| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
DOCUMENTE SIMILARE |
|
TERMENI importanti pentru acest document |
|
Metody molekulárních orbitalů
Perturbační teorie
Interakce dvou orbitalů
Popis molekulárních orbitalů v termínech interakce atomových orbitalů je nazýván perturbační teorie. Představme si, že interagují dva atomové orbitaly - v průběhu tohoto děje se navzájem perturbují a vytvářejí dva molekulární orbitaly, jeden s vyšší energií než kterýkoli z atomových orbitalů a druhý analogicky s nižší energií. Rozsah vzájemné interakce může být popsán třemi parametry. Coulombický integrál a je energie izolovaného atomového orbitalu. Překryvový integrál S je mírou rozsahu překryvu mezi interagujícími atomovými orbitaly a rezonanční nebo vazebný integrál b vystihuje energii této interakce. Energie jsou konvenčně vztahovány k energii elektronu v nekonečnu - a i b jsou záporné. Pomocí těchto parametrů je energie E molekulárního orbitalu tvořeného atomovými orbitaly A a B dána:
aA - E)(aB - E) = (b - SE)2
Jestliže jsou atomové orbitaly degenerované, (např. vodíkové 1s orbitaly při tvorbě molekulového orbitalu molekuly vodíku) platí aA aB a
a - E) = ± (b - SE)
tudíž E = (a + b)/(1 + S) nebo (a- b)/(1- S)
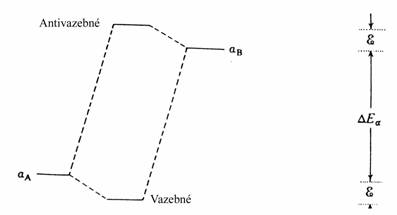
Tato situace je zobrazena na následujícím obrázku (část a)
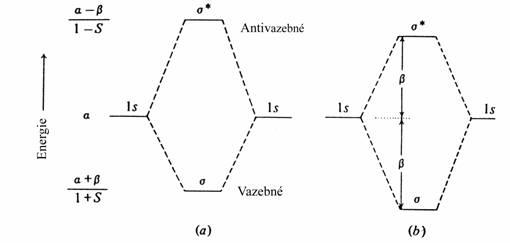
Často se používá zjednodušení spočívající v zanedbání překryvu (S = 0) . To vede k následující rovnici a části b předcházejícího obrázku
E = a ± b
Překryv způsobuje asymetrii v rozštěpení orbitalu. Tedy ačkoliv molekula vodíku je stabilní s oběma elektrony v s orbitalu, interakce dvou heliových atomů, která naplní oba s i s* orbitaly není tak energeticky neutrální, jak by se mohlo jevit z dosavadního výkladu, ale je ve skutečnosti repulzní. Na dalším obrázku je ukázána interakce dvou orbitalů, jejichž energie aA a aB jsou velmi různé.
První aproximace (aB - E) pro vazebný molekulový orbital může být nahrazena výrazem (aB aA a podobně pro antivazebný orbital (aA - E) může být nahrazena výrazem (aA - aB). Jako další zjednodušení můžeme zanedbat S, což je poněkud méně dramatická aproximace než předcházející, protože překryv mezi velmi různými orbitaly je pravděpodobně velmi malý. Rovnice tedy mohou být přepsány do tvaru:
aA - E)(aB aA b2
aA aB aB - E) = b2
Energetický rozdíl e je potom ve vztahu ke vzdálenosti mezi atomovými orbitaly dán výrazem:
e b2/DEa
Interakce mezi degenerovanými orbitaly, která v prvé aproximaci může být vyjádřena jako b, je nazývána poruchou prvního řádu. Analogická interakce mezi velmi rozdílnými orbitaly závisí na b a je nazývána poruchou druhého řádu. Samozřejmě existuje řada přechodných stavů, kde rozštěpení závisí na b komplikovanějším způsobem. Vlnová funkce molekulového orbitalu Y může být vyjádřena jako kombinace atomových orbitalů y podle rovnice:
Y = cA yA + cB yB (vazebný)
Y = cA* yA - cB* yB (antivazebný)
kde koeficienty c jsou mírou příspěvku každého atomového orbitalu k molekulovému orbitalu. Pro normalizované orbitaly (Y2 =1 ) platí cA2 + cB2 = cA*2 + cB*2 = cA2 + cA*2 = cB2 cB*2 = 1. Jestliže jsou atomové orbitaly identické, potom všechny koeficienty musí být shodné a rovny 1/ 2. Jestliže jsou atomové orbitaly velmi různé, potom vazebný molekulový orbital je podobný atomovému orbitalu A s nižší energií a nevazebný molekulový orbital je podobný atomovému orbitalu B s vyšší energií.
Lokalizované dvoucentrické vazby
Přejít od prostého popisu interakce dvouatomových orbitalů k popisu interakcí multielektronových a multiorbitálních systémů je velmi komplikované. Naštěstí pro kvalitativní účely organické chemie mohou být popisy nasycených a nekonjugovaných systémů popsány podobně jednoduše. Skutečnost, že takovéto sloučeniny se téměř ve všech aspektech chovají jako souhrn atomů držených pohromadě lokalizovanými vazbami - vazbami s jasně definovanými vlastnostmi - délka, síla, polarita, polarizabilita, vibrační frekvence a pod., je podstatou toho, že tyto vlastnosti můžeme aplikovat z jedné molekuly na druhou. Např. enthalpie atomizace alkanů může být přesně vyjádřena jako suma příspěvku energií C-C a C-H vazeb, dipólové momenty mohou být vypočteny vektorovým součtem vazebných momentů. Právě toto chování je základní myšlenkou koncepce lokalizovaných, dvoucentrových molekulových orbitalů, tvořených hybridizovanými atomovými orbitaly každého z atomů. Takovýto popis není zcela pravdivý, ale je adekvátní pro většinu chemických účelů a je snadno představitelný a zobrazitelný. Ve své jednoduché formě někdy selhává, např. v předpovědi existence dvou různých typů vazebných molekulových orbitalů v methanu, jak vyplývá z měření fotoelektronovou spektroskopií a jak vyplývá i z exaktnějších delokalizovaných modelů.
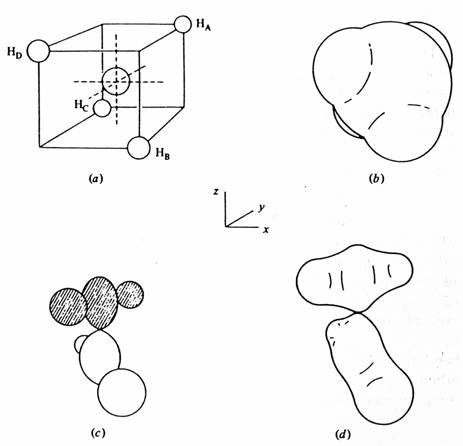
Popis vazeb v methanu pomocí delokalizovaných orbitalů. Část (a) popisuje geometrické relace mezi tetraedrickou symetrií molekuly methanu a kartézským systémem. Sférický 2s orbital uhlíku interaguje ve fázi se všemi čtyřmi 1s orbitaly vodíku za vzniku delokalizovaných vazebných orbitalů (b) s tetraedrickou symetrií. Uhlíkový 2p orbital interaguje s vodíkovým 1s orbitalem, jak je ukázáno v části (c), kde jedna část 2p orbitalu interaguje s HA a HD a druhá část s HB a HC. Tím vzniká delokalizovaný molekulový orbital (d) s vyšší energií než (b) s nodální rovinou procházející uhlíkovým atomem. Další dva podobné orbitaly jsou tvořeny analogickou interakcí orbitalů 2px a 2py.
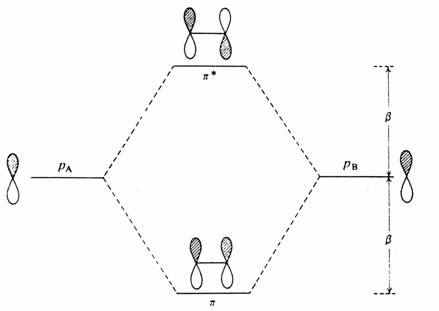
Na druhé straně zobrazení delokalizovaného přístupu nekoresponduje s chemicky přijatelnou koncepcí diskrétní vazby C -H a je dost obtížné ho rozšířit i na větší molekuly - vzhled delokalizovaných molekulových orbitalů se velmi rychle stává komplexnější a zahrnuje interakci mnoha molekulových orbitalů. Nekonjugované p vazby mohou být také považovány za izolované, vzniklé interakcí dvou nehybridizovaných p orbitalů nezávisle na s skeletu a ležících v rovině kolmé k němu. Jejich vzájemná perturbace vede v prvé aproximaci ke vzniku vazebného a antivazebného p orbitalu s energií E = a ± b
Konjugace a delokalizace
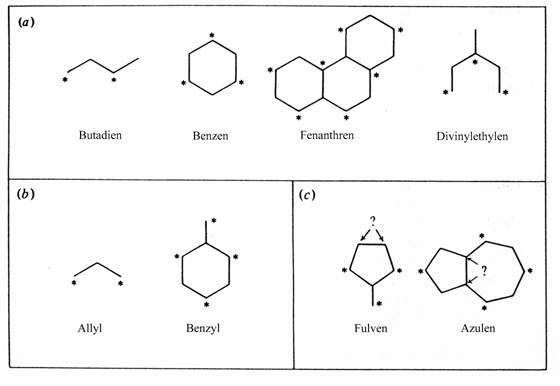
V konjugovaných systémech, kde tři nebo více sousedních atomů interaguje p vazbami, musíme uvažovat rozsáhlejší překryv p orbitalů. Např. butadienový p systém může být zobrazen jako hypotetická konstrukce ze dvou molekul ethylenu a efekt delokalizace může být odhadnut na základě vzájemné perturbace dvou lokalizovaných p systémů . Existují ovšem dva zásadní rozdíly mezi molekulární perturbací a meziatomovou perturbací. Atomové orbitaly jsou zcela lokalizovány na atomu, naproti tomu ethylenové p orbitaly jsou sdíleny dvěma uhlíky, ale jenom jeden z nich se přímo účastní perturbace - překryv je tedy neúplný.
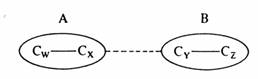
Hypotetická konstrukce butadienu ze dvou ethylenových fragmentů A a B spojených atomy x a y.
Rozsah, jakým se každý orbital zapojí, je dán koeficienty cx a cy, které jsou mírou příspěvku separovaných atomových p orbitalů do celkového p molekulárního orbitalu. V případě butadienu je cx = cy = 1/ 2. Musíme ovšem vzít v úvahu, že v každém ethylenovém fragmentu existují dva orbitaly p a p a že musíme uvažovat všechny čtyři vzájemné perturbace. Interakce dvou vazebných orbitalů v prvním přiblížení poskytuje molekulární orbitaly p a p
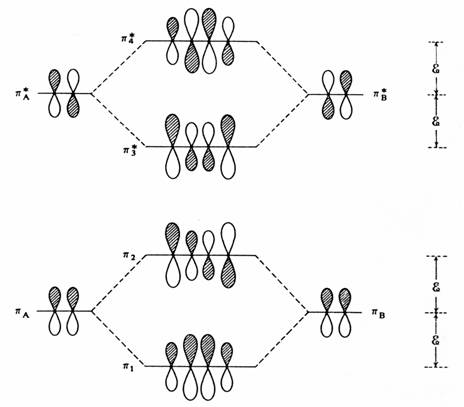
Perturbace prvého řádu p orbitalů ethylenu při konstrukci butadienového p systému
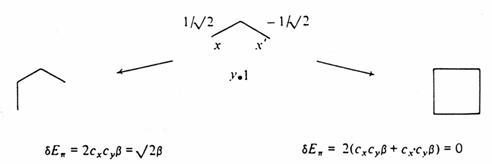
Odpovídající interakce nevazebných orbitalů dává orbitaly p a p . Energetická změna doprovázející tuto interakci je rovna E = a ± cx cy b . Celková energie čtyř vazebných elektronů není změněna - snížení energie p je kompenzováno zvýšením energie p . I když vezmeme v úvahu překryv, je takováto perturbace prvního řádu repulzní. Interakce mezi vazebným a nevazebným orbitalem je druhého řádu. Konvenčně je uvažována jako následná perturbace molekulových orbitalů vzniklých perturbací prvního řádu .
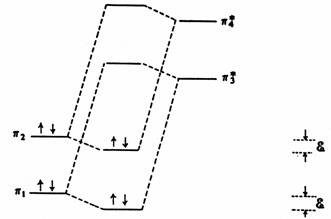
Porucha druhého řádu mezi vazebnými a antivazebnými orbitaly při konstrukci butadienového p systému.
Orbitaly opačné symetrie nemohou vzájemně interagovat. Např. jestliže p interaguje s p , potom překryv C1 a C3 je přesně vykompenzován překryvem C2 a C4 . Avšak p a p , které jsou symetrické vzhledem ke středu molekuly, se mohou vzájemně mísit a podobně tak p a p , které jsou antisymetrické vzhledem ke středu molekuly. Velikost této poruchy druhého řádu je rovna e = (cx cy b DE, a poněvadž DE v tomto případě je rovno 2 b (viz obr.“ Vzájemná perturbace dvou atomových p orbitalů“), e je pouze b/8. Ovšem na rozdíl od perturbace prvního řádu, tato snižuje energii všech vazebných elektronů - delokalizovaný čtyřcentrový p-systém je tedy stabilnější než izolovaná p vazba o 4 e b/2. Není snadné vypočítat koeficienty c, které jsou mírou příspěvku každého atomového p orbitalu k delokalizovanému molekulovému orbitalu. Pro nerozvětvené lineární polyeny s n uhlíkovými atomy pro koeficienty platí
cij = 1/2 sin , kde i je pořadí molekulového orbitalu a j je pozice uhlíkového atomu v řetězci. Pro případ butadienu vznikají tedy tyto molekulové orbitaly:
Y y y y y
Y = 0.60 y y 0.37 y y
Y = 0.60 y y 0.37 y y
Y = 0.37 y 0.60 y y y
Zmiňme se o dvou významných vlastnostech těchto orbitalů:
Pro každý molekulový orbital Y je suma c2 přes všechny přispívající orbitaly rovna jedné a pro každý individuální atomový orbital y je analogická suma přes všechny molekulové orbitaly, do nichž přispívá, opět rovna jedné. Toto je typická vlastnost tzv. normalizovaných orbitalů.
Orbitaly jsou uspořádány v párech a koeficienty uvnitř daného páru se liší pouze znaménky na alternujících atomech. To je obecná vlastnost tzv. alternujících uhlovodíků.
Alternující uhlovodíky
2.1 Párování orbitalů
Při konstrukci p systému butadienu ze dvou lokalizovaných p vazeb zůstává orbitalová struktura symetrická vzhledem k energetické hladině p orbitalu. Podobný výsledek najdeme, jestliže ze dvou butadienů vytvoříme oktatetraen, anebo jestliže butadien interaguje oběma svými konci s ethylenovým fragmentem za vzniku p systému benzenu. V obou těchto případech pracujeme se dvěma symetricky rozloženými soubory orbitalů a jejich vzájemná perturbace je rovněž symetrická. Pro každý vazebný orbital s energií a - e existuje antivazebný orbital s energií a+ e, kde a je energie původního p orbitalu. Podobný obrázek dostaneme, jestliže bude interagovat samotný p orbital A s ethylenovým fragmentem B za vzniku tříčlenného allylového p systému.
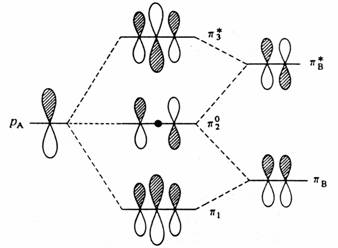
Interakce (porucha) mezi samotným p orbitalem a orbitaly molekuly ethylenu při konstrukci allylového p systému.
Vezmeme-li v úvahu symetrické požadavky, vidíme, že pA a pB interagují za vzniku vazebného orbitalu p a pA a pB interagují za vzniku antivazebného p orbitalu. Třetí molekulární orbital můžeme získat jen kombinací všech tří přispívajících orbitalů (pA- pB) + (pA + pB ), ve kterém se vazebný a antivazebný příspěvek ruší. Výsledkem je nevazebný p orbital, jehož vlnová funkce (znaménko) se mění na centrálním uhlíkovém atomu
- dochází ke změně nodálních vlastností vlnové funkce. Symetrický vzhled je zachován - vazebné a antivazebné orbitaly jsou v párech. Jediná výjimka se vyskytuje u kruhů s lichým počtem atomů. Následující obrázek ukazuje interakci p orbitalu s ethylenovým fragmentem za vzniku cyklopropenylu.
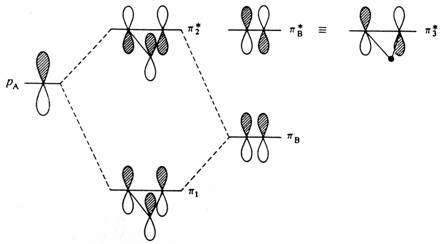
Interakce samostatného p orbitalu s orbitaly
ethylenu při konstrukci cyklopropenylového p systému.
Interakce nyní zahrnuje oba konce ethylenu a pA může interagovat s pB za vzniku buď vazebného p nebo antivazebného p orbitalu. Neexistuje ovšem způsob, jak pA může
interagovat s pB - interakce na jednom konci je rušena interakcí na konci druhém. Třetí molekulový orbital p je ve skutečnosti ekvivalentní původnímu pB - cyklopropenylový p
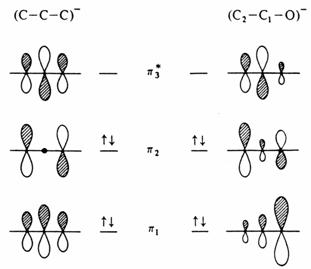
systém má tedy jeden silně vazebný orbital s energií E = a b a dva méně silně antivazebné orbitaly s energií E = a b p systémy, které nemají kruhy s lichým počtem
atomů, se nazývají alternující, protože konjugované atomy se mohou rozdělit na dvě skupiny - konvenčně se značí jedna skupina hvězdičkou.
p systémy konjugovaných uhlovodíků: a) alternující sudý, b) alternující lichý, c) nealternující
Vlastnosti alternujících uhlovodíků
Alternující systémy jsou velice důležité, protože jejich struktura a reaktivita je snadno vypočitatelná.
Elektronová distribuce sudých alternujících uhlovodíků
Pro každý atomový orbital je S c2 přes všechny molekulové orbitaly jednotková. Z toho, že molekulové orbitaly jsou uspořádány symetricky ve dvojici - antivazebný a vazebný - lišící se pouze znaménkem koeficientu, vyplývá, že S c2 přes vazebné orbitaly je rovna 0.5. Každý vazebný orbital obsahuje dva elektrony a tedy celková p elektronová hustota na každém atomu je 2 S c2 = 1 - existuje tedy pravidelná distribuce p elektronů přes celý konjugovaný systém.
Elektronová distribuce lichých alternujících uhlovodíků
Klíč k chování alternujících lichých uhlovodíků leží ve vlastnosti nevazebného molekulového orbitalu, jehož koeficient c0 lze nalézt podle následujícího postupu:
Označme hvězdičkou větší skupinu alternujících pozic
Pro neoznačené pozice považujme koeficienty za nulové
Uvažujme sumu koeficientů všech ohvězdičkovaných atomů spojených s jakýmkoliv neoznačeným atomem za nulovou
Použijme normalizační podmínku pro celý orbital S c02 = 1
Ilustrujme si použití těchto pravidel na příkladu benzylu:
|
|
1. Ohvězdičkujme 4 pozice a 3 ponechme neoznačené
|
|
Neoznačené pozice mají nulové koeficienty
|
|
Jednoduchý výpočet poskytuje relativní hodnoty koeficientů na označených pozicích. Začněme libovolně s hodnotou x pro C4, pak C2 musí být - x, aby suma kolem pozice 3 byla nulová. Proveďme to stejným způsobem pro celou molekulu. U větších systémů můžeme použít další náhodně zvolené hodnoty. V konečném důsledku budeme mít vždy dost rovnic, abychom získali koeficienty jako funkce jediné
proměnné x.
|
|
S c02 = 7x2 = 1, x = 1/ (7)1/2 Poznamenejme, že ačkoliv relativní znaménka c jsou významná, jejich absolutní hodnoty ( + či -) nikoliv.
Rovnice pro nevazebný MO má tedy tvar
Y y2 y4 y y
Neutrální benzylový radikál má jeden elektron v nevazebném molekulovém orbitalu a celková p elektronová hustota je jednotková na každém uhlíkovém atomu. Ionizace benzylového radikálu způsobuje odtržení elektronu a zanechává pozitivní „díru“ na jeho místě. Náboj je pak rozprostřen po systému podle hodnoty c02 na každém atomu. Stejně tak pro případ benzylového aniontu je nevazebný orbital opět ten, který přijme elektron
a rozdělení přijatého náboje je opět řízeno c02.
Delokalizační energie a aromaticita
Energetickou změnu způsobenou vzájemnou perturbací dvou p systémů sudých alternujících uhlovodíků (např. butadien ze dvou ethylenových fragmentů) není jednoduché vypočítat. Efekty prvního řádu, zahrnující plně obsazené vazebné orbitaly, se vzájemně anulují a interakční energie pochází z relativně malého poruchového příspěvku druhého řádu mezi vazebnými a antivazebnými orbitaly. V prvním přiblížení je velikost tohoto efektu druhého řádu pro acyklické sloučeniny nezávislá na aktuální velikosti sudého alternujícícho uhlovodíku, poněvadž jej můžeme budovat postupným přidáváním ethylenového fragmentu, a celková energie se pokaždé mění o stejnou hodnotu. Acyklické polyeny se tedy chovají jako lokalizované systémy a to se projevuje např. v alternujících délkách vazeb. Dvojná vazba je potom přibližně stejné délky jako dvojná vazba v ethylenu, ale jednoduchá vazba je poněkud kratší než v ethanu (cca 148 pm místo 154 pm). To je důsledek p příspěvku druhého řádu. Podobně jako v případě alkanů, použití modelu lokalizovaných vazeb neznamená, že tyto jsou v reálném systému skutečně lokalizovány. Orbitální struktura butadienu zřetelně ukazuje, že delokalizace je významná, ale její efekt na celkovou energii je malý.
Interagují-li spolu dva liché alternující uhlovodíkové fragmenty, potom dominujícím příspěvkem je interakce prvního řádu mezi jejich nevazebnými molekulovými orbitaly, které jsou degenerované. Celková změna p energie je potom dána výrazem
dEp e = 2cx cy b,
kde cx a cy jsou koeficienty nevazebného molekulového orbitalu pro atomy x a y, mezi nimiž existuje interakce. Spojením dvou allylových fragmentů může vzniknout buď hexatrien, anebo mohou uzavřít kruh a vytvořit benzen. Poněvadž poruchové příspěvky prvního řádu jsou aditivní, můžeme aplikovat uvedenou rovnici dvakrát, abychom vypočetli hodnotu dEp pro tvorbu benzenu, ze které vyplývá, že benzen je stabilnější než acyklický trien - stabilnější znamená jinými slovy, že získaná hodnota je různá od hodnoty vypočtené z tabelovaných vazebných energií. Říkáme, že benzen je aromatický.
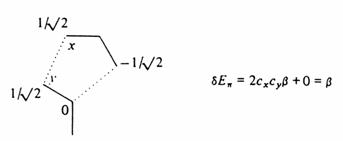
Na následujícím obrázku je ukázána odpovídající interakce allylu s p orbitalem, který můžeme považovat za limitní případ lichých alternujících uhlovodíků.V tomto případě je uzavření kruhu energeticky neutrální, protože cxcy a cx´cy se ruší. Delokalizovaný cyklobutadien je nejen nearomatický, ale je ve skutečnosti antiaromatický, méně stabilní než jeho otevřený analog, anebo než takový, jak to předpovídají výpočty z tabelovaných vazebných energií.
Konstrukce p systémů butadienu a cyklobutadienu z allylového fragmentu a samostatného p orbitalu.
Rozšíření na nealternující uhlovodíky
Metoda popsaná v předcházejícím odstavci může být použita k odhadu delokalizační energie sudých nealternujících uhlovodíků. Obrázek ukazuje tvorbu fulvenu ze dvou allylových zbytků. Výpočet ukazuje, že není stabilnější než otevřený trien, tj. je nearomatický. Na druhé straně azulen, ačkoliv je méně stabilní než naftalen, je ještě pořád aromatický.
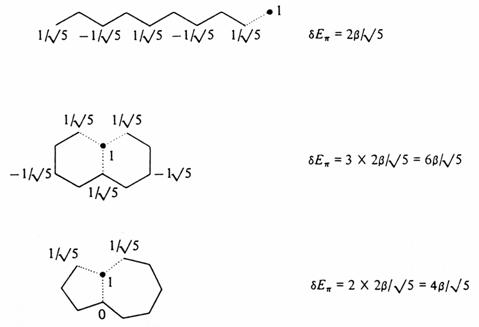
Konstrukce p systémů dekapentaenu, naftalenu a azulenu.
Systémy s heteroatomy
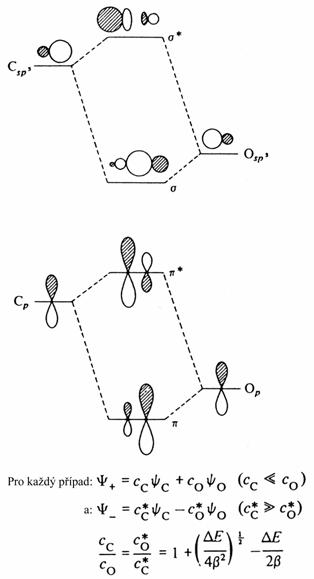
Jestliže
interagují dva energeticky velmi rozdílné orbitaly, potom výsledný vazebný
orbital připomíná původní orbital s nižší energií a antivazebný orbital
připomíná původní orbital s vyšší energií. To se odráží jak
v hodnotách koeficientů molekulových orbitalů, tak i v elektronové
distribuci. Následující obrázek to ilustruje pro případ lokalizovaných C=O s
a p vazeb.
Polarizace vazby uhlík – kyslík. Vazebné elektrony jsou koncentrovány v blízkosti kyslíkového atomu.
Stejné principy platí i pro delokalizované orbitaly, ale efekt záměny uhlíkového atomu je komplexnější a výpočty koeficientů jsou již doménou metod kvantové chemie. Vztah p systému enolátového aniontu vzhledem k allylovému aniontu je ukázán na obrázku:
Porovnání p orbitalů allylu a enolátového aniontu.
Porovnáním s obrázkem interakce p orbitalu s ethylenem (viz dříve) je možno očekávat, že kyslík bude hrát hlavní roli v nejnižším molekulovém orbitalu, menší v orbitalu p a nevýznamnou v p . Kyslík tedy nese největší část negativního náboje. Ovšem největší příspěvek v orbitalu p pochází z p orbitalu uhlíku C2 s velmi zajímavými důsledky (viz dále).
Poruchová teorie a reaktivita
Hraniční molekulové orbitaly a coulombické efekty
Dalším typem systému, který nelze adekvátně popsat v termínech lokalizovaných dvoucentrických vazeb, jsou částečně vázané struktury tranzitních stavů. Např. v SN2 reakci je centrální uhlíkový atom přibližně sp2 hybridizován a zbývající p orbital interaguje jak s nukleofilem, tak s odstupující skupinou. Přesné řešení těchto otázek je věcí metod kvantové chemie a určení reakčních rychlostí a průběhu reakce je velmi specializovanou otázkou. Poruchová teorie nabízí velmi jednoduchou nevýpočetní cestu k získání pohledu na reaktivitu, zvláště na reaktivitu dvou podobných systémů nebo na dvě konkurenční cesty reakce stejného substrátu. V minulém výkladu jsme uvažovali tvorbu molekulových orbitalů pomocí hypotetické interakce atomových orbitalů nebo molekulových orbitalů menších fragmentů. Stejný přístup můžeme aplikovat při interakci dvou reaktantů. Otázka zní, jakým způsobem se změní energie systému, jestliže orbitaly začnou navzájem interagovat. Obecně řečeno, není možné tyto změny sledovat až do tranzitního stavu, protože ten většinou zahrnuje značnou změnu pozic jader a je tedy zřejmě něčím víc než jen elektronovou poruchou. Můžeme jej ovšem aplikovat na začátek reakce. Ten je většinou indikátorem, jak reakce bude pokračovat, pokud ovšem nejsou přehlédnuty některé intervenující faktory - např. sterické. Reakční koordináty dvou podobných reakcí mají tendenci se velmi podobat. Pokud by tomu tak nebylo, bylo by těžké vysvětlit, proč jsou lineární závislosti Gibbsovy energie reakcí tak běžné. Poruchová teorie aplikovaná na chemickou reaktivitu tedy říká, že počáteční perturbace řídí průběh následných reakčních kroků. Jestliže dva reaktanty spolu interagují, potom celkovou změnu energie lze rozdělit do tří příspěvků:
Interakce mezi plně obsazenými orbitaly
V prvním přiblížení se ruší. Jestliže vezmeme do úvahy překryv, potom výsledný efekt bude repulze. A tato repulze je hlavním příspěvkem k aktivační energii reakce. V porovnání relativních reaktivit jsou rozdíly v repulzních termech obvykle malé a v další diskusi budou zanedbány. Toto zanedbání je sice velmi těžce obhajitelné, ale v prvním přiblížení dává užitečné výsledky.
Interakce mezi obsazeným orbitalem reaktantu a neobsazeným orbitalem druhého reaktantu
Obecně řečeno, takovéto interagující orbitaly mají značně rozdílnou energii a je to tedy typický příklad poruchy druhého řádu. Je to stabilizující interakce, protože energie obsazeného orbitalu je snížena, a odpovídající zvýšení energie ostatních orbitalů nemá význam, poněvadž jsou prázdné. Stabilizace získaná touto interakcí je 2e, protože zahrnuje dva elektrony. To v principu není přesné, protože bychom měli uvažovat interakce mezi všemi obsazenými orbitaly reaktantu A a všemi neobsazenými orbitaly reaktantu B se stejnou symetrií a obráceně. Z kvalitativního hlediska je ale zřejmé, že největší příspěvek k energii poruchového příspěvku druhého řádu budou mít interakce mezi obsazenými a neobsazenými orbitaly ležícími co nejblíže u sebe. Proto pro zcela kvalitativní přístup k reaktivitě můžeme
naše úvahy omezit pouze na jeden soubor interakcí - interakce mezi tzv. hraničními molekulovými orbitaly.
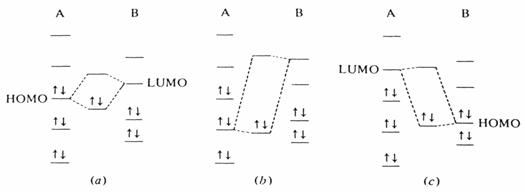
Molekula A má relativně vysokou energii HOMO a molekula B relativně nízkou energii LUMO. Interakce mezi těmito orbitaly je velká -viz obr. Pro interakci druhého níže ležícího obsazeného orbitalu látky A s druhým výše ležícím neobsazeným orbitalem látky B je hodnota DE mnohem větší a interakce je malá. Na třetí části obrázku je interakce mezi HOMO látky B a LUMO látky A, ale ta je také z energetického pohledu nevýznamná. V této reakci je A zřejmě nukleofilem, který elektrofilu B dodává elektrony ze svého HOMO orbitalu . Samozřejmě, že tato diskuse se nemusí týkat jen HOMO orbitalu, ale elektron může pocházet i ze SOMO orbitalu radikálových reaktantů.
Elektrostatické interakce
V reakcích zahrnujících ionty nebo dipolární molekuly existuje coulombická atrakce nebo repulze mezi interagujícími atomy. Její velikost je úměrná výrazu Qx Qy/r, kde Qx a Qy jsou náboje interagujících atomů a r jejich vzdálenost.
Porovnání reaktivit podobných systémů může být učiněno na základě dvou kriterií: rozhodující HOMO - LUMO interakce a příspěvku od elektrostatické atrakce nebo repulze. Mohou být sumarizovány v rovnici
P = 2(cx cyb DEFMO - QxQy/r
kde P je snížení energie při interakci reaktantů a DEFMO je rozdíl energií HOMO nukleofilu - LUMO elektrofilu. Uvažujme např. alternativní cesty reakce enolátového aniontu s elektrofilem
![]()
![]()
Atak na kyslíku se zdá být řízen nábojově, zatímco atak na uhlíku je řízen orbitalově. Kyslík nese největší část negativního náboje, ale HOMO, který řídí kovalentní term, má nejvýznamnější příspěvek od uhlíkového p orbitalu. Tedy proton (pozitivně nabitý, malý tedy i s malým r a s relativně vysoko položeným LUMO) reaguje nejrychleji na kyslíku. Na druhé straně brom (nenabitý, velký a s nízko ležícím neobsazeným orbitalem) reaguje mnohem rychleji na uhlíku.
Měkké a tvrdé kyseliny a báze
Lewisova definice zobecňuje definici Brønstedovy a Lowryho báze jako donoru elektonového páru a kyseliny jako akceptoru elektronového páru. V případě bází jsou tyto dvě definice prakticky shodné, ale Lewisovy kyseliny zahrnují širokou škálu jiných částic, než jsou proton-donory - kovové kationty, karbeniové ionty a jiné elektrondeficitní sloučeniny, jako jsou BF3 a CH2 a sloučeniny s nízko ležícím neobsazeným d orbitalem, jako jsou vyšší halogeny. Jednou z komplikací Lewisovy definice je nemožnost kvantifikace - neexistuje totiž univerzální škála síly Lewisových kyselin analogická pKA škále protonových kyselin. Již řadu let bylo zřejmé, že interakce kov-ligand se dají rozdělit na dvě zcela rozdílné kategorie. Amoniak, voda a fluoridový ion silně komplexují kationty alkalických kovů a alkalických zemin, ale jen velmi slabě komplexují těžší přechodné kovy jako Hg 2+ nebo Pt2+ . Opačná situace je u komplexů s fosfiny, sulfidy nebo těžšími ionty halogenů. Jinými slovy Hg2+ je slabší Lewisovou kyselinou než Mg2+, jestliže je voda referenční bází a naopak silnější, jestliže je referenční bází sirovodík. Pokus o racionalizaci této situace je založen na konceptu měkkých a tvrdých kyselin a bází. Měkké jsou většinou velké, polarizovatelné, s relativně malým nábojem, tvrdé jsou malé, s malou polarizabilitou a obvykle nabité částice. Princip měkkých a tvrdých částic zároveň říká, že tvrdé kyseliny preferují reakce s tvrdými bázemi, jak kineticky tak termodynamicky a podobně měkké kyseliny preferují reakce s měkkými bázemi. Rozdělení do těchto kategorií bylo původně čistě empirické, ale jeho racionalizace je možná na základě poruchové teorie. Měkká - měkká interakce je zřejmě z velké míry kovalentní. Měkké báze mají nízkou elektronegativitu, jejich elektronové páry jsou tedy snadno přístupné (vysoká energie HOMO); měkké kyseliny mají nízko ležící neobsazený orbital (nízká energie LUMO). Vzájemná interakce těchto orbitalů je tedy značná a interakce je preferována. Interakce typu tvrdá - tvrdá je z velké míry elektrostatická, mezi ionty nebo velmi silně dipolárními molekulami; ty jsou obvykle malé a mohou tedy interagovat velmi těsně, s velmi značnou coulombickou atrakcí. Podobně zvýšení náboje obvykle zvyšuje tvrdost: Fe3+ je tvrdší než Fe2+. Tvrdé kyseliny mají obvykle vysoko položený LUMO orbital a tvrdé báze mají nízko ležící HOMO. Interakce tvrdá - měkká je slabá: hraniční orbitaly tvrdé komponenty jsou buďto příliš vysoko nebo příliš nízko pro kovalentní interakce a velikost a nízká polarita měkké komponenty minimalizuje možnost elektrostatických interakcí. Tuto terminologii můžeme rozšířit i do oblasti organické chemie, kde pojmy kyselina a báze můžeme nahradit pojmy elektrofil a nukleofil. Např. ethylen je slabým nukleofilem, protože nemá náboj a má značně vysokou energii p orbitalů. Reaguje tedy rychleji s bromem než s vodnou kyselinou.
Steroelektronické efekty
V reakcích řízených orbitalovou interakcí musí být hraniční orbitaly schopny se efektivně překrývat, aby reakce mohla proběhnout. Běžné příklady jsou SN2 reakce nukleofilů řízené požadavkem, aby HOMO orbital nuklefilu interagoval se s LUMO orbitalem substrátu. Podobně preferovaná stereochemie eliminačních reakcí, kde obě odcházející skupiny leží ve stejné rovině, je dána překryvem mezi hraničními orbitaly, které tvoří nově vznikající p vazbu.
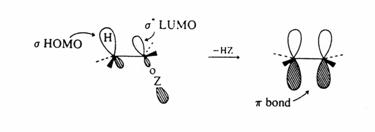
V případě
cyklických reakcí může přítomnost kruhu být překážkou efektivního překryvu interagujících orbitalů. Reakce tedy
může být zpomalena nebo může probíhat jiným alternativním mechanismem. Např.
v acyklických systémech vede příznivá interakce mezi volným elektronovým
párem kyslíku (HOMO ) alkoholu a p (LUMO) orbitalu na b
uhlíku
esteru kyseliny metakrylové k preferenci Michaelovy adice před
trans-esterifikací. Právě možnost
zaujmout konformace s efektivním překryvem je rozhodující pro průběh
reakcí. V následující intramolekulární reakci je produktem lakton a
nikoliv intramolekulární Michaelovský adukt, protože HOMO hydroxylu nemůže
přímo interagovat s LUMO koncové methylenové skupiny.
|
|
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 2148
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved