| CATEGORII DOCUMENTE | ||
|
||
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Drugs Used in the Chemotherapy of Protozoal Infections: Malaria
Overview
|
Malaria, caused by four species of Plasmodium, of which Plasmodium falciparum is the most dangerous, remains the world's most devastating human parasitic infection. This chapter deals with the properties and uses of important drugs used to treat and prevent this infection. Highly effective agents that act against asexual erythrocytic stages of malarial parasites responsible for clinical attacks include chloroquine, quinine, quinidine, mefloquine, atovaquone, and the artemisinin compounds. Less effective, slower-acting drugs in this category are proguanil, pyrimethamine, sulfonamides, sulfones, and the antimalarial antibiotics. Primaquine is the only drug used against latent tissue forms of Plasmodium vivax and Plasmodium ovale that cause relapsing infections. No single antimalarial agent has successfully controlled the spread of increasingly drug-resistant strains of P. falciparum. Instead, multidrug regimens are discussed as the optimal strategy to address this problem. |
Drugs Used in the Chemotherapy of Protozoal Infections: Malaria: Introduction
|
Malaria remains the world's most devastating
human parasitic infection, afflicting more than 500 million people and
causing from 1.7 million to 2.5 million deaths each year (World Health
Organization, 1997). Infection with Plasmodium falciparum causes much
of this mortality, which preferentially affects children less than 5 years of
age, pregnant women, and nonimmune individuals. Although mosquito-transmitted
malaria virtually has been eliminated from North America, Europe, and Practical, inexpensive, effective, and safe drugs, insecticides, and
vaccines still are needed to combat malaria. In the 1950s, attempts to
eradicate this scourge from most parts of the world failed, primarily because
of the development of resistance to insecticides and antimalarial drugs.
Since 1960, transmission of malaria has risen in most regions where the
infection is endemic; chloroquine-resistant and multidrug-resistant strains
of P. falciparum have spread, and the degree of drug resistance has
increased. More recently, chloroquine-resistant strains of P. vivax
also have been documented in Nearly all antimalarial drugs were developed because of their action against asexual erythrocytic forms of malarial parasites that cause clinical illness. Efficacious, rapidly acting drugs in this category include chloroquine, quinine, quinidine, mefloquine, atovaquone, and the artemisinin compounds. Proguanil, pyrimethamine, sulfonamides, sulfones, and antimalarial antibiotics, such as the tetracyclines, are slower acting and less effective. Primaquine is the only drug used clinically to eradicate latent tissue forms that cause relapses of P. vivax and P. ovale infections. Due to the continuing spread of increasingly drug-resistant and multidrug-resistant strains of P. falciparum, no single agent successfully controls infections with these parasites. Instead, use of two or more antimalarial agents with complementary properties is recommended (seeWhite, 1997, 1999). The discovery of techniques for continuous maintenance of P. falciparum in vitro (Trager and Jensen, 1976) led to practical assays of susceptibility of these organisms to antimalarial drugs. This important advance, together with the imminent availability of the sequence of the entire 24.6-megabase P. falciparum genome (Su et al., 1999), should reveal molecular targets for antimalarial drug action and resistance as well as for vaccine development. The biology of malarial infection must be appreciated in order to understand the actions and therapeutic uses of antimalarial drugs. |
Biology of Malarial Infection
|
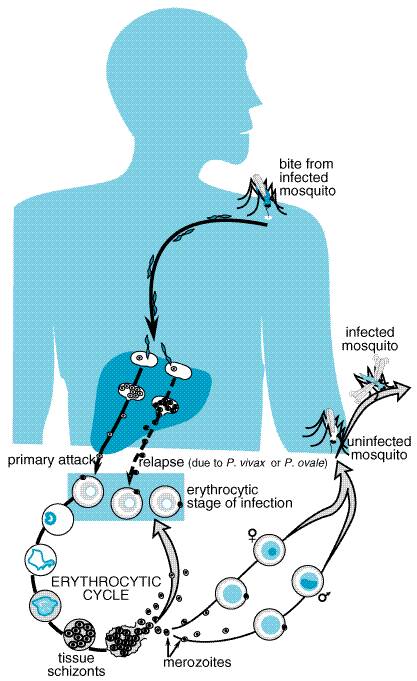
Nearly all human malaria is caused by four species of obligate intracellular protozoa of the genus Plasmodium. Although malaria can be transmitted by transfusion of infected blood and by sharing needles, human beings usually are infected by sporozoites injected by the bite of infected female mosquitoes (genus Anopheles). These parasite forms rapidly leave the circulation and localize in hepatocytes, where they transform, multiply, and develop into tissue schizonts (Figure 401). This primary asymptomatic tissue (preerythrocytic or exoerythrocytic) stage of infection lasts for 5 to 15 days, depending on the Plasmodium species. Tissue schizonts then rupture, each releasing thousands of merozoites that enter the circulation, invade erythrocytes, and initiate the erythrocytic stage of cyclic infection. Once the tissue schizonts burst in P. falciparum and Plasmodium malariae infections, no forms of the parasite remain in the liver. But in P. vivax and P. ovale infections, there persist tissue parasites that can produce relapses of erythrocytic infection months to years after the primary attack. The origin of such latent tissue forms is unclear. Once plasmodia enter the erythrocytic cycle, they cannot invade other tissues; thus, there is no tissue stage of infection for human malaria contracted by transfusion. In erythrocytes, most parasites undergo asexual development from young ring forms to trophozoites and finally to mature schizonts. Schizont-containing erythrocytes rupture, each releasing 6 to 24 merozoites, depending on the Plasmodium species. It is this process that produces febrile clinical attacks. The released merozoites invade more erythrocytes to continue the cycle, which proceeds until death of the host or modulation by drugs or acquired partial immunity. The periodicity of parasitemia and febrile clinical manifestations thus depend on the timing of schizogony of a generation of erythrocytic parasites. For P. falciparum, P. vivax, and P. ovale, it takes about 48 hours to complete this process. Synchronous rupture of infected erythrocytes and release of merozoites into the circulation lead to typical febrile attacks on days 1 and 3, hence the designation 'tertian malaria.' Actually the periodic febrile pattern is less regular in falciparum malaria due to a combination of asynchronous release of parasites and segregation of infected erythrocytes in the periphery. In P. malariae infection, schizogony requires about 72 hours, resulting in malarial attacks on days 1 and 4, or 'quartan malaria.'
Some erythrocytic parasites differentiate into sexual forms known as gametocytes. After infected human blood is ingested by a female mosquito, exflagellation of the male gametocyte is followed by male gametogenesis and fertilization of the female gametocyte in the insect's gut. The resulting zygote, which develops as an oocyst in the gut wall, eventually gives rise to the infective sporozoite, which invades the salivary gland of the mosquito. The insect then can infect another human host by taking a blood meal. Each Plasmodium species causes a characteristic illness and shows distinguishing morphological features in blood smears: (1) P. falciparum causes malignant tertian malaria, the most dangerous form of human malaria. By invading erythrocytes of any age, this species can produce an overwhelming parasitemia, sequestration of infected erythrocytes in the peripheral microvasculature, hypoglycemia, hemolysis, and shock with multiorgan failure. Delay in treatment until after demonstration of parasitemia may lead to a fatal outcome even after the peripheral blood is free of parasites. If treated early, the infection usually responds with 48 hours to appropriate chemotherapy. If treatment is inadequate, recrudescence of infection may result from multiplication of parasites that persist in the blood. (2) P. vivax causes benign tertian malaria. Like the other benign malarias, it produces milder clinical attacks than does P. falciparum, because erythrocytes it infects are not sequestered in the peripheral microvasculature. P. vivax infection has a low mortality rate in untreated adults and is characterized by relapses caused by latent tissue forms. (3) P. ovale causes a rare malarial infection with a periodicity and relapses similar to those of P. vivax, but it is even milder and more readily cured. (4) P. malariae causes quartan malaria, an infection that is common in localized areas of the tropics. Clinical attacks may occur years after infection but are much rarer than after infection with P. vivax. |
Classification of Antimalarial Agents
|
Antimalarials can be categorized by the stage of the parasite that they affect and the clinical indication for their use. Some drugs have more than one type of antimalarial activity. Drugs Used for Causal Prophylaxis These agents act on primary tissue forms of plasmodia within the liver, which are destined within less than a month to initiate the erythrocytic stage of infection. Invasion of erythrocytes and further transmission of infection are thereby prevented. Proguanil (formerly called chloroguanide) is the prototypic drug of this class, which has been extensively used for causal prophylaxis of falciparum malaria. Because of widespread drug resistance, however, it no longer provides reliable protection when used alone. Although primaquine also has such activity against P. falciparum, this potentially toxic drug is reserved for other clinical applications (seePrimaquine). Drugs Used to Prevent Relapse These compounds act on latent tissue forms of P. vivax and P. ovale remaining after the primary hepatic forms have been released into the circulation. Such latent tissue forms eventually mature, invade the circulation, and produce malarial attacks, i.e., relapsing malaria, months or years after the initial infection. Drugs active against latent tissue forms are used for terminal prophylaxis and for radical cure of relapsing malarial infections. For terminal prophylaxis, regimens with such a drug are initiated shortly before or after a person leaves an endemic area. To achieve radical cure, this type of drug is taken either during the long-term latent period of infection or during an acute attack. In the latter case, the agent is given together with an appropriate drug, usually chloroquine, to eradicate erythrocytic stages of P. vivax and P. ovale. Primaquine is the prototypical drug used to prevent relapse, the term reserved to specify recurring erythrocytic infection stemming from latent tissue plasmodia. Drugs (Blood Schizontocides) Used for Clinical and Suppressive Cure These agents act on asexual erythrocytic stages of malarial parasites to interrupt erythrocytic schizogony and thereby terminate clinical attacks (clinical cure). Such drugs also may produce suppressive cure, which refers to complete elimination of parasites from the body by continued therapy. Inadequate therapy with blood schizontocides may result in recrudescence of infection due to erythrocytic schizogony. With the notable exception of primaquine, virtually all antimalarial drugs used clinically were developed primarily for their activity against asexual parasite stages. These agents can be divided into two groups. The rapidly acting blood schizontocides include classical antimalarial alkaloids such as chloroquine, quinine, and their related derivatives quinidine and mefloquine. Atovaquone and the artemisinin antimalarial endoperoxides also are rapidly acting agents. Slower-acting, less effective blood schizontocides are exemplified by the antimalarial antifolate and antibiotic compounds. These drugs most commonly are used in conjunction with their more rapidly acting counterparts. Gametocytocides These agents act against sexual erythrocytic forms of plasmodia, thereby preventing transmission of malaria to mosquitoes. Chloroquine and quinine have gametocytocidal activity against P. vivax, P. ovale, and P. malariae, whereas primaquine displays especially potent activity against gametocytes of P. falciparum. However, antimalarials are not used clinically just for their gametocytocidal action. Sporontocides Such drugs ablate transmission of malaria by preventing or inhibiting formation of malarial oocysts and sporozoites in infected mosquitoes. Although chloroquine prevents normal plasmodial development within the mosquito, neither this nor other antimalarial agents are used clinically for this purpose. Regimens currently recommended for chemoprophylaxis in nonimmune individuals are given in Table 401, whereas regimens for treatment of malaria in nonimmune individuals are given in Table 402. Properties of individual agents are discussed in more detail in a separate section. |
Antimalarial Drugs
|
Artemisinin and Derivatives History Artemisinin
is a sesquiterpene lactone endoperoxide derived from the weed qing hao
(Artemisia annua), also called sweet wormwood or annual wormwood. The
Chinese have ascribed medicinal value to this plant for more than 2000 years
(reviewed by Klayman, 1985). As early as 340 A.D., Ge Hong prescribed tea made from qing
hao as a remedy for fevers, and in 1596 Li Shizhen recommended it to
relieve the symptoms of malaria. By 1972, Chinese scientists had extracted
and crystallized the major antimalarial ingredient, qinghaosu, now
known as artemisinin. They synthesized three derivatives with greater
antimalarial potency than artemisinin itself, namely dihydroartemisinin,
a reduced product, artemether, an oil-soluble methyl ester, and artesunate,
the water-soluble hemisuccinate salt of dihydroartemisinin. In 1979, the
Chinese reported that artemisinin drugs were rapidly acting, effective, and
safe for the treatment of patients with P. vivax or P. falciparum
infections. More than two million people with malaria in
Antiparasitic Activity The endoperoxide moiety is required for antimalarial activity of artemisinin compounds, whereas substitutions on the lactone carbonyl group markedly increase potency. These compounds act rapidly upon asexual erythrocytic stages of P. vivax and chloroquine-sensitive, chloroquine-resistant, and multidrug-resistant strains of P. falciparum. Their potency in vivo is 10- to 100-fold greater than that of other antimalarial drugs (White, 1997). They have gametocytocidal activity but do not affect either primary or latent tissue stage parasites. Thus, artemisinin compounds are not useful either for chemoprophylaxis or for preventing relapses of vivax malaria. The current model of artemisinin action involves two steps. First, intraparasitic heme iron of infected erythrocytes catalyzes cleavage of the endoperoxide bridge. This is followed by intramolecular rearrangement to produce carbon-centered radicals that covalently modify and damage specific malarial proteins (seeMeshnick et al., 1996). Artemisinin and its derivatives also exhibit antiparasitic activity in vitro against several other protozoa including Leishmania major and Toxoplasma gondii and in vivo against schistosomes, but they are not used clinically to treat infections with these parasites. Absorption, Fate, and Excretion The disposition of the artemisinin compounds is incompletely understood due to difficulties with proper preservation of biological samples and reliable analytical assays. Indeed, few pharmacokinetic studies carried out in humans have been published (seeBarradell and Fitton, 1995; de Vries and Dien, 1996). Time to peak plasma levels for the artemisinin compounds varies from minutes to several hours, depending on the drug formulation and its route of administration. Likewise, the profile and extent of drug binding to plasma proteins is variable. Artemether and artesunate are both converted to dihydroartemisinin. Much of the hydrolysis of artesunate to dihydroartemisinin may occur presystemically. Artemisinin itself is metabolized to at least four inactive metabolites, although it is unclear whether dihydroartemisinin is formed as an intermediate (seede Vries and Dien, 1996). The antimalarial effect of artemisinin compounds results primarily from dihydroartemisinin, which rapidly disappears from plasma with a half-life of about 45 minutes. Little or none of the administered drugs or dihydroartemisinin is recovered in urine. Although artemisinin can induce CYP2C19 in humans (Svensson et al., 1998), there is no evidence yet of clinically important drug interactions as a consequence. Therapeutic Uses Artemisinin compounds are the most rapidly acting, effective, and safe drugs for the treatment of severe malaria, including infections due to chloroquine- and multidrug-resistant strains of P. falciparum (seeWhite, 1999). They should not be used for prophylaxis of malaria or treatment of mild attacks (Meshnick et al., 1996). Artemisinin drugs act more rapidly and produce less toxicity than the antimalarial alkaloids; moreover, they are just as effective against cerebral malaria. Although artemisinin and its derivatives can be used as single agents, infections often relapse unless therapy is continued for 5 to 7 days. A brief course of these agents given in tandem with a longer-acting quinoline or antibiotic antimalarial, e.g., mefloquine or doxycycline, usually prevents relapses and may delay the development of drug resistance (White, 1997, 1999). Although optimal dosage regimens have yet to be standardized, one strategy is to give a course of artesunate to reduce parasite burden rapidly, followed by one or two doses of mefloquine to eradicate the infection (White, 1999; Price et al., 1999; seeTable 402). This approach has the advantage of reducing the frequency of side effects while retaining antimalarial efficacy. Individual endoperoxide antimalarials differ in formulation and clinical utility. Dihydroartemisinin can be given only orally. The oil-soluble artemether can be given only orally or intramuscularly. Artemisinin is effective when given orally or as a rectal suppository. Of the various artemisinin compounds, artesunate is perhaps the most versatile, because it is effective when given orally, intramuscularly, intravenously, or rectally. The intravenous formulation is particularly suitable for treating cerebral malaria, whereas suppositories are especially advantageous for treating patients with severe malaria in isolated areas. Toxicity and Contraindications Given for up to 7 days at therapeutic doses, the artemisinin endoperoxides appear to be surprisingly safe in human beings (seede Vries and Dien, 1996). Transient first-degree heart block, dose-related reversible decreases in reticulocyte and neutrophil counts, and temporary elevations of serum aspartate aminotransferase activity have been reported, but their clinical significance is not established. Brief episodes of drug-induced fever in human volunteers were noted in some studies but not in others. Because high doses of artemisinin drugs can produce neurotoxicity, prolongation of the QT interval, bone marrow depression, and fetal reabsorption in experimental animals, the possibility of long-term toxicity in human beings exists (seede Vries and Dien, 1996). But evidence thus far indicates that these effective drugs are remarkably safe for emergency treatment of severe, multidrug-resistant malaria, even in pregnant women (McGready et al., 1998) and in children (Price et al., 1999). Atovaquone History Based on the antiprotozoal activity of certain hydroxynaphthoquinones,
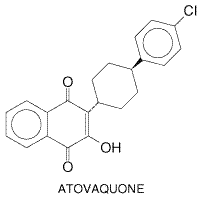
atovaquone (MEPRON) was developed as a promising synthetic derivative with potent
activity against Plasmodium species and opportunistic pathogens (Hudson
et al., 1991). Subsequent clinical studies revealed that atovaquone
produced good responses but high rates of relapse in patients with
uncomplicated falciparum malaria (Looareesuwan et al., 1996). In
contrast, use of proguanil with atovaquone evoked high cure rates with few
relapses and minimal toxicity (Looareesuwan et al., 1996, 1999a). A
fixed combination of atovaquone with proguanil (MALARONE) is now available in the
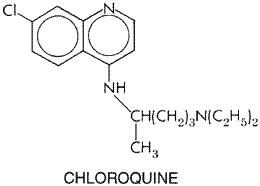
Antiparasitic Effects Atovaquone is a highly lipophilic analog of ubiquinone. In animal models and in vitro systems, it has potent activity against blood stages of plasmodia, tachyzoite and cyst forms of T. gondii, the fungus P. carinii, and Babesia species (Hughes et al., 1990; Hudson et al., 1991; Hughes and Oz, 1995). Atovaquone is highly potent against rodent malaria and P. falciparum, both in culture (IC50 0.7 to 4.3 nM) and in Aotus monkeys (Hudson et al., 1991). This compound selectively interferes with mitochondrial electron transport and related processes, such as ATP and pyrimidine biosynthesis in susceptible malaria parasites. Thus, atovaquone acts selectively at the cytochrome bc1 complex of malaria mitochondria to inhibit electron transport and collapse the mitochondrial membrane potential (seeVaidya, 1998). Synergism between proguanil and atovaquone appears due to the capacity of proguanil as a biguanide to enhance the membrane-collapsing activity of atovaquone (Srivastava and Vaidya, 1999). Atovaquone likewise affects mitochondrial function in permeabilized T. gondii tachyzoites (Vercesi et al., 1998). Absorption, Fate, and Excretion Because of its low water solubility, the bioavailability of atovaquone depends on formulation. A microfine suspension shows twofold greater oral bioavailability than do tablets. Drug absorption after a single oral dose is slow, erratic, and variable; increased by 2- to 3-fold by fatty food; and dose-limited above 750 mg. More than 99% of the drug is bound to plasma protein, so its concentration in cerebrospinal spinal fluid is less than 1% of that in plasma. Plasma leveltime profiles often show a double peak, albeit with considerable variability; the first peak appears in 1 to 8 hours while the second occurs 1 to 4 days after a single dose. This pattern suggests an enterohepatic circulation, as does the long half-life, averaging 1.5 to 3 days. Atovaquone is not significantly metabolized by human beings. It is excreted in bile, and more than 94% of the drug is recovered unchanged in feces; only traces appear in the urine (Rolan et al., 1997). Clearance of atovaquone may vary among different ethnic populations treated for falciparum malaria (Hussein et al., 1997). Therapeutic Uses Atovaquone is used with a biguanide for treatment of malaria to obtain optimal clinical results and avoid emergence of drug-resistant plasmodial strains. A tablet containing a fixed dose of 250 mg of atovaquone and 100 mg of proguanil hydrochloride, taken orally, has been highly effective and safe in a 3-day regimen for treating mild to moderate attacks of chloroquine- and multidrug-resistant falciparum malaria (seeLooareesuwan et al., 1999a and Table 402). The same regimen followed by primaquine produced excellent results in chloroquine-resistant vivax malaria (Looareesuwan et al., 1999b). To delay emergence of drug resistance, atovaquone plus proguanil is not recommended generally for prophylaxis of malaria, even though the combination is highly effective in adults and children. Such resistance develops readily when either drug is used alone. Opportunistic infections due to the fungus P. carinii or the protozoan T. gondii are especially serious threats to immunocompromised patients such as those with HIV infection and AIDS. Atovaquone remains an attractive alternative for prophylaxis and treatment of pulmonary P. carinii infection in patients who can take oral medication but cannot tolerate trimethoprim-sulfamethoxazole or parenteral pentamidine isethionate (seeChapters 44: Antimicrobial Agents: Sulfonamides, Trimethoprim-Sulfamethoxazole, Quinolones, and Agents for Urinary Tract Infections and 49: Antimicrobial Agents: Antifungal Agents and the 9th edition of this textbook). T. gondii infections in these patients, especially cerebral lesions, have shown only limited dose-related positive responses to prolonged regimens of atovaquone (Torres et al., 1997). Toxoplasma chorioretinitis in immunocompetent patients probably responds better to this drug (Pearson et al., 1999). Atovaquone may have potential use in human infections due to Babesia species (Hughes and Oz, 1995). Toxicity and Contraindications Both in patients with acute falciparum malaria and in severely debilitated and immunocompromised patients such as those with AIDS, adverse effects directly attributable to atovaquone have been difficult to distinguish from manifestations of underlying disease. Atovaquone causes few side effects that require withdrawal of therapy. The most common reactions are rash, fever, vomiting, diarrhea, and headache. Vomiting and diarrhea may result in therapeutic failure due to decreased drug absorption. However, readministration of this drug within an hour of vomiting still may evoke a positive therapeutic response in patients with falciparum malaria (Looareesuwan et al., 1999a). Dose-related maculopapular rashes occur in about 20% of treated patients, but most are mild and do not progress even when therapy is continued. Caution would dictate, however, that atovaquone not be given to patients with histories of allergic skin reactions or possible allergy to the drug. Patients treated with atovaquone only occasionally exhibit abnormalities of serum transaminase and amylase levels. Atovaquone lacks proven efficacy against bacterial, viral, and most opportunistic infections that commonly afflict immunocompromised individuals; these infections must be treated separately. On balance, the drug appears to cause few acute adverse effects, but more clinical evaluation is needed, especially to detect possible rare, unusual, or long-term toxicity. An example of the last is the association of reversible vortex keratopathy with highly lipid-soluble antiparasitic drugs like atovaquone (Shah et al., 1995). Precautions and Contraindications While atovaquone seems remarkably safe, the drug needs further evaluation in pediatric patients, older persons, pregnant women, and lactating mothers. Accordingly, the drug should be used with caution in these individuals. Routine tests for carcinogenicity, mutagenicity, and teratogenicity have been negative thus far, although therapeutic doses can cause maternal toxicity and interfere with normal fetal development in rabbits. Atovaquone may possibly compete with certain drugs for binding to plasma proteins, and therapy with rifampin, a potent inducer of cytochrome P450mediated drug metabolism, can substantially reduce plasma levels of atovaquone, whereas plasma levels of rifampin are raised. Until it is known whether atovaquone induces or inhibits the hepatic metabolism or biliary uptake and elimination of other drugs, caution is advised in using the drug in patients with severe liver disease. Chloroquine and Congeners History Chloroquine ARALEN) is one of a large series of
4-aminoquinolines investigated as part of the extensive cooperative program
of antimalarial research in the Chemistry Chloroquine has the following chemical structure:
Chloroquine closely resembles the obsolete 8-aminoquinoline antimalarials, pamaquine and pentaquine. It contains the same side chain as quinacrine but differs from this antimalarial in having a quinoline instead of an acridine nucleus and in lacking the methoxy moiety. The d, l, and dl forms of chloroquine have equal potency in duck malaria, but the d isomer is somewhat less toxic than the l isomer in mammals. A chlorine atom attached to position 7 of the quinoline ring confers the greatest antimalarial activity in both avian and human malarias. Research on the structure-activity relationships of chloroquine and related alkaloid compounds continues in an effort to find new, effective antimalarials with improved safety profiles that can be used successfully against chloroquine- and multidrug-resistant strains of P. falciparum (see below and, for example, Goldberg et al., 1997; O'Neill et al., 1998; Raynes, 1999). Amodiaquine
is a congener of chloroquine that is no longer recommended for
chemoprophylaxis of falciparum malaria because its use is associated with
hepatic toxicity and agranulocytosis. Pyronaridine is a Mannich-base
antimalarial that is structurally related to amodiaquine. This compound,
developed by the Chinese in the 1970s, was shown to be well tolerated and
effective against falciparum and vivax malarias. However, it cannot be
recommended for routine use because of a lack of standardized dosage regimens
and because its possible long-term toxicity has yet to be adequately
evaluated (Naisbitt et al., 1998). Hydroxychloroquine (PLAQUENIL), in which one of the N-ethyl
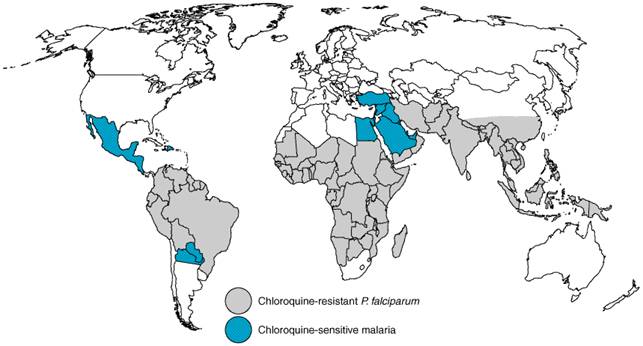
substituents of chloroquine is Pharmacological Effects Antimalarial Actions Chloroquine is highly effective against erythrocytic forms of P. vivax, P. ovale, P. malariae, and chloroquine-sensitive strains of P. falciparum. It exerts activity against gametocytes of the first three plasmodial species but not against those of P. falciparum. The drug has no activity against latent tissue forms of P. vivax or P. ovale and thus cannot cure infections with these species. Other Effects Chloroquine or its analogs are used for therapy of conditions other than malaria. Their use to treat hepatic amebiasis is described in Chapter 41: Drugs Used in the Chemotherapy of Protozoal Infections: Amebiasis, Giardiasis, Trichomoniasis, Trypanosomiasis, Leishmaniasis, and Other Protozoal Infections. Chloroquine and hydroxychloroquine have been used as secondary drugs to treat a variety of chronic diseases, because both alkaloids concentrate in lysosomes and have antiinflammatory properties. Thus, high doses of these compounds, often together with other agents, have clinical efficacy in rheumatoid arthritis, systemic lupus erythematosus, discoid lupus, sarcoidosis, and photosensitivity diseases such as porphyria cutanea tarda and severe polymorphous light eruption (Danning and Boumpas, 1998; Fritsch et al., 1998; Baltzan et al., 1999). Mechanisms of Antimalarial Action of and Resistance to Chloroquine and Other Antimalarial Quinolines Asexual malaria parasites flourish in host erythrocytes by digesting hemoglobin in their acidic food vacuoles, a process that generates free radicals and heme (ferriprotoporphyrin IX) as highly reactive by-products. After nucleation aided by histidine-rich proteins and perhaps by lipids, heme polymerizes into an insoluble unreactive malarial pigment termed hemozoin. Quinoline blood schizontocides that behave as weak bases concentrate in food vacuoles of susceptible plasmodia, where they increase pH, inhibit the peroxidative activity of heme, and disrupt its nonenzymatic polymerization to hemozoin. Failure to inactivate heme then kills the parasites via oxidative damage to membranes, digestive proteases, and possibly other critical biomolecules (reviewed by Foley and Tilley, 1998). Of possible mechanisms for the action of the malarial quinolines, inhibition of heme polymerization appears crucial. Recent kinetic studies indicate that radiolabeled chloroquine, quinidine, and mefloquine bind first to heme and then prevent further heme polymerization by incorporating as heme-quinoline complexes into growing heme polymer chains. This unifying model also may apply to amodiaquine, quinacrine, and quinine but not to primaquine (Sullivan et al., 1996; Mungthin et al., 1998). Whether resulting accumulation of heme, heme-quinoline complexes, or both suffices to kill the parasites or other actions of the antimalarial quinolines are required is unknown (Ginsburg et al., 1998; Bray et al., 1998; Loria et al., 1999). Intrinsic resistance of erythrocytic asexual forms of P. falciparum to antimalarial quinolines, especially chloroquine, has been slow to develop but is now common worldwide, particularly in areas of extensive antimalarial drug use (Figure 402). Chloroquine resistance is emphasized here even though resistance mechanisms probably differ between antimalarial quinoline classes (see below). More than 20 years ago, Fitch and coworkers noted that chloroquine-sensitive falciparum parasites concentrated the drug to higher levels than did chloroquine-resistant organisms (Fitch et al., 1979). Reasons for the relatively reduced levels of chloroquine in food vacuoles of chloroquine-resistant parasites have yet to be completely clarified. These could include differences in plasmodial uptake and transport of chloroquine to food vacuoles as well as differences in vacuolar influx, efflux, and trapping of drug (Goldberg et al., 1997; Bray et al., 1998; Foley and Tilley, 1998).
Chloroquine export initially attracted attention because verapamil, an inhibitor of P-glycoproteinmediated drug efflux by multidrug-resistant (MDR) tumor cells, enhanced chloroquine efflux and partially restored susceptibility of resistant P. falciparum to this drug (seeFoley and Tilley, 1998). Indeed, two homologues of mdr genes, pfmdr1 and pfmdr2, were later identified in P. falciparum (Wilson et al., 1989; Foote et al., 1989). But neither gene was linked to chloroquine resistance in genetic studies (Wellems et al., 1990). One, pfmdr1, encodes a P-glycoproteinlike protein, Pgh1, which, when overexpressed, may even confer relative sensitivity to chloroquine but resistance to antimalarial aminoalcohols such as mefloquine, halofantrine, and quinine (Cowman et al., 1994). Moreover, field studies failed to link chloroquine resistance to alterations in pfmdr1 (von Seidlein et al., 1997; Pvoa et al., 1998; Zalis et al., 1998), and inhibitors of P-glycoproteinmediated transport when given with chloroquine have not proven clinically effective in treating chloroquine-resistant falciparum malaria. Thus, based on evidence accumulated from both laboratory strains and clinical isolates, resistance to chloroquine, mefloquine/halofantrine, and quinine probably involves at least three nonidentical mechanisms (seeZalis et al., 1998; Reed et al., 2000). More recently, the proposed existence of a chloroquine transporter (Sanchez et al., 1997) was supported by an elegant study indicating that chloroquine resistance in a P. falciparum gene cross-mapped to a 36-kb segment of chromosome 7 (Su et al., 1997). This segment contains cg2, a gene that encodes a 330,000-dalton protein with complex polymorphisms, a set of which was associated with the chloroquine-resistant phenotype in 20 of 21 progeny examined; the finding of one chloroquine-sensitive strain with the same set of polymorphisms indicated that this set was necessary but not sufficient to confer chloroquine resistance. The same genetic study supported clinical evidence that South American and Asian/African P. falciparum have separate origins of chloroquine resistance. The CG2 protein (the product of cg2) was located both at the parasite periphery and in association with hemozoin in the food vacuoles, consistent with a role in chloroquine transport (Su et al., 1997). Further studies confirmed an incomplete but positive association of cg2 polymorphisms with chloroquine resistance in clinical isolates from travelers returning from endemic regions (Durand et al., 1999). Indeed, chemical probes to identify CG2 already may have been identified (Goldberg et al., 1997). In summary, resistance to the antimalarial quinolines probably involves multiple mechanisms under complex multigenic control. Absorption, Fate, and Excretion Chloroquine is well absorbed from the gastrointestinal tract and rapidly from intramuscular and subcutaneous sites. The drug distributes relatively slowly into a very large apparent volume (over 100 liters/kg; seeKrishna and White, 1996). This is due to extensive sequestration of chloroquine in tissues, particularly liver, spleen, kidney, lung, melanin-containing tissues, and, to a lesser extent, brain and spinal cord. Chloroquine binds moderately (60%) to plasma proteins and undergoes appreciable biotransformation via the hepatic cytochrome P450 system to two active metabolites, desethylchloroquine and bisdesethylchloroquine (seeDucharme and Farinotti, 1996). These metabolites may reach concentrations in plasma 40% and 10% of that of chloroquine, respectively. The S(+) enantiomer of chloroquine exhibits both greater binding to plasma proteins and a greater metabolic clearance than the R() enantiomer (seeKrishna and White, 1996). The renal clearance of chloroquine is about half of its total systemic clearance. Unchanged chloroquine and its major metabolite account for more than 50% and 25% of the urinary drug products, respectively, and the renal excretion of both compounds is increased by acidification of the urine. Both in adults and children, chloroquine exhibits complex pharmacokinetics such that plasma levels of the drug shortly after dosing are determined primarily by the rate of distribution rather than elimination (seeKrishna and White, 1996). Because of extensive tissue binding, a loading dose is required to achieve effective concentrations in plasma. After parenteral administration, rapid entry together with slow exit of chloroquine from a small central compartment can result in transiently high and potentially lethal concentrations of the drug in plasma. Hence, chloroquine is given either slowly by constant intravenous infusion or in small divided doses by the subcutaneous or intramuscular routes (Foley and Tilley, 1998). Chloroquine is safer when given orally because the rates of absorption and distribution are more closely matched; peak plasma levels are achieved in about 3 to 5 hours after dosing by this route. The half-life of chloroquine increases from a few days to weeks as plasma levels decline, reflecting the transition from slow distribution to even slower elimination from extensive tissue stores. The terminal half-life ranges from 30 to 60 days, and traces of the drug can be found in the urine for years after a therapeutic regimen. Therapeutic Uses Chloroquine is the most versatile antimalarial drug available, but its
usefulness has declined in those parts of the world where strains of P.
falciparum have emerged that are relatively or absolutely resistant to
its action. The compound is superior to quinine in that it is more potent and
less toxic, and it need be given only once weekly as a suppressive agent.
Chloroquine has neither prophylactic nor radical curative value in human P.
vivax or P. ovale malarias. However, except in Toxicity and Side Effects Taken in proper doses, chloroquine is an extraordinarily safe drug. Acute chloroquine toxicity is most frequently encountered when therapeutic or high doses are administered too rapidly by parenteral routes (see above). Toxic manifestations relate primarily to the cardiovascular and central nervous systems. Cardiovascular effects include hypotension, vasodilation, suppressed myocardial function, cardiac arrhythmias, and eventual cardiac arrest. Confusion, convulsions, and coma indicate central nervous system dysfunction. Chloroquine doses of more than 5 g given parenterally usually are fatal. Prompt treatment with mechanical ventilation, epinephrine, and diazepam may be lifesaving. Doses of chloroquine used for oral therapy of the acute malarial attack may cause gastrointestinal upset, headache, visual disturbances, and urticaria. Pruritus also occurs, most commonly among dark-skinned persons. Prolonged medication with suppressive doses occasionally causes side effects such as headache, blurring of vision, diplopia, confusion, convulsions, lichenoid skin eruptions, bleaching of hair, widening of the QRS interval, and T-wave abnormalities. These complications usually disappear soon after the drug is withheld. Rare instances of hemolysis and blood dyscrasias have been reported. Chloroquine may cause discoloration of nail beds and mucous membranes. Chloroquine can interfere with the immunogenicity of certain vaccines (Brachman et al., 1992; Horowitz and Carbonaro, 1992; Pappaioanou et al., 1986). High daily doses (>250 mg) of chloroquine or hydroxychloroquine used for treatment of diseases other than malaria can result in irreversible retinopathy and ototoxicity. Retinopathy presumably is related to drug accumulation in melanin-rich tissues and can be avoided if the daily dose is 250 mg or less (seeRennie, 1993). Prolonged therapy with high doses of either 4-aminoquinoline also can cause toxic myopathy, cardiopathy, and peripheral neuropathy; these reactions improve if the drug is promptly withdrawn (Estes et al., 1987). Rarely, neuropsychiatric disturbances, including suicide, may be related to overdose. Precautions and Contraindications This topic has been briefly reviewed by Diaminopyrimidines History Based on their ability to antagonize folic and folinic acids in
supporting the growth of Lactobacillus casei, a number of
diaminopyrimidines were tested for inhibitory activity against other
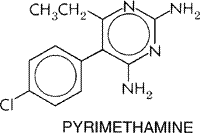
pathogenic organisms. Several 2,4-diaminopyrimidines, including pyrimethamine
(DARAPRIM) and the antibacterial agent trimethoprim,
exhibited significant antimalarial activity in animal models. Pyrimethamine
was later found to be especially effective against plasmodia infecting human
beings (seeSymposium, 1952). The antifolate combination (FANSIDAR) of pyrimethamine and
sulfadoxine, a long-acting sulfonamide, has been used extensively for
prophylaxis and suppression of human malarias, especially those caused by chloroquine-resistant
strains of P. falciparum. Resistance to this formulation rapidly
developed in Indochina and is now widespread except in parts of
Antiprotozoal Effects Antimalarial Actions Pyrimethamine is a slow-acting blood schizontocide with antimalarial effects in vivo similar to those of proguanil (see below). However, pyrimethamine has greater antimalarial potency because it acts directly on malarial parasites, and its half-life is much longer than that of cycloguanil, the active metabolite of proguanil. Unlike proguanil, pyrimethamine does not show marked efficacy against hepatic forms of P. falciparum. At therapeutic doses, pyrimethamine fails to eradicate latent tissue forms of P. vivax or gametocytes of any plasmodial species. The antimalarial effects of both pyrimethamine and proguanil have been reviewed by Davey (1963) and by Hill (1963). Action against Other Protozoa High doses of pyrimethamine given concurrently with sulfadiazine is the preferred therapy for toxoplasmosis, an infection with Toxoplasma gondii that can be particularly severe in infants and immunosuppressed individuals (seeChapter 41: Drugs Used in the Chemotherapy of Protozoal Infections: Amebiasis, Giardiasis, Trichomoniasis, Trypanosomiasis, Leishmaniasis, and Other Protozoal Infections). Mechanisms of Antimalarial Action and Resistance In an elegant series of investigations, the 2,4-diaminopyrimidines were shown to act by inhibiting dihydrofolate reductase of plasmodia at concentrations far lower than those required to produce comparable inhibition of the mammalian enzymes (Ferone et al., 1969). Plasmodial dihydrofolate reductase, unlike its mammalian counterparts, possesses both dihydrofolate reductase and thymidylate synthetase activities. Synergism between pyrimethamine and the sulfonamides or sulfones is explained by inhibition of two steps in an essential metabolic pathway (seeChapter 44: Antimicrobial Agents: Sulfonamides, Trimethoprim-Sulfamethoxazole, Quinolones, and Agents for Urinary Tract Infections). The two steps involved are the utilization of p-aminobenzoic acid for the synthesis of dihydropteroic acid, which is catalyzed by dihydropteroate synthase and inhibited by sulfonamides, and the reduction of dihydrofolate to tetrahydrofolate, which is catalyzed by dihydrofolate reductase and inhibited by pyrimethamine. Inhibition by antifolates is manifested late in the life cycle of malarial parasites by failure of nuclear division at the time of schizont formation in erythrocytes and liver. This mechanism is consistent with the slow onset of action of the antifolate as compared with the quinoline antimalarials. However, resistance to pyrimethamine does develop in regions of prolonged or extensive drug use. Dihydrofolate reductasethymidylate synthetase genes have been cloned and sequenced in strains of P. falciparum that are either sensitive or resistant to pyrimethamine. Several different mutations have been identified that produce single amino acid changes linked to pyrimethamine resistance; these changes are thought to decrease the binding affinity of pyrimethamine for its active site on the dihydrofolate reductase moiety of the parasite enzyme. The primary change associated with pyrimethamine resistance is a substitution of asparagine for serine at position 108 (S108N). Secondary mutations associated with increasing levels of resistance result from amino acid substitutions at Arg50, Ile51, Arg59, and Leu164; of these, the Leu164 change most markedly enhances pyrimethamine resistance when associated with the primary S108N mutation. These and other amino acid changes in various combinations also may contribute to pyrimethamine resistance. However, the pattern of amino acid substitutions differs from that observed for resistance to cycloguanil, even though cross resistance can occur between these structurally related compounds that target plasmodial dihydrofolate reductase (see'Proguanil'; Cowman, 1998; Cortese and Plowe, 1998). Absorption, Fate, and Distribution After oral administration pyrimethamine is slowly but completely absorbed; it reaches peak plasma levels in about 4 to 6 hours. The compound binds to plasma proteins and accumulates mainly in kidneys, lungs, liver, and spleen. It is eliminated slowly with a half-life in plasma of about 80 to 95 hours. Concentrations that are suppressive for responsive plasmodial strains remain in the blood for about 2 weeks, but these are lower in patients with malaria (Winstanley et al., 1992). Several metabolites of pyrimethamine appear in the urine, but their identities and antimalarial properties have not been fully characterized. Pyrimethamine also is excreted in the milk of nursing mothers. Therapeutic Uses Pyrimethamine is not a first-line antimalarial. The drug is virtually
always given with either a sulfonamide or sulfone to enhance its antifolate
activity, but it still acts slowly relative to the quinoline blood
schizontocides, and its prolonged elimination encourages the selection of
resistant parasites. The use of pyrimethamine should be restricted to the
suppressive treatment of chloroquine-resistant falciparum malaria in areas, e.g.,
parts of High doses of pyrimethamine plus sulfadiazine is the treatment of choice for infections with Toxoplasma gondii in immunocompromised adults; if such patients are left untreated, these infections rapidly progress to a fatal outcome (seeKasper, 1998; Chapter 41: Drugs Used in the Chemotherapy of Protozoal Infections: Amebiasis, Giardiasis, Trichomoniasis, Trypanosomiasis, Leishmaniasis, and Other Protozoal Infections). Initial therapy consists of an oral loading dose of 200 mg followed by 50 to 75 mg of pyrimethamine daily for 4 to 6 weeks along with 4 to 6 g of sulfadiazine daily in four divided doses. Leucovorin (folinic acid), 10 to 15 mg daily, should be taken for the same period to prevent bone marrow toxicity (see below). For subsequent long-term suppressive therapy, lower doses of pyrimethamine (25 to 50 mg daily) and sulfadiazine (2 to 4 g daily) may suffice. To deal with toxicity, pyrimethamine often has been used with agents such as clindamycin, spiramycin, or other macrolides (seeKasper, 1998). Infants with congenital, placentally transmitted toxoplasmosis usually respond positively to oral pyrimethamine (0.5 to 1.0 mg/kg daily) and oral sulfadiazine (100 mg/kg daily) given over a one-year period. Toxicity, Precautions, and Contraindications Antimalarial doses of pyrimethamine alone cause little toxicity except occasional skin rashes and depression of hematopoiesis. Excessive doses produce a megaloblastic anemia, resembling that of folate deficiency, that responds readily to drug withdrawal or treatment with folinic acid. At very high doses pyrimethamine is teratogenic in animals, but there is no evidence for such toxicity in human beings. Sulfonamides or sulfones, rather than pyrimethamine, usually account
for the toxicity associated with coadministration of these antifolate drugs (seeChapter
44: Antimicrobial Agents: Sulfonamides, Trimethoprim-Sulfamethoxazole,
Quinolones, and Agents for Urinary Tract Infections). The combination of
pyrimethamine (25 mg) and sulfadoxine (500 mg) (FANSIDAR) is no longer recommended for
antimalarial prophylaxis, because in about 1:5000 to 1:8000 individuals, it
causes severe and even fatal cutaneous reactions, such as erythema
multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis. This
combination also has been associated with serum-sickness- type reactions,
urticaria, exfoliative dermatitis, and hepatitis. Pyrimethaminesulfadoxine
is contraindicated for individuals with previous reactions to sulfonamides,
for lactating mothers, and for infants less than 2 months old. Administration
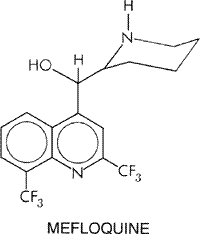
of pyrimethamine with dapsone (MALOPRIM, a drug combination unavailable in the Halofantrine Halofantrine HALFAN) is a phenanthrene methanol antimalarial drug with blood schizontocidal properties similar to those of the quinoline antimalarials. This compound was originally developed and has been used as an alternative to quinine and mefloquine to treat acute malarial attacks caused by chloroquine-resistant and multidrug-resistant strains of P. falciparum. Because halofantrine displays erratic bioavailability, potentially lethal cardiotoxicity, and extensive cross resistance with mefloquine, its use generally is not recommended. Details of the history, pharmacology, and toxicology of halofantrine are presented in the 9th edition of this textbook. Mefloquine History Mefloquine LARIAM) is a product of the Malaria
Research Program established in 1963 by the Walter Reed Institute for Medical
Research to develop promising new compounds to combat emerging strains of
drug-resistant P. falciparum. Of many 4-quinoline-methanols tested
based on their structural similarity to quinine, mefloquine displayed high
antimalarial activity in animal models and emerged from clinical trials as
safe and highly effective against drug-resistant strains of P. falciparum
(Schmidt et al., 1978). Mefloquine was first used to treat chloroquine-resistant
falciparum malaria in
Antimalarial Actions Mefloquine exists as a racemic mixture of four optical isomers with about the same antimalarial potency. It is a highly effective blood schizontocide, especially against mature trophozoite and schizont forms of malarial parasites. Mefloquine has no activity against early hepatic stages and mature gametocytes of P. falciparum or latent tissue forms of P. vivax. The drug may have some sporontocidal activity but is not used clinically for this purpose. Mechanisms of Antimalarial Action and Resistance The exact mechanism of action of mefloquine is unknown (see'Mechanisms of Antimalarial Action of and Resistance to Chloroquine and Other Antimalarial Quinolines,' above). As a blood schizontocide, mefloquine behaves like quinine in many respects but does not intercalate with DNA. The two compounds produce similar morphological changes in early erythrocytic ring stages of P. falciparum and P. vivax (Schmidt et al., 1978). Like quinine, mefloquine competes for accumulation of chloroquine and inhibits chloroquine-induced clumping of pigment in erythrocytic plasmodia (Fitch et al., 1979). Mefloquine causes swelling of the parasitic food vacuoles in P. falciparum. Like chloroquine, low extracellular concentrations of mefloquine raise the intravacuolar pH of plasmodia in excess of that predicted from passive distribution of a weak base. This suggests that mefloquine is concentrated in plasmodia by an unknown mechanism. Mefloquine may act by both inhibiting heme polymerization and forming toxic complexes with free heme that damage membranes and interact with other plasmodial components (seePalmer et al., 1993; Sullivan et al., 1998). The orientation of the hydroxyl and amine groups with respect to each other in mefloquine may be essential for its hydrogen bonding and antimalarial activity (Karle and Karle, 1991). Certain isolates of P. falciparum exhibit resistance to mefloquine, especially those obtained from people exposed to the drug. Individuals harboring resistant parasites generally require larger than the usual doses of mefloquine to control their infections. Depending on their geographic origin and history of exposure to antimalarial drugs, many isolates of P. falciparum also display multidrug-resistant phenotypes. This raises the question of common or overlapping mechanisms responsible for intrinsic or acquired resistance to mefloquine and its structurally related antimalarials (seePalmer et al., 1993). Genes in the multidrug-resistant (MDR) family can play a role in the resistance of P. falciparum to mefloquine. Products of this gene family lower intracellular concentrations of drugs in mammalian cells by increasing their efflux in an ATP-dependent manner; this effect is inhibited by some Ca2+ channel blockers but not by others. In P. falciparum, a gene of this family, pfmdr1, is usually but not always amplified, that is, the gene copy number is increased in parasites unresponsive in vitro to mefloquine and halofantrine (seeWilson et al., 1993; Lim et al., 1996). In contrast, there is no clear-cut association between chloroquine resistance and amplification of pfmdr1 (Mungthin et al., 1999). Patterns of resistance to mefloquine and quinine usually but not always overlap, suggesting that genetic differences aside from pfmdr1 can play a differential role in resistance to these structurally related compounds (Zalis et al., 1998). The stereoselectivity of mefloquine resistance (and action) has yet to be characterized. Absorption, Fate, and Excretion Mefloquine is taken orally because parenteral preparations cause
severe local reactions. The drug is well absorbed, a process enhanced by the
presence of food. Probably due to extensive enterogastric and enterohepatic
circulation, plasma levels of mefloquine rise in a biphasic manner to their
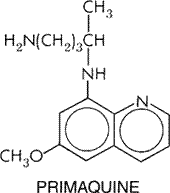
peak in about 17 hours. The drug is widely distributed, highly bound ( Therapeutic Uses Mefloquine should be reserved for the prevention and treatment of malaria caused by chloroquine-resistant and multidrug-resistant P. falciparum. The drug is especially useful as a prophylactic agent for nonimmune travelers who stay for only brief periods in areas where these infections are endemic (seeTable 401); prophylactic use of mefloquine for long-term residents of these regions should be avoided to prevent the selection of mefloquine-resistant parasites. Mefloquine and halofantrine are currently the only agents capable of ensuring suppression and cure of infections with multidrug-resistant P. falciparum. However, both medications can be given only orally, which is a major disadvantage for acutely ill patients, who are best treated with parenteral preparations of quinidine or quinine. Because of possible cross resistance, misuse of either mefloquine or halofantrine is likely to encourage the selection of falciparum parasites resistant to both drugs and possibly to quinine as well (seeWilson et al., 1993). Clinical resistance to mefloquine can be overcome by increasing the dose, but only at the cost of increased drug toxicity. Vomiting frequently occurs when high single or divided doses of mefloquine are used to treat a malarial attack. Patients should be observed and the full dose repeated if vomiting occurs within the first hour. Typical dosage schedules for monotherapy of falciparum malaria with mefloquine are given in Table 402. These may be modified; for example, lower doses than shown are effective for suppression of malarial attacks in partially immune individuals. More information about this topic is provided in the review by Palmer and colleagues (1993). To treat uncomplicated attacks of malaria due to chloroquine- and multidrug-resistant strains of P. falciparum, recent evidence indicates that mefloquine is most effective when used in tandem with an artemisinin compound such as artesunate (see'Artemisinin and Derivatives,' above) (White, 1997, 1999). The artemisinin derivative is given first to reduce the parasite burden followed by mefloquine therapy to enhance parasite clearance and prevent recrudescence of infection. Studies have shown no clinically significant pharmacokinetic or toxic interactions between the artemisinin compounds and mefloquine when therapy with the latter is begun about 36 to 48 hours after the former. The 1250-mg adult dose of mefloquine is usually split; namely, 750 mg is given after food, followed by 500 mg 12 hours later. A similar strategy with 25 mg/kg of mefloquine, given either as a single or split dose on the second day of antimalarial therapy with artesunate, has produced excellent results in children (Price et al., 1999). Toxicity and Side Effects The adverse effects of mefloquine were reviewed in detail by Palmer and colleagues (1993) and more recently by Schlagenhauf (1999). Mefloquine, given orally in single doses up to 1500 mg or in 250- to 500-mg doses each week, is generally well tolerated. Side effects such as nausea, late vomiting, abdominal pain, diarrhea, dysphoria, and dizziness are noted frequently. These tend to be dose-related, self-limiting, and at times difficult to distinguish from the clinical features of malarial illness. Signs of central nervous system toxicity occur in about half of the individuals taking mefloquine. Dizziness, ataxia, headache, alterations in motor function or the level of consciousness, and visual or auditory disturbances are self-limiting and usually mild. Severe neuropsychiatric reactions such as disorientation, seizures, encephalopathy, and a range of neurotic and psychotic manifestations are rare and usually reversible upon drug withdrawal and symptomatic therapy. While such complications occur more frequently in patients receiving therapeutic than prophylactic doses of mefloquine, they appear to be unrelated to plasma levels of the drug. Contraindications and Interactions At very high doses, mefloquine causes teratogenesis and developmental abnormalities in rodents. Prophylactic doses of mefloquine appear to cause little if any toxicity during the second and third trimesters of pregnancy, and they may even provide benefit to the fetus (Schlagenhauf, 1999). However, until more information becomes available or unless the risk of malarial infection outweighs the benefits of prophylaxis, caution dictates that mefloquine should be avoided during pregnancy, especially during the first trimester. Mefloquine should not be used in children weighing less than 5 kg. The drug is contraindicated for patients with a history of seizures, severe neuropsychiatric disturbances, or adverse reactions to quinoline antimalarials such as quinine, quinidine, and chloroquine. Use of mefloquine with these compounds must be avoided because of the increased risk of convulsions and cardiotoxicity. Although mefloquine can be taken safely 12 hours after a last dose of quinine, taking quinine shortly after mefloquine can be very hazardous because the latter is eliminated so slowly. Mefloquine is reported to increase the risk of seizures in epileptic patients controlled by valproate, and it may compromise adequate immunization by live typhoid vaccine. Until more data become available, caution is advised for use of mefloquine along with drugs that can perturb cardiac conduction. Recent studies do not indicate that mefloquine compromises the performance of tasks that require good motor coordination, for example, driving or operating machinery (Schlagenhauf, 1999). Primaquine History The weak plasmodicidal activity of methylene blue, first discovered by Ehrlich in 1891, was later exploited to develop the 8-aminoquinoline antimalarials. From a large series of quinoline derivatives synthesized with methoxy and substituted 8-amino groups, pamaquine was the first introduced into medicine. During World War II the search for more potent and less toxic 8-aminoquinoline antimalarials led to the selection of pentaquine, isopentaquine, and primaquine for further evaluation (see earlier editions of this textbook). These compounds, in contrast with other antimalarials, act on tissue stages (exoerythrocytic) of P. vivax and P. ovale to prevent and cure relapsing malaria. Only primaquine, tested extensively during the Korean War, is widely used now. Unfortunately, hemolysis due to human glucose-6-phosphate dehydrogenase (G6PD) deficiency is notoriously identified with primaquine therapy, so there is a pressing need for alternatives to this important drug. The 8-aminoquinoline tafenoquine, formerly called WR 238605, shows promise but needs more evaluation in this regard (Walsh et al., 1999). The chemical structure of primaquine is shown below:
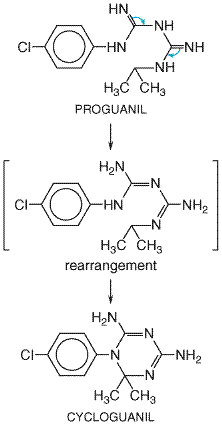
Antimalarial Actions Primaquine destroys late hepatic stages and latent tissue forms of P. vivax and P. ovale and thus has great clinical value for the radical cure of relapsing malarias. The drug by itself will not suppress attacks of vivax malaria, even though it displays activity against erythrocytic stages of P. vivax. Though primaquine has activity against hepatic stages of P. falciparum, the compound is ineffective against erythrocytic stages of this parasite and hence is not used clinically to treat falciparum malaria. The 8-aminoquinolines exert a marked gametocidal effect against all four species of plasmodia that infect human beings, especially P. falciparum. Some strains of P. vivax exhibit partial resistance to the action of primaquine (Smoak et al., 1997), which makes it imperative that the drug not be misused and that other antimalarials with similar properties be developed. Mechanism of Antimalarial Action There are as yet no methods for maintaining P. vivaxin vitro, so little is known about the antimalarial action of the 8-aminoquinolines, especially why they are far more active against tissue forms and gametes than asexual blood forms of plasmodia. Primaquine may be converted to electrophiles that act as oxidation-reduction mediators (see below and Tarlov et al., 1962). Such activity could contribute to antimalarial effects by generating reactive oxygen species or interfering with electron transport in the parasite (Bates et al., 1990). Absorption, Fate, and Excretion Primaquine causes marked hypotension after parenteral administration and therefore is given only by the oral route. Absorption from the gastrointestinal tract is nearly complete. After a single dose the plasma concentration reaches a maximum within 3 hours and then falls with an apparent elimination half-time of 6 hours (Fletcher et al., 1981). The apparent volume of distribution is several times that of total body water. Primaquine is rapidly metabolized; only a small fraction of an administered dose is excreted as the parent drug. Three identified oxidative metabolites of primaquine are 8-(3-carboxyl-1-methylpropylamino)-6-methoxyquinoline, 5-hydroxy primaquine, and 5-hydroxy-6-desmethylprimaquine. The carboxyl derivative is the major metabolite found in human plasma. After a single dose it reaches concentrations in plasma more than 10 times those of primaquine; this nontoxic metabolite also is eliminated more slowly and accumulates with multiple doses (seeSymposium, 1987). The three metabolites of primaquine appear to have appreciably less antimalarial activity than does primaquine. However, except for the carboxyl derivative, their hemolytic activity, as assessed by formation of methemoglobin in vitro, is greater than that of the parent compound (Fletcher et al., 1988). Therapeutic Uses Primaquine is reserved primarily for the terminal prophylaxis and radical cure of vivax and ovale (relapsing) malarias because of its high activity against latent tissue forms of these plasmodial species. The compound is given together with a blood schizontocide, usually chloroquine, to eradicate erythrocytic stages of these plasmodia and reduce the possibility of emerging drug resistance. For terminal prophylaxis, primaquine regimens are initiated shortly before or immediately after the patient leaves an endemic area (seeTable 401). Radical cure of vivax or ovale malarias can be achieved if the drug is given either during the long-term latent period of infection or during an acute attack. Regimens for this purpose are shown in Table 402. Long-term use of primaquine should be avoided because of the risk of toxicity and sensitization. Toxicity and Side Effects Primaquine is fairly innocuous when given to Caucasians in the usual therapeutic doses. Larger doses cause occasional epigastric distress and mild-to-moderate abdominal distress in some individuals; these symptoms often are alleviated by taking the drug at mealtime. Mild anemia, cyanosis (methemoglobinemia), and leukocytosis are less common. High doses (60 to 240 mg of primaquine daily) accentuate the abdominal symptoms and cause methemoglobinemia in most subjects and leukopenia in some. Methemoglobinemia can occur even with usual doses of primaquine, chloroquine, or dapsone and can be severe in individuals with congenital deficiency of nicotinamide adenine dinucleotide (NADH) methemoglobin reductase (Coleman and Coleman, 1996). Hepatic function is unaffected. Granulocytopenia and agranulocytosis are rare complications of therapy and usually are associated with overdosage. Also rare are hypertension, arrhythmias, and symptoms referable to the central nervous system. Therapeutic or higher doses of primaquine, via its oxidative metabolites, may cause acute hemolysis and hemolytic anemia in humans with G6PD deficiency. This X-linked inherited condition, primarily due to amino acid substitutions in the G6PD enzyme, affects more than 200 million people worldwide. More than 400 genetic variants have been identified that are associated with variable responses to oxidative stress. About 11% of African Americans have the A variant of G6PD, which makes them vulnerable to hemolysis caused by prooxidant drugs such as primaquine. Sensitivity of erythrocytes to primaquine can be even more severe in some darker-hued Caucasian ethnic groups, including Sardinians, Sephardic Jews, Greeks, and Iranians. Because primaquine sensitivity is inherited by a gene on the X chromosome, hemolysis often is of intermediate severity in heterozygous females who have two populations of red cells, one normal and the other deficient in G6PD. Due to 'variable penetrance,' such females may be less frequently affected than predicted. Primaquine is the prototype of more than 50 drugs, including antimalarial sulfonamides, and other substances known to cause hemolysis in susceptible individuals with G6PD deficiency. Precautions and Contraindications Patients should be tested for G6PD deficiency before they receive primaquine. If a daily dose of more than 30 mg of primaquine base (more than 15 mg in possibly sensitive patients) is given, repeated blood counts and at least gross examination of the urine for hemoglobin should be undertaken. Primaquine is contraindicated for acutely ill patients suffering from systemic disease characterized by a tendency to granulocytopenia; very active forms of rheumatoid arthritis and lupus erythematosus are examples of such conditions. Primaquine should not be given to patients receiving other potentially hemolytic drugs or agents capable of depressing the myeloid elements of the bone marrow. Proguanil History Proguanil PALUDRINE) is the common name for chloroguanide, a biguanide derivative that emerged in 1945 as a product of British antimalarial drug research. The antimalarial activity of proguanil was ascribed to cycloguanil, an active cyclic triazine metabolite shown to be a selective inhibitor of the bifunctional plasmodial dihydrofolate reductase-thymidylate synthetase. Indeed, investigation of compounds bearing a structural resemblance to cycloguanil resulted in the development of antimalarial dihydrofolate reductase inhibitors such as pyrimethamine. Accrued evidence also indicates that proguanil itself has intrinsic antimalarial activity independent of its effect on parasite dihydrofolate reductasethymidylate synthetase (seeFidock and Wellems, 1997). Chemistry Proguanil and its triazine metabolite cycloguanil have the following chemical structures:
Proguanil has the widest margin of safety of a large series of antimalarial biguanide analogs examined. Dihalogen substitution in positions 3 and 4 of the benzene ring yields chlorproguanil (LAPUDRINE), a more potent prodrug than proguanil that also is used clinically. Cycloguanil is structurally related to pyrimethamine. Antimalarial Actions Through its active metabolite, proguanil exerts causal prophylactic and suppressive activity in sporozoite-induced falciparum malaria, adequately controls the acute attack, and usually eradicates the infection. Proguanil suppresses acute attacks of vivax malaria, but because latent tissue stages of P. vivax are unaffected, erythrocytic forms often appear shortly after the drug is withdrawn. Proguanil treatment does not destroy gametocytes, but fertilized gametes encysted in the gut of the mosquito fail to develop normally. Mechanisms of Antimalarial Action and Resistance The active triazine metabolite of proguanil selectively inhibits the bifunctional dihydrofolate reductase-thymidylate synthetase of sensitive plasmodia, causing inhibition of DNA synthesis and depletion of folate cofactors. This mechanism accounts for the slow antimalarial action of the antifolate biguanides compared to the quinoline antimalarials. By cloning and sequencing dihydrofolate reductase-thymidylate synthetase genes from sensitive and resistant P. falciparum, investigators found that certain amino acid changes near the dihydrofolate reductase binding site are linked to resistance to either the triazine metabolite, to pyrimethamine, or to both antimalarials. Specifically, resistance to cycloguanil (and chlorcycloguanil) can be linked to mutations leading to paired V16/Thr108 substitutions in plasmodial dihydrofolate reductase, such resistance being especially enhanced by an additional substitution at Leu164. This pattern differs from that typically observed for pyrimethamine resistance, where mutations result in a primary Asn108 substitution and secondary Arg50, Ile51, Arg59, and Leu164 substitutions that progressively increase resistance; again resistance is increased most by a Leu164 substitution. However, overlapping resistance to cycloguanil and pyrimethamine indicates that mutation patterns leading to the final resistance phenotype may be quite complex. Thus, genetic analyses of resistant P. falciparum strains together with novel expression and assay systems represent a powerful approach for identifying and monitoring new generations of antimalarials directed at vulnerable plasmodia biochemical targets (see, for example, Fidock and Wellems, 1997; Cowman, 1998; Cortese and Plowe, 1998). The presence of plasmodial dihydrofolate reductase is not required for the intrinsic antimalarial activity of proguanil or chlorproguanil (Fidock and Wellems, 1998), but the molecular basis for this activity is still unknown. Proguanil as the biguanide accentuates the mitochondrial membrane-potential-collapsing action of atovaquone against P. falciparum but displays no such activity by itself (see'Atovaquone,' above) (Srivastava and Vaidya, 1999). In contrast to cycloguanil, resistance to the intrinsic antimalarial activity of proguanil itself, either alone or in combination with atovaquone, has yet to be well documented. Absorption, Fate, and Excretion Proguanil is slowly but adequately absorbed from the gastrointestinal tract. After a single oral dose, peak concentrations of the drug in plasma usually are attained within 5 hours. The mean plasma elimination half-life is about 20 hours or longer depending on the rate of metabolism. Metabolism of proguanil in mammals cosegregates with mephenytoin oxidation polymorphism (Ward et al., 1991) controlled by isoforms in the 2C subfamily of cytochrome P450. Only about 3% of Caucasians are deficient in this oxidation phenotype, as contrasted to about 20% of Asians and Kenyans. Proguanil is oxidized to two major metabolites, cycloguanil and an inactive 4-chlorophenyl-biguanide. On a 200-mg daily dosage regimen, extensive metabolizers develop plasma levels of cycloguanil that are above the therapeutic range, whereas poor metabolizers may not (Helsby et al., 1993). Proguanil itself does not accumulate appreciably in tissues during long-term administration, except in erythrocytes, where its concentration is about three times that in plasma. Accumulation in infected erythrocytes could be critical for the intrinsic antimalarial effects of proguanil, either by itself or together with atovaquone. The inactive 4-chlorophenyl-biguanide metabolite is not readily detected in plasma but appears in increased quantities in the urine of poor proguanil metabolizers. In human beings, from 40% to 60% of the absorbed proguanil is excreted in urine either as the parent drug or the active metabolite. Therapeutic Uses Proguanil together with chloroquine is used as a safe alternative to mefloquine
or other regimens for the prophylaxis of falciparum malaria or mixed vivax
and falciparum infections in parts of eastern, southern, and central Proguanil is effective and well tolerated when given orally once daily
for three days in combination with atovaquone for treatment of malarial
attacks due to chloroquine- and multidrug-resistant strains of P.
falciparum and P. vivax (see'Atovaquone,' above)
(Looareesuwan et al., 1999a,b). Indeed, this drug combination (MALARONE) has been successful in Toxicity and Side Effects In prophylactic doses of 200 to 300 mg daily, proguanil causes few untoward effects except occasional nausea and diarrhea. Large doses (1 g daily or more) may cause vomiting, abdominal pain, diarrhea, hematuria, and the transient appearance of epithelial cells and casts in the urine. Gross accidental or deliberate overdose (as much as 15 g) has been followed by complete recovery. Doses as high as 700 mg twice daily have been taken for more than 2 weeks without serious toxicity. Proguanil is considered safe for use during pregnancy. It is remarkably safe when used in conjunction with other antimalarial drugs such as chloroquine, atovaquone, tetracyclines, and other antifolates. This may bear on the use of proguanil plus atovaquone in conjunction with artemisinin derivatives to reduce parasite burden, impede transmission of infection, and further delay the development of drug resistance (van Vugt et al., 1999). Quinine History The medicinal use of quinine dates back more than 350 years.
Quinine is the chief alkaloid of cinchona, the bark of the South American
cinchona tree, otherwise known as Peruvian, Jesuit's, or Cardinal's bark. In
1633, an Augustinian monk named Calancha, of For almost two centuries the bark was employed for medicine as a powder, extract, or infusion. In 1820, Pelletier and Caventou isolated quinine and cinchonine from cinchona, and the use of the alkaloids as such gained favor rapidly. Quinine, together with secondary antimalarial antifolate or antibiotic drugs, is still the mainstay for treating attacks of chloroquine- and multidrug-resistant falciparum malaria today (seeTable 402). However, multitherapy with other antimalarials may supplant the 7-day quinine regimens because of increasing resistance of P. falciparum to quinine together with its toxicity (see'Prospectus,' below). Chemistry Although quinine has been synthesized, the procedure is complex; quinine and the other alkaloids are, therefore, still obtained entirely from natural sources. Cinchona contains a mixture of more than 20 alkaloids. The most important of these are two pairs of optical isomers, quinine and quinidine, and cinchonidine and cinchonine. Quinine and cinchonidine are levorotatory. Quinine has the following chemical structure:
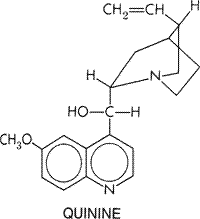
Quinine contains a quinoline group attached through a secondary alcohol linkage to a quinuclidine ring. A methoxy side chain is attached to the quinoline ring and a vinyl to the quinuclidine. Quinidine has the same structure as quinine except for the steric configuration of the secondary alcohol group. Stereoisomerism at this position is relatively unimportant in the structureactivity relationship. Quinidine is both more potent as an antimalarial and more toxic than quinine (seeKrishna and White, 1996). The many natural alkaloids related to quinine and the semisynthetic chemicals derived from quinine differ mainly in the nature of the substitutions on the side chain. These alterations cause quantitative but not qualitative changes in the pharmacological actions of the resulting compounds. The structureactivity relationships of the cinchona alkaloids are detailed in earlier editions of this textbook. Such studies provided the basis for the discovery of more effective and less toxic antimalarials, for example, mefloquine. Pharmacological Effects Antimalarial Actions Quinine acts primarily as a blood schizontocide; it has little effect
on sporozoites or preerythrocytic forms of malarial parasites. The alkaloid
also is gametocidal for P. vivax and P. malariae but not for P.
falciparum. Because of this spectrum of antimalarial activity, quinine is
not used for prophylaxis. As both a suppressive and therapeutic agent,
quinine is more toxic and less effective than chloroquine against malarial
parasites susceptible to both drugs. However, quinine, along with its
stereoisomer quinidine, is especially valuable for the treatment of severe
illness due to chloroquine- and multidrug-resistant strains of P.
falciparum, even though these strains have become more resistant to both
agents in certain parts of Southeast Asia and The mechanism of the antimalarial action of quinine and related quinoline antimalarials is reviewed under ' Chloroquine and Congeners,' above. In part because it is a weak base, quinine concentrates in the acidic food vacuoles of P. falciparum. The drug inhibits the nonenzymatic polymerization of the highly reactive, toxic heme molecule into a nontoxic polymer pigment called hemozoin. This is proposed to occur by a two-step process whereby the quinoline binds first to heme, and the resulting heme-drug complex binds to and saturates the heme-polymer chains (Sullivan et al., 1996, 1998). Whether consequent buildup of heme itself, heme-quinoline complexes, or both kill the parasites is not established. The basis of P. falciparum resistance to quinine is unknown and probably is quite complex. As discussed under 'Chloroquine and Congeners' in this chapter, patterns of P. falciparum resistance to quinine more closely resemble those of resistance to mefloquine and halofantrine rather than to chloroquine. Amplification of pfmdr1 in P. falciparum, implicated in resistance to mefloquine and halofantrine, also can confer resistance to quinine in vitro. However, various field isolates that show resistance to mefloquine are quite sensitive to quinine and vice versa (seeMeshnick, 1998; Zalis et al., 1998). Action on Skeletal Muscle Quinine and related cinchona alkaloids exert effects on skeletal muscle that have clinical implications. Quinine increases the tension response to a single maximal stimulus delivered to muscle directly or through nerves, but it also increases the refractory period of muscle so that the response to tetanic stimulation is diminished. The excitability of the motor end-plate region decreases so that responses to repetitive nerve stimulation and to acetylcholine are reduced. Thus, quinine can antagonize the actions of physostigmine on skeletal muscle as effectively as curare. Quinine also may produce alarming respiratory distress and dysphagia in patients with myasthenia gravis. Quinine may cause symptomatic relief of myotonia congenita. This disease is the pharmacological antithesis of myasthenia gravis, such that drugs effective in one syndrome aggravate the other. Absorption, Fate, and Excretion Quinine and its congeners are readily absorbed when given orally or
intramuscularly. In the former case, absorption occurs mainly from the upper
small intestine and is more than 80% complete, even in patients with marked
diarrhea. After an oral dose, plasma levels of quinine reach a maximum in 3 to
8 hours and, after distributing into an apparent volume of about 1.5 liters
per kg in healthy individuals, decline with a half-life of about 11 hours
after termination of therapy. As reviewed by The cinchona alkaloids are extensively metabolized, especially by CYP3A4 in the liver (Zhao et al., 1996), so that only about 20% of an administered dose is excreted unaltered in the urine. There is no accumulation of the drugs in the body upon continued administration, because their metabolites are excreted in the urine. However, the major metabolite of quinine, 3-hydroxyquinine, retains some antimalarial activity and can accumulate and possibly cause toxicity in patients with renal failure (Newton et al., 1999). Renal excretion of quinine itself is more rapid when the urine is acidic than when it is alkaline. Therapeutic Uses Treatment of Malaria Despite its potential toxicity, quinine remains the prototype blood schizontocide for the suppressive treatment and cure of chloroquine-resistant and multidrug-resistant falciparum malaria. In the severe illness, prompt use of loading doses of intravenous quinine (or quinidine) is imperative and can be lifesaving for nonimmune patients. Oral medication to maintain therapeutic concentrations is then given as soon as it can be tolerated and is continued for 5 to 7 days. Especially for treatment of infections with multidrug- resistant strains of P. falciparum, slower acting blood schizontocides such as a sulfonamide or tetracycline are given concurrently to enhance the action of quinine. Formulations of quinine and quinidine and specific regimens for their use in the treatment of falciparum malaria are shown in Table 402. Recommendations shown in Table 402 are derived from practice and
should be modified as appropriate. In a series of studies over the past two
decades, White and associates derived rational regimens, including the
institution of loading doses, for the use of quinine and quinidine in the
treatment of falciparum malaria in Treatment of Nocturnal Leg Cramps Recumbency leg muscle cramps (night cramps) have been reportedly relieved by quinine in a dose of 200 to 300 mg (available until 1995 in products that did not require a prescription) before retiring. In some individuals, only a brief period of quinine therapy has been said to be required to provide relief, but in others even large doses of the drug were ineffective. In 1995, the FDA issued a ruling that required drug manufacturers to stop marketing over-the-counter quinine products for nocturnal leg cramps. The FDA stated, as the basis for the ruling, that there were inadequate data to support the safety and effectiveness of quinine for use in treating nocturnal leg cramps. Toxicity and Side Effects The fatal oral dose of quinine for adults is about 2 to 8 g. Quinine is associated with a triad of dose-related toxicities when it is given at full therapeutic or excessive doses. These are cinchonism, hypoglycemia, and hypotension. Mild forms of cinchonismconsisting of tinnitus, high-tone deafness, visual disturbances, headache, dysphoria, nausea, vomiting, and postural hypotensionoccur frequently and disappear soon after the drug is withdrawn. Hypoglycemia, too, is common but can be life-threatening if not promptly treated with intravenous glucose. Hypotension is rarer but also serious and most often associated with excessively rapid intravenous infusions of quinine or quinidine. Prolonged medication or high single doses also may produce gastrointestinal, cardiovascular, and dermal manifestations. These and other drug-associated toxicities are discussed in more detail below. Hearing and vision are particularly affected. Functional impairment of the eighth nerve results in tinnitus, decreased auditory acuity, and vertigo. Visual signs consist of blurred vision, disturbed color perception, photophobia, diplopia, night blindness, constricted visual fields, scotomata, mydriasis, and even blindness (Bateman and Dyson, 1986). The visual and auditory effects are probably the result of direct neurotoxicity, although secondary vascular changes may have a role. Marked spastic constriction of the retinal vessels occurs; the retina is ischemic, the discs are pale, and retinal edema may ensue. In severe cases, optic atrophy results. Gastrointestinal symptoms also are prominent in cinchonism. Nausea, vomiting, abdominal pain, and diarrhea result from the local irritant action of quinine, but the nausea and emesis also have a central basis. The skin often is hot and flushed, and sweating is prominent. Rashes frequently appear. Angioedema, especially of the face, occasionally is observed. Quinine and quinidine, even at therapeutic doses, may cause hyperinsulinemia and severe hypoglycemia through their powerful stimulatory effect on pancreatic beta cells. Despite treatment with glucose infusions, this complication can be serious and possibly life-threatening, especially in pregnancy and prolonged severe infection. Quinine rarely causes cardiovascular complications unless therapeutic plasma concentrations are exceeded (Krishna and White, 1996). However, severe hypotension is predictable when the drug is administered too rapidly by the intravenous route. Acute overdosage also may cause serious and even fatal cardiac dysrhythmias such as sinus arrest, junctional rhythms, A-V block, and ventricular tachycardia and fibrillation (Bateman and Dyson, 1986). Quinidine is even more cardiotoxic than quinine; its effects on the heart are discussed in detail in Chapter 35: Antiarrhythmic Drugs. When small doses of cinchona alkaloids cause toxic manifestations, the individual usually is hypersensitive to the drug. Cinchonism may appear after a single dose of quinine, but it is usually mild. Cutaneous flushing, pruritus, skin rashes, fever, gastric distress, dyspnea, ringing in the ears, and visual impairment are the usual expressions of hypersensitivity; extreme flushing of the skin accompanied by intense, generalized pruritus is the most common form. Hemoglobinuria and asthma from quinine may occur more rarely. 'Blackwater fever'the triad of massive hemolysis, hemoglobinemia, and hemoglobinuria leading to anuria, renal failure, and even deathis a rare type of hypersensitivity reaction to quinine therapy that occurs in pregnancy and in treatment of malaria. Quinine may cause milder hemolysis upon occasion, especially in people with glucose-6-phosphate dehydrogenase deficiency. Symptomatic thrombocytopenic purpura by an antibody- and complement-dependent mechanism also is rare, but this reaction can occur even in response to ingestion of tonic water ('cocktail purpura'). Asthma may occur in hypersensitive individuals, and other rare reactions to the drug include hypoprothrombinemia, leukopenia, and agranulocytosis. High doses of quinine used to terminate pregnancy may possibly cause fetal abnormalities. Precautions, Contraindications, and Interactions Quinine must be used with considerable caution, if at all, in patients who manifest hypersensitivity to the drug, especially when this takes the form of cutaneous, angioedematous, visual, or auditory symptoms. Quinine should be discontinued immediately if evidence of hemolysis appears. The drug should not be used in patients with tinnitus or optic neuritis. In patients with cardiac dysrhythmias, the administration of quinine requires the same precautions as for quinidine (seeChapter 35: Antiarrhythmic Drugs). Because parenteral solutions of quinine are highly irritating, the drug should not be given subcutaneously; concentrated solutions may cause abscesses when injected intramuscularly or thrombophlebitis when infused intravenously. Absorption of quinine from the gastrointestinal tract can be delayed by antacids containing aluminum. Quinine and quinidine can delay the absorption and elevate plasma levels of digoxin and related cardiac glycosides (seeChapters 34: Pharmacological Treatment of Heart Failure and 35: Antiarrhythmic Drugs). Likewise, the alkaloid may raise plasma levels of warfarin and related anticoagulants. The action of quinine at neuromuscular junctions will enhance the effect of neuromuscular blocking agents and oppose the action of acetylcholinesterase inhibitors (see above). The renal clearance of quinine can be decreased by cimetidine and increased by acidification of the urine. |
Antibacterial Agents in Antimalarial Chemotherapy
|
Shortly after their introduction into
therapeutics, the sulfonamides were found to have antimalarial activity, a
property investigated extensively during World War II. The sulfones also were
shown to be effective; the first trial of dapsone was against P.
falciparum in 1943. The sulfonamides are used together with pyrimethamine
and often in addition to quinine to treat chloroquine-resistant falciparum
malaria, especially in parts of Sulfonamides and Sulfones The sulfonamides and sulfones are slow-acting blood schizontocides
that are more active against P. falciparum than P. vivax. As p-aminobenzoate
analogs that competitively inhibit the dihydropteroate synthase of P.
falciparum, the sulfonamides are used together with an inhibitor of
parasite dihydrofolate reductase to enhance their antiplasmodial action. For
example, sulfadiazine and the dihydrofolate reductase inhibitor, pyrimethamine,
are effective when used together with quinine to treat acute attacks of chloroquine-resistant
falciparum malaria in The future of the slow-acting antimalarial antifolates appears bleak unless they are used together with more effective, rapidly acting drugs, e.g., an artemisinin derivative. Long-term use of pyrimethamine-sulfadoxine, for example, is no longer recommended for prophylaxis of falciparum malaria because of potentially serious toxicity from the sulfonamide (see'Diaminopyrimidines,' above, and Chapter 44: Antimicrobial Agents: Sulfonamides, Trimethoprim-Sulfamethoxazole, Quinolones, and Agents for Urinary Tract Infections) (Bjorkman and Phillips-Howard, 1991). Moreover, resistance of P. falciparum to the antifolates is prevalent and develops rapidly upon exposure to these agents, rendering them ineffective in many parts of the world. Mutations that cause amino acid substitutions at several different loci in the dihydropteroate synthase of P. falciparum confer resistance to sulfadoxine and raise the Ki values for other sulfonamides and dapsone as well. These and other mutations that accumulate during exposure to sulfonamides, together with dihydrofolate reductase mutations associated with pyrimethamine and cycloguanil resistance, can severely compromise therapy of falciparum malaria with antifolate combinations (reviewed by Cowman, 1998). Tetracyclines Tetracyclines are particularly useful for the treatment of the acute malarial attack due to multidrug-resistant strains of P. falciparum that also show partial resistance to quinine (Chongsuphajaisiddhi et al., 1986). Their relative slowness of action makes concurrent treatment with quinine mandatory for rapid control of parasitemia. Several tetracyclines appear equivalent, but tetracycline or doxycycline usually is recommended. Although tetracycline has shown marked activity against primary tissue schizonts of chloroquine-resistant P. falciparum, its long-term use for prophylaxis is not advised. Instead, doxycycline is used alone by travelers for short-term prophylaxis of multidrug-resistant strains. Dosage regimens for tetracyclines and doxycycline are listed in Tables 401 and 402. Because of their adverse effects on bones and teeth, tetracyclines should not be given to pregnant women or children less than 8 years old. Photosensitivity reactions or drug-induced superinfections may mandate discontinuation of therapy or prophylaxis with these agents (seeChapter 47: Antimicrobial Agents: Protein Synthesis Inhibitors and Miscellaneous Antibacterial Agents). |
Guidelines for Prophylaxis and Chemotherapy of Malaria
|
Pharmacological control of malaria poses a
difficult challenge, because P. falciparum, which causes more than 85%
of the cases and nearly all the deaths from human malaria, has become
progressively more resistant to available antimalarial drugs. Fortunately, chloroquine
is still effective against malarias caused by P. ovale, P. malariae,
most strains of P. vivax (Newton and White, 1999), and chloroquine-sensitive
strains of P. falciparum found in some geographic areas. However,
chloroquine-resistant strains of P. falciparum now prevail in all
endemic areas except Genetic studies indicate that single isolates of P. falciparum from patients in highly endemic areas contain many parasite clones with different drug-resistance phenotypes (Druilhe et al., 1998). Any given clone may exhibit several drug-resistant traits and considerable variation in its infectious behavior. Differing and often complex genetic profiles also may be associated with a particular drug-resistant phenotype. Such findings strongly suggest that using any single agent to treat falciparum malaria encourages the selection and spread of drug-resistant strains that ultimately render such therapy both ineffective and hazardous. Indeed, parasites that had already developed drug-resistant traits might be even more prone to acquiring resistance to new unrelated antimalarial agents (Rathod et al., 1997). These observations, along with evidence from dose-finding pharmacokinetic/pharmacodynamic clinical studies, strongly suggest using regimens consisting of two or more agents with complementary antimalarial actions to treat drug-resistant falciparum malaria (see reviews by White, 1997, 1999). Promising examples of such regimens under evaluation are an artemisinin compound followed by mefloquine; an artemisinin compound, chlorproguanil, and dapsone; and proguanil together with atovaquone. Current recommendations for drugs and dosing regimens for the prophylaxis and therapy of malaria in nonimmune individuals are shown in Tables 401 and 402. These will change and should serve only as general guidelines to be appropriately modified according to the status and habitat of the patient; the geographic origin, species, and drug resistance profile of infecting parasites; and the agents used locally for malaria control. The following section presents an overview of the chemoprophylaxis and
chemotherapy of malaria. For more details about individual drugs and their
clinical applications, the reader should consult the text and references,
especially reviews by Zucker and Campbell (1993) and Importantly, drugs should not replace simple, inexpensive measures for malaria prevention. Individuals visiting malarious areas should take appropriate steps to prevent mosquito bites. Avoiding exposure to mosquitoes at dusk and dawn, usually times of maximal feeding, is one such measure. Others include wearing long-sleeved dark clothes, using insect repellents containing at least 30%N,N'-diethylmetatoluamide (DEET), and sleeping in well-screened rooms or under bed nets impregnated with a pyrethrin insecticide such as permethrin (Zucker and Campbell, 1993). In areas where malaria is endemic, mefloquine is the drug of choice
for prophylaxis. For those individuals who cannot take mefloquine, doxycycline
represents an alternative chemoprophylactic agent. If both mefloquine and
doxycycline are contraindicated, travelers must resort to less-effective
regimens. In certain parts of Africa south of the With the notable exception of infection due to drug-resistant strains
of P. falciparum, the chemotherapy of an acute attack of human malaria
is the same for all species of Plasmodium; only subsequent treatment
depends on the species. A malarial attack should be viewed as a medical
emergency, especially for vulnerable populations such as nonimmune travelers,
pregnant women, or young children. Treatment with a rapidly acting blood
schizontocide must be instituted promptly if falciparum malaria is suspected
from a travel history and clinical findings. One should not wait for a
definitive parasitological diagnosis in such patients because their clinical
status may deteriorate rapidly. Moreover, the clinical presentation may be
atypical, and thick blood smears may fail to reveal plasmodia in early stages
of this infection. Chloroquine is the drug of choice for P. vivax, P.
ovale, P. malariae, or chloroquine-sensitive strains of P.
falciparum. The oral route of administration is used whenever possible,
but chloroquine can be given intramuscularly or even intravenously if
suitable precautions are taken (see text). Within 48 to 72 hours of
initiating therapy, patients should show marked clinical improvement and a
substantial decrease in parasitemia as monitored by daily thick blood smears.
Lack of such a response or failure to clear parasites from the blood by 7
days is indicative of drug resistance. If chloroquine-resistant falciparum
malaria is suspected, either from the travel history or lack of response to
chloroquine, the preferred schizontocide is quinine despite its toxicity. For
multidrug-resistant falciparum malaria, quinine is given together with other
effective but slower-acting blood schizontocides such as antifolates or
tetracyclines; choice of the latter drugs depends on multiple factors (seeTable
402 and text). Again, the oral route of drug administration is preferred (quinine
sulfate), but intravenous preparations (quinine dihydrochloride) should be
given until oral medication can be taken. Quinidine gluconate, available in
most hospitals in the Attacks of malaria may recur during or after a course of antimalarial
chemotherapy, even in the absence of reinfection. Recurrent attacks caused by
P. vivax, P. ovale, or P. malariae usually are well
controlled by another course of chloroquine, combined with or followed by a
course of primaquine in the case of P. vivax or P. ovale. Some
patients with vivax infection may require more than one course to effect a
radical cure. Recrudescence of falciparum malarial attacks or parasitemia
after appropriate treatment with chloroquine usually denotes infection with
chloroquine-resistant plasmodia (for clinical classification of drug
resistance, seeWorld Health Organization, 1981). Quinine together with
a slower-acting drug such as doxycycline in Southeast Asia or with antifolate
antimalarials (e.g., pyrimethamine-sulfadoxine) in Malarial infection, especially with P. falciparum, is a severe threat to children and pregnant women. With appropriate adjustments and safety precautions, the treatment of children is generally the same as for adults. However, tetracyclines should not be given except in an emergency to children less than 8 years old. Pregnant women should be urged not to travel to endemic areas if at all possible. While chloroquine, proguanil, and the artemisinin compounds may be used during pregnancy, antifolates, tetracyclines, and primaquine should be avoided (see text and Tables 401 and 402). |
Prospectus
|
The future of antimalarial chemotherapy, long the mainstay for malaria control, appears bleak unless prompt action is taken. As with antibiotics, extensive use of antimalarials has expedited the selection and spread of increasingly drug-resistant strains of P. falciparum, which cause lethal infections in human beings. Previously effective drugs such as chloroquine rapidly are becoming obsolete. The search for novel antimalarials has suffered from lack of financial resources and commitment. Even development and distribution of lifesaving alternatives such as artemisinin and its derivatives have been seriously delayed. Ironically, the pace of relevant scientific discovery has dramatically increased. Cultivation and isolation of drug-sensitive and drug-resistant clones of P. falciparum from human beings and experimental animals now provide parasite materials essential for biological characterization, drug screening, and identification of molecular targets for drug action and resistance. The sequencing of the entire P. falciparum genome soon will be completed. Together with a rich variety of multidisciplinary experimental approaches, this achievement provides a major advance for antimalarial drug development. Clinical evaluation of antimalarial chemotherapy must keep pace with the identification of promising new agents. To place the chemotherapy of malaria on a rational basis, combined pharmacokinetic and pharmacodynamic studies must continue to determine the optimal indications and appropriate dosing regimens for different parasite and human populations. For further discussion of malaria, seeChapter 195 in Harrison's
Principles of Internal Medicine, 16th ed., |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3904
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved