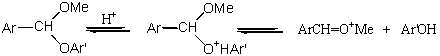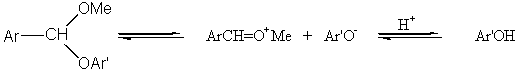| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
DOCUMENTE SIMILARE |
|
TERMENI importanti pentru acest document |
|
Adice na karbonylovou skupinu a příbuzné reakce
Adice na C=O vazbu je z pohledu mechanismu mnohem méně komplikovaná než adice na vazbu C=C. Většina reakcí zahrnuje nukleofilní atak na karbonylový uhlík a elektrofilní atak, obvykle protonaci, na kyslík.
|
|
Nenacházíme zde zdaleka takovou řadu mechanistických typů, jako v případě adicí na alkeny. Reakce ovšem mohou být kysele nebo bazicky katalyzované - báze mohou aktivovat nukleofil, kyseliny mohou aktivovat karbonylovou skupinou a na druhé straně reagovat s nukleofilem. Řada primárních karbonylových aduktů také podléhá dalším reakcím.
Jednoduché adice
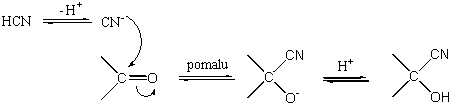
Jednou z prvých podrobně zkoumaných reakcí tohoto typu byla reakce kyanovodíku - tvorba kyanhydrinů. Rychlost reakce závisí na koncentraci aldehydu nebo ketonu i kyanidového iontu. Reakce je katalyzována bázemi, kyselá katalýza je zanedbatelná. Rychlost limitujícím krokem v reakci je atak kyanidového iontu na karbonylovou skupinu.
|
|
Zanedbatelný vliv kyselé katalýzy ukazuje, že protonace karbonylové skupiny nastává až po rychlost limitujícím kroku. Tento předpoklad byl potvrzen i studiem kinetiky zpětné reakce, která je také bazicky katalyzovaná a u níž bylo zjištěno, že hydroxylový proton musí být kompletně odštěpen dříve, než je odtržen kyanidový iont.
|
|
Další podrobně studovanou reakcí je hydratace. Tato reakce je kysele i bazicky katalyzovaná. Kyseliny urychlují reakci protonací karbonylového kyslíku a báze ji urychlují odtržením protonu z vody. Skutečnost, že reakce je kysele i bazicky katalyzovaná ukazuje, že tvorba
vodíkových vazeb je dostatečnou aktivací, a že dochází k přenosu protonu v průběhu rychlost limitujícího kroku.
Obě tyto reakce jsou vratné, rovnováha je značně ovlivňována charakterem skupin navázaných na karbonylový uhlík aldehydu nebo ketonu. Např. formaldehyd je téměř úplně hydratován ve zředěných vodných roztocích, acetaldehyd ze 60% a aceton prakticky vůbec. Příčinou těchto efektů jsou jak elektronické, tak sterické vlivy. Elektrondonory stabilizují kladný náboj na karbonylovém uhlíku, objemné substituenty destabilizují tetraedrický diol.
Obdobnou reakcí k hydrataci je tvorba hemiacetalu. Velmi široce studovanou variantou je mutarotace glukosy nebo jejího 2,3,4,6-tetra-O-methylderivátu, která zahrnuje rovnováhu epimerních hemiacetalů přes otevřené hydroxyaldehydy. Reakce je kysele i bazicky katalyzována. V benzenových roztocích, obsahujících směsný katalyzátor (pyridin + fenol), je reakcí třetího řádu se závislostí na koncentraci substrátu i obou katalyzátorů. Tato skutečnost byla původně interpretována jako existence trimolekulárního tranzitního stavu, ale mnohem pravděpodobněji se jedná o obvyklou bimolekulární katalýzu jednou z komponent iontového páru pyH+PhO-, poněvadž je reakce katalyzována i kvarterními ammoniumfenoxidy, které neobsahují žádnou kyselou funkci. 2-pyridony jsou mnohem efektivnějším katalyzátorem než zmíněná směs, ale řídí se kinetikou druhého řádu. Jedná se zřejmě o skutečný příklad současné kyselé i bazické katalýzy.
|
|
Katalýza, která kombinuje kyselé i bazické vlastnosti analogickým způsobem, se nazývá bifunkcionální a bývá předpokládáno, že je důležitým rysem řady enzymatických reakcí.
Adice následovaná substitucí - acetaly
Většina acyklických hemiacetalů, podobně jako hydrátů, je stabilní pouze v roztocích. Reakce s další molekulou alkoholu vede potom ke stabilním produktům - acetalům.
|
|
Entropický term (3 molekuly nalevo v reakci a 2 napravo) vede k posunu rovnováhy doleva. Většina mechanistických studií byla proto prováděna na zpětné reakci - hydrolýze acetalu.
Přeměna acetalu na hemiacetaly - specificky kysele katalyzovaná reakce
Hydrolýza alkylacetalů je proticky katalyzována, není katalyzována bazicky a i katalýza neprotickými kyselinami je téměř zanedbatelná. To ukazuje, že hydrolýza je zahájena rychlou protonací acetalu. Další krok musí zahrnovat substituci nukleofilu molekulou vody za tvorby hemiacetalu. Můžeme si představit celkem čtyři možné modely průběhu reakce - monomolekulární nebo bimolekulární a s atakem buď na alkoxylovém uhlíkovém atomu, anebo na uhlíku vznikající karbonylové skupiny.
|
|
V případě jednoduchých acetalů probíhají reakce zjevně mechanismem APC1. V této souvislosti musí být nalezena odpověď na několik otázek:
a) která vazba je přerušená
Acetaly jsou hydrolyzovány v 18O značené vodě za vzniku neznačeného ROH a podobně 18O značené aldehydy reagují s alkoholem za vzniku neznačeného acetalu a H218O. Oba výsledky ukazují, že vazba R-O je zachována. Hydrolýza acetalů s chirální alkoxyskupinou poskytuje alkoholy se zachovanou stereochemickou konfigurací. Účast mechanismu AAL2 by vedla k Waldenově zvratu a mechanismu AAL1 k racemizaci. Methanolýza acetalu, která zřejmě probíhá podobným mechanismem, dává v souladu s APC1 O-methylacetal a ROH. Účast AL mechanismu by vedla k tvorbě hemiacetalu a ROMe.
b) monomolekulární nebo bimolekulární průběh
Nepřítomnost bazické katalýzy je prvním argumentem ve prospěch monomolekulárního mechanismu. Jestliže totiž voda může přímo nahradit ROH (APC2), potom by podobně OH- mohl nahradit RO-. Stejně tak efekt substituentu na „karbonylovém uhlíku“ opět podporuje myšlenku monomolekulárního mechanismu - reakce je výrazně urychlována elektrondonory. Entropie aktivace hydrolýzy acetalů jsou malé, ale kladné (cca 30 J/ K mol), což je typické pro disociativní tranzitní stavy. V případě A2 mechanismu, ve kterém participuje molekula vody v tranzitním stavu, by bylo zřejmě oprávněné očekávat velkou negativní hodnotu aktivační entropie. Preferovaným mechanismem je tedy zřejmě APC2.
Přeměna acetalu na hemiacetaly - obecná kyselá katatýza
V případě, že kationtový intermediát je neobvykle stabilizován, může vzniknout i bez předchozí protonace acetalu a reakce může být obecně kysele katalyzována. Příkladem může být hydrolýza ortho-esterů.
|
|
V případě, že odstupující skupina R nese silně elektronakceptorní substituent, potom dochází nejen k usnadnění rozpadu C-OR vazby, ale i ke znevýhodnění protonace snížením bazicity acetalu. Přenos protonu probíhá tedy současně s rozpadem C-O vazby a reakce je obecně kysele katalyzována.
|
|
Příkladem může být i efekt odstupující p-nitrofenoxyskupiny, kde v roztocích s pH větším než 4 je kysele katalyzovaný proces potlačen monomolekulární disociací neprotonovaného acetalu.
|
|
|
|
Podobně v methylfenylacetalu benzaldehydu vede stabilizace jak kationtu PhCH=OMe+, tak i odstupujícího aniontu PhO- k součinnému obecně katalyzovanému mechanismu.
V tomto systému je reakce urychlována elektrondonory substituentu Ar na uhlíku vznikajícího karbonylu a vzrůstající kyselostí katalyzátoru HA. Změna substituentu v aryloxyskupině Ar’O má jiný vliv - elektrondonory i elektronakceptory urychlují reakci. Průběh reakce potom může být popsán v termínech stupně přerušení C-O vazby a míry přenosu protonu z katalyzátoru na acetal. V jednom extrému si můžeme představit, že acetal je úplně protonován před začátkem rozpadu C-O vazby - to odpovídá specifické kyselé katalýze.
|
|
Ve druhém extrému může být fenoxidový aniont odštěpen v nekatalyzovaném kroku a potom protonován na fenol.
|
|
Následující obrázek schematicky ukazuje plochu potenciální energie v rozvinutém stádiu rozpadu C-O vazby a tvorby O-H vazby.

Zobrazení plochy potenciální energie pro obecně kysele katalyzovanou hydrolýzu acetalů: a) perspektivně, b) v rovině. Schéma ilustruje proces současného zániku C-O vazby a přenosu protonu. V centrálním TS je C-O vazba přerušena právě z poloviny a proton je na půli cesty mezi katalyzátorem a aryloxy kyslíkovým atomem.
Část (b) obrázku ukazuje, jak modifikace reakce mění její průběh. Vrchní polovina obrázku odpovídá reakci, ve které je C-O rozpad v tranzitním stavu vyvinut více než přenos protonu a odstupující aryloxyskupina nese částečný záporný náboj. Spodní polovina je obrazem situace, ve které přenos protonu převažuje nad štěpením C-O vazby a odstupující aryloxyskupina nese částečný kladný náboj. Všimněme si, že snížení energie ve směru kolmém k reakční koordinátě posouvá tranzitní stav tímto směrem, ale její snížení ve směru rovnoběžném k reakční koordinátě posouvá tranzitní stav na druhou stranu. Tedy elektrondonorové substituenty v Ar stabilizací intermediátu ArCH=O+Me posouvají tranzitní stav směrem k vrchní části schematu a pryč od pravého rohu. Výsledkem je redukce rozsahu přenosu protonu v tranzitním stavu, jak je ukázáno v části (a) obrázku. Efekt vzrůstající síly kyselé katalýzy se projevuje posunem tranzitního stavu dolů a doleva. Dochází tedy k menší míře rozpadu C-O vazby v tranzitním stavu a tedy k tvorbě méně kladného náboje na vznikající uhlík -kyslík dvojné vazbě. Substituční efekt v aryloxyskupině Ar’O je poněkud komplikovanější. Elektronakceptory stabilizují aniont, zvyšují jeho schopnost odstoupit a snižují tím nároky na katalyzátor. Parciální záporný náboj na kyslíkovém atomu aryloxyskupiny je působením substituentu delokalizován, energie tranzitního stavu je tedy snížena a reakce je urychlena. Elektrondonory Ar’ usnadňují protonaci acetalu. Energie tranzitního stavu se ovšem také snižuje, protože aryloxyskupina nyní nese parciální kladný náboj, který je stabilizován substituentem a reakce je opět urychlována. Zobecnění těchto skutečností potom umožňuje konstruovat následující obrázek, shrnující vlivy různých faktorů na variantní tranzitní stav obecně katalyzované hydrolýzy acetalu.

Variace TS pro obecně katalyzovanou hydrolýzu acetalů. Šipky ukazují vliv následujících změn na centrální TS: a) vliv elektrondonorních substituentů karbonylu, b) vliv vzrůstající kyselosti katalyzátoru, c) přítomnost elektronakceptorů a d) elektrondonorů v odstupující skupině.
Hydrolýza hemiacetalu
Konečným stádiem hydrolýzy acetalu je hydrolýza intermediátu - hemiacetalů. Ta je zřejmě zpětným krokem kysele katalyzované adice. Obecně je tento krok mnohem rychlejší než počáteční rozpad acetalu. Jestliže by tomu tak nebylo, potom by docházelo k vzrůstu koncentrace hemiacetalu v průběhu reakce. Hydrolýza dimethylacetalů s D2O v přítomnosti CD3OD ničemu takovému nenasvědčuje. Vymizení acetalových methoxysignálů v NMR spektru je pravidelné a nevykazuje známky přítomnosti dvou procesů s různými rychlostmi a přesně souhlasí s tvorbou CH3OD signálu. Dosud existuje pouze málo studií hydrolýz hemiacetalu především z důvodu jejich špatné přístupnosti. Příkladem může být velmi rychlá hydrolýza PhCH(OMe)OAc, kde acyloxyskupina odstupuje mnohem snadněji než alkoxyskupina (a obecném případě i aryloxyskupina) a v celkovém průběhu reakce je tedy hydrolýza hemiacetalu rychlost limitujícím krokem. Reakce je sledovatelná NMR spektry a ukazuje růst a následný úbytek signálu hemiacetalového CH protonu. Tato reakce je obecně kysele katalyzovaná a narozdíl od většiny hydrolýz acetalu i obecně bazicky katalyzovaná. To ukazuje, že důvodem rychlé hydrolýzy hemiacetalového intermediátu v průběhu normální kysele katalyzované hydrolýzy acetalů je účast rozpouštědla nebo aniontu kyselého katalyzátoru.

V méně kyselých roztocích může hydrolýza neprotonovaného hemiacetalu vykazovat rysy obecné bazické katalýzy. V případě hemiacetalu formaldehydu HOCH2OR a odpovídajících derivátů deuteroformaldehydu byl nalezen velký sekundární kinetický izotopový efekt odpovídající pokročilému vývoji C-O dvojné vazby a následné CH2 rehybridizaci v tranzitním stavu.
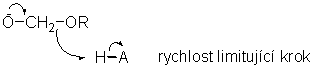
Adice následovaná eliminací: iminy
Primární aminosloučeniny reagují s aldehydy a ketony za vzniku iminoderivátu. Patří sem dobře prostudované reakce vzniku N-aryl- a N-alkyliminů, oximů, semikarbazonů a fenylhydrazonů.

Závislost rychlosti na pH
Závislost reakční rychlosti na pH má charakteristický tvar:

Závislost rychlosti reakce na pH pro případ reakce 0.0005M acetonu s 0.0167M hydroxylaminem. Plná čára představuje experimentální závislost. Tečkované křivky jsou vypočtené průběhy pro jednotlivé kroky reakce.
a) nekatalyzovaná adice: rychlost = k1[Me2CO][NH2OH] – rychlost klesá se zvyšující se kyselostí v důsledku protonace hydroxylaminu, b) kysele katalyzovaná adice: rychlost = k1’[Me2CO][NH2OH][H3O+], c) kysele katalyzovaný dehydratační krok: rychlost = K1k2[Me2CO][NH2OH] ][H3O+].
Dehydratační krok je obvykle kysele katalyzován a při nízkém pH je podstatně rychlejší než krok 1. Pro rychlost potom platí:
rychlost = k1[RNH2][R2’CO]
Koncentrace volné báze klesá se vzrůstající kyselostí v důsledku protonace aminu a tím je i reakce zpomalována. Při nižších koncentracích kyseliny dochází k poklesu účinnosti katalýzy dehydratace a krok 2 se stává rychlost určujícím. Existuje oblast složitého kinetického chování, v níž probíhá počáteční tvorba intermediátového aminohydrinu s poněkud pomalejším rozpadem, ale při vyšších pH, kdy k2 << k -1, je první krok prakticky v rovnováze (k1/k -1 = K1) a rychlostní rovnice se zjednodušuje na tvar:
Rychlost = K1k2[RNH2][R2’CO] [H3O+]
Reakce tohoto typu je možné monitorovat pomocí UV spektroskopie, anebo užitím NMR technik.
Katalýza
a) adiční krok
|
|
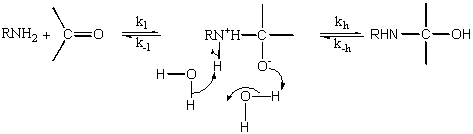
Počátečním atakem aminosloučeniny na karbonylovou skupinu vzniká energeticky bohatý
zwitteriontový intermediát, který se přeměňuje na aminohydrin. Tento proces, zahrnující přenos dvou protonů, může proběhnout intramolekulárně, anebo za účasti dvou molekul vody vázaných vodíkovými vazbami.
Nestabilitu zwitteriontu je možno lépe objasnit na základě zpětné reakce. Slabě bazický dusíkový atom aminohydrinu je protonován a slabě kyselý hydroxyl je ionizován. Důsledkem této skutečnosti je, že jejich disociace na reaktanty může být extrémně rychlá - např. k -1 pro odštěpení methoxyaminu z MeONH2+-CHAr-O- je zhruba 108 s-1 . To je rychlejší proces než přenos protonu a vede k velmi neobvyklému chování, totiž že rychlost reakce je určována přenosem protonu mezi dvěma elektronegativními prvky. Přenos protonu může být urychlen přídavkem kyseliny.
|
|
Brøstedův graf této reakce je nelineární.

Brøstedova závislost pro reakci MeONH2 a p-MeOC6H4CHO. k je katalytická rychlostní konstanta obecně kyselého katalyzátoru o síle pKa. Tečkované linie mají směrnici 0 a 1 a protínají se v oblasti pKa 8-9, tj. velmi blízko odhadnuté hodnotě pKa pro hydroxylovou skupinu p-MeOC6H4CH(OH)NH2+OMe.
Tento průběh je v souladu s tzv. „eigen mechanismem“ protonového přenosu a je vizualizací třístupňového procesu: difuzí kontrolovaná srážka mezi kyselinou a bází, přenos protonu uvnitř tohoto srážkového páru a následná difuzí kontrolovaná separace.
|
|
Jedině za předpokladu, že BH+ a HA jsou stejně silné kyseliny, může být prostřední krok nejpomalejší. Srážka je limitujícím krokem, jestliže HA je podstatně silnější než BH+ a opačně, separace bude rychlost limitujícím krokem, když bude HA slabší. Podporou tohoto mechanismu je i pozorované maximum H/D kinetického izotopového efektu v oblasti D pKA = 0. Záměna HA za DA nemá významný vliv na difuzí kontrolované kroky, ale velmi degraduje katalýzu, jestliže přenos vodíku je sám o sobě rychlost limitujícím krokem. Uvedené katalytické chování velmi závisí na bazicitě aminosloučeniny. Pro báze silnější než methoxyamin je zwitteriontový intermediát méně nestabilní, jeho disociace na reaktanty je pomalejší a jeho tvorba se stává rychlost limitujícím krokem. Tedy např. při reakci s hydroxylaminem nebo zvláště s alifatickými aminy je adiční krok v podstatě nekatalyzován - lépe řečeno: katalýza molekulami vody zdaleka převažuje. Reakce tedy zahrnuje nukleofilní atak na karbonylovou skupinu (neaktivovanou nebo částečně aktivovanou tvorbou vodíkových vazeb s rozpouštědlem) následovaný rychlou protonovou výměnou.
|
|
b) dehydratace
V roztocích až do pH cca 9 jsou dehydratační kroky kysele katalyzovány. Aminohydriny odvozené od silných bází vykazují citlivost k obecné kyselé katalýze zřejmě z důvodu podstatně rozvinutého přenosu protonu v tranzitním stavu.
|
|
|
|
Slabší báze vyžadují úplnou protonaci hydroxylové skupiny a vykazují tedy rysy specifické kyselé katalýzy.
Dehydratace NH2CSNHNH-CH2-OH na thiosemikarbazon formaldehydu při pH = 7 leží zhruba na hranici mezi specifickou a obecnou kyselou katalýzou. Volný protonovaný aminohydrinový iont RNH-CH2-OH2+ zřejmě může existovat za podmínek reakce. Je ovšem zřejmé, že jeho spontánní dehydratace (specifická kyselá katalýza) zahrnuje tranzitní stav s vyšší energií než v případě součinné obecně katalyzované reakce. Tuto skutečnost potvrzuje i značný rozpouštědlový kinetický izotopový efekt. V alkaličtějších roztocích probíhají jiné typy katalytických procesů. Silně bazické aminohydriny mohou dokonce disociovat bez katalýzy.
|
|
Příkladem takového chování jsou hydrolýzy různých iminů benzaldehydu. V případě méně bazických aminohydrinů je bazická katalýza účinná v oblasti pH > 9. Není zřejmé, zdali se jedná o obecnou nebo specifickou katalýzu, ale dosavadní poznatky ukazují na následující reakční průběh:
|
|
Dalším typem katalýzy je nukleofilní katalýza. Příkladem je reakce tvorby oximů a semikarbazonů, která probíhá za katalýzy anilinhydrochloridu mnohem rychleji než za katalýzy kyselinou octovou (i když oba dva jsou přibližně stejně kyselé). Reakce s anilinhydrochloridem probíhá stejnou rychlostí nezávislou na koncentraci hydroxylaminu resp. semikarbazidu. Reakční průběh je vysvětlován rychlou reakcí anilinu za tvorby protonovaného anilu, který je velmi rychle atakován hydroxylaminem nebo semikarbazidem.
|
|
Aldolizace
Aldolizace a jí podobné reakce jsou mechanismem příbuzné tvorbě aminohydrinu nebo iminů. Přistupuje zde ovšem další komplikace - karbonylové sloučeniny jsou nejen elektrofily, ale mohou také být zdrojem nukleofilů. To vede ke vzniku dalšího reakčního kroku, který může být rychlost určující. Kinetická analýza je také komplikována řadou vedlejších reakcí. I klasická bazicky katalyzovaná aldolizace acetaldehydu může být velmi komplikovaná. Měli bychom vzít v úvahu možnost hydratace acetaldehydu a částečnou disociaci vzniklého diolu, což vede k tomu, že ani koncentrace aldehydu ani hydroxidu, nebudou ve stechiometrických poměrech idealizované stechiometrické rovnice. Produktem reakce také nebude jen klasický aldol, ale i trimerní 2- hydroxy-4,6-dimethyl-1,3-dioxan tvořený následnou rychlou reakcí. I přes tyto komplikace je možno stanovit podstatu mechanismu těchto reakcí. V případě bazické katalýzy je nukleofilem enolátový aniont.
|
|

Při vysokých koncentracích aldehydu je ionizace rychlost určujícím krokem (k2[CH3CHO] >> k -1) a kinetické chování se zjednodušuje na k1[OH-][CH3CHO]. Reakce je obecně bazicky katalyzována v souladu s tím, že přenos protonu je rychlost určujícím krokem. Jestliže se reakce provádí v D2O a je vhodně přerušena, nezreagovaný acetaldehyd neobsahuje deuterium, což znamená, že nedochází k protonaci vzniklého enolátu. V případě, že koncentrace aldehydu je nižší, kinetika se stává složitější a vratnost kroku 1 může být demonstrována existencí deuteriové výměny. Při velmi nízkých koncentracích, kdy k -1 >> k2 [CH3CHO], je rychlost určujícím krokem krok 2 a kinetika vykazuje rysy druhého řádu vůči acetaldehydu a katalýza je specifická. Tato různost mechanismů je pozorována obvykle jen u jednoduchých alifatických aldehydů. Většina ostatních bazicky katalyzovaných aldolových reakcí, jako jsou dimerace ketonů či smíšené aldolizace aromatických aldehydů s ketony obvykle ukazují, že adiční krok je podstatně pomalejší než ionizace. Bazicky katalyzovaná dehydratace aldolů je obvykle minoritní vedlejší reakcí v případě alifatických aldehydů, pokud reakce není prováděna v silně alkalických roztocích, anebo za zvýšené teploty. V případě aromatických aldehydů je naopak běžná a nenasycený produkt je normálním produktem. Eliminace obvykle probíhá E1cB mechanismem.
|
|
V tomto případě nám kinetika neposkytuje odpověď na otázku, zdali rychlost určujícím krokem je tvorba aldolu, anebo rychlost jeho dehydratace.
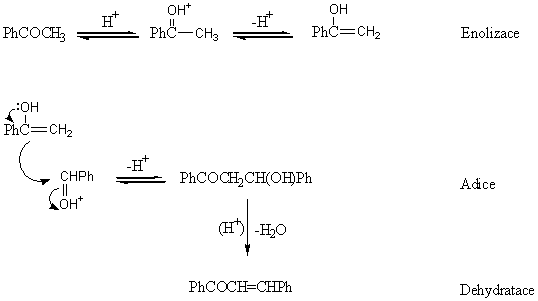
V případě kysele katalyzovaných aldolizací je počátečním krokem enolizace následovaná v rychlost limitujícím kroku atakem enolu na protonovanou karbonylovou skupinu. Kysele katalyzovaná dehydratace je potom velmi rychlá a reakce jen velmi zřídka zůstává ve stádiu aldolu.
|
|
Reakční rychlost je úměrná [PhCHO][CH3COPh][H3O+]. Existují dvě alternativní cesty dehydratace - přímá eliminace (s výrazným E1 charakterem) a reakce probíhající přes enol.
|
|
Rychlost limitujícím krokem může být tedy buď adice nebo dehydratace. Ve většině případů kysele katalyzovaných aldolizací bývá rychlejší dehydratace. Např. smíšená aldolizace benzaldehydu a butanonu může poskytnout tři různé aldoly:
|
|
Ve zředěném alkalickém roztoku všechny tři poskytují jediný nerozvětvený dehydratační produkt, zřejmě v důsledku existence rychlé rovnováhy prostřednictvím retroaldolové reakce, která je podstatně rychlejší než vlastní dehydratace. V kyselých roztocích nedochází k těmto reakcím a dehydratace je rychlejší než disociace. U většiny kysele katalyzovaných reakcí a u těch bazicky katalyzovaných, kde dehydratace je dostatečně rychlá, reakce vede k nenasyceným produktům bez ohledu na rovnováhu intermediátu. Např. mírně bazicky katalyzovaná dimerace acetonu dává v rovnováze jen několik procent diacetonalkoholu. Kysele katalyzovaná dimerace vede k vysokému výtěžku mesityloxidu.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1737
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved