| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
DOCUMENTE SIMILARE |
|
Stereoisomeria
Gli stereoisomeri hanno la stessa connessionedegli atomi e differiscono tra loro solo per il modo in cui i legami sono orientati nello spazio.
Un primo tipo di stereoisomeria È l� isomeria conformazionale. Essa È legata all�interconversione dinamica a cui vanno soggette le molecole quando i loro atomi si muovono l�uno rispetto all�altro per rotazione intorno a legami semplici. Gli isomeri conformazionali, o conformeri, comunemente esistono come miscele in equilibrio e sono soggetti a rapida interconversione.
Un altro tipo di stereosiomeria È l�isomeria configurazionale
Gli isomeri configurazionali differiscono l�uno dall�altro solo per la disposizione dei loro atomi nello spazio ed al contrario degli isomeri conformazionali non possono essere interconvertiti per semplice rotazione intorno ai legami semplici. Gli isomeri configurazionali si distinguono in enantiomeri e diastereoisomeri.
Gli enantiomeri sono isomeri configurazionali che sono l�uno l�immagine speculare non sovrapponibile dell�altro, mentre i diastereoisomeri sono tutti quegli isomeri configurazionali che non sono immagini speculari.
Enantiomeri
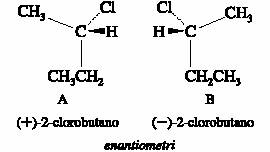
Figura 1
Il (+)-2-clorobutano ed il (�)-2-clorobutano rappresentano due specie molecolari che differiscono l�una dall�altra per l�orientazione degli atomi nello spazio e sono quindi stereoisomeri del 2-clorobutano. Tra le due strutture c�È la stessa relazione che esiste tra un oggetto e la sua immagine speculare. Le due strutture, e quindi anche le due specie molecolari che esse rappresentano, sono immagini speculari non sovrapponibili (Figura 1).
Gli enantiomeri sono composti non simmetrici che si definiscono chirali dal greco �ceir� che significa mano.
Gli enantiomeri, quando si trovano in un contesto
achirale, hanno le stesse proprietÀ fisiche (punto di fusione, punto
di ebollizione, solubilitÀ, assorbimento della luce ordinaria etc.) e
chimiche (velocitÀ di reazione, velocitÀ di formazione etc.)
mentre hanno proprietÀ e
comportamenti differenti in contesti chirali, come per es. i sistemi
biologici e i sistemi ottici che producono
In particolare quando una luce polarizzata che vibra lungo un piano attraversa una soluzione contenente una molecola chirale essa viene deviata di un certo angolo [a] che dipende dalla concentrazione, dalla lunghezza d�onda della luce monocromatica, dalla temperatura, dal solvente usato e dalla lunghezza del percorso ottico.
Per standardizzare i valori delle attivitÀ ottiche si È adottata come unitÀ di misura la rotazione specifica misurata ad una certa temperatura (per es. a 25� C) ed ad una certa lunghezza d�onda (per es. a 589 nm di una lampada al sodio) che È uguale a:
![]()
La misura dell�attivitÀ ottica È molto comune e serve a distinguere gli enantiomeri fra di loro.
Tutti i composti che ruotano il piano della luce polarizzata si definiscono otticamente attivi ed il senso di rotazione del piano della luce polarizzata si indica con il segno piÙ (+) o meno (�).
Il (�)-2-clorobutano ruota il piano della luce polarizzata in senso antiorario mentre il (+)-2-clorobutano ruota il piano della luce polarizzata dello stesso valore ma in senso orario.
Miscele racemiche ed eccessi enantiomerici.
Una miscela racemica contiene un numero uguale di molecole dei due enantiomeri non presenta attivitÀ ottica, e viene contraddistinta dal simbolo (�) posto davanti al nome del composto.
Per es: il 2-bromobutano, ottenuto come prodotto di addizione dell�acido bromidrico alla molecola dell�1-butene (figura 2),
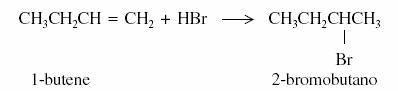
Figura 2
ha un potere rotatorio specifico [a . Non È otticamente attivo e viene indicato come (�)-2-bromobutano.
Se abbiamo una miscela in cui uno dei due enantiomeri È presente in maggiore quantitÀ, la miscela mostrerÀ una rotazione ottica proporzionale alla percentuale della specie in eccesso.
La percentuale dell�enantiomero in eccesso viene indicata come eccesso enantiomerico ed È calcolata mediante una formula che tiene conto della rotazione misurata per la miscela in esame e del potere rotatorio
specifico dell�enantiomero puro.
![]()
Es: Un campione
di 2-bromobutano presenta a 22� C una rotazione specifica [a] = +11.5. Il potere rotatorio specifico del (+)-2-bromobutano a
![]()
La quantitÀ di (+)-2-bromobutano presente in soluzione sarÀ ovviamente del 75% (100+ e.e%)/2. La quantitÀ dell�altro enantiomero sarÀ = (100-ee%)/2.
Una miscela racemica non puÒ essere separata dai suoi componenti mediante i comuni metodi fisici (cristallizzazione, distillazione etc.). Per la separazione si deve prevedere l�uso di specie chirali che interagiscono diversamente con le due molecole di differente chiralitÀ. Nei sistemi biologici, ad esempio, questo ruolo È svolto dagli enzimi che sono in grado di metabolizzare una forma enantiomerica lasciando l�altra inalterata.
Il metodo piÙ usato nei laboratori chimici per la separazione degli enantiomeri o, come si suole dire, per la risoluzione di una miscela racemica, consiste nel trasformare, per reazione con un reagente chirale, gli enantiomeri in composti stereochimicamente differenti (diastereoisomeri) che possiedono cosÌ proprietÀ fisiche diverse.
CONFIGURAZIONE ASSOLUTA
In un composto chirale l�orientazione nello spazio dei gruppi legati allo stereocentro È definita come configurazione assoluta
L�esatta struttura tridimensionale, e quindi la stereochimica di molte molecole organiche viene spesso determinata mediante tecniche di diffrazione ai raggi x
E� importante conoscere che la configurazione di un composto chirale resta inalterata a meno che non venga rotto almeno uno dei legami dello stereocentro. Da ciÒ si evince che per molecole non cristalline in cui non È possibile utilizzare la tecnica ai raggi x la configurazione assoluta puÒ essere accertata per correlazione con composti noti
Per assegnare univocamente il nome e la configurazione degli stereoisomeri si ricorre alle regole di di Cahn-Ingold-Prelog che permettono di descrivere la configurazione di uno stereocentro definendola rispettiva-mennte R (dal latino rectus, quindi �destrorsa�) o S (dal latino sinister, quindi �sinistrorsa�) in base all�ordine con cui i differenti sostituenti sono disposti intorno allo stereocentro.
Le regole sono le seguenti:
1. Si
attribuisce a ciascun gruppo legato allo stereocentro una prioritÀ. La
prioritÀ viene attribuita sulla base del numero atomico dell�atomo legato direttamente allo stereocentro: piÙ alto
È il numero atomico piÙ alta È la prioritÀ del
sostituente. Per esempio: 35Cl > 16O
> 14N >
2. Tra gli isotopi, la prioritÀ spetta all�isotopo con massa atomica maggiore. CosÌ il trizio, l�isotopo dell�idrogeno con massa di 3 uma, ha prioritÀ piÙ alta del deuterio che ha massa atomica di 2 uma. L�idrogeno, che ha numero atomico 1 e massa atomica di 1 uma, ha la piÙ bassa prioritÀ non soltanto rispetto ai suoi isotopi, ma anche rispetto a tutti gli altri elementi della tavola periodica.
3. Se allo stereocentro sono legati due atomi uguali, ad esempio due catene di atomi di carbonio, allora si controllano gli atomi successivi in entrambe le catene, a partire dallo stereocentro fino al punto in cui si
individua una differenza di prioritÀ.
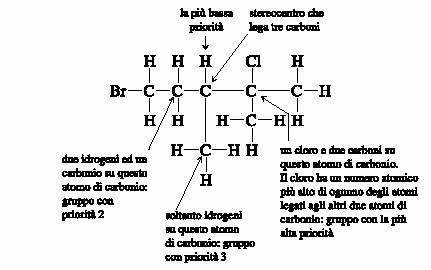
Si noti che all�estremitÀ di una delle catene di atomi di carbonio, È presente un atomo di bromo che, avendo il piÙ alto numero atomico, dovrebbe avere prioritÀ su tutti gli altri atomi presenti. Tuttavia, l�atomo di bromo non influenza l�attribuzione delle prioritÀ in quanto si trova oltre il punto che determina la differenza tra le catene.
4. Un doppio legame viene considerato come due legami semplici per ciascuno degli atomi coinvolti; un triplo legame per tre legami semplici.
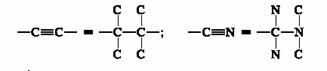
5. Dopo che la prioritÀ È stata assegnata agli atomi e ai gruppi presenti sullo stereocentro, si orienta la molecola in modo tale che il legame tra lo stereocentro ed il sostituente a piÙ bassa prioritÀ sia diretto lontano

dall�osservatore. A questo punto, se per andare dal
gruppo a prioritÀ piÙ alta (prioritÀ 4) a quello a
prioritÀ intermedia (prioritÀ 3) e quindi a quello a
prioritÀ piÙ bassa (prioritÀ 2) si procede in senso
orario, allostereocentro si assegna la configurazione R. Se, invece, si procede in senso antiorario, allo
stereocentro si assegna la configurazione S
È importante mettere in risalto che la designazione della configurazione di un composto come R o S non ha niente a che vedere con il segno della rotazione.Per stabilirlo occorre invece una misura sperimentale di [a
Configurazione relativa.
L�attivitÀ ottica di molti prodotti naturali, in particolare quella degli zuccheri, era nota ai chimici giÀ nel XIX secolo ma per oltre cent�anni la determinazione della configurazione assoluta risultÒ un problema di non semplice soluzione. Per esempio, lo zucchero otticamente attivo piÙ semplice, il 2,3-diidrossipropanale, comunemente chiamato gliceraldeide, ha un solo stereocentro ed esiste come coppia di enantiomeri, la (+)-gliceraldeide e la (�)-gliceraldeide. Non esiste alcun modo diretto per distinguere se la (+)-gliceraldeide corrisponda all�enantiomero R oppure all�enantiomero S
In altre parole, la reale disposizione degli atomi legati allo stereocentro, che corrisponde alla configurazione assoluta di un composto, non puÒ essere assegnata sulla base del segno del potere rotatorio.
Mediante processi di conversione chimica, È possibile stabilire il rapporto di configurazione tra due composti otticamente attivi, trasformando un composto nell�altro tramite reazioni che non implicano la rottura di un legame di un centro chirale. anche senza conoscerne la configurazione assoluta, applicando l�assioma che la configurazione di un composto chirale resta inalterata a meno che non venga rotto almeno uno dei legami dello stereocentro
CosÌ per es. assegnando arbitrariamente alla D(+) gliceraldeide la configurazione riportata nello schema È possibile partendo da essa ottenere mediante sintesi chimica, l�acido (-)-tartarico che È l�acido tartarico meno abbondante in natura. CosÌ facendo si È assegnata la configurazione all�atomo di carbonio C2 e di conseguenza si È assegnata la configurazione relativa all�intera molecola.
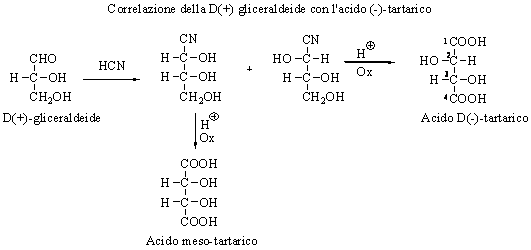
In questo modo sono state stabilite le configurazioni relative di un grande numero di composti organici.
Nel 1951, il chimico olandese J.M. Bijvoet riuscÌ a determinare la configurazione assoluta dell�acido (+)-tartarico, usando la tecnica di diffrazione dei raggi x. Determinata la configuraione di questo acido si È ovviamente assegnata per confronto la configurazione assoluta dell�acido (-)-tartarico e di conseguenza della D(+)gliceraldeide che risulta essere la (R)-(+)-gliceraldeide.
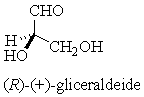
Elementi di simmetria
Un modo facile per verificare la chiralitÀ o meno di una molecola È quello di individuare la presenza o l�assenza di simmetria mediante operazioni di simmetria
In un sistema geometrico sono operazioni di simmetria quelle operazioni (ad esempio, rotazioni, riflessioni etc.) in seguito alle quali si riporta il sistema a coincidere con s� stesso, cioÈ in una posizione indistinguibile
da quella di partenza.
Tali operazioni sono determinate dagli elementi di simmetria (assi per le rotazioni, piani per le riflessioni e loro combinazioni) che sono classificati come primari e secondari
Sono elementi di simmetria primari il piano di simmetria (o piano di riflessione) , il centro di inversione (o centro di simmetria), e l�asse alternante.
Il primo (che si indica con s) È un piano ideale che divide una molecola in due parti uguali l�una immagine speculare dell�altra: alcuni esempi sono mostrati in Figura 3, utilizzando la molecola del butano,
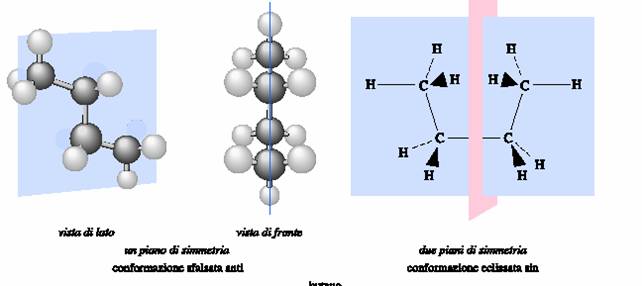
Figura 3
Una molecola possiede un centro di inversione (che si indica con i) quando ad ogni atomo ne corrisponde un altro uguale ed opposto e ad uguale distanza dal centro (figura 4). Esso È quindi un punto ideale per il quale in una molecola passano le congiungenti di tutti gli atomi uguali ed opposti. Es. meso 2,3-diclorobutano
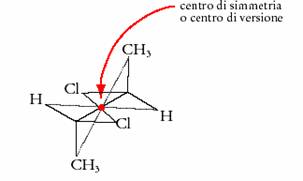
Figura 4
Asse alternante prodotto da una rotazione piÙ una riflessione. Si indicano con la sigla Sn, ove n indica il numero di volte in cui, in una rotazione di 360�, si ripetono le operazioni di simmetria. Es. di asse alternante S4. Figura 5.
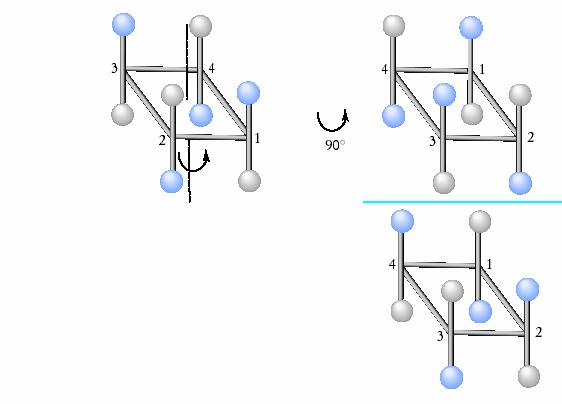
Figura 5
Gli elementi di simmetria secondari sono gli assi semplici di rotazione (o assi di simmetria), assi ideali, per rotazione intorno ai quali di 360�, una molecola si trova n volte in una posizione indistinguibile da quella di partenza. Si indicano con la sigla Cn ed a seconda del valore di n si chiamano binari (C2), ternari (C3), quaternari (C4) e cosÌ via. Un esempio È mostrato in Figura 6.
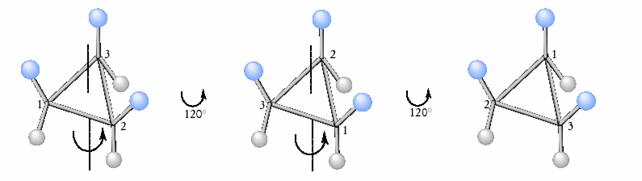
Figura 6
È abbastanza comune che una molecola achirale mostri piÙ di un elemento di simmetria: La presenza di un elemento di simmetria primario È condizione sufficiente per l�assenza di chiralitÀ, mentre la presenza di soli elementi di simmetria secondari, quali gli assi semplici di rotazione, Cn, non È sufficiente a rendere la molecola achirale.
Per esempio, come si puÒ vedere dalla Figura 3, il butano nella conformazione anti non È chirale in quanto presenta un piano di simmetria che passa per i quattro atomi di carbonio e per due degli atomi di idrogeno primari legati ai due atomi di carbonio 1 e 4 e che biseca gli angoli tra le coppie di atomi di idrogeno secondari. Nella conformazione eclissata si possono individuare, cosÌ come È mostrato in figura, addirittura due piani di simmetria.
Come conseguenza della sua simmetria, una molecola di butano nella conformazione anti È sovrapponibile alla sua immagine speculare. Esso È quindi achirale
Il 2-clorobutano Figura 1, al contrario, non ha elementi di simmetria primari. Nessuna conformazione del 2-clorobutano presenta un centro di simmetria o un piano di simmetria e pertanto la molecola È chirale.
A volte non È immediata l�individuazione di un elemento di simmetria primario: ad esempio, le due forme gauche del butano non hanno elementi di simmetria ed infatti rappresentano una coppia di enantiomeri (figura 7).
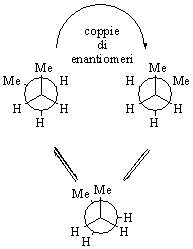
Figura 7
Non si puÒ perÒ parlare di forme otticamente attive del butano in quanto le due forme gauche sono anche conformeri e sono quindi in equilibrio con altri conformeri, invece simmetrici. In generale, È sufficiente che una sola conformazione sia achirale perch� la molecola sia achirale: ovviamente, tale conformazione achirale deve essere raggiungibile e la barriera energetica che la separa dalle conformazioni enantiomere non deve essere cosÌ elevata da impedire la libera rotazione
Nel caso del butano le due forme gauche enantiomere si convertono l�una nell�altra attraverso la forma
sin, conformazione eclissata che È simmetrica (Figura 7). La barriera energetica È piuttosto bassa (3.8 kcal mol�1, 16 kJ mol�1) e l�interconversione a temperatura ambiente È continua.
Esistono casi in cui la barriera energetica È troppo alta e le conformazioni enantiomere non possono interconvertirsi di modo che esse esistono come coppia di enantiomeri che si definiscono enantiomeri conformazionali
PROIEZIONI DI FISCHER

2. Nel proiettare una molecola contenente un solo stereocentro la catena lineare piÙ lunga di atomi di carbonio deve essere riportata sulle valenze verticali, collocando in alto il carbonio al quale spetta il piÙ basso numero di posizione derivante dalla nomenclatura IUPAC.

Acido 2-idrossipropanoico [acido( )-lattico] acido (-)-lattico
3. Nelle proiezioni di Fischer non sono consentiti
spostamenti arbitrari di atomi o gruppi, in quanto lo scambio di due qualsiasi
sostituenti comporta l�inversione della configurazione dello stereocentro. Se
si esamina
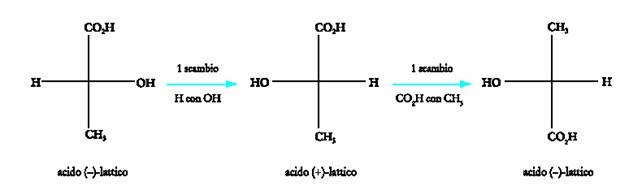
4. Le proiezioni di Fischer non possono essere ruotate arbitrariamente sul piano del foglio. Infatti ogni rotazione di 90� equivale ad un numero dispari di scambi e porta all�inversione della configurazione dello stereocentro, mentre una rotazione di 180� equivale ad un numero pari di scambi e mantiene inalterata la configurazione di partenza. Quanto detto appare chiaro dalla figura seguente in cui si puÒ vedere che partendo dalla proiezione dell�acido (�)-lattico si ottiene, per rotazione di 90� una nuova proiezione
che corrisponde ad un numero dispari (3) di scambi e quindi a quella del suo enantiomero.
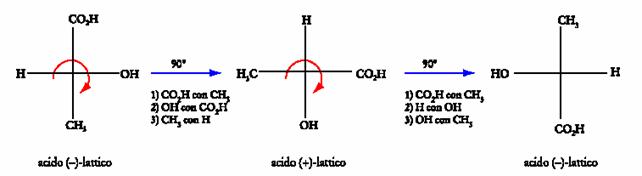
E�possibile assegnare la configurazione R o S ad uno stereocentro, avendo a disposizione una proiezione di Fischer. Per fare ciÒ basti che il gruppo a prioritÀ piÙ bassi si trovi sulla verticale.
Es.

Molecole con due stereocentri
Nel caso di composti con due stereocentri, come ad esempio l�acido tartarico mostrato nella successiva figura, si utilizzano proiezioni di Fischer costituite da una doppia croce. I punti di intersezione tra le linee rappresentano i due stereocentri, le valenze verticali rappresentano i legami diretti dietro il foglio, lontano dall�osservatore, e quelle orizzontali i legami rivolti verso l�osservatore.
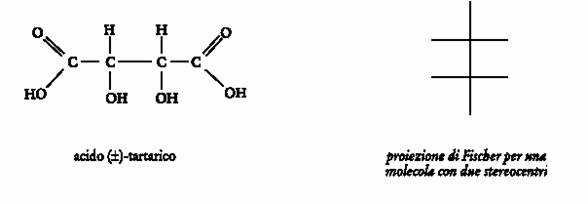
Una proiezione di Fischer ottenuta in questo modo rappresenta la molecola nella sua conformazione eclissata. Volendo rappresentare in proiezione di Fischer uno degli stereoisomeri dell�acido tartarico, si dovranno effettuare le seguenti operazioni. Si rappresenta inizialmente la molecola mediante le formule a cavalletto (rappresentazione spaziale della molecola in prospettiva: visione della molecola dall�alto e da un lato). Quindi si dispone la molecola in una conformazione eclissata in modo da collocare la catena lineare piÙ lunga lontano dall�osservatore e si proietta la molecola sul piano, ricordando di disporre l�atomo di carbonio 1( n.ox. piÙ elevato) in alto.
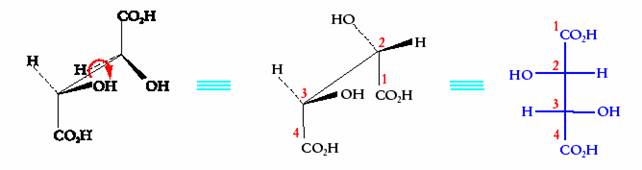
Lo stesso tipo di trasformazione, effettuata su tutti gli stereoisomeri dell�acido tartarico, porta a scrivere le seguenti quattro proiezioni di Fischer:
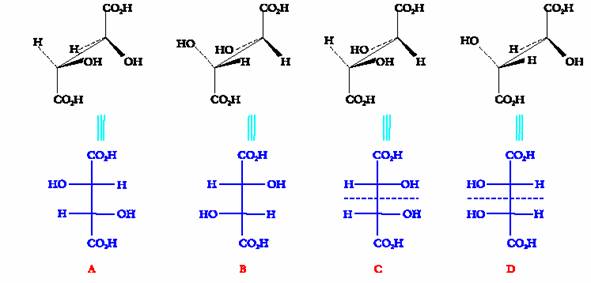
Stereoisomeria dei composti ciclici
Composti cis e trans
La rigiditÀ dell�anello e la mancanza di libera rotazione intorno ai legami carbonio-carbonio sono all�origine della stereoisomeria.
Per es. dell�1,2-dibromociclopentano esistono due isomeri geometrici: uno in cui i due sostituenti sono da parti opposte rispetto al piano dell�anello, ossia trans l�uno rispetto all�altro, ed un altro in cui essi si trovano dalla stessa parte, ossia cis l�uno rispetto all�altro.
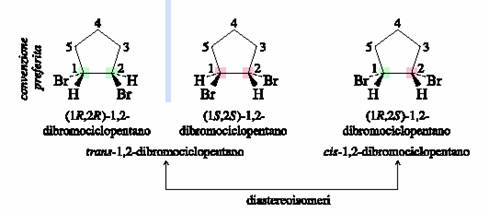
Del trans sono possibili due enantiomeri l�(1R,2R) e l�(1S,2S). Del cis, la cui configurazione assoluta È: (1R,2S) non sono possibili coppie di enantiomeri. Quest�ultima molecola, infatti, presenta un piano di simmetria ed È quindi un composto meso.
La configurazione degli stereocentri si puÒ assegnare utilizzando le proiezioni di Fischer. Per rappresentare, ad esempio, la molecola dell�(1S S)-1,2-dibromociclopentano in proiezione di Fischer bisogna disporre il modello tridimensionale con la catena di atomi di carbonio piÙ lunga, rappresentata dall�anello, che si allontana dall�osservatore. Per ottenere questo risultato È sufficiente ruotare la molecola di 90� attorno all�asse indicato in figura. In questo modo l�anello si dispone perpendicolarmente al foglio mentre i due sostituenti sui carboni 1 e 2 si orientano verso l�osservatore.
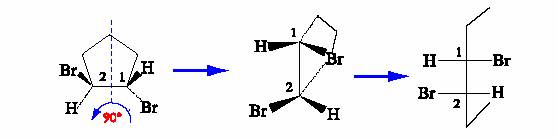
Una volta disposta la molecola secondo le convenzioni delle proiezioni di Fischer essa puÒ essere proiettata sul piano. Conoscendo la prioritÀ dei gruppi È, facile assegnare la configurazione R o S ai carboni asimmetrici in base alle convenzioni di Cahn-Ingold-Prelog.
Configurazione e conformazione dei cicloesani disostituiti
Per un cicloesano con un solo sostituente sull�anello, la conformazione preferita È quella a sedia in cui il sostituente È in posizione equatoriale. Quando sull�anello ci sono due sostituenti, essi possono essere cis o trans l�uno rispetto all�altro. Per esempio, vi sono tre stereoisomeri dell� 1,2-dimetilcicloesano: una coppia di enantiomeri trans e un isomero cis.

Le conformazioni dell�(1S,2S)-1,2-dimetilcicloesano sono:
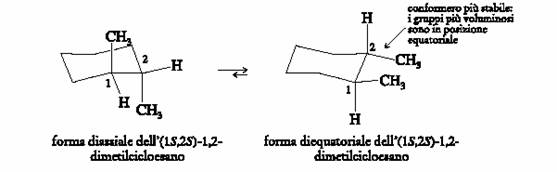
La conformazione diequatoriale conserva tra i due metili la stessa relazione spaziale della conformazione diassiale. Il gruppo metile sull�atomo di carbonio 1 si trova al di sopra del piano dell�anello e quello sull�atomo di carbonio 2 si trova al di sotto.
Se i due composti sono conformeri dello stesso stereoisomero, i metili devono assumere queste orientazioni e il cambio di conformazione non modifica la configurazione degli stereocentri. Ciascun enantiomero del trans-1,2-dimetilcicloesano esiste principalmente in due conformazioni, delle quali la diequatoriale predomina a temperatura ambiente nella miscela di equilibrio.
Nel cis-1,2-dimetilcicloesano, un gruppo metile È equatoriale e l�altro È assiale. L�interconversione dell�anello porta il metile originariamente equatoriale in posizione assiale e quello originariamente assiale in posizione equatoriale.
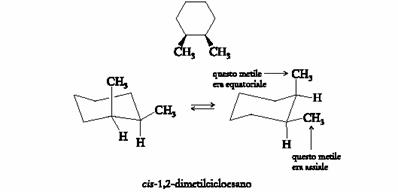
Questi due conformeri hanno uguale energia e quindi sono presenti all�equilibrio in quantitÀ uguale. Essi sono anche enantiomeri e vengono definiti enantiomeri conformazionali. Sebbene ciascuno di essi sia chirale, la loro rapida interconversione a temperatura ambiente ne rende impossibile la separazione e pertanto la miscela, costituita dai due enantiomeri, non È otticamente attiva.
Fra i cicloesani 1,3-disostituiti, il piÙ stabile È l�isomero cis. Il cis-1,3- dimetilcicloesano ha due conformazioni, una in cui entrambi i metili sono equatoriali e l�altra in cui essi sono entrambi assiali.
Il conformero diequatoriale del cis-1,3-dimetilcicloesano È piÙ stabile di quello diassale di circa 5.4 kcal mol-1 (22.6 kJ mol�1) e ciÒ È dovuto principalmente alle interazioni 1,3-diassiali dei due metili.
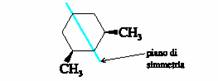
Il cis-1,3-dimetilcicloesano ha un piano di simmetria ed È un composto meso.

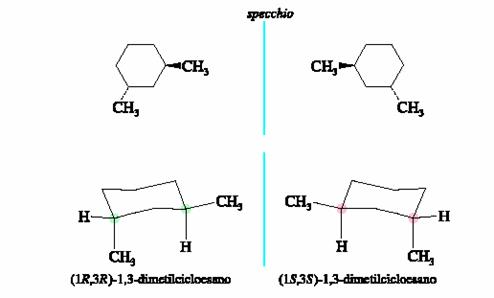
Il trans-1,3-dimetilcicloesano esiste come coppia di enantiomeri e ciascun enantiomero ha un gruppo metile assiale e uno equatoriale.
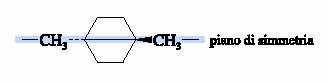
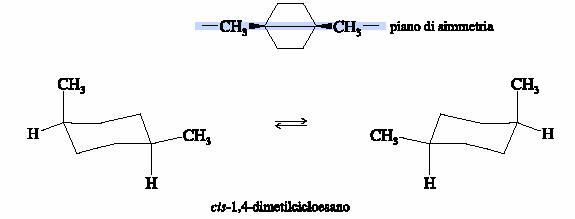
Il cis ed il trans-1,4-dimetilcicloesano non hanno stereocentri e sono entrambi otticamente inattivi. Ciascuna delle due molecole ha un piano di simmetria che passa per gli atomi di carbonio 1 e 4 e per gli atomi di idrogeno ed i gruppi metili ad essi legati.

|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 8633
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved