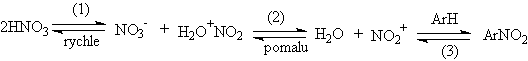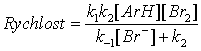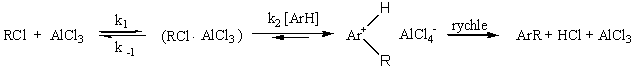| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
DOCUMENTE SIMILARE |
|
TERMENI importanti pentru acest document |
|
Aromatická elektrofilní substituce
Charakteristika elektrofilu
Charakteristika elektrofilu probíhá za nejrůznějších podmínek. Nejpoužívanějším činidlem je kyselina dusičná, ať už ve vodných roztocích, ve směsi s kys. sírovou nebo chloristou, anebo v řadě organických rozpouštědel, jako je kys. octová nebo nitromethan. Jiná činidla obsahují nitroniový kationt v podobě různých solí - příkladem je NO2+BF4- v sulfolanu nebo acetylnitrát. Podobně v přebytku kys. sírové je kys. dusičná kvantitativně přeměňována na nitroniumhydrogensulfát.
|
|
Prokázat existenci tohoto procesu lze Ramanovou spektroskopií z vodivostních měření a velmi snadno i kryoskopicky. Ve vodných roztocích kys. sírové je koncentrace nitroniových iontů velice malá a reakce probíhá pozorovatelnou rychlostí jen s velmi reaktivními substráty. Ale i bez přítomnosti kys. sírové reakce probíhají za účasti nitroniového kationtu. Důkazem existence je např. izotopová výměna 18O izotopu, kdy dochází k výměně mezi kys. dusičnou a vodou rychlostí shodnou s rychlostí nitrace velmi reaktivních substrátů. To ukazuje, že oba procesy musí mít společný pomalý krok.
|
|
V souladu s touto skutečností je pozorovaný nulový řád reakce vzhledem ke koncentraci aromatického substrátu. Méně reaktivní substrát se řídí kinetikou prvního řádu; rychlost limitujícím krokem je substituce. Podobný efekt je pozorován např. při nitraci toluenu vodným roztokem kys. dusičné a sírové. Řád reakce se mění z nulového řádu při nízkých koncentracích kys. sírové k prvnímu řádu při vzrůstající koncentraci. Korelace s funkcemi kyselosti také podporují účast nitroniového iontu - rychlost nitrace velmi těsně souvisí s HR pro systém definovaný rovnicí
|
|
|
|
Tvorba nitroniového kationtu je totiž analogickou reakcí. Na druhé straně, kdyby reaktivní elektrofilní částicí byla protonovaná kys. dusičná, museli bychom očekávat lepší korelace s funkcemi typu HO nebo HA. Podobně reakce s kys.dusičnou v nitromethanu nebo v kyselině octové vykazují změnu od kinetiky nultého ke kinetice prvého řádu v závislosti na reaktivitě substrátu a podmínkách reakce. V reakcích nultého řádu je opět rychlost limitujícím krokem tvorba nitroniového kationtu.
Reakce tohoto typu jsou zpomalovány přídavkem dusičnanového aniontu, který snižuje koncentraci H2ONO2+. Tento přídavek však neovlivňuje celkový obraz kinetického chování. Jestliže je však přidána i voda, dochází ke změně kinetiky z nultého na první řád. Reakce prvého řádu se uplatňuje v situaci, kdy nitroniový kationt je spotřebováván podstatně pomaleji substrátem než vratnou reakcí na kyselinu dusičnou. Voda změní poměr mezi těmito dvěma kroky, ale dusičnanový aniont ovlivňuje pouze první krok. Kinetické chování v organických rozpouštědlech bývá obvykle mnohem složitější.
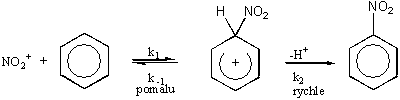
Whelandův intermediát reakce
|
|
Důkaz velmi známého dvoukrokového mechanismu lze nejlépe založit na kinetických izotopových studiích.
Pozorované efekty jsou obvykle velmi malé, s výjimkou, kdy všechny pozice, do nichž je možno zavést nitroskupinu, jsou z obou stran obklopeny objemnými substituenty (2,4-diterc.butyltoluen). Tento intermediát, v němž je kladný náboj nitroniového kationtu rozprostřen do benzenového p systému, se běžně nazývá Whelandův intermediát. Ionty podobného typu se tvoří protonací aromatických uhlovodíků, např. působením HCl/AlCl3 nebo HF/BF3. Výsledná sůl je obvykle barevná a elektricky vodivá. Tato sůl je také zodpovědná za H/D výměnu, ke které dochází působením DCl nebo DF za katalýzy Lewisovými kyselinami.
Orientace a reaktivita
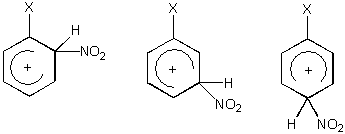
V případě monosubstituovaných benzenů existují tři možné izomerní intermediáty a tři tranzitní stavy, jejichž energie závisí na interakci mezi substituentem a kladným nábojem na kruhu.
|
|
Diskusi reaktivity je zřejmě možno založit na předpokladu, že nábojová distribuce je velmi podobná v intermediátu a odpovídajícím tranzitním stavu. Kladný náboj je soustředěn především do pozic ortho- a para- vzhledem k místu ataku.
|
|
Je tedy zřejmé, že elektrondonory v polohách ortho- a para- stabilizují tranzitní stav a urychlují reakci, elektronakceptory mají potom opačný efekt. Tato skutečnost může být demonstrována na příkladu nitrace toluenu a acetofenonu. Jsou uvedeny parciální rychlostní konstanty, tj. rychlost reakce v dané pozici na daném substrátu vzhledem k benzenu. Např. toluen je nitrován 24x rychleji než benzen a 56.5 % produktu je o-nitrotoluen. Pro ortho-parciální rychlostní konstantu potom platí:
fortho = 24 x 56.5/100 x 6/2 = 41
|
|
První dva členy v rovnici udávají poměr reaktivity obou ortho pozic toluenu vzhledem k šesti pozicím benzenu, třetí člen je statistická korekce.
Selektivita a charakter tranzitního stavu
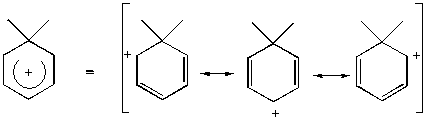
Energie Whelandova intermediátu a protonovaného aromátu jsou si velmi podobné. Reagující elektrofil je oddělen od kladně nabitého p systému tetraedrickým uhlíkem a z toho důvodu je jeho vliv velice omezen. Substituenty na kruhu, zvláště v polohách ortho- a para-, způsobují ovšem na druhé straně značné energetické rozdíly. Jestliže tranzitní stav substituce je velmi podobný Whelandovu intermediátu, potom aktivační energie reakce bude velmi podobná energii protonace uhlovodíku a reakční rychlost substituce bude úměrná bazicitě substrátu. Platnost tohoto předpokladu se skutečně potvrdila v řadě reakcí - např. v korelaci mezi bazicitou polymethylbenzenů a rychlostí jejich bromace.
|
|
Nitrace je mnohem méně citlivá na změnu substituentu. Např. toluen, který je bromován 600x rychleji než benzen, je nitrován jen 20x rychleji. To ukazuje, že tranzitní stav nitrace leží relativně na začátku reakční koordináty a poměrně málo kladného náboje je delokalizováno v kruhu.

Reakční profil pro substituci s tranzitním stavem blízce připomínajícím Whelandův intermediát. Substituční efekt na reakční rychlost koreluje s bazicitou substrátu.

Reakční profil pro reakci s TS podobným reaktantům.
Relativní reaktivity jednotlivých pozic substituovaného benzenového kruhu musí mít také vztah k charakteru tranzitního stavu. Čím více náboje je v samotném kruhu, tím je větší rozdíl mezi meta- a para- pozicemi.
Srážková teorie
Dosavadní poznatky o relativní reaktivitě jednotlivých substrátů v některých případech neplatí. Např. použití nitroniového kationtu v sulfolanu ukazuje, že toluen je 1.7x reaktivnější než benzen a mesitylen jen 2.7x reaktivnější, zatímco relativní rychlosti bromace jsou v poměru asi 1 : 200 milionům. Na druhé straně orientace substituce není nikterak změněna. Interpretace této skutečnosti spočívá ve změně mechanismu obsahujícího dva intermediáty

Ilustrace teorie rychlost limitující tvorby p komplexu
První intermediát, zvaný p komplex, je pouze asociátem nitroniového iontu s p benzenovým systémem, bez existence vazby k jakémukoliv uhlíku. Druhý intermediát, tzv. s-komplex, je již regulérní chemickou sloučeninou a předcházející tranzitní stav TS2 určuje distribuci produktů. Existence těchto komplexů má analogii v tvorbě volně vázaných aduktů aromatických a olefinických uhlovodíků s řadou elektrofilních částic - chinony, kyselinou pikrovou, halogeny, stříbrnými ionty a dalšími. Některé z těchto aduktů jsou izolovatelné nebo detekovatelné spektroskopicky či jinak, např. pomocí rozpustnosti nebo jiných termochemických veličin. Např. rozpustnost halogenovodíků v aprotickém roztoku arenu nebo alkenu vede k poznatku o tvorbě bezbarvého aduktu 1 : 1 se slučovacím teplem asi 10 kJ/mol. Tyto roztoky jsou nevodivé a jejich vlastnosti se velmi liší od s komplexu vznikajícího za přítomnosti Lewisových kyselin. Tyto slabé komplexy jsou v literatuře nazývány jako charge-transferové komplexy, donor-akcepotorové komplexy, p komplexy nebo molekulární komplexy. Tyto pojmy se většinou volně zaměňují, ale musíme si být vždy vědomi rozdílu mezi těmito komplexy, jejichž asociace je dílem diskrétních sil a komplexy, v nichž existují reálné kovalentní vazby. Nejadekvátnější pojem tedy zřejmě představuje p komplex.
|
|
V případě benzen-nitroniového p komplexu můžeme jeho vznik popsat v rámci interakce HOMO benzenu a LUMO nitroniového kationtu. Vznik tohoto komplexu bude zřejmě pomalý, bude tedy rychlost limitujícím krokem celé reakce. Tranzitní stav tohoto procesu TS1 bude velmi podobný volně vázanému molekulárnímu komplexu a bude tedy zřejmě existovat korelace mezi stabilitou tohoto molekulárního komplexu a rychlostí nitrace. Pozorovaný relativně malý substituční efekt na rychlost nitrace je co do velikosti porovnatelný s relativní stabilitou molekulárních komplexů s chlorovodíkem - viz tabulka
Porovnání stabilit molekulárních komplexů polymethylbenzenů s chlorovodíkem a rychlosti nitrace nitroniumfluoroborátem v sulfolanu při 250C
|
Methylové substituenty |
Relativní stability komplexů s HCl |
Relativní rychlosti nitrace |
|
žádný | ||
|
Me | ||
|
1,2-Me2 | ||
|
1,3-Me2 | ||
|
1,4-Me2 | ||
|
1,3,5-Me3 |
Většina nitrací je velice rychlá a konvenční kinetická měření jsou proto obtížná. Velmi důležitou roli hrají i fyzikální charakteristiky reakce, např. míchání. Obvykle jsou měřeny relativní rychlosti konkurenčních reakcí dvou substrátů. Např. relativní reaktivita toluenu a benzenu může být vyjádřena sledováním vznikajícího množství nitrotoluenu a nitrobenzenu, jestliže jejich směs je nitrována za podmínek nedostatku elektrofilu. Jestliže rychlost nitrace je mimořádně velká, potom se limitujícím faktorem stává rychlost, s jakou se reagent
a substrát dostávají do kontaktu. Tato skutečnost byla také nalezena v některých reakcích. Např. při nitraci dibenzylu nitroniumtetrafluoroborátem v sulfolanu byl nalezen abnormálně vysoký podíl dinitroderivátu. Jeho vznik je pravděpodobně důsledkem velmi rychlé reakce v oblasti nehomogenní směsi, kde je lokální koncentrace nitroniového iontu mnohem vyšší než v homogenním roztoku. Podobně ukazují na limitující roli možnosti kontaktu reagentu a substrátu i některé další reakce - toluen je nitrován nitrační směsí 20x rychleji než benzen, ale zavedení dalšího elektrondonoru do molekuly tuto reaktivitu úměrně nezvyšuje. Xyleny a mesitylen reagují přibližně 2x rychleji než toluen, v příkrém kontrastu k rychlosti jejich bromace. Je tedy zřejmé, že limitujícím faktorem není rychlost vlastní reakce, ale rychlosti difuzních procesů v roztoku. Hodnoty aktivační enthalpie těchto difuzí kontrolovaných reakcí jsou v souladu s hodnotami vypočtenými pro dvoustupňový proces, zahrnující disociaci kys. dusičné na nitroniový kationt a následovanou viskozitou determinovanou difuzí nitroniového kationtu k substrátu. Podobné chování bylo nalezeno i v prostředí kyseliny fosforečné. Oba dva faktory, tj. tvorba p komplexu a pravděpodobnost srážky, nejsou v rozporu. Skutečnost, že selektivita reakce je zachována i v reakcích řízených možností srážky ukazuje, že musí ještě existovat jiný intermediát I před Whelandovým s intermediátem.
|
|
Tento intermediát je obvykle nazýván srážkovým párem a vyjadřován vzorcem ArH . NO2+. O struktuře této částice mnoho nevíme. Je možné, že je držena pohromadě prostě jen tlakem okolních molekul rozpouštědla, ale je také možné, že obsahuje kompletní přenos elektronů, tj. že tento intermediát představuje radikál/radikál-kationtový pár ArH.+ NO2.. Mezi těmito extrémy může ovšem existovat řada přechodných forem komplexů.
Nitrace v acetanhydridu.
Nitrace kyselinou dusičnou v acetanhydridu není po stránce mechanismu úplně objasněna. Reakce je zřejmě řízena difuzí - rychlost reakce je úměrná koncentraci aromátu, ale je poměrně nezávislá na jeho druhu. Aktivním elektrofilem je zřejmě NO2+. Jeho tvorba ovšem není nikdy rychlost limitujícím krokem. To vede ke zdánlivě paradoxní situaci. Jestliže reakce zahrnuje sekvenci kroků, které jsou podobné difuzí kontrolované nitraci v kyselině sírové - jako např. v reakci:
|
|
kde kd je rychlostní konstanta difuzí kontrolovaného procesu, pak vzhledem k tomu, že krok 2 může být rychlým krokem, krok 1 není nikdy rychlost limitujícím. k -1 tedy musí být vždycky větší než kd.[ArH] a průměrná doba života nitroniového kationtu musí být extrémně krátká. Výpočty ovšem ukazují, že tato doba je příliš krátká vzhledem k možným rychlostem difuze a nikdy by nebylo možné dosáhnout pozorované rychlosti reakce. Vysvětlení tohoto rozporu je založeno na předpokladu, že částice, které difundují k sobě, musí být přítomny v relativně vysokých koncentracích, a že nitroniový kationt je tvořen v rámci vzniklého srážkového páru. Je tedy navrhován následující mechanismus:
|
|
Prvý srážkový pár je odvozen od neutrálního acetylnitrátu, který je přítomen ve vysoké koncentraci. Ten je potom protonován a vzniklý kationt v tautomerní rovnováze poskytuje částici ArH.NO2+AcOH, která se rozpadá na Whelandův intermediát za ztráty molekuly kyseliny octové. Pro velmi málo reaktivní substráty je tedy rychlost limitující krok 4, ale pro velmi reaktivní areny je to pravděpodobně krok 3. Protonačním činidlem v kroku 2 je pravděpodobně Ac2OH+ a nikoliv HNO3. Je to v důsledku toho, že existence velmi rychlé zpětné reakce elektrofilu na prekursory vyžaduje, aby zpětný krok 2 byl velmi rychlý. Koncentrace NO3- je tedy velmi nízká, než aby mohla významně přispívat k odtržení protonu z (AcONOOH)+ a v důsledku platnosti principu mikroskopické reversibility není tedy kyselina dusičná důležitá pro druhou stranu rovnice.
Nitrace prostřednictvím nitrosace
Řada nitračních reakcí je zpomalována přídavkem dusitanů, jejichž efekt je zřejmě v deprotonaci (H2ONO2)+ dusitanovým aniontem, který je silnější bází než dusičnanový aniont. Velmi reaktivní substráty ovšem vykazují opačný efekt - dusitany urychlují nitrace. Kromě toho řada organických látek je kyselinou dusičnou oxidována a vznikající kyselina dusitá působí jako autokatalyzátor v těchto reakcích. Zřejmě existují dva rozdílné mechanismy těchto procesů. Prvý zřejmě zahrnuje počáteční nitrosaci následovanou oxidací vzniklé nitrososloučeniny. Příkladem může být nitrace fenolu, kde vznikající p-nitrosofenol může být izolován. Kinetická analýza těchto procesů ukazuje, že elektrofilem nitrosace je nitrosoniový kationt NO+ a nikoliv méně aktivní, ale více zastoupený N2O4, který je smíšeným anhydridem kyseliny dusité a dusičné. Poměr mezi přímou nitrací a nitrosooxidací je složitou otázkou. Ve zředěných vodných roztocích, kde je nízká koncentrace nitroniového iontu, reakce probíhá téměř výlučně nitrosační cestou. V silných kyselinách vysoká reaktivita NO2+ způsobuje, že nitrosace je nevýznamná, ale dochází k uplatnění antikatalytického efektu dusitanového aniontu. Nitrace benzenu kyselinou dusičnou v nitromethanu nebo v sulfolanu mohou probíhat až asi 300x rychleji než nitrace benzenu, tj. zjevně rychlostí určenou možností srážek. Některé substráty ovšem reagují ještě rychleji. To může být jedině důsledkem toho, že nitrosace je rychlejší než nitrace a tedy nitrosační elektrofil je přítomen ve vyšší koncentraci než nitroniový iont. Např. nitrace dimethylanilinu v 85% kys. sírové probíhá zřejmě následujícím mechanismem:
|
|
Ipso-nitrace a adičně-eliminační mechanismus
Možnost elektrofilního ataku na substituovaný uhlík byla donedávna považována jen za neproduktivní vedlejší rovnovážný krok v elektrofilních substitucích.
|
|
Ipso-intermediát může ale také být přímým účastníkem reakce. Jestliže skupina X má vhodné vlastnosti, může být vytěsněna jako X+. To je dobře známá reakce v případě, že X je elektropozitivní skupina jako např. Me3Si, který může být nejčastěji nahrazen protonem. Ale i alkylové skupiny mohou být takto nahrazeny – nitrace p-cymenu poskytuje vedle očekávaných produktů až 10% p-nitrotoluenu. Brom a jod jsou také často nahrazovány (na rozdíl od chloru)a samotná nitroskupina ještě snadněji. Druhou možností přeměny
ipso-intermediátu je 1,2-posun, vedoucí přes další intermediát k ortho-produktu.

Není snadné rozhodnout, jaká část ortho-produktu vzniká přímou, anebo touto cestou. Příkladem může být nitrace 1,2,4,5-tetramethylbenzenu (durenu) v kyselině sírové. Ukazuje se, že množství 3-nitrodurenu velmi závisí na koncentraci vodné kyseliny sírové. Při koncentracích vyšších než 85% je jenom jediným produktem, pod 60% koncentrací představuje pouze 12% ze směsi produktů. Zbylých 88% produktů pochází z různých reakcí ipso-intermediátu s rozpouštědlem. To znamená, že zřejmě jen 12% 3-nitrodurenu vzniklého v koncentrované kyselině pochází z přímého ataku polohy 3 a zbytek je tvořen přesmyky následujícími po počátečním ipso-ataku.
|
|
Existuje ovšem také možnost migrace substituentu a nikoliv elektrofilu. V případě disubstituovaného substrátu to vede k přesmyku, který ale není u nitrací běžnou vedlejší reakcí. Konečně také může ipso-intermediát podlehnout nukleofilnímu ataku za vzniku cyklohexadienového kruhu. Jeho vznik je mnohem pravděpodobnější než u normálního Whelandova intermediátu, protože se nemůže přeměnit zpět na aromatický systém odštěpením protonu. Reakce tohoto typu jsou relativně časté u nitrací acetylnitrátem. Např. nitrace p-cymenu acetylnitrátem při 0oC dává směs produktů - dva normální nitrační produkty, p-nitrotoluen a dva stereoizomerní cyklohexadienové adukty tvořené nukleofilním atakem acetátu na ipso-kationt.
|
|
V kyselině sírové tyto adukty podléhají eliminaci s přesmykem za vzniku 4-izopropyl-2-nitrotoluenu, ale v kyselině octové odštěpují kyselinu dusitou za vzniku 3-acetoxyderivátu. Tento typ chování je velmi běžný při nitracích acetylnitrátem.
|
|
Silné elektrondonory usnadňují atak v para-poloze, i když je obsazená jiným substituentem. Např. nitrace p-methyl-N,N-dimethylanilinu v kyselině sírové poskytuje jako hlavní produkt 2-nitro-4-methyl-N,N-dimethylanilin v důsledku dvoustupňového procesu s počátečním atakem v poloze 4 a následným 1,3-přesmykem nitroskupiny.
|
|
Jestliže ortho-pozice k aminoskupině je obsazena, potom takové ipso-intermediáty mohou být dokonce izolovány ve formě hexafluorofosfátových solí. Mechanismus 1,3-přesmyku byl objasněn studiemi nitrací fenolů a fenyletherů. Nitrace p-chloranisolu ve vodné kyselině dává nejen normální o-nitroderivát, ale i p-chloro-o-nitrofenol. Studie pomocí 18O izotopu potvrdily, že fenolická skupina pochází z rozpouštědla a vzniklý ipso-adukt podléhá následnému přesmyku nitroskupiny.
|
|
V souladu s tímto předpokladem byl z reakce izolován i nenitrovaný p-chlorofenol. V koncentrovaných kyselinách je tento přesmyk kysele katalyzován. Při nižších koncentracích kyselin katalytický efekt zaniká. Nekatalyzovaná cesta potom zřejmě zahrnuje disociaci neprotonovaného dienonového intermediátu na fenoxylový radikál a oxid dusičitý. Vznik dimerního produktu při nitraci 4-fluorofenolu je potvrzením tohoto mechanismu.
|
|
Jiné elektrofilní substituce
Halogenace
Chlorace a bromace vykazují mnoho společných rysů s nitrací. Příklady ukazují, že bromace probíhají přes tranzitní stav podobný Whelandovu intermediátu. Vlivem různých podmínek dochází také ke ztrátě selektivity vůči různým substrátům v souladu s tím (podobně jako u nitrace), že reakce je řízena především pravděpodobností srážek. Jsou známy také ipso-ataky halogenů a tvorba aren-halogenových aduktů buď jako vedlejších produktů, nebo intermediátů v adičně-eliminačních mechanismech substitučních reakcí. Určité nejasnosti zůstávají ovšem o charakteru elektrofilu v těchto reakcích. Reaktivní substráty jsou chlorovány např. chlorem v roztocích kys. octové a elektrofil je zřejmě molekulární chlor. Reakce se řídí kinetikou druhého řádu a rychlost není ovlivňována kyselinami, bázemi ani přídavkem chloridového či acetátového iontu. Je tedy zřejmé, že v reakci se neuplatňují takové částice jako Cl+, AcOCl a (AcOHCl)+. Jedná se tedy zřejmě o jednoduchý dvoukrokový mechanismus.
|
|
Podobné chování bylo nalezeno i v ostatních organických rozpouštědlech.
Bromace vykazuje podobné rysy. Z hlediska kinetického chování zůstává reakcí prvého řádu vzhledem k aromatickému substrátu, ale často je mnohem komplikovanější vzhledem k bromu. To je pravděpodobně obrazem snazší reversibility prvého kroku. Aplikujeme-li teorii ustáleného stavu na Whelandův intermediát, dostáváme rychlostní rovnici:
|
|
V případě chlorace je výraz k -1[Cl-] zanedbatelný a vzniká jednoduchá rovnice druhého řádu. V případě bromace jsou oba členy ve jmenovateli porovnatelné a vzniká běžný zpomalující efekt bromidového aniontu. Vedle toho je také koncentrace bromidového iontu nepřímo úměrná koncentraci bromu v důsledku rovnováhy
|
|
Vzhledem k těmto okolnostem vykazují reakce většinou významný H/D kinetický izotopový efekt. Jeho velikost závisí na koncentraci [Br-], přičemž zřejmě k -1[Br-] > k2 a rychlost limitujícím se stává krok 2.
Mnohem problematičtější jsou halogenace bromoniovými či chloroniovými kationty. Reakce s kyselinou bromnou jsou kysele katalyzovány:
Rychlost = k3 [ArH][HOBr][H
Z toho vyplývá, že elektrofilem v této reakci je buď Br+ nebo BrOH2+. V silně kyselých roztocích je to nepochybně pravda, ale v méně kyselých bylo zjištěno, že pozorovaná rychlost reakce je více než milionkrát větší než rychlost srážek mezi substrátem a elektrofilní částicí. Existence lineární korelace mezi rychlostí bromace různých substrátů a rychlostí jejich nitrace
|
|
ukazuje, že nemůže být podstatný rozdíl v charakteru atakujícího elektrofilu. Vysvětlení je založeno na návrhu mechanismu, který zahrnuje dva alternativní předreakční rovnovážné kroky.
Horní cesta je vlastně původně navržený a přijímaný mechanismus reakce zprostředkované molekulárním nebo p komplexem mezi arenem a BrOH2+, který je podporován silně kyselými roztoky a relativně méně kyselým substrátem. Ve slaběji kyselých roztocích a s reaktivnějšími substráty je preferována spodní cesta, která zahrnuje primární komplexaci s neprotonovanou kyselinou bromnou a následnou protonaci komplexu. V obou případech reaktivita i selektivita zůstává zachována, protože rychlost i tvorbu produktu řídí krok rozpadu protonovaného komplexu na Whelandův intermediát. Existenci předreakčních rovnovážných kroků zahrnujících přenos protonu potvrzuje existence značného rozpouštědlového izotopového efektu.
Sulfonace

Většina
sulfonací bývá prováděna vodnou kyselinou sírovou nebo oleem. Kinetické
studie jsou velmi komplikované, protože zahrnují velkou šíři částic přítomných
v různě koncentrovaných roztocích kyseliny sírové. Zahrnují částice jako H2SO4, H3SO4+, H5SO5+, HSO4-,
H3O+, H5O2+,
SO3, S3O9,
H2S2O7. Další problémy jsou spojeny
s vratností sulfonace. Ve vodných roztocích kyseliny sírové do koncentrace
80% koreluje rychlost reakce lineárně s aktivitou H3SO4+,
při koncentracích vyšších než 85% existuje podobná korelace s aktivitou H2S2O7.
Zdá se tedy, že existují dva hlavní elektrofily. Jedná se zřejmě o SO3
solvatovaný H3O+ nebo H2SO4 (v
koncentračním rozmezí 85 - 100% kys. sírové). Mechanismus těchto reakcí je tedy
potom následující:
V koncentracích pod 95% kys. sírové je substrátový H/D kinetický izotopový efekt nevýznamný, ale vzrůstá při vyšších koncentracích kyseliny. To znamená, že druhý krok - přenos protonu - se stává rychlost limitujícím krokem a v souladu s tím rychlost klesá pod hodnotu, která může být předpovězena extrapolací na základě korelace s aktivitou H2S2O7. Při nižších koncentracích kyseliny je proton odtržen HSO4- aniontem (silnější báze než kys. sírová), ale jeho koncentrace velmi rychle klesá se vzrůstající koncentrací kyseliny. V oleu rychlost substituce vzrůstá velmi rychle. Elektrofilem je zřejmě nesolvatovaná molekula SO3. Rychlost reakce závisí na koncentraci SO3 a na funkci kyselosti H0. Pravděpodobný mechanismus je následující:
|
|
Friedel-Craftsovy reakce
a) Alkylace
Kinetické studie Friedel-Craftsových alkylací jsou velmi obtížné - reakční rychlosti jsou velmi citlivé na stopy vlhkosti, jsou často příliš vysoké, než aby mohly být změřeny, reakční směs vykazuje tendence k tvoření nehomogenních vrstev, počáteční reakční produkty snadno izomerizují, dochází k polyalkylacím, kinetické chování závisí na rozpouštědle. I přesto základní rysy těchto reakcí jsou známé. Např. série benzylačních reakcí substituovaných benzylchloridů s chloridem hlinitým v nitrobenzenu se řídí kinetickou rovnicí třetího řádu.
Rychlost = k3[ArH][RCl]AlCl3]
Pravděpodobný mechanismus je
|
|
s krokem 2 jako rychlost určujícím. Pro rychlost potom platí:

a jestliže k -1 >> k2 [ArH], vztah se redukuje na pozorovanou rovnici třetího řádu s k3 = K1k2. Přesnou strukturu intermediátu vyjadřovaného jako RCl . AlCl3 není většinou jednoduché určit. V principu může zahrnovat řadu struktur od volného molekulového komplexu k disociovanému iontovému systému.
|
|
Jeho struktura musí záviset na struktuře R a na rozpouštědle a pochopitelně i na použité Lewisově kyselině. Je nepravděpodobné, že by mnoho reakcí probíhalo až přes stádium volných karbokationtů. Friedel-Craftsovy alkylace vykazují velmi nízkou intermolekulární i intramolekulární selektivitu zřejmě proto, že poměrně velmi malé množství náboje je přeneseno na benzenový kruh v tranzitním stavu více podobném reaktantům. To je chování charakteristické pro vysoce reaktivní elektrofily. Citlivost alkylových skupin k Wagner-Meerweinovu přesmyku v průběhu reakce je také důkazem vysoce polarizovaných C-Hal vazeb. Některé 1:1 komplexy mezi Lewisovými kyselinami a alkylhalogenidy jsou za nízkých teplot izolovatelné. Podléhají pomalé výměně halogenů prokazatelné radioaktivním značením, anebo působením chloridů kovů na alkylbromidy. Rychlost výměny klesá v pořadí terciární > sekundární > primární, což je v souladu s tvorbou a stabilitou iontových párů, ale nevylučuje možnost vzniku koordinovaného aduktu, který podléhá výměně prostřednictvím 1,3-posunu.
|
|
Tato situace není dosud uspokojivě objasněna. Nejpravděpodobnější mechanismus zahrnuje zřejmě postupný přechod od koordinovaného komplexu, např. s methylhalogenidy, až k iontovým párům, např. s terc.butylem.
b) Acylace
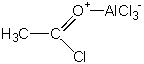
Kinetika Friedel-Craftsovy acylace je často prvého řádu vzhledem k substrátu a acylchloridu, ale vykazuje velmi různé chování vůči koncentraci katalyzátoru. IR spektra směsi acetylchlorid/chlorid hlinitý ukazují na přítomnost volného CH3CO+ a 1:1 a 1:2 komplexů. 1:1 komplex má následující strukturu:
|
|
Struktura 1:2 komplexu není detailněji známa. Reakce tedy zahrnuje v rychlost limitujícím kroku atak kteréhokoliv nebo všech elektrofilů (v závislosti na substrátu, acylchloridu i rozpouštědle) na aromatický substrát. Z toho tedy vyplývá variabilní závislost rychlosti na koncentraci katalyzátoru. Ta je také ovlivněna tvorbou komplexu mezi katalyzátorem a molekulou vzniklého ketonu. NMR spektra reakčních směsí ukazují na tvorbu 1:1:1 komplexu ArH . RCOCl . AlCl3 .
Kopulační reakce diazosloučenin
Slabě elektrofilní diazoniový ion ArN2+ reaguje jen s aktivovanými aromatickými substráty, jako jsou aminy a fenoly. Reakce je bržděna kyselinami, kterými jsou aminy protonovány na nereaktivní amoniové ionty. Skutečnost, že se fenoly chovají podobně, ukazuje, že aktivní částicí je fenolátový aniont a tato skutečnost je potvrzena i tím, že anisol nereaguje s většinou diazoniových solí. V alkalických roztocích je reakce také bržděna, protože diazoniové soli přecházejí na ArN=NOH. I když existují tato specifická působení H3O+ i OH- iontů, ukazuje se, že některé kopulační reakce jsou bazicky katalyzovány. To nastává tehdy, jestliže je z kinetického pohledu důležité odštěpení protonu z Whelandova intermediátu. Pro rychlost reakce
|
|
platí
k = k1k2 [B]/ (k -1 + k2[B]
Jestliže první krok je rychlost určující ( k -1 << k2 [B] ), potom platí k = k1. V případě, že k -1 >> k2 [B], je rychlost úměrná [B]. V obecném případě rychlost závisí nelineárně na [B]. Všechny tři typy chování byly nalezeny, ale poslední z nich je z mechanistického pohledu nejzajímavější, protože malé změny v reagujícím systému způsobují velké kinetické efekty. Efekt bazické katalýzy na elektrofilní aromatickou substituci není omezen jen na diazokopulační reakce, ale tyto jsou velmi výhodné pro kinetická a mechanistická studia z důvodu jejich proveditelnosti ve zředěných vodných roztocích a při mírném pH. Kopulační reakce, které jsou bazicky katalyzované, vykazují také H/D a H/T kinetický izotopový efekt v případě, že přenos vodíku je důležitý z kinetického hlediska. Všimněme si, že k2 je citlivá k izotopovému složení substrátu, zatímco k -1 je na něm nezávislá. V důsledku toho zvýšení faktoru k -1 / k2 [B] přesouvá kinetickou kontrolu na druhý reakční krok a zvyšuje citlivost reakce na bazickou katalýzu i velikost kinetického izotopového efektu. Tuto skutečnost je možno ilustrovat na reakci p-chlorbenzendiazonia s anionty naftalensulfonových kyselin (šipky ukazují místa reakce). Reakce se 4-hydroxynaftalensulfonovou kyselinou nevykazuje bazickou katalýzu ani kinetický izotopový efekt - zřejmě je to reakce, ve které je první reakční krok rychlost limitující. V případě 7-hydroxy-1,3-naftalendisulfonové kyseliny je místo substituce stericky bráněno; to snižuje hodnotu k2 tím, že je bráněno přístupu báze a zabráněno ArN=N skupině zaujmout polohu v rovině naftalenového jádra. Reakce s tímto substrátem je silně katalyzovaná pyridinem a vykazuje značný izotopový efekt. Efektivita katalýzy se vzrůstající koncentrací pyridinu klesá stejně tak, jako velikost izotopového efektu, jak lze očekávat vzhledem k přítomnosti [B] ve jmenovateli zlomku. 2,6-lutidin je horším katalyzátorem než pyridin, protože vede k ještě více bráněnému tranzitnímu stavu. 4-hydroxy-2-naftalensulfonová kyselina je přechod mezi oběma zmíněnými substráty jak v citlivosti ke katalýze, tak i ve velikosti kinetického izotopového efektu. Obsahuje zajímavý případ rozdílného intramolekulárního izotopového efektu. Poloha 1 je více bráněná než poloha 3 a je citlivější na bazickou katalýzu a vykazuje větší kinetický izotopový efekt. Poměr produktů 1:3 je tedy větší pro 1,3-dideuterovanou sloučeninu.
|
|
Poměr ortho/para
Orientační efekty v elektrofilních substitucích na monosubstituovaných benzenech jsou snadno vysvětlitelné na základě efektu substituentů na charakter tranzitního stavu modelovaného Whelandovým intermediátem. Pro meta- a para- řadu je možno substituční efekty kvantifikovat známou Hammettovou rovnicí. Z elektronického pohledu bychom mohli předpokládat podobnost chování ortho- a para- poloh vůči ataku. Hlavní příčinou rozdílnosti jsou sterické efekty v ortho-poloze, jejich velikost vyjádřená reakční rychlostí je uvedená pro nitraci alkylbenzenu acetylnitrátem v následující tabulce.
|
R = Me |
Et |
i-Pr |
t-Bu |
|
|
ortho |
49.7 |
4.5 |
||
|
meta |
1.3 |
2.3 |
2.4 |
3.0 |
|
para |
Objemné substituenty jsou samozřejmě také méně reaktivní vůči více bráněné ortho-poloze. Také interakce mezi rozpouštědlem a substrátem nebo elektrofilem mohou podobně zvýšit sterické pnutí v ortho-tranzitním stavu. Např. Friedel-Craftsova benzoylace toluenu dává v dichlorethanu poměr o/p 0.052, zatímco v nitrobenzenu, který tvoří objemnější komplex s katalyzátorem, je tento poměr 0.039. Existují ovšem i jiné faktory než sterické. Substráty, které obsahují skupiny přitahující p elektrony (nitro, karbonyl, nitril, .) dávají především meta-produkt, ale ortho-poloha vykazuje vždy podstatně vyšší reaktivitu než para. Např. v případě nitrace acetofenonu je poměr reaktivit o/p roven šesti. Nejpravděpodobnější vysvětlení tohoto chování spočívá v tom, že mezomerní interakce mezi substituentem a kruhem je v para-intermediátu prakticky nulová. Následující obrázek ukazuje tuto skutečnost pro názorný případ nitrace nitrobenzenu.

Efekt halogenové substituce je ukázán v následující tabulce pro případ nitrace halogenbenzenů nitrační směsí. Meta-polohy jsou vždy silně deaktivovány. Rozdíly mezi jednotlivými halogeny jsou relativně malé. Ortho-poloha fluorbenzenu je mnohem méně reaktivní než para-poloha; tento rozdíl je méně výrazný u ostatních halogenů a poměr o/p vzrůstá v pořadí Cl < Br < I. Značná část vysvětlení tohoto chování nepochybně spočívá v silném polárním efektu fluoru, který destabilizuje ortho-tranzitní stav. Může také existovat vysvětlení vycházející z ipso-ataku a následného přesmyku - jod je jistě nejméně deaktivující halogen pro ipso-atak, zatímco fluor nejvíce. V současné době není dost informací pro posouzení těchto vlivů, ale zdá se, že vysvětlení řady ortho-reaktivit, a to nejen u halogenových substrátů, je komplikováno právě příspěvkem od zmíněných ipso-reakcí. Navíc ještě přesmyk ipso-intermediátu na ortho-izomer je konkurentem přímého nukleofilního ataku vedoucího k cyklohexadienům a z nich odvozeným dalším produktům. Rovnováha mezi oběma těmito procesy závisí na reakčním mediu.
|
Reakční rychlosti pro polohy |
F |
Cl |
Br |
I |
|
o |
0.045 |
0.067 |
0.077 |
0.17 |
|
m |
0.0021 |
0.0018 |
0.0016 |
0.0049 |
|
p |
0.25 |
0.20 |
0.40 |
Reaktivita polycyklických aromatických substrátů
a) Aproximace izolované molekuly
Odhad reaktivity je založen na vlastnostech p elektronového systému v základním stavu. V podstatě jde o aplikaci poruchové teorie s cílem stanovit, jakým způsobem elektrofil perturbuje p elektronovou distribuci substrátu v počátečních stádiích reakce. Existuje řada příkladů, jak tuto poruchu kvantifikovat. Typickým příkladem je definice Fukuiho superdelokalizability:

Sumace probíhá přes všechny obsazené p molekulové orbitaly, c je rozvojový koeficient každého MO v pozici r a E je energie orbitalu. Přístup je založen na myšlence, že Sr je mírou snadnosti s jakou je p systém perturbován elektrofilem na pozici r, a indikuje tedy schopnost této polohy tvořit novou vazbu s elektrofilem. p elektronová hustota každé polohy je vyjádřena jako c2.
b) Lokalizační metody
V tomto přístupu je užíváno odhadu vlastností Whelandova intermediátu jako modelu tranzitního stavu. Odhad je činěn na základě rozdílu mezi p elektronovými energiemi aromatického substrátu a Whelandova intermediátu. Předpokládá se, že tento rozdíl je úměrný aktivační energii. Vedle některých zjednodušených schémat, jako např. Dewarova NBMO formalismu (viz kapitola 6), je doménou použití těchto metod oblast kvantové chemie.
Slabinou obou metod, a to především v jejich kvalitativních a semikvantitativních podobách, je zanedbání vlivu sterických efektů. V případě přechodu od uhlovodíků ke složitějším systémům je většina těchto teorií jen omezeně aplikovatelná.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 2172
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved