| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
1 Hammettova rovnice
V chemii obvykle hovoříme o vztazích mezi strukturou a reaktivitou. Ačkoliv jsou tyto úvahy založeny na experimentálních výsledcích, jsou pouze kvalitativní povahy - říkají např., že toluen je reaktivnější než benzen v řadě elektrofilních substitucí. I když známe kvantitativní údaj (např. rychlost) pro některou reakci, nelze z ní vyvodit rychlost reakce např. s jiným činidlem, byť by i probíhala analogickým mechanismem. V letech 1920 - 1930 bylo věnováno mnoho úsilí pro nalezení závislosti mezi strukturou a reaktivitou. Velmi úspěšný v tomto snažení byl L.P. Hammett. Zabýval se reakcemi typu:
|
|
|
|
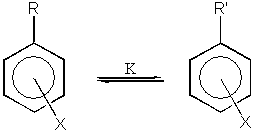
kde R je reakční centrum na řetězci benzenového jádra a X je meta nebo para substituent. Hammett ze svého zkoumání vyloučil ortho substituenty z důvodů existence sterických interakcí mezi reakčním centrem a substituentem, které se velmi obtížně kvantitativně vyjadřují. Na základě zkoumání řady experimentálních výsledků došel ke zjištění, že termodynamické nebo i kinetické chování takovýchto systémů lze popsat rovnicí:
![]()
kde k je buď rovnovážná konstanta nebo rychlostní koeficient, k0 je hodnota odpovídající nesubstituovanému systému. Reakční konstanta r je konstanta pro určitou reakci a pro určité reakční podmínky (teplota, rozpouštědlo) a je nezávislá na substituentu X. Konstanta substituentu s závisí pouze na subst. X a na jeho poloze v jádře (meta, para) a je nezávislá na R. Příkladem takovéto závislosti může být rychlost zmýdelnění esterů kys. benzoové.

Hammettova závislost zmýdelnění ethylesteru kyseliny benzoové.
Relativní rychlosti zmýdelnění substituovaných ethylesterů kys. benzoové při 30oC v 80% vodném ethanolu
|
Substituent X |
Relativní rychlost (k /k0) |
s |
Substituent X |
Relativní rychlost (k /k0) |
s |
|
H |
0 |
m-NO2 |
63 | ||
|
m-CH3 |
p-NO2 | ||||
|
p-CH3 |
m-Br |
8.1 | |||
|
m-NH2 |
p-Br |
4.9 | |||
|
p-NH2 |
m-Cl |
7.4 | |||
|
p-OCH3 |
p-Cl |
4.3 |
Hammettova rovnice je příkladem lineární závislosti Gibbsovy energie ( běžně nazývané „linear free energy relation“ - LFER). Vztahy mezi rovnovážnou konstantou resp. rychlostními koeficienty a Gibbsovou energií jsou dány vztahy :
log K = -DG0 /2.3 RT
log k = log(kBT/h) - DG /2.3 RT
Hammettovu rovnici lze tedy potom psát ve tvaru :
log(kx/k0) = log kx - log k0 = DDGx/2.3 RT = rsX
kde X, resp. 0 značí substituovaný, resp. nesubstituovaný systém. Výraz DDGx je rozdíl mezi DG pro reakci substituovaného a nesubstituovaného substrátu.
Jestliže porovnáme efekty dvou substituentů (A a B) ve shodné reakci za stejné teploty, dostaneme výraz
DDGA/2.3RT) / (DDGB/2.3RT) = rsA rsB
tedy
DDGA =( sA sB DDGB
což je lineární vztah mezi výrazy pro Gibbsovu energii.
Následující obrázek demonstruje schematicky význam Hammettovy rovnice.

Jsou zde zobrazeny dvě reakce, které jsou ovlivněny velmi rozdílnými substituenty. Reakce I je zpomalována substituentem B poněkud více než subst. A, její rovnovážná konstanta je snižována oběma substituenty. Substituenty mají mnohem výraznější vliv na rychlost reakce II, zvyšují velikost rovnovážné konstanty. Hammettova rovnice nám říká, že ačkoliv porovnáváme dvě velmi rozdílné reakce, tak poměr DDGA/ DDGB zůstává konstantní. Na obrázku je tento poměr cca. 0.5, tedy sB je cca 2 sA, a tento poměr platí pro všechny kinetické i termodynamické procesy bez ohledu na to, jsou-li tyto procesy substituenty zvýhodňovány nebo ne.
Rovnice DDGA =( sA sB DDGB říká, že můžeme stanovit poměr mezi dvěma substitučními konstantami, ale ne jejich absolutní hodnoty. Abychom to dokázali, musíme mít k dispozici nějakou vztažnou hodnotu (první volbou je výběr vodíku jako standardního substituentu). Hammett vybral disociaci substituovaných benzoových kyselin ve vodném roztoku při 25 0C a pro tuto rovnováhu definoval r rovnou jedné.
|
|
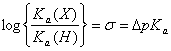
Hammettova s konstanta je tedy jednoduše rozdíl mezi pKa hodnotami benzoové a substituované benzoové kyseliny. Pro jakoukoliv jinou reakci lze patřičnou hodnotu r získat z grafického znázornění.
Disociace benzoových kyselin je usnadňována elektron-akceptory a zpomalována elektron-donory. Poněvadž silná kyselina má malou hodnotu pKa znamená to, že elektron-akceptorové substituenty mají positivní a elektron-donorní subst. negativní s hodnoty. Velikost s je potom mírou velikosti těchto efektů daného substituentu. Jestliže znaménka r a s jsou shodná, potom log(k/k0) je kladný a k je větší než k0. Kladná hodnota r indikuje, že elektron-akceptory reakci urychlují a elektron-donory zpomalují. Velikost r je mírou citlivosti reakce vůči změně substituentu. V hypotetické reakci znázorněné na obrázku (viz hypotetický reakční profil Hammettovy rovnice) je velikost r pro reakci II větší než pro reakci I a znaménko r pro rovnováhu reakce II je opačné než pro ostatní tři procesy. Hodnota r může být použita jako míra efektivity přenosu substitučního efektu různými skupinami. Velikost r je značně závislá na rozpouštědle. Např. pro disociace benzoových kyselin v ethanolu je její hodnota 1.96. Rozdíl této hodnoty oproti vodnému prostředí je zřejmě dán rozdílnou solvatací. Ethanol zřejmě není schopen tak efektivně stabilizovat benzoový aniont solvatací a substituent tedy hraje větší roli. Hodnoty r mohou být využity i při studiích mechanismů. Uvažujme např. zmýdelnění ethylbenzoátu.
|
|
Velká kladná hodnota r ukazuje, že reakce je značně urychlována elektron-akceptory. To znamená, že první krok je rychlost určující : elektron-akceptory usnadňují nukleofilní atak, ale brzdí odstoupení ethoxidu.
Nelineární Hammettovy rovnice
Pro některé reakce není graf závislosti log(kx/k0) na s lineární. To je indikací nesplnění některého z Hammettových předpokladů - buď je r ovlivněno substituentem nebo s reakcí. Mezi těmito případy není jednoduché rozhodnout, ale existují dva zcela zřejmé případy:
a) změna mechanismu - variace r
V dvoukrokové následné reakci o rychlosti rozhoduje pomalejší krok
reaktanty à intermediát à produkty
Naopak jestliže existují dvě alternativní reakční cesty, dominantní je ta, která je rychlejší a na ní závisí celková rychlost reakce.

V některé z těchto variant mechanismu může konkrétní substituent obrátit pořadí relativních rychlostí reakčních kroků a v důsledku tím vede k nelineární Hammettovské závislosti.
Např. tvorba semikarbazonu benzaldehydu je dvoustupňový proces
|
|
Druhý krok je kysele katalyzovaná dehydratace, v silně kyselém prostředí velmi rychlá a rychlost je určena prvním krokem. Ten zahrnuje nukleofilní atak na karbonylový uhlík a je usnadňován přítomností elektronakceptorních substutientů a r je cca 1. V neutrálním prostředí je krok 2 pomalejší a stává se limitujícím. Celková rychlost je nyní úměrná K1k2, přičemž obě veličiny mají velikostí přibližně stejný, ale smyslem opačný vliv na r a rychlost je téměř nezávislá na substituci a pozorovaná hodnota r je přibližně 0. V mírně kyselém prostředí (pH cca 4) pozorujeme přechod mezi těmito stavy. Elektron-akceptorní skupiny urychlují krok 1 a rychlost je dána krokem 2, elektrondonorní skupiny zpomalují krok 1 a ten se stává limitujícím. Výsledek má potom tvar:

Graf Hammettovy závislosti tvorby semikarbazonu benzaldehydu při pH=4
Jiný typ nelineárního chování je hydrolýza ethylbenzoátu v 99.9% k. sírové. Reakce probíhá přes protonovaný intermediát, který se rozpadá dvěma alternativními cestami:
|
|
Obecně se předpokládá, že ArCO+ je stabilnější než ethylový kationt a tím je preferována horní cesta. Ovšem v případě přítomnosti silných elektronakceptorů je acylový iont destabilizován do té míry, že převládne druhá cesta. Tento proces je skutečně podporován přítomností elektronakceptorů v Ar skupině, poněvadž to usnadňuje odštěpení ethylového kationtu.
Výsledný graf má potom tvar
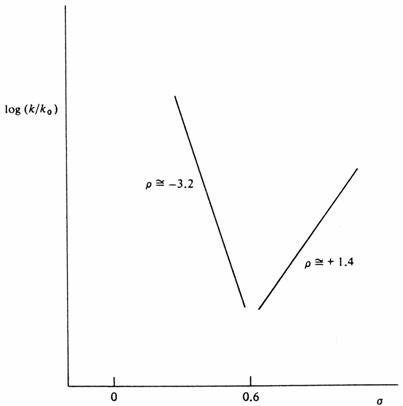
Hammettův graf hydrolýzy ethylesteru kyseliny benzoové v 99.9% H2SO4
b) vliv konjugace
Již v dobách Hammetta byly nalezeny reakce (některé reakce fenolů a anilinů), které vykazovaly pro některé substituenty značné odchylky od linearity. Např. v ionizaci anilinů byla nalezena značná odchylka pro NO2 skupinu. To není ani tak překvapující, poněvadž v případě p-nitroanilinu jistě existuje významná přímá mezomerní interakce mezi NH2 a NO2 skupinami. V případě aniliniového kationtu toto není možné – existuje v podstatě pouze repulzní interakce těchto substituentů a p-nitroanilin je podstatně slabší bází než anilin. Ovšem takovéto rozdílné mezomerní chování není možné v referenční reakci - ionizaci benzoových kyselin. To znamená, že existuje substituční efekt v jedné sérii, zatímco v druhé ne. Hammettův postulát o nezávislosti s je tedy zcela zřejmě porušen.
|
|
Aby byla zachována platnost Hammettovy rovnice i v těchto případech, bylo nutné odvodit speciální sérii parametrů s . Původně byla určena pro studia ionizací anilinů a fenolů, ale ukázala se jejich mnohem širší platnost - např. v reakcích aromatické nukleofilní substituce.
|
|
Analogicky byl vytvořen soubor parametrů s pro reakce, v níž konjugačně interagují elektron-deficitní reakční centrum s para rezonančním donorem - MeO, NH2.
|
|
Značné užití nacházejí tyto parametry v mechanistických studiích. Jestliže např. studovaná reakční série vykazuje lepší korelaci s hodnotami s nebo s ve srovnání s originálními hodnotami s, potom je velmi pravděpodobné, že tranzitní stav reakce vykazuje značnou konjugační interakci. Např. korelace se s hodnotami pro bazicky katalyzovanou dehydrobromaci arylethylbromidů ukazuje, že v tranzitním stavu je vazba C-H přerušena mnohem dříve než vazba C-Br a vzniklý benzylový uhlík nese podstatný náboj, který je citlivý na konjugaci s -M substituentem v para-poloze.
|
|
Je poněkud překvapující, že korelace se s s a s funguje tak dobře, jak funguje. Je totiž zřejmé, že musí existovat řada případů, které leží mezi extrémy . Byla proto definována řada jiných rovnic. Jednou z nich je rovnice Yukawa-Tsunova :
log (k/k0) = r s + r(s s
Myšlenkou je, že (s s) je mírou rozsahu, v jakém je substituent schopen rezonance a r, které se mění s reakcí stejně tak, jako r je mírou, jakou je tato rezonance vyžadována reakcí. Jestliže r = 0, potom je korelace jednoduše se s , je-li roven jedné, jde o korelaci se s . Bylo by možné očekávat, že r bude více či méně úměrné r, protože oba vyjadřují požadavky reakce na substituent. Toto bylo nalezeno pro řadu SEAr reakcí bifenylu.
|
|
Bifenylové kruhy nejsou normálně koplanární. Čím více reakce vyžaduje participaci p elektronů bifenylového systému, tím více se kruhy stáčí do jedné roviny. Výsledný graf je potom parabola, tak jak je předpovězeno Yukawa-Tsunovou rovnicí . Tato idea byla podpořena stejnou reakcí s fluorenovými deriváty, jejichž kruhy jsou koplanární. Výsledná závislost je potom lineární. Je nutno ovšem říci, že v některých případech aplikace Yukawa-Tsunovy rovnice ukazuje na praktickou nezávislost r a r. Je možno říci, že potom zavedení takovéhoto dalšího adjustovatelného parametru umožňuje zlepšení korelace a nemusí mít žádný fyzikální význam. Existuje několik pokusů o definici nové s škály, která by byla oproštěna od konjugačních vlivů. Mezi ně patří i Taftova s0 škála, kterou odvodil na základě reakcí pouze s meta-substituovanými fenyloctovými a 2-fenylpropionovými kyselinami a jejich ethylestery.
Polární a mezomerní efekty
I když je konjugace odseparována tak, jako v Taftově přístupu, přesto existuje řada mechanismů, kterými substituent může uplatňovat svůj vliv v reakci. Můžeme je rozdělit na dvě skupiny:
a) mezomerní efekt, tedy efekt, který zahrnuje přímý přenos náboje mezi kruhem a substituentem prostřednictvím p systému, včetně hyperkonjugace. Je tedy možné zobrazit jej patřičnými mezomerními vzorci.
b) polární efekty, tj. efekty, kde dipól vazby C-X modifikuje rozdělení náboje do zbytku molekuly. Polární efekty se obvykle rozdělují na dvě kategorie - induktivní efekt (I), který působí prostřednictvím sigma vazeb. Závisí na elektronegativitě atomu X, která generuje vznik parciálních nábojů v řetězci připojených uhlíkových atomů.
|
|
Tento efekt byl dlouho považován za dominantní a často se celkovým polárním efektem mínil induktivní efekt. Nyní už je ale zcela zřejmé, že druhý typ polárního efektu, tzv. efekt pole (F), je mnohem významnější. Tento efekt odpovídá přímé polarizaci reakčního místa dipólem substituentu přes prostor. Jasný důkaz této situace vyplývá z porovnání sloučenin, ve kterých jsou substituenty od reakčního místa vzdáleny o stejný počet vazeb (induktivní efekt tedy musí být stejný), ale liší se prostorovou orientací. Efekt pole je potom úměrný kosinu úhlu mezi reakčním místem a dipólem substituentu. Tato situace bývá popisována rovnicí E = q m cos Q e r2. Tento výraz udává energii interakce mezi reakčním místem s nábojem q a substituentem s elektrickým dipólovým momentem m e je dielektrická konstanta molekulární kavity.

2.1 Rozšíření Hammettovy rovnice
Polární efekty substituentů je možné studovat v systémech, které nemají možnost rezonance. Zvláštní důležitost v tomto směru mají 4-substituované bicyklo[2.2.2]oktankarboxylové kyseliny.
|
|
Jejich geometrie je velmi podobná parasubstituovaným benzoovým kyselinám, ale nemohou se v ní uplatňovat mezomerní interakce v důsledku absence p systému. Jejich pKa hodnoty byly použity k definici nové škály substitučních konstant s resp. sI. Velká pozornost byla věnována rozdělení těchto parametrů na meta a para řadu.
Pozorovaný efekt substituentů je součet polárního a mezomerního příspěvku. Pro meta a para polohy je tedy možno psát : sp sI sR sm sI a sR
Podobných snah byla učiněna celá řada s cílem stanovit především velikost mezomerní interakce substituentu s kruhem bez rezonančního příspěvku. Tento přístup ale má svoje kořeny již v samotné Hammettově rovnici. Substituent ovlivňuje reakci charakteristickým způsobem, který lze vyjádřit jediným parametrem s, bereme-li v úvahu polohy meta a para samostatně. Takto získané s konstanty jsou aplikovatelné ve všech reakcích s výjimkou těch, kde podstatnou roli hraje konjugace. Použitím uvedených rovnic je potom možná separace na polární a mezomerní příspěvky.
Víceparametrové rovnice
Alternativní přístup k řešení problému separace efektů je myšlenka, že různé reakce mohou různě odrážet polární a mezomerní efekty. Příklad konjugačního působení může být ukázkou takového přístupu. Výsledkem je rovnice
log(k/k0) = rIsI rRsR
ve které rI a rR jsou nezávislé reakční konstanty, které vyjadřují rozdílnou citlivost reakcí k rozdílným typům substitučního efektu. Nejsou tedy různé hodnoty s pro meta a para substituenty, rozdíly v reaktivitě meta a para isomerů jsou vyjádřeny rozdílným mechanismem přenosu I a R efektů ze substituentu na reakční centrum - tedy rozdílnými hodnotami r. Ve skutečnosti jsou tedy meta a para reakce brány, jako by patřily k rozdílným typům reakcí. Interpretace experimentálních dat podle uvedené rovnice není jednoduchá a vyžaduje nasazení vícenásobné regrese k určení nejlepších odhadů hodnot rI a rR. V nejužívanější verzi této metody - Taftově rovnici - jsou nejprve stanoveny hodnoty sI, stejně tak jako v minulých případech, měřením v systému bez rezonančních vlivů. Hodnoty rR není ovšem snadné stanovit. V praxi bylo užito řady přístupů. Taft analyzoval množství reakčních sérií statisticky a použil tohoto přístupu k získání nejlepšího odhadu sR . Nevýhodou tohoto přístupu je nutnost opakování tohoto postupu při získání nových dat, což může vést k novým hodnotám sR . Alternativou k tomuto postupu je použití nějakého referenčního systému - podobně jak Hammett zvolil disociaci k. benzoové - a použít ho ke stanovení sR . Taft použil hodnot 13C NMR posunů ke stanovení hodnot sR - tedy hodnot sR pro systémy bez konjugace. Získal rovnici
DdpC sI sR
kde DdpC je rozdíl chemických posunů para uhlíku monosubstituovaného benzenu a samotného benzenu.
Druhý způsob odvození hodnot sR použil Katritzky a Topsom a je založen na změně intensity infračerveného pásu (cca 1600 cm-1). Získané hodnoty jsou poněkud odlišné, nicméně poskytují shodnou interpretaci.
Interpretace polárních a mezomerních parametrů
Je nutno říci, že názory na relativní význam různých přístupů oddělujících polární a mezomerní efekt, se velmi různí. Příznivci Taftovy rovnice říkají, že jejich analýzy jsou většinou ve shodě s chováním ve většině reakcí a umožňují tedy realistickou interpretaci. Mají proto i řadu příkladů. Jedním z nich je (viz dále) rozpad diazoniových solí (viz kap. 13). Na druhé straně stejně početnou skupinu příznivců má i původní jednoduchá Hammettova myšlenka. Uznávají sice, že jednoparametrové rovnice nemohou dát odpovídající popis ve všech reakcích, ale poukazují na pozoruhodné množství, ve kterých je splněn. Argumentují tím, že zavedení dalších parametrů do rovnic vede sice většinou ke zlepšení korelace, ale reálný význam použitých parametrů je někdy diskutabilní a těžko obhajitelný z teoretického pohledu na původ substitučních efektů. Oba přístupy ovšem používají separace polárních a rezonančních parametrů v jedné či jiné formě k popisu kvalitativního chování substituentu. Např. substituenty jako Me2N, MeO a F jsou většinou polárními elektronakceptory (pozitivní sI ) a mezomerními donory (negativní sR). Jejich skutečný efekt závisí na relativním poměru těchto efektů a na hodnotě adjustovatelných parametrů, např. a v rovnicích sp sI sR sm sI a sR nebo jim podobných. MeO skupina např. je obvykle elektron-donorem pro para polohy a akceptorem z meta poloh. NO2 skupina je silným elektronakceptorem (sI a sR jsou kladné), ale dominantní mechanismus jejího působení je polární, především v důsledku značného kladného náboje na dusíku a mezomerní interakce, ačkoliv považované za významné, jsou méně důležité.
Značné množství protichůdných názorů existuje na polární a mezomerní (hyperkonjugační) efekt alkylových skupin. Mnoho let převládal názor, že elektrondonorové hyperkonjugační působení klesá v sérii methyl, ethyl, izopropyl, terc.butyl, jinými slovy, že C-H vazba je lepší p-donor než C-C vazba. I když se tento názor zdá být logický, ukázal se být nepravdivý. Ve skutečnosti existuje jen velmi malý rozdíl v p-donaci rozdílných alkylových skupin. Pozorovaný efekt je většinou důsledek sterických interakcí a solvatace.
Dewar-Grisdaleova rovnice
Hammettova rovnice a všechna její rozšířeni jsou empirická. Dewar a Grisdale se pokusili o neempirickou analýzu substitučního efektu. Opět rozdělují s parametr na vliv efektu pole a mezomerního efektu (F resp. M), ale pokoušejí se velikost jejich relativního příspěvku odvodit teoreticky. F-efekt pole působí především přes prostor a bude zřejmě závislý na 1/r, kde r je vzdálenost mezi místem připojení substituentu a místem reakce. M se mění v závislosti na možnosti nábojové delokalizace v systému. Ta je měřena jako náboj vzniklý v místě připojení pobočného řetězce nahrazením substituentu skupinou CH2+ nebo CH2- a je vypočitatelná z vlastností nevazebných molekulových orbitalů (viz dále). Tento přístup se ukázal být poměrně plodný v oblasti polycyklických aromátů. Umožnil stanovit F a M komponenty řady různých substitučních parametrů. Současné teoretické přístupy podporované rozvojem teoretické chemie i výpočetními možnostmi dávají tomuto přístupu široké pole působnosti.
Sterické efekty - Taftova rovnice
Pochopitelnou slabinou dosavadních korelačních rovnic je snaha o vyloučení sterických efektů. Patřičné konstanty jsou odvozeny většinou pro uspořádání analogická meta a para substituovaným benzenům. Přenos těchto substitučních konstant do jiných řad, především alifatických, přináší řadu potíží. Hlavní příčinou je skutečnost, že v alifatických systémech jsou si substituent a reakční místo většinou blíže než v aromátech. Tato skutečnost vedla Tafta ke snaze odvodit sérii odpovídajících substitučních konstant. Tato snaha byla pouze částečně úspěšná, především proto, že v ortho-polohách se uplatňuje velmi složitá rovnováha mezi efekty pole, tj. induktivním, mezomerním, sterickým a rozpouštědlovým. Proto tato snaha nevedla k nalezení šířeji použitelné škály so
Lineární závislost Gibbsovy energie a struktura tranzitního stavu
V kinetických korelacích reakční konstanta r odráží strukturu tranzitního stavu, nebo lépe řečeno rozdíl mezi základním a tranzitním stavem. Vystihuje skutečnost, v jakém rozsahu se mění náboj na reakčním centru a jakým způsobem na to odpovídá substituent. Skutečnost, že rovnice jako např. Hammettova jsou lineární, v sobě obsahuje také lineární závislost Gibbsovy energie. Z toho vyplývá, že r je skutečnou konstantou, charakterizující danou rovnici, a že struktura tranzitního stavu této reakce je nezávislá na substituentu. Tento závěr je však ve značném rozporu se známým Hammondovým postulátem. Ten nám zjednodušeně říká, že je-li reakce endotermní, tranzitní stav se více podobá produktům, je-li exotermní, tranzitní stav se více podobá reaktantům. To vede k závěru, že v sérii podobných reakcí struktura tranzitního stavu závisí na DH a tedy i na DG jako přijatelné aproximaci.
Vezměme jako příklad aromatickou elektrofilní substituci, která probíhá přes energeticky bohatý tranzitní stav TS1, doprovázený rozsáhlou nábojovou delokalizací, která je velmi citlivá na elektronické vlivy substituentů. Hodnota r této reakce je tedy velmi vysoká. Jiná reakce může mít nižší energii tranzitního stavu se strukturou bližší reaktantům a s menší delokalizací náboje méně ovlivňovanou substituentem a hodnota r je malá. Takovéto chování je obvykle nazýváno principem reaktivity a selektivity. Mimo jiné z ní vyplývá, že rychlé reakce mají tranzitní stav většinou podobnější reaktantům. Původní Hammondova myšlenka o zachování struktury tranzitního stavu je často interpretována tak, že malé změny v reaktantech, které zvyšují jejich reaktivitu, vedou ke snížení selektivity a obráceně. Tento závěr je také nazýván jako Hammond-Lefflerův postulát. Zde ovšem existuje určitý problém. Analýzy Hammettova typu jsou užívány pro zkoumání charakteru tranzitního stavu (vyjádřeného pomocí r pomocí variace substituentů a tedy variací reaktivity Hammond říká, že nevyhnutelným důsledkem změny rychlosti je změna charakteru tranzitního stavu. Tato diskuse ovšem souvisí i s diskusí o možném přechodu mezi dvěma různými mechanismy. Uvažujeme např. přechod mechanismu SN1 na SN2 . Otázkou je, co je lepším vystižením - zdali jediný mechanismus s postupně se měnícím tranzitním stavem, nebo dva rozdílné mechanismy, každý charakterizovaný svým tranzitním stavem a uplatňující se paralelně. Pro oba názory najdeme řadu experimentálních důkazů a problém je dosud nedořešen.

Ilustrace Hammondova postulátu. Ve třech příbuzných reakčních krocích, které se liší např. charakterem elektrofilu, se tranzitní stavy TS1, TS2 a TS3 stávají podobnější reaktantům se zmenšujícími se aktivačními entalpiemi
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1765
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved