| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Muscarinic Receptor Agonists and Antagonists
Overview
|
Acetylcholine is the endogenous neurotransmitter at cholinergic synapses and neuroeffector junctions in the central and peripheral nervous systems. Its actions are mediated through nicotinic and muscarinic cholinergic receptors, which transduce signals via distinct mechanisms. Muscarinic receptors in the peripheral nervous system primarily are found on the autonomic effector cells that are innervated by postganglionic parasympathetic nerves. Muscarinic receptors also are present in ganglia and on certain cells, such as endothelial cells of blood vessels, that receive little or no cholinergic innervation. Certain brain regions such as the hippocampus, cortex, and thalamus have high densities of muscarinic receptors. Cholinergic agonists mimic the effects of acetylcholine at these sites. The first section of this chapter describes the pharmacological properties and therapeutic uses of acetylcholine and agonists that stimulate muscarinic receptors; these agonists typically are longer-acting congeners of acetylcholine or natural alkaloids. Several of these agents cross over and confer their cholinomimetic activity by stimulating nicotinic as well as muscarinic receptors. In general, these agonists manifest little selectivity for the various subtypes of muscarinic receptors described below. The clinical uses of the muscarinic receptor agonists, primarily in ophthalmology and to enhance gastrointestinal and bladder tone, are discussed here and also in Chapters 37: Agents Used for Control of Gastric Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease, 39: Agents Used for Diarrhea, Constipation, and Inflammatory Bowel Disease; Agents Used for Biliary and Pancreatic Disease, and 66: Ocular Pharmacology. The last section of this chapter deals with muscarinic receptor antagonists. These drugs inhibit the actions of acetylcholine by blocking receptors at autonomic effector sites innervated by postganglionic cholinergic nerves. They also inhibit the actions of acetylcholine at pre- and postsynaptic muscarinic receptors in ganglia and on central nervous system neurons. Except for the quaternary ammonium-containing compounds, muscarinic receptor antagonists are highly selective for muscarinic over nicotinic receptors. In addition, a growing number of antagonists show selectivity for muscarinic receptor subtypes, thus enhancing selectivity and minimizing unwanted side effects. The therapeutic uses of muscarinic receptor antagonists include treatment of gastrointestinal and urinary tract disorders (see also Chapters 37: Agents Used for Control of Gastric Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease and 39: Agents Used for Diarrhea, Constipation, and Inflammatory Bowel Disease; Agents Used for Biliary and Pancreatic Disease), specific respiratory conditions (see also Chapter 28: Drugs Used in the Treatment of Asthma), motion sickness, parkinsonian symptoms (see Chapter 22: Treatment of Central Nervous System Degenerative Disorders), and poisoning with cholinesterase inhibitors (see also Chapter 8: Anticholinesterase Agents); their use in ophthalmology is discussed fully in Chapter 66: Ocular Pharmacology. |
Muscarinic Receptor Agonists
|
Muscarinic cholinergic receptor agonists can be divided into two groups: (1) acetylcholine and several synthetic choline esters and (2) the naturally occurring cholinomimetic alkaloids (particularly pilocarpine, muscarine, and arecoline) and their synthetic congeners. In addition, the anticholinesterase agents (Chapter 8: Anticholinesterase Agents) and the ganglionic stimulants (Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia) have parasympathomimetic actions; their prominent effects may be indirect or can arise from locations other than the postganglionic cholinergic effector site. Acetylcholine Acetylcholine (ACh), first synthesized by Baeyer in 1867, has virtually no therapeutic applications because its actions are diffuse and its hydrolysis, catalyzed by both acetylcholinesterase (AChE) and plasma butyrylcholinesterase, is rapid. Consequently, numerous derivatives have been synthesized in attempts to obtain drugs with more selective and prolonged actions. Mechanism of Action The mechanisms of action of endogenous ACh at the postjunctional membranes of the effector cells and neurons that correspond to the four classes of cholinergic synapses are discussed in Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems. By way of recapitulation, these synapses are found at (1) autonomic effector sites, innervated by postganglionic parasympathetic fibers; (2) sympathetic and parasympathetic ganglion cells and the adrenal medulla, innervated by preganglionic autonomic fibers; (3) motor end-plates on skeletal muscle, innervated by somatic motor nerves; and (4) certain synapses peripherally and within the central nervous system (CNS), where the actions can be either pre- or postsynaptic. When ACh is administered systemically, it has the potential to act at all of these sites; however, as a quaternary ammonium compound, its penetration into the CNS is limited, and butyrylcholinesterase in the plasma reduces the concentrations of ACh that reach areas in the periphery with low blood flow. The actions of ACh and related drugs at autonomic effector sites are referred to as muscarinic, based on the original observation that muscarine acts selectively at those sites and produces the same qualitative effects as ACh. Accordingly, the muscarinic, or parasympathomimetic, actions of the drugs considered in this chapter are practically equivalent to the effects of postganglionic parasympathetic nerve impulses listed in Table 61; the differences between the actions of the classical muscarinic agonists are largely quantitative, with limited selectivity for one organ system or another. Muscarinic receptors also are present on autonomic ganglion cells and in the adrenal medulla. Muscarinic stimulation of ganglia and the adrenal medulla usually is thought to be modulatory to nicotinic stimulation. All of the actions of ACh and its congeners at muscarinic receptors can be blocked by atropine. The nicotinic actions of cholinergic agonists refer to their initial stimulation, and often in high doses to subsequent blockade, of autonomic ganglion cells, the adrenal medulla, and the neuromuscular junction, actions comparable to those of nicotine. Properties and Subtypes of Muscarinic Receptors Muscarinic receptors were characterized initially by analysis of the responses of cells and tissues in the periphery and the CNS. Differential effects of two muscarinic agonists, bethanechol and McN-A-343, on the tone of the lower esophageal sphincter led to the initial designation of muscarinic receptors as M1 (ganglionic) and M2 (effector cell) (Goyal and Rattan, 1978; see also Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems). The basis for the selectivity of these agonists is unclear, as there is no good evidence that agonists discriminate among the subtypes of muscarinic receptor (see Eglen, et al., 1996; Caulfield and Birdsall, 1998). However, subsequent radioligand binding studies definitively revealed the existence of more than a single population of antagonist binding sites (Hammer et al., 1980). In particular, the muscarinic antagonist pirenzepine was shown to bind with high affinity to sites in cerebral cortex and sympathetic ganglia (M1) but to have lower affinity for sites in cardiac muscle, smooth muscle, and various glands. These data explain the ability of pirenzepine to block agonist-induced responses that are mediated by muscarinic receptors in sympathetic and myenteric ganglia at concentrations considerably lower than those required to block responses that result from direct stimulation of receptors in various effector organs. Newer antagonists that can further discriminate among various subtypes of muscarinic receptors are now available. For example, tripitramine displays selectivity for cardiac M2- relative to M3-muscarinic receptors, while darifenacin is relatively selective for antagonizing glandular and smooth muscle M3 relative to M2 receptors (see Caulfield and Birdsall, 1998; Birdsall et al., 1998; Levine et al., 1999). The cloning of the cDNAs that encode muscarinic receptors identified five distinct gene products (Bonner et al., 1987), now designated as M1 through M5 (see Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems). All of the known muscarinic receptor subtypes interact with members of a group of heterotrimeric guanine nucleotide-binding regulatory proteins (G proteins) that, in turn, are linked to various cellular effectors (see Chapter 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect). Regions within the receptor responsible for the specificity of G protein coupling have been defined, primarily by mutagenesis and receptor subtype chimera studies. In particular, one region at the carboxyl-terminal end of the third intracellular loop of the receptor has been implicated in the specificity of G protein coupling and shows a great deal of homology within M1, M3, and M5 receptors and within M2 and M4 receptors (see Wess, 1996; Caulfield, 1993; Caulfield and Birdsall, 1998). Conserved regions in the second intracellular loop also confer specificity for proper G protein recognition. Although selectivity is not absolute, stimulation of M1 or M3 receptors causes hydrolysis of polyphosphoinositides and mobilization of intracellular Ca2+, as a consequence of interaction with a G protein (Gq) that activates phospholipase C (see Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems); this effect in turn results in a variety of Ca2+-mediated events, either directly or as a consequence of the phosphorylation of target proteins. In contrast, M2- and M4-muscarinic receptors inhibit adenylyl cyclase and regulate specific ion channels (e.g., enhancement of K+ conductance in cardiac atrial tissue) through subunits released from pertussis toxin-sensitive G proteins (G1 and G0) that are distinct from the G proteins used by M1 and M3 receptors (see Chapters 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship between Drug Concentration and Effect and 12: Neurotransmission and the Central Nervous System). Studies using muscarinic receptor subtype-specific antibodies and ligands demonstrate discrete localization of the muscarinic receptor subtypes, for example within brain regions and in different populations of smooth muscle cells (see Levey, 1993; Yasuda et al., 1993; Eglen et al., 1996; Caulfield and Birdsall, 1998). The M1 through M4 subtypes have been disrupted through gene targeting to create null alleles for each of these genes (Hamilton et al., 1997; Gomeza et al., 1999a and b; Matsui et al., 2000). Altered central responses to cholinergic agonists including seizures, hypothermia, tremors, and analgesia are prominent in the phenotypes of M1, M2, and M4 knockout mice. Mice lacking the M3 receptor have more obvious peripheral lesions including altered salivation, pupil constriction, and bladder contraction. Minimal phenotypic alteration accompanying specific receptor deletion suggests redundancy in receptor subtypes in various tissue locations. Pharmacological Properties Cardiovascular System ACh has four primary effects on the cardiovascular system: vasodilation, a decrease in cardiac rate (the negative chronotropic effect), a decrease in the rate of conduction in the specialized tissues of the sinoatrial (SA) and atrioventricular (AV) nodes (the negative dromotropic effect), and a decrease in the force of cardiac contraction (the negative inotropic effect). The last-named effect is of lesser significance in ventricular than in atrial muscle. Certain of the above effects can be obscured by the dampening of the direct effects of ACh by baroreceptor and other reflexes. Although ACh rarely is given systemically, its cardiac actions are of importance because of the involvement of cholinergic vagal impulses in the actions of the cardiac glycosides, antiarrhythmic agents, and many other drugs as well as following afferent stimulation of the viscera during surgical interventions. The intravenous injection of a small dose of ACh produces an evanescent fall in blood pressure owing to generalized vasodilation, accompanied usually by reflex tachycardia. A considerably larger dose is required to elicit bradycardia or block of AV nodal conduction from a direct action of ACh on the heart. If large doses of ACh are injected after the administration of atropine, an increase in blood pressure is observed; the increase is caused by stimulation of the adrenal medulla and sympathetic ganglia to release catecholamines into the circulation and at postganglionic sympathetic nerve endings. ACh produces dilation of essentially all vascular beds, including those of the pulmonary and coronary vasculature. Vasodilation of coronary beds is mediated through release of nitric oxide and may be elicited by baroreceptor or chemoreceptor reflexes or by direct electrical stimulation of the vagus (Feigl, 1998). However, neither parasympathetic vasodilator nor sympathetic vasoconstrictor tone plays a major role in the regulation of coronary blood flow, in comparison with the effects of local oxygen tension and autoregulatory metabolic factors such as adenosine (Berne and Levy, 1997). Dilation of vascular beds by acetylcholine is due to the presence of muscarinic receptors, primarily of the M3 subtype (Bruning et al., 1994; Eglen et al., 1996; Caulfield and Birdsall, 1998), despite the lack of apparent cholinergic innervation of most blood vessels. The muscarinic receptors responsible for relaxation are located on the endothelial cells of the vasculature; when these receptors are stimulated, the endothelial cells release endothelium-derived relaxing factor, or nitric oxide (Moncada and Higgs, 1997), which diffuses to adjacent smooth muscle cells and causes them to relax (Furchgott, 1999; Ignarro et al., 1999; see Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems). Vasodilation also may arise secondarily from inhibition by ACh of norepinephrine release from adrenergic nerve endings. If the endothelium is damaged, ACh can stimulate receptors on vascular smooth muscle cells and cause vasoconstriction. Cholinergic stimulation affects cardiac function directly and by inhibiting the effects of adrenergic activation. The latter depends on the level of sympathetic drive to the heart and results in part from inhibition of cyclic AMP formation and reduction in L-type Ca2+ channel activity (see Brodde and Michel, 1999). Since cholinergic parasympathetic fibers are distributed extensively to the SA and AV nodes and the atrial muscle, vagal impulses have critical actions on most types of specialized cardiac cells. Cholinergic innervation of the ventricular myocardium is sparse, and the parasympathetic fibers terminate predominantly on specialized conduction tissue such as the Purkinje fibers (Kent et al., 1974; Levy and Schwartz, 1994). In the SA node, each normal cardiac impulse is initiated by the spontaneous depolarization of the pacemaker cells (see Chapter 35: Antiarrhythmic Drugs). At a critical levelthe threshold potentialthis depolarization initiates an action potential. The action potential is conducted through the atrial muscle fibers to the AV node and thence through the Purkinje system to the ventricular muscle. ACh slows the heart rate by decreasing the rate of spontaneous diastolic depolarization (the pacemaker current) and by increasing the repolarizing current at the SA node; attainment of the threshold potential and the succeeding events in the cardiac cycle are therefore delayed (DiFrancesco, 1993). In atrial muscle, ACh decreases the strength of contraction. This direct inhibitory effect of ACh results from M2 receptor-mediated activation of G proteinregulated K+ channels (see Wickman and Clapham, 1995). Increased K+ permeability leads to hyperpolarization and shortens the durations of the action potential and the effective refractory period. The rate of impulse conduction in the normal atrium is either unaffected or may increase. The increase is due to the activation of additional Na+ channels in response to the ACh-induced hyperpolarization. The combination of these factors is the basis for the perpetuation or exacerbation by vagal impulses of atrial flutter or fibrillation arising at an ectopic focus. In contrast, primarily in the AV node and to a much lesser extent in the Purkinje conducting system, ACh slows conduction and increases the refractory period. The decrement in AV nodal conduction usually is responsible for the complete heart block that may be observed when large quantities of cholinergic agonists are administered systemically. With an increase in vagal tone, such as is produced by the digitalis glycosides, the increased refractory period can contribute to the reduction in the frequency with which aberrant atrial impulses are transmitted to the ventricle, and thus decrease the ventricular rate during atrial flutter or fibrillation. In the ventricle, ACh, whether released by vagal stimulation or applied directly, also has a negative inotropic effect, although it is much smaller than that observed in the atrium. In human beings and most mammals, direct inhibition is not apparent, unless contractility is enhanced by adrenergic stimulation (Higgins et al., 1973; Levy and Schwartz, 1994; Michel and Brodde, 1999). Automaticity of Purkinje fibers is suppressed, and the threshold for ventricular fibrillation is increased (Kent et al., 1974; Kent and Epstein, 1976). Sympathetic and vagal nerve terminals lie in close proximity, and muscarinic receptors are believed to exist at presynaptic as well as postsynaptic sites (Wellstein and Pitschner, 1988). Inhibition of adrenergic stimulation of the heart arises from the capacity of ACh to modulate or depress the myocardial response to catecholamines as well as from a capacity to inhibit the release of norepinephrine from sympathetic nerve endings. Gastrointestinal and Urinary Tracts Although stimulation of vagal input to the gastrointestinal tract increases tone, amplitude of contraction, and secretory activity of the stomach and intestine, such responses are inconsistently seen with administered ACh. Poor perfusion and rapid hydrolysis by plasma butyrylcholinesterase limit access of ACh to the muscarinic receptors. Parasympathetic sacral innervation causes detrusor muscle contraction, increased voiding pressure, and ureter peristalsis, but for similar reasons these responses are not evident with administered ACh. Miscellaneous Effects The influence of ACh and parasympathetic innervation on various organs and tissues is discussed in detail in Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems. ACh and its analogs stimulate secretion by all glands that receive parasympathetic innervation, including the lacrimal, tracheobronchial, salivary, digestive, and exocrine sweat glands. The effects on the respiratory system, in addition to increased tracheobronchial secretion, include bronchoconstriction and stimulation of the chemoreceptors of the carotid and aortic bodies. When instilled into the eye, they produce miosis (see Chapter 66: Ocular Pharmacology). Synergisms and Antagonisms The muscarinic actions of ACh and all the drugs of this class are blocked selectively by atropine, primarily through competitive occupation of muscarinic receptor sites on the autonomic effector cells and secondarily on autonomic ganglion cells. The nicotinic actions of ACh and its derivatives at autonomic ganglia are blocked by hexamethonium and trimethaphan; their actions at the neuromuscular junction of skeletal muscle are antagonized by tubocurarine and other competitive blocking agents (see Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia). Cholinomimetic Choline Esters and Natural Alkaloids Methacholine
(acetyl- Carbachol CARBOPTIC, others) and bethanechol, which are unsubstituted carbamoyl esters, are resistant to hydrolysis by either AChE or nonspecific cholinesterases; their half-lives are thus sufficiently long that they become distributed to areas of low blood flow. Bethanechol has mainly muscarinic actions, showing some selectivity on gastrointestinal tract and urinary bladder motility. Carbachol retains substantial nicotinic activity, particularly on autonomic ganglia. It is likely that both its peripheral and its ganglionic actions are due, at least in part, to the release of endogenous ACh from the terminals of cholinergic fibers. The three major natural alkaloids in this grouppilocarpine, muscarine, and arecolinehave the same principal sites of action as the choline esters discussed above. Muscarine acts almost exclusively at muscarinic receptor sites, and their classification is derived from this fact. Arecoline also acts at nicotinic receptors. Pilocarpine has a dominant muscarinic action, but it causes anomalous cardiovascular responses, and the sweat glands are particularly sensitive to the drug. Although these naturally occurring alkaloids are of great value as pharmacological tools, present clinical use is restricted largely to the employment of pilocarpine as a sialagogue and miotic agent (see Chapter 66: Ocular Pharmacology). History and Sources Of the several hundred synthetic choline derivatives investigated,
only methacholine, carbachol, and bethanechol have had clinical applications.
The structures of these compounds are shown in Figure 71. Methacholine, the
The
poisonous effects of certain species of mushrooms have been known since
ancient times, but it was not until Schmiedeberg isolated the alkaloid
muscarine from Amanita muscaria in 1869 that its properties could be
systematically investigated. The role played by muscarine in the development
of the neurohumoral theory is recounted in Chapter 6: Neurotransmission: The
Autonomic and Somatic Motor Nervous Systems. Arecoline is the chief alkaloid
of areca or betel nuts, the seeds of Areca catechu. The red-staining
betel nut is consumed as a euphoretic by the natives of the Indian
subcontinent and StructureActivity Relationships The muscarinic alkaloids show marked differences as well as interesting relationships in structure when compared to the quaternary esters of choline (Figure 71). Arecoline and pilocarpine are tertiary amines. Muscarine, a quaternary ammonium compound, shows more limited absorption. The chemistry and pharmacology of many natural and synthetic muscarinic compounds have been reviewed by Bebbington and Brimblecombe (1965). McN-A-343 is an agonist that was originally proposed to stimulate M1 receptors with some selectivity. While it is clear that McN-A-343 can stimulate sympathetic ganglia and inhibitory neurons in myenteric plexus, this is a 'functional' rather than a subtype-specific effect. Indeed, no agonists with subtype specificity are known (Caulfield and Birdsall, 1998). Pharmacological Properties Gastrointestinal Tract All of the muscarinic agonists are capable of stimulating smooth muscle of the gastrointestinal tract, thereby increasing tone and motility; large doses will cause spasm and tenesmus. Carbachol, bethanechol, and pilocarpine, in contrast to methacholine, will stimulate the gastrointestinal tract without significant cardiovascular effects. Urinary Tract The choline esters and pilocarpine contract the detrusor muscle of the bladder, increase voiding pressure, decrease bladder capacity, and increase ureteral peristalsis. In addition, the trigone and external sphincter muscles relax. Selectivity for bladder stimulation relative to cardiovascular activity is evident for bethanechol. In animals with experimental spinal cord lesions, muscarinic agonists promote evacuation of the bladder. Exocrine Glands The choline esters and muscarinic alkaloids stimulate secretion of glands that receive parasympathetic or sympathetic cholinergic innervation, including the lacrimal, salivary, digestive, tracheobronchial, and sweat glands. Pilocarpine (10 mg to 15 mg, subcutaneously), in particular, causes marked diaphoresis in human beings; 2 to 3 liters of sweat may be secreted. Salivation also is increased markedly. Oral pilocarpine appears to cause a more continuous production of saliva. Muscarine and arecoline also are potent diaphoretic agents. Accompanying side effects may include hiccough, salivation, nausea, vomiting, weakness, and, occasionally, collapse. These alkaloids also stimulate the lacrimal, gastric, pancreatic, and intestinal glands, and the mucous cells of the respiratory tract. Respiratory System In addition to tracheobronchial secretions, bronchial smooth muscle is stimulated by the muscarinic agonists. Asthmatic patients respond with intense bronchoconstriction and a reduction in vital capacity. Cardiovascular System Continuous intravenous infusion of methacholine elicits hypotension
and bradycardia just as ACh does, but at 1/200 the dose. Muscarine, at small
doses, also leads to a marked fall in the blood pressure and a slowing or
temporary cessation of the heartbeat. In contrast, carbachol and bethanechol
generally cause only a transient fall in blood pressure at doses that affect
the gastrointestinal and urinary tracts. Likewise, pilocarpine produces only
a brief fall in blood pressure. However, if this is preceded by an
appropriate dose of a nicotinic receptor antagonist, pilocarpine produces a
marked rise in pressure. Both the vasodepressor and pressor responses are
prevented by atropine; the latter effect also is abolished by Eye The muscarinic agonists stimulate the pupillae constrictor and ciliary muscle when applied locally to the eye causing pupil constriction and a loss of accommodation. Central Nervous System The intravenous injection of relatively small doses of pilocarpine, muscarine, and arecoline evokes a characteristic cortical arousal or activation response in cats, similar to that produced by injection of anticholinesterase agents or by electrical stimulation of the brainstem reticular formation. The arousal response to all of these drugs is reduced or blocked by atropine and related agents (Krnjevc, 1974). The choline esters, being quaternary, do not cross the bloodbrain barrier. Therapeutic Uses Bethanechol chloride (carbamyl- Gastrointestinal Disorders Bethanechol can be of value in certain cases of postoperative abdominal distention and in gastric atony or gastroparesis. The oral route is preferred; the usual dosage is 10 to 20 mg three or four times daily. Bethanechol is given by mouth before each main meal in cases without complete retention; when gastric retention is complete and nothing passes into the duodenum, the subcutaneous route is necessary because the drug is not adequately absorbed from the stomach. Bethanechol likewise has been used to advantage in certain patients with congenital megacolon and with adynamic ileus secondary to toxic states. Prokinetic agents with combined cholinergic-agonist and dopamine-antagonist activity (metoclopramide) or serotonin-antagonist activity (see Chapter 38: Prokinetic Agents, Antiemetics, and Agents Used in Irritable Bowel Syndrome) have largely replaced bethanechol in gastroparesis or esophageal reflux disorders. Urinary Bladder Disorders Bethanechol may be useful in combating urinary retention and
inadequate emptying of the bladder when organic obstruction is absent, as in
postoperative and postpartum urinary retention and in certain cases of
chronic hypotonic, myogenic, or neurogenic bladder (Wein, 1991). Xerostomia Pilocarpine is administered orally in 5- to 10-mg doses for the treatment of xerostomia that follows head and neck radiation treatments or that is associated with Sjgren's syndrome (Wiseman and Faulds, 1995). The latter is an autoimmune disorder occurring primarily in women where secretions, particularly salivary and lacrimal, are compromised (Anaya and Talal, 1999; Nusair and Rubinow, 1999). Provided salivary parenchyma maintain a residual function, enhanced salivary secretion, ease of swallowing, and subjective improvement in hydration of the oral cavity are achieved. Side effects typify cholinergic stimulation, with sweating being the most common complaint. Bethanechol offers an alternative oral agent, which some feel produces less diaphoresis (Epstein et al., 1994). Cevimeline (EVOXAC) is a newly approved agonist with activity at M3-muscarinic receptors. These receptors are found on lacrimal and salivary gland epithelia. Cevimeline has a long-lasting sialogogic action and may have fewer side effects than pilocarpine (Anaya and Talal, 1999). Comparative clinical trials with pilocarpine have yet to be conducted. Ophthalmological Pilocarpine also is used in the treatment of glaucoma, where it is instilled into the eye usually as a 0.5% to 4.0% solution. It usually is better tolerated than are the anticholinesterases, and pilocarpine is the standard cholinergic agent for initial treatment of open-angle glaucoma. Reduction of intraocular pressure occurs within a few minutes and lasts 4 to 8 hours. The ophthalmic use of pilocarpine alone and in combination with other agents is discussed in Chapter 66: Ocular Pharmacology. The miotic action of pilocarpine is useful in reversing a narrow-angle glaucoma attack and overcoming the mydriasis produced by atropine; alternated with mydriatics, pilocarpine is employed to break adhesions between the iris and the lens. CNS Agonists that show functional selectivity for M1 and M2 receptors have been targets of development by drug companies, and some have been in clinical trial for use in treating the intellectual impairment associated with Alzheimer's disease. The potential advantage of such agonists would arise from stimulating postsynaptic M1 receptors in the CNS without concomitantly stimulating the presynaptic M2 receptors that inhibit release of endogenous ACh. However, lack of efficacy in improvement of cognitive function has diminished enthusiasm for this approach (Eglen et al., 1999). Precautions, Toxicity, and Contraindications Muscarinic agonists are administered subcutaneously to achieve an acute response and orally to treat more chronic conditions. Should serious toxic reactions to these drugs arise, atropine sulfate (0.5 mg to 1 mg in adults) should be given subcutaneously or intravenously. Epinephrine (0.3 mg to 1 mg, subcutaneously or intramuscularly) also is of value in overcoming severe cardiovascular or bronchoconstrictor responses. Among the major contraindications to the use of the choline esters are asthma, hyperthyroidism, coronary insufficiency, and acid-peptic disease. Their bronchoconstrictor action is liable to precipitate an asthma attack, and hyperthyroid patients may develop atrial fibrillation. Hypotension induced by these agents can severely reduce coronary blood flow, especially if it is already compromised. Other possible undesirable effects of the cholinergic agents are flushing, sweating, abdominal cramps, belching, a sensation of tightness in the urinary bladder, difficulty in visual accommodation, headache, and salivation. Toxicology Poisoning from pilocarpine, muscarine, or arecoline is characterized chiefly by exaggeration of their various parasympathomimetic effects and resembles that produced by consumption of mushrooms of the genus Inocybe (see below). Treatment consists of the parenteral administration of atropine in doses sufficient to cross the bloodbrain barrier and adequate measures to support the respiration and the circulation and to counteract pulmonary edema. Mushroom Poisoning (Mycetism) Mushroom poisoning has been known for centuries. The Greek poet Euripides (fifth century B.C.) is said to have lost his wife and three children from this cause. In recent years the number of cases of mushroom poisoning has been increasing as the result of the current popularity of the consumption of wild mushrooms. Various species of mushrooms contain many toxins, and species within the same genus may contain distinct toxins. Although Amanita muscaria is the source from which muscarine was isolated, its content of the alkaloid is so low (approximately 0.003%) that muscarine cannot be responsible for the major toxic effects. Much higher concentrations of muscarine are present in various species of Inocybe and Clitocybe. The symptoms of intoxication attributable to muscarine develop rapidly, within 30 to 60 minutes of ingestion; they include salivation, lacrimation, nausea, vomiting, headache, visual disturbances, abdominal colic, diarrhea, bronchospasm, bradycardia, hypotension, and shock. Treatment with atropine (1 to 2 mg intramuscularly every 30 minutes) effectively blocks these effects (Kppel, 1993; Goldfrank, 1998). Intoxication produced by A. muscaria and related Amanita species arises from the neurologic and hallucinogenic properties of muscimol, ibotenic acid, and other isoxazole derivatives. These agents stimulate excitatory and inhibitory amino acid receptors. Symptoms range from irritability, restlessness, ataxia, hallucinations, and delirium to drowsiness and sedation. Treatment is mainly supportive; benzodiazepines are indicated when excitation predominates, whereas atropine often exacerbates the delirium. Mushrooms from Psilocybe and Panaeolus species contain psilocybin and related derivatives of tryptamine. They also cause short-lasting hallucinations. Gyromitra species (false morels) produce gastrointestinal disorders and a delayed hepatotoxicity. The toxic substance is acetaldehyde methylformylhydrazone, which is converted in the body to reactive hydrazines. Although fatalities from liver and kidney failure have been reported, they are far less frequent than with amatoxin-containing mushrooms discussed below. The most serious form of mycetism is produced by Amanita phalloides,
other Amanita species, Lepiota, and Galerina species (Goldfrank,
1998). These species account for more than 90% of all fatal cases. Ingestion
of as little as 50 g of A. phalloides (deadly nightcap) can be fatal.
The principal toxins are the amatoxins ( Because the severity of toxicity and treatment strategies for mushroom
poisoning depend on the species ingested, their identification should be
sought. Often symptomatology is delayed, causing gastric lavage and
administration of activated charcoal to be of limited value. Regional poison
control centers in the |
Muscarinic Receptor Antagonists
|
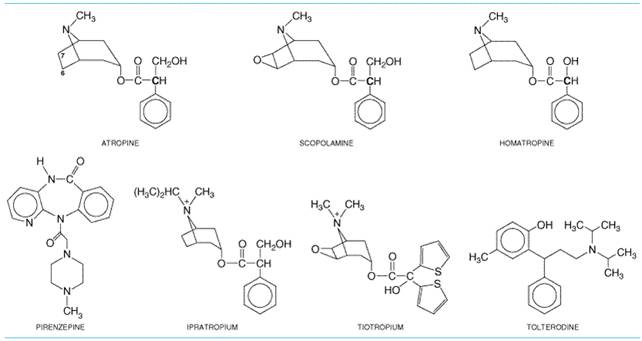
The class of drugs referred to here as muscarinic receptor antagonists includes (1) the naturally occurring alkaloids, atropine and scopolamine; (2) semisynthetic derivatives of these alkaloids, which primarily differ from the parent compounds in their disposition in the body or their duration of action; and (3) synthetic congeners, some of which show selectivity for particular subtypes of muscarinic receptors. Noteworthy agents among the synthetic derivatives include homatropine and tropicamide, which have a shorter duration of action than atropine, and methylatropine, ipratropium, and tiotropium, which are quaternized and do not cross the bloodbrain barrier. The latter two agents are given by inhalation in the treatment of bronchial asthma and chronic obstructive pulmonary disease. The synthetic derivatives possessing partial receptor selectivity include pirenzepine, used in the treatment of acid-peptic disease in some countries, and tolterodine, used in the treatment of urinary incontinence. Muscarinic receptor antagonists prevent the effects of ACh by blocking its binding to muscarinic cholinergic receptors at neuroeffector sites on smooth muscle, cardiac muscle, and gland cells; in peripheral ganglia; and in the central nervous system. In general, muscarinic receptor antagonists cause little blockade of the effects of ACh at nicotinic receptor sites. However, quaternary ammonium analogs of atropine and related drugs generally exhibit a greater degree of nicotinic blocking activity and, consequently, are more likely to interfere with ganglionic or neuromuscular transmission. In the CNS, cholinergic transmission appears to be both muscarinic and nicotinic at spinal, subcortical, and cortical levels in the brain (see Chapter 12: Neurotransmission and the Central Nervous System). At high or toxic doses, the central effects of atropine and related drugs generally consist of CNS stimulation followed by depression. Since quaternary compounds penetrate the bloodbrain barrier poorly, antagonists of this type have little or no effect on the CNS. Parasympathetic neuroeffector junctions in different organs are not equally sensitive to even the nonselective muscarinic receptor antagonists (see Table 72). Small doses of atropine depress salivary and bronchial secretion and sweating. With larger doses, the pupil dilates, accommodation of the lens to near vision is inhibited, and vagal effects on the heart are blocked so that the heart rate is increased. Larger doses inhibit the parasympathetic control of the urinary bladder and gastrointestinal tract, thereby inhibiting micturition and decreasing the tone and motility of the gut. Still larger doses are required to inhibit gastric secretion and motility. Thus, doses of atropine and most related muscarinic receptor antagonists that reduce gastrointestinal tone and depress gastric secretion also almost invariably affect salivary secretion, ocular accommodation, and micturition. This hierarchy of relative sensitivities probably is not a consequence of differences in the affinity of atropine for the muscarinic receptors at these sites, because atropine does not show selectivity toward different muscarinic receptor subtypes. More likely determinants include the degree to which the functions of various end organs are regulated by parasympathetic tone and the involvement of intramural neurons and reflexes. The actions of many clinically available muscarinic receptor antagonists differ only quantitatively from those of atropine, considered below as the prototype of the group. No antagonist in the receptor-selective category, including pirenzepine, is completely selective (i.e., has a distinctive affinity for one relative to all other receptor subtypes). In fact, clinical efficacy of some agents may arise from a balance of antagonistic actions on two or more receptor subtypes. History The naturally occurring muscarinic receptor antagonists atropine
and scopolamine are alkaloids of the belladonna (Solanaceae) plants.
Preparations of belladonna were known to the ancient Hindus and have been
used by physicians for many centuries. During the time of the Accurate study of the actions of belladonna dates from the isolation of atropine in pure form by Mein in 1831. In 1867, Bezold and Bloebaum showed that atropine blocked the cardiac effects of vagal stimulation, and 5 years later Heidenhain found that it prevented salivary secretion produced by stimulation of the chorda tympani. Many semisynthetic congeners of the belladonna alkaloids and a large number of synthetic muscarinic receptor antagonists have been prepared, primarily with the objective of altering gastrointestinal or bladder activity without causing dry mouth or pupillary dilation. Chemistry Atropine and scopolamine are esters formed by combination of an aromatic acid, tropic acid, and complex organic bases, either tropine (tropanol) or scopine. Scopine differs from tropine only in having an oxygen bridge between the carbon atoms designated as 6 and 7 (Figure 72). Homatropine is a semisynthetic compound produced by combining the base tropine with mandelic acid. The corresponding quaternary ammonium derivatives, modified by the addition of a second methyl group to the nitrogen, are methylatropine nitrate, methscopolamine bromide, and homatropine methylbromide. Ipratropium and tiotropium also are quaternary tropine analogs esterified with synthetic aromatic acids.
StructureActivity Relationship An intact ester of tropine and tropic acid is essential for antimuscarinic action, since neither the free acid nor the base exhibits significant antimuscarinic activity. The presence of a free OH group in the acyl portion of the ester also is important for activity. When given parenterally, quaternary ammonium derivatives of atropine and scopolamine are, in general, more potent than their parent compounds in both muscarinic receptor and ganglionic blocking activities; conversion of the nitrogen from a tertiary to a quaternary group increases blockade at nicotinic receptors. The quaternary derivatives lack CNS activity because of poor penetration into the brain. Given orally, they are poorly and unreliably absorbed. Both tropic and mandelic acids have an asymmetrical carbon atom (boldface C in the formulas in Figure 72). Scopolamine is l-hyoscine and is much more active than d-hyoscine. Atropine is racemized during extraction and consists of d,l-hyoscyamine, but antimuscarinic activity is almost wholly due to the naturally occurring l form. The synthetic derivatives show a wide latitude of structures that spatially replicate the aromatic acid and the bridged nitrogen of the tropine. Mechanism of Action Atropine and related compounds compete with ACh and other muscarinic agonists for a common binding site on the muscarinic receptor. The binding site for competitive antagonists and acetylcholine is in a cleft predicted to be formed by several of the receptor's seven transmembrane helices, as shown recently for the position of retinol in the mammalian rhodopsin structure (Palczewski et al., 2000). An aspartic acid present in the N-terminal portion of the third transmembrane helix of all five muscarinic receptor subtypes is believed to form an ionic bond with the cationic quaternary nitrogen in acetylcholine and the tertiary or quaternary nitrogen of the antagonists (see Wess, 1996; Caufield and Birdsall, 1998). Since antagonism by atropine is competitive, it can be overcome if the concentration of ACh at receptor sites of the effector organ is increased sufficiently. Muscarinic receptor antagonists inhibit responses to postganglionic cholinergic nerve stimulation less readily than they inhibit responses to injected choline esters. The difference may be due to release of ACh by cholinergic nerve terminals so close to receptors that very high concentrations of the transmitter gain access to the receptors in the neuroeffector junction. Pharmacological Properties Atropine and scopolamine differ quantitatively in antimuscarinic actions, particularly in their ability to affect the CNS. Atropine has almost no detectable effect on the CNS in doses that are used clinically. In contrast, scopolamine has prominent central effects at low therapeutic doses. The basis for this difference is probably the greater permeation of scopolamine across the bloodbrain barrier. Because atropine has limited CNS effects, it is given in preference to scopolamine for most purposes. Central Nervous System Atropine in therapeutic doses (0.5 to 1 mg) causes only mild vagal excitation as a result of stimulation of the medulla and higher cerebral centers. With toxic doses of atropine, central excitation becomes more prominent, leading to restlessness, irritability, disorientation, hallucinations, or delirium (see discussion of atropine poisoning, below). With still larger doses, stimulation is followed by depression, leading to circulatory collapse and respiratory failure after a period of paralysis and coma. Scopolamine in therapeutic doses normally causes CNS depression manifested as drowsiness, amnesia, fatigue, and dreamless sleep, with a reduction in rapid eye movement (REM) sleep. It also causes euphoria and is therefore subject to some abuse. The depressant and amnesic effects formerly were sought when scopolamine was used as an adjunct to anesthetic agents or for preanesthetic medication. However, in the presence of severe pain, the same doses of scopolamine can occasionally cause excitement, restlessness, hallucinations, or delirium. These excitatory effects resemble those of toxic doses of atropine. Scopolamine also is effective in preventing motion sickness. This action is probably either on the cortex or more peripherally on the vestibular apparatus. The belladonna alkaloids and related muscarinic receptor antagonists have long been used in parkinsonism. These agents can be effective adjuncts to treatment with levodopa (see Chapter 22: Treatment of Central Nervous System Degenerative Disorders). Muscarinic receptor antagonists also are used to treat the extrapyramidal symptoms that commonly occur as side effects of antipsychotic drug therapy (see Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania). Certain antipsychotic drugs are relatively potent muscarinic receptor antagonists (Richelson, 1999), and these cause fewer extrapyramidal side effects. Ganglia and Autonomic Nerves Cholinergic neurotransmission in autonomic ganglia is mediated primarily by activation of nicotinic acetylcholine receptors, resulting in the generation of action potentials (see Chapters 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems and 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia). ACh and other cholinergic agonists also cause the generation of slow excitatory postsynaptic potentials that are mediated by ganglionic muscarinic M1acetylcholine receptors. This response is particularly sensitive to blockade by pirenzepine. The extent to which the slow excitatory response can alter impulse transmission through the different sympathetic and parasympathetic ganglia is difficult to assess, but the effects of pirenzepine on responses of end organs suggest a physiological modulatory function for the ganglionic M1 receptor (Caulfield, 1993; Eglen et al., 1996; Birdsall et al., 1998; Caulfield and Birdsall, 1998). Pirenzepine inhibits gastric acid secretion at doses that have little effect on salivation or heart rate. Since the muscarinic receptors on the parietal cells do not appear to have a high affinity for pirenzepine, the M1 receptor responsible for alterations in gastric acid secretion is postulated to be localized in intramural ganglia (see Eglen et al., 1996). Blockade of ganglionic receptors (rather than those at the neuroeffector junction) also appears to underlie the ability of pirenzepine to inhibit the relaxation of the lower esophageal sphincter. Likewise, blockade of parasympathetic ganglia may contribute to the response to muscarinic antagonists in lung and heart (Barnes, 1993; Wellstein and Pitschner, 1988). Presynaptic muscarinic receptors also are present on terminals of sympathetic and parasympathetic neurons. Blockade of these presynaptic receptors, which are of variable subtype, generally augments transmitter release. Nonselective muscarinic blocking agents may thus augment ACh release, partially counteracting their effective postsynaptic receptor blockade. Since muscarinic receptor antagonists can alter autonomic activity at the ganglion and postganglionic neuron, the ultimate response of end organs to blockade of muscarinic receptors is difficult to predict. Thus, while direct blockade at neuroeffector sites predictably reverses the usual effects of the parasympathetic nervous system, concomitant inhibition of ganglionic or presynaptic receptors may produce paradoxical responses. Eye The muscarinic receptor antagonists block the responses of the pupillae sphincter muscle of the iris and the ciliary muscle of the lens to cholinergic stimulation (see Chapter 66: Ocular Pharmacology). Thus, they dilate the pupil (mydriasis) and paralyze accommodation (cycloplegia). The wide pupillary dilation results in photophobia; the lens is fixed for far vision, near objects are blurred, and objects may appear smaller than they are. The normal pupillary reflex constriction to light or upon convergence of the eyes is abolished. These effects can occur after either local or systemic administration of the alkaloids. However, conventional systemic doses of atropine (0.6 mg) have little ocular effect, in contrast to equal doses of scopolamine, which cause definite mydriasis and loss of accommodation. Locally applied atropine or scopolamine produces ocular effects of considerable duration; accommodation and pupillary reflexes may not fully recover for 7 to 12 days. The muscarinic receptor antagonists used as mydriatics differ from the sympathomimetic agents in that the latter cause pupillary dilation without loss of accommodation. Pilocarpine, choline esters, physostigmine, and isoflurophate (DFP) in sufficient concentrations can partially or fully reverse the ocular effects of atropine. Muscarinic receptor antagonists administered systemically have little effect on intraocular pressure except in patients predisposed to narrow-angle glaucoma, where the pressure may occasionally rise dangerously. The rise in pressure occurs when the anterior chamber is narrow and the iris obstructs flow of aqueous humor into the trabeculae. This interferes with drainage of aqueous humor. The drugs may precipitate a first attack in unrecognized cases of this rare condition. In patients with open-angle glaucoma, an acute rise in pressure is unusual. Atropine-like drugs generally can be used safely in this latter condition, particularly if the patient also is adequately treated with an appropriate miotic agent. Cardiovascular System Heart The main effect of atropine on the heart is to alter the rate. Although the dominant response is tachycardia, the heart rate often decreases transiently with average clinical doses (0.4 to 0.6 mg). The slowing is rarely marked, about 4 to 8 beats per minute, and is usually absent after rapid intravenous injection. There are no accompanying changes in blood pressure or cardiac output. This paradoxical effect once was thought to be due to central vagal stimulation; however, cardiac slowing also is seen with muscarinic receptor antagonists that do not readily enter the brain. Studies in human beings show that pirenzepine is equipotent with atropine in decreasing heart rate; its prior administration can prevent any further decrease by atropine. The data suggest that the decreased heart rate may result from blockade of M1 receptors on postganglionic parasympathetic neurons; this relieves the inhibitory effects of synaptic ACh and increases the release of transmitter (Wellstein and Pitschner, 1988). Larger doses of atropine cause progressively increasing tachycardia by blocking vagal effects on M2 receptors on the SA nodal pacemaker. The resting heart rate is increased by about 35 to 40 beats per minute in young men given 2 mg of atropine intramuscularly. The maximal heart rate (e.g., in response to exercise) is not altered by atropine. The influence of atropine is most noticeable in healthy young adults, in whom vagal tone is considerable. In infancy and old age, even large doses of atropine may fail to accelerate the heart. Atropine often produces cardiac arrhythmias, but without significant cardiovascular symptoms. With low doses of scopolamine (0.1 or 0.2 mg), the cardiac slowing is greater than with atropine. With higher doses, cardioacceleration occurs initially, but it is short lived and is followed within 30 minutes either by a return to the normal rate or by bradycardia. Adequate doses of atropine can abolish many types of reflex vagal cardiac slowing or asystolefor example, from inhalation of irritant vapors, stimulation of the carotid sinus, pressure on the eyeballs, peritoneal stimulation, or injection of contrast dye during cardiac catheterization. It also prevents or abruptly abolishes bradycardia or asystole caused by choline esters, acetylcholinesterase inhibitors, or other parasympathomimetic drugs, as well as cardiac arrest from electrical stimulation of the vagus. The removal of vagal influence on the heart by atropine also may facilitate AV conduction. Atropine also shortens the functional refractory period of the AV node and can increase ventricular rate in patients who have atrial fibrillation or flutter. In certain cases of second-degree heart block (e.g., Wenckebach AV block), in which vagal activity is an etiological factor (such as with digitalis toxicity), atropine may lessen the degree of block. In some patients with complete heart block, the idioventricular rate may be accelerated by atropine; in others it is stabilized. Atropine and scopolamine may improve the clinical condition of patients with early myocardial infarction by relieving severe sinus or nodal bradycardia or AV block. Circulation Atropine, in clinical doses, completely counteracts the peripheral vasodilation and sharp fall in blood pressure caused by choline esters. In contrast, when given alone, its effect on blood vessels and blood pressure is neither striking nor constant. This result is expected, because most vascular beds lack significant cholinergic innervation, and the cholinergic sympathetic vasodilator fibers to vessels supplying skeletal muscle do not appear to be involved to any important extent in the normal regulation of tone. Atropine in toxic, and occasionally therapeutic, doses dilates cutaneous blood vessels, especially those in the blush area (atropine flush). This may be a compensatory reaction permitting the radiation of heat to offset the atropine-induced rise in temperature that can accompany inhibition of sweating. Respiratory Tract The parasympathetic nervous system plays a major role in regulating bronchomotor tone. A diverse set of stimuli cause reflex increases in parasympathetic activity that contribute to bronchoconstriction. Vagal fibers synapse and activate nicotinic and M1-muscarinic receptors in parasympathetic ganglia located in the airway wall; short postganglionic fibers release acetylcholine, which acts on M3-muscarinic receptors in airway smooth muscle. The submucosal glands also are innervated by parasympathetic neurons and have predominantly M3 receptors (see Barnes, 2000). Largely owing to the introduction of inhaled ipratropium and tiotropium, anticholinergic therapy of chronic obstructive pulmonary disease and asthma has been revived (Barnes, 2000; Littner et al., 2000). The belladonna alkaloids inhibit secretions of the nose, mouth, pharynx, and bronchi and thus dry the mucous membranes of the respiratory tract. This action is especially marked if secretion is excessive and is the basis for the use of atropine and scopolamine in preanesthetic medication. The ability of these agents to reduce the occurrence of laryngospasm during general anesthesia appears to be caused by inhibition of respiratory tract secretions that can precipitate reflex laryngospasm. However, the depression of mucous secretion and mucociliary clearance are undesirable side effects of atropine in patients with airway disease. Inhibition by atropine of bronchoconstriction caused by histamine,
bradykinin, and the eicosanoids presumably reflects the participation of
parasympathetic efferents in the bronchial reflexes elicited by these agents.
The ability to block the indirect bronchoconstrictive effects of inflammatory
mediators that are released during attacks of asthma forms the basis for the
use of anticholinergic agents, along with Gastrointestinal Tract Interest in the actions of muscarinic receptor antagonists on the stomach and intestine led to their use as antispasmodic agents for gastrointestinal disorders and in the treatment of peptic ulcer. Although atropine can completely abolish the effects of ACh (and other parasympathomimetic drugs) on the motility and secretions of the gastrointestinal tract, it inhibits only incompletely the effects of vagal impulses. This difference is particularly striking in the effects of atropine on motility of the gut. Preganglionic vagal fibers that innervate the gut synapse not only with postganglionic cholinergic fibers but also with a network of noncholinergic intramural neurons. These neurons, which form the enteric plexus, utilize numerous neurotransmitters including 5-hydroxytryptamine (5-HT) and dopamine. Since therapeutic doses of atropine do not block responses to gastrointestinal hormones or to noncholinergic neurohumoral transmitters, release of these substances from the intramural neurons can still effect changes in motility. Similarly, while vagal activity modulates gastrin-elicited histamine release and gastric acid secretion, the actions of gastrin can occur independent of vagal tone. H2-histamine receptor antagonists, M1-muscarinic receptor antagonists, and K+,H+-ATPase inhibitors (proton pump inhibitors) have replaced atropine and other nonselective antagonists as inhibitors of acid secretion (see Chapter 37: Agents Used for Control of Gastric Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease). Secretions Salivary secretion appears to be mediated through M3 receptors and is particularly sensitive to inhibition by muscarinic receptor antagonists, which can completely abolish the copious, watery, parasympathetically induced secretion. The mouth becomes dry, and swallowing and talking may become difficult. Gastric secretions during the cephalic and fasting phase are reduced markedly by muscarinic receptor antagonists. In contrast, the intestinal phase of gastric secretion is only partially inhibited. The concentration of acid is not necessarily lowered, because secretion of HCO3 as well as of H+ is blocked. The gastric cells that secrete mucin and proteolytic enzymes are more directly under vagal influence than are the acid-secreting cells, and atropine decreases their secretory function. Motility The parasympathetic nerves enhance both tone and motility and relax sphincters, thereby favoring the passage of chyme through the gut. However, the intestine has a complex system of intramural nerve plexuses that regulate motility independent of parasympathetic control; impulses from the CNS only modulate the effects of the intrinsic reflexes (see Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems). Both in normal subjects and in patients with gastrointestinal disease, muscarinic antagonists produce prolonged inhibitory effects on the motor activity of the stomach, duodenum, jejunum, ileum, and colon, characterized by a reduction in tone and in amplitude and frequency of peristaltic contractions. Relatively large doses are needed to produce such inhibition. Other Smooth Muscle Urinary Tract Atropine decreases the normal tone and amplitude of contractions of
the ureter and bladder, and often eliminates drug-induced enhancement of
ureteral tone. However, this inhibition is not achieved in the absence of
inhibition of salivation and lacrimation and blurring of vision. Control of
bladder contraction appears to be mediated by multiple muscarinic receptor
subtypes. Receptors of the M2 subtype appear most prevalent in the
bladder, yet studies with selective antagonists suggest that the M3
receptor mediates detrusor muscle contraction. The M2 receptor may
act to inhibit Biliary Tract Atropine exerts a mild antispasmodic action on the gallbladder and bile ducts in human beings. However, this effect usually is not sufficient to overcome or prevent the marked spasm and increase in biliary duct pressure induced by opioids. The nitrites (see Chapter 32: Drugs Used for the Treatment of Myocardial Ischemia) are more effective than atropine in this respect. Sweat Glands and Temperature Small doses of atropine or scopolamine inhibit the activity of sweat glands innervated by sympathetic cholinergic fibers, and the skin becomes hot and dry. Sweating may be depressed enough to raise the body temperature, but only notably so after large doses or at high environmental temperatures. Absorption, Fate, and Excretion The belladonna alkaloids and the tertiary synthetic and semisynthetic derivatives are absorbed rapidly from the gastrointestinal tract. They also enter the circulation when applied locally to the mucosal surfaces of the body. Absorption from intact skin is limited, although efficient absorption does occur in the postauricular region. Systemic absorption of inhaled quaternary muscarinic receptor antagonists is minimal. The quaternary ammonium derivatives of the belladonna alkaloids also are poorly absorbed after an oral dose (Ali-Melkkila et al., 1993) and penetrate the conjunctiva of the eye less readily. Central effects are lacking, because these agents do not cross the bloodbrain barrier. Atropine has a half-life of approximately 4 hours; hepatic metabolism accounts for the elimination of about half of a dose, and the remainder is excreted unchanged in the urine. The quaternary agents have somewhat longer durations of action. Poisoning by Muscarinic Receptor Antagonists and Other Anticholinergic Drugs The deliberate or accidental ingestion of belladonna alkaloids or other classes of drugs with atropinic properties is a major cause of poisonings. Many H1-histamine receptor antagonists, phenothiazines, and tricyclic antidepressants block muscarinic receptors and, in sufficient dosage, produce syndromes that include features of atropine intoxication. Among the tricyclic antidepressants, protriptyline and amitriptyline are the most potent muscarinic receptor antagonists, with an affinity for the receptor that is approximately one-tenth that reported for atropine. Since these drugs are administered in therapeutic doses considerably higher than the effective dose of atropine, antimuscarinic effects often are observed clinically (see Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders). In addition, overdose with suicidal intent is a danger in the population using antidepressants. Fortunately, most of the newer antidepressants and selective serotonin-reuptake inhibitors are far less anticholinergic (Cusack et al., 1994). In contrast, the newer antipsychotic drugs, classified as 'atypical' and characterized by their low propensity for inducing extrapyramidal side effects, include agents that are potent muscarinic receptor antagonists. In particular, clozapine binds to human brain muscarinic receptors with only fivefold lower affinity than atropine; olanzapine also is a potent muscarinic receptor antagonist (Richelson, 1999). Accordingly, dry mouth is a prominent side effect of these drugs. A paradoxical side effect of clozapine is increased salivation and drooling, possibly the result of selective agonist properties of this drug (Richelson, 1999). Infants and young children are especially susceptible to the toxic effects of atropinic drugs. Indeed, cases of intoxication in children have resulted from conjunctival instillation of atropinic drugs for ophthalmic refraction and for other ocular effects. Systemic absorption occurs either from the nasal mucosa after the drug has traversed the nasolacrimal duct or from the intestinal tract if it is swallowed. Poisoning with diphenoxylate-atropine (LOMOTIL, others), used to treat diarrhea, has been extensively reported in the pediatric literature. Transdermal preparations of scopolamine used for motion sickness have been noted to cause toxic psychoses, especially in children and in the elderly (Wilkinson, 1987; Ziskind, 1988). Serious intoxication may occur in children who ingest berries or seeds containing belladonna alkaloids. Poisoning from ingestion and smoking of jimson weed, or thorn apple, is not uncommon today. Table 72 shows the oral doses of atropine causing undesirable responses or symptoms of overdosage. These symptoms are predictable from the organs receiving parasympathetic innervation. In cases of full-blown atropine poisoning, the syndrome may last 48 hours or longer. Intravenous injection of the anticholinerase agent physostigmine may be used for confirmation. If the typical salivation, sweating, slowing of heart rate, and intestinal hyperactivity do not occur, intoxication with atropine or a related agent is almost certain. Depression and circulatory collapse are evident only in cases of severe intoxication; the blood pressure declines, convulsions may ensue, respiration becomes inadequate, and death due to respiratory failure may follow after a period of paralysis and coma. Measures to limit intestinal absorption should be initiated without delay if the poison has been taken orally. For symptomatic treatment, slow intravenous injection of physostigmine rapidly abolishes the delirium and coma caused by large doses of atropine but carries some risk of overdose in mild atropine intoxication. Since physostigmine is metabolized rapidly, the patient may again lapse into coma within 1 to 2 hours, and repeated doses may be needed (see Chapter 8: Anticholinesterase Agents). If marked excitement is present and more specific treatment is not available, a benzodiazepine is the most suitable agent for sedation and for control of convulsions. Phenothiazines or agents with antimuscarinic activity should not be used, because their antimuscarinic action is likely to intensify toxicity. Support of respiration and control of hyperthermia may be necessary. Ice bags and alcohol sponges help to reduce fever, especially in children. Synthetic and Semisynthetic Substitutes for Belladonna Alkaloids Quaternary Ammonium Muscarinic Receptor Antagonists Ipratropium and Tiotropium Ipratropium bromide ATROVENT) is a
quaternary ammonium compound formed by the introduction of an isopropyl group
to the N atom of atropine. A similar agent, oxitropium bromide, also
is available in Pharmacological Properties Ipratropium produces bronchodilation, tachycardia, and inhibition of secretion similar to that of atropine when it is administered parenterally (Gross, 1988). Although somewhat more potent than atropine, ipratropium and tiotropium lack appreciable action on the CNS but have greater inhibitory effects on ganglionic transmission. An unexpected and therapeutically important property of ipratropium and tiotropium, evident upon either local or parenteral administration, is their minimal inhibitory effect on mucociliary clearance, relative to atropine (see Gross, 1988). Hence, the use of these agents in patients with airway disease avoids the increased accumulation of lower airway secretions encountered with atropine. When ipratropium or tiotropium is inhaled, its action is confined
almost exclusively to the mouth and airways. Dry mouth is the only side
effect reported frequently. Selectivity results from the very inefficient
absorption of the drug from the lungs or the gastrointestinal tract. The
degree of bronchodilation achieved by these agents is thought to reflect the
level of basal parasympathetic tone, supplemented by reflex activation of
cholinergic pathways brought about by various stimuli. In normal individuals,
inhalation of the drugs can provide virtually complete protection against the
bronchoconstriction produced by the subsequent inhalation of such substances
as sulfur dioxide, ozone, or cigarette smoke. However, patients with asthma
or with demonstrable bronchial hyperresponsiveness are less well protected.
Although these drugs cause a marked reduction in sensitivity to methacholine
in asthmatic subjects, more modest inhibition of responses to challenge with
histamine, bradykinin, or prostaglandin F2 Absorption, Fate, and Excretion Ipratropium is administered as an aerosol or solution for inhalation whereas tiotropium is administered as a dry powder. As with most drugs administered by inhalation, about 90% of the dose is swallowed. Most of the swallowed drug appears in the feces. After inhalation, maximal responses usually develop over 30 to 90 minutes, with tiotropium having the slower onset. The effects of ipratropium last for 4 to 6 hours, while tiotropium's effects persist for 24 hours, and the drug is amenable to once-daily dosing (Barnes, 1999; van Noord et al., 2000; Littner et al., 2000). Methscopolamine Methscopolamine bromide PAMINE) is a quaternary ammonium derivative of scopolamine and therefore lacks the central actions of scopolamine. It is less potent than atropine and is poorly absorbed; however, its action is more prolonged, the usual oral dose (2.5 mg) acting for 6 to 8 hours. Its use has been limited chiefly to gastrointestinal diseases. Homatropine Methylbromide Homatropine methylbromide is the quaternary derivative of homatropine. It is less potent than atropine in antimuscarinic activity, but it is four times more potent as a ganglionic blocking agent. It is available in a few combination products intended for relief of gastrointestinal spasm. Propantheline Propantheline bromide PRO-BANTHINE) has been one of the more widely used of the synthetic muscarinic receptor antagonists lacking receptor selectivity. High doses produce the symptoms of ganglionic blockade, and toxic doses block the skeletal neuromuscular junction. Its duration of action is comparable to that of atropine. Glycopyrrolate Glycopyrrolate ROBINUL) is employed orally to inhibit gastrointestinal motility and also is used parenterally to block the effects of vagal stimulation during anesthesia and surgery. Tertiary-Amine Muscarinic Receptor Antagonists Agents useful in ophthalmology include homatropine hydrobromide (ISOPTO HOMATROPINE) (a semisynthetic derivative of atropine; see Figure 72), cyclopentolate hydrochloride (CYCLOGYL), and tropicamide (MYDRIACYL). These agents are preferred to atropine or scopolamine because of their shorter duration of action. Additional information on the ophthalmological properties and preparations of these and other drugs is provided in Chapter 66: Ocular Pharmacology. Tertiary-amine muscarinic receptor antagonists gain access to the CNS and are therefore the anticholinergic drugs used to treat parkinsonism and the extrapyramidal side effects of antipsychotic drugs. Specific agents used primarily for these conditions include benztropine mesylate (COGENTIN) and trihexyphenidyl hydrochloride (ARTANE, others). These drugs are discussed in Chapter 22: Treatment of Central Nervous System Degenerative Disorders. Tertiary amines used for their antispasmodic properties are dicyclomine hydrochloride (BENTYL, others), flavoxate hydrochloride (URISPAS), and oxybutynin chloride (DITROPAN). These agents appear to exert some nonspecific direct relaxant effect on smooth muscle. In therapeutic doses they decrease spasm of the gastrointestinal tract, biliary tract, ureter, and uterus. Flavoxate, oxybutynin, and its more active enantiomer, (S)-oxybutynin, are indicated for overactive bladder. Side effects of dry mouth and eyes limit the tolerability of these drugs with continued use, and patient acceptance declines. Tolterodine (DETROL) is a potent muscarinic antagonist that shows selectivity for the urinary bladder in animal models and in clinical studies. Its selectivity and greater patient acceptance is surprising, since studies on isolated receptors do not reveal a unique subtype selectivity (Chapple, 2000; Abrams et al., 1998, 1999). Inhibition of a particular complement of receptors in the bladder may give rise to synergism and clinical efficacy. Tolterodine's metabolism depends on CYP2B6 to form the 5-hydroxymethyl metabolite. Since this metabolite possesses similar activity to the parent drug, variations in CYP2B6 levels do not affect the duration of action of the drug. Selective Muscarinic Receptor Antagonists Pirenzepine
is a tricyclic drug, similar in structure to imipramine. Pirenzepine has
selectivity for M1-, relative to M2-, and M3-muscarinic
receptors (Caulfield, 1993; Caulfield and Birdsall, 1998). However,
pirenzepine's affinities for M1 and M4 receptors are
comparable, so it does not possess total M1 selectivity. Telenzepine
is an analog of pirenzepine that has higher potency and similar selectivity
for M1-muscarinic receptors. Both drugs are used in the treatment
of acid-peptic disease in Europe, Tripitamine and darifenacin are selective antagonists for M2- and M3-muscarinic receptors, respectively. They are of potential utility in blocking cholinergic bradycardia (M2) and smooth muscle activity or epithelial secretions (M3). Darifenacin is in clinical trial for overactive bladder. While not of current therapeutic value, peptide toxins from the venoms of green and black mambas show the greatest selectivity for specific muscarinic receptor subtypes (M1 and M4) (Caulfield and Birdsall, 1998). Therapeutic Uses of Muscarinic Receptor Antagonists Muscarinic receptor antagonists have been employed in a wide variety of clinical conditions, predominantly to inhibit effects of parasympathetic nervous system activity. The major limitation in the use of the nonselective drugs is often failure to obtain desired therapeutic responses without concomitant side effects. The latter usually are not serious but are sufficiently disturbing to decrease patient compliance, particularly during long-term administration. Selectivity has been achieved by local administration, either by pulmonary inhalation or instillation in the eye. Minimal systemic absorption and dilution from the site of action minimize systemic effects. Subtype-selective muscarinic receptor antagonists hold the most promise for treating specific clinical symptoms; several are in clinical trials. Gastrointestinal Tract Muscarinic receptor antagonists were once the most widely used drugs for the management of peptic ulcer. Although these drugs can reduce gastric motility and the secretion of gastric acid, antisecretory doses produced pronounced side effects, such as dry mouth, loss of visual accommodation, photophobia, and difficulty in urination. As a consequence, patient compliance in the long-term management of symptoms of acid-peptic disease with these drugs was poor. Because of pirenzepine's relative selectivity for M1-muscarinic receptors, it clearly offers a marked improvement over atropine. However, the H2-receptor antagonists and proton pump inhibitors generally are considered to be the drugs of choice to reduce gastric acid secretion (see Chapter 37: Agents Used for Control of Gastric Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease). Most studies indicate that pirenzepine (100 to 150 mg per day) produces about the same rate of healing of duodenal ulcers as the H2 receptor antagonists cimetidine or ranitidine; it also may be effective in preventing the recurrence of ulcers (Carmine and Brogden, 1985; Tryba and Cook, 1997). Although less extensive data are available, similar results have been obtained in the treatment of gastric ulcers. Dry mouth occurred in 14% and blurred vision in 2% to 5% of patients treated with pirenzepine, but these side effects necessitated withdrawal of the drug in fewer than 1% of the patients. Studies in human subjects have shown pirenzepine to be more potent in inhibiting gastric acid secretion produced as a result of neural stimuli than that induced by muscarinic agonists, supporting the postulated localization of M1 receptors at ganglionic sites. The belladonna alkaloids and their synthetic substitutes also have been used and recommended in a wide variety of conditions known or supposed to involve irritable bowel and increased tone ('spasticity') or motility of the gastrointestinal tract. These agents can reduce tone and motility when administered in maximal tolerated doses, and they might be expected to be efficacious if the condition simply involves excessive smooth muscle contraction, a point that is often in doubt. M3-selective antagonists might achieve more selectivity, but M3 receptors also exert a dominant influence on salivation, bronchiolar secretion and contraction, and bladder motility. Alternative agents for treatment of irritable bowel syndrome and its associated diarrhea are discussed in Chapter 38: Prokinetic Agents, Antiemetics, and Agents Used in Irritable Bowel Syndrome. Diarrhea sometimes associated with irritative conditions of the lower bowel, such as mild dysenteries and diverticulitis, may respond to atropine-like drugs. However, more severe conditions such as salmonella dysenteries, ulcerative colitis, and regional enteritis respond poorly. The belladonna alkaloids and synthetic substitutes are very effective in reducing excessive salivation, such as that associated with heavy-metal poisoning or parkinsonism and drug-induced salivation. Uses in Ophthalmology Effects limited to the eye are obtained by local administration of muscarinic receptor antagonists to produce mydriasis and cycloplegia. Cycloplegia is not attainable without mydriasis and requires higher concentrations or more prolonged application of a given agent. Mydriasis often is necessary for thorough examination of the retina and optic disc and in the therapy of iridocyclitis and keratitis. The belladonna mydriatics may be alternated with miotics for breaking or preventing the development of adhesions between the iris and the lens. Complete cycloplegia may be necessary in the treatment of iridocyclitis and choroiditis and for accurate measurement of refractive errors. In instances where complete cycloplegia is required, agents such as atropine or scopolamine, which are more effective, are preferred to drugs such as cyclopentolate and tropicamide. Details of the drugs commonly used are given in Chapter 66: Ocular Pharmacology. Respiratory Tract Atropine and other belladonna alkaloids and substitutes reduce secretion in both the upper and lower respiratory tracts. This effect in the nasopharynx may provide some symptomatic relief of acute rhinitis associated with coryza or hay fever, although such therapy does not affect the natural course of the condition. It is probable that the contribution of antihistamines employed in 'cold' mixtures is due primarily to their antimuscarinic properties, except in conditions with an allergic basis. Systemic administration of belladonna alkaloids or their derivatives for bronchial asthma or obstructive pulmonary disease carries the disadvantage of reducing bronchial secretions and inspissation of the residual secretions. This viscid material is difficult to remove from the respiratory tree, and its presence can dangerously obstruct airflow and predispose to infection. Ipratropium bromide and tiotropium, administered by inhalation, do not
produce adverse effects on mucociliary clearance, in contrast to atropine and
other muscarinic antagonists. Thus, their anticholinergic properties can be
exploited safely in the treatment of reversible airway disease. These agents
often are used with inhalation of long-acting Cardiovascular System The cardiovascular effects of muscarinic-receptor antagonists are of limited clinical application. Generally, these agents are used in coronary care units for short-term interventions or in surgical settings. Atropine may be considered in the initial treatment of patients with acute myocardial infarction in whom excessive vagal tone causes sinus or nodal bradycardia. Sinus bradycardia is the most common arrhythmia seen during acute myocardial infarction, especially of the inferior or posterior wall. Atropine may prevent further clinical deterioration in cases of high vagal tone or AV block by restoring heart rate to a level sufficient to maintain adequate hemodynamic status and to eliminate AV nodal block. Dosing must be done judiciously; doses that are too low can cause a paradoxical bradycardia (see above), while excessive doses will cause tachycardia that may extend the infarct by increasing the demand for oxygen. Atropine occasionally is useful in reducing the severe bradycardia and syncope associated with a hyperactive carotid sinus reflex. It has little effect on most ventricular rhythms. In some patients, atropine may eliminate premature ventricular contractions associated with a very slow atrial rate. It also may reduce the degree of AV block when increased vagal tone is a major factor in the conduction defect, such as the second-degree AV block that can be produced by digitalis. Central Nervous System For many years, the belladonna alkaloids and subsequently the tertiary-amine synthetic substitutes were the only agents helpful in the treatment of parkinsonism. Levodopa or levodopa along with carbidopa now is the treatment of choice, but alternative or concurrent therapy with muscarinic receptor antagonists may be required in some patients (see Chapter 22: Treatment of Central Nervous System Degenerative Disorders). Centrally acting agents such as benztropine have been shown to be efficacious in preventing dystonias or parkinsonian symptoms in patients treated with antipsychotic drugs (Arana et al., 1988; see also Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania). The belladonna alkaloids were among the first drugs to be used in the prevention of motion sickness. Scopolamine is the most effective prophylactic agent for short (4- to 6-hour) exposures to severe motion, and probably for exposures of up to several days. All agents used to combat motion sickness should be given prophylactically; they are much less effective after severe nausea or vomiting has developed. A preparation for the transdermal administration of scopolamine (TRANSDERM SCOP) has been shown to be highly effective when used prophylactically for the prevention of motion sickness. The drug is incorporated into a multilayered adhesive unit that is applied to the postauricular mastoid region. Absorption of the drug is especially efficient in this area. The duration of action of the preparation is about 72 hours, during which time approximately 0.5 mg of scopolamine is delivered. Dry mouth is common, drowsiness is not infrequent, and blurred vision occurs in some individuals. Rare but severe psychotic episodes have also been reported (Wilkinson, 1987; Ziskind, 1988). The use of scopolamine to produce tranquilization and amnesia in a variety of circumstances, including labor, is declining. Given alone in the presence of pain or severe anxiety, scopolamine may induce outbursts of uncontrolled behavior. Uses in Anesthesia The use of anesthetics that are relatively nonirritating to the bronchi has virtually eliminated the need for prophylactic use of muscarinic receptor antagonists. Atropine commonly is given to prevent vagal reflexes induced by surgical manipulation of visceral organs. Atropine or glycopyrrolate is used with neostigmine to counteract its parasympathomimetic effects when the latter agent is used to reverse muscle relaxation after surgery (see Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia). Serious cardiac arrhythmias have occasionally occurred, perhaps because of the initial bradycardia produced by atropine combined with the cholinomimetic effects of neostigmine. Genitourinary Tract Atropine often has been given with an opioid in the treatment of renal colic in the hope that it will relax the ureteral smooth muscle; however, as in biliary colic, it probably does not make a major contribution to the relief of pain. Newer synthetic substitutes of atropine, such as tolterodine, can lower intravesicular pressure, increase capacity, and reduce the frequency of urinary bladder contractions by antagonizing the parasympathetic control of this organ without leading to poorly tolerated side effects. This becomes the basis for the use of such agents in enuresis in children, particularly when a progressive increase in bladder capacity is the objective. These agents also are used to reduce urinary frequency in spastic paraplegia and to increase the capacity of the bladder (Chapple, 2000; Goessl et al., 2000). Anticholinesterase and Mushroom Poisoning The use of atropine in large doses for the treatment of poisoning by anticholinesterase organophosphorus insecticides is discussed in Chapter 8: Anticholinesterase Agents. Atropine also may be used to antagonize the parasympathomimetic effects of neostigmine or other anticholinesterase agents administered in the treatment of myasthenia gravis. It does not interfere with the salutary effects at the skeletal neuromuscular junction, and it is particularly useful early in therapy, before tolerance to muscarinic side effects has developed. As discussed earlier, atropine is useful as an antidote only for the symptoms of mushroom poisoning by the cholinomimetic alkaloid muscarine, found in certain mushroom species. |
Prospectus
|
The availability of cDNAs encoding five distinct muscarinic receptor subtypes has facilitated the development of subtype-selective agents. Functionally selective muscarinic agonists have been in clinical trial for use in treating intellectual impairment associated with Alzheimer's disease; in theory, these agents lack the unwanted effect of concomitant stimulation of presynaptic muscarinic receptors that inhibit release of endogenous ACh. Subtype-selective muscarinic receptor antagonists show promise in several therapeutic settingsfor example, in the treatment of urinary incontinence and for management of irritable bowel syndrome. Therapeutic selectivity may result from targeting unique subsets of receptors that control muscarinic responses within a particular end organ. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3187
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved