| CATEGORII DOCUMENTE |
| Alimentatie nutritie | Asistenta sociala | Cosmetica frumusete | Logopedie | Retete culinare | Sport |
FIZIOPATOLOGIA
METABOLISMULUI PROTEINELOR
Proteinele plasmatice reprezinta un constituent principal al plasmei, alcatuind aproximativ 75% din reziduul uscat al plasmei.
In ultimii ani, s-au realizat mari progrese pe calea identificarii si separarii diverselor componente din plasma. Metoda electroforetica de separare in cinci fractiuni a proteinelor serului este depasita de tehnicile imunochimice prin care se pot evalua cu mare precizie cele peste 90 de componente proteice ale plasmei. Aceste posibilitati au deschis noi perspective pentru o mai buna cunoastere a semnificatiei functionale a diverselor proteine plasmatice si pentru diagnosticul clinic.
Proteinele sunt constituite din unul sau mai multe lanturi polipeptidice, fiecare fiind constituit din mai multi aminoacizi, legati covalent prin legaturi peptidice. Greutatea moleculara a proteinelor variaza intre 500 si mai multe milioane daltoni. Indiferent de functia sau specia de origine, toate proteinele sunt alcatuite din 20 aminoacizi, aranjati in variate secvente specifice.
Spre deosebire de glucide si lipide, proteinele preluate sub forma de aminoacizi din lumenul intestinal, sunt transportate si utilizate la nivelul tuturor tesuturilor in scop predominant plastic. In afara rolului plastic, cei 20 de aminoacizi pot constitui o sursa importanta de cetoacizi, ca urmare a dezaminarii lor oxidative sau a altor interconversiuni metabolice.
O mare parte dintre aminoacizi pot fi sintetizati in organism, suplinind astfel deficitul aportului alimentar. Acesti aminoacizi se numesc neesentiali (glicina, alanina, serina, cisteina, prolina, acid glutamic, acid aspartic, asparagina si glutamina).
Un numar de 10 aminoacizi nu poate fi insa sintetizat la nivelul organismului, singura sursa fiind cea alimentara. Acestia sunt considerati aminoacizi esentiali: triptofan, histidina, treonina, lizina, metionina, arginina, valina, fenilalanina, leucina si izoleucina.
In functie de compozitia lor, proteinele se clasifica in doua mari clase: proteine simple si conjugate:
Proteinele simple sunt cele alcatuite numai din lanturi de aminoacizi, din a caror hidroliza rezulta aminoacizi, fara alte grupari organice sau anorganice.
Proteinele conjugate sunt acele proteine care, in urma hidrolizei, elibereaza atat aminoacizi cat si grupari organice sau anorganice, denumite grupari prostetice.
Proteinele conjugate se clasifica in functie de grupul prostetic:
nucleoproteine,
fosfoproteine,
metaloproteine,
glicoproteine si
lipoproteine.
In stare naturala, molecula proteinelor are o forma tridimensionala denumita conformatie, in functie de care se disting proteine fibrilare si proteine globulare.
Proteinele fibrilare constau din lanturi polipeptidice dispuse in paralel, de-a lungul unui singur ax. Alcatuiesc elementele structurale de baza ale tesuturilor de sustinere ale organismului: colagenul, elastina, cheratina.
Proteinele globulare sunt alcatuite din lanturi polipeptidice impachetate strins in forme moleculare sferice sau globulare. Functional, sunt: enzime, hormoni, proteine imunoefectoare, proteine de transport, proteine contractile, proteine de faza acuta etc.
Cea mai mare parte a proteinelor plasmatice se sintetizeaza in ficat iar imunoglobulinele in celulele imunocompetente (plasmocite). O parte a lipoproteinelor este sintetizata la nivelul mucoasei intestinale; macrofagele contribuie la producerea unor proteine plasmatice (componente ale complementului, inhibitori proteolitici) iar endoteliile vasculare produc activatori ai fibrinolizei, factor von Willebrand, fibronectina.
Hormonii participa la reglarea metabolismului protidic prin efecte anabolizante sau catabolizante.
Hormonii cu efect anabolizant: somatotropul, insulina, estrogenii si androgenii, stimuleaza sinteza proteica din precursori aminati.
Hormonii cu efect catabolizant: tiroidieni, glucocorticoizi, glucagonul, duc la un bilant azotat negativ printr-un catabolism exagerat, cu slabire excesiva si topirea masei musculare, reducerea capacitatii de aparare imunitara.
Proteinele plasmatice indeplinesc importante functii in organism, functii, care pot fi sintetizate astfel:
Functia plastica are o importanta primordiala, mai ales in copilarie, cand procesele de sinteza sunt de o mult mai mare intensitate decat la maturitate. Anabolismul, predominant in perioada de crestere, utilizeaza ca prim substrat proteinele.
Mentinerea presiunii coloidosmotice a plasmei, contribuie la schimburile efectuate de lichidul extracelular (albumina asigura 80% din presiunea coloidosmotica plasmatica). Astfel, 1 g de albumina poate retine 18 ml de apa, fapt ce explica efectul spectaculos al perfuziilor cu solutii concentate de albumina, in terapia sindroamelor nefrotice grave si a insuficientelor hepatice.
Rol in apararea antimicrobiana specifica prin medierea raspunsului imunologic. Functia de aparare este asigurata de fractiunea gama a globulinelor.
Rol de transport al vitaminelor, hormonilor, ionilor, lipidelor, medicamentelor (sulfamide, penicilina, glicozizi digitalici) si a unor metaboliti. Functia de transport se bazeaza pe proprietatea macromoleculelor de a vehicula prin sange o serie de substante insolubile.
Rol in coagulare si fibrinoliza (toti cei XIII factori plasmatici si 9 factori plachetari sunt proteine, precum si antitrombinele ori plasminogenul);
Contribuie la sistemele tampon ale sangelui avand rol in mentinerea pH-ului sanguin (proteinele au caracter amfoter);
Mentinerea constanta a tensiunii arteriale se realizeaza prin interventia proteinelor plasmatice cu proprietati hidrofile si prin faptul ca angiotensinogenul este o proteina circulanta.
Intre proteinele plasmatice sunt incluse enzimele si antienzimele. Unele enzime apar tranzitor in circulatie si in cantitate mica (amilaza pancreatica, pepsinogenul), altele sunt in cantitate mare dar in stare inactiva: angiotensinogen, plasminogen, protrombina.
Asigura fondul comun de proteine care pot fi utilizate drept rezerva de proteine (in procesele de crestere si reparare a tesuturilor).
ELECTROFOREZA PROTEINELOR
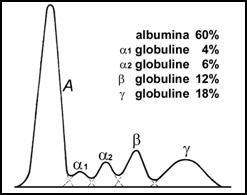
La electroforeza serului se obtin 5 benzi majore.
Curba caracteristica a picurilor serului normal, dupa banda electroforezei pe hartie.
Prima banda (fractiune) este albumina, care reprezinta 60% din totalul proteinelor (3,5-5,0 g/dl).
Fractiunea -globulinica contine mai multi constituenti: eritropoetina, -antitripsina, -lipoproteina, -acid glico-proteina etc; (0,2-0,4 g/dL);
Fractiunea -globulinica cuprinde: haptoglobina, cerulo-plasmina, macroglobulina. Din acest grup mai fac parte kininogenul (substratul eliberator de bradikinina si kalidina) si angiotensinogenul; (0,5-0,9 g/dL);
-globulinele, cuprind: transferina (2/3 din totalul fractiunii), -lipoproteinele, plasminogenul, complementul, hemopexina; (0.6-1,2 g/dL) ;
Gamaglobulinele constituie cea de-a cincea banda, fiind alcatuita din imunoglobulinele IgG, IgA, IgM, IgD si IgE. (0,8-15 g/dL).
Modificarea raportului dintre diferitele fractiuni proteice separate prin electroforeza poarta numele de disproteinemie.
Aceste tulburari se traduc prin: hiperproteinemii, hipoproteinemii, disproteinemii si paraproteinemii.
Reprezinta cresterea valorilor proteinelor plasmatice peste valoarea de 9 g/dL. Acestea pot fi reale, ca urmare a unui catabolism accentuat sau sintezei de proteine monoclonale, ori sunt aparente (false), ca urmare a hemoconcentratiei (in deshidratare). Hiperproteinemiile adevarate sunt rare.
Sunt definite prin scaderea concentratiei proteinelor plasmatice sub valoarea de 6 g/dL. Hipoproteinemiile caracterizeaza sindroamele biologice care au la baza scaderea proteinelor totale cu pastrarea raportului dintre diversele fractii. Ele apar ca urmare a unor deficite de aport, absorbtie, sinteza, prin pierderi excesive (renale, arsuri, hemoragii) sau a unor insuficiente hormonale (hipofizare, corticosuprarenale). Abaterile de la starea de euproteinemie pot fi determinate prin unul din urmatoarele mecanisme sau prin asocierea simultana a mai multor mecanisme.
Hipoproteinemia prin deficit de aport sau de absobtie apare ca o consecinta a inanitiei, sau prin regimuri alimentare sarace in proteine, mai ales de origine animala (ce contin aminoacizi esentiali). Echilibrul dinamic stabilit intre proteinele plasmatice si cele tisulare, dirijat enzimatic la nivelul membranei celulare, este mentinut prin consumul a 30 g proteine tisulare pentru 1 g de proteine plasmatice pierdute.
Aportul necesar pentru un adult este de 1g proteine/kg corp/zi din care cel putin jumatate sa fie de origine animala.
Carenta prelungita de proteine duce la deteriorarea tuturor functiilor organismului. Apar manifestari multiple (sindrom pluricarential) si polimorfe cu cateva exprimari pregnante:
,,edem de foame" datorat hipoalbuminemiei si scaderii consecutive a presiunii coloidosmotice,
scaderea troficitatii tegumentelor si fanerelor,
incetinirea vindecarii plagilor datorita incetinirii formarii tesutului de granulatie,
scaderea rezistentei la infectii,
anemie hipocroma prin deficit de proteine si Fe,
osteoporoza si
incetinirea formarii calusului datorita deficitului in sinteza matricei proteice a oaselor.
Hipoproteinemia prin deficit de aport este rareori pura, cel mai frecvent ea este asociata si cu alte carente: vitaminice, minerale etc.
Hipoproteinemii prin deficit in sinteza, desi de foarte multe ori neglijate, prezinta o deosebita importanta pentru clinician. Sediul principal al proceselor de sinteza a proteinelor se afla la nivelul ficatului.
Sinteze defectuoase proteice pot fi realizate in:
hepatopatii acute sau cronice,
afectiuni diencefalice,
comotii cerebrale, encefalite,
afectiuni ale hipofizei, in special prin dereglari ale hormonilor somatotrop si corticotrop.
Hipoproteinemii prin pierderi excesive pot fi intalnite ca urmare a pierderilor mari de proteine din cursul:
dispepsiilor cronice,
toxicozelor recidivante,
enteritelor si enterocolitelor repetate sau cronice.
hemoragiile cronice si repetate,
plasmoragiile de diverse etiologii,
revarsatele pleurale sau peritoneale,
supuratiile, arsurile, nefrozele etc., sunt insotite frecvent de hipoproteinemii,
boli cronice consumative (t.b.c., lues),
boli endocrine (hipertiroidism, boala Cushing) etc.
In carente proteice organismul face apel, initial, la rezervele glucidice din ficat, pe care le va mobiliza declansand un adevarat ,,stres" metabolic. Sunt apoi mobilizate grasimile neutre din rezervoare, care, dupa transformarea lor in corpi cetonici, vor fi oxidate la nivelul tesuturilor. Dupa epuizarea rezervelor glucido-lipidice, raman proteinele tisulare ca sursa de energie disponibila si in aceste conditii sunt utilizate la inceput proteinele tisulare din ficat, muschi si rinichi.
In denutritia proteo-calorica apar importante modificari fiziopatologice care pot afecta teoretic toate organele si tesuturile. Dintre acestea, cele mai comune sunt slabirea in greutate, scaderea tesutului adipos si a masei musculare. Scaderea masei corporale cu 5-10 % este, de obicei, bine tolerata. Pierderea a 35-40 % din greutate duce la deces.
Dezechilibrele proteice au consecinte dintre cele mai grave asupra organismelor. La copiii cu carente proteice se constata intirzieri in crestere, scaderea capacitatii de aparare, atrofie si dezorganizare a glandelor acinoase pancreatice, diminua acizii nucleici etc. Toate aceste fenomene sunt cu atat mai marcate, cu cat bilantul energetic este mai negativ si s-a instalat mai brusc.
Este posibila aparitia ,,edemelor de foame", edeme moi, albe, pufoase, deplasabile cu pozitia, se accentueaza la efort intens, dispar la repaus, localizate pe fata dorsala a labei piciorulul, in regiunea sacrata, regiunea palpebrala. Caracteristica edemulul de denutritie este instalarea lui dupa o scadere in greutate sau efort fizic intens. Este singurul edem poliuric;
Sistemul osos prezinta leziuni importante, subiectiv dureri osoase, obiectiv prezenta unei osteoporoze sau/si osteomalacie, care pot duce la fracturi diafizare;
Tulburarile psihice: indiferenta, apatie, refuzul alimentar la cei aflati in lagare, desi erau constienti ca mor de foame;
Tulburarile genitale : amenoree la femeie, la barbati impotenta sexuala, hipotrofia organelor genitale pana la disparitia caracterelor sexuale secundare.
La copii, in tarile subdezvoltate, Williams a descris doua forme clinice de denutritie primara:
Kwashiorkor - denutritie proteica si mai putin calorica, cu aparitia leziunilor tegumentare, edeme masive, ascita si atrofii musculare. Temperatura, tensiunea arteriala si pulsul sunt scazute. Exista, de obicei hepatomegalie.
Marasm - denutritie mixta predominant calorica, in care tabloul clinic este dominat de crestere intarziata si atrofii musculare. In contrast cu Kwashiorkorul, nu intalnim edeme, ascita sau leziuni tegumentare. Tesutul celular subcutanat este, de obicei absent, dar apetitul poate fi normal sau crescut.
Albumina este proteina serica cu cea mai mare concentratie. Este sintetizata la nivelul hepatocitelor in cantitate de 20 g/zi. Albumina este prezenta in toate lichidele biologice si in spatiul intercelular. Intreaga cantitate de albumina din organism este de aproximativ 300 g.
Albumina este cea mai importanta proteina de transport din plasma. Astfel, transporta acizi grasi, hormoni, ioni ai metalelor grele, medicamente (sulfamide, digitalice etc.), bilirubina si alti produsi de degradare din circulatie. Un rol important il are ca proteina de rezerva, bogata in aminoacizi esentiali; este utilizabila in conditiile unor procese regenerative ale organismului. Timpul de injumatatire in plasma este de 17-23 zile.
Hiperalbuminemia nu are semnificatie deosebita din punct de vedere practic, aparind doar in marile sindroame de deshidratare sau asociata unor fenomene de regenerare hepatica.
Hipoalbuminemia are multiple semnificatii, putind fi asociata unor variate procese patologice:
deficit in sinteza hepatica (apare relativ tirziu datorita marii rezerve a capacitatii hepatice de sinteza proteica);
malnutritia, cu scaderea marcata a aportulul in aminoacizi esentiali, reprezinta o alta cauza a hipoalbuminemiei de sinteza;
pierderea albuminei la nivel renal (sindromul nefrotic) sau gastrointestinal (gastroenteropatii exudative), cu depasirea capacitatii de sinteza hepatica,
consumul de albumina din infectii cronice exudative, traumatisme majore, arsuri si unele afectiuni dermatologice.
Privitor la valoarea diagnostica a concentratiei serice a albuminei, valori sub 3 g/dL sunt sugestive pentru un proces patologic.
Analbuminemia este o boala genetica transmisa autosomal recesiv. Consta in absenta albuminei la electroforeza. Clinic este rar simptomatica: edeme sau steatoree si constant hipotensiune.
Examenele de laborator evidentiaza: VSH accelerat si teste de disproteinemie pozitive, in conditiile unor probe functionale hepatice normale.
Nivelul altor proteine serice sintetizate hepatic este mult crescut, ca expresie a unei hipersinteze compensatorii, fenomen confirmat de faptul ca perfuzarea cu solutie de albumina umana (la marii denutriti, arsi, sindrom nefrotic) are ca efect normalizarea vitezei de sinteza a celorlalte proteine serice. In aceasta situatie, timpul de injumatatire al albuminei este mult prelungit, ajungind la 55 zile.
Modificarea concentratiilor diverselor proteine plasmatice (disproteinemie), se intalneste in extrem de numeroase stari patologice. Interpretarea datelor de laborator privind comportarea proteinelor plasmatice este mult usurata prin separarea electroforetica a proteinelor plasmatice in cinci fractiuni.
Disproteinemia reactiva din inflamatia acuta se caracterizeaza prin cresterea si mai rar globulinelor. Analiza diferentiata prin metode imunologice releva in aceasta reactie de faza acuta:
cresterea -1 antitripsinei, -1 glicoproteinei acide, a haptoglobinei, ceruloplasminei si proteinei C-reactive;
scaderea albuminei, transferinei, pseudocolinestera-zei, precum si1 si lipoproteinelor.
Se constata totodata o crestere a fibrinogenului si o accelerare corespunzatoare a VSH-ului.
Modificari similare se intalnesc nu numai in infectii acute, dar si postoperator, in procese tumorale sau dupa infarctul miocardic acut si reflecta raspunsul organismului la distructiile tisulare. Detectarea acestor modificari umorale contribuie la diagnosticul diferential al unor afectiuni:
Astfel, in meningitele purulente, globulinele si cresc foarte mult, in timp ce in meningitele virale, aceste modificari sunt diminuate.
Globulinele a cresc de asemenea in pneumopatiile de origine bacteriana, fiind nemodificate in pneumopatiile virale. Este de notat faptul ca in bronhopneumopatii grave, globulinele si fibrinogenul cresc, dar VSH-ul este adesea nemodificat. Aceasta discrepanta poate fi explicata prin existenta unei insuficiente respiratorii cu acidoza consecutiva, fenomen ce duce la marirea de volum a hematiilor si scade astfel viteza lor de sedimentare.
Disproteinemia reactiva din inflamatia cronica este caracterizata prin cresterea reactiva a gamaglobulinelor, consecutiva proliferarii reactive a plasmocitelor. Agentul declansator al acestei cresteri reactive este, de regula, o infectie cu evolutie subacuta sau cronica.
Cresteri deosebit de intense ale gamaglobulinelor sunt intalnite in endocardita bacteriana subacuta, colagenoze, boli hepatice cronice, tuberculoza, sarcoidoza. De regula, cresterea gamaglobulinelor nu depaseste in aceste disproteinemii reactive valorile de 35-38%, iar in cazurile cand se ajunge la 40% se poate banui coexistenta unor leziuni hepatice. Este important sa se faca distinctia intre disproteinemia reactiva, in care cresterea gamaglobulinelor intereseaza mai mult sau mai putin toate sub-fractiunile (IgG, IgA, IgM) si realizeaza un aspect difuz, cu baza larga a fractiunii gama la electroforeza si, pe de alta parte, cresterea in pisc (varf) a unei anumite subfractiuni gama, asa cum se intalneste in gamopatiile monoclonale.
Cresterea gamaglobulinelor se insoteste de o pozitivare a testelor de labilitate coloidala a serului. De mentionat ca fibrinogenul si -globulinele cresc mai ales in cursul puseelor evolutive ale procesului cronic. Merita subliniata observatia ca in endocardita bacteriana subacuta se constata o crestere a gamaglobulinelor, pe cand in endocardita reumatica recidivanta predomina cresterea - globulinelor.
Disproteinemia din pierderile de proteine pe cale renala, intilnita in sindromul nefrotic, se caracterizeaza prin scaderea proteinelor totale, scaderea extrem de accentuata a albuminei si cresterea globulinelor si . In formele severe, scade si concentratia gamaglobulinelor.
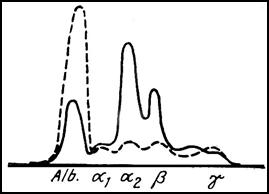
Electroforeza in sindromul nefrotic (punctat: ser normal).
Fibrinogenul plasmatic este, de regula, crescut, constatindu-se totodata o crestere marcata a colesterolului si lipidelor serice.
Toate aceste modificari pot fi in mare masura explicate prin cresterea permeabilitatii filtrului glomerular fata de proteine. De fapt, scaderea intereseaza in primul rind proteinele cu greutate moleculara mica, cum ar fi albumina, -antitripsina si transferina, in timp ce proteinele cu greutate moleculara mare ( -macroglobulina, fibrinogenul, lipoproteinele, IgM) cresc.
Lipsa de eliminare urinara nu poate explica insa cresterea la valori mari a concentratiei unora din aceste proteine cu greutate moleculara mare. S-a dovedit ca in sindromul nefrotic, sinteza unor proteine este mult accelerata. Pseudocolinesteraza serica crcscuta, enzima cu greutate moleculara mare, a cararei sinteza se produce paralel cu sinteza de abumina reprezinta un marker pretios. Sinteza de gamaglobuline pare a fi diminuata in sindromul nefrotic. Se explica astfel susceptibilitatea crescuta la infectii a pacientilor care sufera de aceasta afectiune.
Disproteinemia din pierderea enterala de proteine apare in enteropatia exudativa si reprezinta o manifestare patologica a procesului fiziologic de eliminare, in tractul digestiv, a proteinelor plasmatice. Proteinele eliminate se recupereaza partial sub forma de aminoacizi. Semnificativa este constatarea unei permeabilitati a tractului digestiv pentru substante macromoleculare (albumina). Se ajunge astfel la hipoproteinemie, dar aspectul proteinogramei difera de cel intalnit in sindromul nefrotic. Astfel, si globulinele nu cresc asa de mult ca in sindromul nefrotic, iar hipercolesterolemia si hiperlipemia lipsesc.
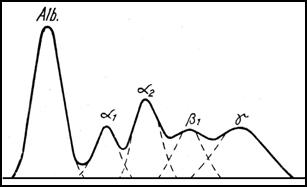
Curba electroforetica a unui caz de icter mecanic prin cancer pancreatic (verificare necropsica
Modificarile proteinogramei in hepatita acuta sunt putin exprimate si necaracteristice si, in consecinta, au o valoare diagnostica mai redusa in comparatie cu cresterea impresionanta a transaminazelor.

Curba electroforetica a unui caz de hepatita acuta.
Desi este vorba de un proces acut, globulinele nu cresc in hepatita virala acuta. Aceasta se explica prin faptul ca -globulinele sunt produse chiar de ficat. In sindromul posthepatitic, nivelul crescut al gamaglobulinelor persista si se poate uneori accentua atragind in acest fel atentia asupra cronicizarii.
In hepatita cronica, aspectul disproteinemiei este caracterizat prin cresterea gamaglobulinelor. Ca si in alte inflamatii cronice, cresterea gamaglobulinelor este difuza si se insoteste de pozitivarea testelor de labilitate coloidala a serului. Disproteinemia este mult mai severa in cirozele hepatice deoarece, in astfel de cazuri, la inflamatia cronica se adauga pierderea de proteine prin lichidul de ascita precum si insuficienta functionala a ficatului (sindromul hepatopriv). Din acest motiv se produce o scadere marcata a albuminelor care, de regula, este asociata cu scaderea sintezei de pseudocolinesteraza si de protrombina. Procesul de degradare a gamaglobulinelor la nivelul ficatului este perturbat. Asa se explica cresterea gamaglobulinelor la valori procentuale de peste 40% (2-2,5 g/dl). Evacuarile frecvente ale lichidului de ascita pot intretine si agrava disproteinemia.
In stadiile de debut ale neoplaziior hepatice, electroforeza proteinelor serice are o valoare diagnostica redusa dar in adenocarcinomul hepatic cu ciroza se produce scaderea albuminelor si cresterea gamaglobulinelor asociata adeseori cu crestere fractiunii -globulinelor. Metode imunologice permit evidentierea la bolnavii cu hepatom prezenta in ser a alfafetoproteinei.
consta in scaderea accentuata a gamaglobulinelor din proteinograma
A. Imunodeficite primare
Dintre acestea, mai importante sunt:
Agamaglobulinemia legata de cromosomul X, a fost descrisa sub numele de agamaglobulinemia Bruton. Se caracterizeaza prin infectii recurente ce debuteaza in primii ani de viata sau in copilarie, prin absenta tuturor claselor de Ig din ser si incapacitatea de a produce anticorpi chiar atunci cand se face stimulare cu un antigen puternic. In tesuturile limfoide lipsesc plasmocitele. Sunt afectati numai copiii de sex masculin. Imunitatea mediata celular este normala. Alaturi de infectiile piogene s-au descris, in astfel de cazuri, si infiectii persistente virale precum si infestari cu paraziti si micoplasme.
Deficitul de imunoglobulime cu IgM crescute (si IgD). Sindromul se caracterizeaza prin infectii piogene severe (septicemii, pneumonii, otite), anemii hemolitice, trombocitopenii. Imunitatea celulara este nemodificata. Stimularea limfocitelor B duce la o sinteza crescuta de IgM si IgD, fara sa se sintetizeze IgG si IgA. Fenomenul se explica prin faptul ca in ontogenia celulei B, IgM se exprima la suprafata celulei, inaintea celorlalte clase de Ig. Se presupune ca defectul se datoreste lipsei de maturare a celulelor B.
Deficitul selectiv de IgA (sub 0,05 mg/ml) are o frecvcnta de 1/500 in populatia generala. O parte din persoanele cu deficit de IgA au fost identificate printre donatorii dc sange care erau sanatosi din punct de vedere clinic. Deficitul de IgA se asociaza frecvent si cu o serie de boli autoimune (LES, poliartrita reumatoida, anemie hemolitica, sindrom Sjogren) si cu infectii recurente respiratorii, gastrointestinale si alergii.
Deficitul in subclase de IgG se asociaza cu cresterea susceptibi1itatii la infectii si boli limfoproliferative.
Hipogamaglobulinemia tranzitorie a noului nascut apare, in general, intre 3 si 6 luni de la nastere si se refera, in principal, la IgG, mai rar la IgA si IgM. Hipogamaglobuline-mia apare ca urmare a faptului ca IgG din serul nou-nascutului, provenite de la mama, sunt catabolizate, iar sinteza propriilor Ig, indeosebi a IgG nu a inceput inca. Acest deficit se remediaza spontan, nefiind necesara administrarea de gamaglobuline decat in cazul unor infectii recurente severe si a unei hipogamaglobulinemii marcate.
Imunodeficitul variabil comun apare la orice varsta, atat la femei cat si la barbati. Sindromul clinic este similar cu cel din agamaglobinemia legata de cromosomul X si se asociaza cu o mare incidenta a LES, anemiei hemolitice, purpurei trombocitopenice idiopatice, infectiilor pulmonare, tulburarilor digestive cu diaree marcata. Tumorile cu localizare digestiva sunt de asemenea intilnite relativ frecvent, ca si anemia Biermer. Mecanismele care duc la imunodeficitul variabil comun sunt: defecte intrinseci ale celulci B, producerea de autoanticorpi fata de celulele T si B precum si dezechilibrul celulelor T imunoreglatoare.
Spre deosebire de agamaglobulinemia legata de cromosomul X, celulele B nu sunt intotdeauna scazute, ele putind fi normale sau crescute. Aceste celule sunt insa relativ imature si nu raspund la antigene sau mitogeni prin diferentiere in plasmocite. La toate grupurile de pacienti, raportul limfocite T-helper/T-supresor este inversat, excesul de limfocite T-supresoare impiedicind diferentierea celulelor B. Cel mai frecvent, cele trei clase majore de imunoglobuline sunt extrem de scazute, iar nivelul IgE este normal sau chiar crescut.
B. Sindroamele de imunodeficienta secundara sunt frecvent intalnite la pacientii cu malnutritie, infectii cronice, cancer, boli renale, boala Hodgkin, sarcoidoza. Starile de imunodeficienta secundara apar frecvent la pacientii tratati cu droguri imunosupresoare pentru prevenirea reactiilor de rejet sau tratamentul bolilor autoimune. Ele sunt mult mai frecvente decat imunodeficientele primare (de cauza genetica). Cea mai de temut imunodeficienta castigata este SIDA, maladie declansata de virusul HIV, ce a capatat in ultimii 10 ani proportie de epidemie.
Sindromul de hiperimunoglobulinemie E (sindromul Job) este o boala complexa care se caracterizeaza prin debut in copilarie, concentratii foarte crescute de IgE si infectii bacteriene recurente, indeosebi cu stafilococ, tegumentare si pleuropulmonare. La nivelul pielii apar abcese subcutanate determinate de stafilococul aureu hemolitic. In cadrul accstui sindrom, alte sedii de localizare a infectiei sunt reprezentate de urechea medie si externa, mastoida, gingii, bronsii si parenchim pulmonar. Mai pot apare dermatite cronice si candidoze.
In afara de IgE mult crescuta, s-a mai descris eozinofilie moderata, nivele crescute ale IgD si deficite ale functiei neutrofilelor, celulelor T supresoare si ale raspunsului la diferite antigene. Aceste anomalii nu explica decat partial susceptibilitatea crescuta la infectii a acestor pacienti.
Gamopatiile monoclonale sunt boli caracterizate prin prezenta in ser sau urina de imunoglobuline monoclonale denumite si paraproteine sau componenta M (monoclonala). Aceasta componenta monoclonala este produsa de o singura clona de celule limfoide, are o mobilitate electroforetica limitata, aparand la electroforeza proteinelor serice sub forma unei bande inguste ,,in pisc". Proteinele monoclonale sunt imunoglobuline normale ca structura, dar produse in exces. Numai in unele cazuri de boala a lanturilor grele s-au semnalat deletii de aminoacizi, fiind vorba, in aceste cazuri, de imunoglobuline anormale.
In mod fiziologic, plasmocitele produc lanturi grele si lanturi usoare in exces fata de lanturile grele. In cazul mielomului IgG, 75% din pacienti prezinta un exces de lanturi usoare care se excreta in urina determinind apantia proteinuriei de tip Bence Jones. In alte cazuri, nu se produc deloc lanturi grele, detectindu-se doar prezenta de lanturi usoare in exces (boala lanturilor usoare). Exista si cazuri rare in care celule mielomatoase nu secreta nici lanturi usoare, nici lanturi grele, datorita fie incapacitatii de sinteza, fie blocarii secretiei, aceste celule fiind nesecretoare (mielom nesecretor). In boala lanturilor grele, portiuni ale lanturilor grele ale IgG, IgA, IgM sau IgD sunt prezente in ser sau urina iar lanturile usoare lipsesc.
Mielomul multiplu apare, in general, la virstnici cu o incidenta de 3 la 100.000 indivizi. Studiul evolutiei naturale a bolii arata ca aceasta devine manifesta clinic atunci cand se ajunge la un numar foarte mare de plasmocite maligne, ceea ce reprezinta aproximativ 1 kg masa tumorala. Perioada latenta a bolii este, in general, de 1-2 ani. Proliferarea plasmocitelor are loc la nivelul maduvei hematotormatoare, difuz, astfel ca tumora nu este palpabila. Plasmocitele proliferate au caractere morfologice modificate, sunt in cantitate mare, si sunt dispuse focal (in cuiburi).
Din punct de vedere clinic, primele semne sunt reprezentate de astenie, anemie si anorexie. Anemia se datoreaza infiltrarii maduvei rosii cu plasmocite, pierderilor de sange si malnutritiei. Semnul clasic al bolii este reprezentat de leziunile osoase, osteolitice, care pot duce la fracturi spontane. Aceste modificari se datoresc inlocuirii tesutului osos cu celule neoplazice proliferate precum si producerii unui factor activator al osteoclastelor de catre plasmocite. Localizarea este predominanta la nivelul craniulul (aspect de "craniu mincat de molii"), sternului, claviculelor si oaselor bazinului.
O alta caracteristica importanta este prezenta de infectii recurente, mai frecvent pulmonare si mai rar renale, determinate de imunodeficienta ce se instaleaza concomitent cu proliferarea unei singure clone de plasmocite si inhibarea celorlalte. Producerea in exces de imunoglobuline monoclonale se asociaza cu tulburari ale functiei neutrofilelor si a limfocitelor T si B.
Producerea de lanturi usoare in exces duce la aparitia acestora in urina, realizand proteinuria de tip Bence Jones. Deoarece lanturile usoare ale Ig sunt catabolizate la nivelul epiteliulul tubilor renali proximali, incarcarea acestora cu cantitati mari de lanturi usoare, asociata cu hipervascozitatea sanguina duce cu timpul la afectarea renala, ce poate evolua pana la insuficienta renala cronica.
Testele de laborator arata existenta unui VSH mult accelerat, prezenta anemiei, eventual a hipercalcemiei in raport de extinderea leziunilor osoase, a hiperproteinemiei (peste 10 g/dL), prezenta de crioglobuline, a hiperviscozitatii si de hematii in fisicuri pe frotiul de sange periferic. La electroforeza serulul se observa aparitia unei benzi inguste monoclonale ce variaza de la regiunea gama pana la alfa 2. Aceasta este determinata de clasa de imunoglobuline care prolifereaza. Se considera, in general, ca 80% din mieloame sunt de tip IgG. La determinarea cantitativa a imunoglobulinelor, aspectul caracteristic este de crestere accentuata a unei clase de Ig si valori scazute ale celorlalte clase.
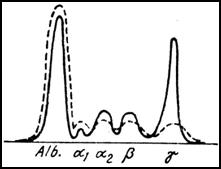
Curba electroforetica in mielomul multiplu ( punctat: ser normal).
Macroglobulinemia Waldenstrm
Este o boala care apare mai frecvent la barbati si difera de mielomul multiplu prin celulele care prolifereaza si care sunt asemanatoare atat cu limfocitele cit si cu plasmocitele (celule limfocitoid-plasmocitare). Aceste celule produc cantitati mari de IgM. Aceasta afectiune include un spectru larg de forme clinice, variind de la forme lent progresive, analoge gamopatiei monoclonale benigne, la forme maligne asemanatoare limfosarcoamelor sau leucemiei limfocitare cronice.
Simptomele clinice sunt reprezentate initial de anemie si hepatosplenomegalie. La acestea se asociaza limfadenopatia periferica. Hiperviscozitatea determinata de IgM monoclonale genereaza tulburari vizuale si la nivelul sistemului nervos central, ce pot conduce la instalarea comei paraproteinemice.
Diagnosticul trebuie suspectat atunci cand, la examenul fundului de ochi, apar vase mult dilatate, hemoragii, exudate asociate cu prezenta de hematii in fisicuri si cu o hiperviscozitate a serului mai mare de 4 ori decat cea a serului normal. Electroforeza serului are aspect monoclonal, cu imunoglobulinele IgM mult crescute.
Reprezinta un grup de afectiuni caracterizate printr-o productie excesiva de fragmente ale lanturilor grele, in special din fragmentul Fc. Fiecare din tipurile de boala a lanturilor grele descrise are manifestari clinice diferite si necesita studii imunoelectroforetice. Caracteristic este insa o accentuata scadere a rezistentei la infectii. Apare mai ales la virstnici si se manifesta prin fatigabilitate, infectii cronice, limfadenopatie, splenomegalie.
Tabloul clinic se aseamana cu cel al unui limfom difuz. Este prezent si un edem al palatului moale si al luetei, deoarece nodulii limfatici Waldeyer sunt frecvent mariti de volum (semn aproape patognomonic). Sunt, de asemenea, prezente: anemia si trombocitopenia iar in maduva hematoformatoare si nodulii limfatici sunt prezente plasmocite in numar mare. Boala este rapid progresiva ducand la moarte.
Diagnosticul trebuie suspectat pornind de la criterii clinice si este confirmat prin demonstrarea prezentei Ig monoclonale atat in ser cat si in urina. Proteina monoclonala are mobilitate electroforetica identica in ser si urina si reprezinta fragmentul Fc al moleculei lantului greu.
De la descoperirea ei de catre Rudolf Virchow in 1854, amiloidoza a fost considerata ca reprezentind un grup de boli caracterizate prin depunerca in tesuturi a unui infiltrat proteic omogen cu proprietati tinctoriale caracteristice. Cu exceptia formelor localizate, amiloidoza este o boala progresiva care duce la moarte prin distrugerea organelor afectate. Rezistenta amiloidului la fagocitoza si proteoliza impiedica indepartarea lui din depozite si determina progresiunea manifestarilor clinice.
Amiloidoza primara se caracterizeaza prin prezenta unor depozite fibrilare de amiloid, formate din fragmente amino-terninale ale lanturilor usoare ale imunoglobulinelor.
Cresteri ale Ig pot fi produse si prin proliferarea mai multor clone de plasmocite asociate cu cresteri ale uneia sau mai multor clase de Ig. Determinarea cantitativa a Ig precizeaza care din clasele de Ig cresc. Cresteri ale Ig se intalnesc in afectiuni cu patogeneza imuna, cum ar fi colagenozele, unele afectiuni inflamatorii hepatice si renale si unele boli infectioase. Aceste cresteri se realizeaza prin mecanisme diferite implicind tulburari ale cooperarii dintre limfocitele T si B. Se incrimineaza fie o hiperreactivitate a limfocitelor B, fie persistenta unor factori care stimuleaza functiile limfocitului B.
SUBSTANTELE AZOTATE NEPROTEICE
Sunt reprezentate de produsii de catabolism ai proteinelor: uree, creatina, creatinina, acid uric, acizi aminati. Separarea lor din plasma sau ser se face dupa precipitarea proteinelor.
Azotul, continut de toate substantele micromoleculare aflate in filtrat, constituie azotul neproteic sau rezidual. Normal este in cantitate de 25-35 mg/dL, cea mai mare parte (80%) revenind azotului ureic. Deoarece principala cale de eliminare a azotului organic din organism este calea renala, insuficienta excretorie a rinichiului determina cresterea azotului neproteic din sange. Uzual, se dozeaza ureea care, desi nu este toxica prin ea insasi, creste o data cu toate celelalte substante azotate neproteice, majoritatea toxice.
Uricemia normala este de in relatie cu sexul: 3,5-7 mg/dL la barbati si de 2,5-6 mg/dL la femei.
De notat ca, dupa menopauza, valorile uricemiei la cele doua sexe se egaleaza.
Uricozuria este de 500-800 mg/24 ore cand regimul alimentar este normal, iar 200 mg se elimina zilnic pe cale digestiva.
Hiperuricemiile (> 7mg/dL), mult mai frecvent intilnite decit hipouricemiile, pot fi insotite de manifestari clinice de guta sau nu. In functie de mecanismul lor de producere, hiperuricemiile pot fi impartite in doua mari categorii: seundare si primare (tabel).
Guta poate rezulta din supraproductia de acid uric, deficit in eliminare sau prin ambele mecanisme.
Pentru a intelege mecanismul producerii gutei, este necesara o prezentare succinta a metabolismului acidului uric. Acidul uric reprezinta produsul final de metabolism al bazelor purinice. Cresterea producerii de acid uric reprezinta, de cele mai multe ori, o consecinta a defectelor de metabolizare a nucleotidelor purinice. Sinteza nucleotidelor purinice urmeaza doua cai metabolice: de novo si prin reciclarea produsilor interni (salvare a purinelor).
Producerea de novo implica sinteza purinelor si a acidului uric din precursori nonpurinici. Substanta de plecare este riboza-5-fosfatul, care este convertit, trecand printr-o serie de produsi intermediari, in nucleotide purinice (acid inozinic, acid guanilic si acid adenilic). Aceasta cale este controlata de mai multe mecanisme de reglare. Cele mai importante sunt:
reglarea prin feed-back negativ realizata de nucleotidele purinice prin doua enzime: amido-fosfo-ribozil-transferaza (amido-PRT) si 5-fosfo-ribozil-1-pirofosfat-sintetaza (PRPP) si
activarea amido-PRP prin substratul sau PRPP.
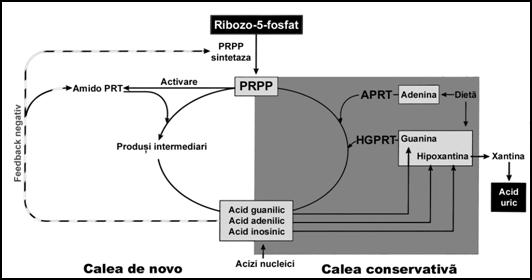
Metabolismul purinelor
Calea a doua de reciclare a produsilor interni reprezinta mecanismul prin care bazele purinice libere (din descompunerea acizilor nucleici si din aportul alimentar) sunt utilizate pentru sinteza nucleotidelor purinice. Aceasta cale presupune condensarea bazelor purinice libere (hipoxantina, guanina si adenina) cu PRPP pentru formarea nucleotidelor purinice (acid inozinic, acid guanilic si acid adenilic). Aceste reactii sunt catalizate de doua transferaze:
hipoxantin-guanin-fosforibozil-transferaza (HGPRT);
adenin-fosforibozil-transferaza (APRT).
In lipsa ori suprasolicitarea HGPRT, hipoxantina si guanina sunt degradate in xantina si apoi in acid uric, substanta responsabila de producerea gutei.
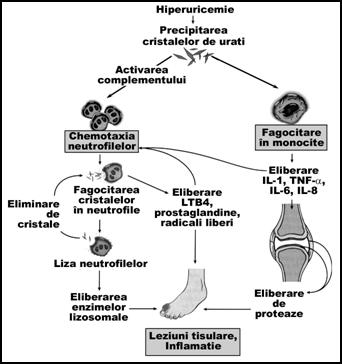
Patogenia artritei gutoase acute (dupa Kumar, Cotran, Robbins, Basic Pathology, sixth edition, 1997)
Cresterea nivelului de acid uric in sange si alte lichide din organism (lichidul sinovial) favorizeaza precipitarea acestuia sub forma de cristale de urati de sodiu. Aparitia acestor cristale la nivelul articulatiilor determina activarea complementului si atragerea de polimorfonucleare neutrofile si macrofage la nivel membranelor sinoviale. Fagocitarea acestor cristale genereaza radicali liberi si leukotriene (mai ales leukotriene B4) cu efect inflamator. Neutrofilele lizate elibereaza enzimele lizosomale, cu efect proteolitic.
Dupa endocitarea uratilor, macrofagele elibereaza mediatori proinflamatori precum: IL-1, IL-6, IL-8 si TNF- . Aceste citokine intensifica procesul inflamator local (articular) si activeaza celulele sinoviale si condrocitele care elibereaza proteaze (colagenaza) ce determina lezarea tesuturilor adiacente. Toate aceste procese determina aparitia tofilor gutosi la nivelul articulatiilor metatarso-falangiene.
Atacul acut de guta apare de obicei dupa varsta de 40 ani si intereseaza in 50 % din cazuri articulatia metatarsofalangiana a halucelul (podagra) unilateral, dar artrita gutoasa se poate localiza la orice articulatie. Debutul este brusc, de obicei nocturn, cu o durere vie a articulatiei interesate.
La examenul local se observa semnele inflamatiei: tumor, rubor, cresterea temperaturii locale. Examenele de laborator evidentiaza VSH crescut, leucocitoza cu neutrofilie. Examenul de certitudine pentru diagnostic il reprezinta identificarea cristalelor de urat monosodic in lichidul sinovial al articulatiei afectate. Durerea poate ceda spontan dupa 7-8 zile.
Criza acuta de guta poate reapare dupa un timp variabil de luni sau ani, ca apoi sa fie din ce in ce mai frecventa, uneori poliarticulara. Factorii declansatori ai ataculul dc guta sunt: microtraumatismele articulare, abuzurile alimentare cu carne, viscere, vanat, alcool, grasimi animale, stresuri psihice, diverse medicamente (ACTH, antibiotice, saluretice), infectii.
Guta cronica se caracterizeaza prin prezenta tofilor gutosi, a artropatiei gutoase cronice, litiazei urice, nefropatiei gutoase. Tofii gutosi se localizeaza si sub tegumente, dar cel mai des la nivel articular si periarticular (cartilaje, epifize osoase, sinoviale si structuri periarticulare). Tofii gutosi cresc in dimensiuni si pot determina deformari articulare, anchiloze, subluxatii. Ei pot ulcera. Reactia articulara cronica la "agresiunea" urica realizeaza aspecte clinice similare cu poliartrita cronica.
Un alt organ care sufera foarte mult in cursul gutei si a carui alterare poate constitui pericolul letal pentru bolnav este rinichiul. Cea mai zgomotoasa forma a afectarii renale in guta este litiaza urica. Litiaza urica apare la aproximativ 25 % din subiectii cu guta. O treime din cazuri au hiperuricozurie, peste 800 mg/24 h si urina acida (pH sub 5,5), ceea ce favorizeaza precipitarea acidului uric in parenchimul renal si aparitia litiazei renale.
Infiltratia intraparenchimatoasa cu cristale de acid uric poate duce la o nefropatie interstitiala, la care contribuie si infectia urinara, consecutiva nefropatiei obstructive. Pe de alta parte, foarte multi gutosi sunt si hipertensivi, ceea ce poate duce de asemeni la modificari renale, mai mult arteriolare. Notiunea de nefropatie gutoasa este rezultanta tuturor acestor procese intricate. Nefropatia gutoasa se poate manifesta prin dureri lombare, proteinurie, hematurie, leucociturie, cilindrurie, eliminare crescuta de cristale de urati, uneori hipertensiune arteriala, cu evolutie spre insuficienta renala cronica progresiva
Un procentaj de 5 % dintre pacientii cu sindrom gutos prezinta boala (sd.) Lesch-Nyhan. Copii cu acesta boala se nasc normali, dar, la aproximativ un an de la nastere incep sa prezinte simptome neurologice ca spasticitate, atetoza si retardare intelectuala; in aproximativ 50 % din cazuri apar crize epileptiforme. Unii copii prezinta si malformatii congenitale si anemie megaloblastica dar simptomul cel mai caracteristic al bolii este automutilarea. Copiii au tendinta sa-si roada falangele si buzele, si uneori, pot deveni agresivi fata de persoanele din anturaj. Cei mai multi manifesta si o guta severa precum si litiaza urica.
Defectul enzimatic, ce sta la baza acestei afectiuni este deficitul (sau lipsa) enzimei HGPRT. Locusul genetic al acestei enzime este pe bratul lung al cromosomului X si, ca atare, boala va fi prezenta la barbati, femeile fiind purtatoare si transmitatoare. Se pare ca existenta doar de 1% din activitatea enzimatica normala poate preveni simptomele neurologice dar nu pe cele gutoase. Explicatie biochimica a modificarilor neuropsihice este dificilia. Se pare ca acestea nu depind de nivelul uricemiei (gutosii nu au fenomene neurologice). Acumularea in SNC a hipoxantinei si xantinei ar putea explica doar partial fenomenele. Se pare insa ca SNC ca si alte tesuturi extrahepatice recurg la sinteza "in situ" de nucleotide direct din purine, prin reactia catalizata de HGPRT. Blocarea acestei cai directe (salvage pathway), in sindromul Lesch-Nyhan, produce astfel o depletie de nucleotide necesare celulelor nervoase. Anemia megaloblastica prezenta uneori in acest sindrom este explicata prin relativa reducere a cantitatii de tetrahidrofolat, utilizat in exces de purinosinteza.
In fiziopatologia terapiei gutei, un rol important revine Alopurinolului care produce cea mai intensa inhibitie a sintezei de acid uric. Fiind un analog chimic al xantinei, el este un inhibitor puternic al xantinoxidazei, interferind deci cu transformarea hipoxantinei in xantina si respectiv formarea de acid uric. Deoarece, prin mecanismul aratat mai sus, se produce cresterea xantinei, alopurinolul poate duce la litiaza xantica, desi xantina este mai solubila decit acidul uric. In acelasi timp, antiinflamatoriile au un rol deosebit de util in terapia gutei.
Hipouricemia (< 2,5 mg/dL) este mult mai rar intilnita comparativ cu hiperuricemia. Deoarece acidul uric se sintetizeaza in special in ficat, in afectiunile parenchimatoase hepatice grave, nivelul uricemiei scade. Rareori, in cursul bolii Wilson, s-a observat hipouricemie. Se pare ca in acest caz este vorba despre o eliminare renala crescuta de acid uric, uneori asociata si cu aminoacidurie.
Administrarea unor medicamente duce de asemeni la hipouricemie: uricozurice (Probenecit) si inhibitori ai sintezei acidului uric (allopurinol) precum si in urma administrarii de substante de contrast iodate.
FIZIOPATOLOGIA METABOLISMULUI ACIZILOR AMINATI
Acizii aminati, alaturi de polipeptidele si proteinele din plasma, constituie forma de circulatie a proteinelor. Proteinele tuturor tesuturilor (atat proteinele de structura cat si proteinele solubile) se degradeaza si se reconstituie in mod continuu. Ele elibereaza acizii aminati proveniti din procesele distructive tisulare, in lichidul interstitial si plasma sanguina, unde se amesteca cu aminoacizii proveniti din alimentatie, formand asa numitul "fond comun". De aici, celulele extrag aminoacizii necesari refacerii proteinelor proprii.
Degradarea acizilor aminati se realizeaza prin dezaminare si transaminare.
Dezaminarea, prima etapa din degradarea aminoacizilor, care se desfasoara la nivelul tesuturilor si mai ales la nivel hepatic, consta in eliberarea gruparii aminice. In urma procesului de dezaminare, se formeaza alfa cetoacizi si amoniac.
Transaminarea, reactie catalizata de transaminaze, consta in transferul unei grupari aminice a unui aminoacid, pe un cetoacid, cu formarea aminoacidului corespunzator (Fig. 24). Reactiile sunt mediate de transaminaza glutamic-piruvica (TGP) si de transaminaza glutamic oxalacetica (TGO). Determinarea activitatii celor doua enzime, in ser, are o semnificatie clinica majora deoarece ele prezinta cresteri insemnate in cursul primelor 48-72 de ore de la producerea unei necroze tisulare. Astfel, TGO din ser creste, in cazurile de infarct miocardic acut, de 2-10 ori si revine la normal dupa cinci zile de evolutie a bolii. Nivelul TGP creste indeosebi in leziunile hepatice; de 10-30 ori in hepatitele acute (este direct proportionala cu gradul de hepatocitoliza) si de 4-10 ori in hepatitele cronice agresive.
Cetoacizii formati in cursul procesului de dezaminare sau transaminare pot intra in ciclul acizilor tricarboxilici (piruvic, oxalacetic, cetoglutaric) unde sufera oxidarea completa pana la CO2 si apa sau pot intra in reactii de transaminare refacand molecule de aminoacizi sau pot constitui material pentru sinteza de glucide (aminoacizi glucoformatori) si lipide.
Posibilitatea sintezei endogene de glucoza pe seama aminoacizilor glucoformatori se explica prin faptul ca procesul de dezaminare oxidativa conduce la formarea de cetoacizi care, in cazul alaninei, acidului glutamic si acidului aspartic sunt totodata si produsi intermediari ai metabolismului glucidic. Aminoacizii glucoformatori (sau glicoformatori) sunt: alanina, arginina, acidul aspartic, acidul glutamic, cisteina, histidina, hidroxiprolina, metionina, valina etc.
La fel, reactiile de beta-oxidare a acizilor grasi care furnizeaza unitati de acid acetic activat, fiind si ele reversibile, exista posibilitatea de sinteza a lipidelor plecand de la cetoacizii rezultati din dezaminari. Aminoacizii care se pot transforma in lipide (cetoformatori) sunt: leucina, izoleucina, lizina, fenilalanina, tirozina si triptofanul. Ultimii cinci sunt, in egala masura glico- si cetoformatori.
Hiperaminoacidemia se poate produce prin aport alimentar hiperproteic, distrugeri tisulare mari, eliminari reduse sau incapacitatii de sinteza proteica a ficatului (sindrom hepatopriv). Starile patologice care sunt insotite de hiperaminoacidemii sunt: insuficientele hepatice, starile de soc, sindromul posttraumatic, starile febrile, post radioterapie, leucozele leucemice, etc.
Hipoaminoacidemia se semnaleaza, de regula in sindroamele nefrotice (prin aminoacidurie accentuata) si in starile grave de denutritie proteica prin lipsa de aport sau malabsorbtie.
O afectiune grava de metabolizare a acizilor aminati este reprezentata de
Hiperfenilalaninemie. Boala este determinata de reducerea activitatii fenil-alanin-hidroxilazei, enzima care transforma fenilalanina in tirozina. Hiperfenilalaninemia este o maladie ereditara, autosomal recesiva, ce apare cu ofrecventa de 1/10 000 de noi nascuti (mai ales la rasa alba si galbena). Concentratia plasmatica de fenilalanina este suficient de ridicata (20 mg/dl sau 1200 mol/L) pentru a activa cai alterne de metabolizare care genereaza fenilpiruvat, fenilacetat, fenillactat si alti derivati care se elimina rapid prin urina. Concentratia plasmatica a altor aminoacizi este moderat scazuta, probabil prin inhibitia secundara a absorbtiei intestinale. Manifestarea clinica majora este reprezentata de retardarea psiho-intelectuala (oligofrenia fenilpiruvica). Afectarea severa a sistemului nervos este o consecinta a acumularii de fenilalanina ce blocheaza competitiv transportul altor aminoacizi necesari sintezei de mielina, formarii de norepinefrina si serotonina. In acelasi timp, fenilalanina este un puternic inhibitor al tirozinazei, enzima cheie in sinteza de melanina, ceea ce genereaza hipopigmentarea parului si tegumentelor (albinism).
Albinismul este consecinta unei tulburari a catabolismului tirozinei, situatie in care nu se mai formeaza melanina (pigmentul pielii). In consecinta, tegumentul si parul sunt decolorate. Deficitul in sinteza pigmentului, de catre celulele melanofore, este atribuit unei insuficiente a tirozinazei, enzima necesara formarii DOPA (dioxifenilalanina) sau/si a DOPA-azei. Depigmentarea poate fi limitata la anumite zone (vitiligo) sau poate fi generalizata (albinism). Bolnavii mai prezinta: fotofobie, nistagmus, astigmatism si uneori cecitate (orbire).
In unele conditii, cu sau fara hiperaminoacidemie, se pot produce aminoacidurii patologice. Acestea pot fi:
Aminoacidurii de cauza renala
- prin cresterea filtrarii glomerulare in glomerulonefrite si sdr. nefrotice;
- prin insuficienta reabsorbtie tubulara renala produsa ca o consecinta a tulburarii transportului activ. Diminuarea re-absorbtiei tubulare genereaza:
- aminoacidurii secundare intoxicatiilor cu plumb, mercur, tetraciclina, salicilati; carentelor alimentare (avita-minoza D); nefropatii dobandite (sindrom nefrotic, necroze tubulare);
- aminoacidurii primare prin defect ereditar de reabsorbtie tisulara: cistinuria, cistinoza, sidromul Fanconi, boala Hartnup.
2. Aminoacidurii de cauza extrarenala (functiile renale sunt normale):
- secundare unor afectiuni generale: boli hepatice cronice, hepatite acute, eclampsie, boli consumptive, leziuni tisulare extinse;
- prin enzimopatii ereditare care se datoresc unor defecte enzimatice in care unul sau mai multi aminoacizi, fiind incomplet degradati, raman in sange in cantitate mare. Excesul se elimina prin urina (fenilcetonuria, boala urinii cu miros de artar)
Cistinoza reprezinta un defect ereditar (autosomal recesiv) de metabolizare (dezaminare) a cistinei. Cistinoza se caracterizeaza prin depunerea in tesuturi (ficat, splina, rinichi, maduva osoasa, conjunctiva, cornee) a cristalelor de cistina. Leziunile renale secundare determina pierderea prin urina a altor aminoacizi. Clinic este prezenta poliuria, polidipsia, fenomene de rahitism si incetinirea cresterii. Investigatiile biologice confirma existenta nefropatiei prin: glucozurie, aminoacidurie, fosfaturie, proteinurie, concomitent cu modificarea unor parametri sanguini: hipofosfatemie, acidoza metabolica, hipopotasemie. Nefrita interstitiala evolueaza spre insuficienta renala ireversibila. Singura solutie terapeutica este transplantul de rinichi.
Sindromul Fanconi reprezinta o entitate clinica caracterizata prin prezenta simultana a hiperaminoaciduriei generalizate, a glucozuriei si hiperfosfaturiei. Boala se asociaza, de cele mai multe ori, cu rahitism sau osteomalacie, cu cistinoza. Aminoacidemia este scazuta. Boala apare in primii ani de viata.
Boala Hartnup se caracterizeaza prin eliminarea urinara crescuta de acizi monoamino-monocarboxilici (mai ales de triptofan). Leziunea ereditara afecteaza transportul tubular si absobtia intestinala a triptofanului. Deoarece, in cursul catabolismului sau, triptofanul genereaza acid nicotinic si nicotinamida (vitamina PP) boala se manifesta la nivelul tegumentelor expuse razelor solare prin leziuni pelagroide. Clinic, mai sunt prezente: leziuni nervoase severe, nistagmus, ataxie c, ataxie cebeloasa si tulburari psihice. Administrarea de vitamina PP amelioreaza simptomatologia.
Alcaptonuria se datoreste tulburarilor ereditare (autosomal recesive) de metabolizare a tirozinei prin lipsa de homogentizic acid - oxidaza. Astfel, acidul homogentizic nu poate fi oxidat, creste in sange si se elimina prin rinichi in cantitate mare colorand urina in brun. Excesul din sange se depune in unele tesuturi (regiunea axilara, inghinala, conjunctiva globului ocular) imprimand acestora o culoare bruna (ocronoza). Depunerea la nivelul cartilajelor articulare (genunchi) poate provoca procese degenerative (artroza ocronotica).
Diagnosticul este usor de stabilit, deoarece urina bolnavilor , care are la emisie o culoare obisnuita, in contact cu aerul se oxideaza si se coloreaza in brun sau negru.
Boala urinii cu miros de artar se produce datorita unui deficit ereditar de alfa-ceto-decarboxilaza. Ca urmare, se acumuleaza in sange si creste eliminarea urinara de aminoacizi ramificati: valina, leucina, izoleucina. Acesti acizi aminati precum si produsii lor de dezaminare confera urinii mirosul caracteristic de artar (zahar caramelizat). Tulburarile apar rapid dupa nastere si se manifesta prin leziuni ale sistemului nervos central (cu crize de apnee) care determina moartea in prima luna de viata.
Amoniacul rezultat din procesele de dezaminare este indepartat rapid, astfel incat concentratia lui in sange nu trece peste 20 g/dl. Indepartarea amoniacului se realizeaza prin fixarea lui de catre acidul glutamic, pentru a forma glutamina si prin formarea de uree. In acest mod, amoniacul cu o toxicitate mare, se transforma in produsi netoxici si este excretat. Glutamina furnizeaza amoniac pentru sinteza de uree in ciclul ureogenetic si pentru secretia de amoniac in rinichi. Rinichiul, fiind bogat in glutaminaza, scindeaza cu usurinta glutamina in acid glutamic si amoniac, care este secretat in tubii renali si serveste pentru neutralizarea surplusului de radicali acizi. Sinteza de uree se face plecand de la CO2 si NH3 prin intermediul ciclului lui Krebs si Henselheit la care participa ornitina, citrulina si arginina. Formarea de uree are loc exclusiv in ficat.
Pe langa un aport alimentar de proteine care intretine un bilant azotat echilibrat, fondul comun de aminoacizi este constituit mai ales din acizii aminati proveniti din alimentele recent ingerate, iar ureea excretata prin urina este dependenta de ingestia de proteine. Un regim alimentar echilibrat determina o excretie de uree de 15-40 g/24 ore.
FIZIOPATOLOGIA METABOLISMULUI Creatinei Si creatininei
Creatina este sintetizata din aminoacizi la nivelul ficatului si rinichilor. O mica cantitate de creatina este eliminata urinar iar cea mai mare parte este captata de muschi, fosforilata si apoi transformata in creatinina, care difuzeaza in plasma si apoi este excretata prin urina. Creatina plasmatica are valori de 0,2-0,8 mg/dl. In conditii normale, creatina nu se elimina prin urina decat in cantitate foarte mica, de 0-50 mg/24 ore. Creatinuria este prezenta la copii si la gravide. Valorile crescute ale creatinei in urina reprezinta un indicator fidel al leziunilor musculare. Astfel, la pacientii cu afectiuni musculare (amiotrofie, distrofie musculara, tireotoxicoza), transformarea creatinei in creatinina este perturbata si se produce o hipercreatinemie cu creatinurie variabila de la o zi la alta. Disparitia creatinei din urina este un indicator fidel al incetarii procesului lezional muscular.
Creatinina, produs al catabolismului muscular, creste in insuficienta renala cronica, fara insa a da nastere la simptome toxice deosebite. Este un semn mai precoce de retentie azotata si un test fidel de apreciere a capacitatii functionale renale pentru ca nivelul sau plasmatic nu depinde de continutul in proteine al regimului alimentar. Creatinina plasmatica are valori de 0,7-1,4 mg/dL (62-132 mol/L).
Cantitatea de creatinina urinara este reprezentativa pentru totalitatea masei musculare active. In conditii normale se elimina o cantitate de 1-2 g/24 ore.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 5716
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved