| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
DOCUMENTE SIMILARE |
|
TÉCNICAS DE AISAMIENTO, PURIFICACIÓN Y CARACTERIZACIÓN DE BIOMOLÉCULAS
Metodos o técnicas de cromatografía:
La cromatografía implica el paso de una solución a través de un medio que presenta una adsorción relativa para los distintos solutos. Puede desarrollarse en papel, capa fina o columna. Las dos primeras no son muy utilizadas en la actualidad, por ello veremos las de columna.
Las columnas suelen ser de vidrio y se rellenan con un material que puede adsorber moléculas de modo selectivo debido a alguna diferencia en su estructura química. Este material se introduce suspendido en una disolución tampón adecuada. A continuación, la muestra la colocamos en la parte superior de la columna, y vamos añadiendo solución tampón lentamente, la cual va pasando lentamente a lo largo de la columna, y recogemos por la parte inferior de la columna fracciones del tampón que salen.
Este proceso es la “ELUCIÓN”. Lo que sale de la columna es el “eluído”, y la disolución tampón que va atravesando la columna es el “eluyente”. Las moléculas que no se adsorben al material con el que rellenamos la columna o que lo hacen débilmente son las primeras en salir, y las recogemos en las primeras fracciones. Las moléculas que se adsorben más fuertemente tardan más en eluír y además en algunas ocasiones incluso es necesario cambiar la composición del tampón de elución para que estas moléculas se suelten y las podamos recoger en la fracción.
Tanto el relleno como la disolución tampón se seleccionan dependiendo de la base que desee usarse para la separación.
Según cual sea el fundamento de la separación, distinguimos 3 grandes tipos de cromatografía en columna:
de intercambio iónico
de afinidad
de exclusión molecular
• también estudiaremos : - HPLC
- cromatografía de gases
Las cromatografías de intercambio iónico
se usan para separar moléculas de acuerdo con sus cargas eléctricas. Aquí el material de relleno de la columna es una resina de intercambio iónico. Estas son polianiones o policationes. Supongamos que separamos 3 clases de moléculas de una mezcla: una con carga negativa, otra con carga positiva débil y otra con positiva fuerte. La resina que debemos usar en este caso es una aniónica, que lleva grupos cargados negativamente por lo que las moléculas de la mezcla que están cargadas negativamente pasarán a través de la columna sin adsorberse y se recogerán en las fracciones iniciales. Las moléculas con carga positiva serán ligadas por la resina y se unirán más fuertemente con las de mayor carga positiva. Para separarlas de la columna y recogerlas en el eluído usamos un tampón con un gradiente de NaCl ya que estas disoluciones salinas rompen las interacciones electrostáticas. Vamos metiendo aumentando gradualmente la concentración de NaCl en el tampón de elución. A una pequeña concentración recogemos en el eluído las partículas de carga positiva débil y a mayor concentración de sal las de cargas positiva fuertes.
Cromatografía de afinidad:
es más específica, se basa en que muchas proteínas interaccionan fuertemente con otras moléculas como por ejemplo sucede con las encimas y análogos de sustratos, o como sucede con los antígenos y anticuerpos. Las moléculas adecuadas (análogo del sustrato o el anticuerpo) se unen covalentemente al material con el que se rellena la columna y van a actuar a modo de anzuelos moleculares para pescar a la proteína adecuada. El resto de las moléculas que no se unen al anzuelo simplemente pasan a través de la columna y salen en las fracciones iniciales.
Para eluír las moléculas que se han unido al anzuelo lo que se hace es eluír con una sal tampón que mantenga moléculas anzuelo libres o bien algún otro reactivo que pueda romper la unión entre la proteína o análogo del sustrato o anticuerpo.
Cromatografía de exclusión molecular:
se denomina también de filtración en gel, y en este caso las moléculas las vamos a separar en función de su tamaño y no de sus propiedades químicas. En este caso la columna se rellena con esferas de un gel poroso. Es muy empleado el “sephadox”, un polímero ramificado de la glucosa. Se dispone de sephadox de distintas porosidades y seleccionamos aquella según el tamaño de las moléculas que queremos purificar de modo que las moléculas más pequeñas de la mezcla pueden penetrar en las esferas mientras que las más grandes no pueden hacerlo. La mezcla se aplica en la parte superior de la columna y se va eluyendo con un disolución tampón. Las moléculas más grandes se mueven más rápido ya que no pueden penetrar en las esferas del gel sino que se cuelan en los intersticios entre ellas, entonces estas saldrán más rápidamente en las primeras fracciones mientras que las moléculas pequeñas pueden entrar en las esferas y tardan más en salir, eluyéndose consecuentemente en orden decreciente de pesos moleculares.
La cromatografía en gel también sirve para determinar pesos moleculares ya que existe una relación lineal entre el logaritmo del peso molecular de las proteínas globulares y el volumen de elución en un rango de pesos moleculares que depende del tipo de gel usado.
Hacemos pasar una serie de proteínas de peso molecular conocido (patrones) y medimos en qué volumen recogemos estas y representamos en unos ejes coordinados el logaritmo del peso molecular frente al volumen de elución (Ve). Observamos una línea de puntos recta. Así podemos calcular el peso molecular a partir del volumen de elución con la gráfica.
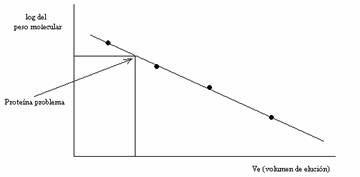
Para construir una recta de calibrado de este tipo necesitamos medir una serie de parámetros de la columna que son :
• Vt o volumen total de la columna. Es el volumen que ocupa el gel más el volumen de los intersticios. Se determina haciendo pasar una molécula con bajo peso molecular como NaCl y midiendo el volumen en que la recogemos.
• Vo o volumen de exclusión. Es el volumen de los huecos que hay entre las partículas de gel. Para calcularlo hacemos pasar a través de la columna una molécula de peso molecular alto, generalmente azul de dextramo.
• Ve o volumen de elución, que es el volumen en el cual recogemos cada una de las fracciones que recuperamos de las proteínas.
En lugar del Ve podemos usar el Kav , que es el coeficiente entre la fase líquida y el gel: ![]()
Cromatografía HPLC
( High Performance Liquid Chromatography o Cromatografía líquida de alta eficacia):
Las cromatografías estudiadas hasta ahora eran lentas, ya que solo se aplicaba una presión hidrostática para que los líquidos se colasen por la columna. Por ello, la elución suele tardar varias horas. En este tiempo las moléculas más frágiles se pueden deteriorar. Otro problema con que nos encontramos es que al ser un proceso lento, la muestra tiende a difundir (esparcirse) a medida que baja a lo largo de la columna. Cuanto más dure la separación peor será la resolución.
La HPCL resuelve estos problemas ya que usa unas presiones entre 5.000 y 10.000 psi para hacer que los líquidos atraviesen más rápidamente la columna, con lo que las separaciones que tardaban horas se reducen a minutos y la resolución es más alta debido a que no hay tanta difusión.
Para poder hacer esto fue necesario desarrollar resinas no compresibles con las que rellenar la columna, y además el cambio de columnas de vidrio por unas metálicas, más resistentes para soportar las presiones.
Este tipo de cromatografía HPLC también puede ser usado como cromatografía de intercambio iónico, de afinidad
Cromatografía de gases:
Hasta ahora el eluyente era un líquido. Aquí es un gas. Esta cromatografía tiene lugar con la columna rellenada de un sólido inerte que está finamente dividido de materiales muy diversos como tierra diatomeas, cromosol. Este material se empapa de un líquido no volátil como un aceite de silicona que constituye la “fase estacionaria”.
El eluyente es un gas inerte como helio, argón o nitrógeno que se hace fluir a velocidad constante a través de la columna.
La muestra debe ser volátil ( para las moléculas no volátiles como aminoácidos, se hacen reacciones químicas que las transformen en derivados volátiles) que se introducen en la columna disueltos con un disolvente orgánico que se evaporan al introducirlos a alta temperatura por calentamiento.
Los productos volátiles se van separando a lo largo de la columna según su reparto entre las dos fases, la estacionaria y la móvil (gas o “gas portador”). Como la muestra se volatiliza a alta temperatura, y es a la que se produce la operación, las columnas están metidas en un horno.
Los productos se van cuantificando a la salida de la columna mediante detectores (muy variados) que están acoplados a registradores que nos van a dar un registro o cromatograma en el que cada componente que hemos separado se va a corresponder con un pico que sale cada cierto tiempo de retención que nos permite identificar a qué molécula corresponde cada tipo, siempre por comparación de patrones conocidos.
El área de cada pico nos permite cuantificar el compuesto que hay en la muestra. Esta es una técnica analítica, cuantificamos y cualificamos. No es una técnica preparativa porque recuperamos cantidades mínimas.
Electroforesis:
Son aquellas en las que aplicamos un campo eléctrico a una solución con moléculas del soluto con carga positiva. Estas se desplazan hacia el cátodo y las de carga negativa hacia el ánodo. A este desplazamiento se le denomina electroforesis.
La velocidad de las moléculas en la electroforesis depende de 2 factores:
En primer lugar, guiando el movimiento está la fuerza producida por el
campo sobre la partícula que se corresponde por ![]() donde “q” es la carga
de la molécula en Culombios, y
donde “q” es la carga
de la molécula en Culombios, y ![]() es la carga del campo
eléctrico en
es la carga del campo
eléctrico en ![]() . Pero resistiendo al
movimiento está la fuerza de fricción que ejerce el entorno sobre la partícula
que es
. Pero resistiendo al
movimiento está la fuerza de fricción que ejerce el entorno sobre la partícula
que es ![]() siendo “v” la
velocidad de la partícula y “F” su coeficiente de fricción, que depende del
tamaño y forma de las partículas (moléculas) de modo que las grandes o
asimétricas presentan coeficientes de fricción mayores que las pequeñas.
siendo “v” la
velocidad de la partícula y “F” su coeficiente de fricción, que depende del
tamaño y forma de las partículas (moléculas) de modo que las grandes o
asimétricas presentan coeficientes de fricción mayores que las pequeñas.
Cuando se activa el campo eléctrico, las
moléculas se aceleran hasta alcanzar una velocidad en la que ambas fuerzas se
igualan, siendo ![]()
A continuación se mueve constantemente a esa
velocidad. Esta igualdad también la podemos expresar así: ![]() o “movilidad
electroforética”, y es la velocidad de movimiento por unidad de fuerza de
campo. Consecuentemente, la movilidad de una molécula es la electroforesis, y
depende de su carga y dimensiones moleculares.
o “movilidad
electroforética”, y es la velocidad de movimiento por unidad de fuerza de
campo. Consecuentemente, la movilidad de una molécula es la electroforesis, y
depende de su carga y dimensiones moleculares.
Aunque la electroforesis se puede llevar a cabo en disolución, normalmente se usa algún soporte. Las dos más corrientes son el papel y el gel.
La de papel se usa para separar moléculas cargadas pequeñas. Se hace que la tira de papel de filtro se extienda humedecida en un tampón entre dos cámaras que son las que contienen los dos electrodos. En el centro del papel se coloca una gota de la muestra a analizar y se conecta el campo eléctrico. Al cabo de una hora, cuando las moléculas se han separado, se seca el papel y se tiñe con un colorante que nos deja ver las moléculas que estamos separando. Cada molécula la identificamos como una mancha, y se habrá desplazado hacia el ánodo o cátodo según su carga, y una determinada distancia que depende de su carga y dimensiones.
Al igual que en la cromatografía cada componente lo identificamos comparándolo con factores conocidos.
Actualmente se usa más la electroforesis en gel, que se emplea para separar proteínas y ácidos nucleicos. Se coloca un gel que contenga la disolución tampón adecuada entre dos placas de cristal. Estos geles son láminas finas de tan solo uno milímetros de espesor. Los geles suelen ser de poliacrilamida o agrarosa. La primera es para separar proteínas y ácidos nucleicos de bajo peso molecular. La agarosa para separar ácidos nucleicos de mayor tamaño. El gel se coloca entre los compartimentos que tienen los electrodos.
La muestra se aplica en la parte superior del gel, en los “pocillos” (hendiduras) y se le añade glicerol y un colorante. El primero es para que la muestra sea densa y se quede en el pocillo y no difunda por el tampón que hay en receptáculo superior. El segundo (colorante) nos sirve para seguir el proceso de la electroforesis. Cuando el colorante llega abajo, desconectamos, y aquí se supone que las moléculas ya están separadas. Con esto realizado retiramos el gel y lo teñimos con un colorante que nos permita visualizar las moléculas que estamos separando, que las veremos como bandas estrechas.
Si la electroforesis se realiza en gel, la movilidad es menor de la que presentan las moléculas en disolución por el efecto de tamizado molecular que ejerce el soporte, que es más fuerte cuanto más concentrado esté el gel.
Para moléculas cuya carga es proporcional a su longitud, como el DNA, que presenta una unidad de carga en cada residuo, presentan una movilidad libre casi independiente de su tamaño. Entonces, en la electroforesis en gel la movilidad de estas moléculas viene determinada por el efecto del tamizado molecular del gel.
Así, mediante electroforesis en gel podemos separar las moléculas casi exclusivamente por su tamaño.
Representamos el logaritmo del peso molecular frente a la distancia recorrida, y obtenemos unos puntos que se ajustan a una línea recta.
Técnicas de Diálisis y Ultracentrifugación
La diálisis y la ultracentrifugación sirven para concentrar y purificar biopolímeros. Se basan en la utilización de membranas semipermeables que permiten el paso de moléculas pequeñas pero no de proteínas o otras macromoléculas.
La diálisis se usa normalmente para eliminar moléculas pequeñas contaminantes o para cambiar las condiciones de una solución tampón.
Habitualmente se coloca la muestra (disolución de una proteína, por ejemplo) en una bolsa cerrada pero con las paredes de membranas semipermeables y se sumerge en un volumen mayor de una disolución tampón dentro de un recipiente. El tampón se somete a una agitación mediante un agitador magnético situado debajo de la muestra en la bolsa.
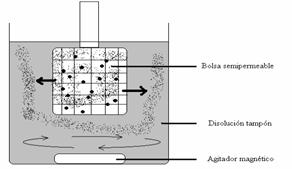
Las moléculas pequeñas “contaminadoras” saldrán de esta bolsa hacia la disolución tampón, y esta tenderá a reemplazar el líquido disolvente en el que tenemos la muestra. Si reemplazamos el tampón varias veces, al final tendremos la proteína disuelta en la disolución tampón exterior y habremos eliminado otras moléculas pequeñas que al principio la acompañaban.
La ultracentrifugación se utiliza para
concentrar disoluciones de macromoléculas. También se usan membranas semipermeables,
pero se aplica presión para hacer salir el disolvente y las moléculas pequeñas.
Hay una gran diversidad de métodos. El más sencillo consta de un recipiente con
una membrana semipermeable al que se le aplica una presión con ![]() gas como se indica en
el dibujo:
gas como se indica en
el dibujo:
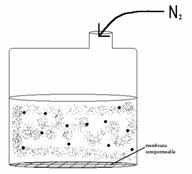
La diálisis purifica, y la ultracentrifugación concentra. Otra técnica es la diafiltración, una combinación de las anteriores, donde se concentra y se eliminan contaminantes al mismo tiempo.
Primero se concentra la muestra por ultracentrifugación y se le añade la disolución tampón (gran volumen) y la volvemos a concentrar hasta que queda un pequeño volumen, y así sucesivas veces.
Técnicas de Radiactividad
Los isótopos radiactivos se empezaron a
utilizar después de la segunda guerra mundial, y presentaron un gran avance ya que amplían en varios órdenes
de magnitud la sensibilidad con la que detectamos una especie química. Los
análisis tradicionales permiten detectar moléculas en cantidades mínimas de
“nanomoles” (![]() moles), mientras que con estas técnicas llegamos a una
sensibilidad de “fentomoles” (
moles), mientras que con estas técnicas llegamos a una
sensibilidad de “fentomoles” (![]() moles).
moles).
Los compuestos marcados radiactivamente son los “trazadores”, ya que se pueden seguir las transformaciones que sufren en presencia de un exceso de material no radiactivo.
En bioquímica se usan dos isótopos, los
radiactivos y los estables. En el caso del hidrógeno, tenemos el tritio (![]() ), radiactivo, y el deuterio (
), radiactivo, y el deuterio (![]() ), estable.
), estable.
La utilidad de los isótopos estables la resumimos en:
- incorporando un isótopo estable aumentamos la densidad de un material, lo que nos proporciona un método físico para separar los compuestos marcados de los no marcados.
Los compuestos marcados con
isótopos estables, en especial el ![]() , se usan mucho en experimentos de resonancia magnética
nuclear para el estudio de la estructura molecular y de los mecanismos de
reacción.
, se usan mucho en experimentos de resonancia magnética
nuclear para el estudio de la estructura molecular y de los mecanismos de
reacción.
- Como trazadores, cuando no se dispone de los
radiactivos adecuados de ese elemento,
como es el caso del ![]() y
y ![]() , que no poseen radiactivos
, que no poseen radiactivos
De entre los isótopos radiactivos, en
bioquímica se usan los que emiten radiaciones ![]() y
y ![]() , pero no con emisiones
, pero no con emisiones ![]() . - Una
. - Una ![]() es un electrón emitido
es un electrón emitido
- Una ![]() es una radiación
electromagnética (fotón con alta energía)
es una radiación
electromagnética (fotón con alta energía)
Las emisiones ![]() se usan sobre todo en
inmunología ya que existen isótopos del yodo que son emisores
se usan sobre todo en
inmunología ya que existen isótopos del yodo que son emisores ![]() y es fácil unir estos
isótopos a anticuerpos. Salvo esta excepción, en bioquímica, los más usados son
los emisores de radiaciones
y es fácil unir estos
isótopos a anticuerpos. Salvo esta excepción, en bioquímica, los más usados son
los emisores de radiaciones ![]() .
.
La radiactividad es un proceso cinético de primer orden: el número de desintegraciones que se producen en un determinado intervalo de tiempo depende solamente del número de isótopos radiactivos presentes.
Este fenómeno da lugar a la ley de desintegración radiactiva que tiene esta expresión:
![]() ,
donde: -
,
donde: - ![]() es el número de átomos radiactivos en el “t” = 0
es el número de átomos radiactivos en el “t” = 0
- N es en ns de átomos que quedan en el tiempo “t”
- ![]() es la constante de desintegración
es la constante de desintegración
La constante de desintegración es
característica para cada isótopo, y está relacionada con otro parámetro que se
emplea más, que se llama “hemivida” o ![]() , que es el tiempo
requerido para que se desintegren la mitad de los núcleos de una muestra, y es
igual a:
, que es el tiempo
requerido para que se desintegren la mitad de los núcleos de una muestra, y es
igual a:
![]() o
o ![]() La hemivida es característica de
cada isótopo.
La hemivida es característica de
cada isótopo.
La unidad básica de desintegración es el
“Curio” (Ci). 1 Ci es la cantidad de radiactividad equivalente a la que existe
en un gramo de radio, es decir, ![]() desintegraciones por
minuto. En bioquímica se trabaja con cantidades de microcurios (
desintegraciones por
minuto. En bioquímica se trabaja con cantidades de microcurios (![]() ).
).
Los detectores de radiactividad de que disponemos no presentan una eficacia del 100%, por lo que la radiactividad se expresa en unas unidades relativas que son las cuentas por minuto (c.p.m.), el número de desintegraciones que detecta el aparato en un minuto (menos que las reales).
Por ejemplo, un contador de radiactividad
con una eficacia del 50% para una muestra de 0,1![]() nos daría un “contage” de
nos daría un “contage” de ![]() c.p.m.
c.p.m.
Para medir la radiactividad disponemos de contadores “GEIGER” y de contadores “de centelleo”, más empleados.
Los contadores geiger se basan en el fenómeno de la ionización que producen las emisiones radiactivas sobre un medio gaseoso. Estos se usan principalmente para saber, de modo relativo, si hay o no radiactividad en una muestra, y para cuantificarla pero de modo no muy aproximado.
Los contadores de centelmo nos ofrecen una
mayor precisión. Se basan en los procesos de excitación. Aquí la muestra se
disuelve o suspende en un disolvente orgánico que incluye uno o dos compuestos
fluorescentes. Una partícula ![]() emitida por la muestra
tiene una elevada probabilidad de contactar con una molécula del disolvente. Al
ocurrir esto, la molécula del disolvente es excitada, haciendo que uno de sus
electrones pase a un orbital de energía superior. Cuando este electrón vuelve a
su estado inicial se emite un fotón de luz. Este fotón es absorbido por una
molécula de flúor que a su vez se excita.
emitida por la muestra
tiene una elevada probabilidad de contactar con una molécula del disolvente. Al
ocurrir esto, la molécula del disolvente es excitada, haciendo que uno de sus
electrones pase a un orbital de energía superior. Cuando este electrón vuelve a
su estado inicial se emite un fotón de luz. Este fotón es absorbido por una
molécula de flúor que a su vez se excita.
La fluorescencia implica la absorción de luz a una determinada energía seguida de la emisión de luz a una energía inferior o longitud de onda mayor .
Un fotomultiplicador detecta este pequeño destelleo de luz y lo convierte en una señal eléctrica que, que es la que se “cuenta”.
Los isótopos emisores ![]() presentan un espectro
electromagnético característico cada uno. Estas diferencias se usan en los
contadores de centelleo líquido para cuantificar simultáneamente dos isótopos
de la misma muestra.
presentan un espectro
electromagnético característico cada uno. Estas diferencias se usan en los
contadores de centelleo líquido para cuantificar simultáneamente dos isótopos
de la misma muestra.
Las aplicaciones de los isótopos radiactivos en bioquímica son muy variadas. Se incluyen el estudio de rutas metabólicas con trazadores, estudios de cinética encimática con sustratos marcados, y la autoradiografía, que se basa en la capacidad de las emisiones radiactivas para impresionar placas fotográficas. Esta técnica combinada con la electroforesis nos permite, por ejemplo, secuenciar el DNA o hacer estudios de la expresión de los genes, entre muchas otras cosas.
Técnicas espectroscópicas
Tanto proteínas, como ácidos nucleicos o hidratos de carbono, son moléculas complejas que pueden absorber radiación en un amplio margen del espectro electromagnético. En la región infrarroja del espectro se estudia la conformación de las moléculas proteicas.
La región visible se usa para la medida de la absorbancia de las soluciones coloreadas, y esto es conocido como “colorimetría”.
En la zona visible la mayoría de los biopolímeros no absorben luz. Aunque algunas proteínas se ven coloreadas, ello es debido a los grupos prostéticos o iones, como el cobre que forman parte del polipéptido. El color rojo de la sangre se debe al grupo “hemo” de la hemoglobina.
Para poder medir las proteínas o ácidos nucleicos por colorimetría, hay que transformarlos en compuestos coloreados mediante reacciones químicas específicas.
En la región ultravioleta absorben luz los ácidos nucleicos y las proteínas, midiéndose respectivamente a 260 y 280 nm. En realidad se mide en las cadenas laterales aromáticas de los aminoácidos Phe, Tyr y Trp.
Las medidas se absorbancia de luz ultravioleta y visible se hacen en unos aparatos llamados “espectrofotómetros”.
La muestra se coloca en una cubeta sobre la que incide una luz monocromática (con una sola longitud de onda) que se consigue con un monocromador. Parte de esta luz incidente va a ser absorbida por la muestra y otra va a atravesarla.
La que la atraviesa es la luz “i”, y es detectada y registrada.
La absorbancia a la longitud de onda “![]() ” se define como el logaritmo del cociente entre Io e I:
” se define como el logaritmo del cociente entre Io e I: ![]() y se relaciona con la concentración mediante
la ley de LAMBERT – BEER que dice que la absorbancia es igual a Epsilon por la
concentración y por el grosor de la cubeta:
y se relaciona con la concentración mediante
la ley de LAMBERT – BEER que dice que la absorbancia es igual a Epsilon por la
concentración y por el grosor de la cubeta: ![]()
![]()
![]()
![]() es el coeficiente de
extinción, característico para cada sustancia. Sus unidades dependen de las
unidades de la concentración que se emplean ya que la absorbancia no tiene
unidades.
es el coeficiente de
extinción, característico para cada sustancia. Sus unidades dependen de las
unidades de la concentración que se emplean ya que la absorbancia no tiene
unidades.
El grosor de la
cubeta suele ser de un centímetro, por lo tanto si la concentración la
expresamos como molaridad, las unidades de ![]() serían
serían ![]()
Esta ley nos permite calcular concentraciones de una sustancia a partir de su absorbancia.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 2606
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved