| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Local Anesthetics
Overview
|
Local anesthetics prevent or relieve pain by interrupting nerve conduction. They bind to a specific receptor site within the pore of the Na+ channels in nerves and block ion movement through this pore. In general, their action is restricted to the site of application and rapidly reverses upon diffusion from the site of action in the nerve. The chemical and pharmacological properties of each drug determine its clinical use. Local anesthetics can be administered by a variety of routes, including topical, infiltration, field or nerve block, intravenous regional, spinal, or epidural, as dictated by clinical circumstances. This chapter covers the mechanism of action of various local anesthetics, their therapeutic use and routes of administration, and individual side effects. The frequency and voltage-dependence of local anesthetics also are properties of antiarrhythmic agents, discussed in Chapter 35: Antiarrhythmic Drugs. |
Local Anesthetics: Introduction
|
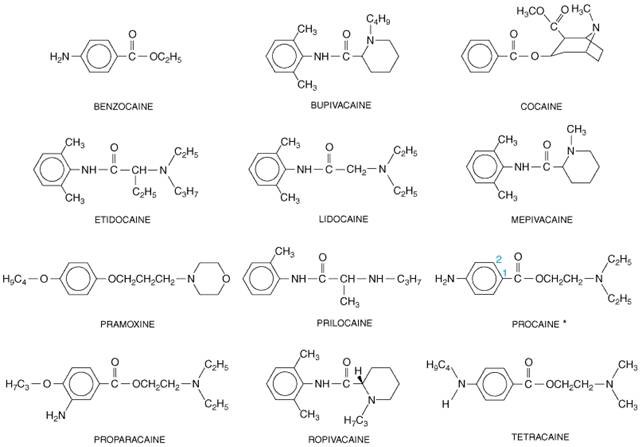
When applied locally to nerve tissue in appropriate concentrations, local anesthetics reversibly block the action potentials responsible for nerve conduction. They act on any part of the nervous system and on every type of nerve fiber. Thus, a local anesthetic in contact with a nerve trunk can cause both sensory and motor paralysis in the area innervated. The necessary practical advantage of the local anesthetics is that their action is reversible at clinically relevant concentrations; their use is followed by complete recovery in nerve function with no evidence of damage to nerve fibers or cells. History The first local anesthetic, cocaine, was serendipitously discovered to have anesthetic properties in the late nineteenth century. Cocaine occurs in abundance in the leaves of the coca shrub (Erythroxylon coca). For centuries, Andean natives have chewed an alkali extract of these leaves for its stimulatory and euphoric actions. Cocaine was first isolated in 1860 by Albert Niemann. He, like many chemists of that era, tasted his newly isolated compound and noted that it caused a numbing of the tongue. Sigmund Freud studied cocaine's physiological actions, and Carl Koller introduced cocaine into clinical practice in 1884 as a topical anesthetic for ophthalmological surgery. Shortly thereafter, Halstead popularized its use in infiltration and conduction block anesthesia. The many local anesthetics used in clinical practice today all stem from these early observations. Chemistry and StructureActivity Relationship Cocaine is an ester of benzoic acid and the complex alcohol 2-carbomethoxy, 3-hydroxy-tropane (Figure 151). Because of its toxicity and addictive properties (see Chapter 24: Drug Addiction and Drug Abuse), a search for synthetic substitutes for cocaine began in 1892 with the work of Einhorn and his colleagues. In 1905, this resulted in the synthesis of procaine, which became the prototype for local anesthetics for nearly half a century. The most widely used agents today are procaine, lidocaine, bupivacaine, and tetracaine.
Figure 151 shows that the structure of typical local anesthetics contains hydrophilic and hydrophobic moieties that are separated by an intermediate ester or amide linkage. A broad range of compounds containing these minimal structural features can satisfy the requirements for action as local anesthetics. The hydrophilic group usually is a tertiary amine, but it also may be a secondary amine; the hydrophobic moiety must be aromatic. The nature of the linking group determines certain of the pharmacological properties of these agents. For example, local anesthetics with an ester link are hydrolyzed readily by plasma esterases. The structureactivity relationship and the physicochemical properties of local anesthetics have been reviewed by Courtney and Strichartz (1987). In brief, hydrophobicity increases both the potency and the duration of action of the local anesthetics. This arises because association of the drug at hydrophobic sites enhances the partitioning of the drug to its sites of action and decreases the rate of metabolism by plasma esterases and liver enzymes. In addition, the receptor site for these drugs on Na+ channels is thought to be hydrophobic (see below), so that receptor affinity for anesthetic agents is increased for more hydrophobic drugs. Hydrophobicity also increases toxicity, so that the therapeutic index actually is decreased for more hydrophobic drugs. Molecular size also influences the rate of dissociation of local anesthetics from their receptor sites (Courtney and Strichartz, 1987). Smaller drug molecules can escape from the receptor site more rapidly. This characteristic is important in rapidly firing tissues, in which local anesthetics bind during action potentials and dissociate during the period of membrane repolarization. Rapid binding of local anesthetics during action potentials allows the frequency- and voltage-dependence of their action (see below). Mechanism of Action Local anesthetics prevent the generation and the conduction of the nerve impulse. Their primary site of action is the cell membrane. Conduction block can be demonstrated in squid giant axons from which the axoplasm has been removed. Local anesthetics block conduction by decreasing or preventing the large transient increase in the permeability of excitable membranes to Na+ that normally is produced by a slight depolarization of the membrane (see Chapter 12: Neurotransmission and the Central Nervous System and Strichartz and Ritchie, 1987). This action of local anesthetics is due to their direct interaction with voltage-gated Na+ channels. As the anesthetic action progressively develops in a nerve, the threshold for electrical excitability gradually increases, the rate of rise of the action potential declines, impulse conduction slows, and the safety factor for conduction decreases. These factors decrease the probability of propagation of the action potential, and nerve conduction eventually fails. In addition to Na+ channels, local anesthetics can bind to other membrane proteins (see Butterworth and Strichartz, 1990). In particular, they can block K+ channels (see Strichartz and Ritchie, 1987). However, since the interaction of local anesthetics with K+ channels requires higher concentrations of drug, blockade of conduction is not accompanied by any large or consistent change in resting membrane potential. Quaternary analogs of local anesthetics block conduction when applied internally to perfused giant axons of squid, but they are relatively ineffective when applied externally. These observations suggest that the site at which local anesthetics act, at least in their charged form, is accessible only from the inner surface of the membrane (Narahashi and Frazier, 1971; Strichartz and Ritchie, 1987). Therefore, local anesthetics applied externally first must cross the membrane before they can exert a blocking action. Although a variety of physicochemical models have been proposed to
explain how local anesthetics achieve conduction block (see Courtney
and Strichartz, 1987), it now is generally accepted that the major mechanism
of action of these drugs involves their interaction with one or more specific
binding sites within the Na+ channel (see Butterworth and
Strichartz, 1990). Biochemical, biophysical, and molecular biological
investigations during the past two decades have led to a rapid expansion of
knowledge about the structure and function of the Na+ channel and
other voltage-gated ion channels (see Catterall, 2000, and Chapter 12:
Neurotransmission and the Central Nervous System). The Na+
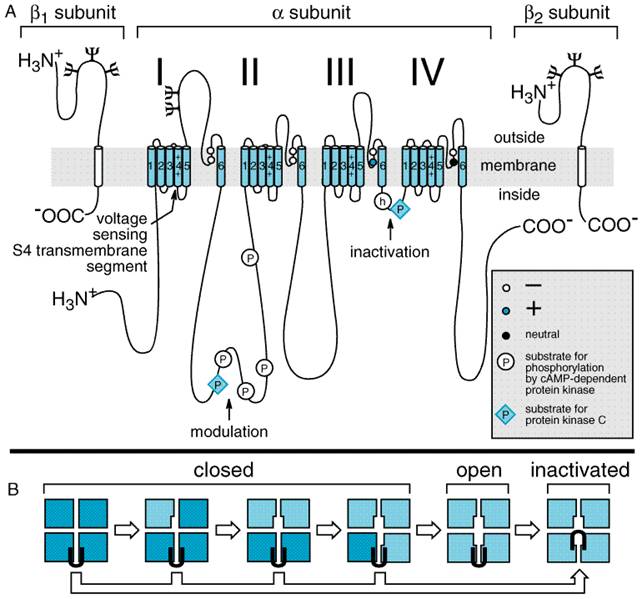
channels of the mammalian brain are heterotrimeric complexes of glycosylated
proteins with an aggregate molecular size in excess of 300,000 daltons; the
individual subunits are designated
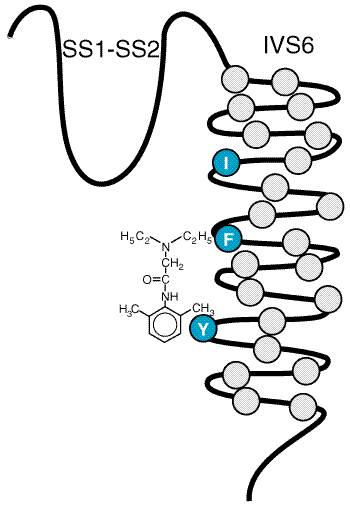
The transmembrane pore of the Na+ channel is thought to be surrounded by the S5 and S6 transmembrane helices and the short membrane-associated segments between them, designated SS1 and SS2. Amino acid residues in these short segments are the most critical determinants of the ion conductance and selectivity of the channel. After it opens, the Na+ channel inactivates within a few milliseconds due to closure of an inactivation gate. This functional gate is formed by the short intracellular loop of protein that connects homologous domains III and IV (Figure 152). The loop folds over the intracellular mouth of the transmembrane pore during the process of inactivation. It may bind to an inactivation gate 'receptor' formed by the intracellular mouth of the pore. Amino acid residues that are important for local anesthetic binding are found in the S6 segment in domain IV (Ragsdale et al., 1994). Hydrophobic amino acid residues near the center and the intracellular end of the S6 segment may interact directly with bound local anesthetics (Figure 153). Experimental mutation of a large hydrophobic amino acid residue (isoleucine) to a smaller one (alanine) near the extracellular end of this segment creates a pathway for access of charged local anesthetic drugs from the extracellular solution to the receptor site. These findings place the local anesthetic receptor site within the intracellular half of the transmembrane pore of the Na+ channel, with part of its structure contributed by amino acids in the S6 segment of domain IV. Frequency- and Voltage-Dependence of Local Anesthetic Action The degree of block produced by a given concentration of local anesthetic depends on how the nerve has been stimulated and on its resting membrane potential. Thus, a resting nerve is much less sensitive to a local anesthetic than one that is repetitively stimulated; higher frequency of stimulation and more positive membrane potential cause a greater degree of anesthetic block. These frequency- and voltage-dependent effects of local anesthetics occur because the local anesthetic molecule in its charged form gains access to its binding site within the pore only when the Na+ channel is in an open state and because the local anesthetic binds more tightly to and stabilizes the inactivated state of the Na+ channel (see Courtney and Strichartz, 1987; Butterworth and Strichartz, 1990). Local anesthetics exhibit these properties to different extents depending on their pKa, lipid solubility, and molecular size. In general, the frequency dependence of local anesthetic action depends critically on the rate of dissociation from the receptor site in the pore of the Na+ channel. A high frequency of stimulation is required for rapidly dissociating drugs so that drug binding during the action potential exceeds drug dissociation between action potentials. Dissociation of smaller and more hydrophobic drugs is more rapid, so a higher frequency of stimulation is required to yield frequency-dependent block. Frequency-dependent block of ion channels is most important for the actions of antiarrhythmic drugs. (see Chapter 35: Antiarrhythmic Drugs). Differential Sensitivity of Nerve Fibers to Local Anesthetics Although there is great individual variation, for most patients
treatment with local anesthetics causes the sensation of pain to disappear
first followed by the sensations of temperature, touch, deep pressure, and
finally motor function (Table 151). Classical experiments with intact nerves
showed that the The precise mechanisms responsible for this apparent specificity of
local anesthetic action on pain fibers are not known, but several factors may
contribute. The initial hypothesis from the classical work on intact nerves
was that sensitivity to local anesthetic block decreases with increasing
fiber size, consistent with high sensitivity for pain sensation mediated by
small fibers and low sensitivity for motor function mediated by large fibers
(Gasser and Erlanger, 1929). However, when nerve fibers are dissected from
nerves to allow direct measurement of action potential generation, no clear
correlation of the concentration dependence of local anesthetic block with
fiber diameter is observed (Franz and Perry, 1974; Fink and Cairns, 1984; Huang
et al., 1997). Therefore, it is unlikely that the fiber size per se
determines the sensitivity to local anesthetic block under steady-state
conditions. However, the spacing of nodes of Ranvier increases with the size
of nerve fibers. Because a fixed number of nodes must be blocked to prevent
conduction, small fibers with closely spaced nodes of Ranvier may be blocked
more rapidly during treatment of intact nerves, because the local anesthetic
reaches a critical length of nerve more rapidly (Franz and Perry, 1974).
Differences in tissue barriers and location of smaller C fibers and A Effect of Ph Local anesthetics tend to be only slightly soluble as unprotonated amines. Therefore, they are generally marketed as water-soluble salts, usually hydrochlorides. Inasmuch as the local anesthetics are weak bases (typical pKa values range from 8 to 9), their hydrochloride salts are mildly acidic. This property increases the stability of the local anesthetic esters and any accompanying vasoconstrictor substance. Under usual conditions of administration, the pH of the local anesthetic solution rapidly equilibrates to that of the extracellular fluids. Although the unprotonated species of the local anesthetic is necessary for diffusion across cellular membranes, it is the cationic species that interacts preferentially with Na+ channels. This conclusion has been supported by the results of experiments on anesthetized mammalian nonmyelinated fibers (Ritchie and Greengard, 1966). In these experiments, conduction could be blocked or unblocked merely by adjusting the pH of the bathing medium to 7.2 or 9.6, respectively, without altering the amount of anesthetic present. The primary role of the cationic form also has been demonstrated clearly by Narahashi and colleagues, who perfused the extracellular and axoplasmic surface of the giant squid axon with tertiary and quaternary amine local anesthetics (Narahashi and Frazier, 1971). However, the unprotonated molecular forms also possess some anesthetic activity (Butterworth and Strichartz, 1990). Prolongation of Action by Vasoconstrictors The duration of action of a local anesthetic is proportional to the
time during which it is in contact with nerve. Consequently, maneuvers that
keep the drug at the nerve prolong the period of anesthesia. Cocaine itself
constricts blood vessels by potentiating the action of norepinephrine (see
Chapters 6 and 10), thereby preventing its own absorption. In clinical
practice, preparations of local anesthetics often contain a vasoconstrictor,
usually epinephrine. The vasoconstrictor performs a dual service. By
decreasing the rate of absorption, it not only localizes the anesthetic at
the desired site but also allows the rate at which it is destroyed in the
body to keep pace with the rate at which it is absorbed into the circulation.
This reduces its systemic toxicity. It should be noted, however, that
epinephrine also dilates skeletal muscle vascular beds through actions at Some of the vasoconstrictor agent may be absorbed systemically, occasionally to an extent sufficient to cause untoward reactions (see below). There also may be delayed wound healing, tissue edema, or necrosis after local anesthesia. These effects seem to occur partly because sympathomimetic amines increase the oxygen consumption of the tissue; this, together with the vasoconstriction, leads to hypoxia and local tissue damage. The use of vasoconstrictors in local-anesthetic preparations for anatomical regions with limited collateral circulation could produce irreversible hypoxic damage, tissue necrosis, and gangrene and therefore is contraindicated. Undesired Effects of Local Anesthetics In addition to blocking conduction in nerve axons in the peripheral
nervous system, local anesthetics interfere with the function of all organs
in which conduction or transmission of impulses occurs. Thus, they have
important effects on the central nervous system (CNS), the autonomic ganglia,
the neuromuscular junction, and all forms of muscle (for review see Covino,
1987; Central Nervous System Following absorption, local anesthetics may cause stimulation of the CNS, producing restlessness and tremor that may proceed to clonic convulsions. In general, the more potent the anesthetic, the more readily convulsions may be produced. Alterations of CNS activity are thus predictable from the local anesthetic agent in question and the blood concentration achieved. Central stimulation is followed by depression; death usually is caused by respiratory failure. The apparent stimulation and subsequent depression produced by applying local anesthetics to the CNS presumably is due solely to depression of neuronal activity; a selective depression of inhibitory neurons is thought to account for the excitatory phase in vivo. Rapid systemic administration of local anesthetics may produce death with no or only transient signs of CNS stimulation. Under these conditions, the concentration of the drug probably rises so rapidly that all neurons are depressed simultaneously. Airway control and support of respiration are essential features of treatment in the late stage of intoxication. Benzodiazepines or rapidly acting barbiturates administered intravenously are the drugs of choice for both the prevention and arrest of convulsions (see Chapter 17: Hypnotics and Sedatives). Although drowsiness is the most frequent complaint that results from the CNS actions of local anesthetics, lidocaine may produce dysphoria or euphoria and muscle twitching. Moreover, both lidocaine and procaine may produce a loss of consciousness that is preceded only by symptoms of sedation (see Covino, 1987). Whereas other local anesthetics also show the effect, cocaine has a particularly prominent effect on mood and behavior. These effects of cocaine and its potential for abuse are discussed in Chapter 24: Drug Addiction and Drug Abuse. Cardiovascular System Following systemic absorption, local anesthetics act on the cardiovascular system (see Covino, 1987). The primary site of action is the myocardium, where decreases in electrical excitability, conduction rate, and force of contraction occur. In addition, most local anesthetics cause arteriolar dilation. Untoward cardiovascular effects usually are seen only after high systemic concentrations are attained and effects on the CNS are produced. However, on rare occasions, lower doses of some local anesthetics will cause cardiovascular collapse and death, probably due to either an action on the pacemaker or the sudden onset of ventricular fibrillation. It should be noted that ventricular tachycardia and fibrillation are relatively uncommon consequences of local anesthetics other than bupivacaine. The effects of local anesthetics such as lidocaine and procainamide, which also are used as antiarrhythmic drugs, are discussed in Chapter 35: Antiarrhythmic Drugs. Finally, it should be stressed that untoward cardiovascular effects of local anesthetic agents may result from their inadvertent intravascular administration, especially if epinephrine also is present. Smooth Muscle The local anesthetics depress contractions in the intact bowel and in strips of isolated intestine (see Zipf and Dittmann, 1971). They also relax vascular and bronchial smooth muscle, although low concentrations initially may produce contraction (see Covino, 1987). Spinal and epidural anesthesia, as well as instillation of local anesthetics into the peritoneal cavity, cause sympathetic nervous system paralysis, which can result in increased tone of gastrointestinal musculature (see below). Local anesthetics may increase the resting tone and decrease the contractions of isolated human uterine muscle; however, uterine contractions seldom are depressed directly during intrapartum regional anesthesia. Neuromuscular Junction and Ganglionic Synapse Local anesthetics also affect transmission at the neuromuscular junction. Procaine, for example, can block the response of skeletal muscle to maximal motor-nerve volleys and to acetylcholine at concentrations where the muscle responds normally to direct electrical stimulation. Similar effects occur at autonomic ganglia. These effects are due to block of the ion channel of the acetylcholine receptor by high concentrations of the local anesthetics (Neher and Steinbach, 1978; Charnet et al., 1990). Hypersensitivity to Local Anesthetics Rare individuals are hypersensitive to local anesthetics. The reaction may manifest itself as an allergic dermatitis or a typical asthmatic attack (see Covino, 1987). It is important to distinguish allergic reactions from toxic side effects and from the effects of coadministered vasoconstrictors. Hypersensitivity seems to occur almost exclusively with local anesthetics of the ester type and frequently extends to chemically related compounds. For example, individuals sensitive to procaine also may react to structurally similar compounds (e.g., tetracaine) through reaction to a common metabolite. Although agents of the amide type are essentially free of this problem, solutions of such agents may contain preservatives such as methylparaben that may provoke an allergic reaction (Covino, 1987). Local anesthetic preparations containing a vasoconstrictor also may elicit allergic responses due to the sulfite contained in them. Metabolism of Local Anesthetics The metabolic fate of local anesthetics is of great practical importance, because their toxicity depends largely on the balance between their rates of absorption and elimination. As noted above, the rate of absorption of many anesthetics can be reduced considerably by the incorporation of a vasoconstrictor agent in the anesthetic solution. However, the rate of destruction of local anesthetics varies greatly, and this is a major factor in determining the safety of a particular agent. Since toxicity is related to the free concentration of drug, binding of the anesthetic to proteins in the serum and to tissues reduces the concentration of free drug in the systemic circulation and, consequently, reduces toxicity. For example, in intravenous regional anesthesia of an extremity, about half of the original anesthetic dose is still tissue bound 30 minutes after release of the tourniquet; the lungs also bind large quantities of local anesthetic (Arthur, 1987). Some of the common local anesthetics (e.g., tetracaine) are esters. They are hydrolyzed and inactivated primarily by a plasma esterase, probably plasma cholinesterase. The liver also participates in hydrolysis of local anesthetics. Since spinal fluid contains little or no esterase, anesthesia produced by the intrathecal injection of an anesthetic agent will persist until the local anesthetic agent has been absorbed into the circulation. The amide-linked local anesthetics are, in general, degraded by the
hepatic endoplasmic reticulum, the initial reactions involving N-dealkylation
and subsequent hydrolysis (Arthur, 1987). However, with prilocaine, the
initial step is hydrolytic, forming o-toluidine metabolites that can
cause methemoglobinemia. Caution is indicated in the extensive use of
amide-linked local anesthetics in patients with severe hepatic disease. The
amide-linked local anesthetics are extensively (55% to 95%) bound to plasma
proteins, particularly |
Cocaine
|
Chemistry As outlined in the introduction above, cocaine occurs in abundance in the leaves of the coca shrub and is an ester of benzoic acid and methylecgonine. Ecgonine is an amino alcohol base closely related to tropine, the amino alcohol in atropine. It has the same fundamental structure as the synthetic local anesthetics (see Figure 151). Pharmacological Actions and Preparations The clinically desired actions of cocaine are the blockade of nerve impulses, as a consequence of its local anesthetic properties, and local vasoconstriction, secondary to inhibition of local norepinephrine reuptake. Toxicity and its potential for abuse have steadily decreased the clinical uses of cocaine. Its high toxicity is due to block of catecholamine uptake in both the central and peripheral nervous systems. Its euphoric properties are due primarily to inhibition of catecholamine uptake, particularly dopamine, at central nervous system synapses. Other local anesthetics do not block the uptake of norepinephrine and do not produce the sensitization to catecholamines, vasoconstriction, or mydriasis characteristic of cocaine. Currently, cocaine is used primarily for topical anesthesia of the upper respiratory tract, where its combined vasoconstrictor and local anesthetic properties provide anesthesia and shrinking of the mucosa with a single agent. Cocaine hydrochloride is used as a 1%, 4%, or 10% solution for topical application. For most applications, the 1% or 4% preparation is preferred to reduce toxicity. Because of its abuse potential, cocaine is listed as a schedule II drug by the United States Drug Enforcement Agency. |
Lidocaine
|
Lidocaine XYLOCAINE, others), introduced in 1948, is now the most widely used local anesthetic. The chemical structure of lidocaine is shown in Figure 151. Pharmacological Actions The pharmacological actions that lidocaine shares with other local anesthetic drugs have been described. Lidocaine produces faster, more intense, longer-lasting, and more extensive anesthesia than does an equal concentration of procaine. Unlike procaine, it is an aminoethylamide and is the prototypical member of the amide class of local anesthetics. It is an alternative choice for individuals sensitive to ester-type local anesthetics. Absorption, Fate, and Excretion Lidocaine is absorbed rapidly after parenteral administration and from the gastrointestinal and respiratory tracts. Although it is effective when used without any vasoconstrictor, in the presence of epinephrine the rate of absorption and the toxicity are decreased, and the duration of action usually is prolonged. In addition to preparations for injection, an iontophoretic, needle-free drug-delivery system for a lidocaine and epinephrine solution (IONTOCAINE) is available. This system generally is used for dermal procedures and provides anesthesia to a depth of up to 10 mm. Lidocaine is dealkylated in the liver by mixed-function oxidases to monoethylglycine xylidide and glycine xylidide, which can be metabolized further to monoethylglycine and xylidide. Both monoethylglycine xylidide and glycine xylidide retain local anesthetic activity. In human beings, about 75% of the xylidide is excreted in the urine as the further metabolite 4-hydroxy-2,6-dimethylaniline (see Arthur, 1987). Toxicity The side effects of lidocaine seen with increasing dose include drowsiness, tinnitus, dysgeusia, dizziness, and twitching. As the dose increases, seizures, coma, and respiratory depression and arrest will occur. Clinically significant cardiovascular depression usually occurs at serum lidocaine levels that produce marked CNS effects. The metabolites monoethylglycine xylidide and glycine xylidide may contribute to some of these side effects. Clinical Uses Lidocaine has a wide range of clinical uses as a local anesthetic; it has utility in almost any application where a local anesthetic of intermediate duration is needed. Lidocaine also is used as an antiarrhythmic agent (see Chapter 35: Antiarrhythmic Drugs). |
Bupivacaine
|
Pharmacological Actions Bupivacaine MARCAINE, SENSORCAINE), introduced in 1963, is a widely used amide local anesthetic; its structure is similar to that of lidocaine except that the amine-containing group is a butyl piperidine (Figure 151). It is a potent agent capable of producing prolonged anesthesia. Its long duration of action plus its tendency to provide more sensory than motor block has made it a popular drug for providing prolonged analgesia during labor or the postoperative period. By taking advantage of indwelling catheters and continuous infusions, bupivacaine can be used to provide several days of effective analgesia. Toxicity Bupivacaine (and etidocaine, below) are more cardiotoxic than equieffective doses of lidocaine. Clinically, this is manifested by severe ventricular arrhythmias and myocardial depression after inadvertent intravascular administration of large doses of bupivacaine. The enhanced cardiotoxicity of bupivacaine probably is due to multiple factors. Lidocaine and bupivacaine both rapidly block cardiac Na+ channels during systole. However, bupivacaine dissociates much more slowly than does lidocaine during diastole, so a significant fraction of Na+ channels remains blocked at the end of diastole (at physiological heart rates) with bupivacaine (Clarkson and Hondeghem, 1985). Thus the block by bupivacaine is cumulative and substantially more than would be predicted by its local anesthetic potency. At least a portion of the cardiac toxicity of bupivacaine may be mediated centrally, as direct injection of small quantities of bupivacaine into the medulla can produce malignant ventricular arrhythmias (Thomas et al., 1986). Bupivacaine-induced cardiac toxicity can be very difficult to treat, and its severity is enhanced in the presence of acidosis, hypercarbia, and hypoxemia. |
Other Synthetic Local Anesthetics
|
The number of synthetic local anesthetics is so large that it is impractical to consider them all here. Some local anesthetic agents are too toxic to be given by injection. Their use is restricted to topical application to the eye (see Chapter 66: Ocular Pharmacology), the mucous membranes, or the skin (see Chapter 65: Dermatological Pharmacology). Many local anesthetics are suitable, however, for infiltration or injection to produce nerve block; some of them also are useful for topical application. The main categories of local anesthetics are given below; agents are listed alphabetically. Local Anesthetics Suitable for Injection Chloroprocaine Chloroprocaine NESACAINE), an ester local anesthetic introduced in 1952, is a chlorinated derivative of procaine (Figure 151). Its major assets are its rapid onset and short duration of action and its reduced acute toxicity due to its rapid metabolism (plasma half-life approximately 25 seconds). Enthusiasm for its use has been tempered by reports of prolonged sensory and motor block after epidural or subarachnoid administration of large doses. This toxicity appears to have been a consequence of low pH and the use of sodium metabisulfite as a preservative in earlier formulations. There are no reports of neurotoxicity with newer preparations of chloroprocaine, which contain calcium EDTA as the preservative, although these preparations also are not recommended for intrathecal administration. A higher-than-expected incidence of muscular back pain following epidural anesthesia with 2-chloroprocaine also has been reported (Stevens et al., 1993). This back pain is thought to be due to tetany in the paraspinus muscles, which may be a consequence of Ca+2 binding by the EDTA included as a preservative; the incidence of back pain appears to be related to the volume of drug injected and its use for skin infiltration. Etidocaine Etidocaine DURANEST), introduced in 1972, is a long-acting amino amide (Figure 151). Its onset of action is faster than that of bupivacaine and comparable to that of lidocaine, yet its duration of action is similar to that of bupivacaine. Compared to bupivacaine, etidocaine produces preferential motor blockade. Thus, while it is useful for surgery requiring intense skeletal muscle relaxation, its utility in labor or postoperative analgesia is limited. Its cardiac toxicity is similar to that of bupivacaine (see above). Mepivacaine Mepivacaine CARBOCAINE, others), introduced in 1957, is an intermediate-acting amino amide (Figure 151). Its pharmacological properties are similar to those of lidocaine. Mepivacaine, however, is more toxic to the neonate and thus is not used in obstetrical anesthesia. The increased toxicity of mepivacaine in the neonate is related to ion trapping of this agent because of the lower pH of neonatal blood and the pKa of mepivacaine rather than to its slower metabolism in the neonate. It appears to have a slightly higher therapeutic index in adults than does lidocaine. Its onset of action is similar to that of lidocaine and its duration slightly longer (about 20%) than that of lidocaine in the absence of a coadministered vasoconstrictor. Mepivacaine is not effective as a topical anesthetic. Prilocaine Prilocaine CITANEST) is an intermediate-acting amino amide (Figure 151). It has a pharmacological profile similar to that of lidocaine. The primary differences are that it causes little vasodilation and thus can be used without a vasoconstrictor, if desired, and its increased volume of distribution reduces its CNS toxicity, making it suitable for intravenous regional blocks (below). It is unique among the local anesthetics for its propensity to cause methemoglobinemia. This effect is a consequence of the metabolism of the aromatic ring to o-toluidine. Development of methemoglobinemia is dependent on the total dose administered, usually appearing after a dose of 8 mg/kg. In healthy persons, methemoglobinemia usually is not a problem. If necessary, it can be treated by the intravenous administration of methylene blue (1 to 2 mg/kg). Methemoglobinemia following prilocaine has limited its use in obstetrical anesthesia, because it complicates evaluation of the newborn. Also, methemoglobinemia is more common in neonates due to decreased resistance of fetal hemoglobin to oxidant stresses and the immaturity of enzymes in the neonate that convert methemoglobin back to the ferrous state. Ropivacaine The cardiac toxicity of bupivacaine stimulated interest in developing a less toxic, long-lasting local anesthetic. The result of that search was the development of a new amino ethylamide, ropivacaine (NAROPIN) (Figure 151), the S-enantiomer of 1-propyl-2',6'-pipecoloxylidide. The S-enantiomer was chosen because, like most local anesthetics with a chiral center, it has a lower toxicity than the R-isomer (McClure, 1996). This is presumably due to slower uptake, resulting in lower blood levels for a given dose. Ropivacaine is slightly less potent than bupivacaine in producing anesthesia. In several animal models, it appears to be less cardiotoxic than equieffective doses of bupivacaine. In clinical studies, ropivacaine appears to be suitable for both epidural and regional anesthesia, with a duration of action similar to that of bupivacaine. Interestingly, it seems to be even more motor-sparing than bupivacaine. Procaine Procaine NOVOCAIN), introduced in 1905, was the first synthetic local anesthetic and is an amino ester (Figure 151). While it formerly was used widely, its use now is confined to infiltration anesthesia and occasionally for diagnostic nerve blocks. This is because of its low potency, slow onset, and short duration of action. While its toxicity is fairly low, it is hydrolyzed in vivo to produce paraaminobenzoic acid, which inhibits the action of sulfonamides. Thus, large doses should not be administered to patients taking sulfonamide drugs. Tetracaine Tetracaine PONTOCAINE), introduced in 1932, is a long-acting amino ester (Figure 151). It is significantly more potent and has a longer duration of action than procaine. Tetracaine may exhibit increased systemic toxicity because it is more slowly metabolized than the other commonly used ester local anesthetics. Currently, it is widely used in spinal anesthesia when a drug of long duration is needed. Tetracaine also is incorporated into several topical anesthesic preparations. With the introduction of bupivacaine, tetracaine is rarely used in peripheral nerve blocks because of the large doses often necessary, its slow onset, and its potential for toxicity. Local Anesthetics Used Primarily to Anesthetize Mucous Membranes and Skin Some anesthetics are either too irritating or too ineffective to be applied to the eye. However, they are useful as topical anesthetic agents on the skin and/or mucous membranes. These preparations are effective in the symptomatic relief of anal and genital pruritus, poison ivy rashes, and numerous other acute and chronic dermatoses. They are sometimes combined with a glucocorticoid or antihistamine and are available in a number of proprietary formulations. Dibucaine NUPERCAINAL) is a quinoline derivative. Its toxicity resulted in its removal from the
Dyclonine hydrochloride DYCLONE) has a rapid onset of action and a duration of effect comparable to that of procaine. It is absorbed through the skin and mucous membranes. The compound is used as 0.5% or 1.0% solution for topical anesthesia during endoscopy, for oral mucositis pain following radiation or chemotherapy, and for anogenital procedures. Pramoxine hydrochloride ANUSOL, TRONOTHANE, others) is a surface anesthetic agent that is not a benzoate ester. Its distinct chemical structure (Figure 151) may help minimize the danger of cross-sensitivity reactions in patients allergic to other local anesthetics. Pramoxine produces satisfactory surface anesthesia and is reasonably well tolerated on the skin and mucous membranes. It is too irritating to be used on the eye or in the nose. Various preparations, usually containing 1% pramoxine, are available for topical application. Anesthetics of Low Solubility Some local anesthetics are poorly soluble in water and, consequently, too slowly absorbed to be toxic. They can be applied directly to wounds and ulcerated surfaces, where they remain localized for long periods of time, producing a sustained anesthetic action. Chemically, they are esters of paraaminobenzoic acid lacking the terminal amino group possessed by the previously described local anesthetics. The most important member of the series is benzocaine (ethyl aminobenzoate; AMERICAINE ANESTHETIC, others). Benzocaine is structurally similar to procaine; the difference is that it lacks the terminal diethylamino group (Figure 151). It is incorporated into a large number of topical preparations. Benzocaine has been reported to cause methemoglobinemia (see text concerning methemoglobinemia caused by prilocaine, above); consequently, dosing recommendations must be carefully followed. Local Anesthetics Largely Restricted to Ophthalmological Use Anesthesia of the cornea and conjunctiva can be obtained readily by topical application of local anesthetics. However, most of the local anesthetics described above are too irritating for ophthalmological use. The first local anesthetic used in ophthalmology, cocaine, has the severe disadvantages of producing mydriasis and corneal sloughing and has fallen out of favor. The two compounds used most frequently today are proparacaine (ALCAINE, OPHTHAINE, others) and tetracaine (Figure 151). In addition to being less irritating during administration, proparacaine has the added advantage of bearing little antigenic similarity to the other benzoate local anesthetics. Thus, it sometimes can be used in individuals sensitive to the amino ester local anesthetics. For use in ophthalmology, these local anesthetics are instilled a single drop at a time. If anesthesia is incomplete, successive drops are applied until satisfactory conditions are obtained. The duration of anesthesia is determined chiefly by the vascularity of the tissue; thus it is longest in normal cornea and least in inflamed conjunctiva. In the latter case, repeated instillations are necessary to maintain adequate anesthesia for the duration of the procedure. Long-term administration of topical anesthesia to the eye has been associated with retarded healing, pitting and sloughing of the corneal epithelium, and predisposition of the eye to inadvertent injury. Thus, these drugs should not be prescribed for self-administration. For drug delivery, pharmacokinetic, and toxicity issues unique to drugs for ophthalmic use, see Chapter 66: Ocular Pharmacology. Tetrodotoxin and Saxitoxin These toxins are two of the most potent poisons known; the minimal
lethal dose of each in the mouse is about 8 |
Clinical Uses of Local Anesthetics
|
Local anesthesia is the loss of sensation in a body part without the loss of consciousness or the impairment of central control of vital functions. It offers two major advantages. The first is that the physiological perturbations associated with general anesthesia are avoided; the second is that neurophysiological reponses to pain and stress can be modified beneficially. As discussed above, local anesthetics have the potential to produce deleterious side effects. The choice of a local anesthetic and care in its use are the primary determinants of such toxicity. There is a poor relationship between the amount of local anesthetic injected and peak plasma levels in adults. Furthermore, peak plasma levels vary widely depending on the area of injection. They are highest with interpleural or intercostal block and lowest with subcutaneous infiltration. Thus, recommended maximum doses serve only as general guidelines. The following discussion concerns the pharmacological and physiological consequences of the use of local anesthetics categorized by method of administration. A more comprehensive discussion of their use and administration is presented in standard anesthesiology texts (e.g., Cousins and Bridenbaugh, 1998). Topical Anesthesia Anesthesia of mucous membranes of the nose, mouth, throat, tracheobronchial tree, esophagus, and genitourinary tract can be produced by direct application of aqueous solutions of salts of many local anesthetics or by suspension of the poorly soluble local anesthetics. Tetracaine (2%), lidocaine (2% to 10%), and cocaine (1% to 4%) typically are used. Cocaine is used only in the nose, nasopharynx, mouth, throat, and ear. Cocaine has the unique advantage of producing vasoconstriction as well as anesthesia. The shrinking of mucous membranes decreases operative bleeding while improving surgical visualization. Comparable vasoconstriction can be achieved with other local anesthetics by the addition of a low concentration of a vasoconstrictor such as phenylephrine (0.005%). Epinephrine, topically applied, has no significant local effect and does not prolong the duration of action of local anesthetics applied to mucous membranes because of poor penetration. Maximal safe total dosages for topical anesthesia in a healthy 70-kg adult are 300 mg for lidocaine, 150 mg for cocaine, and 50 mg for tetracaine. Peak anesthetic effect following topical application of cocaine or lidocaine occurs within 2 to 5 minutes (3 to 8 minutes with tetracaine), and anesthesia lasts for 30 to 45 minutes (30 to 60 minutes with tetracaine). Anesthesia is entirely superficial; it does not extend to submucosal structures. This technique does not alleviate joint pain or discomfort from subdermal inflammation or injury. Local anesthetics are absorbed rapidly into the circulation following topical application to mucous membranes or denuded skin. Thus, it must be kept in mind that topical anesthesia always carries the risk of systemic toxic reactions. Systemic toxicity has occurred even following the use of local anesthetics to control discomfort associated with severe diaper rash in infants. Absorption is particularly rapid when local anesthetics are applied to the tracheobronchial tree. Concentrations in blood after instillation of local anesthetics into the airway are nearly the same as those that follow intravenous injection. Surface anesthetics for the skin and cornea have been described above. The introduction of an eutectic mixture of lidocaine (2.5%) and prilocaine (2.5%) (EMLA) bridges the gap between topical and infiltration anesthesia. The efficacy of this combination lies in the fact that the mixure of prilocaine and lidocaine has a melting point less than that of either compound alone, existing at room temperature as an oil that can penetrate intact skin. EMLA cream produces anesthesia to a maximum depth of 5 mm and is applied as a cream on intact skin under an occlusive dressing, which must be left in place for at least 1 hour. It is effective for procedures involving skin and superficial subcutaneous structures (e.g., venipuncture and skin graft harvesting). The component local anesthetics will be absorbed into the systemic circulation, potentially producing toxic effects (above). Guidelines are available to calculate the maximum amount of cream that can be applied and area of skin covered. It must not be used on mucous membranes or abraded skin, as rapid absorption across these surfaces may result in systemic toxicity. Infiltration Anesthesia Infiltration anesthesia is the injection of local anesthetic directly into tissue without taking into consideration the course of cutaneous nerves. Infiltration anesthesia can be so superficial as to include only the skin. It also can include deeper structures, including intraabdominal organs when these, too, are infiltrated. The duration of infiltration anesthesia can be approximately doubled
by the addition of epinephrine (5 The local anesthetics most frequently used for infiltration anesthesia are lidocaine (0.5% to 1.0%), procaine (0.5% to 1.0%), and bupivacaine (0.125% to 0.25%). When used without epinephrine, up to 4.5 mg/kg of lidocaine, 7 mg/kg of procaine, or 2 mg/kg of bupivacaine can be employed in adults. When epinephrine is added, these amounts can be increased by one-third. The advantage of infiltration anesthesia and other regional anesthetic techniques is that it is possible to provide satisfactory anesthesia without disruption of normal bodily functions. The chief disadvantage of infiltration anesthesia is that relatively large amounts of drug must be used to anesthetize relatively small areas. This is no problem with minor surgery. When major surgery is performed, however, the amount of local anesthetic that is required makes systemic toxic reactions likely. The amount of anesthetic required to anesthetize an area can be reduced significantly and the duration of anesthesia increased markedly by specifically blocking the nerves that innervate the area of interest. This can be done at one of several levels: subcutaneously, at major nerves, or at the level of the spinal roots. Field Block Anesthesia Field block anesthesia is produced by subcutaneous injection of a solution of local anesthetic in such a manner as to anesthetize the region distal to the injection. For example, subcutaneous infiltration of the proximal portion of the volar surface of the forearm results in an extensive area of cutaneous anesthesia that starts 2 to 3 cm distal to the site of injection. The same principle can be applied with particular benefit to the scalp, the anterior abdominal wall, and the lower extremity. The drugs used and the concentrations and doses recommended are the same as for infiltration anesthesia. The advantage of field block anesthesia is that less drug can be used to provide a greater area of anesthesia than when infiltration anesthesia is used. Knowledge of the relevant neuroanatomy obviously is essential for successful field block anesthesia. Nerve Block Anesthesia Injection of a solution of a local anesthetic into or about individual peripheral nerves or nerve plexuses produces even greater areas of anesthesia than do the techniques described above. Blockade of mixed peripheral nerves and nerve plexuses also usually anesthetizes somatic motor nerves, producing skeletal muscle relaxation, which is essential for some surgical procedures. The areas of sensory and motor block usually start several centimeters distal to the site of injection. Brachial plexus blocks are particularly useful for procedures on the upper extremity and shoulder. Intercostal nerve blocks are effective for anesthesia and relaxation of the anterior abdominal wall. Cervical plexus block is appropriate for surgery of the neck. Sciatic and femoral nerve blocks are useful for surgery distal to the knee. Other useful nerve blocks prior to surgical procedures include blocks of individual nerves at the wrist and at the ankle, blocks of individual nerves such as the median or ulnar at the elbow, and blocks of sensory cranial nerves. There are four major determinants of the onset of sensory anesthesia following injection near a nerve. These are the proximity of the injection to the nerve, concentration and volume of drug, the degree of ionization of the drug, and time. Local anesthetic is never intentionally injected into the nerve, as this would be painful and could lead to nerve damage. Instead, the anesthetic agent is deposited as close to the nerve as possible. Thus the local anesthetic must diffuse from the site of injection into the nerve, where it acts. The rate of diffusion will be determined chiefly by the concentration of the drug, its degree of ionization (as ionized local anesthetic diffuses more slowly), its hydrophobicity, and the physical characteristics of the tissue surrounding the nerve. Higher concentrations of local anesthetic will result in a more rapid onset of peripheral nerve block. The utility of using higher concentrations, however, is limited by systemic toxicity as well as direct neural toxicity of concentrated local anesthetic solutions. Local anesthetics with lower pKa values tend to have a more rapid onset of action for a given concentration, because more drug is uncharged at neutral pH. For example, the onset of action of lidocaine occurs in about 3 minutes; 35% of lidocaine is in the basic form at pH 7.4. In contrast, the onset of action of bupivacaine requires about 15 minutes; only 5% to 10% of bupivacaine is in the basic (uncharged) form at this pH. Increased hydrophobicity might be expected to speed onset by increased penetration into nerve tissue. However, it also will increase binding in tissue lipids. Furthermore, the more hydrophobic local anesthetics also are more potent (and toxic) and thus must be used at lower concentrations, decreasing the concentration gradient for diffusion. Tissue factors also play a role in determining the rate of onset of anesthetic effects. The amount of connective tissue that must be penetrated can be significant in a nerve plexus compared to isolated nerves and can serve to slow or even prevent adequate diffusion of local anesthetic to the nerve fibers. Duration of nerve block anesthesia depends on the physical
characteristics of the local anesthetic used and the presence or absence of
vasoconstrictors. Especially important physical characteristics are lipid
solubility and protein binding. In general, local anesthetics can be divided
into three categories: those with a short (20 to 45 minutes) duration of
action in mixed peripheral nerves, such as procaine; those with an
intermediate (60 to 120 minutes) duration of action, such as lidocaine and mepivacaine;
and those with a long (400 to 450 minutes) duration of action, such as bupivacaine,
etidocaine, ropivacaine, and tetracaine. Block duration of the
intermediate-acting local anesthetics such as lidocaine can be prolonged by
the addition of epinephrine (5 The types of nerve fibers that are blocked when a local anesthetic is injected about a mixed peripheral nerve depend on the concentration of drug used, nerve-fiber size, internodal distance, and frequency and pattern of nerve-impulse transmission (see above). Anatomical factors are similarly important. A mixed peripheral nerve or nerve trunk consists of individual nerves surrounded by an investing epineurium. The vascular supply is usually centrally located. When a local anesthetic is deposited about a peripheral nerve, it diffuses from the outer surface toward the core along a concentration gradient (DeJong, 1994; Winnie et al., 1977). Consequently, nerves in the outer mantle of the mixed nerve are blocked first. These fibers usually are distributed to more proximal anatomical structures than are those situated near the core of the mixed nerve and are often motor. If the volume and concentration of local anesthetic solution deposited about the nerve are adequate, the local anesthetic eventually will diffuse inwardly in amounts adequate to block even the most centrally located fibers. Lesser amounts of drug will block only nerves in the mantle and the smaller and more sensitive central fibers. Furthermore, since removal of local anesthetics occurs primarily in the core of a mixed nerve or nerve trunk, where the vascular supply is located, the duration of blockade of centrally located nerves is shorter than that of more peripherally situated fibers. Choice of local anesthetic, as well as the amount and concentration
administered, is determined by the nerves and the types of fibers to be
blocked, the duration of anesthesia required, and the size and health of the
patient. For blocks of 2 to 4 hours, lidocaine (1.0% to 1.5%) can be used in
the amounts recommended above (see'Infiltration
Anesthesia'). Mepivacaine (up to 7 mg/kg of a 1.0% to 2.0% solution)
provides anesthesia that lasts about as long as that from lidocaine. Bupivacaine
(2 to 3 mg/kg of a 0.25% to 0.375% solution) can be used when a longer
duration of action is required. Addition of 5 Peak concentrations of local anesthetics in blood depend on the amount
injected, the physical characteristics of the local anesthetic, and whether
or not epinephrine is used. They also are determined by the rate of blood
flow to the site of injection and the surface area exposed to local
anesthetic. This is of particular importance in the safe application of nerve
block anesthesia, as the potential for systemic reactions also is related to
peak free serum concentrations. For example, peak concentrations of lidocaine
in blood following injection of 400 mg without epinephrine for intercostal
nerve blocks average 7 Intravenous Regional Anesthesia (Bier's Block) This technique relies on using the vasculature to bring the local anesthetic solution to the nerve trunks and endings. In this technique, an extremity is exsanguinated with an Esmarch (elastic) bandage, and a proximally located tourniquet is inflated to 100 to 150 mm Hg above the systolic blood pressure. The Esmarch bandage is removed, and the local anesthetic is injected into a previously cannulated vein. Typically, complete anesthesia of the limb ensues within 5 to 10 minutes. Pain from the tourniquet and the potential for ischemic nerve injury limits tourniquet inflation to 2 hours or less. However, the tourniquet should remain inflated for at least 15 to 30 minutes to prevent toxic amounts of local anesthetic from entering the circulation following deflation. Lidocaine, 40 to 50 ml (0.5 ml/kg in children) of a 0.5% solution, without epinephrine, is the drug of choice for this technique. For intravenous regional anesthesia in adults using a 0.5% solution without epinephrine, the dose administered should not exceed 4 mg/kg. A few clinicians prefer prilocaine (0.5%) over lidocaine because of its higher therapeutic index. The attractiveness of this technique lies in its simplicity. Its primary disadvantages are that it can be used only for a few anatomical regions, sensation (that is, pain) returns quickly after tourniquet deflation, and premature release or failure of the tourniquet can produce toxic levels of local anesthetic (e.g., 50 ml of 0.5% lidocaine contains 250 mg of lidocaine). For the last reason and because their longer durations of action offer no advantages, the more cardiotoxic local anesthetics, bupivacaine and etidocaine, are not recommended for this technique. Intravenous regional anesthesia is used most often for surgery of the forearm and hand but can be adapted for the foot and distal leg. Spinal Anesthesia Spinal anesthesia follows the injection of local anesthetic into the cerebrospinal fluid (CSF) in the lumbar space. This technique was first performed in human beings and described by Bier in 1899. For a number of reasons, including the ability to produce anesthesia of a considerable fraction of the body with a dose of local anesthetic that produces negligible plasma levels, it still remains one of the most popular forms of anesthesia. In most adults, the spinal cord terminates above the second lumbar vertebra; between that point and the termination of the thecal sac in the sacrum, the lumbar and sacral roots are bathed in CSF. Thus, in this region, there is a relatively large volume of CSF within which to inject drug, thereby minimizing the potential for direct nerve trauma. A brief discussion of the physiological effects of spinal anesthesia and those features relating to the pharmacology of the local anesthetics used are presented here. The technical performance and extensive discussion of the physiological consequences of spinal anesthesia are beyond the scope of this text (see Greene and Brull, 1993; Cousins and Bridenbaugh, 1998). Physiological Effects of Spinal Anesthesia Most of the physiological side effects of spinal anesthesia are a consequence of the sympathetic blockade produced by local anesthetic block of the sympathetic fibers in the spinal nerve roots. A thorough understanding of these physiological effects is necessary for the safe and successful application of spinal anesthesia. Although some of them may be deleterious and require treatment, others can be beneficial for the patient or can improve operating conditions. Most sympathetic fibers leave the spinal cord between T1 and L2 (Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems, Figure 61). Although local anesthetic is injected below these levels in the lumbar portion of the dural sac, cephalad spread of the local anesthetic is seen with all but the smallest volumes injected. This cephalad spread is of considerable importance in the practice of spinal anesthesia and potentially is under the control of numerous variables, of which patient position and baricity (density of the drug relative to the density of the CSF) are the most important (Greene, 1983). The degree of sympathetic block is related to the height of sensory anesthesia; often the level of sympathetic blockade is several spinal segments higher, since the preganglionic sympathetic fibers are more sensitive to block by low concentrations of local anesthetic. The effects of sympathetic blockade involve both the actions (now partially unopposed) of the parasympathetic nervous system as well as the response of the unblocked portion of the sympathetic nervous system. Thus, as the level of sympathetic block ascends, the actions of the parasympathetic nervous system are increasingly dominant, and the compensatory mechanisms of the unblocked sympathetic nervous system are diminished. As most sympathetic nerve fibers leave the cord at T1 or below, few additional effects of sympathetic blockade are seen with cervical levels of spinal anesthesia. The consequences of sympathetic blockade will vary among patients as a function of age, physical conditioning, and disease state. Interestingly, sympathetic blockade during spinal anesthesia appears to be inconsequential in healthy children. Clinically, the most important effects of sympathetic blockade during
spinal anesthesia are on the cardiovascular system. At all but the lowest
levels of spinal blockade, some vasodilation will occur. Vasodilation is more
marked on the venous than on the arterial side of the circulation, resulting
in a pooling of blood in the venous capacitance vessels. At low levels of
spinal anesthesia in healthy patients, this reduction in circulating blood
volume is well tolerated. With an increasing level of block, this effect
becomes more marked and venous return becomes gravity-dependent. If venous
return decreases too much, cardiac output and organ perfusion precipitously
decline. Venous return can be increased by modest (10 to 15) head-down tilt
or by elevating the legs. At high levels of spinal blockade, the cardiac
accelerator fibers, which exit the spinal cord at T1 to T4, will be blocked.
This is detrimental in patients dependent on elevated sympathetic tone to
maintain cardiac output (e.g., during congestive heart failure or
hypovolemia), and it also removes one of the compensatory mechanisms
available to maintain organ perfusion during vasodilation. Thus, as the level
of spinal block ascends, the rate of cardiovascular compromise can accelerate
if not carefully observed and treated. Sudden asystole also can occur,
presumably because of loss of sympathetic innervation in the continued
presence of parasympathetic activity at the sinoatrial node (Caplan, et
al., 1988). In the usual clinical situation, blood pressure serves as a
surrogate marker for cardiac output and organ perfusion. Treatment of
hypotension usually is warranted when the blood pressure decreases to about
30% of resting values. Therapy is aimed at maintaining brain and
cardiac perfusion and oxygenation. To achieve these goals, administration of
oxygen, fluid infusion, manipulation of patient position as mentioned above,
and the administration of vasoactive drugs are all options. In particular,
patients typically are administered a bolus (500 to 1000 ml) of fluid prior
to the administration of spinal anesthesia in an attempt to prevent some of
the deleterious effects of spinal blockade. As the usual cause of hypotension
is decreased venous return, possibly complicated by decreased heart rate,
vasoactive drugs with preferential venoconstrictive and chronotropic
properties are preferred. For this reason ephedrine, 5 to 10 mg
intravenously, often is the drug of choice. In addition to the use of
ephedrine to treat deleterious effects of sympathetic blockade, direct-acting
A beneficial effect of spinal anesthesia partially mediated by the sympathetic nervous system is on the intestine. Sympathetic fibers originating from T5 to L1 inhibit peristalsis; thus, their blockade produces a small, contracted intestine. This, together with a flaccid abdominal musculature, produces excellent operating conditions for some types of bowel surgery. The effects of spinal anesthesia on the respiratory system mostly are mediated by effects on the skeletal musculature. Paralysis of the intercostal muscles will reduce a patient's ability to cough and clear secretions, which may be undesirable in a bronchitic or emphysematous patient and may produce dyspnea. It should be noted that respiratory arrest during spinal anesthesia is seldom due to paralysis of the phrenic nerves or to toxic levels of local anesthetic in the CSF of the fourth ventricle. It is much more likely to be due to medullary ischemia secondary to hypotension. Pharmacology of Spinal Anesthesia Currently in the Drug Baricity and Patient Position The baricity of the local anesthetic injected will determine the direction of migration within the dural sac. Hyperbaric solutions will tend to settle in the dependent portions of the sac, while hypobaric solutions will tend to migrate in the opposite direction. Isobaric solutions usually will stay in the vicinity where they were injected, diffusing slowly in all directions. Consideration of the patient position during and after the performance of the block and the choice of a local anesthetic of the appropriate baricity is crucial for a successful block during some surgical procedures. For example, a saddle (perineal) block is best performed with a hyperbaric anesthetic in the sitting position, with the patient remaining in that position until the anesthetic level has become 'fixed.' On the other hand, for a saddle block in the prone, jackknife position, a hypobaric local anesthetic is appropriate. Lidocaine and bupivacaine are marketed in both isobaric and hyperbaric preparations and, if desired, can be diluted with sterile, preservative-free water to make them hypobaric. Complications of Spinal Anesthesia Persistent neurological deficits following spinal anesthesia are extremely rare. Thorough evaluation of a suspected deficit should be performed in collaboration with a neurologist. Neurological sequelae can be both immediate and late. Possible causes include introduction of foreign substances (such as disinfectants or talc) into the subarachnoid space, infection, hematoma, or direct mechanical trauma. Aside from drainage of an abscess or hematoma, treatment usually is ineffective; thus, avoidance and careful attention to detail while performing spinal anesthesia are necessary. High concentrations of local anesthetic can cause irreversible block. After administration, local anesthetic solutions are diluted rapidly, quickly reaching nontoxic concentrations. However, there are several reports of transient or longer-lasting neurological deficits following lidocaine spinal anesthesia, particularly with 5% lidocaine (i.e., 180 mM) in 7.5% glucose (Hodgson et al., 1999). Spinal anesthesia sometimes is regarded as contraindicated in patients with preexisting disease of the spinal cord. No experimental evidence exists to support this hypothesis. Nonetheless, it is prudent to avoid spinal anesthesia in patients with progressive diseases of the spinal cord. However, spinal anesthesia may be very useful in patients with fixed, chronic spinal cord injury. A more common sequela following any lumbar puncture, including spinal anesthesia, is a postural headache with classic features. The incidence of headache decreases with increasing age of the patient and decreasing needle diameter. Headache following lumbar puncture must be thoroughly evaluated to exclude serious complications such as meningitis. Treatment usually is conservative, with bed rest and analgesics. If this approach fails, an epidural blood patch can be performed; this procedure usually is successful in alleviating postdural puncture headaches, although a second blood patch may be necessary. If two epidural blood patches are ineffective in relieving the headache, the diagnosis of postdural puncture headache should be reconsidered. Intravenous caffeine (500 mg as the benzoate salt administered over 4 hours) also has been advocated for the treatment of postdural puncture headache. However, the efficacy of caffeine is less than that of a blood patch, and relief usually is transient. Evaluation of Spinal Anesthesia Spinal anesthesia is a safe and effective technique. Its value is greatest during surgery involving the lower abdomen, the lower extremities, and the perineum. It often is combined with intravenous medication to provide sedation and amnesia. The physiological perturbations associated with low spinal anesthesia often have less potential harm than those associated with general anesthesia. The same does not apply for high spinal anesthesia. The sympathetic blockade that accompanies levels of spinal anesthesia adequate for mid- or upper-abdominal surgery, coupled with the difficulty in achieving visceral analgesia, is such that equally satisfactory and safer operating conditions can be realized by combining the spinal anesthetic with a 'light' general anesthetic or by the administration of a general anesthetic and a neuromuscular blocking agent. Epidural Anesthesia Epidural anesthesia is administered by injecting local anesthetic into the epidural spacethe space bounded by the ligamentum flavum posteriorly, the spinal periosteum laterally, and the dura anteriorly. Epidural anesthesia can be performed in the sacral hiatus (caudal anesthesia) or in the lumbar, thoracic, or cervical regions of the spine. Its current popularity arises from the development of catheters that can be placed into the epidural space, allowing either continuous infusions or repeated bolus administration of local anesthetics. The primary site of action of epidurally administered local anesthetics is on the spinal nerve roots. However, epidurally administered local anesthetics also may act on the spinal cord and on the paravertebral nerves. The selection of drugs available for epidural anesthesia is similar to
that for major nerve blocks. As for spinal anesthesia, the choice of drugs to
be used during epidural anesthesia is dictated primarily by the duration of
anesthesia desired. However, when an epidural catheter is placed,
short-acting drugs can be administered repeatedly, providing more control
over the duration of block. Bupivacaine, 0.5% to 0.75%, is used when a long
duration of surgical block is desired. Due to enhanced cardiotoxicity in
pregnant patients, the 0.75% solution is not approved for obstetrical use.
Lower concentrations0.25%, 0.125%, or 0.0625%of bupivacaine, often with 2 For each anesthetic agent, a relationship exists between the volume of local anesthetic injected epidurally and the segmental level of anesthesia achieved. For example, in 20- to 40-year-old, healthy, nonpregnant patients, each 1 to 1.5 ml of 2% lidocaine will give an additional segment of anesthesia. The amount needed will decrease with increasing age and also will be decreased during pregnancy and in children. The concentration of local anesthetic used determines the type of nerve fibers blocked. The highest concentrations are used when sympathetic, somatic sensory, and somatic motor blockade are required. Intermediate concentrations allow somatic sensory anesthesia without muscle relaxation. Low concentrations will block only preganglionic sympathetic fibers. As an example, with bupivacaine these effects might be achieved with concentrations of 0.5%, 0.25%, and 0.0625%, respectively. The total amounts of drug that can be injected with safety at one time are approximately those mentioned above under 'Nerve Block Anesthesia' and 'Infiltration Anesthesia.' Performance of epidural anesthesia requires a greater degree of skill than does spinal anesthesia. The technique of epidural anesthesia and the volumes, concentrations, and types of drugs used are described in detail in standard anesthesiology texts (e.g., see Cousins and Bridenbaugh, 1998). A significant difference between epidural and spinal anesthesia is
that the dose of local anesthetic used can produce high concentrations in
blood following absorption from the epidural space. Peak concentrations of lidocaine
in blood following injection of 400 mg (without epinephrine) into the lumbar
epidural space average 3 to 4 Another major difference between epidural and spinal anesthesia is
that there is no zone of differential sympathetic blockade with epidural
anesthesia; thus, the level of sympathetic block is close to the level of
sensory block. Because epidural anesthesia does not result in the zone of
differential sympathetic blockade that is observed during spinal anesthesia,
cardiovascular responses to epidural anesthesia might be expected to be less
prominent. In practice, however, this is not the case; this potential
advantage of epidural anesthesia is offset by the cardiovascular responses to
the high concentration of anesthetic in blood that occurs during epidural
anesthesia. This is most apparent when, as is often the case, epinephrine is
added to the epidural injection. The resulting concentration of epinephrine
in blood is sufficient to produce significant High concentrations of local anesthetics in blood during epidural anesthesia are of special importance when this technique is used to control pain during labor and delivery. Local anesthetics cross the placenta, enter the fetal circulation, and at high concentrations may cause depression of the neonate (Scanlon et al., 1974). The extent to which they do so is determined by dosage, acidbase status, the level of protein binding in both maternal and fetal blood (Tucker, et al., 1970), placental blood flow, and solubility of the agent in fetal tissue. These concerns have been lessened by the trend toward using more dilute solutions of bupivacaine for labor analgesia. Epidural and Intrathecal Opiate Analgesia Small quantities of opioid injected intrathecally or epidurally produce segmental analgesia (Yaksh and Rudy, 1976). This observation led to the clinical use of spinal and epidural opioids during surgical procedures and for the relief of postoperative and chronic pain (Cousins and Mather, 1984). As with local anesthesia, analgesia is confined to sensory nerves that enter the spinal cord dorsal horn in the vicinity of the injection. Presynaptic opioid receptors inhibit the release of substance P and other neurotransmitters from primary afferents, while postsynaptic opioid receptors decrease the activity of certain dorsal horn neurons in the spinothalamic tracts (Willcockson, et al., 1986; see also Chapters 6 and 23). Since conduction in autonomic, sensory, and motor nerves is not affected by the opioids, blood pressure, motor function, and nonnociceptive sensory perception typically are not influenced by spinal opioids. The volume-evoked micturition reflex is inhibited, implicating opioid receptors in this reflex pathway. Clinically, this is manifest by urinary retention. Other side effects include pruritus and nausea and vomiting in susceptible individuals. Delayed respiratory depression and sedation, presumably from cephalad spread of opioid within the CSF, occurs infrequently with the doses of opioids currently used. Spinally administered opioids by themselves do not provide satisfactory
anesthesia for surgical procedures. Thus, opioids have found the greatest use
in the treatment of postoperative and chronic pain. In selected patients,
spinal or epidural opioids can provide excellent analgesia following
thoracic, abdominal, pelvic, or lower extremity surgery without the side
effects associated with high doses of systemically administered opioids. For
postoperative analgesia, spinally administered morphine, 0.2 to 0.5 mg,
usually will provide 8 to 16 hours of analgesia. Placement of an epidural
catheter and repeated boluses or an infusion of opioid permits an increased
duration of analgesia. Many opioids have been used epidurally. Morphine, 2 to
6 mg, every 6 hours, commonly is used for bolus injections, while fentanyl,
20 to 50 |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 4921
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved