| CATEGORII DOCUMENTE |
| Astronomie | Biofizica | Biologie | Botanica | Carti | Chimie | Copii |
| Educatie civica | Fabule ghicitori | Fizica | Gramatica | Joc | Literatura romana | Logica |
| Matematica | Poezii | Psihologie psihiatrie | Sociologie |
Cinetica secventelor cu intermediari activi
Simplificarile introduse de aproximatia stationaritatii pentru secventele de reactii cuplate prin intermediari activi au permis abordarea cantitativa a unui mare numar de reactii chimice complexe. Din punct de vedere experimental astfel de reactii au o
evolutie temporala caracteristica reactiilor singulare, dar nu pot fi acceptate ca modele de reactii elementare. O explicatie justificata a comportarii experimentale a acestor reactii se bazeaza pe modelul reactiilor consecutive cuplate prin intermediari activi. Ceea ce nu se poate realiza intr-o etapa elementara, este posibil in urma unor etape succesive, fiecare dintre acestea respectand restrictiile impuse reactiilor elementare.
Timpul de viata al unui intermediar activ este cu mult mai mic decat cel caracteristic reactantilor obisnuiti, dar mult mai mare decat cel caracteristic starilor de tranzitie (de ordinul de marime 10-13 s). In timpul sau de viata, un intermediar activ poate suferi un mare numar de ciocniri cu moleculele din sistem. O parte din aceste ciocniri pot fi reactive si au ca rezultat formarea altor compusi.
Existenta intermediarilor activi de reactie a fost dovedita atat cu ajutorul metodelor moderne de analiza, cat si din concordanta dintre viteza de reactie masurata si cea data de ecuatia cinetica.
Caracteristicile cinetice ale secventelor cu intermediari activi pot fi ilustrate cu ajutorul unei secvente cu trei etape:
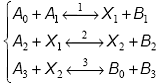
Daca X si X sunt intermediari activi, prin insumarea ecuatiilor stoichiometrice corespunzatoare, se obtine ecuatia stoichiometrica globala: A A A B B B
Viteza globala de reactie este data de:
![]()
Daca vitezele nete ale etapelor componente sunt egale, componentele
acestora ![]() pot fi foarte diferite, conditia
obligatorie fiind ca diferentele dintre ele sa fie egale cu viteza
globala. Printre nenumaratele posibilitati, se pot distinge
trei comportari limita. Acestea pot fi ilustrate cu ajutorul unei
diagrame Tamaru - Boudart, similara cu cea din Figura 9.
pot fi foarte diferite, conditia
obligatorie fiind ca diferentele dintre ele sa fie egale cu viteza
globala. Printre nenumaratele posibilitati, se pot distinge
trei comportari limita. Acestea pot fi ilustrate cu ajutorul unei
diagrame Tamaru - Boudart, similara cu cea din Figura 9.
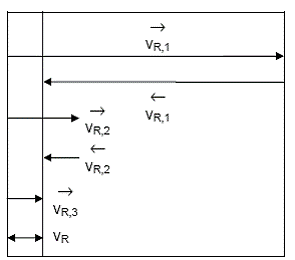
Figura 9. Diagrama Tamaru - Boudart pentru o secventa cu trei
etape cuplate prin intermediari activi
Viteza globala, vR, poate fi comparata cu vitezele componente ale iferitelor etape, gasindu-se si urmatoarele cazuri limita:
1. o etapa poate fi considerata cvasiechilibrata atunci cand itezele etapelor sale componente sunt mult mai mari decat viteza lobala (egala cu diferenta dintre ele).
Daca se noteaza o astfel de etapa cu indicele "e", se poate
scrie conditia ia de cvasiechilibru in forma: ![]() Concentratiile componentilor
participanti la o astfel de etapa indeplinesc conditia de
echilibru cu o buna aproximatie, desi reactia neta are
loc. Viteza globala de reactie, egala cu diferenta celor doua
componente, reprezinta o perturbare slaba a echilibrului (etapa 1 in
figura).
Concentratiile componentilor
participanti la o astfel de etapa indeplinesc conditia de
echilibru cu o buna aproximatie, desi reactia neta are
loc. Viteza globala de reactie, egala cu diferenta celor doua
componente, reprezinta o perturbare slaba a echilibrului (etapa 1 in
figura).
2. o etapa poate fi considerata unilaterala (numita uneori si ireversibila) atunci cand viteza reactiei inverse este neglijabila in comparatie cu viteza reactiei directe (in limitele erorilor experimentale). Daca se noteaza o astfel de etapa cu indicele "u",
se poate scrie conditia de etapa unilaterala in forma: ![]() , sau
, sau ![]() . Rezulta ca
viteza unei etape unilaterale este egala cu viteza globala (etapa 3
din figura).
. Rezulta ca
viteza unei etape unilaterale este egala cu viteza globala (etapa 3
din figura).
3. o etapa este numita limitativa
sau determinanta de viteza (sau uneori, impropriu, etapa
lenta) atunci cand vitezele etapelor sale componente sunt cu mult mai mici
decat etapele componente ale celorlalte etape. Daca se noteaza o
astfel de etapa cu indicele "l", se poate scrie conditia de
etapa limitativa in forma: ![]() . Daca in sistem
exista o etapa limitativa, atunci celelalte etape sunt fie
cvasiechilibrate fie nesemnificative din punct de vedere cinetic. Cand
exista o astfel de etapa, intreaga rezerva de energie
libera Gibbs pentru procesul global seconcentreaza in ea (pentru
celelalte etape cvasiechilibrate variatia energiei libere Gibbs,
. Daca in sistem
exista o etapa limitativa, atunci celelalte etape sunt fie
cvasiechilibrate fie nesemnificative din punct de vedere cinetic. Cand
exista o astfel de etapa, intreaga rezerva de energie
libera Gibbs pentru procesul global seconcentreaza in ea (pentru
celelalte etape cvasiechilibrate variatia energiei libere Gibbs, ![]() , este aproximativ
egala cu zero).
, este aproximativ
egala cu zero).
Deseori, diferitele simplificari in secventele de reactii cuplate prin intermediari activi sunt ilustrate cu ajutorul unor profile de energie potentiala (aceasta fiind unul dintre factorii importanti care controleaza viteza unei reactii chimice). Un exemplu de astfel de
profil este dat in Figura 10.
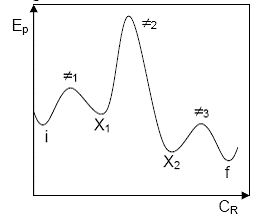
Figura 10. Profil de energie potentiala in functie de coordonata de
reactie pentru o secventa cu trei etape
(i - stare initiala, f - stare finala, X , X - stari cu intermediari activi,
- starile de tranzitie pentru cele trei etape)
Etapele cu bariere mici de energie potentiala sunt cvasiechilibrate. Cea de a doua etapa, cu barierele de energie potentiala cele mai mari si deci cu vitezele componente cele mai mici, este etapa limitativa sau etapa determinanta de viteza. O etapa unilaterala se reprezinta intr-o astfel de diagrama printr-un maxim cu o bariera mica pentru procesul direct si una foarte mare pentru cel invers.
Principalele caracteristici ale secventelor cu intermediari activi pot fi formulate astfel:
1. Viteza de reactie scade monoton in timp (cu exceptia unor reactii cu feedback, cum ar fi reactiile autocatalitice, reactiile exoterme in conditii adiabatice si reactiile in lant ramificat)
2. Ecuatiile cinetice au in general forme nefactorizabile. Numai in anumite cazuri particulare acestea devin factorizabile. Consideram ca exemplu urmatoarea secventa, presupusa cuplata printr-un intermediar activ:
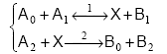
Viteza globala de reactie se poate calcula ca viteza neta pentru oricare dintre etapele componente. Pentru cea de a doua etapa calculul este cel mai simplu, deoarece este unilaterala:
![]()
Concentratia intermediarului X se calculeaza cu ajutorul aproximatiei stationaritatii:
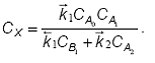
Ecuatia cinetica are forma finala:
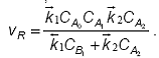
Se observa ca ecuatia obtinuta nu este factorizabila. Exista totusi doua cazuri limita, cand in suma de la numitor numai unul dintre termeni are valori semnificative.
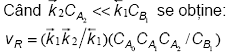
Cea de a doua etapa este limitativa, iar prima este
cvasiechilibrata. Ecuatia obtinuta este factorizabila,
avand o constanta de viteza globala ![]() Reactia este de ordinele unu in raport cu
A0, A1, A2 si de ordinul (-1) in raport
cu B1.
Reactia este de ordinele unu in raport cu
A0, A1, A2 si de ordinul (-1) in raport
cu B1.
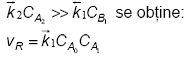
Prima etapa este limitativa, iar cea de a doua este nesemnificativa cinetic (nu mai apare in ecuatia cinetica) Ecuatia obtinuta este de asemenea factorizabila. Numeroase reactii de acest fel au fost intalnite la studiul mecanismelor de reactie in chimia anorganica, in chimia organica ca si la studiul reactiilor catalitice.
3. In ecuatiile cinetice ale unor secvente apar si ordine de reactie fractionare. Sa analizam de exemplu secventa:
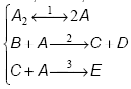
Ecuatia stoichiometrica globala este data de A + B = D + E. Aplicand aproximatia stationaritatii pentru intermediarul C si considerand ca prima etapa este cvasiechilibrata se obtine:
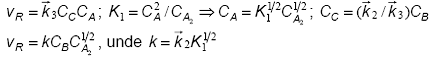
Astfel de comportari au fost gasite pentru reactiile de halogenare, incluzand reactia bromului cu hidrogenul discutata anterior.
4. Dependenta de temperatura a vitezei de reactie este de regula de tip Arrhenius pentru reactiile cu ecuatii cinetice factorizabile. Parametrii de activare, A si Ea, pot avea semnificatii mai simple sau mai complexe. Pentru celelalte reactii dependenta de temperatura a vitezei de reactie este mai complexa.
5. Ecuatiile cinetice factorizabile pot fi exprimate in forma generala:
![]()
unde aj sunt ordinele partiale de reactie, care pot lua valori intregi, semiintregi, pozitive sau negative.
Dupa cum se vede, exista diferente importante intre forma ecuatiei cinetice pentru o reactie elementara si pentru o secventa cuplata prin intermediari activi: ecuatii cinetice nefactorizabile, ordine partiale de reactie negative, nule, semiintregi, etc.
Ordinele de reactie se pot determina experimental utilizand fie metode integrale, fie diferentiale. Metodele integrale presupun cunoasterea curbei cinetice si incercarea tuturor ordinelor in ecuatia generala. Compararea rezultatelor experimentale cu cele calculate pentru diferite ordine da posibilitatea selectarii celui mai bun ordin. Utilizarea ecuatiei cinetice integrale si determinarea timpului de injumatatire permite de asemenea o evaluare a ordinului global de reactie.
Metodele diferentiale presupun in plus derivarea curbei experimentale si utilizarea ecuatiei cinetice diferentiale. Metoda izolarii presupune de exemplu utilizarea unui exces din componentii reactanti, mai putin unul. In acest caz se determina ordinul partial de reactie in raport cu reactantul minoritar j cu ajutorul ecuatiei:
![]()
Se poate proceda in principiu la fel si cu ceilalti reactanti. Deseori, pentru a elimina influenta produsilor de reactie, se utilizeaza metoda vitezelor initiale (cand conversia este sub
Secvente deschise si inchise de reactii
In urma producerii reactiilor care implica participarea intermediarilor activi, pot rezulta fie numai produsi stabili, fie produsi stabili si alti intermediari activi. Ca urmare, secventele cuplate prin intermediari activi pot fi de doua feluri: deschise si inchise.
Cand produsii uneia dintre etape sunt numai substante stabile, secventa este numita deschisa. Un exemplu de secventa deschisa cu trei etape poate fi ilustrat cu ajutorul modelului:
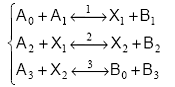
O astfel de secventa poate fi vizualizata cu ajutorul schemei din Figura 11.
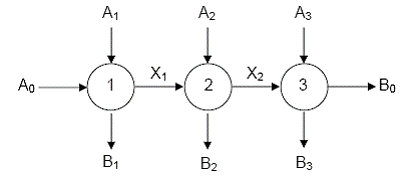
Figura 11. Secventa deschisa de trei etape (1, 2, 3) cuplate prin
intermediari activi (X si X
Fiecare reactie este reprezentata printr-un cerc in care intra reactantii si din care ies produsii. Etapele componente pot fi bimoleculare (ca in schema) sau monomoleculare. In ultima etapa se formeaza numai produsi stabili.
Cand in fiecare etapa se formeaza un intermediar activ, procesul capata un caracter ciclic:
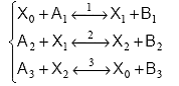
Componentul X0, consumat in prima etapa, se regenereaza in ultima, conferind procesului un caracter ciclic. O astfel de secventa poate fi vizualizata cu ajutorul schemei din Figura12.
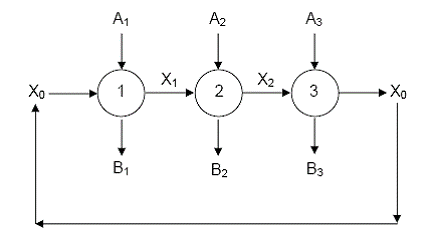
Figura 12. Secventa inchisa de trei etape (1, 2, 3) cuplate prin intermediari activi
Cele doua reprezentari schematice justifica notiunile de secventa deschisa si secventa inchisa. Etapele componente sunt liniare, in fiecare formandu-se cel mult un intermediar activ. Exista si etape neliniare, caracteristice reactiilor in lant ramificat, in care dintr-un intermediar activ si un component stabil se formeaza doi intermediari activi.
In secventele inchise, primul intermediar activ, X , se poate forma in doua moduri distincte:
a) Printr-o reactie de initiere dintr-o substanta stabila (fie reactant, fie initiator). Intermediarii activi astfel obtinuti (de regula radicali liberi) participa la un numar foarte mare de cicluri datorita existentei etapei de regenerare, pana la epuizarea reactantilor sau pana la scoaterea intermediarilor din sistem, ca urmare a formarii unor produsi stabili. Astfel de reactii sunt numite reactii in lant.
b) Prin introducerea in sistem a unui component stabil capabil sa formeze intermediari activi in urma interactiei cu componentii reactanti si sa se regenereze in ultima etapa a secventei. Un astfel de component este numit catalizator, iar reactiile rezultate au fost numite reactii catalitice.
Reactiile in lant radicalic au loc cu viteze mai mari decat reactiile dintre moleculele obisnuite, care poseda un numar par de electroni. Radicalii liberi sunt prezenti in sistem in concentratii foarte mici si au reactivitati diferite in functie de structura lor. Cei mai reactivi (cu mase moleculare mici) reactioneaza cu moleculele practic la fiecare ciocnire cu acestea.
Reactiile de aparitie in sistem a radicalilor
liberi, numite reactii de
initiere a lantului, au loc fie prin disocierea termica
homolitica a unor molecule cu legaturi slabe numite initiatori
(de exemplu peroxizii), ROOR 2RO , fie prin fotodisocierea unui initiator sau chiar a unui reactant ca
de exemplu ![]() in urma
absorbtiei unei
in urma
absorbtiei unei
cuante de energie luminoasa, hn
Avand in vedere diferentele mari dintre concentratiile radicalilor liberi si cele ale moleculelor obisnuite, reactiile radical - molecula, numite reactii de propagare a lantului, sunt mult mai probabile decat cele dintre radicali.
Daca in urma reactiei radical - molecula se formeaza un singur radical si o alta molecula, procesul global este numit reactie in lant drept. Reactiile de recombinare radicalica, numite reactii de intrerupere, au ca rezultat scaderea concentratiilor acestor specii reactive din sistem. Speciile care se pot combina cu radicalii liberi eliminandu-i din sistem sunt numiti inhibitori. Inhibitorii pot avea un efect modulator, in sensul reglarii vitezei de reactie. Numeroase reactii de halogenare si de polimerizare au loc conform unui astfel de mecanism. Ecuatiile lor cinetice se obtin cu ajutorul aproximatiei de stationaritate a concentratiilor radicalilor liberi. Reactiile care au loc prin intermediul lanturilor drepte au vitezele maxime la inceputul reactiei si scad pe masura ce concentratiile reactantilor scad.
Cinetica reactiilor catalitice poate sa fie ilustrata cu ajutorul unei secvente in doua etape de forma:
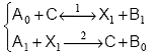
unde catalizatorul C poate sa fie un component al unui sistem omogen, o enzima, sau un centru catalitic de pe o suprafata solida, iar X este un intermediar activ.
Ecuatia cinetica poate sa fie obtinuta utilizand aproximatia de stationaritate pentru intermediarul activ X1:
![]() (105)
(105)
si legea de conservare a centrilor catalitici:
![]() (106)
(106)
unde ![]() reprezinta concentratia totala de
centri catalitici (in mol L-1 pentru sisteme omogene si
enzimatice si mol m-2 pentru sistemeeterogene
fluid-solid).
reprezinta concentratia totala de
centri catalitici (in mol L-1 pentru sisteme omogene si
enzimatice si mol m-2 pentru sistemeeterogene
fluid-solid).
Tinand cont ca vitezele nete ale celor doua etape sunt egale in stare stationara, ecuatia cinetica se obtine plecand de la:
![]()
Din cele trei ecuatii se obtine in final:
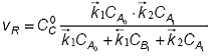
Se observa ca viteza procesului este direct proportionala cu concentratia totala a catalizatorului. In functie de natura sistemului, ecuatia cinetica generala (108) poate sa devina mai simpla.
In cataliza omogena catalizatorul poate sa fie un acid (in particular ionul de hidroniu, H3O+), o baza (in particular ionul hidroxid, HO-), un ion metalic sau un complex al acestuia etc.
In cataliza enzimatica enzimele, cu structuri complexe de proteina, sunt catalizatorii cu cea mai mare eficacitate si specificitate. Cand o reactie de forma S = P (cu S substrat si P produs) este catalizata de o enzima E, conform modelului:
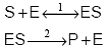
aproximatia stationaritatii pentru complexul
enzima substrat ES si legea de conservare a centrilor catalitici ![]() , conduc la obtinerea
ecuatiei Michaelis-Menten:
, conduc la obtinerea
ecuatiei Michaelis-Menten:
![]()
In cataliza in sisteme eterogene fluid - solid centrii catalitici sunt situati pe suprafata solidului. Catalizatorii solizi prezinta multe avantaje practice legate de separarea componentilor. Deoarece reactia catalitica are loc pe suprafata, viteza de reactie este dependenta si de gradul de dispersie a solidului catalitic. Ecuatia cinetica este mai complexa intrucat, pe langa reactia de pe suprafata catalizatorului, viteza de reactie este dependenta si de etapele de difuzie ale reactantilor catre suprafata solida si ale produsilor dinspre aceasta suprafata catre faza fluida, ca si de procesele de absorbtie - desorbtie ale reactantilor si produsilor pe suprafata solida.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1224
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved