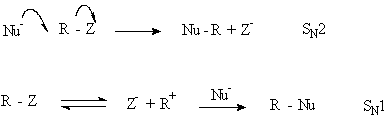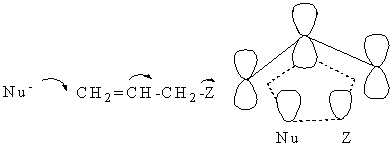| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
DOCUMENTE SIMILARE |
|
TERMENI importanti pentru acest document |
|
Alifatická nukleofilní substituce
SN1 a SN2 mechanismus
Alifatická nukleofilní substituce může být založena na dvou mechanismech: jednostupňovém SN2 a disociačním SN1
|
|
Důkazy pro SN2 mechanismus
Kinetika druhého řádu ukazující závislost na koncentraci nukleofilu i substrátu je běžně pozorována u nukleofilních substitucí. To je sice nezbytným kriteriem pro důkaz SN2 mechanismu, ale samo o sobě to nestačí. Mnohem přesvědčivější důkaz leží ve stereochemii reakce. Inverze konfigurace, která doprovází atak nukleofilu, vynucuje jeho atak z opačné strany, než leží odcházející skupina a stereochemická korelace mezi substrátem
a produktem ukazuje, že jsou spojeny společným tranzitním stavem.
|
|
Tato inverze konfigurace byla poprvé pozorována v r. 1890 Paulem Waldenem, podle kterého se i nazývá. Typickým příkladem může být tvorba enantiomerních etherů z chirálního alkoholu v následné serii reakcí:
R=MeCH*CH2Ph; [a]D je uvedeno v závorce
|
|
Ve všech těchto reakcích, s výjimkou reakce značené # zůstává vazba R - O zachována.
Každá jednotlivá SN2 substituce je tedy doprovázena inverzí konfigurace. Velmi důležitý důkaz o průběhu SN2 reakce představuje reakce chirálního 2-jodoktanu s radioaktivním jodidovým aniontem. Vzhledem k tomu, že jodid působí zároveň jako nukleofil a zároveň je
odstupující skupinou, nedochází k chemické změně, ale reakce může být sledována měřením rychlosti zavádění radioaktivity do molekuly. Při 30 oC v acetonu je rychlost 1.36 . 10-3 l/mol s. Pro stejnou reakci je rychlost stereochemické inverze (polovina pozorované rychlosti racemizace) shodná. To dokazuje, že každý nukleofilní atak je doprovázený inverzí. Původní vysvětlení tohoto chování bylo založeno na elektrostatické repulzi mezi nukleofilem a odcházející skupinou. Byla nalezena řada případů, u nichž hovořit o repulzi je obtížné. Současné vysvětlení je založeno na interakci orbitalů. Orbitaly, které vstupují do reakce, jsou HOMO nukleofilu a LUMO substrátu, což je C - Z s* orbital.
|
|
Důkazy pro existenci SN1 mechanismů
Studia SN1 mechanismů jsou založena na kinetických měřeních. Jestliže použijeme aproximaci ustáleného stavu na SN1 reakční schéma
|
|
můžeme odvodit následující výraz
|
|
V počátcích reakce je koncentrace [Z-] malá a reakce se řídí kinetikou prvního řádu
s rychlostí rovnou k1[RZ] nezávislou na koncentraci a charakteru nukleofilu. Takovéto chování lze např. pozorovat pro reakci benzhydrylchloridu (Ph2CHCl) s různými nukleofily v kapalném SO2. Jak v průběhu reakce vzrůstá koncentrace [Z-], tak reakční rychlost klesá podle uvedené rovnice. Vedle toho i přídavek Z- zpomaluje reakci běžným iontovým
efektem. Bohužel, většina SN1 reakcí se nedá provádět za takto kontrolovaných podmínek. Obvykle jsou to solvolýzy, kde dobré ionizující rozpouštědlo usnadňuje ionizaci a působí také jako nukleofil ve druhém stupni reakce. Koncentrace nukleofilu zůstává prakticky konstantní, a i když jde o bimolekulární proces, řídí se většinou kinetikou pseudoprvního řádu. Podstatná změna koncentrace nukleofilu znamená tedy nepochybně zásadní změnu celého systému a změna reakční rychlosti je nejednoznačná. Např. hydrolýza terc.butylbromidu ve vodném acetonu se zrychlí 40x, jestliže obsah vody vzroste z 10 na 30%. Je tedy nemožné rozhodnout, zdali se jedná o reakci druhého řádu doprovázenou třináctinásobným zrychlením v důsledku změny solvatace, anebo jestli jde o reakci prvého řádu se čtyřicetinásobným zrychlením v důsledku změny rozpouštědla. Situaci nám pomůže objasnit přídavek solí do této
solvolýzy. Přídavek elektrolytu zvyšuje iontovou sílu a přispívá tedy ke stabilizaci polárního tranzitního stavu. Na druhé straně, podle uvedené kinetické rovnice, zvýšení koncentrace iontů reakci zpomaluje. Tyto efekty mohou být někdy velmi výrazné. Např. solvolýza nebo hydrolýza tritylhalogenidů je přídavkem NaCl na výslednou koncentraci 0.01M zpomalena ve vodném acetonu 4x. Terc.butylhalogenidy ovšem na druhé straně nevykazují žádnou citlivost na přídavek iontů a jejich solvolýzy nejsou zpomalovány ani vzrůstající koncentrací halogenidového iontu v průběhu reakce. To je vysvětlitelné klasickým SN1 mechanismem zahrnujícím karbkationt. Tento kationt je velice reaktivní a poměrně neselektivní, tedy rychlosti k-1 a k2 jsou porovnatelné a převládající koncentrace rozpouštědla dominuje. Podstatně stabilnější karbokationty mohou rozlišovat mezi koncentrovaným, ale slabě nukleofilním rozpouštědlem a zředěným, ale mnohem nukleofilnějším roztokem iontu. Předchozí rovnici potom můžeme přepsat pro rozpouštědlo S jako nukleofil do tvaru

Výraz k-1/k2[S] vypočtený z rozsahu zpomalení reakce přídavkem Z- je mírou selektivity mezi rozpouštědlem a iontem. Pro různé substráty v 80 - 90% acetonu má tyto hodnoty:
|
Ph3CCl |
p-Tol2CHCl |
Ph2CHCl |
t-BuBr |
|
cca 400 |
< 2 |
Pořadí selektivity velmi dobře souhlasí s očekávanou stabilitou karbokationtu. Přídavek jiného iontu (tedy než toho, který vzniká v reakci), který je sám o sobě silně nukleofilní, může poskytnout vhodnou metodu k rozlišení preference substrátu vůči SN1 a SN2 mechanismu. Takovým obvyklým iontem bývá azidový ion.
Iontové páry
Výsledky řady reakcí, u nichž se předpokládá SN1 ukazují, že existence volného karbokationtu není nijak samozřejmá. V případě řady reakcí s chirálním substrátem nedochází totiž k úplné racemizaci. Vysvětlení by mohlo spočívat v tom, že oba mechanismy se uplatňují paralelně, zatímco jedna molekula disociuje na ionty, druhá podléhá přímé SN2 substituci. Ale bohužel experimentální výsledky tomu vždy nenasvědčují. Např. acetolýza 1-fenylethylchloridu není urychlována přídavkem octanu sodného a ani se nezvyšuje podíl inverze. Je tedy zřejmé, že inverze není důsledkem SN2 procesu. Často používané vysvětlení spočívá ve skutečnosti, že velmi reaktivní primárně vzniklý karbokationt je z jedné strany velmi rychle atakován reagentem, zatímco z druhé strany je ještě stíněn odstupující skupinou. I když je to logické vysvětlení, zasluhuje si pečlivou analýzu. Úplná ionizace chirálního substrátu za vzniku karbokationtu musí vést k úplné ztrátě chirality. Dva alternativní tranzitní stavy (atak zprava a zleva) jsou enantiomerní a mají stejnou energii. Částečná racemizace ovšem napovídá, že musí existovat dva tranzitní stavy s různou energií. Vysvětlení, že jedna strana je stíněna odstupující skupinou, musí znamenat, že tato je ještě asociována s tranzitním stavem. Rychlost určující ionizační krok nemůže tedy dát volný karbokationt, protože odstupující skupina je stále asociována s následným tranzitním stavem - viz následující obr.

Zajímavá otázka je struktura intermediátu I. Ještě než se pokusíme najít na ni odpověď, zabývejme se jinou situací, která zřejmě vyžaduje podobný mechanismus. Solvolýza 2- adamantylsulfonátu ve vodném ethanolu dává směs adamantanolu a adamantylethyletheru. Jejich poměr závisí na substituci sulfonové skupiny.
|
|
Ze studia vyplývá, že se v tomto případě téměř neuplatňuje SN2 reakční mechanismus a reakce zřejmě probíhá podobným mechanismem, ovšem v tomto případě s chemicky různými produkty. Strukturou intermediátu I se zabýval již Hammett. Definoval jej jako částečně disociovaný systém, v němž kovalentní vazby jsou z velké míry nahrazeny elektrostatickými interakcemi, ale s oběma ionty v těsné blízkosti uvnitř rozpouštědlové kavity.Tuto strukturu nazval iontovým párem. Platnost této myšlenky byla potvrzena a rozšířena na to, aby ukázala, že přinejmenším v některých případech musí existovat dva rozdílné typy iontových párů na cestě mezi kovalentní sloučeninou a úplně disociovanými ionty. To bylo ukázáno např. při acetolýze (+)-threo-threo- anisyl-but-2-yl brosylátu.
|
|
Reaktanty jsou racemizovány v průběhu reakcí rychleji, než je rychlost solvolýzy. Volný anisylbutylkationt má cyklickou strukturu a je achirální, což svádí k vysvětlení rychlé racemizace SN1 mechanismem s rychlou vratnou disociací následovanou pomalým solvolytickým atakem. To ovšem nemůže být pravda, protože solvolýza není zpomalována generovanými ionty, jak by měla být, jestliže v rovnici
je k-1 velké.
|
|
Mechanismy zahrnující iontový pár nabízejí lepší alternativu
Racemizace probíhá v důsledku rychlé rovnováhy s iontovým párem. Ve zpětné reakci se brosylátový anion může přesunout na druhou stranu uhlíkového atomu, který právě opustil. To je ovšem intramolekulární proces, kdy anion není nikdy zcela volný a přídavek stejného aniontu tedy nemá vliv na kinetiku tohoto kroku. Racemizace i solvolýza jsou poněkud urychlovány normálním efektem elektrolytu, např. přídavkem lithiumperchlorátu do roztoku, solvolýza vykazuje velmi neobvyklý a velmi značný elektrolytický efekt při nízké koncentraci perchlorátu - viz obr.

Efekt koncentrace LiClO4 na acetolýzu ( )-threo-3-anisylbut-2-yl brosylátu
Perchlorátový anion je zanedbatelně nukleofilní, tento efekt tedy musí mít příčinu v nějaké elektrostatické interakci. Jedno z možných vysvětlení je, že perchlorát působí tím, že vyměňuje brosylátový anion v iontovém páru a přerušuje tedy rychlost brzdící zpětný proces. Ale to nemůže být stejný iontový pár, který je používán k vysvětlení rychlosti racemizace: jestliže existuje efektivní konkurence pro k-1, potom racemizace, stejně jako solvolýza, budou zpomaleny, ovšem racemizace nevykazuje žádný zvláštní elektrolytický efekt. K vysvětlení proto bylo navrženo následující schéma:
|
|
Přídavek perchlorátu vytváří další cestu vedoucí k produktům (k4), která urychluje reakci. Při koncentraci perchlorátu cca 0.02 M je tato cesta tak efektivní, že téměř každý separovaný
iontový pár je přeměněn na produkty tou rychlostí, jakou se tvoří. Vyšší přídavek perchlorátu potom už způsobuje jenom normální elektrolytický efekt. Rychlá racemizace (k-1 > k2) ovšem pokračuje i nadále bez ohledu na působení chloristanu v dalších krocích. V souladu s tímto schématem přídavek lithiumbrosylátu, který vykazuje jen velmi zanedbatelný ion-retardační efekt na solvolýzu, sníží rychlost perchlorátově katalyzované reakce snížením hodnoty k -4 . Další důkaz pro existenci dvou různých typů iontových párů poskytla hydrolýza 18O značeného p-chlorbenzhydryl p-nitrobenzoátu ClC6H4-CHPh-OC18OC6H4NO2 v 90% vodném acetonu. V průběhu reakce je kyslík 18O distribuován mezi oba karboxylové kyslíky a ester podléhá částečné racemizaci. Oba procesy jsou prvního řádu. Výměna mezi esterem a 14C značenou p-nitrobenzoovou kyselinou za stejných reakčních podmínek probíhá mnohem pomaleji, což ukazuje, že disociace neprobíhá až do stádia úplně separovaných iontů. Přídavek azidu sodného do reakce potlačí racemizaci, ale nemá vliv na 18O výměnu. To opět ukazuje, že existují dva rozdílné intermediáty - iontové páry. V těsném iontovém páru se stávají oba kyslíkové atomy karboxylátového aniontu ekvivalentní a zpětný proces vede k pomíchání kyslíků. Racemizace ovšem vyžaduje, aby anion migroval z jedné strany benzhydrylového kationtu na druhou. To je ovšem značně obtížné v případě těsného iontového páru a je to možné až v okamžiku, kdy jsou ionty volněji asociovány v rozpouštědlově separovaném iontovém páru, tomu je však rychle zabráněno silně nukleofilním azidovým iontem. Přesná struktura iontových párů není známá a není ani jisté, zda existují jen dvě energetická minima na cestě mezi kovalentně vázaným RZ a plně disociovanými ionty. Představu o tom, co se děje, můžeme získat pomocí zpětného procesu. Oba separované ionty budou plně solvatovány rozpouštědlem. Při jejich přiblížení dojde k první interakci mezi molekulami rozpouštědla v solvatační sféře. Nepochybně existuje řada uspořádání této solvatační sféry, eventuálně až takové uspořádání, které vytváří strukturu s jedinou molekulou rozpouštědla separující oba přibližující se ionty. To posléze vede až k situaci, kdy jsou oba ionty orientovaně uspořádány v rozpouštědlové kavitě a mohou konečně vytvořit i kovalentní vazbu. I když ovšem uvažujeme jen dva typy iontových párů, existuje celá řada reakčních typů:
|
|
Reakční kroky 1, 2, 3 jsou monomolekulární a značíme je SN1 (A), SN1 (B), SN1 (C), reakční kroky 4 - 7 jsou bimolekulární a jsou značeny SN2 (A) až SN2 (D). Ani tyto všechny varianty nevystihují všechny možnosti mechanismů. SN1 (A) reakce může být ukončena rychlým nukleofilním atakem v těsném iontovém páru (k5 > k2), anebo pokračovat v další reakční cestě ve smyslu disociace (k2 > k5) s produkčními kroky 6 nebo Ve smyslu této analýzy klasický SN2 mechanismus je SN2 (A) a klasický SN1 mechanismus (předpokládající úplnou disociaci) může zahrnovat kterýkoliv ze tří SN1 mechanismů s produkčním krokem Původní SN1 definice zahrnuje všechna stereochemická chování od totální racemizace k totální inverzi. Tyto rozdíly můžeme nyní odlišit pomocí rozdílných produkčních kroků. Reakce, která obvykle neprobíhá dále než k tvorbě těsného iontového páru, probíhá obvykle s inverzí konfigurace, tak jak jsme viděli v azidem modifikované hydrolýze
p-chlorbenzhydrylnitrobenzoátu. Všechny reakční stavy, které probíhají přes úplně disociované ionty, musí probíhat s totální racemizací. Parciální racemizace může probíhat přes krok 6, nebo jako výsledek participace více produkčních kroků.
Mechanismy a jejich hranice
Uvedený předpoklad existence dvou různých iontových párů ponechává velký prostor k úvahám o konkrétním mechanismu mezi oběma extrémy. Velmi obtížné je rozhodnout, zdali hraniční chování některého případu má příčinu v tom, že jeden mechanismus se velice přibližuje druhému, nebo zdali oba mechanismy probíhají současně. Zaměříme se na několik případů.
Solvolýza alkylderivátů
Není pochybnost, že primární alkylderiváty reagují klasickým SN2 mechanismem. Terciární alkylderiváty na druhé straně téměř za všech okolností reagují SN1 mechanismem. Např. reakce terc.butylbromidu s radioaktivním bromidovým aniontem v acetonu je reakcí prvního řádu. Na druhé straně se jeví velmi nepravděpodobné, aby jednoduché terciární alkylderiváty disociovaly až na volné ionty. Často pozorovaná částečná racemizace a závislost na charakteru odstupující skupiny ukazuje, že reakce obvykle probíhá přes rozpouštědlově separovaný iontový pár. I solvolýza např. terc.butylSMe2+ může zahrnovat určité párování iontů s perchlorátovým aniontem, ale to nabízí jenom velmi hrubý model chování volných kationtů. Údaje v následující tabulce ukazují, že ve vodě probíhá disociace do značné míry, protože prakticky všechny substráty se chovají jako sulfoniová sůl. V jiných rozpouštědlech může ovšem odstupující skupina hrát významnou roli v produkčním kroku a disociace je tedy omezena.
Poměr eliminace/substituce při solvolýze t-Bu substrátů: efekt odstupující skupiny
|
% tvorby olefinů |
|||
|
Rozpouštědlo |
|||
|
t-BuZ |
H2O |
AcOH |
EtOH |
|
Cl | |||
|
Br | |||
|
I | |||
|
SMe2+, ClO4- | |||
Není zcela jasné, jestli jsou rychlost limitujícími kroky reakce 1 nebo 2 (viz předchozí schéma). Zpětné kroky -1 a -2 jsou určitě pomalé - projevuje se u nich jen velmi malý zpomalující efekt aniontu a je velmi zanedbatelná výměna radioaktivního chloru z přídavku značeného chloridu sodného v průběhu acetolýzy terc.butylchloridu.
Sekundární alkylové substráty se klasifikují mnohem obtížněji a bylo věnováno mnoho úsilí při poznání hranic SN1/ SN2 mechanismu. Existují přinejmenším dva myšlenkové proudy. Představitelem prvého z nich je Schiner a jeho kolegové, kteří přijímají standardní Winsteinův model iontových párů s kterýmkoliv z kroků 1, 2, 5 a 6 závisejících na struktuře alkylové skupiny, na ustupující skupině a na rozpouštědle. Hlavní důkazy pro jejich závěry pocházejí ze studia velké šíře velikosti sekundárního izotopového efektu v a a b deuterovaných substrátech. Hlavní příspěvek přispívající k a kH/kD je oslabení vibračního módu H-Ca-Z při přechodu k tranzitnímu stavu, který je podobný karbokationtu. Jestliže vaznost Ca-Z v tranzitním stavu je minimální, potom a kinetický izotopový efekt je maximální a maximálně závisí na charakteru Z. Platnost těchto podmínek můžeme očekávat v případě, že krok 2 je rychlost limitující. Jestliže rychlost určující krok je 1, potom vazba Ca-Z je v tranzitním stavu méně přerušena, a jestliže kroky 5 nebo 6 jsou rychlost určujícími, potom oslabení Ca-Z vazby je částečně kompenzováno vytvořením HS-Ca (HS je nukleofilní rozpouštědlo). b kinetický izotopový efekt má původ zřejmě v hyperkonjugační interakci ve vznikajícím H-Cb-Ca karbokationtu a je tedy také mírou rozpadu Ca-Z vazby. Nicméně proti této interpretaci byly nalezeny určité argumenty. Jejich vysvětlení je předmětem přístupu Schleyera a Bentleyho. Říkají, že existuje plynulý přechod mezi mechanismem SN1(A) a SN2(A). 2-adamantylsubstráty jsou brány jako modely SN1 konce mechanistického spektra. Jejich solvolýza probíhá přes tranzitní stav s žádnou nebo minimální nukleofilní solvatací. Solvolýza 2-adamantylbromidu je 108 pomalejší než terciárního 2-methyl-2-adamantylbromidu. Tento poměr je víceméně nezávislý na rozpouštědle a blíží se k hodnotám očekávaným na základě relativních rychlostí ionizace sekundárních a terciárních substrátů z měření relativní stability karbokationtů v plynné fázi nebo v superkyselinách. Podobně nukleofilita rozpouštědla má jen nevýznamný efekt na relativní reaktivitu 2-adamantyltosylátu a 1-bicyklooktyltosylátu, který nevykazuje ani stopy SN2 charakteru v tranzitním stavu. Další důkaz pro absenci nukleofilní solvatace v solvolýze 2-adamantylsubstrátů ukazuje přídavek azidového iontu. I přes silnou nukleofilitu azidu nedochází k SN2 reakci s 2-adamantylsubstrátem, pravděpodobně ze sterických důvodů. Jak by potom mnohem méně nukleofilní rozpouštědlo mohlo participovat na takovémto tranzitním stavu?
|
|
Schleyer a Bentley použili 2-adamantylsulfonáty jako základ pro porovnání chování jiných sekundárních, ale i primárních alkylsulfonátů. Našli pravidelnou závislost v řadě různých mechanistických kriterií: velikost a deuteriového izotopového efektu, který odráží změnu vazby v místě substituce; Grunwald-Winsteinův parametr m, který měří citlivost substrátu k ionizujícím schopnostem rozpouštědla; efekt nukleofility rozpouštědla na rychlost solvolýzy. Všechny tyto parametry vykazují změnu od methylu přes ethyl, izopropyl a další objemnější sekundární systémy. Tyto změny mají původ v inverzní závislosti mezi hodnotou m (která zhruba odráží SN1 charakter) a citlivosti k nukleofilitě rozpouštědla (SN2).
Speciálně významná korelace je taková, která umísťuje všechny sekundární tosyláty na škále mezi 2-adamantyl a izopropyl pomocí rovnice
log(k/k0)R = QR log (k/k0)2-Ad + (1-QR) log (k/k0)i-Pr
kde k0 je rychlostní konstanta solvolýzy v referenčním rozpouštědle . Parametr QR je ve skutečnosti nezávislý na rozpouštědle a naznačuje, že každému substituentu přísluší určité místo v mechanistickém spektru. Příklady některých hodnot QR jsou následující:
|
R |
Me-CH-Me |
Et-CH-Me |
Et-CH-Et |
i-Pr-CH-Me |
t-Bu-CH-Me |
2-Ad |
|
QR |
|
|
Toto zřetelně ukazuje na vyloučení účasti rozpouštědla v tranzitním stavu, pravděpodobně v důsledku sterických repulzí. Schleyer a Bentley toto považují za postupné propojování mechanismu solvolýzy od klasické součinné SN2 přes nukleofilně solvatovaný iontový pár, až např. k 2-adamantyliontovému páru, ve kterém neexistuje žádná nukleofilní solvatace.
Následující obrázek ukazuje změnu reakčního profilu. Tranzitní stav je charakterizován zmenšujícím se kontaktem mezi nukleofilním rozpouštědlem a elektrofilním uhlíkovým atomem substrátu.

Hranice mechanismů SN1 SN2
Reakce primárních benzylových substrátů
Řada reakcí benzylových substrátů je urychlována jak elektrondonory na benzenovém kruhu, tak i elektronakceptory. Avšak a deuteriový kinetický izotopový efekt monotónně roste s rostoucí silou elektrondonoru a naznačuje postupné oslabení vazby Ca - Z v tranzitním stavu. Toto chování je obvykle interpretováno jako postupná změna z těsného SN2 tranzitního stavu, ve kterém elektronakceptorová arylová skupina nutí nukleofil být blízko reakčního centra, až k volnějšímu více SN1 podobnému tranzitnímu stavu.
|
|
Reakce anilinu s benzylhalogenidy v ethanolu je bimolekulární. Změna v benzylovém substrátu (Ar) vede ke vzniku nelineárního Hammettova grafu - viz obr.

Hammetova závislost pro reakci PhNH2 + ArCH2Cl v EtOH při 50o C
To je v souladu s myšlenkou stupňovitě se měnícího tranzitního stavu. Ačkoliv reakce probíhá s nenabitým nukleofilem, není urychlována elektronakceptory přítomnými v Ar. Změna v anilinové skupině Ar, dává lineární Hammettovu závislost a její směrnice velmi závisí na charakteru benzylové skupiny. V případě volného tranzitního stavu, jako např. s p-methoxybenzylchloridem, je anilinový dusík velmi málo nabitý a velikost r je malá. V těsném tranzitním stavu s p-nitrobenzylchloridem nukleofil jeho přítomnost více pociťuje a dusíkový atom nese podstatný kladný náboj. Nukleofil sám o sobě může také ovlivnit strukturu tranzitního stavu, jak může být ukázáno na reakci dvou různých nukleofilů se dvěma para-substituovanými benzylchloridy (p-PhO, p-MeO). Bez přídavku nukleofilu je poměr reakčních rychlostí prvního řádu jejich solvolýzy v 70% acetonu k1(PhO)/k1(MeO) = 135. Tyto reakce téměř určitě probíhají SN1 mechanismem. Jestliže do směsi přidáme nukleofil, potom je reakce bimolekulární, ale změna v poměru k2(PhO)/k2(MeO) ukazuje na měnící se strukturu tranzitního stavu. Silné nukleofily, jako např. azid, dávají těsný tranzitní stav s poměrně málo narušenou vazbou C - Cl, generují velmi malý náboj na uhlíkovém benzylovém atomu a jsou velmi málo ovlivněny arylovým substituentem. Slabší nukleofily jako NO3- tvoří mnohem volnější tranzitní stav se značným kladným nábojem na benzylovém uhlíkovém atomu. Tato myšlenka těsnějších a volnějších tranzitních stavů byla v nedávné době znovu zkoumána a poněkud zpochybněna. Byly nalezeny závažné rozpory: aktivační parametry pro reakci bromidového iontu s ArCHBrCH3 byly získány z kinetiky racemizace: viz tabulka.
Kinetická a aktivační data racemizace p-X-C6H4-CHBrCH3 v acetonu při 30oC
|
X |
k/10-3 l mol-1 s-1 |
DH / kJ mol -1 |
DS /J K-1 mol -1 |
|
Me | |||
|
H |
-80 |
||
|
NO2 |
16.75 |
-50 |
Výsledky poskytují nelineární Hammettovu závislost, přičemž reakce je urychlována jak elektrondonorním methylem, tak i elektronakceptorní nitroskupinou. V případě těsného tranzitního stavu (p-nitro) můžeme očekávat vyvinutější vazby než ve volném tranzitním stavu a tedy menší hodnoty DH‡ a negativnější DS‡. Byl ovšem nalezen opak. V případě disociačního iontově-párového mechanismu by měla být nejnižší hodnota DH‡ nalezena pro nejstabilnější kation (p-CH3).
Shrnutí
Zmíněné studie solvolýzy sekundárních alkylderivátů a nukleofilní substituce benzylových substrátů nám ukázaly značnou šíři mechanismů v regionu SN1 - SN2. Ale je nutno konstatovat, že povaha hranic mezi nimi je zatím nejasná. Není zřejmě možné pochybovat o přirozené variabilitě SN2 tranzitního stavu, ale je to skutečně ta vlastnost, která jej nepozorovaně spojuje s SN1 mechanismem, jako důsledek zmenšující se účasti nukleofilu na tvorbě tranzitního stavu? Skutečně je to možno předpokládat, avšak variabilní SN2 tranzitní stav musí vést ke konečnému mechanistickému zlomu na SN1 proces. Každá zdánlivá kontinuita musí být příznakem, že v určitém bodě oba mechanismy probíhají současně. To se může i nemusí jevit jako logicky kontroverzní. Ať tranzitní stav zahrnuje nebo nezahrnuje molekulu nukleofilu, nemůže existovat pozvolný přechod od jedné molekuly ke druhé - představa kontinuální změny mechanismu není totiž v souladu s tím, že u molekul můžeme hovořit jen o nekontinuální sérii diskrétních látek. Diskuse na toto téma je více či méně filosofická a v současnosti se zdá, že čím více se snažíme upřesňovat hranice, tím se nám jeví skrytější, přikryté mlhou neurčitosti a statistické mechaniky.
Vliv struktury a rozpouštědla na mechanismus
Struktura substrátu
Methyl a primární alkyly reagují mechanismem SN2, terciární mechanismem SN1; sekundární sloučeniny reagují někde na rozhraní obou mechanismů. Jsou dva důvody k tomuto chování. SN2 tranzitní stav má pentakoordinovaný uhlíkový atom, takže reakce je stericky bráněná v přítomnosti objemných substituentů; důsledky jsou uvedeny v následující tabulce. Výsledky ukazují, že přítomnost další skupiny v alfa nebo beta poloze reakci zpomaluje.
Rychlostní konstanty reakce druhého řádu RBr + Cl- à RCl + Br- v dimethylformamidu při 25oC (k2/10-5 l mol-1 s-1)
|
a substituce |
b - substituce |
||
|
R |
k2 |
R |
k2 |
|
Me | |||
|
Et |
1 300 |
Et | |
|
i-Pr |
25 |
n-Pr |
930 |
|
t-Bu |
1.1 |
i-Bu |
45 |
|
Neopentyl |
0.0085 |
||
Tyto závěry velmi dobře korelují s vypočítanými energiemi sterického pnutí. Uvedená reakce byla provedena v DMF, jen velmi špatně ionizujícím rozpouštědle, aby byla potlačena co nejvíce SN1 reakce.
SN1 tranzitní stav je stericky méně bráněn než základní stav a reakce tedy může vykazovat efekt sterické akcelerace. Většinou je tento efekt malý, s výjimkou extrémních případů, kdy je ovšem těžké rozhodnout mezi polárním a sterickým vlivem rozdílných alkylových skupin. Mnohem významnější je stabilizace terciárních karbokationtů induktivními a hyperkonjugačními vlivy okolních substituentů. Velikost této stabilizace může být ukázána na příkladu 108 násobného rozdílu v reaktivitě sekundárních a terciárních 2-adamantylových systémů. Mnohem menší rozdíl v reakčních rychlostech ( typicky 104) bývá nalézán při porovnání solvolýzy t-butylu a isopropylderivátů v důsledku podstatného příspěvku SN2 reakce u isopropylderivátů.
Efekt sterického pnutí je významný u cyklických systémů. Např. cyklopentylové deriváty reagují vždy rychleji než cyklohexylová analoga jak v SN1, tak i v SN2 reakcích. Cyklohexanový kruh je více méně nenapnutý s rovnoměrně rozloženými vazbami a tvorba trigonálního uhlíku jak v SN2 tranzitním stavu, tak i v karbokationtu způsobuje vznik napětí. U cyklopentylových derivátů je situace opačná.Vznik trigonálního centra způsobuje určité uvolnění napětí oproti základnímu stavu. Substráty s odstupující skupinou na rozvětvení kruhů jsou většinou velmi nereaktivní. SN2 reakce je prakticky vyloučena v důsledku nemožného přístupu z druhé strany TS a tvorba karbokationtu je znemožněna nemožností tvorby planární konfigurace a její možnou solvatací jen z jedné strany. Nicméně tvorba iontových párů, jak ukazuje následující tabulka solvolýzy v 80% ethanolu je sice obtížná, ale možná.
|
|
1.0 0.46 0.0012 0.00000024 0.000000000002
Relativní rychlosti solvolýzy v 80% etanolu při 25o C.
Každý strukturní rys, který může stabilizovat karbokationty, vede k posílení uplatnění SN1 mechanismu. Viděli jsme, že u benzylových systémů se ionizace majoritně uplatňuje u sekundárních typů, ale může být možná i u typů primárních. Silné nukleofily dávají SN2 reakci s primárními benzylovými substráty. Reakční rychlosti bývají často podobné analogickým methylderivátům. To ukazuje, že existuje určitá rezonanční stabilizace SN2 tranzitního stavu, přinejmenším dostatečná ke kompenzaci sterické destabilizace. Korelace Hammettova typu v těchto případech vykazují mělká minima a oba typy substutice (elektrondonor i akceptor) reakci poněkud urychlují. Tato skutečnost je v souladu s diskutovanou variabilitou SN2 tranzitního stavu: elektrondonory stabilizují volný TS s malým kladným nábojem na benzylovém uhlíku, elektronakceptory stabilizují těsný TS se záporně nabitým benzylovým uhlíkem.
Efekt odstupující skupiny
V obou tranzitních stavech SN1 i SN2 s sebou odstupující skupina odnáší i svoje vazebné elektrony. Není tedy nerozumné předpokládat, že by měla existovat určitá závislost na stabilitě Z- vyjádřené její bazicitou. Kvalitativně byla tato skutečnost pozorována, ale kvantitativní korelace existují jen řídce - např. v sérii velmi podobných odstupujících skupin různě substituovaných arensulfonátů.
Je zřejmé, že významnou roli mohou hrát i jiné faktory - především efekt rozpouštědla. Ačkoliv efekt odstupující skupiny by měl být stejný v SN1 i SN2 reakci, je pravdou, že sama odstupující skupina má vliv na převahu určitého mechanismu. Vazba C - Z je přerušena ve velké míře v SN1 mechanismu a vhodná odstupující skupina může tedy tuto reakci podpořit.
Reakce může být katalyzována modifikací odstupující skupiny a to ve svém důsledku vede k modifikaci mechanismu. Např. alkylfluoridy, normálně inertní vůči nukleofilům, mohou být hydrolyzovány v kys. sírové, kde odstupující skupinou je pravděpodobněji HF než F-. Podobně alkoholy a ethery mohou reagovat v silně kyselém prostředí, jako je tomu v Zeiselově reakci:
I- + Me-O+RH à Me- I + ROH
V aprotických rozpouštědlech může tato katalýza probíhat prostřednictvím vodíkových vazeb. t-butylbromid reaguje SN1 mechanismem v nitromethanu při nízkých koncentracích stejnou rychlostí s celou řadou nukleofilů ( Br-, Cl-, NO2-, H2O, EtOH, PhOH). Při vyšších koncentracích HO-nukleofilů reakce vykazuje kinetiku druhého řádu, ale rychlost reakcí není úměrná nukleofilitám, ale aciditám reagujících nukleofilů. Z toho je zřejmé, že rychlost určující krok je sice pořád ionizace, ale doprovázená tvorbou vodíkové vazby: t-Bus Brs H-OR.
Takováto katalýza musí být zřejmě přítomna i v protických rozpouštědlech, není ovšem přímo detekovatelná. Zde má zřejmě efekt částečného nivelizování reaktivity halogenidů, poněvadž jejich schopnost tvorby vodíkových vazeb je nepřímo úměrná jejich ochotě odstupovat jako aniont. Relativní rychlosti reakce methylhalogenidů s azidem jsou pro Cl, Br, I v DMF 1 : 250 : 2000 a v methanolu 1 : 60 : 100.
Katalytický efekt Lewisových kyselin je také běžný. Příkladem může být Friedel-Craftsova reakce s alkylchloridem a chloridem hlinitým jako katalyzátorem. Brom a jod jsou měkčími anionty než chlor a jejich reakce je lépe katalyzována měkčími Lewisovými kyselinami - solemi těžkých kovů - Ag, Hg. Každý katalytický efekt odstupující skupiny má za důsledek posunutí mechanismu ve prospěch SN1.
Vliv nukleofilu
a) Nukleofilita
Nukleofilita i bazicita mají společný základ v elektron-donorních vlastnostech. Bazicita je jasně definovaná termodynamická veličina, nukleofilita na druhé straně je spíš kinetickým konceptem nemajícím univerzální kvantitativní definici. Vzhledem k různým substrátům a rozpouštědlům tedy existují různé škály nukleofilit. Mohou být vysledovány dva trendy. V případě, že nukleofily reagují prostřednictvím stejného atomu, potom existuje kvantitativní korelace mezi bazicitou a nukleofilitou - např. HO- > PhO- > AcO- > H2O > ClO4-. Druhý trend, většinou zřetelnější, je, že velké, polarizovatelné nukleofily jsou reaktivnější než malé, většinou tvrdší:
I > Br- > Cl- > F-; PhSe- > PhS- > PhO- . Není pochybnost, že v tomto chování hraje solvatace hlavní roli. V protických rozpouštědlech jsou nukleofilní anionty stabilizovány vodíkovou vazbou. Ta je silnější u malých aniontů, u nichž je záporný náboj více koncentrován. Nukleofil se musí zbavit podstatné části této slupky dříve, než může reagovat. V bipolárních aprotických rozpouštědlech jsou anionty slaběji solvatovány a v důsledku toho reagují rychleji než v protických rozpouštědlech. Tento efekt je u neutrálních nukleofilů podstatně menší. Jejich reakce je jen zřídka urychlována víc než 10x při přechodu od methanolu k DMF. Ovšem i aprotická rozpouštědla mohou mít velký vliv. Měření SN2 reakcí v plynné fázi dává rychlosti řádově 1011 l/mol s, enormně vyšší než v kapalné fázi. Pořadí nukleofilit je ovšem zcela jiné než v kapalné fázi.
b) a efekt
V paralele mezi nukleofilitou a bazicitou systémů, které reagují stejným atomem, existuje jedna důležitá výjimka. Látky, které mají dva sousední elektronegativní prvky (HONH2, N2H4, R2S2 ), jsou mnohem nukleofilnější, než bychom mohli očekávat na základě jejich bazicity. Hydroperoxidový aniont HOO- je 104x méně bazický než OH- aniont, ale reaguje 50x rychleji s benzylbromidem. Příčinou tohoto chování známého jako a efekt je interakce mezi volnými elektronovými páry sousedních atomů.
Tato interakce vytváří dva nové orbitaly, jeden s nižší a jeden s vyšší energií. Takto vzniklý abnormálně vysoko ležící HOMO orbital potom silně reaguje s nízko ležícím LUMO orbitalem relativně měkkých nukleofilů, jako jsou alkylhalogenidy.

c) ambidentní nukleofily
Řada nukleofilů může reagovat dvojím způsobem. Některé potenciálně ambidentní nukleofily reagují (při substituční reakci na uhlíku) jen jediným způsobem - siřičitanový aniont dává sulfonáty místo esterů kyseliny siřičité. Řada jiných dává směs produktů, jejichž poměr závisí na substrátu a reakčních podmínkách. Zdá se, že ambidentní nukleofily reagují svým elektronegativnějším prvkem při reakcích s převahou SN1 mechanismu a méně elektronegativním prvkem při SN2 reakcích. Primární alkylhalogenidy reagují s dusitanem stříbrným za vzniku především nitroalkanů, zatímco terciární halogenidy dávají většinou estery kyseliny dusité. Podobně kyanid sodný normálně poskytuje nitrily, ale např. kyanid stříbrný dává přednostně izonitrily. Vysvětlení tohoto chování spočívá v porovnání relativní tvrdosti karbokationtu ve srovnání s měkčím alkylhalogenidem. Elektrostatické interakce v SN1 reakci podporují atak v místě s větším záporným nábojem, v SN2 reakci dominuje překryv orbitalů a atak nastává na místě, kde je lokalizován HOMO orbital, obvykle na méně elektronegativním atomu. To je v souladu i s obvyklým místem protonace takovýchto ambidentních částic. Proton jako tvrdý elektrofil vždycky reaguje nejrychleji na nejelektronegativnějším atomu. Siřičitany a dusitany se tedy protonují na kyslíku. Kinetický produkt ovšem nemusí být vždy termodynamicky nejstabilnější - např. enoly se vytvářejí rychlou protonací svých aniontů na kyslíku, ale ketotautomer je stabilnější.
Poměr tvrdosti a měkkosti je ovšem jen jedním z faktorů. Dalším je nepochybně rozpouštědlový efekt. Data v následující tabulce ukazují vliv rozpouštědla na místo benzylace ambidentního 2-naftolátového aniontu, který může reagovat na C1 uhlíku (Friedel-Craftsova alkylace) nebo na kyslíku za vzniku benzylnaftyletheru. Můžeme rozeznat dva proti sobě stojící efekty. V aprotických rozpouštědlech vzrůstající polarita preferuje SN1 mechanismus a tím atak na tvrdší pozici - kyslíku. Protická rozpouštědla mohou podobně preferovat SN1 reakci, ale tento efekt je značně zastíněn tím, že záporně nabitý kyslíkový atom je značně deaktivován solvatací vodíkovými vazbami.
Distribuce produktů reakce 2-naftolátu sodného s benzylbromidem
|
Rozpouštědlo |
O alkylace |
C alkylace |
Rozpouštědlo |
O alkylace |
C alkylace |
|
Me2NCHO |
MeOH | ||||
|
Me2SO |
EtOH | ||||
|
MeOCH2CH2OMe |
H2O | ||||
|
tetrahydrofuran |
CF3CH2OH |
Jiné SN mechanismy
SNi mechanismus
|
|
Reakce alkoholu s thionylchloridem za vzniku alkylchloridů probíhá často za zachování stereochemické konfigurace alkylové skupiny. Vysvětlení je založeno na intramolekulární substituci v meziproduktu. Původně byla reakce chápána jako SN2 intramolekulární proces, ale vzhledem k tomu, že reakce vykazuje charakteristické rysy ionizace - je usnadňována dobře ionizujícími rozpouštědly a substráty, které poskytují stabilní karbokationt a je často doprovázena Wager-Meerweinovým přesmykem, jde spíše o iontově párový mechanismus.
V přítomnosti báze, např. pyridinu, vznikají volné chloridové ionty a reakce probíhá jako konvenční SN2 substituce s inverzí konfigurace.
Allylový přesmyk SN ´
Allylové substráty podléhají nukleofilní substituci rychleji než nasycená analoga. Reakce je často doprovázena přesmykem označovaným SN ´.
Allylový přesmyk v nukleofilní substituci
|
|
|
|
První příklad v tabulce je zřejmě konvenční SN2 reakce. Ostatní reakce probíhají přes iontový pár:
|
|
Úplná disociace na volné karbokationty dává ovšem shodnou směs produktů z obou reaktantů. Průběh allylového přesmyku byl jeden z prvních důkazů existence iontových párů. Racemizace MeCH=CHC*HZMe (Z = p-nitrobenzoát) je 4x rychlejší než solvolýza v 90% acetonu. Izomerace CH2=CHC(Me)2Cl na ClCH2CH=CMe2 je také rychlejší než acetolýza. V žádném případě přesmyk nemůže mít původ v úplné disociaci, protože reakce není zpomalována přídavkem vznikajícího iontu, ale může být považována za typický proces SN2 (B). Přesto ještě zůstává možnost, že allylové přesmyky probíhají součinným způsobem SN2´. MO výpočty ukazují, že v takovémto procesu nenabitý nukleofil atakuje allylový systém syn k odstupující skupině za tvorby delokalizovaného šestielektronového tranzitního stavu. Takovýto stereochemický průběh byl skutečně pozorován, ale nelze vyloučit, že je to i důsledek uplatnění SN2 (B) mechanismu - viz obr.
|
|
Účast sousedních skupin
Yperit - S(CH2CH2Cl)2 je ve vodném roztoku hydrolyzován několikatisíckrát rychleji než primární alkylchloridy. Alkalická hydrolýza chirálního 2-brompropanoátu na hydroxykyselinu probíhá se zachováním konfigurace. Hydrolýza chloraminu Et2NCH2CHEtCl poskytuje přesmyknutý alkohol Et2NCHEtCH2OH. Toto jsou tři příklady typické účasti sousedních skupin v reakci. Každá z těchto reakcí probíhá přes tříčlenný cyklický intermediát.
|
|
Intramolekulární krok je upřednostňován před přímou intermolekulární substitucí v důsledku nízké entropie aktivace. Vzniklý cyklický intermediát je napnutý a reaguje rychle v následujícím kroku. Obecně řečeno, reakce zahrnující účast vedlejší skupiny musí být rychlejší než odpovídající přímé reakce. Pokud by tomu tak nebylo, probíhala by jen přímá reakce. Není snadné odhadnout, jaké jsou rychlosti obou těchto reakcí. Jedna z použitých metod byla porovnání chování cis- a trans-disubstituovaných cyklohexanů. Účast vedlejší skupiny je možná jen u trans-izomeru a jejím výsledkem je anomální stereochemické chování - trans-konfigurace je zachována, ale enantiomerní reaktant je racemizován, poněvadž můstkový intermediát je v rychlé rovnováze se svým enantiomerem v důsledku inverze kruhu. Vzájemná orientace obou skupin vynucuje u cis-izomeru intramolekulární průběh a normální substituci. Jestliže předpokládáme, že cis-izomer je dobrým modelem pro elektronické efekty mezi sousedními skupinami, potom poměr obou izomerů je měřítkem urychlení reakce.
|
|
Pro uvedenou reakci jsou nalezené poměry tyto:
|
X |
OBs |
Cl |
Br |
OAc |
|
k1/k2 |
Brosylátová skupina je sama o sobě příliš slabě nukleofilní, než aby vstupovala do interakce mezi sousedními skupinami. Blízkost cis- a trans-rychlostí pro dibrosylátový derivát ukazuje bez ohledu na zřejmé konformační rozdíly, že cis-izomer je vhodným modelem pro trans-reakci bez interakce. Reaktivita halogenů vykazuje paralelu s nukleofilitou halogenidových iontů. Jodidový ion je dokonce jednou z nejefektivnějších participujících skupin. Acetoxyskupina je také reaktivnější, než by mohlo být očekáváno na základě její nukleofility. Její reakce ovšem probíhá přes málo napnutý pětičlenný cyklický intermediát.
|
|
Toto chování bylo skutečně potvrzeno experimentem s 18O značeným kyslíkem. Jestliže byl původně značen karbonylový kyslík, potom jeho polovina byla nalezena v diolu vzniklém hydrolýzou diacetátu vzniklého v acetolýze. Účast vedlejší arylové skupiny byla demonstrována solvolytickou racemizací threo-3-anisylbut-2-yl sulfonátu. Erythro-izomer za
podobných podmínek neracemizuje. Neexistuje také interkonverze mezi erythro-
a threo-epimery. To je v souladu s existencí cyklického iontu jako intermediátu.
|
|
Elektronakceptorní substituenty na benzenovém kruhu snižují stabilitu cyklického fenoniového iontu a upřednostňují reakci přes necyklický iontový pár. Tak acetolýza threo-p-nitroderivátu dává 7% threo- a 93% erythro-produktu. Graf závislosti log k na s pro tuto solvolýzu dává přímku pro elektronakceptory a křivku pro donory.

Hammettova závislost pro acetolýzu threo-3-arylbut-2-yl brosylátu při 75o C
Extrapolace lineární části nám poskytuje odhad rychlosti reakce bez účasti sousední skupiny. Příbuzný 2-arylethylový systém ArCH2CH2OTos se chová podobným způsobem a přímá reakce je nyní pravděpodobně SN2. Užití 14C značeného PhCH2CH2OTos umožňuje měřit rychlost přesmyku a z ní potom stanovit poměr mezi přímou cestou a cestou přes fenoniový iont. Údaje v tabulce ukazují důležitost rozpouštědla pro uplatnění SN1 nebo SN2 mechanismu. Ethanol jako slabě ionizující rozpouštědlo a relativně silný nukleofil reaguje téměř úplně SN2 cestou, zatímco kyselina trifluoroctová s opačnými vlastnostmi reaguje prostřednictvím fenoniového iontového páru. Důkaz tohoto chování vychází ze stereochemie threo-1,2-dideuterifenethyltosylátu, který nevykazuje epimerizaci při solvolýze trifluoroctovou kyselinou.
Reakční konstanty prvního řádu pro přímou solvolýzu a solvolýzu s účastí sousední skupiny pro PhCH2CH2OTos při 75oC (k1/10-6s-1):
|
Rozpouštědlo |
Přímo |
Spoluúčast |
|
EtOH |
0.042 |
|
|
AcOH |
0.19 |
0.22 |
|
HCOOH |
35 |
|
|
CF3COOH |
0.05 |
p vazba může také působit jako sousední skupina. 3-b-cholesterylchlorid podléhá nukleofilní substituci se zachováním konfigurace. Ethanolýza odpovídajícího tosylátu dává 3-b-ether v nepufrovaném roztoku, ale v přítomnosti octanu draselného je produkt tzv. i-cholesterylether.
|
|
Acetolýza podobně dává i-acetát v reakci, která je zhruba 1000x rychlejší než acetolýza tosylátu a v kyselině i-acetát rychle izomeruje na 3-b-acetát. Vysvětlení tohoto chování je založeno na účasti p orbitalu cholesterylové dvojné vazby na odpuzování odstupující skupiny s tvorbou intermediátu s delokalizovaným kladným nábojem. To poněkud připomíná delokalizovaný p systém allylového kationtu. Proto bývá často nazýván jako homoallylový a přerušená p konjugace, kterou obsahuje, jako homokonjugace. Podobně jako allylový systém podléhá ataku na svých koncích a tvorba i-produktu je analogií allylového SN´ přesmyku. Ještě výraznějším příkladem stejného chování je acetolýza anti-7-norbornenyltosylátu. Jeho reakce je 1011 rychlejší než nasyceného derivátu a probíhá se zachováním konfigurace. Intermediát této reakce je symetrický a p elektrony jsou výhodně orientovány pro účast v reakci. Kationtové intermediáty těchto reakcí jsou poněkud rozdílné od normálních trikoordinovaných karbokationtů.
|
|
V tomto kationtu je C(7) pentakoordinován a zbylé dva účastnící se uhlíky jsou tetrakoordinované. Tetra-
a pentakoordinované uhlíkaté kationty jsou potom nazývány jako neklasické karbokationty. O jejich existenci se vedla řada diskusí. Např. zdali jejich zdánlivá existence není výsledkem rychlé rovnováhy mezi klasickými karbokationty.
|
|
Důkaz můžeme najít opět v experimentálním chování. Zavedení methylové skupiny do polohy 2 urychluje reakci 13x, zavedení dalšího methylu do polohy 3 zvyšuje rychlost 11x. To ukazuje, že kladný náboj je zřejmě sdílen oběma pozicemi. Jestliže by reakce probíhala přes klasický iont, potom by první methylová skupina urychlovala reakci generováním terciárního karbokationtu, ale druhá methylová skupina by zřejmě měla jen velmi malý efekt, protože obě by současně nemohly stabilizovat vznikající kladný náboj. Jestliže zavedeme do polohy 7 p-anisyl, tak jakýkoliv vzrůst reakční rychlosti vymizí.
|
|
|
|
Reakce je pořád ještě rychlejší, než je možno předpovědět z Hammettovy závislosti pro ostatní 7-arylderiváty, ale není rychlejší než v případě nasyceného analogu. To je také důkazem dříve uvedeného tvrzení, že participace sousedních skupin musí vést k rychlejší reakci. V tomto konkrétním případě reakce probíhá přes klasický stabilní karbokationt.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 2111
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved