| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
General Anesthetics
Overview
|
General anesthetics are a class of drugs used to depress the central nervous system to a sufficient degree to permit the performance ofsurgery and other noxious or unpleasant procedures. Not surprisingly, general anesthetics have very low therapeutic indices and thus are dangerous drugs that require great care in administration. Indeed, an entire specialty of medicine has grown around the administration of this class of drugs. General anesthetics can be administered by a variety of routes, but intravenous or inhalational administration is preferred because effective doses can be more accurately administered and the time course of action more carefully controlled. While all general anesthetics produce a relatively similar anesthetic state, the drugs are quite dissimilar in their secondary actions (side effects) on other organ systems. The selection of specific drugs and routes of administration to produce general anesthesia is based on their pharmacokinetic properties and on the secondary effects of the various drugs, in the context of the individual patient's age, pathophysiology, and medication use. This chapter will review basic aspects of anesthetic action and then will focus on the specific properties of inhalational and intravenous anesthetics as well as on practical aspects of their use. |
Introduction
|
Definition of General anesthetics are a structurally diverse class of drugs that produce a common end pointa behavioral state referred to as general anesthesia. In the broadest sense, general anesthesia can be defined as a global but reversible depression of central nervous system (CNS) function resulting in the loss of response to and perception of all external stimuli. While this definition is appealing in its simplicity, it is not useful for two reasons: First, it is inadequate because anesthesia is not simply a deafferented state; for example, amnesia is an important aspect of the anesthetic state. Second, not all general anesthetics produce identical patterns of deafferentation. Barbiturates, for example, are very effective at producing amnesia and loss of consciousness but are not effective as analgesics. An alternative way of defining the anesthetic state is to consider it as a collection of 'component' changes in behavior or perception. The components of the anesthetic state include amnesia, immobility in response to noxious stimulation, attenuation of autonomic responses to noxious stimulation, analgesia, and unconsciousness. It is important to remember that general anesthesia is useful only insofar as it facilitates the performance of surgery or other noxious procedures. The performance of surgery requires an immobilized patient who does not have an excessive autonomic response to surgery (blood pressure, heart rate) and who has amnesia for the procedure. Thus the essential components of the anesthetic state are immobilization, amnesia, and attenuation of autonomic responses to noxious stimulation. Indeed, if an anesthetic produces profound amnesia, it can be difficult, in principle, to determine if it also produces either analgesia or unconsciousness. Measurement of Anesthetic Potency Given the essential requirement that a general anesthetic agent provide an immobilized patient who does not move in response to surgical stimulation, the potency of general anesthetic agents usually is measured by determining the concentration of general anesthetic that prevents movement in response to surgical stimulation. As described in Chapter 13: History and Principles of Anesthesiology, anesthetic potency is measured in MAC units, with 1 MAC defined as the minimum alveolar concentration that prevents movement in response to surgical stimulation in 50% of subjects. The strengths of MAC as a measurement are that (1) it can be monitored continuously by measuring end-tidal anesthetic concentration using infrared spectroscopy or mass spectrometry; (2) it provides a direct correlate of the free concentration of the anesthetic at its site(s) of action in the central nervous system; (3) it is a simple-to-measure endpoint that reflects an important clinical goal. End points other than immobilization also can be used to measure anesthetic potency. For example, the ability to respond to verbal commands (MACawake) (Stoelting et al., 1970) and the ability to form memories (Dwyer et al., 1992) also have been correlated with alveolar anesthetic concentration. Interestingly, verbal response and memory formation are both suppressed at a fraction of MAC. Furthermore, the ratio of the anesthetic concentrations required to produce amnesia and immobility vary significantly among different inhalational anesthetic agents (nitrous oxide vs. isoflurane, Table 141), suggesting that anesthetic agents may produce these behavioral end points via different cellular and molecular mechanisms. The potency of intravenous anesthetic agents is somewhat more difficult to measure, because there is not an available method to measure blood or plasma anesthetic concentration continuously and because the free concentration of the drug at its site of action cannot be determined. Generally, the potency of intravenous agents is defined as the free plasma concentration (at equilibrium) that produces loss of response to surgical incision (or other end points) in 50% of subjects (Franks and Lieb, 1994). Mechanisms of Anesthesia The molecular mechanisms by which general anesthetics produce their effects have remained one of the great mysteries of pharmacology. For most of the twentieth century, it was theorized that all anesthetics act by a common mechanism (the unitary theory of anesthesia) and that anesthesia is produced by perturbation of the physical properties of cell membranes. This thinking was based largely on observations made in the late nineteenth century that the potency of a gas as an anesthetic correlated with its solubility in olive oil. This correlation, referred to as the Meyer-Overton rule, was interpreted as indicating the lipid bilayer as the likely target of anesthetic action. In the past decade, clear exceptions to the Meyer-Overton rule have been noted (Koblin et al., 1994). For example, it has been shown that inhalational and intravenous anesthetics can be enantioselective in their action as anesthetics (etomidate, steroids, isoflurane) (Tomlin et al., 1998; Lysko et al., 1994; Wittmer et al., 1996). The fact that enantiomers have unique actions but identical physical properties indicates that properties other than bulk solubility are important in determining anesthetic action. This realization has focused thinking on identification of specific protein binding sites for anesthetics. One impediment to understanding the mechanisms of anesthesia has been the difficulty in precisely defining anesthesia. It has now become clear that an anesthetic agent produces different components of the anesthetic state via actions at different anatomic loci in the nervous system and may produce these component effects via different cellular and/or molecular actions. It also is becoming clear that different anesthetic agents can produce a specific component of anesthesia via actions at different molecular targets. Given these insights, the unitary theory of anesthesia has been largely discarded. The ensuing section will focus on the identification of specific anatomic, cellular, and molecular targets of anesthetic action. The complete mechanism(s) of anesthetic action have not been defined. The most difficult issue is mapping the effects of anesthetics on specific molecular targets to the complex component behaviors that compose anesthesia. This is a particularly vexing problem for poorly understood behaviors such as consciousness (see Chapter 13: History and Principles of Anesthesiology). Anatomic Sites of Anesthetic Action General anesthetics could, in principle, interrupt nervous system
function at myriad levels, including peripheral sensory neurons, the spinal
cord, the brain stem, and the cerebral cortex. Delineation of the precise
anatomic sites of action is difficult because many anesthetics diffusely
inhibit electrical activity in the CNS. For example, isoflurane at 2 MAC can
cause electrical silence in the brain (Newberg et al., 1983)! Despite
this, in vitro studies have shown that specific cortical pathways
exhibit markedly different sensitivities to both inhalational and intravenous
anesthetics (MacIver and Roth, 1988; Richards and White, 1975; Nicoll, 1972).
This suggests that anesthetics may produce specific components of the
anesthetic state via actions at specific sites in the CNS. Consistent
with this possibility, studies by Rampil (1994) and Antognini and Schwartz
(1993) have demonstrated that immobilization in response to a surgical
incision (the end point used in determining MAC) results from inhalational
anesthetic action in the spinal cord. It is unlikely that amnesia or
unconsciousness are the result of anesthetic actions in the spinal cord; thus
different components of anesthesia are produced at different sites in the
CNS. One intravenous anesthetic, dexmedetomidine (an Physiological Mechanisms of Anesthesia General anesthetics produce two important physiologic effects at the cellular level. First, the inhalational anesthetics can hyperpolarize neurons (Nicoll and Madison, 1982). This may be an important effect on neurons serving a pacemaker role and on pattern-generating circuits. It also may be important in synaptic communication, since reduced excitability in a postsynaptic neuron may reduce the likelihood that an action potential will be initiated in response to neurotransmitter release. Second, both inhalational and intravenous anesthetics have substantial effects on synaptic function. In this regard it is noteworthy that anesthetics appear to have minimal effects on action-potential generation or propagation at concentrations that affect synapses (Larrabee and Posternak, 1952). The inhalational anesthetics have been shown to inhibit excitatory synapses and enhance inhibitory synapses in various preparations. It seems likely that these effects are produced by both pre- and postsynaptic actions of the inhalational anesthetics. There is clear evidence that the inhalational anesthetic isoflurane can inhibit neurotransmitter release (Perouansky et al., 1995; MacIver et al., 1996); this may be mediated via an effect on the neurosecretory machinery (van Swinderen et al., 1999). It also is abundantly clear that inhalational anesthetics can act postsynaptically, altering the response to released neurotransmitter. These actions are thought to be due to specific interactions of anesthetic agents with neurotransmitter receptors. The intravenous anesthetics produce a narrower range of physiological effects. Their predominant actions are at the synapse, where they have profound but relatively specific effects on the postsynaptic response to released neurotransmitter. Most of the intravenous agents act predominantly by enhancing inhibitory neurotransmission, whereas ketamine predominantly inhibits excitatory neurotransmission at glutamatergic synapses. Molecular Actions of General Anesthetics The electrophysiological effects of general anesthetics at the cellular level suggest several potential molecular targets for anesthetic action. There is strong evidence supporting ligand-gated ion channels as important targets for anesthetic action. Chloride channels gated by the inhibitory neurotransmitter gamma-aminobutyric acid (GABAA receptors; see Chapter 17: Hypnotics and Sedatives) are sensitive to clinical concentrations of a wide variety of anesthetics, including the halogenated inhalational agents and many intravenous agents (propofol, barbiturates, etomidate, and neurosteroids) (Krasowski and Harrison, 1999). At clinical concentrations, general anesthetics increase the sensitivity of the GABAA receptor to GABA, thus enhancing inhibitory neurotransmission and depressing nervous system activity. It appears likely that action of anesthetics on the GABAA receptor is mediated by binding of the anesthetics to specific sites on the GABAA-receptor protein, as point mutations on the receptor can eliminate the effects of the anesthetic on ion channel function (Mihic et al., 1997). It also seems likely that there are specific binding sites for at least several classes of anesthetics, as mutations in various regions (and subunits) of the GABAA receptor selectively affect the actions of various anesthetics (Belelli et al., 1997; Krasowski and Harrison, 1999). It should be noted that none of the general anesthetics competes with GABA for its binding site on the receptor. Which components of anesthesia are mediated by actions of anesthetics on GABAA receptors remains a subject of conjecture. The fact that GABA mimetics themselves can produce unconsciousness suggests a role for GABAA receptors in mediating the hypnotic effects of general anesthetics (Cheng and Brunner, 1985). Closely related to the GABAA receptors are other ligand-gated ion channels including glycine receptors and neuronal nicotinic acetylcholine receptors. Clinical concentrations of the inhalational anesthetics enhance the ability of glycine to activate glycine-gated chloride channels (glycine receptors), which play an important role in inhibitory neurotransmission in the spinal cord and brain stem. Propofol (Hales and Lambert, 1988), neurosteroids, and barbiturates also potentiate glycine-activated currents, whereas etomidate and ketamine do not (Mascia et al., 1996). Glycine receptors may play a role in mediating inhibition by anesthetics of responses to noxious stimuli. Subanesthetic concentrations of the inhalational anesthetics inhibit some classes of neuronal nicotinic acetylcholine receptors (Violet et al., 1997, Flood et al., 1997). The neuronal nicotinic receptors may play a role in mediating the analgesic effects of inhalational anesthetic agents. The only general anesthetics that do not have significant effects on GABAA or glycine receptors are ketamine, nitrous oxide, and xenon. All of these agents have been shown to inhibit a different type of ligand-gated ion channel, the N-methyl-D-aspartate (NMDA) receptor (see Chapter 12: Neurotransmission and the Central Nervous System). NMDA receptors are glutamate-gated cation channels that are somewhat selective for calcium and are involved in long-term modulation of synaptic responses (long-term potentiation) and glutamate-mediated neurotoxicity. Ketamine inhibits NMDA receptors by binding to the phencyclidine site on the NMDA-receptor protein (Lodge et al., 1982; Anis et al., 1983; Zeilhofer et al., 1992). The NMDA receptor is thought to be the principal molecular target for the anesthetic actions of ketamine. Recent studies also show that nitrous oxide (Mennerick et al., 1998; Jevtovic-Todorovic et al., 1998) and xenon (Franks et al., 1998; de Sousa et al., 2000) are potent and selective inhibitors of NMDA-activated currents, suggesting that these agents also may produce unconsciousness via actions on NMDA receptors. Inhalational anesthetics have two other identified molecular targets that may be important in some of their actions. Some members of a class of potassium channels known as two-pore domain channels are activated by inhalational anesthetics (Gray et al., 1998; Patel et al., 1999). These channels are important in setting the resting membrane potential of a neuron and may be the molecular locus through which these agents hyperpolarize neurons. A second target is the molecular machinery involved in neurotransmitter release. Recent evidence shows that the action of inhalational anesthetics requires a protein complex (syntaxin, SNAP-25, synaptobrevin) involved in synaptic neurotransmitter release (van Swinderen et al., 1999). These molecular interactions may explain the ability of inhalational anesthetics to cause presynaptic inhibition in the hippocampus, and could contribute to the amnesic effect of inhalational anesthetics. Summary Current evidence supports the view that most of the intravenous general anesthetics act predominantly through GABAA receptors and perhaps through some interactions with other ligand-gated ion channels. The halogenated inhalational agents have a variety of molecular targets, consistent with their status as complete (all components) anesthetics. Nitrous oxide, ketamine, and xenon constitute a third category of general anesthetics that are likely to produce unconsciousness via inhibition of the NMDA receptor. |
Parenteral Anesthetics
|
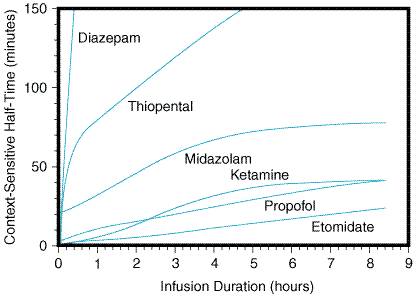
Pharmacokinetic Principles Parenteral anesthetics are small, hydrophobic, substituted aromatic or heterocyclic compounds (Figure 141). Hydrophobicity is the key factor governing the pharmacokinetics of this class of drugs (Bischoff and Dedrick, 1968; Burch and Stanski, 1983; Shafer and Stanski, 1992). After a single intravenous bolus, each of these drugs preferentially partitions into the highly perfused and lipophilic brain and spinal cord tissue where it produces anesthesia within a single circulation time. Subsequently, blood levels fall rapidly, resulting in redistribution of anesthetic out of the central nervous system back into the blood, where it then diffuses into less-well-perfused tissues such as muscle, viscera and, at a slower rate, into the poorly perfused but very hydrophobic adipose tissue. Termination of anesthesia after single boluses of parenteral anesthetics is primarily by redistribution out of the nervous system rather than by metabolism (for example, see Figure 142). After redistribution, anesthetic blood levels fall according to a complex interaction between the metabolic rate and the amount and lipophilicity of the drug stored in the peripheral compartments (Hughes et al., 1992; Shafer and Stanski, 1992). Thus, parenteral anesthetic half-lives are 'context-sensitive,' and the degree to which a half-life is contextual varies greatly from drug to drug as might be predicted based on their markedly different hydrophobicities and metabolic clearances (Table 142; and Figure 143). For example, after a single bolus of thiopental, patients usually emerge from anesthesia within 10 minutes; however, a patient may require more than a day to awaken from a prolonged thiopental infusion. The majority of individual variability in sensitivity to parenteral anesthetics can be accounted for by pharmacokinetic factors (Wada et al., 1997; Wulfsohn and Joshi, 1969). For example, in patients with lower cardiac output, the relative perfusion of and fraction of anesthetic dose delivered to the brain is higher; thus, patients in septic shock and those with cardiomyopathy usually require lower doses of anesthetic. Elderly patients typically require a smaller anesthetic dose, primarily because of a smaller initial volume of distribution (Arden et al., 1986; Homer and Stanski, 1985). As described below, similar principles govern the pharmacokinetics of the hydrophobic inhalational anesthetics with the added complexity of drug uptake by inhalation.
Barbiturates Chemistry and Formulations Anesthetic barbiturates are derivatives of barbituric acid (2,4,6-trioxohexahydropyrimidine), with either an oxygen or sulfur at the 2-position (Figure 141). The three barbiturates used for clinical anesthesia are sodium thiopental (PENTOTHAL), thiamylal (SURITAL), and methohexital (BREVITAL). Sodium thiopental is the most frequently used of the barbiturates for inducing anesthesia. All three barbiturate anesthetics are supplied as racemic mixtures despite enantioselectivity in their anesthetic potency (Andrews and Mark, 1982; Christensen and Lee, 1973; Nguyen et al., 1996). Barbiturates are formulated as the sodium salts with 6% sodium carbonate and reconstituted in water or isotonic saline solution to produce 1% (methohexital), 2% (thiamylal), or 2.5% (thiopental) alkaline solutions with pHs of 10 to 11. Once reconstituted, the thiobarbiturates are stable in solution for up to 1 week and methohexital for up to 6 weeks if refrigerated. Mixing with more acidic drugs commonly used during anesthetic induction can result in precipitation of the barbiturate as the free acid; thus, standard practice is to delay the administration of other drugs until the barbiturate has cleared the intravenous tubing. Pharmacokinetics Pharmacokinetic parameters for each drug are given in Table 142. As discussed above, the principal mechanism limiting anesthetic duration after single doses is redistribution of these hydrophobic drugs from the brain to other tissues. However, after multiple doses or infusions, the duration of action of the barbiturates varies considerably depending on their clearances. Methohexital differs from the other two barbiturates in its much more rapid clearance; thus, it accumulates less during prolonged infusions (Schwilden and Stoeckel, 1990). Prolonged infusions or very large doses of thiopental and thiamylal can produce unconsciousness lasting several days because of their slow elimination and large volume of distributions (Stanski et al., 1980). Even single induction doses of thiopental and, to a lesser degree, methohexital can produce psychomotor impairment lasting up to 8 hours (Beskow et al., 1995; Korttila et al., 1975). Methohexital had been used frequently for outpatient procedures where rapid return to an alert state is particularly desirable, but this role now has been filled largely by the anesthetic propofol (see below). All three drugs are eliminated primarily by hepatic metabolism and renal excretion of inactive metabolites (Broadie et al., 1950); a small fraction of thiopental undergoes a desulfuration reaction to the longer-acting hypnotic pentobarbital (Chan et al., 1985). Each drug is highly protein bound (Table 142). Hepatic disease or other conditions that reduce serum protein concentrations will decrease the volume of distribution and thereby increase the initial free concentration and hypnotic effect of an induction dose (Ghoneim and Pandya, 1975). Clinical Use Recommended intravenous doses for all three drugs in a healthy young
adult are given in Table 142. The typical induction dose of thiopental (3 to
5 mg/kg) produces unconsciousness in 10 to 30 seconds with a peak effect in
one minute and duration of anesthesia of 5 to 8 minutes (Dundee et al.,
1982). Neonates and infants usually require a higher induction dose (5 to 8
mg/kg), whereas elderly and pregnant patients require less (1 to 3 mg/kg) (Gin
et al., 1997; Homer and Stanski, 1985; Jonmarker et al., 1987).
Dosage calculation based on lean body mass reduces individual variation in
dosage requirements. Doses can be reduced 10% to 50% after premedication with
benzodiazepines, opioids, and/or Side Effects Nervous System Besides producing general anesthesia, barbiturates dose-dependently reduce cerebral metabolic rate as measured by cerebral oxygen utilization (cerebral metabolic rate for oxygen; CMRO ). Induction doses of thiopental reduce CMRO about 25% to 30% with a maximal decrease of 55% occurring at about 2 to 5 times the induction dose (Pierce et al., 1962; Stullken et al., 1977). As a consequence of the decrease in CMRO , cerebral blood flow and intracranial pressure are similarly reduced Nussmeier et al., 1986). Thiopental also reduces intraocular pressure (Joshi and Bruce, 1975). Presumably because of their CNS-depressant activity, barbiturates are effective anticonvulsants (see Chapter 21: Drugs Effective in the Therapy of the Epilepsies). Thiopental in particular is of proven value in the treatment of status epilepticus (Modica et al., 1990). Cardiovascular System The anesthetic barbiturates produce dose-dependent decreases in blood
pressure. The effect primarily is caused by vasodilation, particular
venodilation, and to a lesser degree by a mild direct decrease in myocardial
contractility (Elder et al., 1955; Etsten and Li, 1955; Fieldman et
al., 1955). Typically, heart rate increases as a compensatory response to
a lower blood pressure, although barbiturates do blunt the baroreceptor
reflex (Bristow et al., 1969). Drops in blood pressure can be severe
in patients with impaired ability to compensate for venodilation, such as
those with hypovolemia, cardiomyopathy, valvular heart disease, coronary
artery disease, cardiac tamponade, or Respiratory System Barbiturates are respiratory depressants. Induction doses of thiopental decrease minute ventilation and tidal volume with a smaller and inconsistent reduction in respiratory rate (Grounds et al., 1987). Reflex responses to hypercarbia and hypoxia are diminished by anesthetic barbiturates (Gross et al., 1983; Hirshman et al., 1975), and apnea can result at higher doses or in the presence of other respiratory depressants such as opioids. With the exception of uncommon anaphylactoid reactions, these drugs have little effect on bronchomotor tone and can be used safely in asthmatic patients (Kingston and Hirshman, 1984). Other Side Effects Short-term administration of barbiturates has no clinically significant effects on the hepatic, renal, or endocrine systems. A single induction dose of thiopental does not alter gravid uterine tone but produces mild, transient depression of activity of the newborn (Kosaka et al., 1969). True allergies to barbiturates are rare (Baldo et al., 1991); however, drug-induced histamine release occasionally is seen (Hirshman et al., 1982; Sprung et al., 1997). Barbiturates can induce fatal attacks of porphyria in patients with acute intermittent or variegate porphyria and are contraindicated in such patients (Dundee et al., 1962). Unlike inhalational anesthetics and succinylcholine, barbiturates and all other parenteral anesthetics do not appear to trigger malignant hyperthermia (Rosenberg et al., 1997). Propofol Chemistry and Formulations Along with thiopental, propofol (DIPRIVAN) is the most commonly used
parenteral anesthetic. Propofol, 2,6-diisopropylphenol, is essentially
insoluble in aqueous solutions and is formulated only for intravenous
administration as a 1% (10 mg/ml) emulsion in 10% soybean oil, 2.25%
glycerol, and 1.2% purified egg phospholipid. In the Pharmacokinetics The pharmacokinetics of propofol are governed by the same principles that apply to barbiturates. Onset and duration of anesthesia after a single bolus are similar to those of thiopental (Langley and Heel, 1988). However, recovery after multiple doses or infusion has been shown to be much faster after propofol than after thiopental or even methohexital (Doze et al., 1986; Langley and Heel, 1988). The rapid rate of recovery after infusion of propofol can be explained by its very high clearance coupled with the slow diffusion of drug from the peripheral to the central compartment (Figure 143). The rapid clearance of propofol explains its less severe hangover compared to barbiturates and may allow for a more rapid discharge from the recovery room (Bryson et al., 1995). Propofol is metabolized primarily in the liver to less-active metabolites that are renally excreted (Simons et al., 1988); however, its clearance exceeds hepatic blood flow, and extrahepatic metabolism has been demonstrated (Veroli et al., 1992). Propofol is highly protein bound, and its pharmacokinetics, like that of the barbiturates, may be affected by conditions that alter serum protein levels (Kirkpatrick et al., 1988). Clinical Use The induction dose of propofol in a healthy adult is 1.5 to 2.5 mg/kg.
Propofol has an onset and duration of anesthesia similar to those of thiopental
(Table 142). As with barbiturates, dosages should be reduced in elderly
patients and in the presence of other sedatives and increased in young children
(Aun et al., 1992; Dundee et al., 1986). Propofol often is used
for maintenance of anesthesia as well as induction. For short procedures,
small boluses (10% to 50% of the induction dose) every 5 minutes or as needed
are effective. Because they produce more stable drug levels, propofol
infusions (100 to 300 Side Effects Nervous System The central nervous system effects of propofol are similar to those of barbiturates. Propofol decreases CMRO , cerebral blood flow, and intracranial and intraocular pressures by about the same amount as does thiopental (Langley and Heel, 1988; Ravussin et al., 1988; Vandesteene et al., 1988). Like thiopental, propofol has been used in patients at risk for cerebral ischemia (Ravussin and de Tribolet, 1993); however, no human outcome studies have been performed to determine propofol's efficacy as a neuroprotectant. Results from studies on the anticonvulsant effects of propofol have been mixed, with some data even suggesting that it has proconvulsant activity when combined with other drugs (Modica et al., 1990). Thus, unlike thiopental, propofol is not a proven acute intervention for seizures. Cardiovascular System Propofol produces a dose-dependent decrease in blood pressure that is significantly greater than that produced by thiopental (Grounds et al., 1985; Langley and Heel, 1988). The fall in blood pressure can be explained by both vasodilation and mild depression of myocardial contractility (Claeys et al., 1988; Grounds et al., 1985). Propofol appears to blunt the baroreceptor reflex and/or is directly vagotonic, because smaller increases in heart rate are seen for any given drop in blood pressure after doses of propofol (Claeys et al., 1988; Langley and Heel, 1988). As with thiopental, propofol should be used with caution in patients at risk for or intolerant of decreases in blood pressure. Respiratory and Other Side Effects At equianesthetic doses, propofol produces a slightly greater degree of respiratory depression than does thiopental (Blouin et al., 1991; Taylor et al., 1986). Patients given propofol should be monitored to ensure adequate oxygenation and ventilation. Propofol appears to be less likely than barbiturates to provoke bronchospasm (Eames et al., 1996; Pizov et al., 1995). It has no clinically significant effects on hepatic, renal, or endocrine organ systems. Unlike thiopental, propofol appears to have significant antiemetic action and is a good choice for sedation or anesthesia in patients at high risk for nausea and vomiting (Gan et al., 1996; McCollum et al., 1988). Propofol provokes anaphylactoid reactions and histamine release at about the same low frequency as does thiopental (Bryson et al., 1995; Laxenaire et al., 1992). Although propofol does cross placental membranes, it is considered safe for use in pregnant patients and transiently depresses activity of the newborn similarly to thiopental (Abboud et al., 1995). Etomidate Chemistry and Formulation Etomidate AMIDATE) is a substituted imidazole that is supplied as the active D-isomer (Figure 141). Etomidate is poorly soluble in water and is formulated as a 2-mg/ml solution in 35% propylene glycol. Unlike thiopental, etomidate does not induce precipitation of neuromuscular blocking agents or other drugs frequently given during anesthetic induction (Hadzija and Lubarsky, 1995). Pharmacokinetics An induction dose of etomidate has a rapid onset and redistribution-limited duration of action (Table 142). Metabolism of etomidate occurs in the liver, where it is primarily hydrolyzed to inactive compounds (Gooding and Corssen, 1976; Heykants et al., 1975). Elimination is both renal (78%) and biliary (22%). Compared to thiopental, the duration of action of etomidate increases less with repeated doses (Figure 143). The plasma-protein binding of etomidate is high but less than that of barbiturates and propofol (Table 142). Clinical Use Etomidate primarily is used for anesthetic induction of patients at
risk for hypotension. Induction doses of etomidate (0.2 to 0.4 mg/kg) have a
rapid onset and a short duration of action (Table 142); they are accompanied
by a high incidence of pain on injection and myoclonic movements (Giese and
Stanley, 1983). As with propofol, lidocaine effectively reduces the pain of
injection (Galloway et al., 1982). The myoclonic movements can be
reduced by premedication with either benzodiazepines or opioids (Zacharias et
al., 1979). Etomidate is pharmacokinetically suitable for infusion for
anesthetic maintenance (10 Side Effects Nervous System The effects of etomidate on cerebral blood flow, metabolism, and intracranial and intraocular pressures are similar to those of thiopental (Modica and Tempelhoff, 1992; Renou et al., 1978; Thomson et al., 1982). Etomidate has been tried as a protective agent against cerebral ischemia (Batjer, 1993). However, animal studies have failed to show a consistent beneficial effect (Drummond et al., 1995; Guo et al., 1995), and no controlled human trials have been performed. Etomidate has been shown in some studies to be a proconvulsant and is not a proven treatment for seizures (Modica et al., 1990). Cardiovascular System Cardiovascular stability after induction is a major advantage of etomidate over either barbiturates or propofol. Induction doses of etomidate typically produce a small increase in heart rate and little to no decrease in blood pressure or cardiac output (Criado et al., 1980; Gooding and Corssen, 1977; Gooding et al., 1979). Etomidate has little effect on coronary perfusion pressure and reduces myocardial oxygen consumption (Kettler et al., 1974). Thus, of all induction agents, etomidate is best suited to maintain cardiovascular stability in patients with coronary artery disease, cardiomyopathy, cerebral vascular disease, and/or hypovolemia. Respiratory and Other Side Effects The degree of respiratory depression by etomidate appears to be less than that by thiopental (Colvin et al., 1979; Morgan et al., 1977). Like methohexital, it sometimes induces hiccups but does not significantly stimulate histamine release (Doenicke et al., 1973; Zacharias et al., 1979). Despite minimal cardiac and respiratory effects, etomidate does have two major drawbacks. First, etomidate has been associated with a significant increase in nausea and vomiting (Fragen and Caldwell, 1979). A second problem was discovered when an increase in the mortality of intensive-care-unit patients sedated with etomidate infusions was observed (Ledingham and Watt, 1983). The increased mortality was linked to suppression of the adrenocortical stress response (Ledingham et al., 1983). Indeed, etomidate inhibits certain adrenal biosynthetic enzymes required for the production of cortisol and some other steroids. Even single induction doses of etomidate may mildly and transiently reduce cortisol levels (Allolio et al., 1985; Fragen et al., 1984; Wagner et al., 1984), but no significant differences in outcome after short-term administration have been found even for variables specifically known to be associated with adrenocortical suppression (Wagner et al., 1984). Thus, while etomidate is not recommended for long-term infusion, it appears to be safe for anesthetic induction and has some unique advantages in patients prone to hemodynamic instability. Ketamine Chemistry and Formulation Ketamine KETALAR) is an arylcyclohexylamine, a congener of phencyclidine (Figure 141). It is supplied as a racemic mixture, despite the (S)-isomer being more potent and having less side effects than the (R)-isomer (White et al., 1982). Although more lipophilic than thiopental, ketamine is water-soluble and available as 10, 50, and 100 mg/ml in sodium chloride solution plus the preservative benzethonium chloride. Pharmacokinetics The onset and duration of an induction dose of ketamine is determined by the same distribution/ redistribution mechanism operant for all the other parenteral anesthetics. Ketamine is hepatically metabolized to norketamine, which has reduced CNS activity; norketamine is further metabolized and excreted in the urine and bile (Chang and Glazko, 1974). Ketamine has a large volume of distribution and rapid clearance that makes it suitable for continuous infusion without the drastic lengthening in duration of action seen with thiopental (Table 142 and Figure 143). Protein binding is much lower with ketamine than with the other parenteral anesthetics (Table 142). Clinical Use Ketamine has unique properties that make it useful for certain
pediatric procedures and for anesthetizing patients at risk for hypotension
or bronchospasm. However, it has significant side effects that limit its
routine use. Ketamine rapidly produces a hypnotic state distinct from that of
other anesthetics. Patients have profound analgesia, unresponsiveness to
commands, and amnesia but may have their eyes open, move their limbs
involuntarily, and usually have spontaneous respiration. This cataleptic
state has been termed dissociative anesthesia. Ketamine is typically
administered intravenously but also is effective by intramuscular, oral, and
rectal routes. The induction doses are 0.5 to 1.5 mg/kg intravenously, 4 to 6
mg/kg intramuscularly, and 8 to 10 mg/kg rectally (White et al., 1982).
The onset of action after an intravenous dose is similar to that of the other
parenteral anesthetics, but the duration of anesthesia of a single dose is
longer (Table 142). For anesthetic maintenance, ketamine occasionally is
continued as an infusion (25 to 100 Side Effects Nervous System As mentioned, ketamine has behavioral effects distinct from those of other anesthetics. The ketamine-induced cataleptic state is accompanied by nystagmus with pupillary dilation, salivation and/or lacrimation, and spontaneous limb movements with increased overall muscle tone. Although ketamine does not produce the classic anesthetic state, patients are anesthetized in that they are amnestic and unresponsive to painful stimuli. Indeed, ketamine produces profound analgesia, a distinct advantage over other parenteral anesthetics (White et al., 1982). Unlike other parenteral anesthetics, ketamine increases cerebral blood flow and intracranial pressure with minimal alteration of cerebral metabolism (Gardner et al., 1971; Takeshita et al., 1972; Wyte et al., 1972). These effects can be attenuated by concurrent administration of thiopental and/or benzodiazepines along with hyperventilation (Belopavlovic and Buchthal, 1982; Mayberg et al., 1995). However, given that other anesthetics actually reduce intracranial pressure and cerebral metabolism, ketamine is relatively contraindicated for patients with increased intracranial pressure or those at risk for cerebral ischemia. In some studies, ketamine has been shown to increase intraocular pressure, and its use for induction of patients with open eye injuries is controversial (Whitacre and Ellis, 1984). The effects of ketamine on seizure activity appear to be mixed, with neither strong pronor anticonvulsant activity (Modica et al., 1990). Emergence delirium characterized by hallucinations, vivid dreams, and illusions is a frequent complication of ketamine that can result in serious patient dissatisfaction and can complicate postoperative management (White et al., 1982). Delirium symptoms are most frequent in the first hour after emergence and occur less frequently in children (Sussman, 1974). Benzodiazepines reduce the incidence of emergence delirium (Dundee and Lilburn, 1978). Cardiovascular System Unlike other anesthetics, induction doses of ketamine typically increase blood pressure, heart rate, and cardiac output (Stanley et al., 1968). The cardiovascular effects are indirect and are most likely mediated by inhibition of both central and peripheral catecholamine reuptake (White et al., 1982). Ketamine has direct negative inotropic and vasodilating activity, but these effects usually are overwhelmed by the indirect sympathomimetic action (Pagel et al., 1992). Thus, ketamine is a useful drug in patients at risk for hypotension during anesthesia. While not arrythmogenic, ketamine increases myocardial oxygen consumption and is not an ideal drug for patients at risk for myocardial ischemia (Reves et al., 1978). Respiratory System The respiratory effects of ketamine are perhaps the best indication for its use. Induction doses of ketamine produce small and transient decreases in minute ventilation, but respiratory depression is less severe than with other general anesthetics (White et al., 1982). Ketamine is a potent bronchodilator due to its indirect sympathomimetic activity and perhaps some direct bronchodilating activity (Hirshman et al., 1979; Wanna and Gergis, 1978). Thus, ketamine is particularly well suited for anesthetizing patients at high risk for bronchospasm. Summary of Parenteral Anesthetics Parenteral anesthetics are the most commonly used drugs for anesthetic induction of adults. Their lipophilicity coupled with the relatively high perfusion of the brain and spinal cord results in a rapid onset and short duration after a single bolus dose. However, these drugs ultimately accumulate in fatty tissue, prolonging the patient's recovery if multiple doses are given, particularly for drugs with lower rates of clearance. Each anesthetic has its own unique set of properties and side effects (summarized in Table 143). Thiopental and propofol are the two most commonly used parenteral agents. Thiopental has a long-established track record of safety. Propofol is advantageous for procedures where rapid return to a preoperative mental status is desirable. Etomidate usually is reserved for patients at risk for hypotension and/or myocardial ischemia. Ketamine is best suited for patients with asthma and/or for children undergoing short, painful procedures. |
Inhalational Anesthetics
|
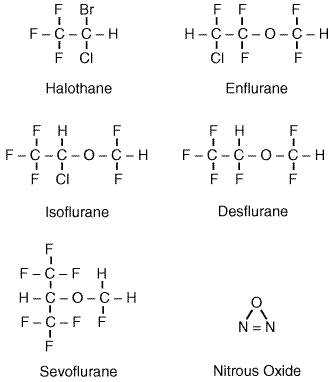
Introduction A wide variety of gases and volatile liquids can produce anesthesia. The first widely used inhalational anesthetic was diethyl ether (see Chapter 13: History and Principles of Anesthesiology). Subsequently, a variety of structurally unrelated compounds have been used as inhalational anesthetics including cyclopropane, elemental xenon, nitrous oxide, and more recently, short-chain halogenated alkanes and ethers. The structures of the currently used inhalational anesthetics are shown in Figure 144. One of the troublesome properties of the inhalational anesthetics is their low safety margin. The inhalational anesthetics have therapeutic indices (LD50/ED50) that range from 2 to 4, making these among the most dangerous drugs in clinical use. The toxicity of these drugs is largely a function of their side effects, and each of the inhalational anesthetics has a unique side-effect profile. Hence, the selection of an inhalational anesthetic often is based on matching a patient's pathophysiology with drug side-effect profiles. The specific adverse effects of each of the inhalational anesthetics are emphasized in the following sections. The inhalational anesthetics also vary widely in their physical properties. Table 141 lists the important physical properties of the inhalational agents in clinical use. These properties are important because they govern the pharmacokinetics of the inhalational agents. Ideally, an inhalational agent would produce a rapid induction of anesthesia and a rapid recovery following discontinuation. The pharmacokinetics of the inhalational agents is reviewed in the following section.
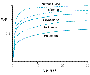
Pharmacokinetic Principles The inhalational agents are some of the very few pharmacological agents administered as gases. The fact that these agents behave as gases rather than as liquids requires that different pharmacokinetic constructs be used in analyzing their uptake and distribution. It is essential to understand that inhalational anesthetics distribute between tissues (or between blood and gas) such that equilibrium is achieved when the partial pressure of anesthetic gas is equal in the two tissues. When a person has breathed an inhalational anesthetic for a sufficiently long time that all tissues are equilibrated with the anesthetic, the partial pressure of the anesthetic in all tissues will be equal to the partial pressure of the anesthetic in inspired gas. It is important to note that while the partial pressure of the anesthetic may be equal in all tissues, the concentration of anesthetic in each tissue will be different. Indeed, anesthetic partition coefficients are defined as the ratio of anesthetic concentration in two tissues when the partial pressures of anesthetic are equal in the two tissues. Blood:gas, brain:blood, and blood:fat partition coefficients for the various inhalational agents are listed in Table 141. These partition coefficients show that inhalational anesthetics are more soluble in some tissues (e.g., fat) than they are in other (e.g., blood), and that there is significant range in the solubility of the various inhalational agents in such tissues. In clinical practice, one can monitor the equilibration of a patient with anesthetic gas. Equilibrium is achieved when the partial pressure in inspired gas is equal to the partial pressure in end-tidal (alveolar) gas. This defines equilibrium, because it is the point when there is no net uptake of anesthetic from the alveoli into the blood. For inhalational agents that are not very soluble in blood or any other tissue, equilibrium is achieved quickly, as illustrated for nitrous oxide in Figure 145. If an agent is more soluble in a tissue such as fat, equilibrium may take many hours to reach. This occurs because fat represents a huge reservoir for the anesthetic, which will be filled slowly because of the modest blood flow to fat. This is illustrated by the slow approach of halothane alveolar partial pressure to inspired partial pressure in Figure 145.
In considering the pharmacokinetics of anesthetics, one important parameter is the speed of anesthetic induction. Anesthetic induction requires that brain partial pressure be equal to MAC. Because the brain is well perfused, anesthetic partial pressure in brain becomes equal to the partial pressure in alveolar gas (and in blood) over the course of several minutes. Therefore, anesthesia is achieved shortly after alveolar partial pressure reaches MAC. While the rate of rise of alveolar partial pressure will be slower for anesthetics that are highly soluble in blood and other tissues, this limitation on speed of induction can be overcome largely by delivering higher inspired partial pressures of the anesthetic. Elimination of inhalational anesthetics is largely the reverse process of uptake. For agents with low blood and tissue solubility, recovery from anesthesia should mirror anesthetic induction, regardless of the duration of anesthetic administration. For inhalational agents with high blood and tissue solubility, recovery will be a function of the duration of anesthetic administration. This occurs because the accumulated amounts of anesthetic in the fat reservoir will prevent blood (and therefore alveolar) partial pressures from falling rapidly. Patients will be arousable when alveolar partial pressure reaches MACawake, a partial pressure somewhat lower than MAC (see Table 141). Halothane Chemistry and Formulation Halothane FLUOTHANE) is 2-bromo-2-chloro-1,1,1-trifluoroethane (see Figure 144). Halothane is a volatile liquid at room temperature and must be stored in a sealed container. Because halothane is a light-sensitive compound that also is subject to spontaneous breakdown, it is marketed in amber bottles with thymol added as a preservative. Mixtures of halothane with oxygen or air are neither flammable nor explosive. Pharmacokinetic Halothane has a relatively high blood:gas partition coefficient and high blood:fat partition coefficient (see Table 141). Induction with halothane therefore is relatively slow, and the alveolar halothane concentration remains substantially lower than the inspired halothane concentration for many hours of administration. Because halothane is soluble in fat and other body tissues, it will accumulate during prolonged administration. Therefore, the speed of recovery from halothane is lengthened as a function of duration of administration (Stoelting and Eger, 1969). Approximately 60% to 80% of halothane taken up by the body is eliminated unchanged via the lungs in the first 24 hours after its administration. A substantial amount of the halothane not eliminated in exhaled gas is biotransformed in the liver by cytochrome P450 enzymes. The major metabolite of halothane is trifluoroacetic acid, which is formed by removal of bromine and chlorine ions (Gruenke et al., 1988). Trifluoroacetic acid, bromine, and chlorine all can be detected in the urine. Trifluoroacetylchloride, an intermediate in oxidative metabolism of halothane, can trifluoroacetylate covalently several proteins in the liver. An immune reaction to these altered proteins may be responsible for the rare cases of fulminant halothane-induced hepatic necrosis (Kenna et al., 1988). There also is a minor reductive pathway accounting for approximately 1% of halothane metabolism and generally observed only under hypoxic conditions (Van Dyke et al., 1988). Clinical Use Halothane, introduced in 1956, was the first of the modern,
halogenated inhalational anesthetics used in clinical practice. It is a
potent agent that usually is used for maintenance of anesthesia. It is not
pungent and is therefore well tolerated for inhalation induction of
anesthesia. This is most commonly done in children, where preoperative
placement of an intravenous catheter can be difficult. Anesthesia is produced
by halothane at end-tidal concentrations of 0.7% to 1.0% halothane. The
end-tidal concentration of halothane required to produce anesthesia is
substantially reduced when it is coadministered with nitrous oxide. The use
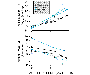
of halothane in the Side Effects Cardiovascular System The most predictable side effect of halothane is a dose-dependent reduction in arterial blood pressure. Mean arterial pressure decreases about 20% to 25% at MAC concentrations of halothane. This reduction in blood pressure primarily is the result of direct myocardial depression leading to reduced cardiac output (see Figure 146). Myocardial depression is thought to result from attenuation of depolarization-induced intracellular calcium transients (Lynch, 1997). Halothane-induced hypotension usually is accompanied by either bradycardia or a normal heart rate. This absence of a tachycardic (or contractile) response to reduced blood pressure is thought to be due to an inability of the heart to respond to the effector arm of the baroceptor reflex. Heart rate can be increased during halothane anesthesia by exogenous catecholamine or by sympathoadrenal stimulation. Halothane-induced reductions in blood pressure and heart rate generally disappear after several hours of constant halothane administration. This is thought to occur because of progressive sympathetic stimulation (Eger et al., 1970).
Halothane does not cause a significant change in systemic vascular resistance. Nonetheless, it causes changes in the resistance and autoregulation of specific vascular beds leading to redistribution of blood flow. The vascular beds of the skin and brain are dilated directly by halothane, leading to increased cerebral blood flow and skin perfusion. Conversely, autoregulation of renal, splanchnic, and cerebral blood flow is inhibited by halothane, leading to reduced perfusion of these organs in the face of reduced blood pressure. Coronary autoregulation is largely preserved during halothane anesthesia. Finally, halothane does inhibit hypoxic pulmonary vasoconstriction, which leads to increased perfusion to poorly ventilated regions of the lung and an increased alveolar:arterial oxygen gradient. Halothane also has significant effects on cardiac rhythm. Sinus
bradycardia and atrioventricular rhythms occur frequently during halothane
anesthesia but are usually benign. These rhythms result mainly from a direct
depressive effect of halothane on sinoatrial node discharge. Halothane also
can sensitize the myocardium to the arrythmogenic effects of epinephrine (Sumikawa
et al., 1983). Premature ventricular contractions and sustained
ventricular tachycardia can be observed during halothane anesthesia when
exogenous administration or endogenous adrenal production elevates plasma
epinephrine levels. Epinephrine-induced arrhythmias during halothane
anesthesia are thought to be mediated by a synergistic effect on Respiratory System Spontaneous respiration is rapid and shallow during halothane anesthesia. This produces a decrease in alveolar ventilation resulting in an elevation in arterial carbon dioxide tension from 40 mm Hg to >50 mm Hg at 1 MAC (see Figure 147). The elevated carbon dioxide does not provoke a compensatory increase in ventilation, because halothane causes a concentration-dependent inhibition of the ventilatory response to carbon dioxide (Knill and Gelb, 1978). This action of halothane is thought to be mediated by depression of central chemoceptor mechanisms. Halothane also inhibits peripheral chemoceptor responses to arterial hypoxemia. Thus, neither hemodynamic (tachycardia, hypertension) nor ventilatory responses to hypoxemia are observed during halothane anesthesia, making it prudent to monitor arterial oxygenation directly. Halothane also is an effective bronchodilator, producing direct relaxation of bronchial smooth muscle (Yamakage, 1992) and has been effectively used as a treatment of last resort in patients with status asthmaticus (Gold and Helrich, 1970).
Nervous System Halothane dilates the cerebral vasculature, increasing cerebral blood flow under most conditions. This increase in blood flow can increase intracranial pressure in patients with space-occupying intracranial masses, brain edema, or preexisting intracranial hypertension. For this reason, halothane is relatively contraindicated in patients at risk for elevated intracranial pressure. Halothane also attenuates autoregulation of cerebral blood flow. For this reason, cerebral blood flow can decrease when arterial blood pressure is markedly decreased. Modest decreases in cerebral blood flow generally are well tolerated, because halothane also reduces cerebral metabolic consumption of oxygen. Muscle Halothane causes some relaxation of skeletal muscle via its central-depressant effects. Halothane also potentiates the actions of nondepolarizing muscle relaxants (curariform drugs; see Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia), increasing both their duration of action and the magnitude of their effect. Halothane also is one of the triggering agents for malignant hyperthermia, a syndrome characterized by severe muscle contraction, rapid development of hyperthermia, and a massive increase in metabolic rate in genetically susceptible patients. This syndrome frequently is fatal and is treated by immediate discontinuation of the anesthetic and administration of dantrolene. Uterine smooth muscle is relaxed by halothane. This is a useful property for manipulation of the fetus (version) in the prenatal period and for delivery of retained placenta postnatally. Halothane, however, does inhibit uterine contractions during parturition, prolonging labor and increasing blood loss. Halothane therefore is not used as an analgesic or anesthetic for labor and vaginal delivery. Kidney Patients anesthetized with halothane usually produce a small volume of concentrated urine. This is the consequence of halothane-induced reduction of renal blood flow and glomerular filtration rate; these parameters may be reduced by 40% to 50% at 1 MAC. (Mazze et al., 1963). Halothane-induced changes in renal function are fully reversible and are not associated with long-term nephrotoxicity. Liver and Gastrointestinal Tract Halothane reduces splanchnic and hepatic blood flow as a consequence of reduced perfusion pressure, as discussed above. This reduced blood flow has not been shown to produce detrimental effects on hepatic or gastrointestinal function. Halothane can produce fulminant hepatic necrosis in a small number of patients. This syndrome generally is characterized by fever, anorexia, nausea, and vomiting developing several days after anesthesia and can be accompanied by a rash and peripheral eosinophilia. There is a rapid progression to hepatic failure, with a fatality rate of approximately 50%. This syndrome occurs in about 1 in 10,000 patients receiving halothane and is referred to as halothane hepatitis (Subcommittee on the National Halothane Study, 1966). Current thinking is that halothane hepatitis is the result of an immune response to trifluoracetylated proteins on hepatocytes (see'Pharmacokinetics,' above). Isoflurane Chemistry and Physical Properties Isoflurane FORANE) is 1-chloro-2,2,2-trifluoroethyl difluoromethyl ether (see Figure 144). It is a volatile liquid at room temperature and is neither flammable nor explosive in mixtures of air or oxygen. Pharmacokinetics Isoflurane has a blood:gas partition coefficient substantially lower than that of halothane or enflurane (see Table 141). Consequently, induction with isoflurane and recovery from isoflurane are relatively rapid. Changes in anesthetic depth also can be achieved more rapidly with isoflurane than with halothane or enflurane. More than 99% of inhaled isoflurane is excreted unchanged via the lungs. Approximately 0.2% of absorbed isoflurane is oxidatively metabolized by cytochrome P450 2E1 (Kharasch et al., 1993). The small amount of isoflurane degradation products produced are insufficient to produce any renal, hepatic, or other organ toxicity. Isoflurane does not appear to be a mutagen, teratogen, or carcinogen (Eger et al., 1978). Clinical Use Isoflurane is the most commonly used inhalational anesthetic in the Side Effects Cardiovascular System Isoflurane produces a concentration-dependent decrease in arterial blood pressure. Unlike halothane, cardiac output is well maintained with isoflurane, and hypotension is the result of decreased systemic vascular resistance (see Figure 146). Isoflurane produces vasodilation in most vascular beds, with particularly pronounced effects in skin and muscle. Isoflurane is a potent coronary vasodilator, simultaneously producing increased coronary blood flow and decreased myocardial oxygen consumption. In theory, this makes isoflurane a particularly safe anesthetic to use for patients with ischemic heart disease. However, concern has been raised that isoflurane may produce myocardial ischemia by inducing 'coronary steal' (i.e., the diversion of blood flow from poorly perfused to well-perfused areas) (Buffington et al., 1988). This concern has not been substantiated in subsequent animal and human studies. Patients anesthetized with isoflurane generally have mildly elevated heart rates, and rapid changes in isoflurane concentration can produce transient tachycardia and hypertension. This is the result of direct isoflurane-induced sympathetic stimulation. Respiratory System Isoflurane produces concentration-dependent depression of ventilation. Patients spontaneously breathing isoflurane have a normal rate of respiration but a reduced tidal volume, resulting in a marked reduction in alveolar ventilation and an increase in arterial carbon dioxide tension (see Figure 147). Isoflurane is particularly effective at depressing the ventilatory response to hypercapnia and hypoxia (Hirshman et al., 1977). While isoflurane is an effective bronchodilator, it also is an airway irritant and can stimulate airway reflexes during induction of anesthesia, producing coughing and laryngospasm. Nervous System Isoflurane, like halothane, dilates the cerebral vasculature, producing increased cerebral blood flow and the risk of increased intracranial pressure. Isoflurane also reduces cerebral metabolic oxygen consumption. Isoflurane causes less cerebral vasodilation than do either enflurane or halothane, making it a preferred agent for neurosurgical procedures (Drummond et al., 1983). The modest effects of isoflurane on cerebral blood flow can be reversed readily by hyperventilation (McPherson et al., 1989). Muscle Isoflurane produces some relaxation of skeletal muscle via its central effects. It also enhances the effects of both depolarizing and nondepolarizing muscle relaxants. Isoflurane is more potent than halothane in its potentiation of neuromuscular blocking agents. Isoflurane, like other halogenated inhalational anesthetics, relaxes uterine smooth muscle and is not recommended for analgesia or anesthesia for labor and vaginal delivery. Kidney Isoflurane reduces renal blood flow and glomerular filtration rate. This results in a small volume of concentrated urine. Changes in renal function observed during isoflurane anesthesia are rapidly reversed, and there are no long-term renal sequelae or toxicity associated with isoflurane. Liver and Gastrointestinal Tract Splanchnic (and hepatic) blood flow is reduced with increasing doses of isoflurane, as systemic arterial pressure decreases. Liver function tests are minimally affected by isoflurane, and there is no described incidence of hepatic toxicity with isoflurane. Enflurane Chemical and Physical Properties Enflurane ETHRANE) is 2-chloro-1,1,2-trifluoroethyl difluoromethyl ether (see Figure 144). It is a clear colorless liquid at room temperature with a mild, sweet odor. Like other inhalational anesthetics, it is volatile and must be stored in a sealed bottle. It is nonflammable and nonexplosive in mixtures of air or oxygen. Pharmacokinetics Because of its relatively high blood:gas partition coefficient, induction of anesthesia and recovery from enflurane are relatively slow (see Table 141). Enflurane is metabolized to a modest extent, with 2% to 8% of absorbed enflurane undergoing oxidative metabolism in the liver by cytochrome P450 2E1 (Kharasch et al., 1994). Fluoride ions are a by-product of enflurane metabolism, but plasma fluoride levels are low and nontoxic. Patients taking isoniazid exhibit enhanced metabolism of enflurane with significantly elevated serum fluoride concentrations (Mazze et al., 1982). Clinical Use Surgical anesthesia can be induced with enflurane in less than 10 minutes with an inhaled concentration of 4% in oxygen. Anesthesia can be maintained with concentrations from 1.5% to 3%. As with other anesthetics, the enflurane concentrations required to produce anesthesia are reduced when it is coadministered with nitrous oxide or opioids. Use of enflurane has decreased substantially in recent years with the introduction of newer inhalational agents with preferable pharmacokinetic and side-effect profiles. Side Effects Cardiovascular System Enflurane causes a concentration-dependent decrease in arterial blood pressure. Hypotension is due, in part, to depression of myocardial contractility with some contribution from peripheral vasodilation (see Figure 146). Enflurane has minimal effects on heart rate and produces neither the bradycardia seen with halothane nor the tachycardia seen with isoflurane. Respiratory System The respiratory effects of enflurane are similar to those of halothane. Spontaneous ventilation with enflurane produces a pattern of rapid, shallow breathing. Minute ventilation is markedly decreased, and a PaCO of 60 mm Hg is seen with 1 MAC of enflurane (see Figure 147). Enflurane produces a greater depression of the ventilatory responses to hypoxia and hypercarbia than do either halothane or isoflurane (Hirshman et al., 1977). Enflurane, like other inhalational anesthetics, is an effective bronchodilator. Nervous System Enflurane is a cerebral vasodilator and thus can increase intracranial pressure in some patients. Like other inhalational anesthetics, enflurane reduces cerebral metabolic oxygen consumption. Enflurane has an unusual property of producing electrical seizure activity. High concentrations of enflurane or profound hypocarbia during enflurane anesthesia result in a characteristic high-voltage, high-frequency electroencephalographic (EEG) pattern, which progresses to spike-and-dome complexes. The spike-and-dome pattern can be punctuated by frank seizure activity, which may or may not be accompanied by peripheral motor manifestations of seizure activity. The seizures are self-limited and are not thought to produce permanent damage. Enflurane is not thought to precipitate seizures in epileptic patients. Nonetheless, enflurane is generally not used in patients with seizure disorders. Muscle Enflurane produces significant skeletal muscle relaxation in the absence of muscle relaxants. It also significantly enhances the effects of nondepolarizing muscle relaxants. As with other inhalational agents, enflurane relaxes uterine smooth muscle. It thus is not widely used for obstetrical anesthesia. Kidney Like other inhalational anesthetics, enflurane reduces renal blood
flow, glomerular filtration rate, and urinary output. These effects are
rapidly reversed with discontinuation of the drug. Enflurane metabolism
produces significant plasma levels of fluoride ions (20 to 40 Liver and Gastrointestinal Tract Enflurane reduces splanchnic and hepatic blood flow in proportion to reduced arterial blood pressure. Enflurane does not appear to alter liver function or to be hepatoxic. Desflurane Chemistry and Physical Properties Desflurane SUPRANE) is difluoromethyl 1-fluoro-2,2,2-trifluoromethyl ether (see Figure 144). It is a highly volatile liquid at room temperature (vapor pressure = 681 mm Hg) and thus must be stored in tightly sealed bottles. Delivery of a precise concentration of desflurane requires the use of a specially heated vaporizer that delivers pure vapor that is then diluted appropriately with other gases (oxygen, air, nitrous oxide). Desflurane is nonflammable and nonexplosive in mixtures of air or oxygen. Pharmacokinetics Desflurane has a very low blood:gas partition coefficient (0.42) and also is not very soluble in fat or other peripheral tissues (see Table 141). For this reason, the alveolar (and blood) concentration rapidly rises to the level of inspired concentration. Indeed, within five minutes of administration, the alveolar concentration reaches 80% of the inspired concentration. This provides for a very rapid induction of anesthesia and for rapid changes in depth of anesthesia following changes in the inspired concentration. Emergence from anesthesia also is very rapid with desflurane. The time to awakening following desflurane is half as long as with halothane or sevoflurane and usually does not exceed 5 to 10 minutes (Smiley et al., 1991). Desflurane is metabolized to a minimal extent, and more than 99% of absorbed desflurane is eliminated unchanged via the lungs. A small amount of absorbed desflurane is oxidatively metabolized by hepatic cytochrome P450 enzymes. Virtually no serum fluoride ions are detectable in serum after desflurane administration, but low concentrations of trifluoroacetic acid are detectable in serum and urine (Koblin et al., 1988). Clinical Use Desflurane is a widely used anesthetic for outpatient surgery because of its rapid onset of action and rapid recovery. Desflurane is irritating to the airway in awake patients and can provoke coughing, salivation, and bronchospasm. Anesthesia therefore usually is induced with an intravenous agent, with desflurane subsequently administered for maintenance of anesthesia. Maintenance of anesthesia usually requires inhaled concentrations of 6% to 8%. Lower concentrations of desflurane are required if it is coadministered with nitrous oxide or opioids. Side Effects Cardiovascular System Desflurane, like all inhalational anesthetics, causes a
concentration-dependent decrease in blood pressure. Desflurane has a very
modest negative inotropic effect and produces hypotension primarily by
decreasing systemic vascular resistance ( Respiratory System Similar to halothane and enflurane, desflurane causes a concentration-dependent increase in respiratory rate and a decrease in tidal volume. At low concentrations (less than 1 MAC) the net effect is to preserve minute ventilation. At desflurane concentrations greater than 1 MAC, minute ventilation is markedly depressed, resulting in elevated arterial carbon dioxide tension (see Figure 147) (Lockhart et al., 1991). Patients spontaneously breathing desflurane at concentrationsgreater than 1.5 MAC will have extreme elevations of arterial carbon dioxide tension and may become apneic. Desflurane, like other inhalational agents, is a bronchodilator. It is also a strong airway irritant, however, and can cause coughing, breath-holding, laryngospasm, and excessive respiratory secretions. Because of its irritant properties, desflurane is not used for induction of anesthesia. Nervous System Desflurane decreases cerebral vascular resistance and cerebral metabolic oxygen consumption. Under conditions of normocapnia and normotension, desflurane produces an increase in cerebral blood flow and can increase intracranial pressure in patients with poor intracranial compliance. The vasoconstrictive response to hypocapnia is preserved during desflurane anesthesia, and increases in intracranial pressure thus can be prevented by hyperventilation. Muscle Desflurane produces direct skeletal muscle relaxation as well as enhancing the effects of nondepolarizing and depolarizing neuromuscular blocking agents (Caldwell et al., 1991). Kidney Desflurane has no reported nephrotoxicity. This is consistent with its minimal metabolic degradation. Liver and Gastrointestinal Tract Desflurane is not known to affect liver function tests or to cause hepatotoxicity. Sevoflurane Chemistry and Physical Properties Sevoflurane ULTANE) is fluoromethyl 2,2,2-trifluoro-1-[trifluoromethyl]ethyl ether (see Figure 144). It is a clear, colorless, volatile liquid at room temperature and must be stored in a sealed bottle. It is nonflammable and nonexplosive in mixtures of air or oxygen. Pharmacokinetics The low solubility of sevoflurane in blood and other tissues provides for rapid induction of anesthesia, rapid changes in anesthetic depth following changes in delivered concentration, and rapid emergence following discontinuation of administration (see Table 141). Approximately 3% of absorbed sevoflurane is biotransformed. Sevoflurane is metabolized in the liver by cytochrome P450 2E1, with the predominant product being hexafluoroisopropanol (Kharasch et al., 1995). Hepatic metabolism of sevoflurane also produces inorganic fluoride. Serum fluoride concentrations reach a peak shortly after surgery and decline rapidly. Interaction of sevoflurane with soda lime also produces decomposition products. The major product of interest is referred to as compound A and is pentafluoroisopropenyl fluoromethyl ether (see'Side Effects''Kidney,' below) (Hanaki et al., 1987). Clinical Use Sevoflurane has been widely used in Side Effects Cardiovascular System Sevoflurane, like all other halogenated inhalational anesthetics, produces a concentration-dependent decrease in arterial blood pressure. This hypotensive effect primarily is due to systemic vasodilation, although sevoflurane also produces a concentration-dependent decrease in cardiac output (see Figure 146). Unlike isoflurane or desflurane, sevoflurane does not produce tachycardia and thus may be a preferable agent in patients prone to myocardial ischemia. Respiratory System Sevoflurane produces a concen-tration-dependent reduction in tidal volume and increase in respiratory rate in spontaneously breathing patients. The increased respiratory frequency is not adequate to compensate for reduced tidal volume, with the net effect being a reduction in minute ventilation and an increase in arterial carbon dioxide tension (Doi and Ikeda, 1987) (see Figure 147). Sevoflurane is not irritating to the airway and is a potent bronchodilator. Because of this combination of properties, sevoflurane is the most effective clinical bronchodilator of the inhalational anesthetics (Rooke et al., 1997). Nervous System Sevoflurane produces effects on cerebral vascular resistance, cerebral metabolic oxygen consumption, and cerebral blood flow that are very similar to those produced by isoflurane and desflurane. While sevoflurane thus can increase intracranial pressure in patients with poor intracranial compliance, the response to hypocapnia is preserved during sevoflurane anesthesia, and increases in intracranial pressure thus can be prevented by hyperventilation. Muscle Sevoflurane produces direct skeletal muscle relaxation as well as enhancing the effects of nondepolarizing and depolarizing neuromuscular blocking agents. Its effects are similar to those of other halogenated inhalational anesthetics. Kidney Controversy has surrounded the potential nephrotoxicity of compound A, the degradation product produced by interaction of sevoflurane with the carbon dioxide absorbant soda lime. There has been a report showing transient biochemical evidence of renal injury in studies with human volunteers but no evidence of permanent renal injury (Eger et al., 1997). Large clinical studies have showed no evidence of increased serum creatinine, blood urea nitrogen, or any other evidence of renal impairment following sevoflurane administration (Mazze et al., 2000). The current recommendation of the U.S. Food and Drug Administration is that sevoflurane be administered with fresh gas flows of at least 2 liters/minute to minimize accumulation of compound A. Liver and Gastrointestinal Tract Sevoflurane is not known to cause hepatotoxicity or alterations of hepatic function tests. Nitrous Oxide Chemical and Physical Properties Nitrous oxide (dinitrogen monoxide; N2O) is a colorless, odorless gas at room temperature (see Figure 144). It is sold in steel cylinders and must be delivered through calibrated flow meters provided on all anesthesia machines. Nitrous oxide is neither flammable nor explosive, but it does support combustion as actively as oxygen does when it is present in proper concentration with a flammable anesthetic or material. Pharmacokinetics Nitrous oxide is very insoluble in blood and other tissues (see Table 141). This results in rapid equilibration between delivered and alveolar anesthetic concentrations and provides for rapid induction of anesthesia and rapid emergence following discontinuation of administration. The rapid uptake of nitrous oxide from alveolar gas serves to concentrate coadministered halogenated anesthetics; this effect (the 'second gas effect') speeds induction of anesthesia. On discontinuation of nitrous oxide administration, nitrous oxide gas can diffuse from blood to the alveoli, diluting oxygen in the lung. This can produce an effect called diffusional hypoxia. To avoid hypoxia, 100% oxygen rather than air should be administered when nitrous oxide is discontinued. Nitrous oxide is almost completely eliminated by the lungs, with some minimal diffusion through the skin. Nitrous oxide is not biotransformed by enzymatic action in human tissue, and 99.9% of absorbed nitrous oxide is eliminated unchanged. Nitrous oxide can be degraded by interaction with vitamin B12 in intestinal bacteria. This results in inactivation of methionine synthesis and can produce signs of vitamin B12 deficiency (megaloblastic anemia, peripheral neuropathy) following long-term nitrous oxide administration (O'Sullivan et al., 1981). For this reason, nitrous oxide is not used as a chronic analgesic or as a sedative in critical care settings. Clinical Use Nitrous oxide is a weak anesthetic agent and produces reliable surgical anesthesia only under hyperbaric conditions. It does produce significant analgesia at concentrations as low as 20% and usually produces sedation in concentrations between 30% and 80%. It is used frequently in concentrations of approximately 50% to provide analgesia and sedation in outpatient dentistry. Nitrous oxide cannot be used at concentrations above 80%, because this limits the delivery of an adequate amount of oxygen. Because of this limitation, nitrous oxide is used primarily as an adjunct to other inhalational or intravenous anesthetics. Nitrous oxide substantially reduces the requirement for inhalational anesthetics. For example, at 70% nitrous oxide, MAC for other inhalational agents is reduced by about 60%, allowing for lower concentrations of halogenated anesthetics and a lesser degree of side effects. One major problem with nitrous oxide is that it will exchange with nitrogen in any air-containing cavity in the body. Moreover, nitrous oxide will enter the cavity faster than nitrogen escapes, thereby increasing the volume and/or pressure in this cavity. Examples of air collections that can be expanded by nitrous oxide include a pneumothorax, an obstructed middle ear, an air embolus, an obstructed loop of bowel, an intraocular air bubble, a pulmonary bulla, and intracranial air. Nitrous oxide should be avoided in these clinical settings. Side Effects Cardiovascular System Although nitrous oxide produces a negative inotropic effect on heart muscle in vitro, depressant effects on cardiac function generally are not observed in patients. This is because of the stimulatory effects of nitrous oxide on the sympathetic nervous system. The cardiovascular effects of nitrous oxide also are heavily influenced by the concomitant administration of other anesthetic agents. When nitrous oxide is coadministered with halogenated inhalational anesthetics, it generally produces an increase in heart rate, arterial blood pressure, and cardiac output. In contrast, when nitrous oxide is coadministered with an opioid, it generally decreases arterial blood pressure and cardiac output. Nitrous oxide also increases venous tone in both the peripheral and pulmonary vasculature. The effects of nitrous oxide on pulmonary vascular resistance can be exaggerated in patients with preexisting pulmonary hypertension (Schulte-Sasse et al., 1982). Nitrous oxide, therefore, is not generally used in patients with pulmonary hypertension. Respiratory System Nitrous oxide causes modest increases in respiratory rate and decreases in tidal volume in spontaneously breathing patients. The net effect is that minute ventilation is not significantly changed and arterial carbon dioxide tension remains normal. However, even modest concentrations of nitrous oxide markedly depress the ventilatory response to hypoxia (Yacoub et al., 1975). Thus it is prudent to monitor arterial oxygen saturation directly in patients receiving or recovering from nitrous oxide. Nervous System When nitrous oxide is administered alone, it can produce significant increases in cerebral blood flow and intracranial pressure. When nitrous oxide is coadministered with intravenous anesthetic agents, increases in cerebral blood flow are attenuated or abolished. When nitrous oxide is added to a halogenated inhalational anesthetic, its vasodilatory effect on the cerebral vasculature is slightly reduced. Muscle Nitrous oxide does not relax skeletal muscle and does not enhance the effects of neuromuscular blocking drugs. Unlike the halogenated anesthetics, nitrous oxide is not a triggering agent for malignant hyperthermia. Kidney, Liver, and Gastrointestinal Tract Nitrous oxide is not known to produce any changes in renal or hepatic function and is neither nephrotoxic nor hepatotoxic. Xenon Xenon is an inert gas that was first identified as an anesthetic agent
in 1951 (Cullen and Gross, 1951). It is not approved for use in the Xenon is extremely insoluble in blood and other tissues, providing for rapid induction and emergence from anesthesia (see Table 141). It is sufficiently potent to produce surgical anesthesia when administered with 30% oxygen. Most importantly, xenon has minimal side effects. It has no effects on cardiac output or cardiac rhythm and is not thought to have a significant effect on systemic vascular resistance. It also does not affect pulmonary function and is not known to have any hepatic or renal toxicity. Finally, xenon is not metabolized at all in the human body. Xenon is an anesthetic that may be available in the future if limitations on its availability and its high cost can be overcome. |
Anesthetic Adjuncts
|
General anesthetics rarely are given as the sole agent. Rather, anesthetic adjuncts usually are used to augment specific components of anesthesia, permitting lower doses of general anesthetics with fewer side effects. Because they are such an integral part of general anesthetic drug regimens, why and how they are utilized as anesthetic adjuncts will be described briefly here. The detailed pharmacology of each drug is covered in other chapters. Benzodiazepines Benzodiazepines (see Chapter 17: Hypnotics and Sedatives) are commonly used for sedation rather than general anesthesia because of the prolonged amnesia and sedation that may result from anesthetizing doses. As adjuncts, benzodiazepines are used for anxiolysis, amnesia, and sedation prior to induction of anesthesia or for sedation during procedures not requiring general anesthesia. The benzodiazepine most frequently used in the perioperative period is midazolam (VERSED) followed distantly by diazepam (VALIUM) and lorazepam (ATIVAN). Midazolam is water soluble and is typically administered intravenously but also can be given orally, intramuscularly, or rectally; oral midazolam is particularly useful for sedation of young children. Midazolam produces minimal venous irritation as opposed to diazepam and lorazepam, which are formulated in propylene glycol and are painful on injection, sometimes producing thrombophlebitis. Midazolam has the pharmacokinetic advantage, particularly over lorazepam, of being more rapid in onset and shorter in duration of effect. Sedative doses of midazolam (0.01 to 0.07 mg/kg intravenously) reach peak effect in about 2 minutes and provide sedation for about 30 minutes (Reves et al., 1985). Elderly patients tend to be more sensitive to and have a slower recovery from benzodiazepines (Jacobs et al., 1995); thus, titration of smaller doses in this age group is prudent. Midazolam is hepatically metabolized with a clearance (6 to 11 ml/min per kg) similar to that of methohexital and about 20 and 7 times higher than those of diazepam and lorazepam, respectively (Greenblatt et al., 1981; Reves et al., 1985). Either for prolonged sedation or for general anesthetic maintenance, midazolam is more suitable for infusion than are other benzodiazepines, although its duration of action does significantly increase with prolonged infusions (Figure 143). Benzodiazepines reduce both cerebral blood flow and metabolism but at equianesthetic doses are less potent in this respect than are barbiturates. They are effective anticonvulsants and are sometimes given to treat status epilepticus (Modica et al., 1990). Benzodiazepines modestly decrease blood pressure and respiratory drive, occasionally resulting in apnea (Reves et al., 1985). Thus, blood pressure and respiratory rate should be monitored in patients sedated with intravenous benzodiazepines. Analgesics With the exception of ketamine, neither parenteral nor currently
available inhalational anesthetics are effective analgesics. Thus, analgesics
typically are administered with general anesthetics to reduce anesthetic
requirement and minimize hemodynamic changes produced by painful stimuli.
Nonsteroidal antiinflammatory drugs, including cyclo-oxygenase-2
inhibitors or acetaminophen, sometimes provide adequate analgesia
for minor surgical procedures. However, because of the rapid and profound
analgesia produced, opioids are the primary analgesics used during the
perioperative period. Fentanyl SUBLIMAZE), sufentanil (SUFENTA), alfentanil (ALFENTA), remifentanil (ULTIVA), meperidine (DEMEROL), and morphine are the major parenteral opioids used in the
perioperative period. The
primary analgesic activity of each of these drugs is produced by agonist
activity at The choice of a perioperative opioid is based primarily on duration of
action, given that, at appropriate doses, all produce similar analgesia and
side effects. Remifentanil has an ultra-short duration of action ( During the perioperative period, opioids often are given at induction
to preempt responses to predictable painful stimuli (e.g.,
endotracheal intubation and surgical incision). Subsequent doses either by
bolus or infusion are titrated to the surgical stimulus and the patient's
hemodynamic response. Marked decreases in respiratory rate and heart rate
with much smaller reductions in blood pressure are seen to varying degrees
with all opioids (Bowdle, 1998). Muscle rigidity that can impair ventilation
sometimes accompanies larger doses of opioids. The incidence of sphincter of
Oddi spasm is increased with all opioids, although morphine appears to be
more potent in this regard (Hahn et al., 1988; Thune et al.,
1990). After emergence from anesthesia, the frequency and severity of nausea,
vomiting, and pruritus are increased by all opioids to about the same degree
(Watcha and White, 1992). A useful side effect of meperidine is its ability
to reduce shivering, a common problem during emergence from anesthesia (Pauca
et al., 1984); other opioids are not as efficacious against shivering
perhaps due to less Neuromuscular Blocking Agents The practical aspects of the use of neuromuscular blockers as anesthetic adjuncts are briefly described here. The detailed pharmacology of this drug class is presented in Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia. Depolarizing (e.g., succinylcholine) and nondepolarizing muscle relaxants (e.g., pancuronium) often are administered during the induction of anesthesia to relax muscles of the jaw, neck, and airway and thereby facilitate laryngoscopy and endotracheal intubation. As mentioned above, barbiturates will precipitate when mixed with muscle relaxants and should be allowed to clear from the intravenous line prior to injection of a muscle relaxant. Following induction, continued muscle relaxation is desirable for many procedures to aid surgical exposure and/or to provide additional insurance of immobility. Of course, muscle relaxants are not by themselves anesthetics and should not be used in lieu of adequate anesthetic depth. The action of nondepolarizing muscle relaxants is usually antagonized, once muscle paralysis is no longer desired, with an acetylcholine esterase inhibitor such as neostigmine or edrophonium (see Chapter 8: Anticholinesterase Agents) combined with a muscarinic receptor antagonist (see Chapter 7: Muscarinic Receptor Agonists and Antagonists) (e.g., glycopyrrolate or atropine to offset the muscarinic activation of the esterase inhibitors). Other than histamine release by some agents, nondepolarizing muscle relaxants have little in the way of side effects. However, succinylcholine has multiple serious side effects (bradycardia, hyperkalemia, severe myalgia) including induction of malignant hyperthermia in susceptible individuals (see Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia). |
Prospectus
|
Two drug classes, For some time, certain steroids have been known to produce sedation
and anesthesia. In 1971, the neurosteroid alphaxalone (ALTHESIN) was approved in Acknowledgment The authors wish to acknowledge Drs. Bryan E. Marshall and David E. Longnecker, the authors of this chapter in the ninth edition of Goodman and Gilman's The Pharmacological Basis of Therapeutics, some of whose material has been retained in this edition. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 2041
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved