| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Antimicrobial Agents: Antiretroviral Agents
Overview
|
The acquired immunodeficiency syndrome (AIDS) epidemic is one of the greatest challenges facing the medical community today. Infection with human immunodeficiency virus (HIV) is a dynamic process characterized by vigorous viral replication, CD4 lymphocyte depletion, and profound immunodeficiency. The error-prone nature of HIV reverse transcriptase promotes rapid evolution of genetic diversity and a remarkable propensity to develop resistance to antiretroviral agents. Improved understanding of viral pathogenesis as well as the genetic basis of resistance has fueled the rapid and rational development of numerous effective drugs that target either HIV reverse transcriptase or HIV protease. Various multidrug regimens have been shown to inhibit viral replication effectively, reverse CD4 cell depletion, and reduce morbidity and mortality dramatically. Despite much progress, many patients do not benefit from antiretroviral therapy due to emergence of viral resistance, adverse effects of chronic therapy, or inability to adhere to complex regimens. In addition, current agents generally are not available in developing countries, where HIV has its greatest impact. This chapter addresses the pathophysiological rationale for HIV therapy, considers general treatment principles, and reviews individual agents. |
Overview of HIV Infection
|
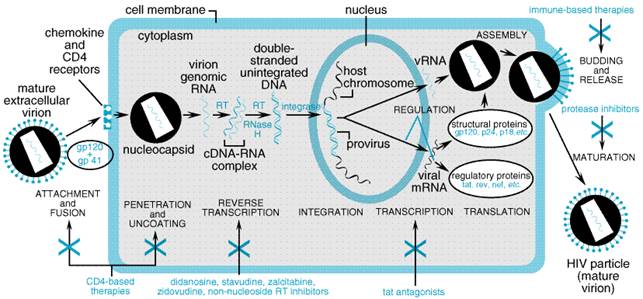
The hallmark of human immunodeficiency virus (HIV) infection is depletion of CD4 lymphocytes, leading to cellular immunodeficiency. Since the first reports of acquired immunodeficiency syndrome (AIDS) appeared in 1981 (Gottlieb et al., 1981; Masur et al., 1981), the vast majority of cases worldwide has been caused by HIV-1. Another retrovirus, HIV-2, is a prevalent cause of AIDS in west Africa. Mature virions contain two single-stranded RNA molecules surrounded by a nucleocapsid and an outer lipid envelope. Like all retroviruses, HIV contains three major genes (gag, pol, and env). Although HIV-1 and HIV-2 share approximately 50% amino-acid homology, individual antiretroviral agents may be more active against one than the other. An entire drug class (nonnucleoside reverse transcriptase inhibitors) is not active against HIV-2. Improved understanding of HIV pathogenesis has led to rational drug development, sound treatment principles, and decreased morbidity and mortality due to AIDS (Palella et al., 1998). Therapeutic strategies evolve rapidly, and treatment errors may have dire and irreversible consequences. It is therefore recommended that HIV infection be managed only by practitioners with specific training and expertise in treating the disease (HIV/AIDS Treatment Information Service, 2000). HIV Pathogenesis Development of AIDS is characterized by susceptibility to various infections and malignancies. Early in the AIDS epidemic, it was erroneously assumed that HIV had a prolonged latent phase without viral replication and that eventual reactivation of viral replication triggered disease progression. However, sensitive viral culture and nucleic acid detection methods showed that almost all untreated patients experience continuous plasma viremia (Ho et al., 1989). Landmark studies demonstrated that the plasma HIV-1 RNA concentration predicts time for progression to AIDS and death (Mellors et al., 1996) and that CD4 cell counts are independently prognostic (Mellors et al., 1997). Such discoveries have focused research on ways to achieve durable control of HIV replication. The development of highly effective antiretroviral agents made it possible to probe viral pathogenesis. Administering such agents disrupts the steady-state equilibrium between virion production and clearance. Studies of treatment-nave patients demonstrated that plasma HIV-1 RNA concentrations decline by 10- to 100-fold within one week of initiating treatment with potent inhibitors of either HIV protease or reverse transcriptase. Mathematical modeling of such data established that HIV infection is extremely dynamic, with daily production of an estimated 109 virions (Ho et al., 1995; Perelson et al., 1996; Wei et al., 1995). Approximately 99% of plasma HIV arises from recently infected CD4+ lymphocytes, which have an average life span of 2.2 days (Perelson et al., 1997). A second source of virus (presumably macrophages) decays with a life span of 2 weeks (Perelson et al., 1996, 1997). It was predicted that complete inhibition of HIV replication for 2 to 3 years might allow all infected cells to be eradicated if these were the only reservoirs for HIV, and that infection would be cured in some patients. Unfortunately, there is an additional long-lived pool of resting CD4+ lymphocytes cells that harbor replication-competent HIV (Chun et al., 1997; Chun et al., 1998; Finzi et al., 1997). Although there are relatively few such cells in the body, their average life span may be months or even years. Based on the previous strategy, greater than 100 years of complete control of viral replication would be needed to eradicate these cells! In fact, discontinuing efficacious antiretroviral therapy after years of apparently complete response invariably leads to relapse of viremia (Davey et al., 1999). The Life Cycle of HIV HIV is an RNA retrovirus that infects CD4+ lymphocytes, macrophages, and dendritic cells. Its cellular life cycle suggests many potential drug targets (Figure 511). Currently approved agents target either reverse transcriptase or protease, but drugs with different targets are being developed.
The
initial step in infection involves attachment and membrane fusion. Virus
enters cells through interactions between HIV envelope glycoproteins (gp41
and gp120) and cell receptors (CD4 and chemokine receptors such as CCR5 and
CXCR4) (He et al., 1997; Sodroski, 1999). In general, only cells
expressing both CD4 and chemokine receptors are susceptible. The envelope
glycoprotein spike is a trimeric structure consisting of three outer gp120
molecules and three transmembrane gp41 molecules. Attachment of gp120 to CD4
and chemokine receptors brings HIV very close to the cell. Before gp120 binds
to the receptor (the prefusogenic state), a region of the gp41 molecule (N36)
is an After entering the cytoplasm and uncoating, viral RNA serves as a template from which complementary DNA strands are transcribed. This reverse transcription is the distinguishing feature of retroviruses and is catalyzed by the HIV RNA-dependent DNA polymerase (reverse transcriptase). Two classes of antiretroviral agents (nucleoside reverse transcriptase inhibitors and nonnucleoside reverse transcriptase inhibitors) target this enzyme. Following reverse transcription, the double-stranded DNA circularizes and enters the nucleus. Integration of this proviral DNA into the host chromosome is mediated by a second essential viral enzyme, integrase. Although integrase seems an attractive target for drug treatment, the discovery of inhibitors for clinical use has been difficult because of the complex interactions between host and viral molecules during integration. After being incorporated into the host chromosome, proviral DNA can be transcribed into HIV RNA by the cellular transcription machinery. This RNA then may be translated into viral polyproteins (including gag-pol and env) or be incorporated into immature virions during virion assembly. Immature virions then undergo a process of maturation and budding from the cell membrane. Maturation requires that the gag-pol polyprotein be cleaved by protease, the third essential enzyme of HIV. Mature virions may then infect other susceptible cells. Exposure of infected cells to inhibitors of HIV protease results in production of immature virions that lack a typical nucleocapsid and that are noninfectious. |
General Principles of Antiretroviral Therapy
|
The clinical benefit of antiretroviral therapy is determined by the magnitude and duration of plasma HIV RNA suppression (Marschner et al., 1998; O'Brien et al., 1996). A central principle of therapy is to inhibit viral replication as completely and durably as possible while avoiding toxicity as much as possible. This requires administering multiple drugs simultaneously (HIV/AIDS Treatment Information Service, 2000). Adherence to complex regimens is difficult for many patients, and nonadherence is a major cause of therapeutic failure and death. In a study of patients for whom HIV protease inhibitors had been prescribed (mostly indinavir, nelfinavir, or saquinavir/ritonavir) plus nucleoside reverse transcriptase inhibitors, at least 95% adherence was necessary for optimal response (Paterson et al., 2000). Drug Resistance The HIV reverse transcriptase is highly error prone. Like all polymerases that use RNA as a template, it lacks the 3' exonuclease activity needed to correct transcription errors. With an error rate of 3.4 x 105 per base pair during each replication cycle and a genome of 104 base pairs, every mutation at every nucleotide probably occurs many times each day in untreated patients, and every double mutation may arise within 100 days (Coffin, 1995; Wain-Hobson, 1993). (The latter prediction assumes that single point mutations do not impair replication efficiency, which is likely not the case.) Incomplete therapeutic control of replication inevitably selects for drug-resistant mutants (Havlir and Richman, 1996; Molla et al., 1996). Although nonnucleoside reverse transcriptase inhibitors are very effective antiretroviral agents, a single mutation at reverse transcriptase codon 103 confers high-level resistance. Since all patients infected with HIV likely harbor such mutants prior to therapy, monotherapy with a nonnucleoside reverse transcriptase inhibitor will cause an initial plasma HIV RNA decline (inhibition of susceptible virus), followed within weeks by virologic failure (emergence of resistant virus). In contrast, nonnucleoside reverse transcriptase inhibitors can provide durable control of HIV in multidrug regimens (Staszewski et al., 1999). Since only replicating HIV can accumulate mutations, complete suppression of replication will both reverse CD4 lymphocyte depletion and prevent resistance (Havlir and Richman, 1996). Many patients who initiate antiretroviral therapy experience emergence of multidrug-resistant virus. Future treatment options always must be kept in mind when antiretroviral therapy is begun or when a regimen is changed (HIV/AIDS Treatment Information Service, 2000). Virologic response to therapy may be improved by using HIV-resistance assay results to guide prescribing (Durant et al., 1999). However, resistance testing is only one factor to consider when making treatment decisions. At least as important are prior treatment regimens, previous plasma HIV RNA responses to each regimen, comorbidities, anticipated adherence to complex regimens, and patient preference. Although virus in plasma is replaced by sensitive strains if selective drug pressure is not maintained, resistant strains may remain in tissues indefinitely (Hirsch et al., 2000; Romanelli and Pomeroy, 2000). Therefore, resistance testing may falsely indicate susceptibility if the patient is no longer on the drug that selected for resistant virus. In addition, available resistance assays do not reliably detect minor resistant subpupulations. The role of HIV resistance testing is being evaluated in clinical trials. Deciding When to Initiate Therapy The decision to initiate treatment must be individualized and should involve consideration of both the plasma HIV-1 RNA concentration and CD4 cell count. Treatment probably should be offered to all patients with greater than 20,000 plasma HIV-1 RNA copies per milliliter (by polymerase-chain-reaction assay) or less than 350 CD4 cells per cubic millimeter. It is reasonable to defer treatment for other patients, since the short-term prognosis is excellent (Harrington and Carpenter, 2000; Mellors et al., 1996; HIV/AIDS Treatment Information Service, 2000). Monitoring Treatment Response Periodic plasma HIV-1 RNA determinations are used to monitor therapeutic response (Marschner et al., 1998). Due to biological and inherent assay variability, results of two assays using samples obtained on different days generally should be used to define the pretreatment baseline. It is essential that the patient fully understand the importance of adherence to long-term therapy and that nonadherence will promote drug resistance, limiting future options. After starting therapy, plasma HIV RNA levels should be quantified within 2 to 4 weeks to assure at least a 1.0 log10 HIV RNA decline, and every 3 to 4 months thereafter to assure that undetectable plasma HIV RNA levels are achieved and maintained (HIV/AIDS Treatment Information Service, 2000). Extraordinary patients with HIV infection have no evidence of viral replication or immunodeficiency in the absence of therapy. These 'long-term nonprogressors' have robust cellular immune responses to HIV, and it has been suggested that their immune systems can control HIV. If this is true, then boosting HIV-specific immunity in other patients who are receiving antiretroviral therapy might allow immunological control of virus without the need for continued medications. Current clinical trials are testing this hypothesis. |
Antiretroviral Drug Development
|
The very rapid discovery of effective antiretroviral agents was made possible by drug-development programs already established for other diseases including cancer, hypertension, and other viral infections. Pharmaceutical companies previously had produced many nucleoside analogs as potential antiviral and anticancer agents. The ability to grow HIV in cell culture allowed these agents to be screened for activity. Through a labor-intensive process, thousands of compounds were tested, and promising lead compounds were modified to enhance anti-HIV activity, minimize toxicity, and improve bioavailability. Zidovudine arose from this process, and in 1987 it was the first drug approved by the United States Food and Drug Administration (FDA) for treating HIV infection. By 2000, five additional nucleoside reverse transcriptase inhibitors had been approved. Although nucleoside reverse transcriptase inhibitors remain cornerstones of antiretroviral therapy, early agents had limited efficacy. They could delay only temporarily progression to AIDS and death (Fischl et al., 1987). The search for more effective agents with different mechanisms of action produced the nonnucleoside reverse transcriptase-inhibitor class of drugs. Nevirapine, one of the first such agents, inhibited HIV-1 replication far more completely than either zidovudine or didanosine, another nucleoside analog. However, enthusiasm for reverse transcriptase inhibitors temporarily waned when resistance emerged rapidly when the drugs were administered in suboptimal regimens. Concurrently with the development of nonnucleoside reverse transcriptase inhibitors, pharmaceutical companies sought inhibitors of HIV protease. The sequence of HIV-1 protease was first published in 1985, its enzymatic activity reported the following year, and its crystalline structure determined in 1989 (Navia et al., 1989; Wlodawer et al., 1989). Although there were no known inhibitors of HIV protease when the enzyme was first identified, the protease was known to be an aspartyl protease like renin. Hypertension researchers previously had developed nonhydrolyzable substrate analogs as renin inhibitors (transition state analogs); this approach was taken to develop HIV protease inhibitors (Dreyer et al., 1989). By 1990, several groups had reported peptidic inhibitors of HIV protease. Saquinavir, the first HIV-1 protease inhibitor, entered clinical trials in 1992 and received FDA approval three years later. Elucidating the three-dimensional structure of HIV protease has facilitated structure-based 'rational drug design' (Frecer et al., 1998; Thaisrivongs and Strohbach, 1999). Although lead protease inhibitors designed via these strategies were poorly absorbed due to high lipophilicity and poor water solubility, these limitations were overcome. Clinical trials demonstrated that protease inhibitors are effective inhibitors of HIV replication and that they select for resistance more slowly than do nonnucleoside reverse transcriptase inhibitors. Measuring Antiretroviral Efficacy in Clinical Trials The effectiveness of early antiretroviral agents was assessed by determining whether or not treatment, often as monotherapy, delayed or prevented AIDS and death in HIV-infected patients. Once it was demonstrated that plasma levels of HIV RNA and CD4 cell responses could predict clinical benefit, these laboratory tests became preferred surrogate markers of efficacy. Drugs that lowered HIV RNA the most were considered to be most efficacious. Once the basis of drug resistance became appreciated, it was no longer acceptable to administer prolonged monotherapy. Efficacy of newer drugs thus was tested in multidrug regimens, making it difficult to assess the relative contributions of individual agents in multidrug regimens, since many regimens appear to inhibit viral replication completely based on available assays. In addition, because multidrug therapies so effectively prevent opportunistic infections, it became impractical to perform 'clinical end point' studies. At present, the usual way to assess antiretroviral efficacy in phase III clinical trials is by determining the proportion of patients with plasma HIV RNA below limits of detection at 24 weeks and/or 48 weeks. The complexity of individual treatment regimens in terms of the number of pills, frequency of dosage, and dietary requirement varies. Patient adherence rather than inherent drug efficacy may explain apparent differences among regimens in comparative studies. A review of available results of clinical trials of antiretroviral agents is beyond the scope of this chapter, and this area is evolving rapidly. Although selected important studies are cited in the text, the reader is referred elsewhere for an overview of major clinical trials (Tavel et al., 1999). |
Nucleoside Reverse Transcriptase Inhibitors
|
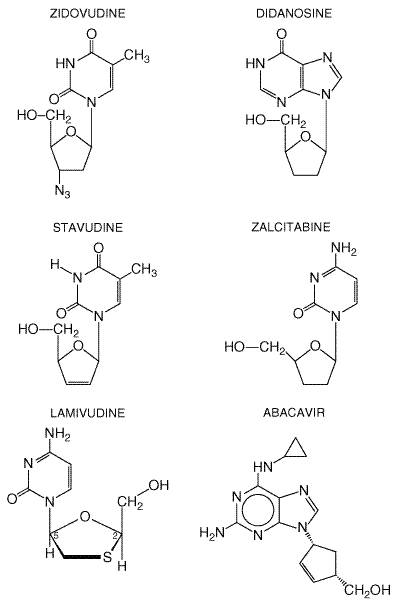
Reverse transcriptase converts viral RNA into proviral DNA before its incorporation into the host cell chromosome. Because agents in this class act at an early and essential step in HIV replication, they prevent acute infection of susceptible cells but have little effect on cells already infected with HIV. All drugs of the nucleoside reverse transcriptase-inhibitor class are substrates for reverse transcriptase. To become active, these drugs first must be phosphorylated by host cell enzymes in the cytoplasm. Since nucleoside reverse transcriptase inhibitors lack a 3'-hydroxyl group, incorporation into DNA terminates chain elongation. There currently are six FDA-approved nucleoside reverse transcriptase inhibitors (Table 511). Their chemical structures are shown in Figure 512. These agents differ in phosphorylation pathways utilized and in untoward effects. This class of drugs includes both early (e.g., zidovudine and didanosine) and recently approved (e.g., abacavir) drugs for treating HIV infection. Although initially evaluated as monotherapy or in dual combinations, these agents now are most important as components of efficacious three- and four-drug regimens. Recently recognized complications include lactic acidosis and severe hepatomegaly with steatosis. A summary of the pharmacokinetic properties of these agents is presented in Table 512.
Zidovudine Chemistry and Antiviral Activity Zidovudine
(3'-azido-3'-deoxythymidine; AZT) is a synthetic thymidine analog active
against HIV-1, HIV-2, and human T-cell lymphotrophic virus (HTLV) I and II (McLeod
and Hammer, 1992). Its in vitro 90% inhibitory concentration (IC90)
against laboratory and clinical isolates of HIV-1 ranges from 0.03 to 0.3 Mechanisms of Action and Resistance After entering the host cell, zidovudine is phosphorylated by
thymidine kinase to a monophosphate, then by thymidylate kinase to the
diphosphate, and finally by nucleoside diphosphate kinase to active
zidovudine, 5-triphosphate (Furman et al., 1986). High concentrations
of the monophosphate may accumulate in the cell, and the intracellular
half-life of zidovudine 5-triphosphate is approximately 3 hours. Zidovudine
5-triphosphate terminates viral DNA chain elongation by competing with
thymidine triphosphate for incorporation into DNA. Zidovudine 5-triphosphate
also weakly inhibits cellular DNA polymerase- Resistance to zidovudine is associated with the mutations at reverse transcriptase codons 41, 67, 70, 215, and 219. Those at codons 41, 215, and 219 are most important. Mutations accumulate gradually, and resistance develops in one-third of patients after one year of zidovudine monotherapy. Cross-resistance to multiple nucleoside analogs has been reported following prolonged therapy and has been associated with mutations at codons 62, 75, 77, 116, and especially 151 (Richman, 1993). Absorption, Distribution, and Elimination Zidovudine is absorbed rapidly from the gastrointestinal tract, with
peak serum levels achieved within about one hour ( Zidovudine undergoes rapid first-pass hepatic metabolism by conversion to 5-glucuronyl zidovudine. This metabolite has an elimination half-life of 1 hour. Total urinary recovery of zidovudine and its major metabolite is approximately 90%. The pharmacokinetics of zidovudine generally is unaffected by pregnancy, and drug concentrations in the newborn approach those of the mother (Watts et al., 1991). Untoward Effects Common adverse effects include anorexia, fatigue, headache, malaise, myalgia, nausea, and insomnia. Anemia may develop as early as four weeks, occurs in 7% of patients with advanced HIV disease, and probably is due to toxic effects on erythroid stem cells (Walker et al., 1988). Evaluation may reveal depletion of bone marrow red-cell precursors, elevated serum erythropoietin levels, and normal serum folate and vitamin B12 levels. Management involves replacing zidovudine with another antiretroviral agent or administering recombinant human erythropoietin. Erythrocytic macrocytosis occurs in approximately, 90% of patients but is not necessarily associated with anemia. Neutropenia also can occur within four weeks of initiating zidovudine
and is more frequent (37%) during advanced HIV disease. Management may
involve replacing zidovudine with another agent or administering recombinant
granulocyte or granulocyte/macrophage colony-stimulating factors (see Chapter
54: Hematopoietic Agents: Growth Factors, Minerals, and Vitamins). Chronic
zidovudine administration may cause nail hyperpigmentation, myopathy (Dalakas
et al., 1990), hepatic toxicity with or without steatosis, and lactic
acidosis (Chattha et al., 1993). Zidovudine can cause muscle damage
associated with reduced amounts of mitochondrial DNA, possibly by inhibiting
mitochondrial DNA polymerase- Drug Interactions and Precautions Since zidovudine may cause bone marrow suppression, it should be used cautiously in patients with preexisting granulocytopenia or anemia. Concurrent administration with other potentially marrow-suppressive agents such as ganciclovir, interferon-alfa, dapsone, flucytosine, vincristine, or vinblastine increases the risk of toxicity. Probenecid, fluconazole, atovaquone, and valproic acid administration may increase plasma zidovudine levels, but the clinical significance of these interactions is unknown, because intracellular triphosphate levels may be unchanged. Both stavudine and ribavirin compete with zidovudine for intracellular activation by common pathways. Zidovudine has been shown to decrease the efficacy of stavudine in clinical trials. Concomitant use of these drugs should be avoided. Therapeutic Uses Zidovudine is FDA-approved for treating adults and children with HIV infection, as monotherapy or in combination with other antiretroviral agents. It also is approved for preventing prenatal transmission of virus in pregnant women with HIV infection and is recommended for postexposure chemoprophylaxis in HIV-exposed health-care workers. Zidovudine was the first antiretroviral agent to show clinical efficacy in treating HIV infection. Since its release in, 1987, the effectiveness of zidovudine has been established in numerous clinical trails. An early monotherapy trial in patients with advanced disease showed that zidovudine improved survival over 24 weeks (Fischl et al., 1987). A later study confirmed decreased risk of disease progression among patients with symptomatic and asymptomatic disease over a 12-month period but did not show a survival benefit (Fischl et al., 1990). Zidovudine plus other nucleoside reverse transcriptase inhibitors provides greater clinical benefits than zidovudine alone. Zidovudine combined with lamivudine produced a 66% reduction in disease progression. More effective regimens have combined zidovudine with two nucleoside analogs (Saag et al., 1998), with a protease inhibitor and a nucleoside analog (Hammer et al., 1997), or with a nonnucleoside reverse transcriptase inhibitor and a nucleoside analog (Staszewski et al., 1999). These regimens control viremia, substantially increase CD4 counts, and decrease morbidity and mortality. When given to pregnant HIV-infected mothers and to their newborns, zidovudine reduces the relative risk of perinatal transmission by two-thirds (Connor et al., 1994). Administration to health-care workers soon after exposure to contaminated body fluids can prevent HIV transmission (Cardo et al., 1997). Didanosine Chemistry and Antiviral Activity Didanosine (2',3'-dideoxy-inosine; ddI) is a purine nucleoside analog active against HIV-1, HIV-2, and HTLV-1 (Hitchcock, 1993; McGowan et al., 1990). Its 50% inhibitory concentration (IC50) for HIV-1 is 0.24 to 0.6 mg/l in T-cell cultures and, 0.002 to 0.02 mg/l in monocycte/macrophage cultures (Perry and Balfour, 1996). Mechanisms of Action and Resistance The active intracellular metabolite of didanosine, 2',3'-dideoxyadenosine, 5'-triphosphate (ddATP), competes with cellular dATP for incorporation into viral DNA. Didanosine enters the cell via a nucleobase carrier and undergoes monophosphorylation by a 5'-nucleotidase. This metabolite then is converted to dideoxyadenosine monophosphate by adenylosuccinate synthetase and adenylosuccinate lyase. Phosphorylation produces the diphosphate and ultimately the active triphosphate, which accumulates with an intracellular half-life of many hours (Shelton et al., 1992). Decreased susceptibility to didanosine is associated with mutations at reverse transcriptase codon 74. This causes a 5- to 26-fold decrease in didanosine susceptibility. Additional mutations at codons 184, 65, 135, and 200 also have been associated with resistance (St. Clair et al., 1991). Absorption, Distribution, and Elimination Didanosine is acid labile and is degraded at low gastric pH (Burger et al., 1995b). Some oral preparations include buffers to minimize degradation; delayed-release capsules do not. The chewable tablets contain calcium carbonate and magnesium hydroxide, while the powder form contains citrate-phosphate buffer. A pediatric powder formulation is available without buffer and can be reconstituted with purified water and mixed with liquid antacid preparations. Oral bioavailability of didanosine is variable and dose-dependent (Shelton et al., 1992). It is approximately 40% for the chewable tablet and somewhat less for the powder form (Cooney et al., 1987). Percentage bioavailability decreases with increasing dose and in children. Food may decrease the drug's absorption (Perry and Balfour, 1996), so oral dosing should be at least one hour before or two hours after meals. Peak plasma concentrations occur approximately one hour after oral administration of chewable tablets or powder formulations and two hours after delayed-release capsules. The plasma elimination half-life ranges from 0.76 to 2.74 hours (Perry and Balfour, 1996). Didanosine is excreted both by glomerular filtration and tubular secretion (Knupp et al., 1993). Purine nucleoside phosphorylase converts the active drug to hypoxanthine, which is ultimately converted to uric acid. The mean CSF-to-plasma ratio is 0.20, but variable ratios have been reported in children (Burger et al., 1995a). Didanosine has been detected in placental and fetal circulation at a small fraction of concentrations in maternal circulation (Dancis et al., 1993). Untoward Effects Although diarrhea is a frequent side effect, peripheral neuropathy and pancreatitis are the most serious dose-limiting toxicities of didanosine. Diarrhea has been attributed in part to the buffer in oral preparations and was reported in 16% of AIDS patients receiving didanosine through the expanded access program. However, in some controlled trials, rates of diarrhea in patients receiving chewable didanosine tablets did not differ from rates in patients receiving zidovudine (Dolin et al., 1995; Kahn et al., 1992). Didanosine-associated peripheral neuropathy is dose-related and is more frequent in patients with underlying neuropathy or receiving neurotoxic drugs. It is a symmetrical, distal, sensory polyneuropathy that most frequently involves the feet and lower extremities. Patients report numbness, tingling, and painful dysthesias. The incidence of peripheral neuropathy increases with addition of stavudine and hydroxyurea to didanosine-containing regimens (Moore et al., 2000). Acute pancreatitis is a rare but potentially fatal complication of didanosine. Lactic acidosis and severe hepatomegaly with steatosis are other potentially fatal complications. In early monotherapy trials, pancreatitis occurred in 3% to 4% more patients receiving didanosine than receiving zidovudine (Rozencweig et al., 1990). This is more common with advanced HIV disease. Risk factors include a previous history of pancreatitis, alcohol or illicit drug use, and hypertriglyceridemia (Hammer et al., 1996). Other untoward effects include elevated liver function tests, headache, and retinal pigmentation in children (Pike and Nicaise, 1993). Drug Interactions and Precautions Didanosine should be used cautiously in patients with a history of pancreatitis or neuropathy. Concurrent administration of drugs that cause pancreatitis (e.g., ethambutol, pentamidine) or neuropathy (e.g., ethambutol, isoniazid, vincristine, cisplatin) should be avoided. Oral ganciclovir can increase plasma didanosine concentrations by twofold. Regimens containing stavudine and/or hydroxyurea increase the risk of neuropathy (Moore et al., 2000). Concurrent administration with zalcitabine is contraindicated. Therapeutic Use Didanosine is FDA-approved for treating adults and children with HIV infection in combination with other antiretroviral agents. Initial studies that compared didanosine to zidovudine monotherapy demonstrated that didanosine reduced clinical progression and death (Dolin et al., 1995; Kahn et al., 1992; Spruance et al., 1994). Several trials showed decreased clinical progression and greater viral suppression with dual nucleoside regimens containing didanosine than with zidovudine monotherapy (Anonymous, 1996; Hammer et al., 1996; Katzenstein et al., 1996; Saravolatz et al., 1996). Multidrug regimens that include didanosine and nonnucleoside reverse transcriptase inhibitors and protease inhibitors have produced effective control of viremia and increased CD4 cell counts. Studies of the use of didanosine in children also have revealed beneficial effects (Englund et al., 1997; Luzuriaga et al., 1997). Stavudine Chemistry and Antiviral Activity Stavudine
(2',3'-didehydro-2',3'-dideoxythymidine; d4T) is a thymidine analog reverse
transcriptase inhibitor that is active in vitro against HIV-1 and
HIV-2. Its IC50 in various in vitro cell systems ranges
from 0.002 to 0.9 Mechanisms of Action and Resistance After passive diffusion into the cell, stavudine must be phosphorylated to the active form, stavudine triphosphate. It is first phosphorylated by thymidine kinase. Unlike zidovudine monophosphate, stavudine monophosphate does not accumulate in the cell. Subsequent phosphorylation events are catalyzed by thymidylate kinase and pyrimidine diphosphate kinase. Stavudine triphosphate inhibits reverse transcriptase by competing with cellular 2'-deoxythymidine-5'-triphosphate, resulting in chain DNA termination (Ho and Hitchcock, 1989). Because thymidine kinase has a higher affinity for zidovudine than for stavudine, zidovudine antagonizes the effect of stavudine (Merrill et al., 1996). Unlike the situations with other nucleoside analogs, the genetic basis of stavudine resistance is poorly understood. Exposure of HIV-1. to stavudine in vitro selects for mutations at codon 75, which confers modest (sevenfold) resistance, and at codon 50. However, in vivo resistance is not clearly associated with these mutations. Strains that are highly resistant to multiple nucleoside reverse transcriptase inhibitors are consistently resistant to stavudine (Lin et al., 1994). Absorption, Distribution, and Elimination Stavudine has very high oral bioavailability (Dudley et al., 1992) and reaches peak plasma concentrations within 1 hour (Zhu et al., 1990). The ratio of the CSF area under the concentrationtime curve (AUC) to the plasma AUC is approximately 0.40 in adults (Haas et al., 2000b). Stavudine has been detected in human placental tissue and in the fetal circulation of pregnant macaques (Odinecs et al., 1996). Untoward Effects The major adverse effect of stavudine is dose-related peripheral neuropathy. In early high-dose studies, the cumulative incidence of peripheral neuropathy exceeded 60% among patients receiving greater than 4 mg/kg daily. At the current daily dosage of approximately 1 mg/kg, the incidence of neuropathy of any grade is 12% (Skowron, 1995). Neuropathy causes numbness, tingling, and pain of the feet and usually resolves after stopping therapy, although temporary worsening may occur. Neuropathy is more common with advanced HIV disease, preexisting neuropathy, or administration of other neurotoxic compounds (Moore et al., 2000; Spruance et al., 1997a). Lactic acidosis has been associated with stavudine administration (Mokrzycki et al., 2000). Moderate transaminase elevations are common during stavudine administration but rarely require discontinuation of therapy. Other, less frequent adverse events include headache, nausea, and rash. An increased risk of pancreatitis has not been demonstrated clearly (Spruance et al., 1997). Drug Interactions and Precautions Medications that cause neuropathy (e.g., ethambutol, isoniazid, phenytoin, vincristine) should be used cautiously in patients receiving stavudine. Regimens containing stavudine, didanosine, and/or hydroxyurea are associated with an increased risk for neuropathy. Zidovudine and stavudine should not be used concomitantly because zidovudine antagonizes the effect of stavudine (see above). Therapeutic Use Stavudine is FDA-approved for treating patients with HIV infection, in combination with other antiretroviral agents. In initial clinical trials, zidovudine-experienced patients were randomized to either continue zidovudine monotherapy or switch to stavudine. Stavudine administration delayed clinical progression compared to continued zidovudine therapy (Spruance et al., 1997a). Stavudine plus lamivudine has yielded plasma HIV RNA decreases of up to 1.6 log10 copies per milliliter. Stavudine, in combination with didanosine and hydroxyurea, has produced median plasma HIV RNA decreases ranging from 1.2 to 1.9 log10 copies per milliliter. Durable suppression of viremia has been reported in three- and four-drug regimens that include stavudine plus other nucleoside reverse transcriptase inhibitors in combination with nonnucleoside reverse transcriptase inhibitors or protease inhibitors (Gisolf et al., 2000; Roca et al., 2000). Zalcitabine Chemistry and Antiviral Activity Zalcitabine
(2',3'-dideoxy-cytidine; ddC) is a cytosine analog reverse transcriptase
inhibitor. It was the first antiretroviral agent licensed through the FDA's
accelerated approval process and is active against HIV-1, HIV-2, and
hepatitis B virus (Mitsuya and Broder, 1986; Yokota et al., 1991). It
is especially active against macrophage-tropic strains of HIV-1 in
monocyte/macrophage cell lines. Zalcitabine inhibits HIV in peripheral blood
lymphocytes at 0.5 Mechanisms of Action and Resistance Zalcitabine enters the cell through carrier-mediated and
noncarrier-mediated mechanisms (Cooney et al., 1986; Plagemann and
Woffendin, 1989; Ullman et al., 1988). It is first phosphorylated by
deoxycytidine kinase and further by cellular kinases to its active
metabolite, dideoxycytidine 5'-triphosphate (Broder, 1990). Unlike other
nucleoside analogs, zalcitabine is most efficiently triphosphorylated in
resting peripheral blood mononuclear cells (Gao et al., 1993). The
triphosphate terminates viral DNA elongation. Zalcitabine decrease the
intracellular pool of deoxycytidine triphosphate and binds somewhat to host High-level resistance to zalcitabine has not been reported. Low-level to moderate resistance has been associated with mutations at reverse transcriptase codons 65, 69, 74, and 184. Strains resistant to multiple nucleoside analogs including zalcitabine have demonstrated mutations at codon 151. Resistance mutations related to zalcitabine exposure develop slowly, although phenotypic cross-resistance with didanosine and lamivudine has been reported (Craig and Moyle, 1997). Absorption, Distribution, and Elimination Zalcitabine has high oral bioavailability and is recovered mostly unchanged in urine (Gustavson et al., 1990; Klecker et al., 1988). The CSF-to-plasma ratio is approximately 0.2. Pharmacokinetic properties in adults and children are similar. Untoward Effects The major adverse events of zalcitabine administration are peripheral neuropathy, stomatitis, rash, and pancreatitis. Zalcitabine-associated peripheral neuropathy is dose-related and more common with preexisting neuropathy and advanced disease. Alcohol consumption, diabetes, and low vitamin B12 levels also are associated with an increased risk of zalcitabine-induced neuropathy (Fichtenbaum et al., 1995; Fischl et al., 1995). Severe neuropathy occurs in up to 15% of patients. Symptoms include numbness, burning, and tingling of the feet. Symptoms may worsen after stopping the drug, then slowly improve. Stomatitis with ulcerations of buccal mucosa, soft palate, tongue, or pharynx occurs in 3% of patients (Fischl et al., 1995) and may resolve with continued therapy. Mild and self-limited rash is common but rarely necessitates discontinuing therapy (Yarchoan et al., 1988). Pancreatitis is a rare complication of zalcitabine therapy (Saravolatz et al., 1996). Other toxicities include arthragias, myalgias, and elevated serum transaminase levels (Fischl et al., 1995; Saravolatz et al., 1996; Yarchoan et al., 1988). Drug Interactions and Precautions Concurrent administration of agents that cause neuropathy or pancreatitis should be avoided in patients receiving zalcitabine. Coadministration with didanosine is contraindicated. Cimetidine and probenecid decrease zalcitabine elimination, and dosage adjustment may be needed with their coadministration. Therapeutic Use Zalcitabine has been approved by the FDA for use in combination with other antiretroviral agents to treat HIV infection in adults. Early trials that evaluated zalcitabine monotherapy in advanced disease showed moderate increases in CD4 cell counts and decreases in HIV antigenemia (Yarchoan et al., 1988). Zidovudine monotherapy was clearly superior to zalcitabine monotherapy (Fischl et al., 1993). Several studies showed clinical benefit of zalcitabine in combination with zidovudine compared with zidovudine monotherapy (Anonymous, 1996; Saravolatz et al., 1996). A few trials have evaluated zalcitabine in efficacious three-drug combinations. A regimen of zalcitabine, zidovudine, and saquinavir was superior to two-drug regimens (Collier et al., 1996). Since the antiretroviral efficacy of zalcitabine is less than that of other agents, this drug is prescribed relatively infrequently. Lamivudine Chemistry and Antiviral Activity Lamivudine (2'-deody-3-thiacytidine; 3TC) is a pyrimidine analog reverse transcriptase inhibitor active against HIV-1, HIV-2, and hepatitis B virus. It was first prepared as a racemic mixture (BCH-189). Lamivudine, the () enantiomer, has less cytotoxicity and greater antiviral activity than the (+) enantiomer (Skalski et al., 1993). The IC50 of lamivudine against laboratory strains of HIV-1 ranges from 4.0 to, 670 nM (Coates et al., 1992). It is synergistic with other antiretroviral agents including zidovudine, stavudine, didanosine, nevirapine, and delavirdine. Lamivudine antagonizes zalcitabine by interfering with its phosphorylation (Bridges et al., 1996; Merrill et al., 1996; Veal et al., 1996). Mechanism of Action and Resistance Lamivudine enters cells by passive diffusion and is phosphorylated to
its active metabolite, lamivudine triphosphate. Lamivudine triphosphate competes
with deoxycytidine triphosphate for binding to reverse transcriptase, and
incorporation into DNA results in chain termination. Lamivudine has very low
affinity for human High-level resistance to lamivudine, unlike other nucleoside analogs, develops rapidly (Schinazi et al., 1993; Schuurman et al., 1995). A single mutation at codon 184 causes high-level resistance and also may inhibit viral growth somewhat (Larder et al., 1995). This codon lies in an essential amino acid motif in the active site of the enzyme. Importantly, the codon mutation restores zidovudine susceptibility to zidovudine-resistant HIV. Cross-resistance to didanosine and zalcitabine occurs, but the decreased susceptibility to these drugs is less than for lamivudine (Schinazi et al., 1993). Absorption, Distribution, and Elimination Lamivudine has high oral bioavailability with or without food and reaches peak plasma levels within approximately 1 hour. The long intracellular half-life of the triphosphate suggests than once-daily dosing may be effective (Gao et al., 1994; Heald et al., 1996; Yuen et al., 1995). Lamivudine is excreted primarily unchanged in the urine. The CSF-to-plasma AUC ratio is 0.15 (Haas et al., 2000b; Lewis et al., 1996). Lamivudine crosses the placenta and has been detected in the fetal circulation. Untoward Effects Significant adverse effects of lamivudine are rare. Headache and nausea have been reported at higher-than-recommended doses (Bartlett et al., 1996). Pancreatitis has been reported in pediatric patients, but this has not been confirmed in controlled trials of adults or children. Drug Interactions and Precautions Lamivudine and zalcitabine may be antagonistic and should not be used concomitantly. Trimethoprimsulfamethoxazole increases plasma lamivudine concentrations, but this does not require dose adjustment. Therapeutic Use Lamivudine is FDA-approved for treating HIV infection in adults and children, in combination with other antiretroviral agents. Before the rapid emergence of resistance was appreciated with lamivudine monotherapy, a limited number of monotherapy trials were performed (Pluda et al., 1995; Schuurman et al., 1995). Lamivudine in combination with zidovudine produced substantial but incomplete decreases in plasma HIV-1 RNA (Eron et al., 1995; Katlama et al., 1996). Many trials have confirmed the antiretroviral activity of lamivudine in three-drug regimens with other nucleoside analogs, protease inhibitors, and/or nonnucleoside reverse transcriptase inhibitors. Lamivudine has been effective in combination with other antiretroviral compounds for treating experienced or nave patients (Gulick et al., 1997; Hammer et al., 1997). Abacavir Chemistry and Antiviral Activity Abacavir sulfate(1S,cis)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl)]-2-cyclopentene-1-methanol
sulfate (salt) (2:1)is a carbocyclic nucleoside analog that contains a novel
6-cyclopropylamino-substituted purine. The active metabolite of abacavir,
carbovir triphosphate, is a potent HIV-1 reverse transcriptase inhibitor. The
IC50 of abacavir against clinical isolates is 0.26 Mechanisms of Action and Resistance Abacavir undergoes intracellular phosphorylation by enzymes that do not phosphorylate other FDA-approved nucleoside reverse transcriptase inhibitors. It is monophosphorylated by a pathway involving adenosine phosphotransferase and is then di- and triphosphorylated. Carbovir triphosphate accumulates and has an intracellular half-life of 3 hours (Daluge et al., 1997; Faletto et al., 1997). In vitro passage of HIV-1 in the presence of abacavir selects for modest resistance (up to 10-fold) with mutations at reverse transcriptase codons 184, 65, 74, and 115. Resistance in clinical isolates from patients who had received prior therapy with nucleoside reverse transcriptase inhibitors is associated with multiple mutations. Strains that are resistant to all other nucleoside reverse transcriptase inhibitors also are resistant to abacavir (Tisdale et al., 1997), and may contain mutations at codons 41, 210, 215, or 151 or an insertion mutation at codon 69. Increasing numbers of mutations increase the likelihood of abacavir resistance (Tisdale et al., 1997). Absorption, Distribution, and Elimination The oral bioavailability of abacavir is high with or without food (Daluge et al., 1997; Chittick et al., 1999); the CSF-to-plasma AUC ratio is approximately, 0.3. Abacavir is partially metabolized by alcohol dehydrogenase (to form the 5'-carboxylic acid) and glucuronidation (to form the, 5'-glucuronide) (Chittick et al., 1999). Untoward Effects The most common adverse events are gastrointestinal symptoms, neurologic complaints, and a unique hypersensitivity syndrome. Nausea, vomiting, diarrhea, and abdominal pain were common in one large clinical trial, but only 10% of patients withdrew because of adverse events. Neurological complaints, including headache, dizziness, and insomnia, were less frequent. Asthenia has been reported in, 40% of patients (Harrigan et al., 2000). Abacavir Hypersensitivity Reaction A unique and potentially fatal hypersensitivity reaction occurs in 2% to 5% of patients receiving abacavir. Symptoms typically occur within the first six weeks of therapy and include fever, rash, nausea, malaise, and respiratory complaints, in various combinations. Symptoms initially may be mild but increase in severity with continued administration. Discontinuation of the medication usually resolves all signs and symptoms, but rechallenge may cause rapid onset of severe reactions, hypotension, and death. Once an abacavir hypersensitivity reaction is suspected or confirmed, it is recommended that the patient never be rechallenged with abacavir. Drug Interactions and Precautions Ethanol increases plasma levels of abacavir by 41% (McDowell et al., 2000). Patients who begin abacavir must be educated regarding the hypersensitivity reaction (see above) and instructed about what to do if this occurs. This generally involves seeking medical help immediately if they develop symptoms of hypersensitivity. Therapeutic Use Abacavir is FDA-approved for treating HIV infection in adults and children in combination with other antiretroviral agents. Several studies have evaluated abacavir in combination with other nucleoside analogs, nonnucleoside reverse transcriptase inhibitors, and protease inhibitors. The combination of abacavir, zidovudine, and lamivudine has efficacious antiretroviral activity in both adults and pediatric patients. Substantially greater decreases in plasma HIV-l RNA occurred in patients receiving this three-drug regimen versus zidovudine plus lamivudine. Adding abacavir to stable antiretroviral therapy may exert significant antiviral effect, but not if there are multiple, preexisting zidovudine resistance mutations. Abacavir also has been used in three- and four-drug regimens for heavily experienced patients failing previous therapies (Deeks et al., 1999; Falloon et al., 2000). |
Nonnucleoside Reverse Transcriptase Inhibitors
|
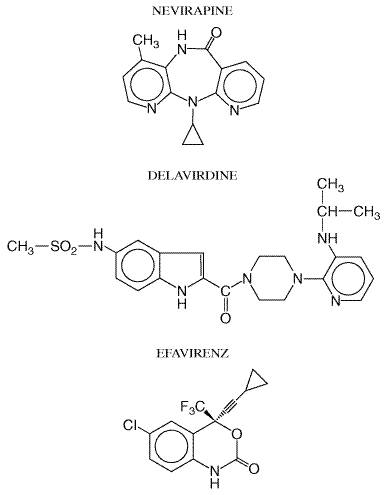
The nonnucleoside reverse transcriptase inhibitors are a class of chemically distinct synthetic compounds that block reverse transcriptase activity by binding adjacent to the enzyme's active site, inducing conformational changes in this site. These agents share not only a common mechanism of action but also some toxicities and resistance profiles. Unlike nucleoside analogs, nonnucleoside reverse transcriptase inhibitors do not undergo phosphorylation. In addition, they are active against only HIV-1, not HIV-2. All compounds in this class are metabolized by the CYP450 system and thus are prone to drug interactions. Three nucleoside reverse transcriptase inhibitors are FDA-approved (Table 511). Their chemical structures are shown in Figure 513 and selected pharmacokinetic parameters in Table 513.
Nevirapine Chemistry and Antiviral Activity Nevirapine is a nonnucleoside reverse transcriptase inhibitor with potent activity against HIV-1. It is active in several cell lines including T lymphocytes and macrophages. Infectivity of extracellular virions also can be decreased by exposure to nevirapine. The IC50 of nevirapine ranges from 10 to 100 nM (Zhang et al., 1996). Like other compounds in this class, nevirapine does not have significant activity against HIV-2 and other retroviruses. Mechanism of Action and Resistance Nevirapine diffuses into the cell and binds to reverse transcriptase adjacent to the catalytic site. This induces conformational changes that inactivate the enzyme. Resistance develops rapidly in cells exposed to nevirapine. High-level resistance is associated with mutations at reverse transcriptase codons 101, 103, 106, 108, 135, 181, 188, and 190. A single mutation at either codon 103 or 181 decreases susceptibility more than 100-fold. Cross-resistance may extend to all FDA-approved nonnucleoside reverse transcriptase inhibitors, especially with the codon 103 mutation (Casado et al., 2000). Absorption, Distribution, and Elimination Nevirapine is well absorbed orally. Neither food nor antacids affect bioavailability (Cheeseman et al., 1993, 1995). Nevirapine readily crosses the placenta and has been found in breast milk. The CSF-to-plasma ratio for nevirapine is approximately 0.45 (Mirochnick et al., 1998; Zhou et al., 1999). Oxidative metabolism of nevirapine in the liver by cytochrome P450 isoforms CYP3A4 and CYP2B6 produces several metabolites including, 2-, 3-, 8-, and 12-hydroxynevirapine. Glucuronidation of metabolites and urinary excretion constitute the primary elimination pathway (Erickson et al., 1999; Riska et al., 1999). Nevirapine also induces the synthesis of CYP3A4, which decreases the plasma half-life of nevirapine from 45 hours following the first dose to 25 hours after 2 weeks. To compensate for this induction, initiation of nevirapine includes a 14-day initial dosing period, at the end of which the dose is increased if no adverse reactions have occurred. Untoward Effects The most frequent adverse events associated with nevirapine include rash, fever, fatigue, headache, somnolence, nausea, and elevated liver enzymes. Rash occurs in approximately 16% of patients. This usually is a mild macular or papular eruption involving the trunk, face, and extremities and generally occurs within the first six weeks of therapy. Pruritus is common. Approximately 7% of patients discontinue therapy due to rash, and preemptive administration of glucocorticoids paradoxically may cause a more severe rash. StevensJohnson syndrome occurs with an incidence of approximately 0.3%. The incidence of nevirapine-induced hepatitis approaches 1%. Drug Interactions and Precautions Since nevirapine induces CYP3A4, coadministration with agents metabolized by this system may lower their plasma levels. Methadone withdrawal has been reported in patients receiving nevirapine (Altice et al., 1999). Rifampin and ketoconazole are contraindicated in patients receiving nevirapine. Plasma ethinyl estradiol levels decrease significantly with nevirapine coadministration, and alternative methods of birth control are advised. Although nevirapine can lower plasma concentrations of protease inhibitors, most such combinations do not require dose adjustment. Nevirapine levels generally are unaffected by concomitant protease inhibitors (Table 514). Therapeutic Use Nevirapine is FDA-approved for treating HIV-1 infection in adults and children, in combination with other antiretroviral agents. It can be very effective during long-term administration in multidrug regimens. Nevirapine was evaluated originally in small clinical trials as monotherapy or alternating monotherapy with zidovudine administration. Only modest, short-term improvements in laboratory markers of HIV infection were reported because of rapid emergence of resistance (Havlir et al., 1995). More recent trials have evaluated nevirapine in three-drug regimens. In one large trial that examined a three-drug regimen of nevirapine, zidovudine, and didanosine in antiretroviral-nave adults, 52% of patients had plasma HIV-1 RNA below, 400 copies per milliliter (Montaner et al., 1998). Many trials currently are evaluating nevirapine-containing regimens in treatment of nave and experienced patients. Clinical trials also are addressing whether or not lipodystrophy (see section on Untoward Effects of protease inhibitors, below) resolves when a protease inhibitor is replaced by nevirapine (Raboud et al., 1999; Raffi et al., 1998). Nevirapine has been evaluated in pregnant HIV-infected women. In a
landmark study performed in Delavirdine Chemistry and Antiviral Activity Delavirdine
is a bisheteroarylpiperazine nonnucleoside reverse transcriptase inhibitor
that selectively inhibits HIV-1 in several in vitro cell systems. Its
median inhibitory concentration is, 0.006 Mechanism of Action and Resistance After entering the cell, delavirdine binds to a hydrophobic pocket in the p66 subunit of reverse transcriptase. This causes a conformational change to a stable, inactive form of the enzyme. The delavirdine-reverse transcriptase complex is stabilized by hydrogen bonds at residue Lys-103 and strong hydrophobic interactions wxith residue Pro-236 (Spence et al., 1995). Much higher concentrations of delavirdine are required to inhibit cellular polymerase than reverse transcriptase (Dueweke et al., 1993a). As with other nonnucleoside reverse transcriptase inhibitors, high-level resistance to delavirdine can emerge rapidly. In vitro passage of HIV-1 in the presence of delavirdine selects for a mutation at reverse transcriptase codon 236, which does not confer cross-resistance to other compounds in this class. However, in vivo resistance rarely is associated with the 236 mutation. Most clinically derived resistant strains have mutations at reverse transcriptase codons 181 and/or 103. Resistance also has been associated with mutations at codons 100, 101, 106, and 188 (Dueweke et al., 1993b). There is evidence that resistance to delavirdine may restore zidovudine susceptibility to zidovudine-resistant HIV. Absorption, Distribution, and Elimination Delavirdine is well absorbed, especially at pH less than 2.0. Antacids, histamine H2-receptor antagonists, achlorhydria, and high-fat meals may decrease its absorption (Barry et al., 1999). Its plasma half-life increases with increasing doses (Cheng et al., 1997; Freimuth, 1996). Delavirdine binds extensively to plasma proteins and primarily is metabolized by CYP3A4. The major metabolic pathway results in N-dealkylation. There is considerable intersubject variability in plasma delavirdine concentrations related to differences in CYP3A activity. The CSF-to-plasma ratio is 0.02. Untoward Effects The most common side effect of delavirdine is rash, which develops in 18% to 36% of subjects. This typically occurs in the first few weeks of administration and often resolves despite continued therapy. The rash may be macular, papular, erythematous, or pruritic and usually involves the trunk and extremities. Rash causes discontinuation of delavirdine in less than 5% of patients and generally is less severe than the rash associated with nevirapine. Severe dermatitis, including StevensJohnson syndrome, is rare. Elevated liver function tests and rare cases of neutropenia have been reported (Para et al., 1999). Drug Interactions and Precautions Delavirdine is both a substrate and inhibitor of CYP3A4, and it can alter the metabolism of other CYP3A4 substrates. Such drugs include rifampin, rifabutin, ergot derivatives, triazolam, midazolam, and cisapride. Delavirdine also inhibits CYP2C9. Carbamazepine, phenobarbital, phenytoin, rifabutin, and rifampin may decrease delavirdine levels by inducing CYP3A4. Compounds metabolized by CYP3A4 should be administered cautiously to patients receiving delavirdine. Delavirdine increases plasma levels of saquinavir, indinavir, nelfinavir, and ritonavir (Table 514). Therapeutic Use Delavirdine is approved for the treatment of HIV-1 infection in adults in combination with other antiretroviral agents. Although delavirdine may be highly effective in multidrug regimens, initial monotherapy studies showed only transient decreases in plasma HIV-1 RNA levels due to rapid emergence of resistance. Later studies of delavirdine in combination with nucleoside analogs showed sustained decreases in HIV-1 RNA levels. Delavirdine increases plasma concentrations of indinavir, which may increase the efficacy of this combination. Current studies are evaluating the efficacy of delavirdine in three-drug regimens (Friedland et al., 1999; Para et al., 1999). Efavirenz Chemistry and Antiviral Activity Efavirenz is a 1,4-dihydro-2H-3,1-benzoxazin-2-one nonnucleoside reverse transcriptase inhibitor. Efavirenz inhibits HIV-1 reverse transcriptase both in vitro and in vivo, with an IC90 ranging from 3 to 9 nM (Young et al., 1995). Like other compounds in this class, efavirenz does not have significant activity against HIV-2 or other retroviruses. Mechanisms of Action and Resistance Efavirenz diffuses into the cell where it binds adjacent to the active site of reverse transcriptase. This produces a conformational change in the enzyme that inhibits function. High-level resistance to efavirenz can develop in vitro and in vivo. Passage of HIV-1 in the presence of efavirenz yielded a strain with a greater than 300-fold decreased susceptibility. This strain had mutations at reverse transcriptase codons 100, 179, and 181. High-level in vitro resistance also was associated with mutations at codon 188 (Winslow et al., 1996). The most frequent resistance-associated mutation in patients receiving efavirenz is at codon 103. Additional mutations have been reported at codons 100, 106, 188, and 190. Absorption, Distribution, and Elimination Efavirenz is well absorbed from the gastrointestinal tract and reaches peak plasma concentrations within 3 to 4 hours. The proportion absorbed decreases with increasing doses; bioavailability is increased by a high-fat meal. Its long half-life allows once-daily dosing of efavirenz. Its low CSF-to-plasma ratio of 0.01 may reflect the drug being highly bound to plasma proteins (Tashima et al., 1999; Villani et al., 1999). Efavirenz is a substrate for cytochrome P450 isoforms, particularly CYP3A4 and CYP2B6. The 8-hydroxy metabolite is excreted in the urine, and the glucuronide conjugate of 8-hydroxy-efavirenz is present in plasma and urine. Sixty percent of the dose is excreted in urine as the glucuronide conjugate (Villani et al., 1999). Untoward Effects Common side effects of efavirenz include headache, dizziness, abnormal dreams, impaired concentration, and rash. Central nervous symptoms usually occur with the first dose and may last for hours. More severe symptoms usually resolve over several weeks. As many as 52% of patients report some central nervous system or psychiatric side effects, but less than 5% discontinue drug for this reason. Rash develops in up to 27% of patients, usually within the first 1 or 2 weeks; it is mild and rarely requires drug discontinuation. Serious rashes are uncommon. Increased liver enzymes, elevated lipid levels, and false positive screening tests for marijuana metabolites have been reported (Adkins and Noble, 1998). Although teratotoxicity studies in rats and rabbits demonstrated no significant effects, when efavirenz was administered to pregnant cynomolgus monkeys, 25% of fetuses developed malformations. Women of childbearing potential should use two methods of birth control to avoid pregnancy when taking efavirenz. Drug Interactions and Precautions Efavirenz may decrease levels of phenobarbital, phenytoin, carbamazepine, and methadone by inducing cytochrome P450 isoforms. Rifampin levels are unchanged by concurrent administration, but it may reduce levels of efavirenz. Rifabutin levels are reduced by efavirenz. The effect of efavirenz on protease inhibitor concentrations is variable (Table 514). Indinavir, saquinavir, and amprenavir levels are reduced by efavirenz, but ritonavir and nelfinavir levels are increased (Adkins and Noble, 1998). Drugs that induce CYP3A4 (e.g., phenobarbital, phenytoin, carbamazepine) would be expected to increase the clearance of efavirenz and lower its plasma levels. Therapeutic Use Efavirenz is approved for treating HIV-1 infection in combination with other antiretroviral agents. It was the first antiretroviral agent approved by the FDA for once-daily administration. Initial short-term monotherapy studies showed significant antiviral effects of efavirenz. Later studies evaluated efavirenz in multidrug combinations in HIV-infected adults and children. In antiretroviral-nave patients, suppression of HIV-1 RNA to undetectable levels occurred in 70% of patients receiving efavirenz, zidovudine, and lamivudine and in 48% of those receiving indinavir plus zidovudine and lamivudine (Staszewski et al., 1999). Much of this difference reflected greater patient compliance with the former regimen. Efavirenz prescribed in multiple-drug combinations also has demonstrated activity in patients failing prior regimens (Falloon et al., 2000; Piketty et al., 1999). When children failing prior therapy with a nucleoside analog reverse transcriptase inhibitor were treated with a combination of efavirenz, nelfinavir, and a nucleoside analog, 60% had a sustained antiviral benefit at 48 weeks (Starr et al., 1999). |
Protease Inhibitors
|
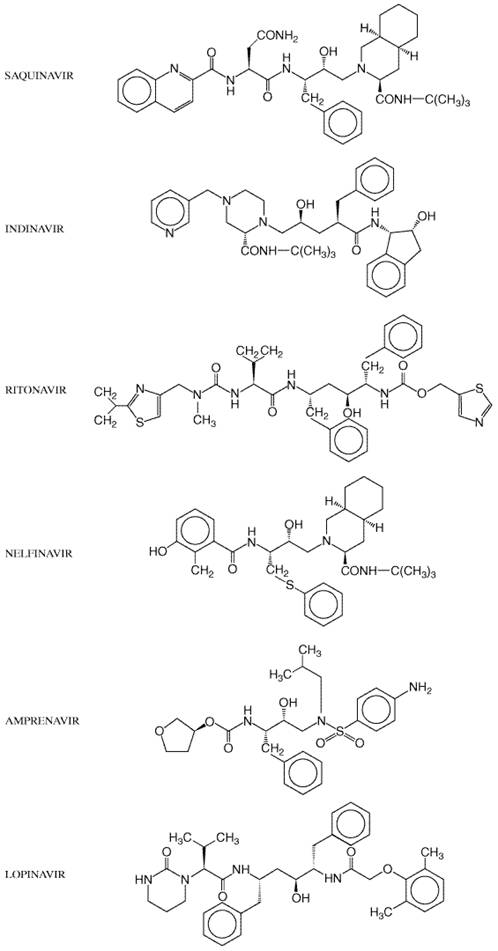
A review of available clinical trials of HIV-1 protease inhibitors is beyond the scope of this chapter (Flexner, 2000). Although selected important studies are cited in the text, the reader is referred elsewhere for a summary of many important clinical trials (Tavel et al., 1999). Shared Features Mechanism of Action and Resistance The HIV-1 protease is a dimer consisting of two, 99-amino acid monomers; each monomer contributes an aspartic acid to form the catalytic site (Pearl and Taylor, 1987). In contrast, human aspartyl proteases (renin, gastricsin, and cathepsin D/E) contain only one polypeptide chain. Such structural differences allow the HIV protease inhibitors to have greater than 1000-fold higher affinity for HIV protease than for human aspartyl proteases. The HIV protease is essential for viral infectivity (Kohl et al., 1988) and cleaves the viral polyprotein (gag-pol) into active viral enzymes (reverse transcriptase, protease, and integrase) and structural proteins (p17, p24, p9, and p7). Its preferred cleavage site is the N-terminal side of proline residues, especially between phenylalanine and proline. All six available protease inhibitors (amprenavir, indinavir, nelfinavir, ritonavir, nelfinavir, and lopinavir) act by binding reversibly to the active site of HIV protease. This prevents the protease from cleaving the viral precursor polypeptide and blocks subsequent viral maturation. Cells incubated in the presence of HIV protease inhibitors produce viral particles that are immature and noninfectious. As with all antiretroviral agents, viral replication in the presence HIV protease inhibitors ultimately will select for drug-resistant virus. Suboptimal plasma protease inhibitor concentrations predispose to viral breakthrough and resistance (Schapiro et al., 1996). Although each drug selects for different mutations of the protease gene, resistance to one HIV protease inhibitor often predicts less-favorable clinical responses to other subsequently prescribed protease inhibitors regardless of resistance test results. Resistance to protease inhibitors generally occurs by stepwise accumulation of mutations of the protease gene. It is thought that initial mutations confer low-level resistance that allows the virus to replicate inefficiently in the presence of drug. Additional mutations may be compensatory, restoring more efficient replication and conferring high-level resistance. Resistance mutations also may alter the cleavage sites of gag-pol. Almost all approved agents of this class are potent inhibitors of HIV-1 replication, and monotherapy lowers plasma HIV-1 RNA levels approximately 100- to 1000-fold within 4 to 12 weeks. The exception is a hard-gelatin capsule dosage form of saquinavir, which has poor bioavailability unless administered with ritonavir. Otherwise, relative efficacies of individual agents are uncertain, because comparative studies generally are lacking. When any potent protease inhibitor has been administered with two nucleoside analogs during clinical trials, 60% to 95% of antiretroviral-nave patients have achieved plasma HIV RNA levels below limits of detection. In this situation, many (if not most) therapeutic failures likely are due to poor patient adherence. Absorption, Distribution, and Elimination Most HIV protease inhibitors have poor systemic bioavailability. Amprenavir, indinavir, ritonavir, nelfinavir, saquinavir, and lopinavir all undergo oxidative metabolism by CYP3A4, and additional CYP isoforms metabolize individual protease inhibitors. Metabolism occurs predominantly in the liver, but metabolism by intestinal epithelial cells also may decrease bioavailability. The greater than 10- to 20-fold range in activity of various isoforms among individuals may contribute to variable drug pharmacokinetics (Rendic and Di Carlo, 1997). Only nelfinavir has an active metabolite, which is generated by CYP2C19. All HIV protease inhibitors also are substrates for P-glycoprotein, the multidrug efflux pump encoded by MDR 1. This may limit cellular penetration and tissue delivery. P-glycoprotein in capillary endothelial cells of the bloodbrain barrier may limit drug penetration into the brain (Kim et al., 1998). P-glycoprotein also is present in bile canaliculi, small intestine epithelia, renal tubules, testes, and the placenta. Most protease inhibitors penetrate less well into semen than do nucleoside reverse transcriptase inhibitors and nonnucleoside reverse transcriptase inhibitors, although virologic response in plasma and semen usually are concordant (Taylor et al., 1999). Since little drug is excreted unchanged by the kidneys, dose adjustments for renal dysfunction are generally unnecessary (Jayasekara et al., 1999). The six FDA-approved HIV protease inhibitors are listed in Table 511, and their chemical structures are shown in Figure 514. Selected pharmacokinetic parameters for individual protease inhibitors are summarized in Table 515.
Protein Binding The HIV protease inhibitors bind extensively to normal plasma
proteins, especially Untoward Effects Toxicities of protease inhibitors include nausea, vomiting, diarrhea, and paresthesias. These agents also may cause glucose intolerance, diabetes, hypercholesterolemia, and hypertriglyceridemia. Prolonged administration of HIV protease inhibitors has been associated with fat redistribution, especially central fat accumulation, in some patients. This includes increased visceral fat and abdominal girth, an enlarged fat pad at the base of the neck ('buffalo hump'), breast enlargement, and/or subcutaneous lipomas. Current studies are defining the epidemiology, mechanisms, and optimal management of this complication. Precautions and Interactions Since all HIV protease inhibitors are both substrates and inhibitors
of CYP isoforms, drug interactions are common. The rank order of CYP3A4
inhibitory potency is ritonavir A summary of interactions between individual protease inhibitors is presented in Table 516; interactions between protease inhibitors and nonnucleoside reverse transcriptase inhibitors are summarized in Table 514. Saquinavir Chemistry and Antiviral Activity Saquinavir is a peptidomimetic HIV protease inhibitor of the hydroxyethylamine class developed by rational drug design (Roberts et al., 1990). Once it was recognized that some sites cleaved by HIV-1 protease were unique compared to sites cleaved by eukaryotic protease, peptides were synthesized to mimic the transition state of the phenylalanine-proline cleavage site in the viral polyprotein. Saquinavir is a potent inhibitor of both HIV-1 and HIV-2 proteases. In peripheral blood lymphocytes, its IC50 ranges from 3.5 to 10 nM (Craig et al., 1991). Mechanisms of Action and Resistance HIV protease cleaves the viral polyprotein (gag-pol) into active enzymes and structural proteins. Saquinavir reversibly binds to the active site of HIV protease, preventing polypeptide processing and subsequent viral maturation. Viral particles produced in the presence of saquinavir are immature and noninfectious. Viral replication in the presence of saquinavir selects for drug-resistant virus. Among patients treated with saquinavir, resistance has been associated with progressive accumulation of resistance mutations over time. The most common mutation associated with saquinavir resistance is at protease codon 90, followed in frequency by codon 48 (Ives et al., 1997). With prolonged administration, additional mutations at positions 36, 46, 82, and 84 occur and are associated with cross-resistance to other protease inhibitors. Absorption, Distribution, and Elimination Oral bioavailability of the hard-gelatin capsule formulation of saquinavir (saquinavir mesylate, INVIRASE) is only, 4% due to limited absorption and extensive first-pass metabolism, with considerable interpatient variability (Noble and Faulds, 1996). Limited antiviral efficacy is achieved at the approved dosage, yielding only a fivefold reduction in plasma HIV-1 RNA. A greater effect is achieved by administering 7200 mg daily in six divided doses, four times the approved dose, with a strong correlation between plasma drug concentration and therapeutic effect (Schapiro et al., 1996). A soft-gelatin capsule formulation (FORTOVASE) with approximately threefold increased oral bioavailability became available in 1997 (Perry and Noble, 1998). Absorption of saquinavir may be enhanced when the drug is taken with a high-calorie, high-fat meal. Saquinavir's short half-life requires administration every 8 hours. In addition, saquinavir demonstrates a greater than dose-proportional increase in exposure. For example, tripling the oral dose of saquinavir is associated with an eightfold increase in exposure. Substances that inhibit intestinal but not hepatic CYP3A4, such as grapefruit juice, increase the saquinavir AUC by threefold at most (Flexner, 2000). Saquinavir is metabolized primarily by hepatic CYP3A4 (Fitzsimmons and Collins, 1997). The metabolites of saquinavir are not active against HIV-1. Saquinavir and its metabolites are eliminated from the body primarily through the biliary system and feces (more than 95% of drug), with minimal urinary excretion (less than 3% of administered drug). Untoward Effects The most frequent side effects of both saquinavir formulations are gastrointestinal, and include nausea, vomiting, diarrhea, and abdominal discomfort. Most side effects of saquinavir are mild. A causal relationship between other adverse effects and saquinavir is less clear. Precautions and Interactions Although saquinavir is a weak inhibitor of CYP3A4, it should not be prescribed with ergot derivatives, cisapride, triazolam, or midazolam. Decreased clearance of these agents because of CYP3A4 inhibition may cause life-threatening cardiac arrhythmias or prolonged sedation, depending on the drug. Among the HIV protease inhibitors, saquinavir is particularly susceptible to increased clearance due to CYP3A4 induction. Coadministration of nevirapine or efavirenz lowers saquinavir levels considerably, so coadministration of these drugs should be avoided. The ability of nevirapine or efavirenz to lower saquinavir levels may be overcome when ritonavir is coadministered. Coadministering ritonavir with saquinavir markedly increases plasma saquinavir concentrations, presumably by inhibiting CYP3A4 (Flexner, 2000). In a single-dose study, ritonavir increased the saquinavir AUC by 50- to 132-fold (Hsu et al., 1998). This is a commonly prescribed dual protease inhibitor combination. Therapeutic Use In 1995, saquinavir became the first protease inhibitor approved by the FDA for treating HIV infection. Among patients with susceptible strains, soft-gelatin capsules of saquinavir substantially lower plasma HIV-1 RNA concentrations. In early clinical trials, hard-gelatin capsules of saquinavir mesylate (600 mg 3 times daily) demonstrated only modest virologic effect because of poor oral bioavailability. Greater activity was achieved with high-dose therapy of 1200 mg given 6 times daily (Schapiro et al., 1996). The soft-gelatin-capsule formulation (1200 mg every 8 hours) appears to have antiviral activity comparable to other HIV protease inhibitors. Saquinavir is commonly prescribed in combination with ritonavir because of a favorable pharmacokinetic interaction (see'Precautions and Interactions,' above). Indinavir Chemistry and Antiviral Activity Indinavir is a peptidomimetic hydroxyethylamine HIV protease inhibitor (Plosker and Noble, 1999). It is formulated as the sulfate salt to yield more consistent plasma concentrations than with the free base following oral administration. Indinavir is tenfold more active against the protease of HIV-1 than that of HIV-2, and its 95% inhibitory concentration (IC95) for wild-type HIV-1 ranges from 25 to 100 nM. The lead developmental compound for indinavir was a renin inhibitor that mimicked the phenylalanine-proline cleavage site in the viral polyprotein. Chemical modifications enhanced its antiretroviral activity and oral absorption, ultimately leading to the discovery of indinavir (Vacca et al., 1994). Mechanisms of Action and Resistance Indinavir reversibly binds to the active site of HIV protease, preventing viral polypeptide processing and viral maturation. Viral particles produced in the presence of indinavir are immature and noninfectious. Viral replication in the presence of indinavir selects for drug-resistant virus. Clinical indinavir resistance has been associated with progressive accumulation of mutations at protease codons 10, 20, 24, 46, 54, 63, 71, 82, 84, and 90. The mutations of primary importance are at positions 46 and 82. High-level resistance requires multiple mutations. Absorption, Distribution, and Elimination Indinavir is rapidly absorbed after oral administration, with peak levels achieved in approximately 0.8 hour. Its aqueous solubility is much greater at pH 3.5 than at 7.4 (Lin et al., 1995). Absorption is unaffected by light, low-fat meals, but high-calorie, high-fat, high-protein meals reduce absorption by, 75%. Therefore, indinavir must be taken while fasting or with a low-fat meal (e.g., corn flakes with skim milk and sugar). Its short half-life makes three-times-daily (every 8 hours) dosing necessary. Dietary restrictions and frequent dosing make patient compliance a challenge. Coadministration with ritonavir overcomes the effect of food and may allow twice-daily dosing. Indinavir undergoes extensive hepatic metabolism by CYP3A4. Glucuronidation, oxidation, and N-acetylation produce at least six major metabolites. These achieve appreciable plasma concentrations, but none has documented antiretroviral activity. Indinavir and its metabolites are eliminated primarily in feces (81% of unchanged drug and metabolites) and urine (19% of unchanged drug and metabolites). Plasma indinavir levels increase with moderate liver disease, and dose reduction may be required. Indinavir is only about 60% bound to plasma proteins, considerably less than other available protease inhibitors. The fact that only free drug is directly available for transfer to other compartments may explain the relatively high CSF-to-plasma AUC ratio of 0.07 for total indinavir, and 0.15 for free drug (Haas et al., 2000c). Only 5% of drug is protein bound in CSF. Untoward Effects A unique adverse effect of indinavir is crystalluria. Precipitation of indinavir and its metabolites in urine may cause renal colic. Nephrolithiasis occurs in approximately 3% of patients. To reduce this risk, patients must drink sufficient fluids to maintain dilute urine. Prolonged administration of indinavir may be associated with fat redistribution in some patients. Hair and skin problems also have been observed with indinavir, including hair loss, dry skin, dry and cracked lips, and ingrown toenails (Bouscarat et al., 1999, 1998). Gastrointestinal disturbances and central nervous system symptoms (headache, insomnia) also can occur. Precautions and Interactions Persons receiving indinavir should drink at least 72 fluid ounces of fluid daily. Indinavir-induced renal colic usually resolves with vigorous hydration and without discontinuing therapy. Some cases require drug discontinuation. Since indinavir solubility markedly decreases at higher pH, antacids or other buffering agents should not be taken at the same time. Didanosine is coformulated with a buffer and should not be taken within one hour of indinavir. Since indinavir is metabolized by CYP3A4 and also is a modest inhibitor of the enzyme, drugs most prone to interactions with indinavir either induce, inhibit, or are metabolized by CYP3A4. Drugs that induce CYP3A4 activity may lower indinavir levels. Rifampin lowers the indinavir AUC by 90%, and is contraindicated. Efavirenz, nevirapine, and rifabutin lower indinavir levels less substantially (25% to 35%) and necessitate an increased indinavir dose. Indinavir can inhibit the metabolism of other CYP3A4 substrates. Indinavir raises rifabutin concentrations, increasing toxicity. The rifabutin daily dose in regimens containing indinavir therefore should be reduced by 50%. Indinavir may inhibit metabolism of cisapride, triazolam, and midazolam, causing cardiac arrhythmias or prolonged sedation, depending on the drug (Plosker and Noble, 1999). Ritonavir increases the indinavir plasma AUC and trough concentrations by inhibiting CYP3A4. This may allow indinavir to be administered twice daily rather than three times daily, and may allow lower total daily doses. Delavirdine increases indinavir plasma concentrations, unlike other nonnucleoside reverse transcriptase inhibitors. Therapeutic Use Indinavir is indicated for treating HIV infection in adults and children. Among patients with susceptible strains of HIV-1, indinavir therapy substantially lowers plasma HIV-1 RNA levels. Large clinical trials have demonstrated both virologic and survival benefit when patients with AIDS received indinavir three times daily in combination with zidovudine and lamivudine (Gulick et al., 1997; Hammer et al., 1997). However, twice-daily administration of the same total daily dose was less efficacious (Haas et al., 2000a). Various clinical trials currently are evaluating the use of indinavir in combination with ritonavir, since this combination is predicted to yield at least equal efficacy with less frequent administration of indinavir. Ritonavir Chemistry and Antiviral Activity Ritonavir is a peptidomimetic hydroxyethylamine HIV protease inhibitor. Ritonavir is more active against HIV-1 than against HIV-2, and its IC50 for wild-type HIV variants in MT4 cells in the presence of, 50% human serum is approximately 45 nM. Mechanisms of Action and Resistance Ritonavir reversibly binds to the active site of HIV protease, preventing polypeptide processing and subsequent viral maturation. Viral particles produced in the presence of ritonavir are immature and noninfectious. In patients treated with ritonavir, viral replication in the presence of drug selects for progressive accumulation of drug-resistance mutations (Molla et al., 1996). The first ritonavir resistance mutation is usually at protease codon 82. Additional mutations associated with increasing resistance occur at codons 20, 32, 46, 54, 63, 71, 84, and 90. High-level resistance requires multiple mutations. Absorption, Distribution, and Elimination Absorption of ritonavir is only slightly affected by diet, and this is somewhat dependent on the formulation. The overall absorption of ritonavir from the capsule formulation may increase by 15% when taken with meals. Its half-life allows twice-daily administration. There is greater than sixfold variability in drug trough concentrations among patients given 600 mg of ritonavir every 12 hours (Danner et al., 1995). Ritonavir is metabolized primarily in the liver by cytochrome P450 isoforms, especially CYP3A and less so by CYP2D6. Ritonavir and its metabolites are eliminated from the body predominantly in the feces (86% of unchanged drug and metabolites), with minor urinary elimination (11%, mostly metabolites). Ritonavir in Dual-Protease-Inhibitor Regimens Ritonavir initially was developed as a potent protease inhibitor and produced clinical benefit in patients with advanced AIDS. Its pronounced inhibitory effect on CYP3A4 was viewed as a detriment, because this would reduce elimination of many drugs. For example, delayed clearance of midazolam by ritonavir causes prolonged sedation and respiratory depression. However, coadministration with ritonavir also increases the plasma AUCs, half-lives, and trough concentrations of saquinavir, indinavir, amprenavir, and lopinavir (Table 516) (Flexner, 2000). This effect can allow lower doses of a second protease inhibitor to be used. Using ritonavir as a 'pharmacokinetic enhancer' to increase plasma concentrations of other HIV protease inhibitors has become common practice, and many clinical trials are in progress. For drugs with limited oral bioavailability, such as saquinavir and lopinavir, ritonavir markedly increases plasma drug levels and enhances antiretroviral effect, and lopinavir is available only as a coformulation with ritonavir. Ritonavir also overcomes the deleterious effect of food on indinavir bioavailability. Untoward Effects Side effects of ritonavir are dose-dependent, and include gastrointestinal complaints, such as nausea, diarrhea, anorexia, abdominal pain, and taste perversion. Peripheral and perioral paresthesias also are common. Ritonavir induces its own metabolism, and gradual dose escalation over the first two weeks minimizes early intolerance when the drug is prescribed at full dose. Ritonavir causes dose-dependent elevations in serum cholesterol and triglycerides, raising the concern that ritonavir therapy will increase the long-term risk of atherosclerosis. Precautions and Interactions To minimize intolerance during the initial weeks of therapy when prescribed at maximal approved doses for adults and adolescents, ritonavir should be initiated at 300 mg every 12 hours, and gradually escalated to 600 mg every 12 hours by day 14 of therapy. It is better tolerated if taken with meals. Ritonavir potently inhibits CYP3A4, markedly increasing plasma levels
of many drugs including amiodarone, propafenone, ergot derivatives, pimozide,
cisapride, triazolam, and midazolam. Coadministration of ritonavir with these
agents is contraindicated because it may cause life-threatening cardiac
arrhythmias or prolonged sedation, depending on the drug (Plosker and Noble, 1999).
Ritonavir may increase rifabutin concentrations markedly and cause toxicity.
This combination therefore should be avoided. Concomitant administration of
agents that induce CYP3A4 activity such as rifampin may lower ritonavir
levels and also should be avoided. Therapeutic Use Ritonavir is indicated for treating HIV infection in adults and children. Among patients with susceptible strains of HIV-1, ritonavir substantially lowers plasma HIV-1 RNA levels. Clinical trials have demonstrated both virologic benefit and improved survival when patients with advanced disease were treated with ritonavir. Ritonavir was the first HIV protease inhibitor to have survival benefit (Tavel et al., 1999). Numerous clinical trials have demonstrated the efficacy of ritonavir in various dual-protease-inhibitor regimens (Gisolf et al., 2000). Nelfinavir Chemistry and Antiviral Activity Traditional development of antiviral agents was based on screening thousands of compounds for viral activity in the laboratory. In contrast, nelfinavir was developed by rational drug design (Roberts et al., 1990). Nelfinavir is a nonpeptide protease inhibitor that is active against both HIV-1 and HIV-2. and is formulated as the mesylate salt of a basic amine (Bardsley-Elliot and Plosker, 2000). The mean IC95 for HIV-1 in various in vitro assays is 59 nM. Mechanisms of Action and Resistance Nelfinavir inhibits protease by reversibly binding to the active site, preventing polypeptide processing and subsequent viral maturation. Viral particles produced in the presence of nelfinavir are immature and noninfectious. Viral replication in the presence of nelfinavir can select for drug-resistant virus. The central resistance mutation associated with resistance to nelfinavir is at protease codon 30. Isolates with only this mutation still may be inhibited by other protease inhibitors. However, additional mutations are associated with decreased susceptibility to nelfinavir and to other protease inhibitors, including those at positions 35, 36, 46, 71, 77, 88, and 90. Absorption, Distribution, and Elimination Nelfinavir is absorbed more slowly than are other HIV-1 protease inhibitors, with peak levels achieved in 2 to 4 hours. In addition, intraindividual and interindividual variability in plasma nelfinavir levels is considerable. Administration with food increases the plasma AUC of nelfinavir two- to threefold. The drug is FDA-approved for clinical use as a three-times-daily agent, although pharmacokinetic and some clinical data suggest that it may be effective at a dose of 1250 mg given twice daily. Nelfinavir undergoes oxidative metabolism in the liver primarily by CYP3A4, but also by CYP2C19 and CYP2D6. Its major hydroxy-t-butylamide metabolite (M8) has in vitro antiretroviral activity comparable to that of the parent drug but achieves plasma levels that are only 40% of nelfinavir levels. The M8 metabolite is generated primarily by CYP2C19. Nelfinavir and its metabolites are eliminated primarily in feces, with less than 2% of drug being excreted in the urine. Moderate or severe liver disease may prolong the half-life and increase plasma concentrations of the parent drug while lowering plasma concentrations of M8. Nelfinavir induces CYPs involved in its metabolism, and average trough plasma concentrations after one week of therapy are approximately one-half of those at day 2 of therapy. Steady-state plasma levels may be achieved at about one week. Nelfinavir inhibits CYP3A4 less than does ritonavir and does not appear to inhibit other CYP isoforms. Nelfinavir is greater than 98% bound to plasma proteins, mostly to albumin
and Untoward Effects A common side effect of nelfinavir is diarrhea or loose stools. This is generally mild, and less than 2% of patients discontinue drug due to diarrhea. Other side effects associated with nelfinavir as well as other HIV protease inhibitors include diabetes, glucose intolerance, elevated triglycerides, and elevated cholesterol levels. Precautions and Interactions Since nelfinavir is metabolized by CYP3A4, concomitant administration of agents that induce CYP3A4 may be contraindicated (e.g., rifampin) or may necessitate an increased nelfinavir dosage (e.g., rifabutin). In addition, nelfinavir may alter plasma concentrations of drugs that are metabolized through CYP3A4. Nelfinavir lowers the delavirdine plasma AUC by 42%. In addition, levels of midazolam, triazolam, ergot derivatives, amiodarone, and quinidine may be increased by nelfinavir, and coadministration of these drugs with nelfinavir should be avoided. Therapeutic Use Nelfinavir is indicated for the treatment of HIV infection in adults and children. Among patients with susceptible strains of HIV-1, initial studies of nelfinavir monotherapy produced substantial decreases in plasma HIV-1 RNA concentrations. Large clinical trials have demonstrated both virologic and clinical benefit when patients nave to HIV protease inhibitors and lamivudine received nelfinavir in combination with zidovudine and lamivudine. Amprenavir Chemistry and Antiviral Activity Amprenavir is an N, N-disubstituted (hydroxyethyl) amino sulfonamide nonpeptide HIV protease inhibitor (Adkins and Faulds, 1998). It was developed from a structure-based drug design program that utilized the known crystal structure of HIV-1 to create progressively smaller and more potent inhibitors, and it is the only available HIV protease inhibitor that is a sulfonamide. Amprenavir is active against both HIV-1 and HIV-2, with an IC90 for wild-type HIV-1 of approximately 80 nM. Mechanisms of Action and Resistance Amprenavir acts by reversibly binding to the active site of HIV protease. This prevents polypeptide processing and subsequent viral maturation. Viral particles produced in the presence of lopinavir are immature and noninfectious. Viral replication in the presence of amprenavir selects for drug-resistant virus. A mutation at protease codon 50 is central to amprenavir resistance and confers twofold decreased susceptibility. Accumulation of additional mutations at codons 46 and 47 greatly increase resistance (Partaledis et al., 1995; Tisdale et al., 1995). Absorption, Distribution, and Elimination Amprenavir is absorbed rapidly after oral administration. Taking amprenavir with a standard meal reduces the plasma AUC by only about 13%, but high-fat meals may have greater effects and should be avoided. Ritonavir increases amprenavir concentrations considerably by inhibiting CYP3A4 and allows lower amprenavir doses to be administered. Coadministration with low doses of ritonavir may allow the total daily dose of amprenavir to be reduced by one-half. Amprenavir undergoes limited hepatic metabolism by CYP3A4 and is
excreted primarily via the biliary route. Amprenavir also is a modest
inhibitor of CYP3A4 and CYP2C19. In plasma, amprenavir is 90% bound to
proteins, mostly Untoward Effects The most common adverse effects associated with amprenavir are nausea, vomiting, diarrhea or loose stools, hyperglycemia, fatigue, paresthesias, and headache. In one study of amprenavir monotherapy, rash occurred in 5 of 35 patients over 24 weeks, and began within 7 to 12 days of starting therapy. Precautions and Interactions Drugs that induce CYP3A4 activity (e.g., rifampin and efavirenz) will lower plasma amprenavir levels, whereas plasma concentrations of some agents metabolized by CYP3A4 will increase if the drugs are coadministered with amprenavir (e.g., ketoconazole, carbamazepine, sildenafil, midazolam, triazolam). Therapeutic Use Amprenavir is indicated for the treatment of HIV infection in adults and children, in combination with other agents. Among patients with susceptible strains of HIV-1, amprenavir monotherapy substantially lowers plasma HIV-1 RNA levels. Clinical trials have demonstrated virologic benefit when patients nave to HIV protease inhibitors and lamivudine were treated with amprenavir (1200 mg every 12 hours) in combination with zidovudine and lamivudine. A number of clinical trials are evaluating amprenavir in combination with ritonavir. Lopinavir Chemistry and Antiviral Activity Lopinavir (ABT-378) is a peptidomimetic HIV protease inhibitor that is structurally similar to ritonavir, but is tenfold more potent against HIV-1 in vitro in the presence of human serum. Lopinavir is active against both HIV-1 and HIV-2, with an IC50 for wild-type HIV variants of approximately 100 nM. Mechanisms of Action and Resistance Lopinavir acts by binding reversibly to the active site of HIV protease, thus preventing polypeptide processing and subsequent viral maturation. Viral particles produced in the presence of amprenavir are immature and noninfectious. As with all antiretroviral agents, viral replication in the presence of lopinavir will select for drug-resistant virus. Lopinavir resistance mutations have not been well defined. Absorption, Distribution, and Elimination When administered orally without ritonavir, lopinavir plasma concentrations are very low. However, metabolism of lopinavir by CYP3A4 is exquisitely sensitive to inhibition by ritonavir. To overcome its inherent poor bioavailability, lopinavir is coformulated with ritonavir in a fixed dosage combination. (Lopinavir is available only in this coformulation, which will be referrred to hereafter as lopinavirR.) Lopinavir is rapidly absorbed after oral administration. Although the capsules contain lopinavir/ritonavir in a fixed 4:1 ratio, the observed plasma concentration ratio following oral administration is nearly 20:1, reflecting the sensitivity of lopinavir to the inhibitory effect of ritonavir on CYP3A4. Lopinavir undergoes extensive hepatic oxidative metabolism by CYP3A4. Most metabolites are eliminated in feces, and 89% of total drug in plasma is the parent compound. Although lopinavir is a weak inhibitor of CYP3A4, the ritonavir in the coformulated capsule strongly inhibits CYP3A4 activity. Untoward Effects LopinavirR generally is very well tolerated. The most common adverse events have been gastrointestinal and include abnormal stools, diarrhea, and nausea. The most common laboratory abnormalities include elevated cholesterol and triglycerides. Precautions and Interactions Since lopinavir is metabolized predominantly by CYP3A4, concomitant
administration of agents that strongly induce CYP3A4, such as rifampin, may
lower plasma lopinavir concentrations considerably, and such drugs should be
avoided. Coadministration of nevirapine or efavirenz may require increasing
the dose of lopinavirR. Therapeutic Use LopinavirR has been approved by the FDA for the treatment of HIV infection in adults and children. In clinical trials, lopinavirR has had antiretroviral activity at least comparable to that of other potent HIV protease inhibitors. It also has had considerable and sustained antiretroviral activity in patients who were not responding to other HIV protease inhibitors. In one study, 70 subjects who had failed therapy with one previous HIV protease inhibitor were treated for 2 weeks with lopinavirR, after which nevirapine was added. These patients had relatively low baseline plasma HIV RNA levels. At 48 weeks, 60% of subjects had plasma HIV-1 RNA levels of less than 50 copies/ml, despite substantial phenotypic resistance to other HIV protease inhibitors. It has been postulated that the very high plasma levels of lopinavir overcome such resistance. |
Prospectus
|
Emergence of multidrug-resistant virus among patients treated with current anti-AIDS agents has created an urgent need for new antiretroviral agents, including ones that target molecules other than reverse transcriptase and protease. A number of agents in development are shown in Table 517. Potential steps to target include binding, fusion, integration, and assembly (see Figure 511). In addition, antagonists of regulatory molecules such as tat possibly could be developed. It is hoped that at least some agents directed at these novel targets will control replication of drug-resistant HIV, have fewer side effects, and/or require less frequent dosing and fewer capsules or tablets than currently available agents. Drugs that target HIV fusion with the cell membrane are furthest in clinical development. Investigators serendipitously discovered that peptides corresponding to short sequences of gp41 inhibited HIV entry into cells (Jiang et al., 1993; Wild et al., 1994). These peptides disrupt the interaction of the N36 and C34 sequences of the gp41 glycoprotein. One such 36-amino acid peptide known as T20 (also called DP178) binds to the hydrophobic groove on the N36 coiled coil, and effectively blocks HIV-1 entry into target cells with an IC50 of 50 to 60 nM (Sodroski, 1999). In the first treatment trial, administration of T20 monotherapy to 16 HIV-infected individuals reduced plasma HIV-1 RNA by a median of 2 log10 copies per milliliter after two weeks (Kilby et al., 1998), which compares favorably with standard combination therapy (Richman, 1998; Rimsky et al., 1998; Sodroski, 1999). Resistance to T20 emerged due to mutations in gp41 (Rimsky et al., 1998). Studies of T20 in combination with other agents are in progress. Since T20 is a large peptide, it is not orally bioavailable and must be administered by subcutaneous or intravenous injection twice daily. Efforts to develop small, orally bioavailable HIV fusion inhibitors are under way. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1335
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved