| CATEGORII DOCUMENTE |
| Alimentatie nutritie | Asistenta sociala | Cosmetica frumusete | Logopedie | Retete culinare | Sport |
Mecanismele etiopatogenice ale DZ tip 1 si ale DZ tip 2 sunt foarte diferite si variate in cele doua tipuri, nefiind inca complet cunoscute.
Etiopatogenia DZ tip 1 este extrem de complexa, multifactoriala, necesitand interrelatia predispozitiei genetice cu fenomene autoimune si factori de mediu. Pana la aceasta ora nu s-a demonstrat nici un agent diabetogen care sa reprezinte un factor major unic de aparitie a DZ tip 1.
Argumente: asocierea DZ tip 1 cu unele afectiuni sigur genetice, agregarea familiala, studiul gemenilor, evidentierea asocierii DZ tip1 cu genele sistemului H.L.A.
Modul de transmitere genetica: este necunoscut, nici transmiterea dominanta, nici cea recesiva neputand fi sustinuta. S-a demonstrat o interactiune stransa intre factorii de mediu si bagajul genetic, dar si natura poligenica a afectiunii. Peste 20 de gene sunt responsabile de dezordinile ce pot sa apara in diabet. Acestea sunt gene de susceptibilitate care, prin definitie, cresc sau modifica riscul de boala. Genele de susceptibilitate nu sunt nici necesare nici suficiente pentru ca boala sa apara.. Prevalenta DZ tip 1 este de 30-50 % la gemeni, de 20 % la fratii sau surorile cu antigen HLA identic, de 5% la fratii si surorile haploidentici, de 1 % la fratii si surorile neidentice. Riscul de a dezvolta diabet este mai mare la copiii din tata diabetic (8,6%) decat la copiii din mama diabetica (3%). Riscul de a face diabet in cursul vietii pentru rudele de gradul unu ale unui diabetic este de 5-8 %, functie de haplotipurile HLA. Antigenele leucocitare umane (HLA- Humane Leukocyte Antigen) - sunt controlate de pe bratul scurt al cromozomului 6. Aceasta regiune include clasele I, II, III. Genele din clasa I sunt exprimate in toate celulele nucleare si servesc ca antigene clasice la transplant. Aceste proteine reprezinta peptide antigenice la celulele CD8T. Clasa II de gene duce la expresia restrictiva a unor antigene la nivelul celulelor b, macrofagelor, celulelor dendridice si functioneaza prin prezentarea peptidelor catre celulele CD4T. Produsii clasei III de gene include proteinele complementului (ex. B, C2 si C4) si tumor necrosis factor. ( Figura 7).Figura de mai jos reprezinta secventele genetice care codifica sinteza unor polipeptide, care alcatuiesc moleculele HLA. situate pe membranele celulare si care initiaza si/sau amplifica raspunsul imun, dar asigura printr-o colaborare complexa cu macrofagele si limfocitele si toleranta imunologica pentru sructurile proprii (self).
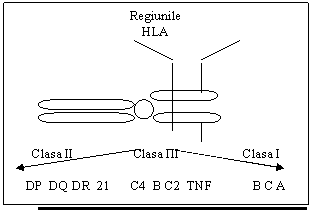
Figura 7. Regiunile HLA
Persoanele cu HLA - DR si HLA-DQ reprezinta 30-50 % din riscul de a dezvolta DZ tip1. Aproximativ 95% din albii cu DZ tip 1 au fie HLA-DR3 fie HLA-DR4. Totusi susceptibilitatea pare sa rezide predominant in HLA-DQ, sub influenta HLA-DR. Riscul poate fi modificat de prezenta unei susceptibilitati puternice sau alele de rezistenta la boala, cum ar fi DQB1 0602, DR2, DR5.
Inafara genelor sistemului HLA, care moduleaza raspunsul imun al organismului, mai sunt implicate si alte gene in etiopatogenia DZ tip 1, cum ar fi: gena insulinei, alelele properdinei si complementului etc.
Riscul pentru DZ tip 1 este de 0,4 % in populatia generala. Totusi aproape 85% dintre pacientii diabetici nu au in momentul depistarii bolii DZ tip 1 in antecedentele familiale.
1.2. Autoimunitate.
DZ tip 1 este considerat actual ca o afectiune autoimuna cu etiologie multifactoriala, caracterizata prin distructia progresiva a celulelor beta pancreatice (care sintetizeaza insulina), ajungandu-se in timp la deficit absolut de insulina endogena. Fenomenele imune s-au demonstrat insa la 90% din persoanele cu DZ tip 1, multi specialisti considerand ca numai forma de etiologie imuna se incadreaza in aceasta clasa. Totusi 10% din pacientii cu DZ tip 1 nu prezinta nici unul din markerii procesului autoimun, dar prezinta deficit absolut de insulina (mai ales negrii din Africa), cu tabloul clinic al DZ tip 1. Acesti pacienti se incadreaza in DZ tip 1 idiopatic.
Argumente ale implicarii autoimune
argument clinic: asocierea DZ cu boli autoimune: boala Basedow, mixedemul, tiroidita Hashimoto, boala Addison, vitiligo, anemia pernicioasa
argumente serologice, citochimice:
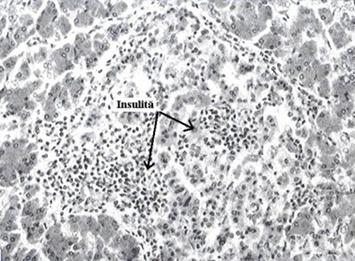
Figura 8 Insulita la un pacient cu DZ tip 1
Insulita (descrisa in 1964) este o leziune specifica DZ tip 1 care dispare dupa un an de la instalarea clinica a acestuia. Consta in infiltratia difuza cu limfocite a insulelor pancreatice. Pe masura progresiei bolii in insulele Largerhans nu mai exista decat celule a si d (figura 8) Autoanticorpii - anti celule insulare pancreatice.
De la prima descriere, in 1974, a anticorpilor anticelule insulare - de suprafata (ICAs), s-au identificat numerosi autoanticorpi la pacientii cu DZ tip 1, markeri ai imunitatii antiinsulare. Identificandu-se titrul acestora in momentul depistarii diabetului, multi din acesti markeri s-au dovedit utili in identificarea persoanelor susceptibile, in perioada presimptomatica, cu luni sau ani de zile inaintea debutului clinic al diabetului. Dintre acestia 4 markeri s-au dovedit a avea senzitivitate si specificitate: I.C.A (Islet Cell Cytoplasmic Autoantibodies - anticorpi anticitoplasma de celule insulare), IAA ( Insulin Autoantibodies = autoanticorpi antiinsulina), anticorpi anti GAD (Glutamic Acid Decarboxilaze = decarboxilaza acidului glutamic) si IA-2 A ( Insulinoma Asociated 2 Autoantibody)
ICA - fac parte din clasa IgG. Sunt prezenti la 70-90% dintre pacientii cu DZ tip 1, in momentul diagnosticarii (valoare diagnostica), uneori cu multi ani inaintea debutului (valoare predictiva). Ei se gasesc si la rudele de gradul 1 ale diabeticilor cu DZ tip 1. In DZ tip 2 se gasesc la 9-16% din cazuri, la majoritatea lor demonstrand de fapt existenta LADA ( Latent Autoimun Diabetes of Adult= diabet autoimun cu debut tardiv al adultului).Dupa 5 ani de la diagnostic sunt prezenti la 5% dintre pacienti. In populatia generala sunt prezenti la 0,2% din aceasta.
Anticorpii anti GAD Sunt prezenti la 55-85% din cazurile nou diagnosticate (valoare diagnostica), la 3-4% din rudele de gradul 1 ale pacientilor cu DZ tip 1, la 10% din pacientii cu DZ gestational, la 10-15% din DZ tip 2, la 1-2% din pacientii cu alte dezordini endocrine autoimune si la 0,5% din populatia de control sanatoasa. Prezenta lor prezice un risc de 50% de aparitie a DZ tip 1 in urmatorii 5 ani
Anticorpi antiinsulina - IAA. Sunt prezenti la 50% dintre pacientii cu DZ tip 1 nou descoperit si netratat inca.
IA-2A sunt prezenti la 60% din pacientii cu DZ tip 1 in momentul diagnosticarii
Combinatia GADA+IA-2A prezice cel mai bine dezvoltarea DZ tip 1.
Testul inhibitiei migrarii leucocitelor la antigen pancreatic este pozitiv.
Alterarea raportului dintre limfocitele supresoare si helper. S-a elaborat un model al citotoxinelor " bune si rele " in patogenia DZ tip 1. Activarea celulelor Th2 duce la producerea de citokine "bune" (IL-4 si IL-10) care vor bloca actiunea distructiva a raspunsului imun celular. Dimpotriva, raspunsul imun caracterizat prin activarea celulelor Th1 duc la producerea de citokine "rele", promotorii distructiei celulelor beta. Th1 duc la cresterea productiei de IFN-g (interferon g), IL-2 (interleukina 2), IL-12 (interleukina 12). Aceste citokine duc la cresterea substantelor oxidante (H2O2-, O2-) cu oxidarea lipidelor, proteinelor, nucleotidelor, carbohidratilor, la cresterea oxidului nitric cu nitrozilarea proteinelor si scaderea NAD+, conducand in final la disfunctia celulelor beta si apoptoza.
1.3. Factorii de mediu
Implicarea factorilor de mediu in aparitia DZ tip 1 a fost impusa de numeroase studii epidemiologice. Exista mari variatii geografice ale prevalentei DZ tip 1 , aceasta fiind, de exemplu, de 18 ori mai mare in Finlanda decat in Japonia; existenta unei variatii sezoniere a incidentei DZ tip 1, aceasta fiind mai mare toamna si iarna etc. Nu s-a demonstrat insa un agent diabetogenic unic care ar putea induce cu siguranta DZ tip 1. Iata cativa factori de mediu implicati:
virusurile- sunt cel mai frecvent incriminate, in special virusul rubeolei congenitale, Coxackie B, virusul urlian, dar si alte virusuri: Cytomegalovirus, virusul Epstein-bar, virusul encefalomiocarditic, retrovirusuri. Ele ar actiona ca factor declansator (trigger) al procesului autoimun sau ca factor citolitic direct al celulei beta sau ca factor aditiv la actiunea distructiva a altor agenti. Alteori ele induc expresia moleculelor HLA-DR pe suprafata celulei b
Expunerea la substante toxice: vacor ,streptozotocina, rodenticide.
Durata alaptarii, consumul laptelui de vaca
Consumul de cafea la varste mici
Stresul
Istoria naturala a procesului diabetogen in DZ tip 1
Exista 5 stadii in evolutia DZ tip 1 (figura 9).
Stadiul 1 include susceptibilitatea genetica (HLA si non HLA), cu masa celulelor b intacta.
Stadiul 2 este indus de o actiune a factorilor de mediu in primele luni-ani de viata, declansandu-se fenomenele imunologice .
Incepe procesul de distructie a celulelor beta. Apar autoanticorpi anti celule insulare, marcand procesul autoimun al bolii. Totusi, in aceasta perioada nu apare o disfunctie masurabila a celulelor beta.
In stadiul 3 apare o scadere a masei celulelor beta, cu o panta foarte variabila de la o persoana la alta (luni-ani). Inceperea distructiei beta celulare este prima data detectabila printr-un test de toleranta la glucoza intravenos anormal, cu deficit al raspunsului insulinic in prima faza.
In stadiul 4 apare un grad avansat de distrugere a celulelor beta, cu hiperglicemie simptomatica, rezultand debutul DZ tip 1( cu secretie minima de peptid C) si dependenta de insulina exogena.
In stadiul 5 este distructie completa a celulelor beta, C peptidul devine indetectabil, autoanticorpii markeri ai bolii dispar.

|
|
Factori precipitanti de mediu) abnormalitati imunologice. Eliberare normala a insulinei |
Descresterea eliberarii insulinei TTG-iv alterat |
Diabet clinic peptid C prezent |
Diabet clinic peptid C absent |
|||
|
| |||||||
|
|
HIPERGLICEMIE | ||||||
|
|
| ||||||
|
|
Remisie partiala |
|
|||||
|
Ani |
Figura 9 Istoria naturala a DZ tip 1 (dupa Eisenbarth modificat).
In general, dupa 10 ani distructia celulara beta este completa. Perioada de remisiune ("luna de miere") poate sa dureze cateva luni, ca exceptie ani, dar totdeauna este tranzitorie. De aceea se va scadea insulina exogena initial, apoi se vor creste dozele sub control strict al glicemiei, pastrandu-se regimul alimentar.
In DZ tip 2 se oscileaza intre predominanta insulinorezistentei cu deficit relativ de insulina pana la predominanta unui deficit in secretia insulinei cu insulinorezistenta. Acesti pacienti nu necesita initial insulina pentru a su pravietui. Exista multe cauze diferite ale acestei forme de diabet si este posibil ca pacientii clasati actual in acest tip de diabet sa scada simtitor in viitor, prin identificarea unor procese patogenice specifice si a unor defecte genetice, care va permite o mai buna diferentiere si o alta subclasificare a diabetului. Atat insulinorezistenta cat si insulinosecretia sunt programate genetic. Aceste programari sunt modificate de o multime de factori de mediu, in special de dieta si activitatea fizica (figura 10) Homeostazia glicemiei a jeune este men tinuta datorita unui echilibru intre productia hepatica de glucoza si utilizarea acesteia de catre muschi si tesutul adipos.
Desi reglarea fina a metabolismului glucozei este influentata de multi factori, majoritatea hormoni si metaboliti intermediari, totusi patru factori influenteaza prioritar variatiile glicemice:
1.Abilitatea pancreasului de a secreata insulina,atat in faza acuta (secretia rapida), cat si in faza sustinuta (secretia bazala)
2.Abilitataea insulinei de a inhiba productia hepatica de glucoza.
3.Abilitatea glucozei de a patrunde in tesuturile insulinodependente - insulinosensibilitate( tesut adipos, muschi)
4.Abilitatea glucozei de a patrunde in celule in absenta insulinei- glucozosensibilitate (celula cerebrala, renala, hematii) (figura 11)
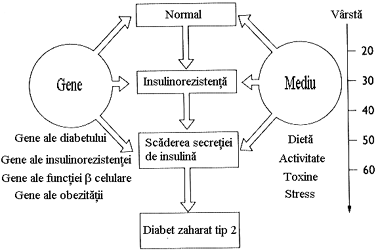
Figura 10 Rolul factorilor genetici si de mediu in aparitia diabetului zaharat
(dupa Kahn - modificat).
In patogenia DZ tip 2 par sa fie implicate cel putin 2 defecte din acest sistem de reglare. Cea mai precoce modificare detectabila este insulino rezistenta din tesuturile periferice. Celulele beta pancreatice detecteaza insu linorezistenta si raspund prin cresterea secretiei de insulina- detectabila prin cresterea insulinemiei a jeune. Pe masura ce starea prediabetica progreseaza creste insulinorezistenta si nivelul insulinemiei. Cand pancreasul isi pierde abilitatea de a suplini insulinorezistenta, nivelul insulinemiei incepe sa scada si apare DZ tip 2. In acest stadiu celula beta este desensibilizata la stimularea de catre glucoza, dar raspunde inca la alte secretogage de insulina (figura 12, figura 13 a, 13 b) Mecanismul exact nu este cunoscut , dar poate fi implicata descresterea transportorilor de glucoza din pancreas- GLUT 2
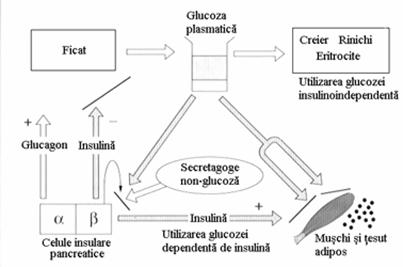
Figura 11 Reglarea homeostazei glicemice.
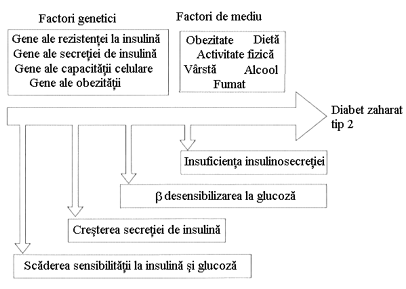
Figura 12 Patogenia diabetului zaharat tip 2 (Dupa Kahn modificat)
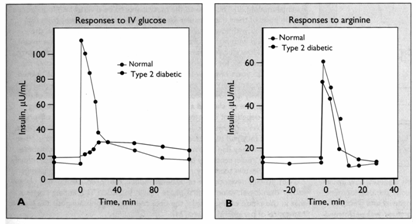
Figura 13 Secretia insulinica in diabetul zaharat tip 2 comparata cu persoanele nediabetice, dupa stimularea cu glucoza i.v.(a) si dupa stimularea cu arginina,
administrata i.v.(b) (Dupa Kahn).
Studii recente sugereaza ca actiunea insulinei la nivelul celulelor beta poate fi importanta in sensibilitatea acestora la glucoza si ca acesta este un alt loc al insulinorezistentei in DZ-insulinorezistenta pancreatica.
2.1.Factori genetici implicati in patogenia DZ tip 2
Predispozitia genetica in DZ tip 2 este mai importanta decat in DZ tip 1, fara a fi implicate insa genele sistemului HLA. Se intalneste la 50% dintre diabetici, iar la gemenii monozigoti concordanta este de 100%. DZ tip 2 este o afectiune heterogena din punct de vedere genetic. Pentru cei mai multi pacienti afectiunea este poligenica. Cand este monogenica boala apare precoce (MODY) (Tabelul 3).
In general perturbarile genetice afecteaza sinteza si secretia insulinica, sau actiunea insulinei, prin : a) afectarea legarii insulinei de receptor; b) modificarea receptorilor; c) modificari intracelulare, post receptor.
Tabelul 3. Clasificarea genetica a diabetului zaharat tip 2.
Defecte in sinteza sau secretia insulinei MODY 1: HNF -4a MODY 2: Glucokinaza MODY 3: HNF - 1a MODY 4: PDX - 1 (IPF-1)(PDX factor de
dezvoltare pancreatica; IPF factor promotor al insulinei). MODY 5: HNF - 1b Mutatii in AND mitocondrial, care codifica t-ARN pentru
leucina. Se asociaza cu surditate. Defecte la nivelul receptorilor insulinei
(in actiunea insulinei) -Leprechaunism -Insulinorezistenta tip A , cu acanthosis nigricans - Sindrom Rabson Mendenhall DZ tip 2
Forme monogenice ale
diabetului zaharat tip 2
Forme poligenice de diabet
zaharat tip 2
Celulele beta capteaza glucoza si aminoacizii cu ajutorul unor transportori specifici, de la nivelul membranei celulare, cum ar fi GLUT- 2 transportorii de glucoza. Acesti transportori sunt exprimati numai la nivelul celulei beta pancreatice si a celulei hepatice. Odata patrunsa in celula glucoza este fosforilata de glucokinaza, apoi este catabolizata, ducand la cresterea raportului ATP/ADP in celula, care activeaza canalele de K -ATP dependente. Celula se depolarizeaza prin iesirea K din celula, are loc un influx de Ca2 , care duce la eliberarea insulinei din granulele secretorii. Nivelul expresiei mai multor molecule implicate in detectarea glucozei, incluzand transportorii de glucoza GLUT 2 si dezvoltarea celulei beta sunt controlate de mai multi factori de transcriptie nucleara. Cei mai studiati dintre acestia sunt HNF-1 alfa, HNF-1beta, HNF-4 alfa si PDX-1( IPF-1). DZ tip MODY poate rezulta din defecte genetice la oricare dintre acesti factori de transcriptie sau ca defect la nivelul glucokinazei.
In DZ tip 2 locul exact al defectiunii la nivelul detectarii glucozei nu este cunoscut, dar studii pe animale sugereaza existenta "down regulation" la nivelul transportorilor de glucoza GLUT 2 (figura 14)
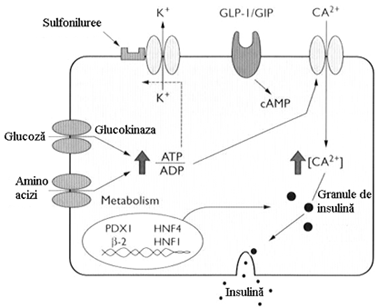
Figura 14. Secretia insulinei de catre celulele beta (dupa Kahn modificat)
Alte defecte genetice identificate in DZ tip 2
gena insulinei (cromozomul 11)
gena receptorilor insulinei (cromozomul 19)
gena transportorilor de glucoza (GLUT 2, GLUT 4- cromozomul 17)
gena peptidului amiloidogen numit amilina a canalelor ionice de conductanta mare pentru K (cromozomul 10)
gena glicogen sintetazei (cromozomul 9) s.a.
De cele mai multe ori aceste perturbari se asociaza, prima secventa fiziopatologica constand in rezistenta periferica la actiunea insulinei, urmata de scaderea consumului periferic de glucoza. Defectul apare ,cel mai probabil, in tesuturile insulinodependente, in teritoriul post receptor. Exemplu: scaderea glicogen-sintetazei in tesutul muscular si a hexokinazei in celulele hepatice. Initial secretia de insulina pancreatica este normala, ulterior apare un hiperinsulinism functional, urmat de ingestia crescuta de alimente datorita hipoglicemiilor repetate, cu instalarea obezitatii.
Insulinorezistenta poate sa apara insa datorita unor defecte la nivel de prereceptor, receptor sau postreceptor insulinic:
La nivel de prereceptor
i secretia unei insuline cu structura anormala
i cresterea proinsulinei, prin perturbarea clivarii ei in insulina si C peptid
i Anticorpi antiinsulinici, ce impiedica legarea insulinei de receptor
i Degradarea crescuta a insulinei
i Cresterea in sange a antagonistilor hormonali (glucagon, cortizol, STH)
La nivel de receptor:
iScaderea numarului de receptori pe suprafata membranelor celulare
iReceptori anormali, ce au afinitate scazuta fata de insulina
i alterarea unor functii ale receptorului, precum acaderea activitatii tirozinkinazei sau autofosforilarea receptorului
La nivel postreceptor
i Alterari ale sistemului efectorilor, in special al transportorilor glucozei. Activitatea proteinkinazei celulare, prima veriga intracelulara declansata dupa unirea insulinei cu receptorul specific, este scazuta in DZ tip 2. Ea pare a fi corelata cu scaderea numarului sau functiei transportorilor intracelulari de glucoza (GLUT 4) raspunzatori de intrarea in celula a glucozei.
i Defecte enzimatice intracelulare implicate in metabolismele inter-mediare cum ar fi scaderea glicogen sintetazei musculare sau a hexokinazei hepatice.
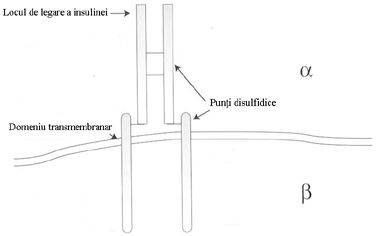
Receptorul insulinic este necesar pentru a media actiunea insulinei. Gena receptorului insulinic este localizata pe bratul scut al cromozomului 19. In forma lui matura este un homodimer, compus din 2 lanturi a si 2 lanturi b a b ). Subunitatile a sunt complet extracelular si sunt legate intre ele prin punti disulfidice. Subunitatile b traverseaza membrana si sunt legate de lanturile a prin punti disulfidice si interactiuni noncovalente. Portiunea intra-celulara a lanturilor b contine o proteina specifica, numita tirozinkinaza.
Insulina se leaga la nivelul portiunii extracelulare a receptorului si induce modificari intracelulare, declansand autofosforilarea.
Insulina actioneaza in tesuturile periferice prin intermediul receptorilor insulinici. Pentru a se media actiunea insulinei este necesara fosforilarea proteinelor. Insulina stimuleaza turnoverul glucozei, favorizand transportul glucozei prin membrana plasmatica, urmat de catabolizarea oxidativa sau nonoxidativa a acesteia, ultima fiind asociata cu sinteza de glicogen. Trans-portul intracelular al glucozei are loc mediat de insulina numai in muschii scheletici, celulele adipoase si inima. Insulina stimuleaza sintezele proteice in aproape toate tesuturile. Insulina are un efect mitogenic, efect mediat prin cresterea sintezei ADN si prevenirea mortii celulare programate sau apoptozei. Ea stimuleaza transportul ionilor prin membrana plasmatica a numeroase tesuturi. Insulina stimuleaza sinteza de lipide in celulele grase, muschii scheletici si ficat; ea previne lipoliza, inhiband lipaza hormonsensibila. Exista tot mai multe dovezi pentru o actiune directa a insulinei prin receptorul de insulina , in reglarea eliberarii de insulina de catre celula b. S-au identificat peste 100 de mutatii ale receptorului insulinic.Aceste mutatii apar pe toata lungimea receptorului. Pacientii cu mutatii la nivelul receptorilor prezinta de obicei insulinorezistenta severa, cum ar fi leprechaunismul, insulinorezistenta tip A si acanthosis nigricans sau sindromul Rabson -Mendenhall
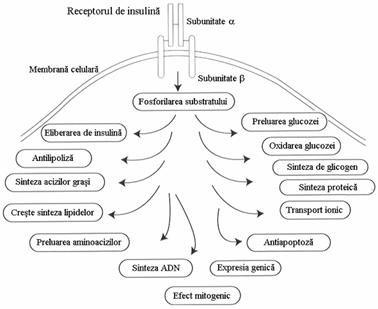
Este cunoscut faptul ca fiecare tesut raspunde diferit la actiunea insulinei. Sensibilitatea tisulara la insulina se coreleaza cu nivelele. receptorilor de insulina exprimati pe membrana plasmatica. Totusi, este tot mai cunoscut ca ansamblul diferitelor componente ale cailor semnalizate de insulina sunt de asemeni responsabile pentru conferirea specificitatii semnalului insulinei asupra celulelor tinta. Astfel, transportul insulinodependent al glucozei este observat numai in muschii scheletici si celulele adipoase, pentru ca aceste tesuturi poseda GLUT 4, transportori de glucoza insulinodependenti. Pe de alta parte, efectul insulinic de a inhiba gluconeogeneza este specific ficatului si rinichiului. In contrast, efectul asupra transportului ionic, sintezei de ADN si sintezelor proteice pare a fi ubiquitar.
Exista doua cai principlae de propagare a semnalului generat de receptorul insulinic:
1 - IRS/PI 3 - K =insulin receptor substrat/phosphatydilinositol 3-kinase. Fosforilarea IRS este stimulata de insulina, factori de crestere asemanatori insulinei (IGF) si interleukina .
2 - Calea MAPK = Ras/mitogen - activated proteinkinase
Calea IRS/PI3-K duce la generarea de PI3-fosfat si apoi la activarea kinazelor dependente de PI, cum ar fi:PDK1, PDK2 si PKC . Unele dintre aceste kinase pot fi necesare pentru activarea kinaselor din aval, cum ar fi serine/ treonine kinase AKT. Se crede ca AKT poate fosforila si inactiva direct GSK3((Glicogen Synthase Kinase 3), conducand astfel la cresterera sintezei de glicogen.
Calea Ras/MAPK are rol in stimularea actiunii de crestere si dezvoltare.
Transportorii de glucoza ( GLUT )
Transportul de glucoza este mediat de o familie a transportorilor de glucoza (glucose carriers). Aceste proteine au o structura tipica, continand 12 domenii transmembranare (figura 17). Se cunosc 8 tipuri diferite de GLUT:
GLUT 1- sunt exprimate in toate tesuturile si liniile celulare. Au rol in captarea glucozei non insulinodependent.
GLUT 2-sunt exprimati in principal in ficat si celulele b pan-creatice. Ei au afinitate relativ mica pentru glucoza dar capacitanta crescuta, servesc la asigurarea unui flux constant de glucoza in aceste organe, la concentratii fiziologice ale glicemiei plasmatice. In celula b acest mecanism cupleaza transportorul de glucoza la fosforilarea glucozei prin intermediul glucokinazei cu afinitate scazuta. Prin modificarea raportului ATP/ADP se inchid canalele de K- ATP dependente, cu patrunderea ionului de Ca in celula si eliberarea insulinei.
GLUT 3 - sunt exprimati ubiquitar, au afinitate crescuta pentru glucoza. Sunt abundenti in sistemul nervos central, unde con-centratia de glucoza este sub nivelul glucozei sanguine si datorita afinitatii crescute pentru glucoza asigura o eficienta maxima in captarea glucozei de catre neuroni.
GLUT 4 - este prototipul transportorilor de glucoza insulinodependenti. Ei se gasesc in organite intracelulare specializate, ca vezicule GLUT 4. Se gasesc in : muschi, celulele adipoase, cord. Exista evidente actuale ca ar exista si un GLUT 8 aditional tot insulinodependent.
GLUT 5 - este exprimat in membrana epiteliala intestinala si in rinichi. Este un transportor de glucoza dependent de sodiu.
GLUT 6 - este o pseudogena nonfunctionala
GLUT 7 este un transportor de glucoza microsomal. Este o proteina a complexului glucozo -6- fosfataza, care joaca un rol in eliberarea glucozei din reticulul endoplasmatic.
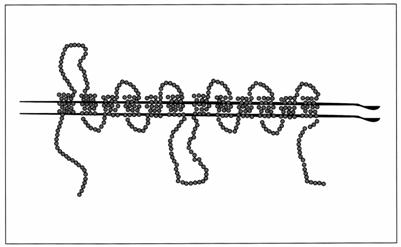
Figura 17. Schema structurii transportorului de glucoza.
In conditii bazale transportul glucozei prin membrana plasmatica se efectueaza insulino-independent, catalizat de GLUT 1. In majoritatea tesu-turilor aceasta este singura cale pentru captarea glucozei. In tesuturile ce prezinta insulino-dependenta in captarea glucozei exista un pool secundar al transportorilor de glucoza, care in conditii bazale este localizat intracelular. Acesti transportori de glucoza sunt localizati in organite intracelulare specializate. (Figura 18)
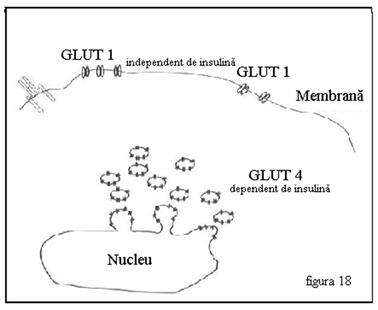
Figura 18. Transportorii de glucoza.
In momentul in care insulinemia creste, apare translocarea GLUT 4. Insulina elibereaza GLUT 4 din vezicule, imbogatind suprafata celulara in GLUT care mediaza capatarea glucozei. (Figura 19)
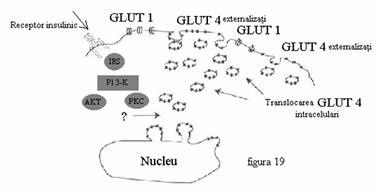
Figura 19. Actiunea insulinei in transportul glucozei.
Mecanismele hiperglicemiei in diabetul zaharat
Homeostazia glicemica.
Glicemia este mentinuta in limite normale, atata timp cat balanta dintre consumul de glucoza pe de o parte si productia si aportul de glucoza pe de alta parte este echilibrata.
iProductia hepatica de glucoza este fin reglata de un complex enzi-matic si hormonal. Sunt implicate doua mecanisme: glicogenoliza (eliberarea rapida a glucozei din glicogen) si glicogeneza ( sinteza de glucoza pornind de la precursori neglucidici: alanina, glicerol, lactat).
i Absorbtia de glucoza post prandial este raspunzatoare de cresterile glicemice post prandiale.
Homeostazia glicemica este mentinuta, in ciuda unui aport si al unui consum foarte variabil al glucozei, prin interventia insulinei, singurul hormon hipoglicemiant si a altor hormoni hiperglicemianti:glucagonul, catecolaminele, cortizolul, hormonul de crestere (tabelul 4)
Scaderea glicemiei apare in starile postabsorbtive (preprandial) sau in caz de efort fizic si sub actiunea insulinei.
Homeostazia glicemica se coreleaza strans cu alte sectoare metabolice: lipoliza, cetogeneza. Lipoliza si cetogeneza sunt inhibate de insulina, dar stimulate de toti ceilalti hormoni amintiti, numiti de contrareglare. Prin lipoliza se elibereaza acizi grasi liberi din tesutul adipos, ducand la cresterea acestora in sange. Din AGL la nivelul ficatului se sintetizeaza trigliceride, ce duc la cresterea in sange a VLDL, si corpi cetonici (cetogeneza). AGL sunt consumati preferential de tesutul muscular.
Tabelul 4. Insulina si hormonii de contrareglare in reglarea principalelor linii metabolice (dupa Hancu - modificat)
|
Linii metabolice |
Insulina |
Glucagon |
Catecol-amine |
Cortizol |
Hormonul de crestere |
|
Captarea glucozei Glicogenoliza Gluconeogeneza Glicogeneza Lipoliza Lipogeneza Cetogeneza Glicemia preprandiala Glicemia postprandiala |
¯ ¯ |
Glicemia bazala (glicemia a jeune sau fasting) este valoarea glicemiei la cel putin 8 ore de la ultima ingestie calorica. Glicemia bazala reprezinta echilibrul dintre secretia bazala de insulina, insulinorezistenta si hormonii de contrareglare, care controleaza productia hepatica de glucoza.
Glicemia preprandiala este asemanatoare din punct de vedere calitativ, dar difera din punct de vedere cantitativ.
Glicemia postprandiala defineste valorile glicemice ce apar la 30-90 minute dupa ingestia calorica, fiind determinata de:
aportul, digestia si absorbtia glucidelor alimentare
captarea glucozei de catre tesuturile periferice, controlata de secretia prandiala. a insulinei, dar si de sensibilitatea tesuturilor la insulina.
Modificari fiziopatologice in diabetul zaharat tip 1
In DZ tip 1 creste atat glicemia bazala (prin cresterea productiei hepatice de glucoza) cat si glicemia post prandiala, in principal prin scaderea captarii glucozei in muschi si tesutul adipos. Deficitul de insulina duce la cresterea lipazei, cu cresterea AGL in sange care duc la insulinorezistenta cu scaderea captarii glucozei in muschi, iar la nivel hepatic AGL duc la sinteza de TG si VLDL, cetogeneza, gluconeogeneza, agravand hiperglicemia si inducand instalarea cetoacidozei
Cele doua meccanisme patogenice implicate in aparitia DZ tip 2.se interconditioneaza reciproc: insulinorezistenta induce deficitul insulinei, care agraveaza insulinorezistenta. Apare astfel hiperglicemia bazala, prin cresterea productiei hepatice de glucoza si hiperglicemia postprandiala, in special prin insulinorezistenta. Insulinorezistenta mai induce hiperlipoliza, prin rezistenta la insulina a lipazei hormonosensibile. Apare cresterea AGL in sange, care agra-veaza insulinorezistenta hepatica si din muschii scheletici. Apare si scaderea activitatii lipoproteinlipazei, care devine "rezistenta" cu inducerea hiper TG,hiper VLDL si scaderea col HDL . Este binecunoscut faptul ca peste 80% dintre pacientii cu DZ tip 2 au fost sau sunt obezi. Mecanismele insu-linorezistentei in obezitate sunt multiple. Obezitatea se adauga predispozitiei genetice la insulinorezistenta. Este foarte posibil ca insulinorezistenta in obezitate sa apara prin productia catorva inhibitori ai actiunii insulinei de catre celula adipoasa. Acesti inhibitori ar putea include: AGL, TNF-alfa (Tumor Necrosis Factor-alfa) si leptina ,hormon regulator al apetitului. TNF-alfa inhiba kinaza receptorului insulinic.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3287
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved