроксидикарбоксилови киселини, наречени –арови или захарни киселини. Например D-глюкозата дава D-глюкарова киселина :
![]()
От много силни окислители /KmnO
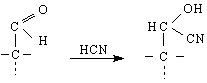
3/ С циановодород HCN алдозите и кетозите дават цианхидрини :
4/ С хидроксиламин NH OH
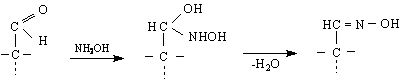

7/ Метилиране. От хидроксилните групи на монозахаридите най-лесно се метилира ОН-групата, произлязла от карбонилната. Достатъчно е да се действа на α- или β-монозахарид с метилалкохолна солна киселина (CH OH HCl ат съответно α- или β-производни, които са аномери. Общото наименование на тези съединения е гликозиди. Метилгликозидите са най-простите представители на тази важна група съединения, които са много разпространени в природата. Останалите ОН-групи са представители на същинските алкохоли и като алкохолите се метилират с CH J Ag O
8/ С ацетон монозахаридите, подобно на някои полихидроксисъединения се кондензират в присъствие на водоотнемащи средства /киселини, двуфосфорен пентаоксид, цинков дихлорид и др./
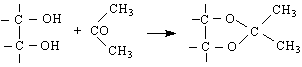
9/ Естерифициране. Тази реакция става много лесно и се извършва главно с оцетен анхидрид. Извършва се пълно естерифициране и се получава пентаацетилглюкоза. /Не трябва да се забравя, че естерификацията се извършва в слабокисела среда ! /. Много разпространени в природата естери на глюкозата с галовата /шикълчена / киселина са дъбилните вещества – танините. Това са аморфни вещества, способни да превръщат суровата кожа в обработена кожа, да коагулират лепливите разтвори и да дават неразтворими утайки с алкалоиди и оловни соли. Танините биват китайски и турски танин. Различават се по галовите остатъци в молекулите си. Китайския е с 9 галови остатъка, а турския – с 5-6 остатъка.
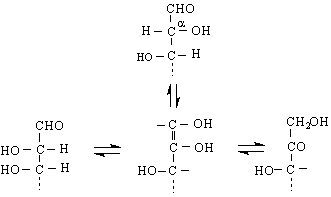
маноза ендиол фруктоза
Глюкозата и манозата се различават само по конфигурацията на α-въглеродния атом. Такива изомери се наричат епимери / епи – от гр.против/.Следователно, за да се превърне един епимер с друг, той претърпява α-инверсия.
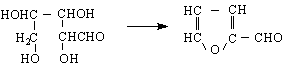
12/ Синтези. а/превръщането на един монозахарид в негов по-висш хомолог става по комбинирания метод на Килиани-Фишер (действа се с циановодород и получаващия се цианхидрин се хидролизира ; следва редукция ) б/ разпадане на монозахарида в по-низши хомолози става по методите на Руф, метод на Веерман и др.
С6Н12О6
![]() 2С2Н5ОН + 2СО2
2С2Н5ОН + 2СО2
D
D
D
Производно на D-глюкозата е витамин С.Той се намира в много растения – шипки, цитруси, пиперки, магданоз и др.Вит.С е противоскорбутен препарат. За синтеза на вит.С се излиза от D-глюкоза :
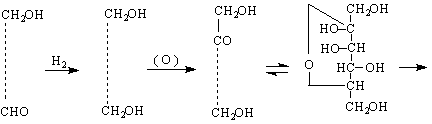
D L-сорбит L-сорбоза полуацетална форма
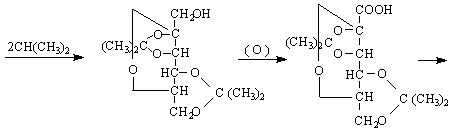
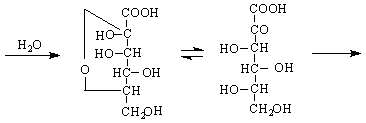
2-кето-L-гулонова киселина
γ-лактон на L – аскорбинова киселина
трехалоза /свързване от трехалозов тип /
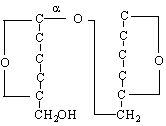
малтоза /свързване от малтозов тип /
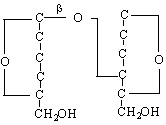
гентиобиоза /свързване от гентиобиозов тип /
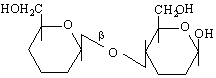
целобиоза /свързване от целобиозов тип /
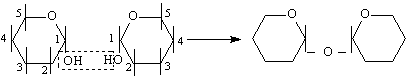
трехалозов тип / 1-1 свързване / с участие на 2 гликозидни ОН-групи
малтозов тип /1-4 свързване / с участие на 1 гликозидна и 1 алкохолна ОН-групи
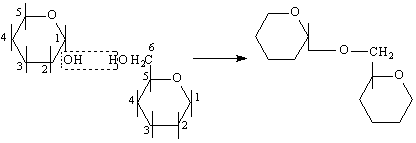
гентиобиозов тип /1-6 свързване / с участие на 1 гликозидна и 1 алкохолна ОН-групи
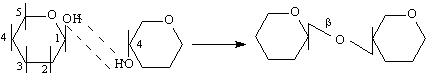
целобиозов тип /1-4 свързване/, но гликозидната ОН-група е в β-положение
Тя е един α-D-глюкопиранозил-β-D-фруктофуранозид. Молекулата и е образувана чрез 1-2 свързване на две гликозидни С-атоми - на α-D-глюкопираноза и β-D- фруктофураноза :
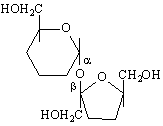
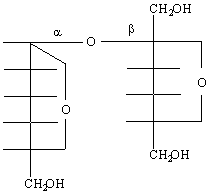
α-D-глюкоза β-D-фруктоза
Захарозата се хидролизира лесно в присъствие на слаби киселини или под действието на ензима инвертаза до D (+)-глюкоза и D (-)-фруктоза, еквимоларната смес от които се нарича инвертна захар. Захарозата има специфично въртене [ D . Тъй като въртенето на хидролизните продукти е : D (+) глюкозата [α D
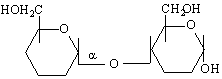
малтоза
целобиоза
, скорбяла (Amylum )
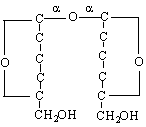
Молекулите на нищестето са изградени от α-D-глюкопиранозни остатъци (звена). Свързването на тези звена става чрез поликондензация от малтозов тип /1-4 свързване / и гентиобиозов тип /1-6 свързване /. Пръв френския учен Maquenne ства : амилоза и амилопектин. Количеството на амилозата е около 20%, а на амилопектина – 80%. Амилозата е разтворима във вода, а амилопектина не се разтваря а само набъбва.
Молекулата на амилозата е изградена чрез 1-4 свързване на α-D-гликозидни остатъци. Тя съдържа около 200 – 1000 подобни остатъци и има спираловидна или т.нар.helix-структура в права линия :
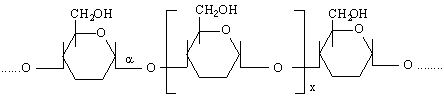
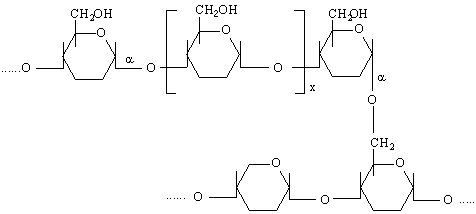
Частичната хидролиза на амилопектина дава декстрини – достатъчно едри молекули, съдържащи 8-12 глюкозни остатъци. По-нататъшната хидролиза дава дизахариди – главно малтоза. При пълната хидролиза се получава α-D-глюкоза.
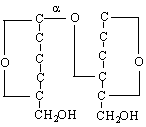
Името и произлиза от cellula – клетка. Целулозата образува стените на клетките у висшите растения. Тя има линейна структура на молекулите си и те са изтрадени от β-D-глюкопиранозни остатъци, свързани както в целобиозата /1-4 свързване както в малтозата, но аномерния атом е в β-положение / :
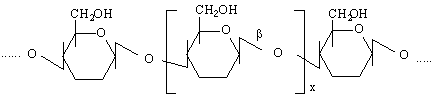
При частична хидролиза на целулозата се получават олигозахариди – главно целобиоза, а по-нататък – β-D-глюкоза. Поради наличието на алкохолни ОН-групи целулозата проявява реакционна способност на поливалентни алкохоли. Тя се разтваря в разтвор на медно(II)-амонячен комплекс /Швайцеров реактив/ поради образуването на комплексни съединения с Cu (II)-йоните. В разтвор на алкални основи целулозата дава алкохолати. Тя образува естери с различни киселини и етери с алкилхалогениди.
При взаимодействието на алкална целулоза със CS се получава Na-сол на естера на ксантогеновата киселина /целулозен ксантогенат /, чиито алкален разтвор се нарича вискоза. Целофанът е регенерирана въ вид на листове. Ако се накъдри вискозата се получава т.нар.изкуствена вълна или целволе.
Освен същинската целулоза са познати още хидратна целулоза, хемицелулоза, пектин, хитин и др.
глюкозамин
КАРБОКСИЛОВИ КИСЕЛИНИ
Производни на въглеводородите, които съдържат в молекулата си функционалната група -СООН се наричат карбоксилови киселини. Карбоксиловата функционална група е съставена от две по-прости функционални групи : карбонилна ( С = О ) и хидроксилна (-ОН ), откъдето произлиза и наименованието и /карбоксилна/

Б. Според вида на въглеводородния остатък, свързан с –СООН и наличието на други функционални групи /заместители / в него : алканови, алкенови, аренови; хидрокси-, кето-, халогено- и т.н.
Н-СООН СН3-СООН СН3СН2-СООН и т.н. R
етанова пропанова алканова киселина
( оцетна ) (пропионова)
Според IUPAC наименованията се образуват като към името на съответния алкан със същия брой С-атоми се прибави окончанието -ова киселина. При разклонените алкилови вериги се избира главна /права/ верига, в единия край на която е -СООН-групата и чиито С-атом носи № 1. Оттук почва броенето.
СН3СН
= СН-СООН ![]()
2 - бутенова киселина пропинова киселина
(акрилова ) ( кротонова ) (пропионова )
![]()
циклохексанкарбоксилова циклопропанкарбоксилова
киселина киселина киселина
аренови /ароматни / монокарбоксилови киселини :
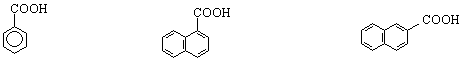
бензоена нафтален-1-карбоксилова нафтален-2-карбоксилова
(α-нафталенкарбоксилова ) (β-нафталенкарбоксилова )
Киселинния
остатък ![]() се
нарича
ацилов
остатък /R е
алкил,
алкенил, арил
и т.н. /. Напр. :
се
нарича
ацилов
остатък /R е
алкил,
алкенил, арил
и т.н. /. Напр. :
![]()
формилов ацетилов бензилов
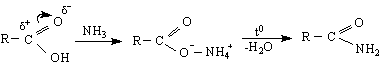
Когато заменяме атоми или атомни групи във функционалната група –СООН с други атоми или атомни групи, тогава казваме, че се получават техни функционални производни : ако заместим Н-ат.от хидроксилната група на карбоксилова киселитна с метален атом, тогава получаваме сол на съответната киселина ; ако водородния атом се замени с алкилов или арилов радикал (R, Ar), тогава се получават естери ; ако заместим водорода с ацилов остатък (RCO), получаваме анхидрид ;ако заменим цялата хидроксилна група с халогенен елемент, тогава получаваме киселинни халогениди ; ако вместо ОН-групата имаме аминогрупа, тогава са получени киселинни амиди ; когато заместваме с хидразин, получаваме хидразиди ; при заместване на всичките функционални групи в карбоксиловата киселина (ОН-групата и карбонилния кислороден атом) с азотен атом се получават нитрили.
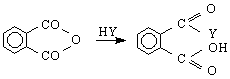
1/ От киселинни производни /естери, халогениди, анхидриди, амиди, нитрили / чрез хидролиза :
![]()
където Y = OR′, Cl, NH2, CN, OCOR и др.
2/ Чрез окисление на въглеводороди, алкохоли и алдехиди :
![]()
![]()
3/ Чрез присъединяване на органомагнезиеви съединения към СО2 :
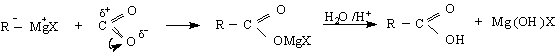
4/ Метод на карбонилиране. Смес от алкен, СО и вода се нагряват в присъствие на катализатор Ni (CO )
![]()
![]()
5/ От ацетиленови въглеводороди /алкини/ чрез хидролиза до алдехид и последващо окисление . Напр.познатата реакция на Кучеров :
![]()
6/ Чрез ферментация на алкохол с помощта на ензима acetobacter :
![]()
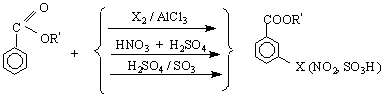
Основните методи за получаване на аренови карбоксилови киселини са дадени на базата на бензоената киселина.
7/ От бензен и фосген с последваща хидролиза :
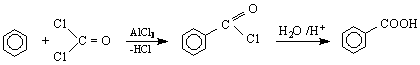
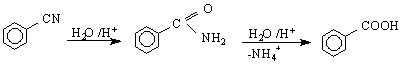
9/ Чрез окисление на нафтален /промишлен метод / :
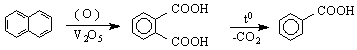
![]()
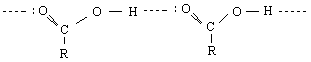
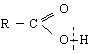
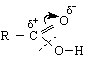
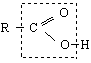
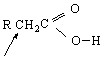
Тези реакции стават с едновременното участие и на функционалната група на карбоксиловата киселина (-СООН ).
Започваме от първия тип реакции - разкъсване на връзката О – Н. На този тип разкъсване се дължат много важни свойства на карбоксиловите киселини. Една от тях е, че карбоксиловите киселини имат киселинно-основен характер. Те притежават ясно изразени киселинни свойства, които се проявяват във възможността за дисоциацията им, напр.във водна среда :
![]()
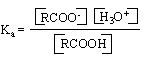
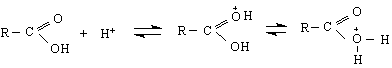 Дисоциацията
на
монокарбоксиловите
киселинив
разтвор
протича в
различна
степен в
зависимост
от вида на
въглеводородните
остатъци,
свързани с
карбоксилната
група. Алкановите
и ареновите
киселини
имат рКа ~ 4-5 и
във всички
случаи са
по-слаби от
най-известните
неорганични
киселини.
Обаче са по-силни
киселини от
алкохолите
/рКа = 16 / и
фенолите.
По-голямата
дисоциация
на карбоксиловите
киселини се
дължи на
по-добрата резонансна
стабилизация
на
карбоксилатния
анион RCOO за
разлика от
алкохолатния
анион RO
Дисоциацията
на
монокарбоксиловите
киселинив
разтвор
протича в
различна
степен в
зависимост
от вида на
въглеводородните
остатъци,
свързани с
карбоксилната
група. Алкановите
и ареновите
киселини
имат рКа ~ 4-5 и
във всички
случаи са
по-слаби от
най-известните
неорганични
киселини.
Обаче са по-силни
киселини от
алкохолите
/рКа = 16 / и
фенолите.
По-голямата
дисоциация
на карбоксиловите
киселини се
дължи на
по-добрата резонансна
стабилизация
на
карбоксилатния
анион RCOO за
разлика от
алкохолатния
анион RO
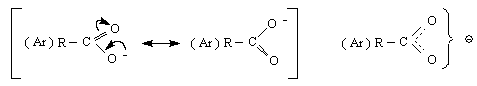
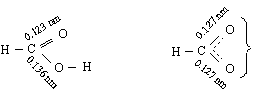
заместители / с +I и +М-ефект/ повишават електронната плътност при кислородния атом в ОН-групата и затрудняват дисоциацията, а след дисоциация увеличават отрицателния (-) заряд на карбоксилатния анион и го дестабилизират :
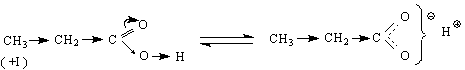
заместители /с –I и –М-ефекти/ оказват обратно влияние върху дисоциацията на карбоксилната група и стабилизацията на карбоксилатния анион, като повишават силата на киселините :
Освен дисоциацията характерна реакция от първия тип е и получаването на соли на карбоксиловите киселини при взаимодействието им с основи /неутрализация/ или метали :
RCOOH + NaOH ![]() RCOO-Na+ + H2O
RCOO-Na+ + H2O
ArCOOH + K ![]() ArCOO-K+ + 1/2H2
ArCOO-K+ + 1/2H2
/разкъсване на връзката С – О и заместване на ОН-групата в карбоксиловите киселини/ са заместителни реакции – нуклеофилни /SN/ и електрофилни /SE
В този случай реакцията протича като “присъединяване – елиминиране” и се нарича ацилно нуклеофилно заместване, т.е.АсSN
Видът на нуклеофилния агент Н-В:, който може да бъде алкохол (ROH), киселина (RCOOH) и др., определя характера на реакционните продукти - производни на киселините, съответно естери, анхидриди и др.
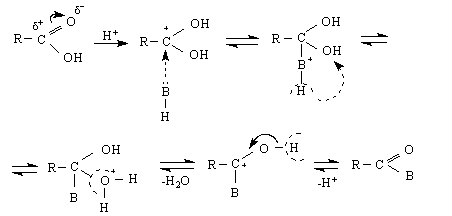
Напр.реакцията естерификация : това е реакция между киселина и алкохол. Ролятя на нуклеофила в случая се изпълнява от алкохола /по-точно от свободната електронна двойка на хидроксилната група в алкохола /. Това е слаб нуклеофил. Още повече, че и товарите в карбоксилната група е частичен. Ето защо реакцията се подпомага от присъствието на Н+, който превръща частичния товар при С-атом в карбоксилната група в С+ - способен да взаимодейства със слабия нуклеофил ROH
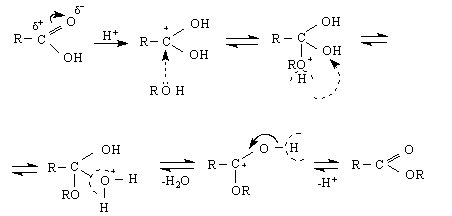
Б. Електрофилни заместителни реакции (SE ) се извършват чрез атака на електрофили, напр.киселинни хлориди, SOCl , PCl
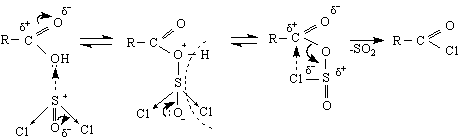
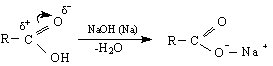
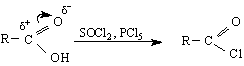
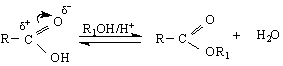
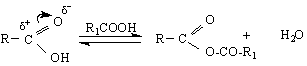
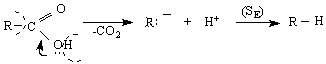
В първия стадий на реакцията се образува карбаниона R по SE-реакция и се получава алкан. Декарбоксилирането по този механизъм протича най-трудно при незаместените алканови киселини. Заместените карбоксилови киселини с електроноакцепторни заместители, съседни на -СООН –групата се декарбоксилира по-лесно.
![]()
![]()
![]()
2Р + 3Br ![]() 2PBr3
2PBr3

При ареновите киселини /бензеновото ядро играе ролята на въглеводороден остатък / особено характерни са заместителните електрофилни реакции (SE , които се извършват в ароматното ядро, Това са нитриране, сулфониране, халогениране /с катализатор / по механизма, който е разглеждан при ароматните въглеводороди. Трябва да се знае, че карбоксилната група е деактивиращ m-ориентант и затова този тип реакции протичат по-трудно, отколкото при незаместените арени :
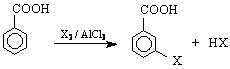
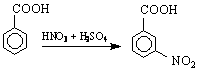
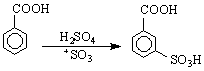
Друг тип реакции /тип 5 / на карбоксиловите киселини е редукцията им до алдехиди и алкохоли. Тя протича трудно чрез каталитично хидриране и по успешно под действието на литиево-алуминиев хидрид /LiAlH
![]()
![]()
, Ac.formicum – името произлиза от червената мравка /Formica rufa
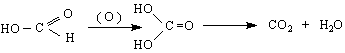
, Ac.aceticum. Тя е разпространена в растенията в свързано или в свободно състояние. Намира се също в някои животински секрети като пот, урина и др. От древни времена тя се получава като се изложат на действието на въздуха алкохолни разтвори. Окислението на алкохола става от кислорода на въздуха при каталитичното действие на ензимите на оцетнокиселите бактерии (Mucoderma aceti Виланд опитно е установил, че ензимите увеличават реактивоспособността на водородните атоми и затова се извършва не окислителен, а дехидриращ процес, при което се образува ацеталдехид :
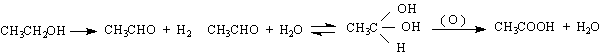
Тя е безцветна течност с остра миризма с т.к.1180.Безводната киселина образува кристали, подобни на лед /Ac aceticum glaciale
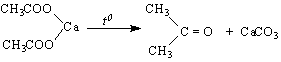
Солите и се наричат ацетати. Някои ацетати се използват в лабораториите, други в медицината, трети – в промишлеността.. Напр.Са-ацетат се използва главно за получаване на ацетон, Pb-ацетат, нар.още “оловна захар”/защото има сладък вкус/ се използва за лекуване на очи, а също и за оловно белило.Cu-ацетат има зелен цвят и е разтворим във вода. Тази сол служи за добиване на швайнфутово зелено, което е двойна сол от меден ацетат и за лекуване на рани.Разтвор на Al-ацетат под името Liquor aluminii acetici служи като антисептично и адстрингиращо /свиващо/ средство в медицината за лекулане на рани.
, Ac.butyricum. Има два изомера – нормална и изомаслена. Нормалната маслена киселина се намира в кравето масло, а в други мазнини се намира като глицерид. В свободно състояние се среща в потта на животните и човека, а също и в мускулния сок.Тя се образува при ферментацията на захар или нишесте под действието на Bacillus butylicus
. Те са четири изомера. Имат миризма на развалено сирене. Най-важната от тях е изовалериановата киселина, която се намира в растението Valeriana officinalis /котешка билка/. Корените на това растение от древни времена се използват като лечебно средство за успокоение на нервите.
От средните киселини – капроновата (С6) и каприновата (С10) са с миризма на пръч /caper/. Силната миризма на сиренето “Рокфор” се дължи на тази киселина.
, Ac.benzoicum.
MgCl
![]()
Някои химични свойства са общи за функционалните производни. В тези съединения С-ат. е сравнително активен електрофилен център. Ето защо и тук /както при карбоксиловите киселини/ е характерна нуклеофилната заместителна реакция AcSN илно нуклеофилно заместване/. Тази реакция се извършва чрез атака на нуклеофила (база) В: върху С-атом на ацилната група R-C = O и след отцепване на Y се получава заместения продукт RCOB. Механизмът на реакцията е следния :
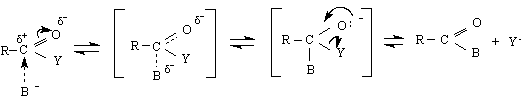
преходно състояние тетраедричен
субстрат продукт
хлориди > анхидриди > естери > амиди, като
надясно ![]() нараства
нуклеофилността
на Y:
нараства
нуклеофилността
на Y: ![]() намалява
реакционната
способност
при AcSN
намалява
реакционната
способност
при AcSN
Нуклеофилната заместителна реакция при функционалните производни може да се катализира от Н+ както при карбонилните съединения и протича подобно на AcSN – реакцията като отначало Н+ се присъединява към карбонилния О-атом, облекчава атаката на В:- и спомага за отделяне на Y: H Y
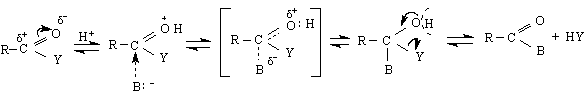
- свойства :
![]()
![]()
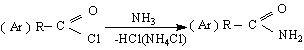
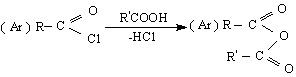
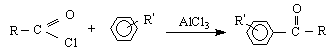
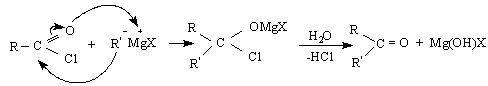
до алдехиди /по Розенмунд с Н2 и Pd/BaSO
![]()
![]()
Анхидридите биват прости ( RCO O RCO O COR . За анхидридите също са характерни реакциите AcSN
![]()
![]()
![]()
![]()
6/ Ацилиране по Фридел-Крафтс – анхидридите много лесно дават ацилиеви йони RC O SE
![]()
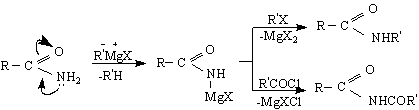
Амидите биват незаместени RCONH N RCONHR RCONR
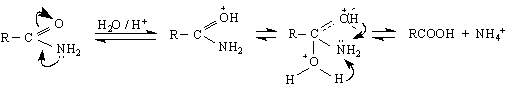
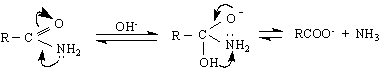
Незаместените амиди са слаби NH /по силни от NH
2/ С Гринярови реактиви амидите дават N-заместени производни :
3/ С алкални хипохалогениди NaOX протича Хофманово разпадане :
![]()
![]()
![]()
COOR
А. Реакции с участие на естерната група - както и при другите карбоксилови производни и при естерите са характерни AcSN
- тъй като киселините катализират реакцията естерификация, то те катализират и обратната реакция - хидролизата :
- използват се разтвори на алкални основи и нагряване :
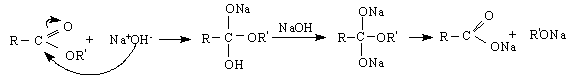
2/ Алкохолиза /преестерификация/ - катализира се от Н+ :
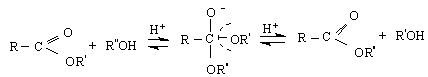
3/ Амонолиза - протича в алкална среда като AcSN B
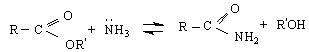
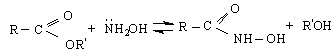
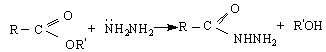
6/ С Гриняров реактив - върви по АN
![]() първични
алкохоли
първични
алкохоли
8/ Хидриране под действието на системата Na/алкохол или LiAlH
![]() първични
алкохоли
първични
алкохоли
1/ Алдолни реакции (Клайзенова кондензация) - катализира се от основи /бази/ като алкокси анион, амиден анион и др.:
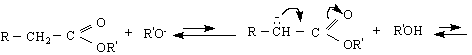
2/ Електрофилни заместителни реакции /SE Тези реакции протичат по-трудно, отколкото при арените поради деактивиращото действие на групата COOR, която е m-ориентант :
1/ Естери с плодова миризма - това са естерите на низшите и средни карбоксилови киселини с низши и средни алкохоли : напр.етилацетат, i-амилацетат, i-амилизовалерианат и др.
2/ Восъците са естери на висшите мастни киселини и висши алкохоли. Восъците биват : а)животински – пчелен, китайски насекомов, китов спермацет, вълнен восък ; б) растителен – карнаубов, планински и др.
3/ Мазнини. Те биват масти и масла. Мастите са твърди или полутвърди, а маслата - течни. В състава на мазнините има само един алкохол – глицерол. Така че те са глицериди на висшите мастни киселини. И те се разделят на животински (млечни масла, лой, свинска мас, рибено масло и др) и растителни (маслиново, слънчогледово, кокосово, памучно, ленено и др.)
4/ Фосфатиди или фосфолипиди - влизат в състава на човешкия организъм /съдържат се в мозъчната тъкан/. Биват естерни и ацетилни.
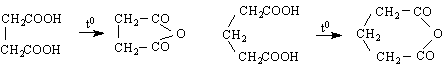
Наситени дикарбоксилови киселини
. По IUPAC наименованията се образуват от името на въглеводородите със същия брой С-атоми и се прибавя наставката “-диова киселина”. Много често обаче се използват и тривиални наименования. Напр.:
![]()
/оксалова киселина/
![]()
/малонова киселина/
![]()
/янтална киселина/
![]()
пентандиова киселина /глутарова киселина
![]()
/адипинова киселина
![]()
/пимелинова
киселина / 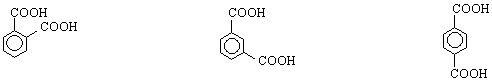
бензен-1,2-дикар- бензен-1,3-дикарбок- бензен-1,4-дикарбоксилова
боксилова к-на силова киселина киселина (терефталова )
(изофталова киселина)
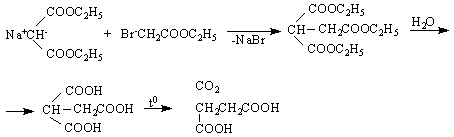
1/ Технически метод - от СО2 и натрий :
Na + CO2 + CO2 + Na ![]() NaOOC – COONa
NaOOC – COONa
KCN
ClCH COONa + KCN ![]() CNCH COONa
CNCH COONa ![]() HOOCCH COOH
HOOCCH COOH
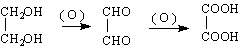
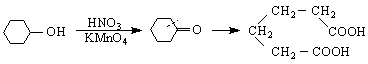
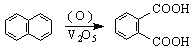
Във воден разтвор алкановите и ароматните дикарбоксиловите киселини претърпяват двустепенна дисоциация :
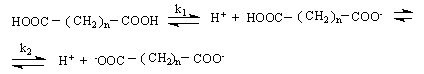
Най-общо дисоциационната константа к1 е двойно по-голяма в сравнение с ка на аналогичните монокарбоксилови киселини, тъй като едната СООН-група (като електроноакцепторен заместител ) благоприятства дисоциацията на другата. Дисоциацията на втората СООН-група обаче протича по-трудно /к2 е малка /, тъй като получения карбоксилатен анион (електронодонорен заместител) затруднява дисоциацията не само по индукционен път, но и чрез ефекта на полето.
А. Реакции на карбоксиловите групи - образуване на функционални производни : соли, хлориди, анхидриди, амиди, естери, нитрили.
Б. Реакции на въглеводородния остатък – проява на СН-киселинност или пък SE
НООС
- СООН![]() СО2 + НСООН
СО2 + НСООН ![]() СО + Н2О
СО + Н2О
НООС-СН2-СООН
![]() СО2 + СН3СООН
СО2 + СН3СООН
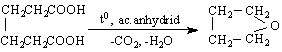
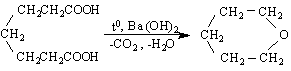
[ O (CH2)8CO ]n O (CH2)nCOOH
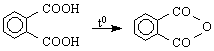
7/ Цикличните анхидриди проявяват същата реакционна способност, както и анхидридите на монокарбоксиловите киселини. Така напр.AcSN
където Y = ROH (RO-), H O OH NH NH
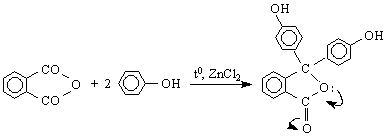
8/ При взаимодействието на фталов анхидрид с фенол в присъствие на ZnCl
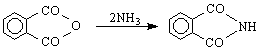 (
фталимида е
една
NH-киселина )
(
фталимида е
една
NH-киселина )
Имидите са по-силни NH-киселини от амидите поради електроноакцепторното влияние на двете карбонилни СО групи. Така имидите много лесно се бромират или метилират (имат подвижен Н-атом) :

. Намира се в спанака, доматите, гроздето и др. В киселеца (Oxalis acetocella) и в други растения тя е застъпена като кисела калиева сол. В стените на растителните клетки се намира неразтворимия Са-оксалат. В някои растения като лишеи, водорасли и гъби тази сол се намира в голямо количество (понякога 60% от сухото вещество напр.на лишеите ).У човека също се намират оксалати. Напр.в урината, а също и в камъните в пикочния мехур се намират калциеви оксалати. В организма оксаловата киселина се образува при окисление на много органични вещества (глюкоза, захароза, нишесте, целулоза и др.).
Ac malonicum Името си носи от това, че е била открита при окисление на ябълчена киселина (malum – ябълка). Намира се в захарното цвекло и в захарния сироп. Получава се от хлороцетна киселина и KCN с последваща хидролиза. Нейния диетилов естер /малонов естер/ се използва за много органични синтези.
/Ac.succcinicum/. Получава се от етиленбромид и две молекули KCN ; от малонов естер и бромоцетна киселина и последващо декарбоксилиране. Солите и естерите и се наричат сукцинати. Напр.сукцинимид.
. Тази киселина още при топене се превръща в анхидрид, който е важно изходно вещество за органични синтези. Напр.фталимидът, който се образува от фталов анхидрид и амоняк притежава NH-кисели свойства. Калиевият фталимид се използва за получаване на първични амини по метода на Габриел. Фталовата киселина и фталовия анхидрид са необходими за производството на багрила, полиестери и пластификатори. Естерите на фталовата киселина с по-висшите алкохоли са пластификатори за синтетични полимери.
Напр.адипинова
киселина + хексаметилендиамин
![]() полиамид
(найлон 66 ) ;
фталов
анхидрид + глицерол
полиамид
(найлон 66 ) ;
фталов
анхидрид + глицерол ![]() глифталов
полиестер
(глифталови
смоли) ; диметилтерефталат + етиленгликол
глифталов
полиестер
(глифталови
смоли) ; диметилтерефталат + етиленгликол
![]()
Последното вещество има т.т.2290 и се нарича захарин. Подобно на фталимида това е една NH-киселина / Н-атом може да се заменя с метали/. Захаринът е 500 пъти по сладък от захарта. Не се усвоява от организма, а се отделя с урината непроменен. В малки количества не е вреден за здравето
малиенова фумарова
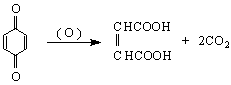
Двете киселини показват различни дисоциационни константи. Малеиновата киселина е по-силна от фумаровата, което се дължи на благоприятното разположение на карбоксиловите групи, които си влияят взаимно. Фумаровата киселина е по-стабилна поради транс-разположението на СООН-групите.
При каталитично хидриране по-лесно се хидрира малеиновата киселина. Същата и по-лесно дава анхидрид - малиенов анхидрид, който е важен изходен /или междинен/ реагент в синтетичната органична химия.
При хидриране двете киселини дават янтарна киселина, а при присъединяване на бромоводород - бромянтарна киселина.
Присъединяването на атом или атомни групи съм двойната С=С връзка става по различен начин. Напр.с KMnO , с който се вкарват две ОН-групи, малеиновата киселина дава мезо-винена киселина /цис-присъединяване/, а фумаровата - гроздена, т.е.D L
Малеиновата киселина се употребява в хранителната промишленост. Тя предотвратява гранясването на мазнини, масла и мляко на прах. Анхидридът на малеиновата киселина служи като диенофил в реакциите на Дийлс-Алдер или като изходно вещество за получаване на полиестери .
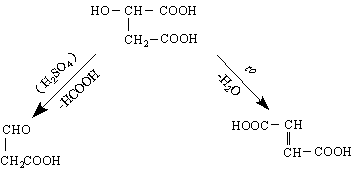
Наименованията се образуват по няколко начина. Според вида на въглеводородния остатък те биват алканови, алкенови, аренови и др. Според броя на хидроксилните групи могат да бъдат моно-, ди-, три- и т.н.хидроксипроизводни.А според отдалечеността на хидроксилната група от карбоксиловата група те биват α, β, γ, δ и т.н. По IUPAC α-хидроксипропионовата киселина е пропанол-2-кисделина, а β-хидроксипропионовата киселина е пропанол-3-киселина.
HOCH COOH CH CH OH COOH – млечна киселина.
1/ От цианхидрини - алдехиди и кетони се превръщат в цианхидрини, които се хидролизират до α-хидроксимонокарбоксилови киселини :
![]()
3/ От халогенхидрини на гликоли като им се действа с KCN и получения хидроксинитрил се хидролизира :
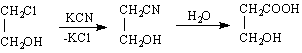
![]()
![]()
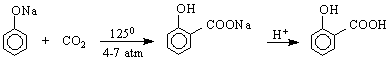
5/ Метод на Колбе - получаване на салицилова киселина. Реакцията се извършва чрез карбоксилиране на натриев фенолат с СО2 в автоклав :
Хидроксимонокарбоксиловите киселини са по-силни от съответните незаместени киселини поради влиянието на ОН-групата /има –I- От друга страна, поради -I-ефект на СООН-групата, хидроксилната група тук проявява по-голяма киселинност от тази на алкохолите и фенолите.
Химичните свойства се определят от реакционната способност на двете функционални групи - СООН и ОН-групата.
2/ β- Хидроксиалкановите киселини се дехидратират като реакции на елиминиране до α, β-ненаситени киселини :
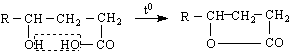
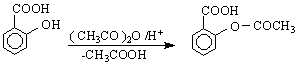
Хидроксиди- и хидрокситрикарбоксилови киселини
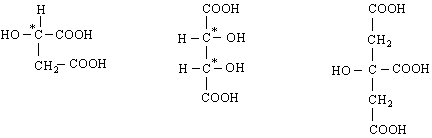
винена киселина лимонена киселина
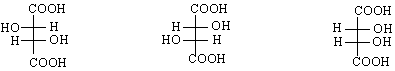
D (- )-винена киселина L мезо –винена киселина
Хидроксиди- и хидрокситрикарбоксиловите ктиселини имат аналогична реакционна способност както хидроксимонокарбоксиловите киселини. Те образуват функционални карбоксилови производни – соли, естери, анхидриди и др. Присъщи са им и реакции с едновременното участие на СООН-групата и ОН-групата. Солите на винената киселина се наричат тартарати. Киселият К-тартарат /моносолта/ се нарича винен камък (бакпулвер), а калиево- натриевия тартарат - сегнетова сол. Някои преходни метали (Сu Fe Ni
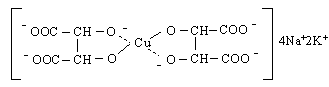
Реактив за алдехиди
При ябълчената киселина съществува т.нар.Валденово обръщане.При взаимодействие на S (-)-ябълчена киселина с фосфорен пентахлорид Валден (1896 г) е открил обръщането на конфигурацията при SN
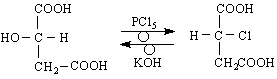
L- ябълчена D-хлорянтарна
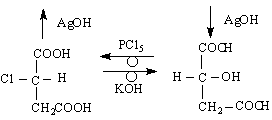
L-хлорянтарна D-ябълчена
. Хидроксикиселините образуват помежду си естери, наречени депсиди, които са природни продукти с дъбилни свойства. Тези свойства се изразяват в обработка на кожи с цел белтъчните им вещества да образуват водонеразтворими високомолекулни съединения. Според броя на молекулите фенолни киселини, те биват : дидепсиди, тридепсиди и т.н.
![]()
тридепсид
Когато се свързва 3,4,5-трихидроксибензоена киселина /галова киселина/ с втора молекула, получава се m-галоилгалова киселина :
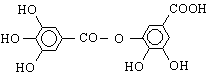
Трябва да се отбележи, че галовата киселина се навързва с ОН-групата на m-място спрямо СООН-групата. Този дидепсид на галовата киселина е в основата на съединения, наречени танини.
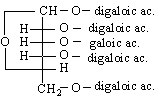
китайски танин
Оксомонокарбоксилови киселини /алдехид- и кетокарбоксилови киселини /
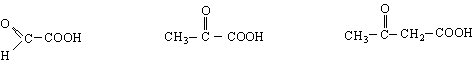
етаналова пропанонова 3-бутанонова
( глиоксалова ) ( пирогроздена ) ( ацетоцетна киселина)
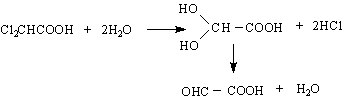
1/ Глиоксаловата киселина - чрез озонолиза на малеинова или фумарова киселина :
![]()
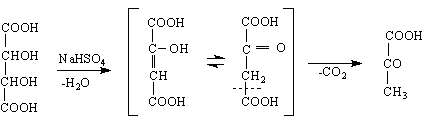
Освен това може да се получи по общия метод с ацетилхлорид, взаимодействие с KCN и последваща хидролиза :
![]()
![]()
Технически ацетоцетов естер се получава от дикетен и етилов алкохол :
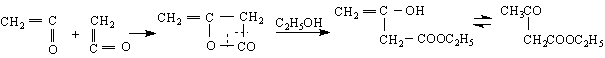 енолна
форма
енолна
форма
Оксокиселините са по-силни от съответните алканови киселини поради индукционното (-I
Химичните свойства се определят от реакционната способност на СООН- и СО-групата. Взаимното влияние на двете функционални групи обуславя и някои специфични свойства. Подобно на карбонилните съединения оксокиселините могат да образуват производни на С=О-групата - цианхидрини, оксими, фенилхидразони и др.
![]()
оксалова к-на гликолова к-на
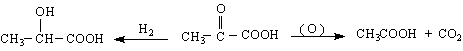
![]()
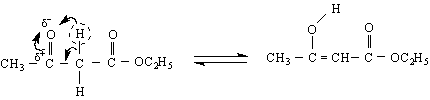
кето-форма енолна форма
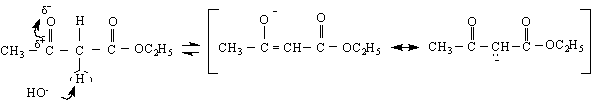
енолат анион кето-анион
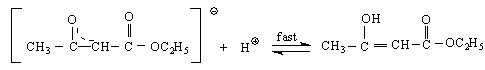
амбидентен анион енол
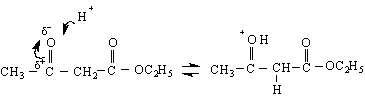
Върху устойчивостта на енолната форма оказват влияние главно два фактора : 1) Спрежението на С=С връзката с естерната С=О-група и 2) Образуването на стабилен шестатомен хелатен пръстен чрез вътрешномолекулна водородна връзка. Както бе споменато при дикетоните, тук стабилизирането се извършва поради разсредоточаване на електронната плътност вътре в шестатомния пръстен :
цис-енолна хелатна форма
Немският учен Лудвиг Кнор (1911 г) доказал наличието на двете форми, т.е.той опитно е установил съществуването на кето-енолната тавтомерия : енолната форма реагира с PCl5 като замества ОН-групата с халогенен атом, реагира и с FeCl3 и дава положителна реакция за хидроксилна група при двойна връзка /червено-виолетово оцветяване/, реагира с бром /бромира се двойната връзка/, взаимодейства с метален натрий и дава енолати ; кето-формата дава с циановодород цианхидрини, с кисел натриев сулфит – бисулфитно съединение, с хидроксиламин - оксими.
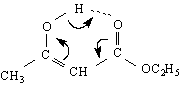
Реакционната способност на ацетоцетовия естер се проявява чрез т.нар.ацетоцетови синтези - реакции, определящи се от СН-киселинните му свойства. Под действието на протоноакцептори (бази, металиращи агенти) се получава карбанион , който може да реагира напр.с алкилиращи R X RCOX реагенти :
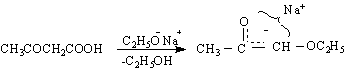
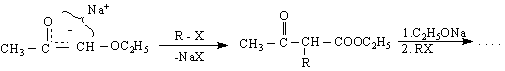
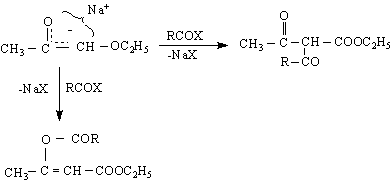
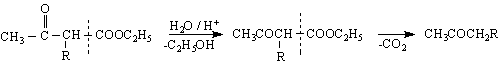
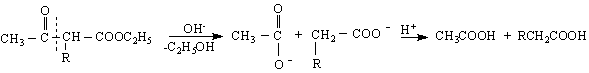
киселинно разпадане
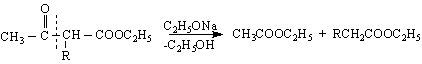
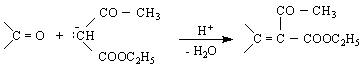
АЗОТСЪДЪРЖАЩИ ОРГАНИЧНИ СЪЕДИНЕНИЯ
Отначало ще разгледаме някои основни групи ациклени /нециклични/ азотсъдържащи съединения. В тях азотът може да бъде тривалентен или четиривалентен положително зареден /т.нар.”амониево” валентно състояние/. Основното валентно състояние на азотния атом в неговите съединения е : 2s px py pz При хибридизацията трите р-електрона образуват връзките, а остават свободни двата s-електрона / Помнете: при азотния атом винаги има два свободни електрона/. Валентните връзки на азота в неговите съединения преминават в sp -хибридно състояние - формират се 4 sp
Органични нитросъединения
Това са съединения, които съдържат нитрогрупата NO
CH3NO2 – нитрометан, CH CH(NO2)CH3 – изонитропропан
Следва да се приеме, че в нитрогрупата азотния атом е четиривалентен с (+) заряд поради образуването на семиполярна връзка с единия О-атом. С другия О-атом връзката трябва да бъде двойна. Спектрални и рентгеноструктурни данни обаче са показали, че структурата на групата NO O N O , а дължината на двете N-O връзки е еднаква, около 0.120 нм. Това може да се обясни с допускането на sp NO
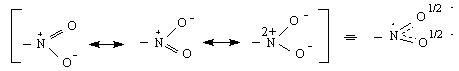
Тази електронна структура на нитрогрупата обяснява и силния и електроноакцепторен характер / -I и –M
Нитропарафини се образуват в малки количества при пряко взаимодействие на азотна киселина и съответния парафин, при което NO -групата отива при вторичен или третичен С-атом. Трябва да се отбележи, че общо взето концентрираната азотна киселина действа върху парафините като окислител, но разредена /~12%/ азотната киселина под налягане действа нитриращо /метод на Коновалов/ :
![]()
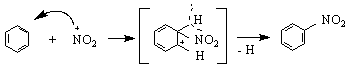
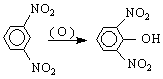
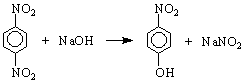
Нитрирането при аломатните съединения е много по-лесно. Използва се т.нар. “нитрирна смес” :
![]()
Реакцията е една електрофилна субституция SE N O
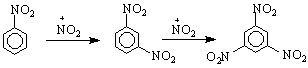
Нитрирането продължава по-нататък като групата NO ориентира другите нитрогрупи на m-място /тя е m-ориентант/. Най-големият брой нитрогрупи, които могат да встъпят в бензеновото ядро е 3, като третата нитрогрупа влиза много трудно чрез дирактно нитриране поради пространствено пречене.
Димяща азотна киселина при хомолозите на бензена може да действа заместително и в страничната верига. Напр.така се получава C H CH NO
R-I + AgNO ![]() RNO + AgI
RNO + AgI
![]()
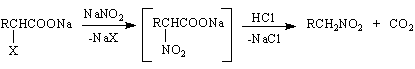
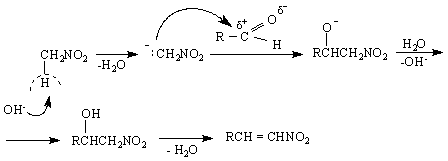
Нитропарафини се образуват като на воден разтвор на α-халогенирани мастни киселини се действа с натриев нитрит NaNO
Те са полярни съединения и имат сравнително високи т.к. и специфични тегла.Високата температура на кипене се обяснява с високия диполен момент (μ≈3.5 – 4 D )
1/ Реакциите с участието само на NO

![]()
2/ Основните отнасяния на първичните и вторичните нитросъединения се дължат главно на подвижността на α-водородния атом, т.е.на тяхната СН-киселинност. Това се дължи на силния електроноакцепторен ефект на нитрогрупата. Ще илюстрираме този факт с един пример.Установено е, че 2-нитропропана, който практически е почти неразтворим във вода, поддържа киселинност на водата рН = 4.3. Следователно трябва да се приеме, че става изамеризиране вследствие на прескачане на един Н-атом като се образува съответната тавтомерна форма, която съдържа хидроксилна група - годна да се дисоциира и да дава соли. :
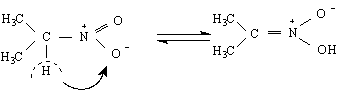
нитронова киселина
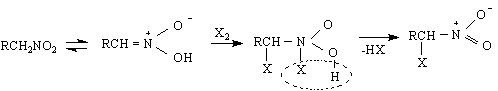
Тъй като полученото съединение съдзържа още един α-водороден атом, то халогенирането може да продължи. Така напр.бойната отрова хлорпикрин Cl3CNO
4/ Друга реакция, която се дължи на активиращото действие на нитрогрупата, е нитрирането на първичните и вторични нитропарафини с азотиста киселина HNO
![]()
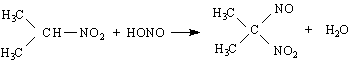
нотьол
По същия начин динитробензените взаимодействат с NH като се измества едната група с аминогрупа, а таках също и с натриев метилат CH ONa NO
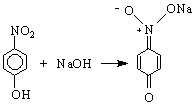
Тя е една силна киселина. Във воден разтвор е силно дисоциирана. Киселината е силен експлозив с бризантно действие. С органични основи пикриновата киселина образува трудноразтворими соли, наречени пикрати. Тези соли се използват в аналитичната органична химия за охарактеризиране на някои съединения .
Амините са най-важните съединения на азота. Ще разгледаме засега относително по-простите амини - алкиламините и ариламините.Амините могат да се разглеждат като производни на амоняка, т.е.като алкил- или арилзаместени амоняци.
Класификацията на амините съответства на броя на алкиловите или арилови групи, свързани с азота. Така че амоняк, в който само един водороден атом е заместен с алкилова или арилова група, се нарича първичен амин ; ако са заместени два Н-атома - вторичен амин ; при третичните амини са заместени всичките три водородни атома на амоняка.с алкилови (арилови) групи. Още един тип съединения влизат в тази класификация - четвъртичните амониеви соли / R N X
Названията на алкиламините се образуват от името на алкиловата група, която е свързана с азотния атом + думата “амин”. Словосъчетанието е едно, т.е.пишат се заедно. Напр :

метиламин метилетиламин триетиламин метиламониев хлорид
първичен/ /вторичен/ /третичен/
По аналогичен начин се образуват имената и на ариламините, като почти всички могат да се разглеждат като производни на анилина - най-простия ароматен амин.
NH
NH CH CH OH - аминоетанол или пък
![]() - р-аминобензоена
киселина.
- р-аминобензоена
киселина.
а) редукция на нитросъединения :
![]()
б/ редукция на нитрили :
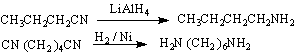
1,6-хексаметилендиамин
![]()
фенилацетонитрил β-фенилетиламин
/бензилцианид/
в/ редукция на амиди :
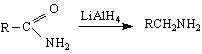
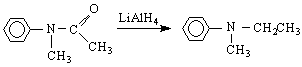
N-метилацетанилид N-метил-N-етиланилин
г/ редукция на оксими :
![]()
CH CH J + NH ![]() CH CH NH J
CH CH NH J
CH CH NH J + NH ![]() CH CH NH + NH J
CH CH NH + NH J
![]()
![]()
3/ Синтез на Габриел - използва се фталимид. Алкилирането на калиевия фталимид с алкилхалогениди дава N-алкилфталимиди, които могат лесно да се хидролизират до съответния амин :
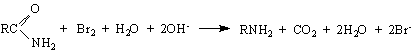
Метиламините са лесновъзпламеняващи се газове с мирис на амоняк. Характерния мирис на риба се обуславя от присъствието на по-високомолекулни амини. Т.нар.”трупна миризма” в значителна степен се предизвиква от наличието на два диамина - путресцин /putrescere се, гниещ/ и кадаверин /cadaverosus – трупен/. Путресцина и кадаверина са отрови и спадат към групата на токсичните амини, наречени птомаини. Отравянето, предизвикано от развалено месо, се дължи на наличието на птомаини.
Най-характерните химични свойства на амините се определят : 1) от тяхната основност и нуклеофилност и 2) от тяхната NH-киселинност. Тези свойства характеризират реакционната им група - аминогрупата, NH
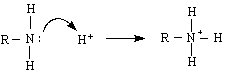
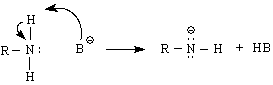
R NH < ( R NH < ( R N
Това се дължи на +I ефектна алкиловите заместители, което води до увеличаване на електронната плътност при азотния атом. Алкиламините са по-силни основи от амоняка, от алкохолите, от етерите, а така също и от ароматните амини. Последното се обяснява с теорията на резонанса. Извършва се делокализация на свободния чифт електрони при азотния атом в бензеновото ядро :
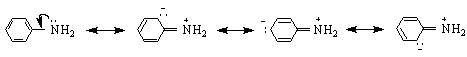
NH RNH < ArNH . Това се обяснява с влиянието на същите електронни фактори, които в случая благоприятстват по-добрата NH-киселинност при ариламините.
присъединителни и заместителни реакции със съединения, съдържащи С=О, N=O и др.групи ;
![]()
по-слаба
основа основа
![]()
по-силна по-слаба
основа основа
, която определя степента на пренос на протон от киселината към водата при дисоциация. По аналогия и при амините за удобство и за сравнение се определя степента на пренос на протона от водата към основата /амина/. Константата на равновесие на тази реакция се нарича константа на основност Кb
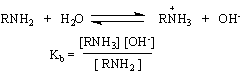
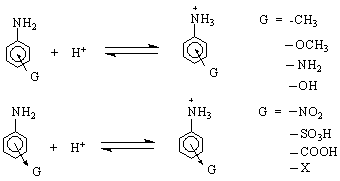
Колкото Кb
Установено е, че електронодонорни заместители от типа –СН3 увеличават основността на анилина, а електроноакцепторни заместители от типа на халогените Х или NO - намаляват основността.Тези ефекти са напълно понятни.Подаването на електрони способства за разсредоточаване на положителния заряд на анилиниевия йон ArN H
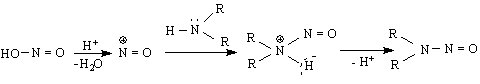
1/ Амините, освен с различни киселини /НХ/, образуват соли и с полярни реагенти /R X /. С алкилхалогенидите взаимодействието може да протече като SN
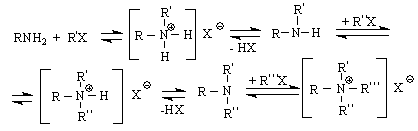
![]()
N
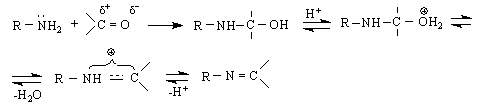
азометинов азометинови
катион съединения
Ацилирането на амини протича като реакция АсSN
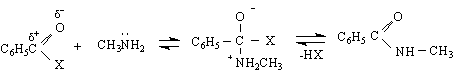
N
![]()
ацетанилид
![]()
Тук е мястото да се отбележи, че заместване може да се извърши и в бензеновото ядро при ароматните амини. Групите NH NHR NR са мощни активатори и о- и р-ориентанти при реакциите на електрофилно заместване ( SE роматното ядро. Ацетамидната група /NHCOCH
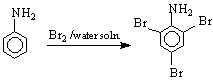
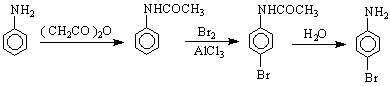
Ако обаче реакцията се проведе като халогениране по Фридел-Крафтс в присъствие на AlCl , тогава е необходимо аминогрупата да се защити, тъй като ако е свободна тя ще реагира с Люисовата киселина /AlCl
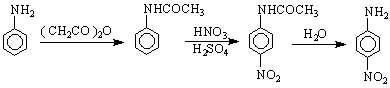
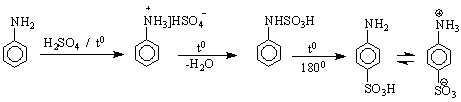
анилинсулфат фенилсул- сулфани- бетаинова
фаминова к-на лова к-на структура
3/ Първичните алкил-и ариламини взаимодействат с азотиста киселина в кисела среда.. Реакцията се нарича диазотиране. Образуват се диазониеви соли. Тези соли, получени от първични амини са нестабилни, а от първични ариламини - сравнително по-стабилни :
![]()
![]()
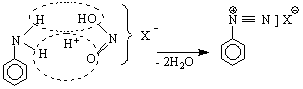
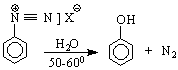
N
![]()
N
![]()
N-диалкилхидроксиламин
![]()
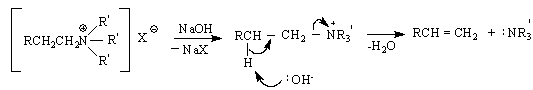
По-интересен е случая с аминооксида, който съдържа лабилен /подвижен/ β-водороден атом и при нагряване до около 1500 се разлага до олефин и производно на хидроксиламина :
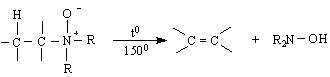
. Получават се при нагряване на амониев хлорид със съответното количество формалдехид. В промишлени условия се получават от амоняк и метанол при 4500 под налягане и над катализатор Al O
а/ без
неорганичен
остатък с
обща формула ![]() ,
чиито
представителе
диазометана CH N N ]
,
чиито
представителе
диазометана CH N N ]
с неорганичен остатък ( Y = OH, X ) с обща формула
![]()
диазониева сол
Първият тип реакции / с отделяне на N
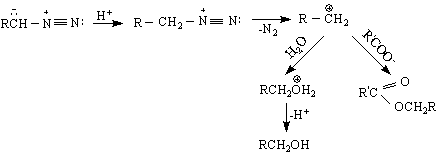
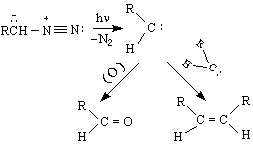
N или CH N N N N
Напоследък засилено развитие търпи биохимията - гранична наука, изучаваща явленията в живите организми чрез химични методи. Възникна и нова наука биоорганична химия - област от органичната химия, изучаваща химичните основи на жизнените процеси. Обект на биоорганичната химия са органичните съединения, на чиято основа се гради животът. Напр.аминокиселините, ензимите, протеините, нуклеиновите киселини и др.
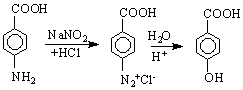
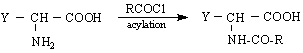
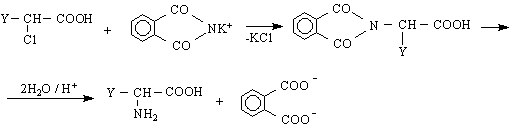
Карбоксилови киселини, във въглеродния остатък на които се съдържа аминогрупа /-NH Следователно техните функционални групи са СООН и NH
Класификацията може да се изгради по различни структурни признаци : вида на въглеводородния остатък, броя на СООН- и NH Така че имаме различни групи аминокиселини. Важна група са моноалканкарбоксиловите аминокиселини. Те биват различни според положението на двете функционални групи в алкановата верига : 2- или α-аминокарбоксилова киселина, 3- или β-аминокарбоксилова киселина, 4- или γ- и т.н.
![]()
3- аминокарбоксилова киселина
/ α – аминокиселина / /β – аминокиселина /
![]()
където Y = алкил, фенилметил, хидроксил, аминоалкил, карбоксиметил, меркаптометил и др.
Днес са известни повече от 20 такива аминокиселини. Наименованията /освен тези по IUPAC/ са обикновено тривиални, които се използват по-често. Тривиалните им наименования са свързани с някои техни свойства или произхождат от наименованията на продуктите, от които са били изолирани. Напр.глицин от гр.glykys сладък, левцин - от гр.leukos – бял /от белтъка на млякото/, цистин – от гр.kystis
Всички α-аминокиселини, освен първата /глицин, аминооцетна киселина/, имат асиметричен С-атом и притежават по два енантиомера D и L /или R и S / :
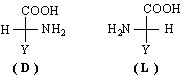
В природата са разпространени L-енантиомерите на α-аминокиселините. Стойностите на специфичното им въртене варират в широки граници. Аминокиселините, съдържащи хетеропръстен /хистидин, триптофан, пролин и др./ имат значителна отрицателна стойност на специфично въртене.
α – Аминокиселините са амфолити /в разтвор/, а иначе са амфотерни - те притежават киселинни /-СООН / и основни / -NH
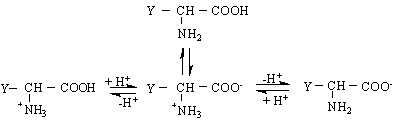
амониева сол биполярен йон аминокарбоксилат
/ киселина/ /основа /
рКа ~ 2 рКа ~ 9
В кисела среда /рН < 7 / аминокиселините се отнасят като киселини, а в алкална среда / рН > 7 / - като основи. При определени стойности на рН всяка аминокиселина съществува само като биполярен йон. Тогава казваме, че се достига т.нар.изоелектрична точка . В областта на изоелектричната точка разтворимостта на аминокиселините е най-ограничена. В изоелектричната точка аминокиселината не се движи между катода и анода в електролизна вана.
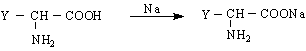
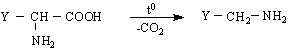
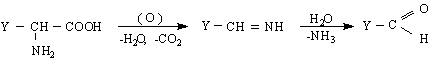
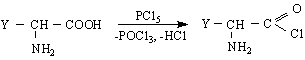
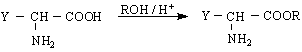
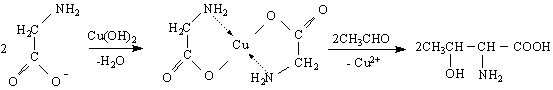
Освен тези реакции трябва да се знае, че α-аминокиселините образуват стабилни хелатни комплекси с преходни метали. Този вид комплексообразуване се използва в количествения анализ на аминокиселините, а така също и за синтетични цели. Така напр.от Cu/II
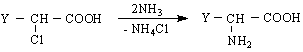
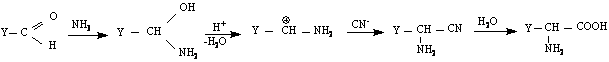
От α-кетокиселина като се мине през оксим, който се хидрира :
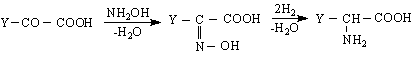
Хетероциклени се наричат органични съединения, чиито молекули са изградени от свързани в пръстен освен въглеродни атоми и атоми на други елементи. Молекулните пръстени на тези съединения се наричат хетеропръстени /хетероцикли./, а атомите на съответните елементи - хетероатоми.
От множеството хетероциклени съединения най-добре са изучени тези, съдържащи в молекулните си пръстени О-, N- и S-. С някои представители на тези съединения вече се запознахме : етиленов оксид, диоксан, лактоните, лактимите, анхидридите и др. Пръстените на тези съединения обаче са нетрайни, докато хетероциклените съединения, които предстои да разглеждаме, притежават стабилна устойчивост. Ще разгледаме кислород-, азот- и сярасъдържащи хетероциклени съединения главно с ароматна структура. Много от тези съединения имат важно значение за биологията, медицината, фармацията и селското стопанство.Така напр. в състава на жизнено важни биологични обекти като кръв, нуклеинови киселини и др. участват хетероциклени съединения. Почти цялата фармацевтична химия е химия на хетероциклите.
- по общия брой на атомите в пръстена, т.е.вида на цикъла - триатомни, четириатомни /четиричленни/, петатомни /петчленни/ и т.н.
- по вида и броя на хетероатомите в пръстена - един, два, три и т.н.кислородни, азотни, серни или др.атоми в пръстена ;
- по броя на пръстените в молекулата - еднопръстенни /моноциклични/, двупръстенни /бициклични/ и т.н.хетероциклени съединения.
Повечето хетероциклени съединения имат тривиални наименования. За еднопръстенните /моноциклични/ съединения номенклатурата на IUPAC въвежда правила, с които се определят наименованията, от които се вижда вида на хетероатома в пръстена, неговата големина и “ненаситеност”(броя на двойните връзки в пръстена). Хетероатомите се означават в наименованията на пръстените с представките : окса- за О-атом, тиа- за S-атом и аза- за N-атом, съответно диокса-, дитиа- и диаза- за два еднакви хетероатома. Ако атомите са различни тогава : оксаза- за кислород и азот, тиаза- за сяра и азот и т.н.
S а той пред N-ат. Номерирането при няколко хетероатома се извършва така че те да получат възможно най-малки номера. Напр. :
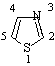
1,3 – тиазол
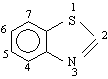
бензотиазол
Когато и двата пръстена са хетероциклени, предимство в избора на основното наименование има N-съдържащия, пред О- и S-съдържащите хетеропръстени. Ако двата хетеропръстена имат ръзличен брой хетероатоми, основното наименование дава този с повече хетероатоми.
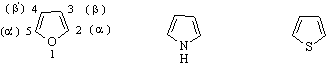
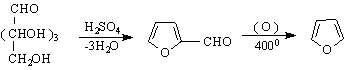
фуран пирол тиофен
Установено е, че в пръстените на трите съединения всички атоми лежат в една равнина, т.е.имаме копланарност. Въглеродните атоми са в sp -връзките) заедно с р-орбиталата на свободната двойка електрони при хетероатома образуват π-електронен секстет, подобно на бензена. Така че четирите π-електрона + р-електрона на хетероатома правят съединението ароматно.
![]() където Х = NH, O, S
където Х = NH, O, S
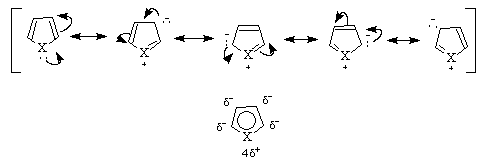
Фуранът има 0.8 D, тиофенът – 0.5 D и пиролът – 1.8 D диполни моменти. ЛКАО-МО метода показва, че най-голям
Както се вижда, почти всички гранични структури са биполярни и в резонансния хибрид електронната плътност при С-атомите е повишена. Това е причина за появата на диполен момент в молекулата за разлика от бензена. Различната електроотрицателност на кислорода, азота и сярата се отразява върху диполния момент и върху стойността на енергията на резонансната стабилизация /Е/. От тези стойности може да се подредят трите хетероциклени съединения по реда на нарастване на тяхната ароматност фуран < пирол < . Но общо взето ароматния характер на тези три съединения е по-слабо изразен от бензена, който има Е ~ 150 кДж/мол /фурана – 80, пирола- 110 и тиофена – 120 кДж/мол /.
Различието в ароматния характер между фурана и пирола се дължи на по-голямата електроотрицателност на О-ат .в сравнение с N-ат., в резултат на което свободната двойка електрони при О-ат.е “локализирана” в по-голяма степен при него и по-малко ефективно участва в π-спрежението.

Трите съединения са течни : фуранът с т.к.310С, тиофенът с т.к.810С и пиролът с т.к.1290С.
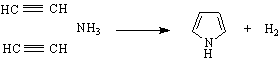
1.Създаден е промишлен метод /на Юрьев/ за получаване на пирол и тиофен чрез пропускане на фуранови пари и NH и H S Al O
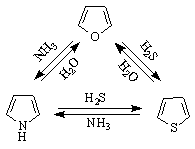
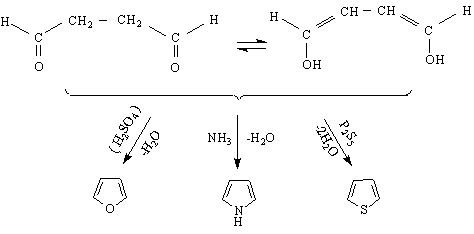
![]()
Химичните свойства на фурана, тиофена и пирола са три вида : 1/ Основни и киселинни ; 2/ Ароматни - участие в S Е-реакции, подобно на бензена и 3 / Свойства на ненаситени съединения /присъединителни реакции /.
1.Основните свойства на тези съединения се определят от наличната свободна двойка електрони при хетероатома. При N-атома в пирола липсва свободна двойка електрони. Оказва се обаче, че ясно изразени основни свойства притежават фурана и пирола /след нарушаване на π-електронния секстет/1 докато тиофена практически не проявява такива свойства. Основността на фурана и пирола е сравнително слаба. Те присъединяват Н+ при хетероатомите, или при α-С-атом след нарушаване на π-електронния секстет :
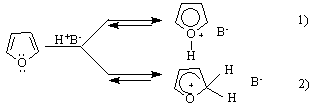
2. Киселинни свойства. И трите съединения проявяват киселинни свойства. Пиролът се отнася като NH-киселина (рКа~15), а фуранът и тиофенът - като сравнително слаби СН-киселини :
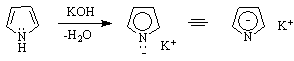
Пиролът е по-силна киселина от NH RNH
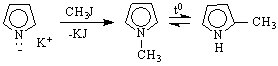
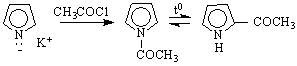
3. За тези съединения са характерни SE Те протичат чрез междинно получаващ се σ-комплекс /-ониев йон/. Реакционната способност на трите съединения спрямо електрофилни реагенти е по-голяма в сравнение с тази на бензена. Това се побяснява с повишената електронна плътност при С-атомите в пръстените на трите съединения, особено на α-място. Поради това при SE
![]()
2,5-дибромофуран
![]() тиофенсулфонова
киселина
тиофенсулфонова
киселина
![]() α-нитротиофен
α-нитротиофен
![]() ацилтиофен
ацилтиофен
![]()
Към π-системата на трите съединения могат да се присъединят различни съединения, напр.Н2 /вж.по-горната схема/, молекула халоген, напр.Br
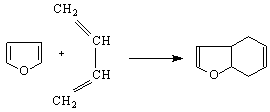
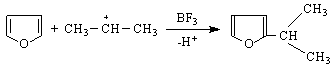
6. При окисление на фуран с въздух в присъствие на V O
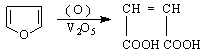
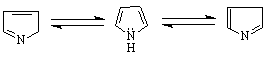
фенилазопирол дифенилдиазопирол
Друг природен продукт, който съдържа в молекулата си тетрахидротиофен е
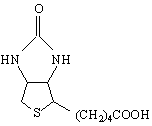
/вит.Н/. Той се среща в жълтъка на яйцето и в черния дроб. Необходим е за растежа на микроорганизмите. Има три хирални атома и съществува само в цис-положение, тъй като транс-кондензирането на двата пръстена създава голямо напрежение. Участва в организма в процеса карбоксилиране /вкарване на СООН /. Тази карбоксипреносителна роля се подпомага от АТФ и АДФ.
Според вида на хетероатомите съединенията от тази група са различни видове. Когато единия от хетероатомите е N, адругите са N, O или S, то групата се нарича азолова група . Към нея принадлежат следните съединения :
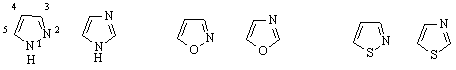
пиразол имидазол изоксазол оксазол изотиазол тиазол
Получаване
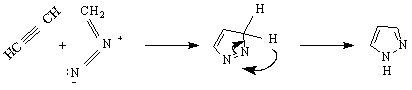
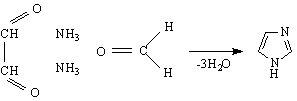
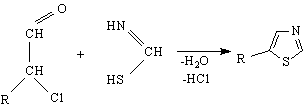
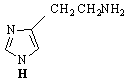
От азолите по-голям интерес представляват тези, които бяха дадени при методите за получаване - пиразола, имидазола и тиазола. Азолите имат свойства на основи. Основността им се дължи на свободната двойка електрони при азотния атом. Тиазола и пиразола имат приблизително еднаква основност, а имидазола - малко по-голяма.
Пиразола и имидазола са NH-киселини /подобно на пирола и притежават аналогични свойства./.
Пиразола, имидазола и тиазола са по-слабореактивоспособни при SE В резултат на това електронната плътност при С-ат.в пръстена е намалена - при тях по-лесно протичат SN
Електрофилните заместителни реакции /SE
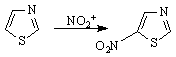
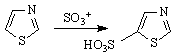
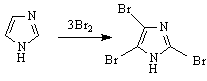 /
извършва се
пълно
халогениране
/
/
извършва се
пълно
халогениране
/
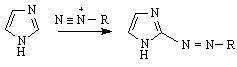
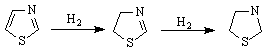
тиазолин тиазолидин
SN
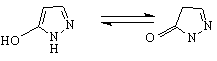
5-пиразолон
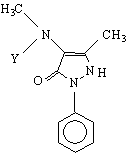 когато
Y = CH e
когато
Y = CH e
когато Y = CH SO Na e
Производни на тиазола са ефикасните лекарствени препарати сулфазоли и пеницилини :
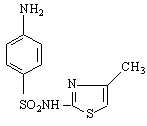 сулфатиазол /ултласептил,
сулфазол /
сулфатиазол /ултласептил,
сулфазол /
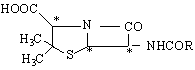
β-лактимен пръстен /
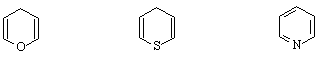
пиран тиопиран пиридин

Петте С-атома и азота са в sp Електроните на р-орбиталите на С-атомите и N-ат.образуват π-електронен секстет, а на sp Поради по-голямата електроотрицателност на N-ат π-електронната плътност в пръстена е частично локализирана към него. Диполен момент μ = 2.3 D.
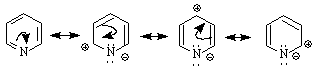
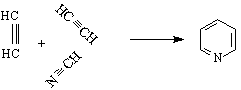
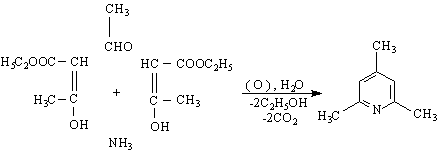
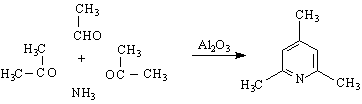
Пиридинът проявява основни свойства (рКа ~ 9 ). Със силни киселини образува соли :
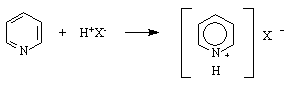 пиридиниева сол
пиридиниева сол
Реакциите SE
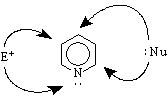
Реакцията SN
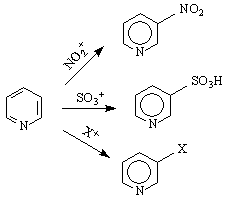
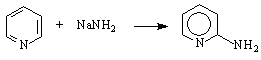 2-аминопиридини
2-аминопиридини
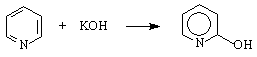
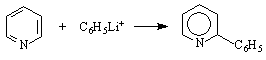
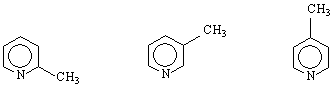
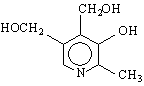
α – пиколин β – пиколин γ – пиколин
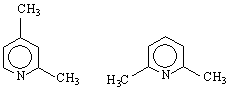
2,4- и 2,6-лутидини
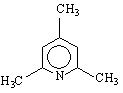
2,4,6 – колидин
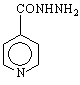
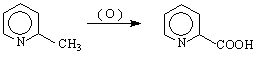 пиколинова
киселина
пиколинова
киселина
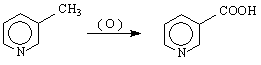
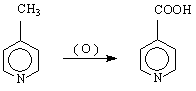
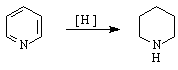
- амидът на тази киселина е съставна част на дехидриращия ензим кодехидраза или коцимаза / NAD NADH
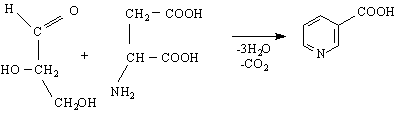
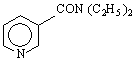
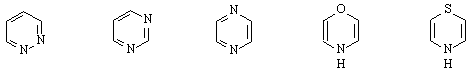
пиримидин пиразин оксазин тиазин
/1,2-диазин/ /1,3-диазин/ /1,4-диазин/
В сравнение с пиридина наличието на втори N-атом в диазините понижава още повече електронната плътност при С-атоми в пръстена. Така напр.в пиримидина с най-ниска плътност са С-атоми на 2-, 4- и 6-място :
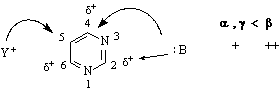
Така, че пиримидина е значително реактивоспособен в нуклеофилните заместителни реакции /SN Електрофилните заместителни реакции /SЕ/ протичат с атака към С5-място. При това реакционната способност на пиримидина е по-малка от тази на пиридина.
Диазините могат да участват и в присъединителни реакции, напр.хидриране, при което се нарушава ароматната структура.
По-голям интерес представляват пиримидиновите производни, тъй като участват в състава на биологични вещества - нуклеинови киселини, витамини и др.
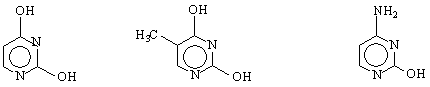
2,4-дихидрокси- 2,4-дихидрокси-5-метил- 2-хидрокси-4-амино –
пиримидин пиримидин пиримидин
/ урацил/ /тимин / /цитозин /
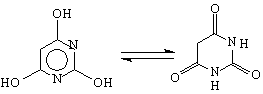
барбитурова к-на кето-форма
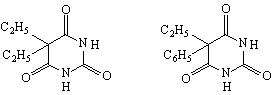
веронал луминал /
Недостигът на вит.В1 уврежда нервната система /полиневрит/ и причинява болеста бери-бери. Установеният лечебен ефект на оризовите люспи за тази болест е дал основание да се въведе понятието “витамин” /от vita – живот/ от Функ.
Производно на пиримидина и по-точно на тимина е лекарството азидотимидин /AZT /, което в момента е най-мощното анти-HIV лекарство. Това лекарство модифицира части на ДНК.

бензофуран индол тионафтен
/кумарон/
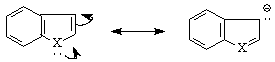
Така, че на това място /β / протичат характерните за ароматните съединения реакции /халогениране, нитриране, сулфониране/. Известни са и хидроксилни производни, напр.на индола :
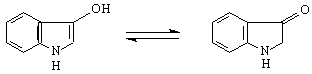
3-хидроксииндол кето-форма
/индоксил/
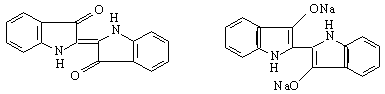
бяло индиго /левкоиндиго/
Indigoferia tinctoria Isatic tinctoria ![]()
![]()
![]()
Добива се от охлюва Murex brandaris. Около 10 000 охлюва съдържат 1.5 г безцветно вещество, което на въздуха се окислява до пурпурночервено багрило.
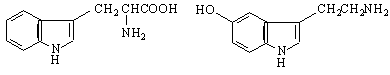
Друго производно на индола е псилоцина/4-хидрокси-N,N’
Това е психотропно вещество, изолирано от мексиканската “вълшебна” гъба Teonancatl
Други индолови алкалоиди са стрихининът и LSD /диетиламид на лизергиновата киселина / :
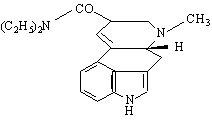 LSD
LSD
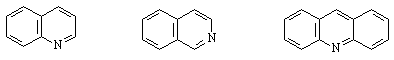
хинолин изохинолин акридин
Молекулите и на трите съединения съдържат пиридинов пръстен, кондензиран с бензенови ядра. Те имат по-слабо изразен ароматен характер от този на бензена и пиридина, а тяхната основност е сходна с тази на пиридина, т.е.те са силни бази. В тези съединения електронната плътност в бензеновите ядра е по-голяма от тази в пиридиновото ядро. Това означава, че SE итано в бензеновите ядра, а SN-реакциите - в пиридиновото ядро.
При SE
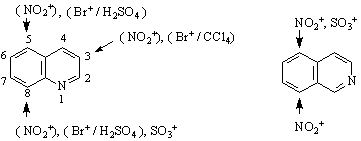
Протичат и SN
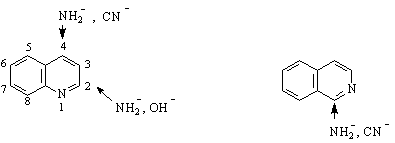
При окисление и трите съединения дават N-оксиди. При по-нататъшно окисление с KmnO
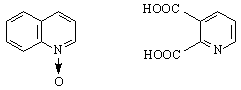
хинолин-N хинолинова киселина
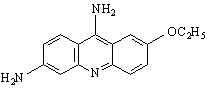
риванол
И трите съединения се намират в каменовъгления катран, откъдето могат да се изолират. Иначе се получават /напр.хинолина/ синтетично – по метода на Скрауп от анилин и акролеин :
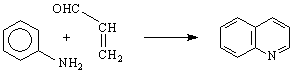
Пуринът се състои от петчленен и шестчленен пръстени с по два азотни хетероатома. Единият пръстен /петчленния/ е имидазолов, а другия /шестчленния / - пиримидинов :
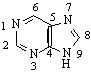
Тези съединения имат ароматен характер, обусловен от π-електронния децет / 10 π-електрона/ в молекулата им, подобно на нафталена.
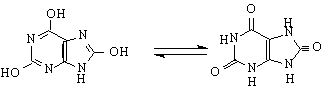
енолна форма кето-форма
Пикочната киселина е краен продукт от белтъчната обмяна в организма на птиците. Тя е и изходен продукт за получаване на други производни на пурина. Чрез хлорирането и с POCl се получават 2,6,8-трихлорпурини, чиито хлорни атоми се заместват лесно с нуклеофилни реагенти по SN
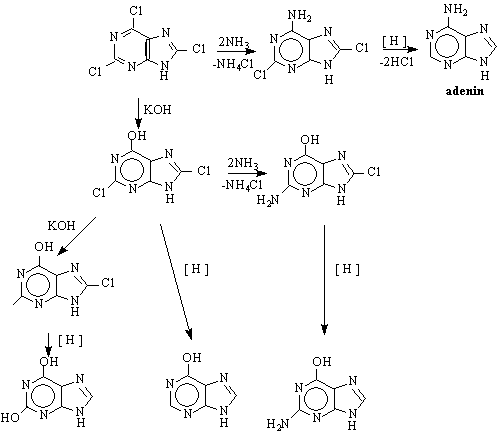
ксантин хипоксантин гуанин
/2,6-дихидрокси- /6-хидроксипурин / /2-амино-6-хидрокси-
пурин / пурин /
Тези пуринови производни съществуват в две форми - енолна (хидрокси-формите) и кето-форми, подобни на пикочната киселина. Заместителни реакции с участието на фенолатните аниони на тези производни водят до получаването на О- или N-заместени продукти. Напр.N-производните на ксантина са следните съединения
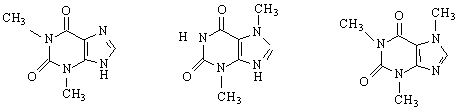
теофилин теобромин кофеин
/1,3-диметилксантин / /3,7-диметилксантин/ /1,3,7-триметилксантин/
Теоброминът се съдържа в какаовите зърна (teo-бог , broma- храна) в количество до 1.8%. Действа по-силно диуретично от теофилина.
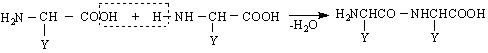
Характерна функционална група в молекулите на пептидите е амидната група СО – NH - , наречена още пептидна връзка.
В зависимост от броя на α-аминокиселинните остатъци в молекулите на пептидите те биват : олигопептиди - дипептиди, трипептиди,декапептиди и полипептиди – с повече от 10 аминокиселинни остатъци. Дългите повтарящи се пептидни връзки –СО-NH- построяват непрекъсната верига, наречена протиенова основа /база/. Общоприето е пептидите винаги да се изписват като N-терминални аминокиселини отляво и като С-аминокиселини отдясно.
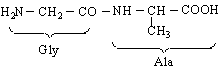 глицилаланин
глицилаланин
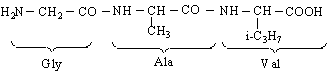
Според данни от рентгеноструктурния анализ пептидните вериги имат плоска /планарна/ конфигурация. Въглеродният атом в амидната група е в sp
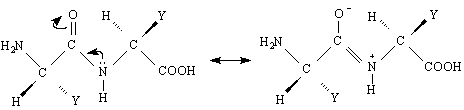
Оказва се, че пептидната връзка е по-малка от простата С-N-връзка /0.132 и 0.147 нм съотв./, което е резултат от р-π-спрежението в амидната група. В молекулната верига разположението на двата α-С-атома при всяка пептидна връзка са в транс-положение по отношение на амидната С-N-връзка.
В резултат на спрежението в амидната връзка, около простата С – N-връзка ще има ограничено въртене.
Накрая трябва да се изясни последователността на свързване на аминокиселините, т.е.да се покаже структурната формула на пептида.За целта се използва комбинация от два метода - определяне на крайните групи и частична хидролиза.
N N N
N N
- защита на някои от NH и СООН-групи в изходните аминокиселини. Това е необходимо по следната причина : ако проведем кондензация, напр.между два вида аминокиселини, могат да се получат 4 различни дипептида ; за получаване само на единия от тях /съдържащ остатъци от двете аминокиселини/ трябва да защитим по една от функционалните групи /NH NH
![]()
![]()
- активиране на N
![]()
Z Cl
- образуване на пептидната връзка между една защитена и една активирана аминокиселина.
- отцепване на защитните групи главно чрез хидролиза.
Cl
NH
Белтъчни вещества /протеини и протеиди /
Белтъчните вещества - това са високомолекулни природни съединения, молекулите на които са изградени главно от α-аминокиселинни остатъци, свързани с пептидни връзки. Белтъчните вещества, чиито молекули имат само пептидна структура и молекулна маса над 10 000, се наричат протеини. Белтъчни вещества, чиито молекули са изградени от полипептидна част и други (органични или неорганични ) остатъци, наречени простетични групи, се наричат протеиди. За живата клетка тези вещества имат първостепенно значение /от гр.protos пръв/. Молекулната маса на белтъчните вещества /протеини и протеиди / варира в широки граници - от 10 000 до 50 млн.
белтъчни вещества - неразтворими във вода, с нишкообразна, влакнеста /фибрилна/ структура ;
белтъчни вещества - лесноразтворими във вода, със сферична, кълбообразна форма.
Фибрилните белтъчни вещества са основен строителен материал на живата тъкан /както целулозата при растенията /. Общото им наименование е склеропротеини Например : кератина /роговото вещество / се намира в рогата, в космите /вълната/, ноктите, копитата, перата ; съдържа около 5% сяра, която идва от цистеина ; колагена – белтъчното вещество на костите, хрущялите, сухожилията и кожата ; съдържа много гликокол /глицин/ и при хидролиза дава желатин /избелен/ или туткал /ако е неизбелен/.
/прости белтъци /
- това е последователността на свързване на аминокиселините в полипептидна верига ;
- вторична структура - конфигурацията на полипептидната верига в молекулата
- третична структура - пространствената структура на протиеновата молекула като цяло ;
- четвъртична структура - асоциация на повече протеинови молекули в определени комплекси /агрегати/ с по-голямо молекулно тегло.
Вторичната структура на протеините показва каква е конфигурацията /ориентацията в пространството/ на пептидната верига. Тя се определя от първичната структура и е фиксирана посредством вътрешномолекулни водородни връзки между карбонилния кислород и азотния водород в една и съща верига. Чрез рентгеноструктурен анализ Полинг /1951 г/е установил, че чрез такива връзки се създават спирални структури. Това са устойчиви конформационни структури, наречени α-спирали или α-helix.
Съществуват няколко начина за откриване ва белтъчни вещества в разтвори. По-важни от тях са утаечните и цветните реакции.Утаечните реакции се извършват при наличие на : а) соли на тежки метали като живак, олово, кадмий, цинк и др.; б) киселини ; в) алкохол, ацетон и др. Цветните реакции за откриване на белтъчните вещества са следните :
биуретова реакция - добавя се меден сулфат към силно алкален разтвор на белтъчното вещество ; важи за съединения с повече от две аминогрупи;
ксантопротеинова реакция - извършва се с конц.азотна киселина ; извършва се нитриране в ароматно ядро /напр.при аминокиселини, съдържащи бензеново ядро като тирозин, триптофан, хистидин / ;
милонова реакция - работи се с меркуринитрат в кон.HNO
реакция на Паули - Na CO
реакция на Адамкевич, Хопкинс, Коле - глиоксалова киселина + сярна киселина ; дава синьовиолетово оцветяване поради кандензиране на алдехидната група с индоловия пръстен на триптофана ;
оловносулфидна реакция - работи се с оловен ацетат в алкална среда ; появява се тъмносива утайка от PbS ; сярата е от сярасъдържащите аминокиселини ;
нинхидринова реакция - с разтвор на нинхидрин белтъците дават синьо оцветяване
фосфопротеиди - съдържат фосфорна киселина, която е свързана солеобразно или естерно ; напр.казеина на млякото или вителина в яйчния жълтък ;
хромопротеиди - съдържат цветни групи, напр.хемоглобина и др.;
гликопротеиди - съдържат въглехидратна група, напрмуцините в слюнката ;
липопротеиди - съдържат мазнини ;
нуклеопротеиди - съдържат нуклеинови киселини, свързани солеобразно с белтъчни молекули. Нуклеопротеидите са главна съставна част на клетъчното ядро. Те имат кисел характер. Тук спадат вирусите,напр.на полиомиелита на тютюневите листа и др.
Макар че химически нуклеиновите киселини рязко да се отличават от белтъците, те все пак са сходни в едно - в молекулите на всички нуклеинови киселини се намира еднаква по природа /но не и по големина/ дълга верига, явяваща се скелет на молекулата, а към този скелет са прикрепени различни групи, последователността и разновидността на които са специфични за всяка нуклеинова киселина.
Два са въглехидратите, които се намират в нуклеиновите киселини : D-рибоза – за класа нуклеинови киселини, наречени рибонуклеинови киселини /РНК/ и D-2-дезоксирибоза – за класа, наречен дезоксирибонуклеинови киселини /ДНК/.Въглехидратите се намират във фуранозна форма и са свързани с фосфорната киселина чрез С3- и С5-хидроксилни групи.
Към всеки С1 /гликозидния С-атом/ от въглехидратите е присъединена азотна база. Обикновено това са хетероциклени съединения. Звеното база-въглехидрат се нарича нуклеозид, а звеното база-въглехидрат-фосфорна киселина - нуклеотид :


Фрагмент от молекула на ДНК
, съдържащ ОН-група от фуранозния остатък. Броят на нуклеотидните звена във веригата на ДНК може да бъде от 1000 до 30 000. За последователността на свързване на четирите вида нуклеозиди /първична структура/ е установено, че броят на А-нуклеотидите е равен на броя на Т-нуклеотидите, а броя на Г-нуклеотидите на този на Ц-нуклеотидите. Този факт, както и рентгеноструктурен анализ дават основание да се твърди, че макромолекулата на ДНК е изградена от две спираловизни полиестерни вериги /вторична структура/, разположени антипаралелно. В кухината на двойната спирала се разполагат нуклеотидните остатъци А, Т, Г, Ц от двете вериги. Между А от едната верига и Т от другата, както и между Г и Ц се образуват водородни връзки /напр.О---Н-N или N---H-N /.
По такъв начин структурата на нуклеиновите киселини определя структурата на молекулата на белтъка. С други думи, нуклеиновите киселини играят важна роля в жизнените процеси. В последно време се появиха експериментални данни, показващи, че нуклеиновите киселини са отговорни за химията на паметта - от най-простите червеи до висшите бозайници.
По същество липидите /от гр.lipos тлъстина и eidos- вид/ са естери или амиди на висши алканови или алкенови карбоксилови киселини. Липидите са полифункционални съединения - съдържат или само алкоксикарбоксилови групи, или алкоксикарбонилни и алкоксифосфорни, или алкоксифосфорилни и амидни. Според химичния си състав и физиологичните функции те се делят на две групи :
- такива, приличащи на мазнини и восъци и които съдържат естерни връзки и могат да бъдат хидролизирани.Това са простите липиди /мазнини и восъци / и
- такива, приличащи на холестерола и други стероиди, ксоито нямат естерни връзки и не се хидролизират. Това са сложните липиди – фосфолипиди и гликолипиди
С15Н31СООН палмитинова киселина
С17Н35СООН стеаринова ксиселина
С17Н33СООН олеинова киселина /с една двойна връзка/
С17Н31СООН линолова киселина /с две двойни връзки/
Триглицеридите биват два вида : прости триестери /на една и съща киселина/ и смесени триестери /на различни киселини/. Имената им се образуват от основата на името на съответната киселина и окончанието –ин :
тристеарин бутиропалмитоолеин
/ /смесен глицерид/
Едно от най-важните свойства на мазнините е хидролизата - реакция с промишлено значение. Когато хидролизата се извършва с алкална основа процесът се нарича осапунване, тъй като продукти на реакцията са соли на висшите мастни киселини /сапуни/ и глицерол :
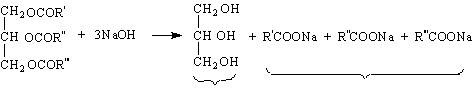
глицерол натриеви сапуни
На доброто им емулгиращо действие върху мастната фаза /която всъщност е онечистването/ във вода поради факта, че неполярните части на сапуна / - R / се разтварят в мастната фаза, а карбоксилните групи - във водата :

При обработка на сапуните с минерални киселини или под действието на ензима липаза се получават смеси от свободни карбоксилови киселини. При редукция на тези киселини се получават висши алкохоли с С12-С18-атоми, които след естерификация със сярна киселина дават кисели алкилсулфати. Техните Na-соли също са добри миещи вещества, наречени детергенти.
Мазнините с алкенови, алкадиенови и алкатриенови карбоксилови остатъци / напр.лененото, памучното и др.растителни масла/ са известни като съхливи масла и са важен компонент на блажните бои и лакове. Съхненето на маслата /съответно на боите и лаковете/ е химичен процес на окисление и полимеризация на глицеридите под действие на кислорода от въздуха. Процесът се ускорява от катализатори /сикативи/ и протича вероятно по радикалов механизъм. Такъв процес на полимеризация на съхливи масла се среща при приготвянето на линолеуми - памучен плат, импрегниран с окислено съхливо масло, смесено с оцветители, колофон и пълнители.
Ni и налягане/, при което се превръщат в твърди мазнини - масти. Този процес е в основата на производството на маргарина - емулсия на хидрирани растителни масла във вода и малко мляко.
Восъците представляват естери на висши монокарбоксилови киселини с висши едновалентни алкохоли. Биват животински и растителни восъци. Животински са : 1) Пчелен восък – изграждащ пчелните килийки в кошерите ; 2) Китайски насекомов восък-добива се в големи количества ; 3) Китов спермацет – намира се в черепната кухина на китовете; употребява се главно за приготвяне на луксозни свещи ; 4) Вълнен восък – като съставна част на вълнената мас; в него се съдъпжа и алкохола холестерол. Растителни восъци са : 1) Карнаубов восък – образува се под формата на прах върху листата на палмата Copernica cerifera; поради високата т.к. и водонепропускливост този восък е един от ценните компоненти на подовите восъци и копирните хартии ; 2) Планински восък – намира се в някои каменни въглища и катрани като парафин ; употребявал се е главно като електроизолатор ;
Фосфолипиди /фосфатиди/
По състав и структура наподобяват мазнините. Делят се на две групи - естерни и ацетални.
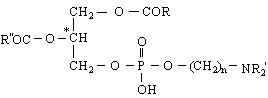
R , R’ R n Лецитините съдържат остатък от аминоалкохола холин [ HO-(CH N CH OH , а цефалините - естерно свързан 2-аминоетанол /коламин/ или аминокиселината серин / HOCH CH NH COOH
Лецитините имат амфотерни свойства и при хидролиза се разпадат до висши мастни киселини, холин и глицерофосфорна киселина. В организма те играят важна биологична роля - с въглехидрати, гликозиди, белтъци и алкалоиди образуват адсорбционни и абсдорбционни “съединения”, които имат добри проникващи способности през клетъчните мембрани. Лецитините притежават добри емулгиращи свойства спрямо мазнините.
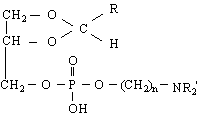
където R е палмитинов или стеаринов остатък, а R
Терпените са особен род органични съединения. Общо взето те се изолират от етерични масла /обикновено чрез дестилация с водна пара /. Много от етеричните масла са се използвали като парфюми, ароматизатори и лечебни средства много преди химията да започне да ги изучава. През 1818 г беше намерено, че природен продукт, изолиран от борова смола има съотношение С : Н = 5 : 8. Този природен продукт е познат като терпен /оттам и терпентин – също изолиран от борова смола или евкалиптово масло /.
През 1877 г германския химик Ото Валах определя структурата на терпените и открива общ структурен белег - всички те се състоят от две или повече изопренови единици /изопрен – 2-метил-1,3-бутадиен/. Свързването на тези изопренови единици става или “глава” с “опашка”, или “глава” с “глава”, или “опашка” с “глава”.
“глава” “опашка”
- димери на изопрена с С10-атома ;
Сескитерпени -
n
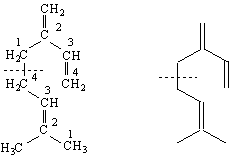
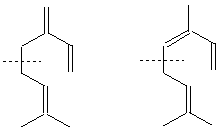
мирцен оцимен

гераниол нерол цитронелол
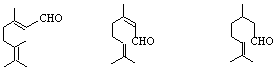
гераниал нерал цитронелал
Моноциклични монотерпени
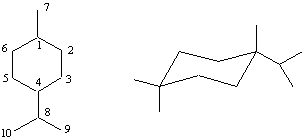

Δ1,3-ментадиен Δ1(7),3-ментадиен Δ1,4(5)-ментадиен Δ1,5-ментадиен
/α-терпинен / /β-терпинен / /γ-терпинен/
Δ1(7),5-ментадиен Δ1,4(8)-ментадиен Δ1,8(9)-ментадиен
/лимонен /
ментол ментон карвон
туйан каран пинан камфан
α-туйен β-туйен
![]()
Δ3-карен Δ4-карен
α-пинен β-пинен борнилен
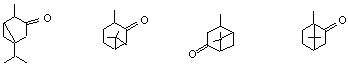
туйон карон вербенон камфор
trans
ликопен
![]()
![]()
![]()
![]()
![]()
ретинол /вит.А/
e
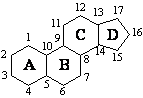
стеран
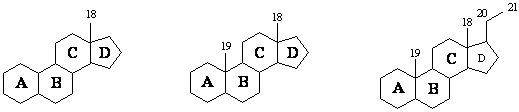
естран андростан прегнан

холан холестан
холестан - основен въглеводород на стеролите ;
- основен въглеводород на жлъчните киселини ;
- основен въглеводород на гестагените и хормоните на надбъбречната жлеза ;
- основен въглеводород на андрогените /мъжки полови хормони/ ;
- основен въглеводород на естрогените /женски полови хормони /.
Стероидите са биологично важни природни съединения и играят съществена роля в жизнената дейност на животни и растения. Въз основа на биологичното действие стероидите се делят на : стероли /стерини/, жлъчни киселини, полови хормони, хормони на надбъбречната жлеза, сърдечни агликони, дигиталисови /жабешки/ отрови и стероидни алкалоиди.
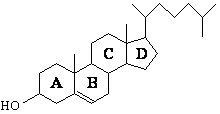
Изследвания са показали, че в организма съществуват липопротеини с ниска плътност /LDL’s / и липопротеини с висока плътност /HDL’s Съществуват доказателства,че LDL’s HDL s Ако LDL’s HDL s

7-дехидрохолестерол ергостерол
Първият се съдържа в нервните тъкани на животните, а ергостерола - в дрожди и гъби. В организма чрез биотрансформация или при облъчване с ултравиолетова светлина тези стероли се превръщат в свои изомери. Например :
 калциферол
/vit D
калциферол
/vit D
Също и холекалциферол /вит.D /. Следователно тези стероли /ергостерола и 7-дехидрохолестерола/ са предшественици или провитамини на витамините от групата D ай-активни от тази група са именно витамините D и D . Те са необходима съставна част от храната на човека и регулират усвояването на калция и съответно растежа на костите. Недостигът на витаминвите ог групата D води до болестта на костната система рахит . Богати на вит.D са рибените масла, яйцата, млякото и други продукти.
Половите хормони са стероиди, които се образуват в гонадите /яйчници и тестикули / и стимулират на свой ред хормоните с пептидна структура,които се отделят в кръвния поток от предната част на хипофизата. Тези стероиди са отговорни за развитието на половите белези и половите чувства у мъжките и женските индивиди. Хормоните на половите жлези съ съответно мъжки и женски и са производни на андростана и естрана. Женските полови хормони се наричат естрогени, а мъжките - андрогени. По-важни андрогени са :

андростерон тестостерон
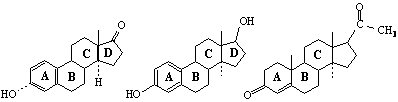
естрон естрадиол прогестерон
/фоликулин/ /лутеостерон/
Вторият тип женски полови хормони, нар.прогестерон, се явява секрет на жълтото тяло (corpus luteum) – един от тъканите на яйчниците.Този хормон участва в подготовката и запазването на бременността.

кортикостерон кортизон
Кортизонът играе важна роля за въглехидратната обмяна в организма, а именно в синтезата на гликоген /животинското нишесте/ и превръщането на белтъците във въглехидрати. Участва също така в извеждането на водата от организма. Кортикостеронът пък поддържа нормалния солеви баланс в организма / съотношението K Na
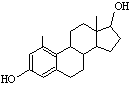
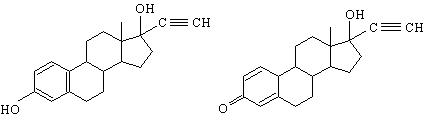
етинестрадиол норетиндрон
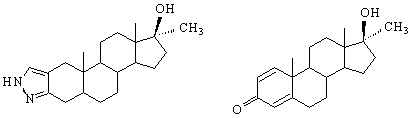
Станозолол Дианабол
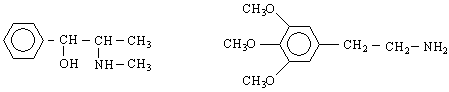
ефедрин мескалин
се съдържа в тропическия храст Ephedra vulgaris. Той действа съдоразширяващо на сърдечно-съдовата система и бронхите. Почти всички лекарства, успокояващи кашлицата, съдържат ефедрин. Мескалинът се намира в кактусите от рода Anhalonium. Този алкалоид предизвиква цветни халюцинации и причинява смущения в зрението. В медицината се използва за лекуване на болезнена меланхолия

никотин кониин
се съдържа с още десетина алкалоида в листата на тютюна /Nicotiana tabacum / в количество от 0.6 до 2 %. В махорката /Nicotiana rustica D
се съдържа в петнистия бучиниш /Conium maculatum / и също е силно отровен. Той парализира краищата на двигателните и сетивни нерви. В древна Атина с воден извлек от бучиниш са привеждали в изпълнение смъртните присъди. През 399 г пр.н.е.Сократ е бил принуден да изпие чаша с тази отрова. Също както и никотина притежава оптическа активност.Кониинът е първият синтетично получен алкалоид.
. Този алкалоид се съдържа в растението лобелия /Lobelia inflata /, което вирее в прериите на Североамериканските щати. Лобелинът възбужда дишането и се прилага в медицината при задушаване, особено когато е причинено от вземане на наркотици.
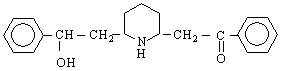
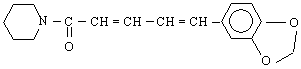
пиперин
Атропин Atropa belladonna/ и в татула /Datura stramonium/. N
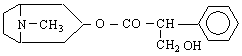
. Той е главният алкалоид на перуанския храст Erythroxylon coca
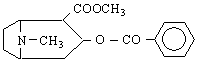
кокаин
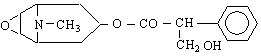
Алкалоиди от хинолинов тип
Към алкалоидите от този тип спадат около 30 алкалоида, наречени хиналкалоиди. Те се съдържат в корите на южноамериканските растения Cinchona
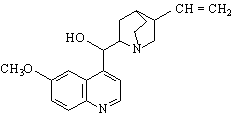
хинин
1820 г и е използван като антипиретик. Дълго време той е бил единственото средство срещу остра малария /маларийна треска/. От 1926 г в употреба се вкарва и синтетичния противомалариен препарат плазмохин. Той превъзхожда по своето действие хинина, като унищожава половите форми на причинителя на маларията – маларийния плазмодий.
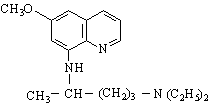
Източник на около 25 алкалоиди от този тип е познатия от III век пр.н.е.опиум, който представлява изсушен млечен сок от неузрели макови главички ( Papaver somniferum).

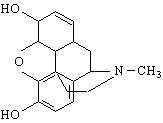
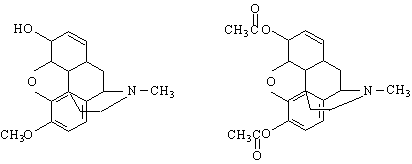
Кодеин Хероин
– намира се в растението Corynanthe yohimbe
Strychnos nux vomica
/LSD/- психоактивно вещество. Предизвиква силни шизофренични цветни халюцинации.
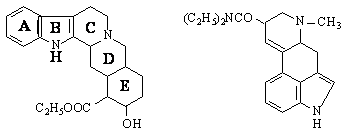
Йохимбин LSD
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 29879
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved