| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders
Overview
|
Drugs with demonstrated efficacy in a broad range of severe psychiatric disorders have been developed since the 1950s, leading to development of the subspecialty of psychopharmacology. Knowledge of the actions of such agents has greatly stimulated research in biological psychiatry aimed at defining pathophysiological changes. This chapter reviews current knowledge of the pharmacology of antidepressants and the treatment of depression and anxiety disorders. Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania covers antipsychotic and antimanic agents and the treatment of psychotic and manic-depressive illness. The treatment of depression relies on a varied group of antidepressant therapeutic agents, in part because clinical depression is a complex syndrome of widely varying severity. The first agents used successfully were tricyclic antidepressants, which elicit a wide range of neuropharmacological effects in addition to their presumed primary action of inhibiting norepinephrine and, variably, serotonin transport into nerve endings, thus leading to sustained facilitation of noradrenergic and perhaps serotonergic function in the brain. Inhibitors of monoamine oxidase, which increase the brain concentrations of many amines, also have been used. Currently, a series of innovative agentsmost notably the selective serotonin-reuptake inhibitors (see Chapter 11: 5-Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists)dominate the treatment of depressive disorders and are widely used to treat severe anxiety disorders. In addition to the widespread use of antidepressants, the pharmacological treatment of anxiety disorders commonly employs benzodiazepine sedativeantianxiety agents, which facilitate neuronal hyperpolarization through the gamma-aminobutyric acid (GABA)-receptorCl-channel macromolecular complex. Potent benzodiazepines are effective in panic disorder as well as in generalized anxiety disorder. Their long-term risk:benefit ratio remains controversial. Serotonin 5-HT1Areceptor partial agonists such as buspirone also have useful anxiolytic and other psychotropic activity and less likelihood of inducing sedation or dependence. Specialized uses of antidepressants discussed in this chapter include the treatment of anxiety disorders, including obsessive-compulsive disorder, panic-agoraphobia, and social phobias. |
Introduction: Psychopharmacology
|
The use of drugs with demonstrated efficacy
in psychiatric disorders has become widespread since the mid-1950s. Today,
about 10% to 15% of prescriptions written in the Psychotropic agents can be placed into four major categories. Antianxiety-sedative agents, particularly the benzodiazepines, are those used for the drug therapy of anxiety disorders; their pharmacology is reviewed in Chapter 17: Hypnotics and Sedatives. Antidepressants (mood-elevating agents) and antimanic or mood-stabilizing drugs (notably, lithium salts and certain anticonvulsants; seeChapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania) are those used to treat affective or mood disorders and related conditions. Antipsychotic or neuroleptic drugs (seeChapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania) are those used to treat very severe psychiatric illnessesthe psychoses and mania; they have beneficial effects on mood and thought, but many standard neuroleptic agents carry the risk of producing characteristic side effects that mimic neurological diseases, whereas modern antipsychotics are associated with weight gain and adverse metabolic effects such as diabetes. The use of drugs in the treatment of psychiatric disorders is becoming
more precise as psychiatric diagnoses continue to gain objectivity,
coherence, and reliability. Searches for biological bases of psychiatric
illnesses have been stimulated by knowledge of the mechanisms of action of
psychotropic agents and the emergence of a medical discipline commonly known
as biological psychiatry (Baldessarini, 2000). The diagnostic
terminology and criteria for psychiatric disorders currently employed in the History Modification of behavior, mood, and emotion by drugs always has been a
favorite practice of human beings. The use of psychoactive drugs evolved
along two related paths: the use of substances to modify normal behavior and
to produce altered states of feeling for religious, ceremonial, or
recreational purposes, and their use to alleviate mental ailments.
Fascinating accounts of the early history and characteristics of many
psychoactive compounds, particularly those derived from natural products, are
presented by Lewin (1931) and Efron and associates (1967) (seeAyd and
Blackwell, 1970; Baldessarini, 1985; Caldwell, 1978). In 1845, Moreau
proposed that hashish intoxication provided a model psychosis useful in the
study of insanity. Three decades later, Freud presented his study of cocaine
and suggested its potential uses in pharmacotherapy. Soon thereafter,
Kraepelin founded the first laboratory of clinical psychopharmacology in The first modern report on the treatment of psychotic excitement or
mania with lithium salts was that of Cade in 1949 (seeChapter
20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania).
Because of concerns about the toxicity of lithium, this discovery was slow in
gaining general acceptance by the medical community. In 1950, chlorpromazine
was synthesized in A report on meprobamate by Berger (1954) marked the beginning of investigations of modern sedatives with useful antianxiety properties. An antitubercular drug, iproniazid, was introduced in the early 1950s and soon recognized as a monoamine oxidase inhibitor and antidepressant (Kline, 1958); in 1958, Kuhn recognized the antidepressant effect of imipramine. The first of the antianxiety benzodiazepines, chlordiazepoxide, was developed by Sternbach in 1957 (seeChapter 17: Hypnotics and Sedatives). In the following year, Janssen discovered the antipsychotic properties of haloperidol, a butyrophenone (seeChapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania), and thus still another class of antipsychotic agents became available. During the 1960s, the expansion of psychopharmacological research was rapid, and many new theories of psychoactive drug effects were introduced. The clinical efficacy of many of these agents was firmly established during that decade. For many years, the role of biogenic amines and their receptors in the central nervous system (CNS) in mediating effects of psychotropic drugs has been emphasized and has stimulated searches for the causes of mental illness (seeBaldessarini, 2000). In addition, increasing attention has been paid to the liabilities of treatment with psychotherapeutic drugs, especially their limited efficacy in severe or chronic mental illnesses, their risk of sometimes serious adverse effects, and the limitations, conservatism, and basic circularity of screening and testing methods used to develop new agents (seeBaldessarini, 2000). The antipsychotic, mood-stabilizing, and antidepressant agents used to treat the most severe mental illnesses have had a remarkable impact on psychiatric practice and theoryan impact that legitimately can be called revolutionary and one that is experiencing continued innovation. Nosology The different classes of psychotropic agents are selective in their ability to modify symptoms of mental illnesses. The optimal use of such drugs thus requires familiarity with the differential diagnosis of psychiatric conditions (seeSadock and Sadock, 2000; American Psychiatric Association, 2000). A few salient aspects of psychiatric classification are summarized briefly here, and additional information is provided in the discussion of specific classes of drugs. Basic distinctions are made among the cognitive disorders, psychotic disorders, mood disorders, anxiety disorders, and disorders of personality. The cognitive disorder syndromes of delirium and dementia commonly are associated with definable neuropathological, metabolic, or toxic (including drug-induced) changes and are characterized by confusion, disorientation, and memory disturbances as well as behavioral disorganization. In general, the effectiveness of pharmacological treatment of the core cognitive impairment in the dementias remains limited, despite extensive efforts to develop effective treatments. These have included use of stimulants, so-called notropics (e.g., periacetam), cholinesterase inhibitors, putative cerebral vasodilators (e.g., ergot alkaloids, papaverine, isoxuprine), and the calcium channel blockers, such as nimodipine (seeChapters 32: Drugs Used for the Treatment of Myocardial Ischemia and 33: Antihypertensive Agents and the Drug Therapy of Hypertension; Knapp et al., 1994; Marin and Davis, 1998). This topic is not specifically covered in this chapter. The psychoses are among the most severe psychiatric disorders, in which there is not only marked impairment of behavior but also a serious inability to think coherently, to comprehend reality, or to gain insight into the presence of these abnormalities. These common disorders (affecting perhaps 0.5% to 1.0% of the population at some age) typically include symptoms of false beliefs (delusions) and abnormal sensations (hallucinations). The psychotic disorders are suspected of having a neurobiological basis but usually are distinguished from the cognitive disorders. The etiological basis of the psychotic disorders remains unknown, although genetic and neurodevelopmental as well as environmental causative factors have been proposed. Representative syndromes in this category include schizophrenia, brief psychoses, and delusional disorders, although psychotic features also are not uncommon in the major mood disorders, particularly mania and severe melancholic depression. Psychotic illnesses are characterized by disorders of thinking processes, as inferred from illogical or highly idiosyncratic communications, with disorganized or irrational behavior and varying degrees of altered mood that can range from excited agitation to severe emotional withdrawal. Idiopathic psychoses characterized mainly by chronically disordered thinking and emotional withdrawal and often associated with delusions and auditory hallucinations are called schizophrenia. Acute or recurrent idiopathic psychoses also occur that bear an uncertain relationship to schizophrenia or the major affective disorders. In addition, more or less isolated delusions can arise in delusional disorder or paranoia. Antipsychotic drugs exert beneficial effects in many types of psychotic illness and are not selective for schizophrenia. Their beneficial actions are found in disorders ranging from postsurgical delirium and amphetamine intoxication to paranoia, mania, and psychotic depression, and they can be beneficial against the agitation of Alzheimer's dementia. They are especially beneficial in severe depression and possibly other conditions marked by severe turmoil or agitation. This class of agents is discussed in Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania. The major disorders of mood or affect include the syndromes of major depression (formerly including melancholia) and bipolar disorder (formerly manic-depressive disorder). These disorders are quite prevalent, affecting several percent of the population at some time. They commonly include disordered autonomic functioning (e.g., altered activity rhythms, sleep, and appetite) and behavior, as well as persistent abnormalities of mood. These disorders parallel an increased risk of self-harm or suicide as well as increased mortality from stress-related general medical conditions, medical complications of commonly comorbid abuse of alcohol or drugs, or from accidents. Bipolar disorder is marked by a high likelihood of recurrences of severe depression and manic excitement, often with psychotic features. Major depression is usually treated with a variety of agents generally considered to be antidepressants, which have beneficial effects on the symptoms of major depression as well as on those of anxiety disorders. They are discussed further in this chapter. Bipolar disorder usually is treated primarily with lithium, certain anticonvulsants, or other agents with mood-stabilizing effects, as discussed in Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania. The less pervasive psychiatric disorders include conditions formerly termed psychoneuroses, which currently are viewed as anxiety-associated disorders. Whereas the ability to comprehend reality is retained, suffering and disability sometimes are severe. Anxiety disorders may be acute and transient or, commonly, recurrent or persistent. Their symptoms may include mood changes (fear, panic, dysphoria) or limited abnormalities of thought (obsessions, irrational fears or phobias) or of behavior (avoidance, rituals or compulsions, pseudoneurological or 'hysterical' conversion signs, or fixation on imagined or exaggerated physical symptoms). In such disorders, drugs may have some beneficial effects, particularly by modifying associated anxiety and depression and so facilitating a more comprehensive program of treatment and rehabilitation. Currently, antidepressants as well as sedative-antianxiety agents commonly are used to treat anxiety disorders, which are considered later in this chapter. Other typically lifelong conditionsincluding the personality disorders, substance-use disorders, and hypochondriasismay or may not respond appreciably to pharmacological intervention. Personality disorders have prominent avoidant, antisocial, paranoid, withdrawn, dependent, or unstable characteristics. Other disorders involve patterns of behavior (e.g., abuse of alcohol or other substances, deviant eating, exaggerated somatic preoccupations, or other abnormal behaviors). Typically, psychotropic drugs alone are not effective in such long-term conditions except when anxiety or depression occur. Pharmacological treatment also is an important component of the medical management of withdrawal from addicting substances and in supporting their avoidance (seeChapter 24: Drug Addiction and Drug Abuse; Cornish et al., 1998). Biological Hypotheses in Mental Illness The introduction in the 1950s of relatively effective and selective drugs for the management of schizophrenic and manic-depressive patients encouraged formulation of biological concepts of the pathogenesis of these major mental illnesses. In addition, other agents were discovered that mimic some of the symptoms of severe mental illnesses. These include LSD, which induces hallucinations and altered emotional states; antihypertensive agents such as reserpine, which can induce depression; and stimulants that can induce manic or psychotic states when taken in excess. A leading hypothesis that arose from such considerations was based on observations indicating that antidepressants enhance the biological activity of monoamine neurotransmitters in the CNS and that antiadrenergic compounds may induce depression. These observations led to speculation that a deficiency of aminergic transmission in the CNS might be causative of depression or that an excess could result in mania. Further, since antipsychotic agents antagonize the actions of dopamine as a neurotransmitter in the forebrain, it was proposed that there may be a state of functional overactivity of dopamine in the limbic system or cerebral cortex in schizophrenia or mania. Alternatively, an endogenous psychotomimetic compound might be produced either uniquely or in excessive quantities in psychotic patients. This 'pharmacocentric' approach to the construction of hypotheses was appealing and gained strong encouragement from studies of the actions of antipsychotic and antidepressant drugs while also encouraging further development of similar agents. In turn, the plausibility of such biological hypotheses has encouraged interest in genetic studies as well as in clinical biochemical studies. Despite extensive efforts, attempts to document metabolic changes in human subjects predicted by these hypotheses have not, on balance, provided consistent or compelling corroboration (Baldessarini, 2000; Bloom and Kupfer, 1995; Musselman et al., 1998). Moreover, results of genetic studies have provided evidence that inheritance can account for only a portion of the causation of mental illnesses, leaving room for environmental and psychological hypotheses. The antipsychotic, antianxiety, antimanic, and antidepressant drugs have effects on cortical, limbic, hypothalamic, and brainstem mechanisms that are of fundamental importance in the regulation of arousal, consciousness, affect, and autonomic functions. It is quite possible that physiological and pharmacological modification of these brain regions have important behavioral consequences and useful clinical effects regardless of the fundamental nature or cause of the mental disorder in question. The lack of symptomatic or even syndromal specificity of most psychotropic drugs tends to reduce the chances of finding a discrete metabolic correlate for a specific disease conceived simply on the actions of therapeutic agents. Finally, the technical problems associated with attempts to study changes in the in vivo metabolism or the postmortem chemistry of the human brain are formidable. Among these are artifacts introduced by drug treatment itself. In summary, the available information does not permit a conclusion as to whether or not discrete biological lesions are the crucial basis of the most severe mental illnesses (other than the deliria and dementias). Moreover, it is not necessary to presume that such a basis is operative to provide effective medical treatment for psychiatric patients. Furthermore, it would be clinical folly to underestimate the importance of psychological and social factors in the manifestations of mental illnesses or to overlook psychological aspects of the conduct of biological therapies (Baldessarini, 2000). Identification and Evaluation of Psychotropic Drugs Although rational, predictive development and assessment of the efficacy of any drug is problematic, the difficulties in evaluating psychoactive drugs are particularly challenging. The essential characteristics of human mental disorders cannot be reproduced in animals. Cognition, communication, and social relationships in animals are difficult to compare with human conditions. Thus, screening procedures in animals are of limited utility for the discovery of unique therapeutic agents. Contemporary pharmacology has provided many techniques for characterizing the actions of known psychotropic and other CNS agents at the cellular and molecular levels. Characteristics such as affinity for specific receptors or transporters can lead to the identification of new agents. Further innovation has been emerging slowly from the rapid recent progress in identifying novel subtypes of classical neurotransmitter receptors, effectors, and many other macromolecular target sites in brain tissue for potential new drugs (Baldessarini, 2000). In addition, clinical evaluation of new drugs is hampered by the lack of homogeneity within diagnostic groups and difficulty in application of valid, sensitive measurements of the effects of therapy. As a consequence, the results of clinical trials of psychotropic agents sometimes seem equivocal or inconsistent. |
Treatment of Depressive and Anxiety Disorders
|
Major depression is characterized by clinically significant depression of mood and impairment of functioning as its primary clinical manifestations. Its clinical manifestations and current treatment overlap the anxiety disorders, including panic-agoraphobia syndrome, severe phobias, generalized anxiety disorder, social anxiety disorder, posttraumatic stress disorder, and obsessive-compulsive disorder. Extremes of mood may be associated with psychosis, manifested as disordered or delusional thinking and perceptions, often congruent with the predominant mood. Conversely, psychotic disorders may have associated or secondary changes in mood. This overlap of disorders may lead to errors in diagnosis and clinical management (American Psychiatric Association, 2000). Each with a lifetime morbid risk of perhaps 10% in the general population, major mood and anxiety disorders are the most common mental illnesses (Kessler et al., 1994). Clinical depression is distinguished from normal grief, sadness, disappointment, and the dysphoria or demoralization often associated with medical illness. The condition is underdiagnosed and frequently undertreated (McCombs et al., 1990; Suominen et al., 1998). Major depression is characterized by feelings of intense sadness and despair, mental slowing and loss of concentration, pessimistic worry, lack of pleasure, self-deprecation, and variable agitation. Physical changes also occur, particularly in severe, vital, or 'melancholic' depression. These include insomnia or hypersomnia; altered eating patterns, with anorexia and weight loss or sometimes overeating; decreased energy and libido; and disruption of the normal circadian and ultradian rhythms of activity, body temperature, and many endocrine functions. As many as 10% to 15% of individuals with this disorder, and up to 25% of those with bipolar disorder, display suicidal behavior during their lifetime (Baldessarini and Jamison, 1999). Depressed patients usually respond to antidepressant drugs or, in severe or treatment-resistant cases, to ECT (seeRudorfer et al., 1997). The decision to treat with an antidepressant is guided by the presenting clinical syndrome and its severity and by the patient's personal and family history. Most antidepressants exert important actions on the metabolism of monoamine neurotransmitters and their receptors, particularly norepinephrine and serotonin (Buckley and Waddington, 2000; Owens et al., 1997). Their therapeutic effectiveness and actions, together with strong evidence for genetic predisposition, have led to speculation that the biological basis of major mood disorders may include abnormal function of monoamine neurotransmission. However, the direct evidence for this view is limited and inconsistent (seeBaldessarini, 2000; Bloom and Kupfer, 1995; Heninger and Charney, 1987; Musselman et al., 1998). Diagnosis and treatment of the severe anxiety disorders has advanced
recently, stimulated by the discovery that selective serotonin-reuptake
inhibitors, which are effective antidepressants, also are powerful
antianxiety agents. Disorders including panic-agoraphobia, social and other
phobias, generalized anxiety, and obsessive-compulsive disorder as well as
apparently related disorders of impulse control all appear to be responsive
to treatment with serotonin-reuptake inhibitors ( Mania and the alternation or admixture of mania and depression (bipolar disorder) are less common than nonbipolar major depression. Mania and its milder form (hypomania) are treated with antipsychotic drugs, anticonvulsants, or lithium salts, sometimes supplemented with a potent sedative in the short term and lithium salts or certain anticonvulsants with mood-stabilizing properties (seeChapters 17: Hypnotics and Sedatives and 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania) for longer-term prevention of recurrences. Mania is characterized by excessive elation, typically tinged with dysphoria or marked by irritability, severe insomnia, hyperactivity, uncontrollable speech and activity, impaired judgment, and risky behaviors, and sometimes by psychotic features. The selection and administration of appropriate treatment for depression and anxiety disorders are discussed below. Antidepressants Imipramine, amitriptyline, their N-demethyl derivatives, and other similar compounds were the first successful antidepressants and, since the early 1960s, have been widely used for the treatment of major depression. Because of their structures (seeTable 191), these agents often are referred to as the 'tricyclic' antidepressants (Frazer, 1997). Their efficacy in alleviating major depression is well established, and they also have proved useful in a number of other psychiatric disorders. Just prior to the discovery of the antidepressant properties of imipramine in the late 1950s, the ability of monoamine oxidase (MAO) inhibitors to cause mania was noted, and during the early 1960s, both types of agents were studied intensively in the treatment of clinical depression. Early MAO inhibitors appeared to be limited in efficacy at the doses used and presented both toxic risks and potentially dangerous interactions with other agents, thus limiting their acceptance in favor of the tricyclic agents. After decades of limited progress, a series of innovative antidepressants has emerged. Mostlike citalopram, fluoxetine, fluvoxamine, paroxetine, sertraline, and venlafaxineare inhibitors of the active reuptake (transport) of serotonin (5-hydroxytryptamine, 5-HT) into nerve terminals (seeChapter 11: 5-Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists). Othersincluding bupropion, nefazodone, and mirtazapinehave a less well defined neuropharmacology and can be considered 'atypical.' Whereas the efficacy of the newer agents is not superior to that of the older agents, their relative safety and tolerability has led to their rapid acceptance as the most commonly prescribed antidepressants. History Monoamine Oxidase Inhibitors In 1951, isoniazid and its isopropyl derivative, iproniazid, were developed for the treatment of tuberculosis. Iproniazid had mood-elevating effects in tuberculosis patients. In 1952, Zeller and coworkers found that iproniazid, in contrast to isoniazid, was capable of inhibiting the enzyme MAO. Following investigations by Kline and by Crane in the mid-1950s, iproniazid was used for the treatment of depressed patients; historically, it is the first clinically successful modern antidepressant (Healy, 1997). Tricyclic Antidepressants Hfliger and Schindler in the late 1940s synthesized a series of more than 40 iminodibenzyl derivatives for possible use as antihistamines, sedatives, analgesics, and antiparkinsonism drugs. One of these was imipramine, a dibenzazepine compound, which differs from the phenothiazines only by replacement of the sulfur with an ethylene bridge to produce a seven-membered central ring analogous to the benzazepine antipsychotic agents (seeChapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania). Following screening in animals, a few compounds, including imipramine, were selected on the basis of sedative or hypnotic properties for therapeutic trial. During clinical investigation of these putative phenothiazine analogs, Kuhn (1958) fortuitously found that, unlike the phenothiazines, imipramine was relatively ineffective in quieting agitated psychotic patients, but it had a remarkable effect on depressed patients; indisputable evidence of its effectiveness in these patients has accumulated (seeBaldessarini, 1989; Hollister, 1978; Potter et al., 1998; Thase and Nolen, 2000). Older tricyclic antidepressants with a tertiary-amine side chain (including amitriptyline, doxepin, and imipramine) block neuronal uptake of both serotonin and norepinephrine, and clomipramine is relatively selective against serotonin (seeTable 192). Following this lead, even more selective serotonin-reuptake inhibitors were developed in the early 1970s, arising from observations by Carlsson that antihistamines including chlorpheniramine and diphenhydramine inhibited the transport of serotonin or norepinephrine. Chemical modifications led to the earliest selective serotonin-reuptake inhibitor, zimelidine, soon followed by development of fluoxetine and fluvoxamine (Carlsson and Wong, 1997; Fuller, 1992; Masand and Gupta, 1999; Tollefson and Rosenbaum, 1998; Wong and Bymaster, 1995). Zimelidine was first in clinical use, but withdrawn due to association with febrile illnesses and cases of Guillain-Barr ascending paralysis, leaving fluoxetine and fluvoxamine as the first widely used selective serotonin-reuptake inhibitors (dubbed SSRIs). The development of these agents was paralleled by identification of compounds with selectivity for norepinephrine reuptake and others effective against both serotonin and norepinephrine reuptake (see'Prospectus,' below). Chemistry and Structure-Activity Relationships Tricyclic Antidepressants The search for compounds related chemically to imipramine yielded multiple analogs. In addition to the dibenzazepines, imipramine and its secondary-amine congener (and major metabolite) desipramine, as well as its 3-chloro derivative clomipramine, there are amitriptyline and its N-demethylated metabolite nortriptyline (dibenzocycloheptadienes), as well as doxepin (a dibenzoxepine) and protriptyline (a dibenzocycloheptatriene). Other structurally related agents are trimipramine (a dibenzazepine, with only weak effects on amine transport); maprotiline (containing an additional ethylene bridge across the central six-carbon ring); and amoxapine (a piperazinyldibenzoxazepine with mixed antidepressant and neuroleptic properties). Since these agents all have a three-ring molecular core and most share pharmacological (norepinephrine-reuptake inhibition) and clinical (antidepressant, anxiolytic) properties, the trivial name 'tricyclic antidepressants' can be used for this group. Structures and other features of antidepressant compounds are given in Table 191. Selective Serotonin-Reuptake Inhibitors Most are aryl or aryloxyalkylamines. Several (including citalopram, fluoxetine, and zimelidine) are racemates; sertraline and paroxetine are separate enantiomers. The (S)-enantiomers of citalopram and of fluoxetine and its major metabolite norfluoxetine are highly active against serotonin transport and also may have antimigraine effects not found with the (R)-enantiomer of fluoxetine. The (R)-enantiomer of fluoxetine also is active against serotonin transport and is shorter-acting than the (S)-enantiomer. (R)-Norfluoxetine is virtually inactive (Wong et al., 1993). Structure-activity relationships are not well established for serotonin-reuptake inhibitors. However, it is known that the para-location of the CF3 substituent of fluoxetine (seeTable 191) is critical for serotonin transporter potency. Its removal and substitution at the ortho-position of a methoxy group yields nisoxetine, a highly selective norepinephrine-uptake inhibitor. Monoamine Oxidase Inhibitors The first MAO inhibitors to be used in the treatment of depression were derivatives of hydrazine, a highly hepatotoxic substance. Phenelzine is the hydrazine analog of phenethylamine, a substrate for MAO; isocarboxazide is a hydrazide derivative that probably is converted to the corresponding hydrazine to produce long-lasting inhibition of MAO. Subsequently, compounds unrelated to hydrazine were found to be potent MAO inhibitors. Several of these agents were structurally related to amphetamine and were synthesized in an attempt to enhance central stimulant properties. Cyclization of the side chain of amphetamine resulted in tranylcypromine, which also produces long-acting inhibition of MAO without covalent bonding. Selegiline and several experimental MAO inhibitors are propargylamines containing a reactive acetylenic bond that interacts irreversibly with the flavin cofactor of MAO (Cesura and Pletscher, 1992). Short-acting, reversible MAO inhibitors include brofaromine (a piperidylbenzofuran), moclobemide (a morpholinobenzamide), and toloxatone (an oxazolidinone). Moclobemide has at least moderate antidepressant activity (Lotufo-Neto et al., 1999). Pharmacological Properties: Central Nervous System Tricyclic Antidepressants and Other Norepinephrine-Reuptake Inhibitors Knowledge of the pharmacological properties of antidepressant drugs remains incomplete, and its coherent interpretation is limited by a lack of a compelling psychobiological theory of mood disorders. The actions of imipramine-like tricyclic antidepressants include a range of complex, secondary adaptations to their initial actions as inhibitors of neuronal transport (reuptake) of norepinephrine and variable blockade of serotonin transport (seeTable 192; Barker and Blakely, 1995; Beasley et al., 1992; Heninger and Charney, 1987; Leonard and Richelson, 2000; Potter et al., 1998; Wamsley et al., 1987). Tricyclic type antidepressants with secondary amine side chains or the N-demethylated (nor) metabolites of agents with tertiary-amine moieties (e.g., amoxapine, desipramine, maprotiline, norclomipramine, nordoxepin, nortriptyline) are relatively selective inhibitors of norepinephrine transport. Most tertiary-amine tricyclic antidepressants also inhibit the uptake-inactivation of serotonin. The amine transportinhibiting effects of antidepressants occur immediately and are sustained indefinitely. It is likely that selective inhibitors of norepinephrine reuptake, including reboxetine, share many of the actions of older norepinephrine-transport inhibitors like desipramine (Delgado and Michaels, 1999). Among the tricyclic antidepressants, trimipramine is exceptional in that it lacks prominent inhibitory effects at monoamine transport (seeTable 192), and its actions remain unexplained. The tricyclic and other norepinephrine-active antidepressants do not block dopamine transport (seeTable 192) and in that way differ from CNS stimulants, including cocaine, methylphenidate, and the amphetamines (seeChapter 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists). Nevertheless, they may have indirect dopamine-facilitating effects through interactions of increased perisynaptic abundance of norepinephrine, particularly in cerebral cortex, where adrenergic terminals exceed those releasing dopamine. Tricyclic antidepressants also can desensitize D2 dopamine autoreceptors, perhaps indirectly enhancing forebrain dopaminergic mechanisms, and so contribute to elevation of mood and behavioral activity (Potter et al., 1998). In addition to their transport-inhibiting effects, tricyclic
antidepressants have variable interactions with adrenergic receptors (seeTable
193). The presence or absence of such receptor interactions appears to be
critical for subsequent responses to increased availability of extracellular norepinephrine
in or near synapses. Most tricyclic antidepressants have at least moderate
and selective affinity for The Postsynaptic Postsynaptic Additional neuropharmacological changes that may contribute to the
clinical effects of tricyclic antidepressants include indirect facilitation
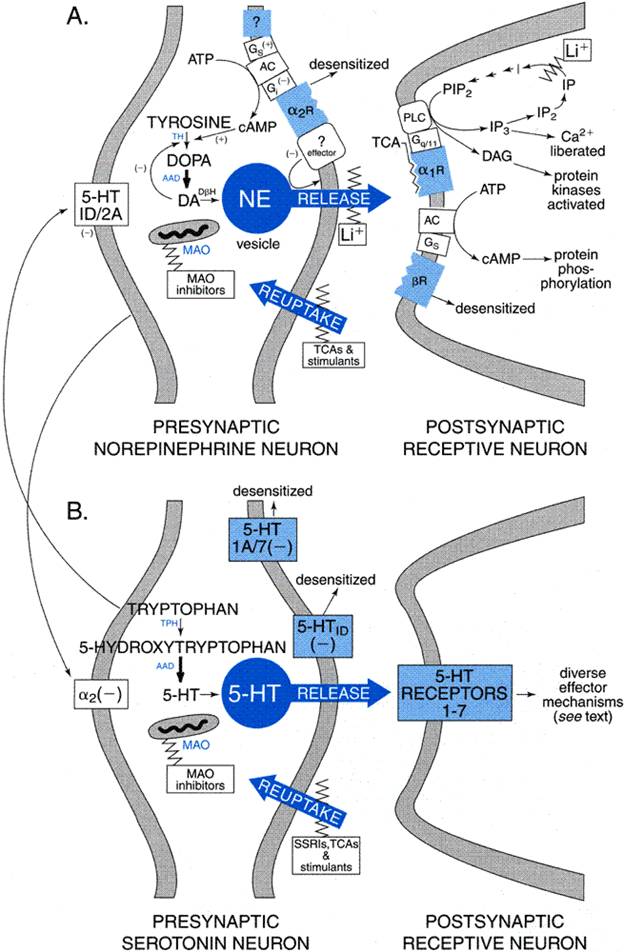
of serotonin and perhaps dopamine neurotransmission through excitatory Other adaptive changes have been observed in response to long-term treatment with tricyclic antidepressants. These include altered sensitivity of muscarinic acetylcholine receptors as well as decreases of GABAB gamma-aminobutyric acid receptors and possibly also NMDA glutamate receptors (Kitamura et al., 1991; Leonard and Richelson, 2000). In addition, there is a net gain in cyclic AMP production and altered activity of protein kinases in some cells, including those acting on cytoskeletal and other structural proteins that may alter neuronal growth and sprouting (Racagni et al., 1991; Wong et al., 1991). Nuclear genetic-regulatory factors also are affected, including the cyclic AMPresponse-element binding protein (CREB) and brain-derived neurotrophic factor (BDNF) (Duman et al., 1997; Siuciak et al., 1997). Additional changes may be indirect effects of antidepressant treatment or may reflect recovery from depressive illness. These include normalization of corticosteroid release and the sensitivity of corticosteroid receptors, as well as shifts in the production of prostaglandins and cytokines and in lymphocyte functions (Kitayama et al., 1988; Leonard and Richelson, 2000). Understanding of the physiological and psychobiological implications of these many molecular and cellular changes during repeated antidepressant treatment remains incomplete. Nevertheless, their occurrence underscores the important concept that repeated administration of neuroactive or psychotropic agents sets off a complex series of adaptive processes. Specifically regarding the tricyclic antidepressants, their neuropharmacology is not accounted for simply by blocking the transport-mediated removal of norepinephrine, even though this effect is no doubt a crucial initiating event leading to a cascade of important secondary adaptations (Duman et al., 1997; Hyman and Nestler, 1996; Leonard and Richelson, 2000). Interactions of antidepressants with monoaminergic synaptic transmission are illustrated in Figure 191.
Selective Serotonin-Reuptake Inhibitors (SSRIs) Understanding of the late and indirect actions of this very commonly used class of antidepressant and antianxiety agents remains much less well developed than does that of the actions of tricyclic antidepressants. However, there are striking parallels between responses in the noradrenergic and serotonergic systems. Like tricyclic antidepressants, which block norepinephrine reuptake, the serotonin-reuptake inhibitors block neuronal transport of serotonin immediately, and apparently indefinitely, leading to complex secondary responses (seeTable 192). Increased synaptic availability of serotonin stimulates a large number of postsynaptic 5-HT receptor types (Azmitia and Whitaker-Azmitia, 1995; seeChapter 11: 5-Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists). Stimulation of 5-HT3 receptors is suspected to contribute to common adverse effects characteristic of this class of drugs, including gastrointestinal (nausea, vomiting) and sexual effects (delayed or impaired orgasm). In addition, stimulation of 5-HT2C receptors may contribute to risk of agitation or restlessness sometimes induced by serotonin-reuptake inhibitors. An important parallel in responses of serotonin and norepinephrine neurons is that negative feedback mechanisms rapidly emerge to restore homeostasis (Azmitia and Whitaker-Azmitia, 1995). In the serotonin system, 5-HT1 subtype autoreceptors (types 1A and 7 at raphe cell bodies and dendrites, type 1D at terminals) suppress serotonin neurons in the raphe nuclei of the brainstem, including inhibition of tryptophan hydroxylase (again, probably through reduced phosphorylation-activation) and neuronal release of serotonin. Repeated treatment leads to gradual downregulation and desensitization of autoreceptor mechanisms over several weeks (particularly of 5-HT1D receptors at nerve terminals), with a return or increase of presynaptic activity, production, and release of serotonin (Blier et al., 1990; Chaput et al., 1991; Tome et al., 1997). Additional secondary changes include gradual down-regulation of postsynaptic 5-HT2A receptors that may contribute to antidepressant effects, as well as influencing the function of other neurons via serotonergic 'heteroceptors.' Many other postsynaptic 5-HT receptors presumably remain available to mediate increased serotonergic transmission and contribute to the mood-elevating and anxiolytic effects of this class of drugs. As in responses to norepinephrine-transport inhibitors, complex late adaptations to repeated treatment with serotonin-reuptake inhibitors occur. These may include indirect enhancement of norepinephrine output by reduction of tonic inhibitory effects of 5-HT2A heteroceptors. Finally, similar nuclear and cellular adaptations occur as with the tricyclic antidepressants, including a net gain of intraneuronal cyclic AMP and of nuclear regulatory factors including CREB and BDNF (Azmitia and Whitaker-Azmitia, 1995; Hyman and Nestler, 1996). Atypical Antidepressants Several antidepressants have effects on both noradrenergic and serotonergic neurotransmission. These include the older tricyclic antidepressants, particularly the tertiary amines including amitriptyline, clomipramine, doxepin, and imipramine (seeTable 192). However, even relatively potent serotonin-transport inhibitors like clomipramine and amitriptyline produce N-dealkylated metabolites with potent norepinephrine-uptakeinhibiting effects. Venlafaxine also has some effect on norepinephrine transport, and a series of novel agents with mixed effects on both transport systems is emerging (e.g., duloxetine and milnacipran). Drugs with significant dopamine-uptakeinhibiting actions include the older psychostimulants (seeChapter 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists; Fawcett and Busch, 1998). These agents provide only limited benefits in major depression and may worsen agitation, psychosis, insomnia, and anorexia associated with severe depressive illness. Nomifensine is an effective antidepressant that inhibits the uptake of both norepinephrine and dopamine (seeTable 192). The aromatic aminoketone bupropion (amfebutamone) and its amphetamine-like active metabolites also affect dopamine and norepinephrine transport (Ascher et al., 1995). The MAO inhibitor tranylcypromine is amphetamine-like in structure but interacts only weakly at dopamine transporters. The phenylpiperazine nefazodone and, to a lesser extent, the
structurally related trazodone have at least weak inhibitory actions
on serotonin transport, and nefazodone also may have a minor effect on norepinephrine
transport. This agent also has a prominent direct antagonistic effect at 5-HT2A
receptors that may contribute to antidepressant and anxiolytic activity. Both
drugs also may inhibit presynaptic 5-HT1 subtype autoreceptors to
enhance neuronal release of serotonin, though they probably also exert at
least partial agonist effects on postsynaptic 5-HT1 receptors (seeTable
193; Golden et al., 1998). Trazodone also blocks cerebral Finally, the structurally similar atypical antidepressants mirtazapine
and mianserin have potent antagonistic effects at several postsynaptic
serotonin receptor types (including 5-HT2A, 5-HT2C, and
5-HT3 receptors) and can produce gradual downregulation of 5-HT2A
receptors (Golden et al., 1998). Mirtazapine also limits the
effectiveness of inhibitory Monoamine Oxidase Inhibitors MAO is a flavin-containing enzyme localized in mitochondrial membranes found in nerve terminals, the liver, intestinal mucosa, and other organs (Cesura and Pletscher, 1992). MAO differs biochemically from nonspecific amine oxidases in plasma. It is closely linked functionally with an aldehyde reductase and an aldehyde dehydrogenase. The products of these reactions can be carboxylic acids or alcohols, depending on the substrate and the tissue. MAO regulates the metabolic degradation of catecholamines and serotonin in the CNS or peripheral tissues. Hepatic MAO has a crucial defensive role in inactivating circulating monoamines or those, such as tyramine, that are ingested or originate in the gut and are absorbed into the portal circulation. Of the two major molecular species of MAO, type A is selectively inhibited by clorgyline and prefers serotonin as a substrate; type B is inhibited by selegiline (-deprenyl) and prefers phenethylamine as a substrate. Both types are found in liver and brain of most species. Serotonin and norepinephrine nerve terminals contain mainly MAO-A; human gut, MAO-A; and blood platelets, MAO-B. Except for selegiline (in low doses), clinically employed MAO inhibitors (phenelzine and tranylcypromine) inhibit both MAO-A and -B. Selective inhibitors of MAO-A usually are more effective in treating major depression than are type B inhibitors (Murphy et al., 1987, 1995; Krishnan, 1998). The MAO-B inhibitor selegiline is approved for treatment of early Parkinson's disease and acts by potentiating remaining dopamine in degenerating nigrostriatal neurons and possibly reducing neuronal damage due to reactive products of the oxidative metabolism of dopamine or other potential neurotoxins (seeChapter 22: Treatment of Central Nervous System Degenerative Disorders). Selegiline also may have antidepressant effects, particularly at higher doses that also may inhibit MAO-A or yield amphetaminelike metabolites (Murphy et al., 1987). Several short-acting selective inhibitors of MAO-A [e.g., brofaromine, moclobemide (MANERIX, in Canada)] have at least moderate antidepressant effects and are much less likely to potentiate the pressor actions of tyramine and other indirectly acting sympathomimetic amines than do the nonselective, irreversible MAO inhibitors (Delini-Stula et al., 1988; Lotufo-Neto et al., 1999). MAO inhibitors in clinical use are site-directed and irreversible, as reactive hydrazines (phenelzine, isocarboxazide) or acetylenic agents (pargyline, clorgyline, selegiline) that attack and inactivate the flavin prosthetic group following their oxidation to reactive intermediates by MAO (Krishnan, 1998). Inhibition by the cyclopropylamine tranylcypromine may involve the reaction of a sulfhydryl group in the active center of MAO following formation of a reactive imine intermediate by the action of MAO. In the clinical setting, maximal inhibition usually is achieved within a few days, although the antidepressant effect of these drugs may be delayed for several weeks, as with most antidepressants. Evaluation of MAO activity in human subjects taking these drugs has led to the impression that favorable clinical responses are likely to occur when human platelet MAO-B is inhibited by at least 85% (Robinson et al., 1978). This relationship is best established for phenelzine, but it suggests the need to use aggressive dosages of MAO inhibitors to achieve their maximal therapeutic potential. Due to the irreversible actions of clinically used MAO inhibitors
(other than moclobemide), up to 2 weeks may be required to regenerate
fresh MAO enzyme and restore amine metabolism to normal after discontinuation
of the drugs (Singer et al., 1979). Nevertheless, optimal therapeutic
benefit appears to require daily dosing. The capacity of MAO inhibitors to
act as antidepressants usually is assumed to reflect increased availability
of monoamine neurotransmitters in the CNS or sympathetic nervous system, but
this assumption is difficult to prove. MAO inhibition occurs rapidly, but
clinical benefits are usually delayed for several weeks. This delay of
therapeutic effects remains unexplained. The delay may reflect secondary
adaptations already described for tricyclic and serotonin-reuptakeinhibitor
antidepressants, including downregulation of Pharmacological Screening for Novel Antidepressants Despite their clinical mood-elevating effects, most antidepressants lack the behavioral-arousal inducing actions of stimulant drugs (seeChapter 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists; Fawcett and Busch, 1998). Nevertheless, several behavioral models have been widely employed in laboratory screening for potential antidepressants. Most are based on the ability of antidepressants to support animal behavior in stressful situations that ordinarily lead to diminished behavioral responsiveness ('learned helplessness'), such as repeated noxious shocks, forced swimming, or separation from other animals; other models involve increasing aggression toward an intruder or shifting dominance hierarchies in animal social settings (seeHenn and McKinney, 1987; Weiss and Kilts, 1998). Such testing can detect both norepinephrine- and serotonin-reuptake inhibitors (Page et al., 1999). Behavioral models sometimes are used following initial biochemical screening of novel agents with potential antidepressant activity. Such initial screening has relied increasingly on molecular techniques that include assessing potency for cellular transport of radiolabeled monoamines or the binding of selective radioligands to specific monoamine transporter proteins in animal brain tissue or to human transporters encoded by cDNAs expressed in transfected cell lines. Absorption, Distribution, Fate, and Excretion Most antidepressants are fairly well absorbed after oral administration. Although they usually are used initially in divided doses, their relatively long half-lives and rather wide range of tolerated concentrations permit a gradual transition toward a single daily dose given at bedtime. With the tricyclic antidepressants, dosing is most safely done with single doses up to the equivalent of 150 mg of imipramine. High doses of the strongly anticholinergic tricyclic agents can slow gastrointestinal activity and gastric emptying time, resulting in slower or erratic drug absorption and complicating management of acute overdosages. Serum concentrations of most tricyclic antidepressants peak within several hours. Intramuscular administration of some tricyclic antidepressants (notably amitriptyline and clomipramine) can be performed under special circumstances, particularly with severely depressed, anorexic patients who may refuse oral medication or ECT, but most antidepressants are available only in oral form (seeTable 191; DeBattista and Schatzberg, 1999). Once absorbed, tricyclic antidepressants, relatively lipophilic drugs,
are widely distributed. They are strongly bound to plasma protein and to
constituents of tissues, leading to large apparent volumes of distribution,
which can be as high as 10 to 50 liters per kilogram with some
antidepressants. The tendency of tricyclic antidepressants
and their relatively cardiotoxic, ring-hydroxy metabolites to accumulate in
cardiac tissue add to their cardiotoxic risks (Pollock and Perel, 1989;
Prouty and Anderson, 1990; Wilens et al., 1992). Serum concentrations
of antidepressants that correlate meaningfully with clinical effects are not
securely established except for a few tricyclic antidepressants (particularly
amitriptyline, desipramine, imipramine, and nortriptyline), typically at
concentrations of approximately 100 to 250 ng/ml (Perry et al., 1994; seeTable
194). Toxic effects of tricyclic antidepressants can be expected at serum
concentrations above 500 ng/ml, and levels above 1 The utility of therapeutic drug monitoring in the routine clinical use of antidepressants is limited, and the relative safety of modern antidepressants has led to a diminished interest in this approach to guiding clinical dosing. Individual variance in tricyclic antidepressant levels in response to a given dose is as high as 10- to 30-fold and is due largely to genetic control of hepatic microsomal oxidative enzymes (DeVane and Nemeroff, 2000). Predictable relationships between initial disposition of a relatively small test dose of nortriptyline or desipramine and doses required to achieve theoretically optimal serum concentrations have been proposed as a guide to clinical dosing of individual patients (Nelson et al., 1987). Serum concentrations of antidepressants, by themselves, are not reliable predictors of the course and outcome of toxic overdoses, and they can be misleading when obtained postmortem for forensic purposes (Prouty and Anderson, 1990). Tricyclic antidepressants are oxidized by hepatic microsomal enzymes, followed by conjugation with glucuronic acid. The major route of metabolism of imipramine is to the active product desipramine; biotransformation of either compound occurs largely by oxidation to 2-hydroxy metabolites, which retain some ability to block the uptake of amines and may have particularly prominent cardiac depressant actions. In contrast, amitriptyline and its major demethylated by-product, nortriptyline, undergo preferential oxidation at the 10 position; the 10-hydroxy metabolites may have some biological activity, but they may be less cardiotoxic than the 2-hydroxy metabolites of imipramine or desipramine (Pollock and Perel, 1989). The conjugation of ring-hydroxylated metabolites with glucuronic acid extinguishes any remaining biological activity. Although the demethylated metabolites of several tricyclic antidepressants are pharmacologically active and may accumulate in concentrations approaching or exceeding those of the parent drug, it is not known to what extent they account for the activity of the parent drugs. Amoxapine is oxidized predominantly to the 8-hydroxy metabolite, with some production of the 7-hydroxy metabolite; the former is pharmacologically active, probably including antagonistic interactions with D2dopamine receptors. There is some risk of extrapyramidal side effects, including tardive dyskinesia, reminiscent of those of the N-methylated congener loxapine, a typical neuroleptic (seeChapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania). Mirtazapine is also N-demethylated and undergoes aromatic hydroxylation. Trazodone and nefazodone are both N-dealkylated and yield meta-chlorophenylpiperazine (mCPP), an active metabolite with serotonergic activity. Bupropion yields active metabolites that include amphetamine-like compounds. The serotonin-reuptake inhibitors clomipramine, fluoxetine, sertraline, and venlafaxine all are N-demethylated to norclomipramine, norfluoxetine, norsertraline, and desmethylvenlafaxine, respectively (DeVane and Nemeroff, 2000; van Harten, 1993). As occurs with the tertiary-amine tricyclic antidepressants, the N-demethylated serotonin-reuptake inhibitor metabolites also are eliminated more slowly, and some are pharmacologically active. Norclomipramine contributes noradrenergic activity. Norfluoxetine is a very long-acting (elimination half-life approximately 10 days; seeTable 194) inhibitor of serotonin transport, particularly the (S)-enantiomer (Wong et al., 1993). Norfluoxetine also competes with other agents for hepatic oxidases to elevate circulating concentrations of other agents, including tricyclic antidepressants, days after administration of the parent drug has been stopped. Norsertraline, though also eliminated relatively slowly (half-life of 60 to 70 hours), appears to contribute limited pharmacological activity or risk of drug interactions. Nornefazodone contributes little to the biological activity or duration of action of nefazodone. Inactivation and elimination of most antidepressants occurs over a period of several days, but there are some notable exceptions. Generally, secondary-amine tricyclic antidepressants and the N-demethylated derivatives of serotonin-reuptake inhibitors have elimination half-lives about twice those of the parent drugs (van Harten, 1993). Nevertheless, most tricyclics are almost completely eliminated within 7 to 10 days. An exceptionally long-acting tricyclic antidepressant is protriptyline (half-life of about 80 hours). Whereas the half-life of fluoxetine is about 50 hours, its N-demethylated by-product may require several weeks for elimination. Also, most MAO inhibitors are long acting, and recovery from their effects requires the synthesis of new enzyme over a period of 1 to 2 weeks; several experimental inhibitors of MAO-A (e.g., brofaromine, moclobemide) are reversible and short acting (Danish University Antidepressant Group, 1993; Delini-Stula et al., 1988; Murphy et al., 1987). At the other extreme, trazodone, nefazodone, and venlafaxine have short half-lives (about 3 to 6 hours), as does the active 4-hydroxy metabolite of venlafaxine (half-life of about 11 hours). The half-life of bupropion is about 14 hours. The bioavailability of nefazodone is only about 20%, and its half-life is very short (about 3 hours), owing to rapid aromatic hydroxylation. The shorter duration of action of these agents usually implies the need for multiple daily doses. Some short-acting antidepressants have been prepared in slow-release preparations (notably bupropion and venlafaxine), to extend absorption time, but without an effect on elimination half-life. As with many other drugs, antidepressants are metabolized more rapidly by children and more slowly by patients over 60 years of age as compared with young adults (seeBaldessarini 1985; Wilens et al., 1992), and dosages are adjusted accordingly, sometimes to mg/kg daily doses that far exceed those typically given to adults (seeWilens et al., 1992). The MAO inhibitors are absorbed readily when given by mouth and produce maximal inhibition of MAO within 5 to 10 days (Murphy et al., 1987). Little information is available on their pharmacokinetics. Although their biological activity is prolonged because of the characteristics of their interaction with the enzyme, their clinical efficacy appears to be reduced when the drug is given less frequently than once daily. The hydrazide MAO inhibitors are thought to be cleaved, with resultant liberation of active products (e.g., hydrazines). They are inactivated primarily by acetylation. About one-half the population of the United States and Europe (and more in certain Asian countries) are 'slow acetylators' of hydrazine-type drugs, including phenelzine, and this may contribute to the exaggerated effects observed in some patients given standard doses of phenelzine (seeChapters 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination and 4: Principles of Toxicology and Treatment of Poisoning). The metabolism of most antidepressants is greatly dependent on the activity of isozymes of the hepatic microsomal cytochrome P450 (CYP) system (seeChapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination). Most tricyclic antidepressants are extensively oxidized by the CYP1A2 isozyme; citalopram, imipramine, the m-chlorophenylpiperidine metabolite of trazodone and nefazodone are substrates for CYP2C19; mirtazapine, paroxetine, trazodone, and some tricyclics are substrates for CYP2D6; and nefazodone as well as some tricyclic and serotonin-reuptakeinhibitor antidepressants are oxidized by CYP3A3/4 (DeVane and Nemeroff, 2000; van Harten, 1993). In general, CYP enzymes 1A2 and 2D6 mediate aromatic hydroxylation, and 3A3/4 mediate N-dealkylation and N-oxidation reactions in the metabolism of antidepressants. Glucuronidation is effected by a non-CYP system. Some antidepressants not only are substrates for metabolism by the CYP system but also can inhibit the metabolic clearance of other drugs, sometimes producing clinically significant drug-drug interactions (see below, 'Interactions with Other Drugs'). Notable inhibitory interactions include fluvoxamine with CYP1A2; fluoxetine and fluvoxamine with CYP2C9, and fluvoxamine with CYP2C19; paroxetine, fluoxetine and, less actively, sertraline with CYP2D6; and fluvoxamine and nefazodone with CYP3A3/4 (seeDeVane and Nemeroff, 2000; Hansten and Horn, 2000; Preskorn, 1997; Weber, 1999). Potentially clinically significant interactions include the tendency for fluvoxamine to increase circulating concentrations of oxidatively metabolized benzodiazepines, clozapine, theophylline, and warfarin. Fluoxetine and nefazodone also can increase levels of terfenadine and astemizole, and sertraline and fluoxetine can increase levels of warfarin, benzodiazepines, and clozapine. Paroxetine increases levels of theophylline and warfarin. Fluoxetine also potentiates tricyclic antidepressants and some class IC antiarrhythmics with a narrow therapeutic index (including flecainide, encainide, and propafenone; seeChapter 35: Antiarrhythmic Drugs). Nefazodone potentiates benzodiazepines other than lorazepam and oxazepam (glucuronidated). Tolerance and Physical Dependence Some tolerance to sedative and autonomic effects tends to develop with continued use of tricyclic antidepressants and to the initial nausea commonly associated with serotonin-reuptake inhibitors. However, it is important to emphasize that various types of antidepressants have been used for months or years by patients with severe recurring depression with limited risk of loss of their desirable effects, though perhaps more often with serotonin-reuptake inhibitors than with older agents (seeCohen and Baldessarini, 1985; Frank et al., 1990; Viguera et al., 1998). Occasionally, patients show physical dependence on the tricyclic antidepressants, with malaise, chills, coryza, muscle aches, and sleep disturbance following abrupt discontinuation, particularly of high doses (Shatan, 1966). Similar reactions, along with gastrointestinal and sensory symptoms (paresthesias) and irritability, also occur with abrupt discontinuation of serotonin-reuptake inhibitors, particularly short-acting agents including paroxetine and venlafaxine (Schatzberg et al., 1997; Tollefson and Rosenbaum, 1998). Some of these effects may reflect increased cholinergic activity following its inhibition by such agents as amitriptyline, imipramine, and paroxetine, but serotonergic mechanisms may contribute to the effects of discontinuing serotonin-reuptake inhibitors. Some of these reactions can be confused with clinical worsening of depressive symptoms. Emergence of agitated or manic reactions also has been observed after abrupt discontinuation of tricyclics (Mirin et al., 1981). Such physiological reactions to antidepressant discontinuation indicate that it is wise to discontinue antidepressants gradually over at least a week, or longer when feasible. Another type of reaction to treatment discontinuation is suspected with several psychotropic agents, involving a period of risk of recurrence of morbidity that is greater than would be predicted by the natural history of untreated illness, particularly if long-term maintenance medication is withdrawn rapidly (Baldessarini et al., 1999; Viguera et al., 1998). This risk probably extends over several months. Evidence for the occurrence of this phenomenon is particularly strong for lithium in bipolar disorder, but it also may occur with antidepressants (Viguera et al., 1998). Such risk may be reduced by gradual discontinuation of long-term medication over at least several weeks (seeChapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania). Toxic Reactions and Side Effects Significant side effects of antidepressants are common. Tricyclic antidepressants routinely produce adverse autonomic effects, in part related to their relatively potent antimuscarinic effects. These include dry mouth and a sour or metallic taste, epigastric distress, constipation, dizziness, tachycardia, palpitations, blurred vision (poor accommodation, with increased risk of glaucoma), and urinary retention. In addition, cardiovascular effects include orthostatic hypotension, sinus tachycardia, and variable prolongation of cardiac conduction times, with the potential of arrhythmias, particularly with overdoses. In the absence of cardiac disease, the principal problem associated
with imipramine-like agents is postural hypotension, probably related to
anti Weakness and fatigue are attributable to central effects of tricyclic antidepressants, particularly tertiary amines, and mirtazapine, which have potent central antihistaminic effects. Trazodone and nefazodone also are relatively sedating. Other CNS effects include variable risk of confusion or delirium, in large part owing to atropine-like effects of tricyclic antidepressants. Epileptic seizures also occur; this is especially likely with doses of bupropion above 500 mg, maprotiline above 250 mg per day, or acute overdoses of amoxapine or tricyclics (Johnston et al., 1991). Risk of cerebral or cardiac intoxication can increase if such agents are given in relatively high doses, with some serotonin-reuptake inhibitors capable of inhibiting their metabolism (seeTable 194). MAO inhibitors can induce sedation or behavioral excitation and have a high risk of inducing postural hypotension, sometimes with sustained, mild elevations of diastolic blood pressure. Miscellaneous toxic effects of tricyclic antidepressants include jaundice, leukopenia, and rashes, but these are very infrequent. Weight gain is a common side effect of most antidepressants, less likely with the serotonin-reuptake inhibitors, and rare with bupropion (seeTable 191). Excessive sweating also is common, but its pathophysiology is not known. Newer antidepressants generally present fewer or different side effects and toxic risks than older tricyclics and MAO inhibitors. The selective serotonin-reuptake inhibitors, as a group, have a high risk of nausea and vomiting, headache, and sexual dysfunction, including inhibited ejaculation in men and impaired orgasm in women. Adverse sexual effects also occur with tricyclic antidepressants but are much less common with bupropion, nefazodone, and mirtazapine. Trazodone can produce priapism in men, presumably due to antiadrenergic actions. Some serotonin-reuptake inhibitors, and perhaps fluoxetine in particular, have been associated with agitation and restlessness that resembles akathisia (seeChapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania; Hamilton and Opler, 1992). Bupropion can act as a stimulant, with agitation, anorexia, and insomnia. Serotonin-reuptake inhibitors, while generally less likely to produce adverse cardiovascular effects than older antidepressants, can elicit electrophysiological changes in cardiac tissue, including interference with Na+ and Ca2+ channels (Pacher et al., 1999). Another risk of antidepressants in vulnerable patients (particularly those with unrecognized bipolar depression) is switching, sometimes suddenly, from depression to hypomanic or manic excitement or mixed, dysphoric-agitated, manic-depressive states. To some extent, this effect is dose-related, and it seems to be somewhat more likely with tricyclic antidepressants than with serotonin-reuptake inhibitors or bupropion and perhaps MAO inhibitors. Risk of mania with newer sedating antidepressants, including nefazodone and mirtazapine, also may be relatively low, but some risk of inducing mania can be expected with any treatment that elevates mood (Sachs et al., 1994). Safety Through the Life Cycle Most antidepressants appear to be generally safe during pregnancy, in that proposed teratogenic associations in newborns exposed to several tricyclic antidepressants and some newer antidepressants (particularly fluoxetine) are not convincing (McGrath et al., 1999; Wisner et al., 1999). Most antidepressants and lithium are secreted in breast milk, at least in small quantities, and their safety in nursing infants is neither established nor safely assumed (Birnbaum et al., 1999). For severe depression during pregnancy and lactation, ECT may be a relatively safe and effective alternative. Children are vulnerable to cardiotoxic and seizure-inducing effects of high doses of tricyclic compounds (Kutcher, 1997). Deaths have occurred in children after accidental or deliberate overdosage with only a few hundred milligrams of drug, and several cases of unexplained sudden death have been reported in children treated with desipramine (Biederman et al., 1995). Children are relatively protected by vigorous hepatic metabolic clearing mechanisms that eliminate many drugs rapidly. Indeed, attaining serum concentrations of desipramine in children like those encountered in adults (seeTable 194) may require doses of 5 mg/kg of body weight or more in some school-age children compared to only 2 to 3 mg/kg in adults (Wilens et al., 1992). Risk/benefit considerations of antidepressants in pediatric populations remain uncertain, particularly since many trials of antidepressants in children have failed to show substantial superiority to a placebo (Kutcher, 1997). Among geriatric patients, dizziness, postural hypotension, constipation, delayed micturition, edema, and tremor are found commonly with tricyclic antidepressants; these patients are much more likely to tolerate serotonin-reuptake inhibitors and other modern antidepressants (Catterson et al., 1997; Flint, 1998; Newman and Hassan, 1999; Oshima and Higuchi, 1999; Small, 1998). Their risks are increased due to less-efficient metabolic clearance of antidepressants and less ability to tolerate them. Acute Overdoses Acute poisoning with tricyclic antidepressants or MAO inhibitors is potentially life threatening. Such fatalities are much less common since modern antidepressants have widely replaced these drugs, but suicide rates probably have not declined (Baldessarini and Jamison, 1999). Deaths have been reported with doses of approximately 2 g of imipramine, and severe intoxication can be expected at doses above 1 g, or about a week's supply. If a patient is severely depressed, potentially suicidal, impulsive, or has a history of substance abuse, prescribing a relatively safe antidepressant agent with close clinical follow-up is an appropriate step. If a potentially lethal agent is prescribed, it is best dispensed in small, sublethal quantities, with a risk that sustained adherence to recommended treatment may be compromised. Acute poisoning with a tricyclic antidepressant often is clinically complex (Nicotra et al., 1981). A typical pattern is brief excitement and restlessness, sometimes with myoclonus, tonic-clonic seizures, or dystonia, followed by rapid development of coma, often with depressed respiration, hypoxia, depressed reflexes, hypothermia, and hypotension. Antidepressants that have relatively strong antimuscarinic potency commonly induce mydriasis, flushed dry skin and dry mucosae, absent bowel sounds, urinary retention, and tachycardia or other cardiac arrhythmias. A tricyclic antidepressantintoxicated patient must be treated early in an intensive care unit. Gastric lavage with activated charcoal sometimes is useful, but dialysis and diuresis are ineffective. Coma abates gradually over 1 to 3 days. Excitement and delirium are then typical. Risk of life-threatening cardiac arrhythmias continues for at least several days, requiring close medical supervision (Boehnert and Lovejoy, 1985). Cardiac toxicity and hypotension in such poisonings can be especially
difficult to manage. The heart is usually hyperactive, with supraventricular
tachycardia and a high output, and with electrocardiographic conduction times
reduced (prolonged QT interval). Cardiac glycosides and antiarrhythmic drugs such
as quinidine or procainamide are contraindicated, but phenytoin has been
given safely and also can suppress seizure risk, as can diazepam. In
addition, Toxic reactions from overdosage of an MAO inhibitor may occur in a matter of hours despite the long delay in onset of a therapeutic response. Effects of overdosage include agitation, hallucinations, hyperreflexia, hyperpyrexia, and convulsions. Both hypotension and hypertension also occur. Treatment of such intoxication is problematic, but conservative treatment is often successful. Interactions with Other Drugs Antidepressants are involved in several clinically important drug
interactions (seeHansten and Horn, 2000; Conversely, the tendency for several serotoninreuptake inhibitors to compete for the metabolism of other drugs can lead to significant and potentially dangerous drug-drug interactions. For example, when using combinations of such agents with tricyclic antidepressants, as is sometimes done to attempt to achieve more rapid therapeutic effect or to manage otherwise treatment-resistant depressed patients, serum concentrations of the tricyclic drug may rise to toxic levels, and these may persist for days after discontinuing fluoxetine, due to the prolonged elimination of norfluoxetine (Nelson et al., 1991). Several serotonin-reuptake inhibitors are potent inhibitors of human hepatic CYP microsomal oxidases in vitro (Crewe et al., 1992), as was discussed above regarding antidepressant drug metabolism. Venlafaxine, citalopram, and sertraline appear to have relatively low risk of such interactions (Caccia, 1998; Ereshevsky et al., 1996; Preskorn, 1997, 1998). Significant interactions may be most likely in persons who are relatively rapid metabolizers through the microsomal oxidase system, perhaps including children (DeVane and Nemeroff, 2000; Preskorn, 1997, 1998). Examples of drug interactions with serotonin-reuptake inhibitors
include potentiation of agents metabolized prominently by CYP1A2 (e.g.,
Antidepressants potentiate the effects of alcohol and probably other sedatives. The anticholinergic activity of tricyclic antidepressants can add to that of antiparkinsonism agents, antipsychotic drugs of low potency (especially clozapine and thioridazine), or other compounds with antimuscarinic activity to produce toxic effects. Tricyclic antidepressants have prominent and potentially dangerous interactions with biogenic amines, such as norepinephrine, which normally are removed from their site of action by neuronal uptake. However, drugs that inhibit norepinephrine transport also block the effects of indirectly acting amines, such as tyramine, which must be taken up by sympathetic neurons to release norepinephrine. Presumably by a similar mechanism, tricyclic antidepressants prevent the antihypertensive action of adrenergic neuron blocking agents such as guanadrel. Tricyclic agents and trazodone also can block the centrally mediated antihypertensive action of clonidine. Serotonin-reuptake inhibitors and virtually any agent with
serotonin-potentiating activity can interact dangerously or even fatally with
MAO inhibitors (particularly long-acting MAO inhibitors). Other agents also
have been implicated in dangerous interactions with MAO inhibitors (notably meperidine
and perhaps other phenylpiperidine analgesics, as well as pentazocine, dextromethorphan,
fenfluramine, and infrequently, tricyclic antidepressants) (seeWhite
and Simpson, 1981). The resulting reactions have been referred to as a
'serotonin syndrome.' This syndrome typically includes
akathisia-like restlessness, muscle twitches and myoclonus, hyperreflexia,
sweating, penile erection, shivering, and tremor as a prelude to more severe
intoxication, with seizures and coma (Sternbach, 1991). The reaction often is
self-limiting if the diagnosis is made quickly and the offending agents are
discontinued. The precise pathophysiological mechanisms underlying these
toxic syndromes remain ill-defined. Newer MAO inhibitors (e.g., selegiline,
moclobemide) also should be considered to have some risk of such interactions
(Sternbach, 1991). MAO inhibitors also can potentiate effects of bupropion (seeWeber,
1999; Hansten and Horn, 2000). These reactions are distinguished from the
hypertensive interaction of MAO inhibitors with indirectly acting pressor
phenethylamines such as tyramine. This reaction requires scrupulous avoidance
of many potentially interacting agents, including over-the-counter cold
remedies containing indirectly acting sympathomimetic agents (seeAyd
and Blackwell, 1970; Gardner et al., 1996; Healy, 1997; Therapeutic Uses The clinical use of antidepressants in depressed patients is discussed below. In addition to their use in adult major depression syndrome, the various antidepressant agents have found broad utility in other psychiatric disorders that may or may not be related psychobiologically to the mood disorders. Encouragement to find new indications has increased with the advent of newer agents that are less toxic, simpler to use, and often better accepted by both physicians and patients (Edwards, 1995; Edwards et al., 1997; Tollefson and Rosenbaum, 1998). Current applications include rapid but temporary suppression of enuresis in children and in geriatric patients by uncertain mechanisms; prebedtime doses as low as 25 mg of imipramine or nortriptyline have been found to be safe and effective. Major affective disorders are being recognized more often in children, and antidepressants are being used increasingly in that age group, despite an inexplicable lack of demonstrable efficacy of tricyclic antidepressants in pediatric depression per se, even at sufficiently high doses (up to 5 mg/kg) to provide plasma concentrations accepted as therapeutic in adults (Hazel, 1996). Serotonin-reuptake inhibitors also have limited evidence of efficacy in depressed children, and other antidepressants have received little assessment in juveniles with various disorders (Emslie et al., 1999; Kutcher, 1997; Steingard et al., 1995). Antidepressants have a growing role in other disorders, including attention deficithyperactivity disorder in children and adults, for which imipramine, desipramine, and nortriptyline appear to be effective, even in patients responding poorly to or intolerant of the stimulants (e.g., methylphenidate) that have been the standard agents for this disorder. Newer norepinephrine-selective-uptake inhibitors also may be useful in this disorder. Utility of serotonin-reuptake inhibitors in this syndrome is not established, and bupropion, despite its similarity to stimulants, appears to have limited efficacy (Kutcher, 1997; Spencer et al., 1993; Wilens et al., 1992). Antidepressants tend to provide a more sustained and continuous improvement of the symptoms of attention deficithyperactivity disorder than do the stimulants, and they do not induce tics or other abnormal movements sometimes associated with the use of stimulants. Indeed, desipramine and nortriptyline even may effectively treat tic disorder, either in association with the use of stimulants, or arising in patients with both attention disorder and Tourette's syndrome (Spencer et al., 1993). The future of tricyclic antidepressant use in children is uncertain due to the difficulty of demonstrating the efficacy of these agents in pediatric major depression (Hazell, 1996) and because of reports of several cases of unexplained sudden death during use of desipramine in preadolescent children (Biederman et al., 1995). Antidepressants also are leading choices in the treatment of severe anxiety disorders, including panic-agoraphobia syndrome, generalized anxiety disorder, social phobia, and obsessive-compulsive disorder (Bennett et al., 1998; Feighner, 1999; Masand and Gupta, 1999; Pigott and Seay, 1999; Roerig, 1999; Uhlenhuth et al., 1998), including the common comorbidity of anxiety in depressive illness (Boerner and Moller, 1999; Hoehn-Saric et al., 2000). Antidepressants, especially serotonin-reuptake inhibitors, also are employed in the management of posttraumatic stress disorder, marked by anxiety, startle, painful recollection of the traumatic events, and disturbed sleep (see American Psychiatric Association, 1994; Roerig, 1999). Nonsedating antidepressants often are poorly tolerated initially by anxious patients, requiring slowly increased doses. Their beneficial actions typically are delayed for several weeks in anxiety disorders as in major depression. For panic disorder, tricyclic antidepressants and MAO inhibitors, as
well as high-potency benzodiazepines (notably alprazolam, clonazepam, and lorazepam;
seeChapter 17: Hypnotics and Sedatives) are effective in blocking the
autonomic expression of panic itself, thus facilitating a comprehensive
rehabilitation program (Argyropoulos and Nutt, 1999; Bennett et al.,
1998; Nagy et al., 1993; Uhlenhuth et al., 1998). Imipramine
and phenelzine are well-studied antidepressants for panic disorder. The
serotonin-reuptake inhibitors also may be effective, but The serotonin-reuptake inhibitors are agents of choice in obsessive-compulsive disorder, as well as in possibly related syndromes of impulse dyscontrol or obsessive preoccupations, including compulsive habits, bulimia (but usually not anorexia) nervosa, and body dysmorphic disorder (Agras, 1998; Geller et al., 1998; Hoehn-Saric et al., 2000; Pigott and Seay, 1999; seeSadock and Sadock, 2000). While their benefits may be limited, serotonin-reuptake inhibitors offer an important advance in the medical treatment of these often chronic and sometimes incapacitating disorders for which no other medical treatment, by itself, has been consistently effective. The effectiveness of pharmacological treatment for these commonly treatment-resistant disorders is greatly enhanced by use of behavioral treatments (Miguel et al., 1997). In addition to the wide use of modern antidepressants to treat depression commonly associated with general medical illnesses (Schwartz et al., 1989), several psychosomatic disorders may respond at least partly to treatment with antidepressants of the tricyclic, MAO inhibitor, or serotoninreuptake inhibitor types. These include chronic pain disorders, including diabetic and other peripheral neuropathic syndromes (for which tertiaryamine tricyclics are probably superior to fluoxetine); fibromyalgia; peptic ulcer and irritable bowel syndrome; chronic fatigue; cataplexy; tics; migraine; and sleep apnea (Baldessarini, 1989; Gruber et al., 1996; Masand and Gupta, 1999; Max et al., 1992; Spencer et al., 1993). These disorders may have some psychobiological relationship to mood or anxiety disorders (Hudson and Pope, 1990). Drug Treatment of Mood Disorders Disorders of mood (affective disorders) are extremely common in
general medical practice as well as in psychiatry. The severity of these
conditions covers an extraordinarily broad range, from normal grief reactions
and dysthymia to severe, incapacitating illnesses that may result in death.
The lifetime risk of suicide in severe forms of major affective disorders is
10% to 15%, but this statistic does not begin to represent the morbidity and
cost of this group of notoriously underdiagnosed and undertreated illnesses.
Perhaps one-fourth to one-third of these cases are
diagnosed, and a similar proportion of these are adequately treated (Isaacson
et al., 1992; Katon et al., 1992; Kind and Sorensen, 1993; McCombs
et al., 1990; Suominen et al., 1998). Clearly, not all types of
human grief, misery, and disappointment are indications for medical
treatment, and even severe affective disorders have a high rate of
spontaneous remission provided that sufficient time (often a matter of
months) passes. The antidepressant agents thus generally are reserved for the
more severe and otherwise incapacitating depressive disorders, and the most
satisfactory results tend to occur in patients who have moderately severe
illnesses with 'endogenous' or 'melancholic'
characteristics without psychotic features (see American Psychiatric
Association, 1994; Baldessarini, 1989; A somewhat surprising fact is that clinically employed antidepressants, as a group, have outperformed inactive placebos in only about two-thirds to three-fourths of controlled comparisons (seeBaldessarini, 1989; Healy, 1997), with a similar proportion of depressed adult subjects rated as showing clinically significant responses. Moreover, assessment-based changes in clinical ratings of depressive symptoms, rather than categorization as 'treatment-responsive,' often yield surprisingly small average differences between active antidepressants and placebo in contemporary outpatient trials involving patients with depressive illness of only moderate severity (Healy, 1997; Kahn et al., 2000). With pediatric and geriatric depression, results are typically even less clear. Pediatric studies often have failed to show superiority of drug over a placebo (Hazell, 1996). Geriatric depression includes an excess of chronic and psychotic illnesses, which tend to respond less well to antidepressant treatment alone but may do better with ECT or when an antipsychotic agent is added, or with amoxapine, a mixed antidepressant-neuroleptic (Schatzberg and Rothschild, 1992). Despite their potential for less favorable responses to simple antidepressant therapy, patients with severe, prolonged, disabling, psychotic, suicidal, or bipolar depression require vigorous and prompt medical intervention. Underdiagnosis arises, in part, from the sometimes misleading presentation to physicians of many depressed patients with nonspecific somatic complaints, anxiety, or insomnia. Undertreatment, in part, has arisen in the past from the reluctance of many physicians to prescribe potentially toxic or pharmacologically complicated tricyclic or MAO inhibitor antidepressants, especially to medically ill patients. This pattern is changing with the availability of less-toxic and better-accepted antidepressants among the serotonin-reuptake inhibitors and atypical agents (Olfson and Klerman, 1993). Another major problem with antidepressant agents is that, because placebo response rates tend to be as high as 30% to 40% in research subjects diagnosed with major depression and possibly even higher in some anxiety disorders, statistical and clinical distinctions between active drug and placebo are difficult to prove (Fairchild et al., 1986; Kahn et al., 2000). Separation of response rates to active antidepressants from placebo improves when patients are selected for moderate severity, presence and persistence of classic melancholic or endogenous symptoms, and absence of psychotic features or of mixed bipolar states. The incorporation of various metabolic, endocrinological, or other physiological testing procedures to predict antidepressant treatment responses has been found to have only marginal predictive power and clinical utility (Arana et al., 1985; Baldessarini, 2000). This situation stresses the importance of continued reliance on placebo-controlled studies in the development of new agents, since comparisons of a new versus a standard agent can risk an erroneous inference of equal efficacy. In addition, information on special depressed populations (particularly, pediatric, geriatric, medically ill, hospitalized, and recurrently or chronically ill patients, as well as bipolar depressed patients) continues to be limited, despite the medical need for such information. Moreover, evidence concerning clinical dose-response and dose-risk relationships is especially limited with this class of drugs. Based on the limited information available, it is evident that increasing doses of a standard agent, such as imipramine, to 200 mg daily or more, with plasma concentrations above 200 ng/ml, yields antidepressant benefit superior to that of lower doses and levels in both short- and long-term treatment; however, tolerance may be limited, and rates of treatment refusal are high (seeDeBattista and Schatzberg, 1999; Mavissakalian and Perel, 1989). Accordingly, the selection of a dose is based on the attempt to exceed a lower limit of perhaps 150 mg of imipramine or its daily equivalent in an otherwise healthy adult depressed patient. Typically this is done by gradually increasing the dose over several days, with an attempt to attain higher doses, as tolerated, if little progress has been made within several weeks of treatment. Although 4 to 8 weeks are required to determine whether an antidepressant trial is successful or not, some indications of improvement should be evident within the first 2 weeks. From 1960 to 1990, the imipramine-like tricyclics were the standard antidepressants on which most of the research and clinical practice in the field have been based. However, the newer, less toxic serotonin-reuptake inhibitors and other atypical agents now are accepted broadly as agents of first choice, particularly for medically ill or potentially suicidal patients and in the elderly (Brown and Khan, 1994; Flint, 1998; Oshima and Higuchi, 1999; Small, 1998). MAO inhibitors commonly are reserved for patients who fail to respond to vigorous trials of at least one of the newer agents and a standard tricyclic antidepressant, administered alone or with lithium (lithium is covered in Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania), in an attempt to potentiate the antidepressant response, or with a low dose of triiodothyronine, also in an effort to enhance overall therapeutic effectiveness (seeAustin et al., 1991; Bauer and Dpfmer, 1999; Lasser and Baldessarini, 1997). Even before the wide acceptance of the newer antidepressants, the somewhat less anticholinergic secondary-amine tricyclics, particularly nortriptyline and desipramine, had come to be preferred, particularly for elderly or medically ill patients; they still can be considered as an alternative or a second choice, particularly if administered in moderate, divided doses (seeTable 191). Despite the general safety of newer agents, they are not without limitations, side effects, and interactions with other agents (see above). They also are relatively expensive, and prices for a day's supply of antidepressants can vary by more than tenfold among agents (seeBaldessarini, 1985, 1989). Moreover, their relative efficacy in the most severely depressed patients, those with psychotic features, and the elderly remains to be further evaluated. The natural history of major depression is that individual episodes tend to remit spontaneously over 6 to 12 months; however, there is a high risk of relapse of depression for at least several months following discontinuation of a successful trial of antidepressant treatment. This risk is estimated at 50% within 6 months and 65% to 70% at one year of follow-up, rising to 85% by 3 years (seeBaldessarini and Tohen, 1988; Viguera et al., 1988). To minimize this risk, it is best to continue antidepressant medication for not less than 6 months following apparent full clinical recovery. Continued use of initially therapeutic doses is recommended, although tolerability and acceptance by patients may require flexibility in this regard. Many depressed patients follow a recurring course of episodic illness, often with lesser levels of symptoms and disability between major episodes, and so require consideration of long-term maintenance medication to reduce the risk of recurrence, particularly in patients with more than three relatively severe episodes or chronic depressive or dysthymic disorders (seeKeller et al., 1998; Viguera et al., 1998). Such treatment has been tested for as long as 5 years, using relatively high doses of imipramine, with evidence that early dose reduction led to a higher risk of relapse (Frank et al., 1990, 1993; Kupfer et al., 1992). Long-term supplementation of an antidepressant with lithium may enhance the result (seeBaldessarini and Tohen, 1988). Prolonged maintenance treatment of patients with recurring major depression for more than a year has not been well evaluated with any antidepressant drug other than imipramine, and dose-response data are very limited (Frank et al., 1993; Keller et al., 1998). The decision to recommend indefinitely prolonged maintenance treatment with an antidepressant is guided by the past history of multiple, and especially severe or life-threatening, recurrences and the impression that recurrence risk is greater in older patients. Due to evidence that rapid discontinuation of antidepressants and lithium may contribute to excess early recurrence of illness, very gradual reduction and close clinical follow-up over many weeks are recommended when maintenance treatment is to be discontinued and, ideally, even when stopping continuation therapy within the months following recovery from an acute episode of depression (seeGreden, 1998; Viguera et al., 1998). The medical literature contains case reports of possible 'tolerance' to the therapeutic effects of antidepressants after prolonged use. Sometimes this loss of benefit may be overcome by increasing the dose of antidepressant, by temporary addition of lithium or perhaps a small dose of an antipsychotic agent, or by changing to an antidepressant in a different class (Cohen and Baldessarini, 1985). Other forms of biological treatment of depression have not been well established or are no longer regularly employed with the important exception of ECT. This remains the most rapid and effective treatment for severe acute depression and is sometimes lifesaving for acutely suicidal patients (seeRudorfer et al., 1997). The MAO inhibitors generally are considered drugs of late choice for the treatment of severe depression, even though the evidence for efficacy of adequate doses of tranylcypromine or phenelzine is convincing. Despite the favorable results obtained with tranylcypromine and with doses of phenelzine above 45 mg per day (Davis et al., 1987; Krishnan, 1998), the possibility of unwanted reactions has limited their acceptance by many clinicians and patients. Nevertheless, MAO inhibitors sometimes are used when a vigorous trial of one or more standard antidepressants has been unsatisfactory and when ECT is refused. In addition, MAO inhibitors may have selective benefits for conditions other than typical major depression, including illnesses marked by phobias and anxiety or panic as well as dysphoria (Liebowitz, 1993). Similar benefits, however, may be found with imipramine-like agents or serotonin-reuptake inhibitors; thus, indications for the MAO inhibitors are limited and must be weighed against their potential toxicity and their complex interactions with many other drugs. Innovative MAO inhibitors selective for types MAO-A and MAO-B enzyme now are available. Selegiline [(R)-()-deprenyl], an MAO-B-selective inhibitor, was introduced for the treatment of Parkinson's disease, but it also may have some antidepressant or other useful psychotropic effects, and its convenience and possibly its safety have been enhanced by recent development of an experimental transdermal preparation (seeTable 191; Mann et al., 1989; Kuhn and Muller, 1996). For consistent beneficial effects, however, daily doses above 10 mg probably are required, which may compromise MAO-B selectivity, especially following repeated use; also, selegiline may be converted in vivo to by-products with amphetamine-like structure and neuropharmacology. The MAO-Aselective inhibitor clorgyline is an effective antidepressant (seeMurphy et al., 1987). Other short-acting, reversible inhibitors of MAO-A (e.g., brofaromine, moclobemide) appear to be moderately effective antidepressants with reduced risk of inducing hypertension when combined with indirectly acting sympathomimetic pressor amines (seeChapter 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists). Stimulants, with or without added sedatives, are an outmoded and ineffective treatment for severe depression. However, some clinicians continue to find utility and safety in the short-term treatment of selected patients with a stimulant such as methylphenidate or amphetamine (Fawcett and Busch, 1998). These include patients with mild dysphoria, temporary demoralization, or lack of energy associated with medical illnesses as well as some geriatric patients; however, none of these possible indications has been investigated systematically (Chiarello and Cole, 1987). A particularly difficult clinical challenge is the safe and effective treatment of bipolar depression (seeChapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania). This condition sometimes is misdiagnosed in states of mixed, dysphoric-agitated moods in patients with bipolar disorder and then inappropriately treated with an antidepressant without a mood-stabilizing agent for protection from worsening agitation or mania (Kukopulos et al., 1983; Wehr and Goodwin, 1987). For this reason, the management of manic, mixed, and depressive mood states in bipolar disorder best relies on lithium or other putative mood-stabilizing agents as the primary treatment (seeChapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania). An antidepressant can be added cautiously and temporarily to treat depression, but the additional benefit and safety of sustained combinations of an antidepressant with a mood stabilizer have not been proven (Prien and Kocsis, 1995). The choice of antidepressant in bipolar depression remains uncertain. Moderate doses of desipramine or nortriptyline have been used in the past; currently, the short-acting serotonin-reuptake inhibitors, bupropion, nefazodone, or mirtazapine often are employed despite a lack of formal research support for a rational choice of agent, dose, or timing (seeZornberg and Pope, 1993). Some of the newer antidepressants like bupropion may have a reduced tendency to induce cycling. Drugs Used in the Treatment of Anxiety Anxiety is a cardinal symptom of many psychiatric disorders and an
almost-inevitable component of many medical and surgical conditions. Indeed,
it is a universal human emotion, closely allied with appropriate fear and
often serving psychobiologically adaptive purposes. A most important clinical
generalization is that anxiety is rather infrequently a 'disease'
in itself. Anxiety that is typically associated with the former
'psychoneurotic' disorders is not readily explained in biological
or psychological terms; contemporary hypotheses implicate overactivity of
adrenergic systems or dysregulation of serotonergic systems in the CNS (Stein
and Uhde, 1998). In addition, symptoms of anxiety commonly are associated
with depression and especially with dysthymic disorder (chronic depression of
moderate severity), panic disorder, agoraphobia and other specific phobias,
obsessive-compulsive disorder, eating disorders, and many personality
disorders (Boerner and Moller, 1999; Liebowitz, 1993). Sometimes, despite a
thoughtful evaluation of a patient, no treatable primary illness is found,
or, if one is found and treated, it may be desirable to deal directly with
the anxiety at the same time. In such situations, antianxiety medications are
frequently and appropriately used ( Currently, the serotonin-reuptake inhibitors discussed above and the benzodiazepines (seeChapter 17: Hypnotics and Sedatives) are the most commonly employed medicinal treatments for the common clinical anxiety disorders. Some high-potency benzodiazepines (alprazolam, clonazepam, and lorazepam) are effective in severe anxiety with strong autonomic overactivity (panic disorder), as are several antidepressant agents, as discussed above. For generalized or nonspecific anxiety, the benzodiazepine selected seems to make little difference (Roerig, 1999). In the elderly or in patients with impaired hepatic function, oxazepam in small, divided doses is sometimes favored due to its brief action and direct conjugation and elimination. The latter property is shared by lorazepam, but not by alprazolam (seeChapter 17: Hypnotics and Sedatives). Benzodiazepines sometimes are given to outpatients presenting with anxiety mixed with symptoms of depression, although the efficacy of these agents in altering the core features of severe major depression has not been demonstrated (Argyropoulos and Nutt, 1999; Boerner and Moller, 1999; Liebowitz, 1993). Potent benzodiazepines also commonly are employed adjunctively in the short-term management of acutely psychotic or manic patients (seeChapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania). The most favorable responses to the benzodiazepines are obtained in situations that involve relatively acute anxiety reactions in medical or psychiatric patients who have either modifiable primary illnesses or primary anxiety disorders. However, this group of anxious patients also has a high response rate to placebo and is likely to undergo spontaneous improvement. Antianxiety drugs also are used in the management of more persistent or recurrent primary anxiety disorders; guidelines for their appropriate use are less clear in these situations (Hollister et al., 1993; Uhlenhuth et al., 1998). Although there has been concern about the potential for habituation and abuse of sedatives, some studies suggest that physicians tend to be conservative and may even undertreat patients with anxiety. They may either withhold drug unless symptoms or dysfunction are severe or cease treatment within a few weeks, with a high proportion of relapses. Patients with personality disorders or a past history of abuse of sedatives or alcohol may be particularly at risk of dose escalation and dependence on benzodiazepines. Benzodiazepines carry some risk of producing impairment of cognition and skilled motor functions, particularly in the elderly, in whom they are a common cause of confusion, delirium (sometimes mistaken for primary dementia), and falls with fractures (Ray et al., 1987). Risk of fatality on acute overdose of benzodiazepines is limited in the absence of other cerebrotoxins or alcohol; risk of suicide with buspirone is very low. A particularly controversial aspect of the use of benzodiazepines, especially those of high potency, is in long-term management of patients with sustained or recurring symptoms of anxiety (Argyropoulos and Nutt, 1999; Hollister et al., 1993; Uhlenhuth et al., 1998). Clinical benefit has been found for at least several months in such cases, but it is unclear to what extent the long-term benefits can be distinguished from nonspecific ('placebo') effects following development of tolerance, on the one hand, or prevention of related withdrawal-emergent anxiety on the other. Many other classes of drugs that act on the CNS have been used for daytime sedation and the treatment of anxiety, but their use for these conditions is now virtually obsolete. Such drugs include the propanediol carbamates (notably, meprobamate and tybamate), the barbiturates (seeChapter 17: Hypnotics and Sedatives), and many other pharmacologically similar nonbarbiturates. The demise of older sedative agents in modern psychiatric practice is due primarily to their tendency to cause unwanted degrees of sedation or frank intoxication at doses required to alleviate anxiety; meprobamate and the barbiturates are likely to produce tolerance, physical dependence, severe withdrawal reactions, and life-threatening toxicity with overdosage. Other drugs that have been used in the treatment of anxiety include
certain anticholinergic agents and antihistamines. Among these is hydroxyzine,
an antihistamine that is not an effective antianxiety agent unless given in
doses (400 mg per day) that produce marked sedation (seeChapter 25:
Histamine, Bradykinin, and Their Antagonists). Propranolol and other Another class of agents with beneficial effects in disorders marked by anxiety or dysphoria of moderate intensity are the azapirones (azaspirodecanediones), currently represented clinically by buspirone (BUSPAR; Ninan et al., 1998). Originally developed as a potential antipsychotic agent with weak antidopaminergic activity, buspirone has pharmacological properties distinct from those of both neuroleptics and sedatives including the benzodiazepines. The antidopaminergic actions of azapirones are limited in vivo, and they do not induce clinical extrapyramidal side effects. Also, they do not interact with binding sites for benzodiazepines or facilitate the action of GABA, are not anticonvulsant (and may even lower seizure threshold weakly), do not appear to cause tolerance or withdrawal reactions, and do not show cross-tolerance with benzodiazepines or other sedatives. Buspirone and several experimental congeners (e.g., gepirone, ipsapirone, tiospirone) have selective affinity for serotonin receptors of the 5-HT1A type, for which they appear to be partial agonists (seeChapter 11: 5-Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists). Buspirone has beneficial actions in anxious patients, particularly those with generalized anxiety of mild or moderate severity (Ninan et al., 1998; Taylor, 1998). Unlike potent benzodiazepines and antidepressants, buspirone lacks beneficial actions in severe anxiety with panic attacks. It also does not share with serotonin-reuptake inhibitors their efficacy as a monotherapy in obsessive-compulsive disorder, although it may have useful antiobsessional activity when added to serotonin-active antidepressants. A lack of cross-tolerance is consistent with a lack of clinical protection against withdrawal-emergent anxiety when changing abruptly from treatment with a benzodiazepine to buspirone; a gradual transition between these classes of antianxiety agents is more likely to be tolerated (Lader, 1987). |
Prospectus
|
Major affective and anxiety disorders continue to represent the most common psychiatric illnesses; they include the most prevalent disorders of unknown cause with psychotic features and represent enormous costs to society in morbidity, disability, and premature mortality (Kessler et al., 1994). Rates of diagnosis and appropriate treatment of major mood disorders have improved somewhat in recent years with the advent of better accepted modern mood-altering medicines. Nevertheless, the majority of patients with depression and bipolar disorder are diagnosed after years of delay, if at all, and many remain inadequately treated (McCombs et al., 1990; Newman and Hassan, 1999). Given these unmet needs, the clinical needs and economic incentives for developing additional, improved mood-altering medicines are clear. Several groups of depressed patients continue to be particularly inadequately treated or studied. They include children and the elderly, those with bipolar depression, and those with severe, chronic, or psychotic forms of depression (seeShulman et al., 1996; Kutcher, 1997). Whereas ambulatory depressed patients are much greater in number, have the highest likelihood of improvement and recovery, and represent the largest potential market, they also are most likely to respond to a placebo or other nonspecific treatment and thus represent a special challenge for the development of clinically useful and cost-effective treatments. A major limitation of efforts to develop new mood-altering agents is the lack of compelling rationales other than imitation or modification of successful precedents. The fundamental problem is the continued lack of a coherent pathophysiology, let alone an etiology, of major depression, bipolar disorder and the common anxiety disorders despite decades of important and useful contributions to the description of the syndromes. Major depression may well represent a spectrum of disorders, varying in severity from relatively mild and self-limited disorders that approach everyday human distress to extraordinarily severe, psychotic, incapacitating, and deadly illnesses. It remains difficult to conceive of a mood-altering agent that does not affect central monoaminergic synaptic neurotransmission, particularly that mediated by either norepinephrine or serotonin, which limits the identification of novel therapeutic targets for these disorders (Murphy et al., 1995; Bloom and Kupfer, 1995; Healy, 1997). Novel Treatments for Major Depression and Anxiety Disorders Many of the large number of potential antidepressants in development continue to exploit interactions with either noradrenergic or serotonergic systems in proven ways (Evrard and Harrison, 1999; Kent, 2000). These agents conceptually are remarkably similar to the tricyclic-type antidepressants and include several relatively selective inhibitors of the neuronal transport of norepinephrine (e.g., oxaprotiline, levoprotiline, lofepramine, nisoxetine, reboxetine, (R)-thionisoxetine, tomoxetine, and viloxazine) that are less cardiotoxic or lethal on overdose than traditional tricyclic antidepressants. Development of additional serotonin-reuptake inhibitors has slowed, although their range of approved indications continues to expand, particularly into the anxiety, impulsive or compulsive, and eating disorders. However, several novel serotonin-reuptake inhibitors including Ro-15-8081 and aryl- or naphthylpiperidines and thiodiphenyls have been developed; tianeptine has complex effects on the storage and release of serotonin and may facilitate its uptake in vivo. Also, some substituted phenyltropane analogs of cocaine are serotonin transporterselective and much less potent at dopamine transporters than are cocaine and older phenyltropanes (Robertson, 1999). Some of these agents have been evaluated as potential antidepressants in clinical trials (seeLeonard, 1994; Murphy et al., 1995). The clinical efficacy of the mixed serotonin/norepinephrine transport antagonist, venlafaxine, and the interesting beneficial properties of an older, similar agent, clomipramine (see above) have encouraged further exploration of the principle of mixed aminergic potentiation (Kent, 2000). This strategy has led to such drugs as duloxetine (LY-248686), milnacipran, and analogs of bupropion. These developments arose conceptually from pursuing the transport-blocking activities of the original tricyclic antidepressants, with efforts to avoid their familiar toxic properties (including potent antimuscarinic and cardiac depressant activities). Surprisingly underexplored are agents with dopamine-potentiating activity (Murphy et al., 1995). Nomifensine is a mixed antagonist of norepinephrine and dopamine transporters (seeTable 192; Zahniser et al., 1999). An effective antidepressant, it was withdrawn from clinical use due to association with febrile illnesses and ascending paralysis of the Guillain-Barr type. The stimulant-antidepressant bupropion also has mixed antagonistic effects on the same amine transporters. Other dopamine-receptor agonists also may have antidepressant effects (Murphy et al., 1995; Mattox et al., 1986; Wells and Marken, 1989; D'Acquila et al., 1994). Additional agents whose actions include inhibition of dopamine transport are the tricyclic-like agent amineptine, medifoxamine, and a series of potent piperazine derivatives (GBR-12909 and others). Antiparkinsonism, directly acting dopamine-receptor agonistsincluding bromocriptine, pramipexole, lisuride, and roxindole (the latter two also serotonergic)also have been reported to have mood-elevating properties (Murphy et al., 1995; seeChapter 22: Treatment of Central Nervous System Degenerative Disorders). Novel approaches to enhancing central adrenergic function include the
use of Interest in MAO inhibitors has continued despite their risks of
dangerous interactions with other substances. Selective irreversible
propargyl inhibitors of MAO-A (e.g., clorgyline) with mood-elevating
activity, as well as inhibitors of MAO-B (e.g., selegiline) with dopamine
sparing, antiparkinsonism, and antidepressant activities (at least, at high
doses probably not selective for MAO-B), represent potentially important
leads to novel psychotropic or neurotropic agents. In addition, several
short-acting reversible inhibitors of MAO-A have at
least moderate antidepressant activity and limit the risk of inducing acute
hypertension by potentiating pressor amines. Such short-acting MAO-A inhibitors include brofaromine, moclobemide (MANERIX), pirlindole, and toloxatone
( The large number of serotonin receptor subtypes provides many
opportunities for developing novel agonists, partial agonists, antagonists,
and negative antagonists (or inverse agonists), some of which may alter mood
or treat anxiety disorders. Pindolol, a mixed An opposite strategy is to evaluate 5-HT1Areceptor agonists as possible mood-altering or antianxiety agents. Such agents may act in part by enhancing release of norepinephrine (Cohen et al., 1999). An example of a 5-HT1A partial agonist with anxiolytic effects is flesinoxan (Albert et al., 1999). Several partial agonists of 5-HT1A receptors have been explored for potential utility both in anxiety disorders and in milder cases of mixed anxiety-depression (Dubovsky and Buzan, 1995; Murphy et al., 1995). Some 5-HT1Areceptor partial agonists that may have antidepressant activity (e.g., gepirone, ipsapirone, and zalospirone) are azapirones related chemically to buspirone. An additional opportunity for modifying serotonin function is to antagonize the 5-HT1A receptor subtypes that serve as autoreceptors; several such antagonists are known, including GR-127935 (Robertson, 1999). Some recently developed antidepressants, notably nefazodone, combine activity as serotonin reuptake inhibitors and 5-HT2A antagonists. Several innovative agents, including YM-35992, have followed this precedent. The 5-HT2C serotonin receptor is prominent in limbic forebrain and cerebral cortex. This receptor subtype has been postulated to be a reasonable therapeutic target for depression or anxiety (Murphy et al., 1995). Nefazodone and the trazodone metabolite m-chlorophenylpiperazine (mCPP), as well as Ro-60-0175, Ro-60-0332, Org-12962, and Org-8484 all have 5-HT2C agonist or partial-agonist properties. Norfluoxetine also interacts potently with 5-HT2C receptors. There also are selective ligands for the 5-HT6 receptor (Ro-63-0563, SB-171046) and emerging compounds for 5-HT7 receptors (Robertson, 1999). However, the potential psychotropic properties of such agents remain obscure, requiring substantial investment in preclinical and exploratory investigations to evaluate their clinical utility in treatment of depression or anxiety. Agents acting at amino-acid neurotransmission systems also provide leads to potential psychotropic drugs. For example, certain analogs of progesterone interact with a distinct allosteric regulatory site in the GABAA-receptor complex to activate hyperpolarizing chloride channels, and so may have anxiolytic properties (Robertson, 1999; Nabeshima and Muraoka, 1999). Agents that interact with N-methyl-D-aspartate (NMDA) glutamate receptors have antidepressantlike activity in some animal behavioral models. They include the NMDA antagonist dizolcipine (MK-801) and NMDA-receptor partial agonist AP-7 (Murphy et al., 1995). Receptors for cerebral peptides also provide targets for psychotropic
drug development. Opioids may have mood-elevating effects, but exploration of
drugs acting at opioid receptors as potential antidepressants has been
limited (Tejedor-Real et al., 1995). Cerebral sigma receptors ( Another neuropeptide-based strategy for developing antidepressant or antianxiety drugs derives from pronounced behavioral effects of intracerebral administration of the large corticotrophin (ACTH)-releasing peptide (CRF). Observed responses include suggestions of fear or anxiety, increased startle response, loss of interest in food or sex, altered sleep, and eventually epileptic seizures. Small-molecule antagonists of receptors for CRF and related peptides can penetrate the blood-brain barrier and reverse these effects. A growing list of CRF1 receptorselective antagonists includes antialarmin, CP-154,526, SP-904, NBI-30545, DNP-606, DNP-695, CRA-1000, and SC-241. Some of these agents also interact with CRF2 receptors, but no highly selective CRF2 antagonists have been identified. The precise cerebral localization and function of the two CRF receptors should yield to exploration with site-selective ligands. Some CRF1 antagonists are in clinical testing (Mansbach et al., 1997; McCarthy et al., 1999; Steckler and Holsboer, 1999). There also is a search for natural products for the treatment of
depression and anxiety disorders (Wong et al., 1998). Hypericum
or New Treatments for Anxiety Disorders Innovative prospects for the treatment of anxiety disorders include
extensions of the pharmacology of benzodiazepines (seeChapter 17:
Hypnotics and Sedatives). Advances in a molecular understanding of the GABAA
receptorbenzodiazepine receptor-Cl channel complex indicate that
this ring-shaped collection of transmembrane proteins includes
representatives of at least 16 subunit proteins in five groups ( A particularly encouraging approach is the development and clinical
testing of benzodiazepine-receptor ligands with agonist activity intermediate
between a full agonist, such as diazepam, and an antagonist, such as flumazenil
(seeChapter 17: Hypnotics and Sedatives; Browne and Shaw, 1991 Potokar
and Nutt, 1994). Benzodiazepines and Elucidation of a growing number of serotonin-receptor subtypes and agents that interact with them has strongly encouraged development of additional psychotropic agents acting on the serotonin system. One approach includes further development of azapirone analogs as 5-HT1A receptor ligands. Another is the use of 5-HT3 receptor antagonists; some of these modulate dopamine synthesis and release, and others have shown properties in animal tests that suggest antianxiety activity. Agents with anti-5-HT3selective activity include the short-term antiemetic compound ondansetron and the benzamide zacopride; many others are known but have been subjected to only limited clinical testing in psychiatric disorders including psychosis and anxiety. Other approaches to the pharmacotherapy of anxiety disorders have
included the use of antiadrenergic compounds usually employed for hypertension
or other cardiovascular indications, including the It is reasonable to anticipate that the expansion of novel macromolecular target sites for CNS-active drug development may lead to innovative principles and agents for treating depressive and anxiety disorders in the future. For further discussion of psychiatric disorders, seeChapter 371
in Harrison's Principles of Internal Medicine, 16th ed., |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1648
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved