| CATEGORII DOCUMENTE |
| Astronomie | Biofizica | Biologie | Botanica | Carti | Chimie | Copii |
| Educatie civica | Fabule ghicitori | Fizica | Gramatica | Joc | Literatura romana | Logica |
| Matematica | Poezii | Psihologie psihiatrie | Sociologie |
Originea si aparitia prionilor. Proteina neuronala PrPc
Atat in creierul de hamster infectat experimental cu mojarat de creier de oaie cu maladie prionica scrapie, cat si in creierul pacientilor umani cu maladii prionice s-au identificat prin metode fizice, doua izoforme de proteine prionice.
Proteina prionica neuronala normala (PrPc) este sintetizata in neuronii si in celulele gliale ale SNC, in leucocite si in alte tesuturi in cantitati limitate, la nivelul reticulului endoplasmic, este modificata in aparatul Golgi si transportata la suprafata neuronilor, unde este "ancorata" de membrana celulara printr-o "ancora" glicolipidica (glicozil-fosfatidil-inozitol) si nu are domeniu citoplasmatic. De la acest nivel, PrPc este endocitata si probabil recirculata, fiind final degradata de proteaze, ca parte a metabolismului celular. PrPc este nepatogena si netransmisibila. Structura spatiala este de tip α-helix, iar functia, necunoscuta.
Proteina PrPc umana contine 253 aminoacizi (254 la soarece si 264 la bovine). Rolul ei este incert. Localizarea la suprafata celulei neuronale ar putea sa reflecte rolul in aderenta celulara, in legarea unui ligand sau o posibila functie de semnalizare. Partea N-terminala contine o secventa de 5 octapeptide repetate, bogate in glicina si prolina, care in vitro, leaga Cu si Zn. In vivo, PrPc ar putea avea rol in transportul si metabolismul Cu.
Cu pare sa aiba un rol esential in protectia SNC fata de stresul oxidativ si fata de maladiile degenerative.
*Cu este un oligoelement esential pentru viata. Are doua grade de oxidare: +1 si +2. Cu2+ este mai stabil. Forta oxidanta a cuplului Cu+/Cu2+ este utilizata in diferite reactii de oxidoreducere, catalizate de enzime dependente de Cu. {n exces, Cu este toxic.
Cu poate reactiona cu H2O2 pentru a genera radicalul HO., un radical liber foarte reactiv, care produce leziuni importante ale macromoleculelor (ADN, proteine, lipide).
Dereglarea metabolismului Cu are ca rezultat cresterea ratei de producere a radicalilor liberi.
SNC este foarte sensibil la leziunile cauzate de radicalii liberi, pentru ca este un tesut sarac in enzime antioxidante (SOD Cu/Zn, catalaza si peroxidaze) si bogat in substraturi usor oxidabile (acizi grasi polinesaturati).
Perturbarea echilibrului Cu genereaza un stres oxidant care determina moartea neuronilor.
Neurodegenerescenta asociata maladiei Alzheimer rezulta din depunerea placilor de amiloid in neuroni, in care s-a gasit un procent important de Cu. Precursorul proteinei amiloid (PPA)contine mai multe situsuri de fixare a Cu. S-a demonstrat ca PPA reduce Cu2+ la Cu+.
In scleroza laterala amiotrofica familiala (SLAF) - o maladie neurodegenerativa caracterizata prin degenerarea neuronilor motori, s-a identificat o gena anormala ca fiind codificatoare a SOD Cu/Zn.
Chelarea experimentala a Cu cu cuprizon induce leziuni de tip spongiform, care regreseaza dupa oprirea administrarii chelatorului. Maladia epuizarii cronice (chronic wasting disease), o maladie prionica a cervidelor, are incidenta crescuta in regiunile in care solul este sarac in Cu.
In SNC, PrPc si ARNm sunt larg distribuite, dar se gasesc in special in neocortex, hipocampus, celule Purkinje si in neuronii motori spinali.
In neuroni PrPc este transportata prin axon, spre terminatia nervoasa si a fost localizata la nivelul sinapselor prin metoda imunoelectron-microscopiei.
PrPc este reciclata intre membrana plasmatica si compartimentul endocitar.
A II-a forma este proteina prionica patologica (PrPsc).
Prionii au 2 origini: endogena si exogena (infectioasa).
Originiea exogena a prionilor a fost demonstrata experimental de Bassler (l986), prin inocularea intracerebrala a hamsterilor cu mojarat de creier de oaie, cu boala scrapie. Proteina patologica PrPsc endogena ar putea avea doua proveniente:
- prin reciclarea proteinei celulare, proces in care se face conversia PrPc la PrPsc ;
- prin mutatia punctiforma a genei Prn-p, codificatoare a proteinei normale.
Mutatiile punctiforme, extrem de rare la nivelul genei Prn-p, au drept consecinta formarea unei proteine prionice modificate, denumita dupa numele bolii la care a fost studiata, PrP scrapie sau PrPsc. Denumirea a fost pastrata pentru agentii patogeni (prioni) ai tuturor bolilor din aceasta familie, deoarece mecanismul a fost demonstrat ca fiind comun, inclusiv in cazul maladiilor prionice umane.
Studiul secventei aminoacizilor la cele doua tipuri de proteine a demonstrat prezenta a peste l8 tipuri de mutatii punctiforme. Cele mai frecvente sunt cele care determina inlocuirea aminoacidului numarul 66 (glicocol din PrPc, cu prolina in PrPsc, sau a aminoacidului l02 (leucina) din PrPc, cu prolina in PrPsc.
Prusiner si Hsiao (l988) au stabilit existenta unei relatii directe intre mutatii si maladie, evidentiind prezenta acestora in structura PrPsc la toti pacientii cu maladii prionice.
Exista o relatie directa intre proteina normala si cea patologica. Proteina Prpsc, mutanta sau de origine infectioasa, actioneaza asupra echivalentului sau celular normal PrPc, in sensul ca, venind in contact cu aceasta, determina deplierea ei de la forma normala de α-helice si replierea ei intr-o conformatie noua, de tip β-pliere. In timpul infectiei prionice are loc o interactiune fizica inalt specifica intre PrPc si PrPsc si se produce conversia conformationala a proteinei, care face PrPsc rezistenta la degradarea cu proteaze si ii confera caracterul infectios si patogen.
Conversia PrPc in PrPsc este conformationala. Cele doua forme au aceiasi secventa de aminoacizi. Schimbarea conformatiei presupune o crestere cantitativa substantiala a structurii β-pliere, cu o usoara descrestere a structurii α-helix. PrPc are circa 42% structura α-helix si 3%, β-pliere, iar PrPsc are 30% structura α-helix si 43%, β- pliere.
"Replicarea" prionilor s-a studiat in vivo, prin inocularea experimentala a animalelor sensibile cu dilutii foarte mari de omogenat de creier infectat cu scrapie, sau de la bolnavi cu maladia Creutzfeldt-Jakob, in creierul acestora se acumuleaza mari cantitati de prioni (pana la l08 DL5o unitati infectioase). In vitro, numai unele linii celulare neuronale sunt sensibile la infectia cu prioni scrapie.
Mecanismul formarii PrPsc poate fi diferit pentru formele infectioase si pentru cele genetice. In primul caz, PrPsc exogen are rolul de catalizator pentru conversia PrPc endogen, la starea PrPsc. Prionii de provenienta exogena (prin infectie), interactioneaza cu moleculele normale de proteina prionica celulara (PrPc), pe masura ce acestea sunt sintetizate, imprimandu-le propria lor conformatie speciala(tip β-pliere), transformandu-le in proteine prionice (PrPsc). Fiind rezistente la degradarea enzimatica, proteinele prionice se acumuleaza in neuroni, determinand vacuolizarea si necroza lor. Prionii eliberati pe aceasta cale, sunt adsorbiti pe neuronii normali adiacenti, determinand extinderea procesului patologic. Locul neuronilor lizati este luat de celulele gliale.
In al II-lea caz, PrPc mutant(rezultat prin mutatii punctiforme in gena Prn-p) este transformat spontan la PrPsc.
Transmiterea prionilor. Prionii reprezinta forma modificata a unei proteine celulare normale (PrPc). Prusiner (l986, l993) a demonstrat originea exogena a proteinei prionice prin transmiterea experimentala a maladiilor prionice. Pasajul prionilor intre organismele diferitelor specii este un proces intamplator, cu o perioada lunga de incubatie (de ordinul anilor). Prin pasaje succesive la indivizi ai aceiasi specii, timpul de incubatie se scurteaza progresiv de la un pasaj la altul si propagarea devine predictibila. Bariera de specie nu functioneaza (sau nu functioneaza totdeauna) si are importanta practica pentru evaluarea riscului ca omul sa dezvolte maladia Creutzfeldt-Jakob, dupa consumul creierului de oaie infectata cu scrapie sau de bovine infectate cu prionul encefalopatiei spongiforme.
Dupa inoculare, perioada de incubare este de 1-3 ani. Odata declansate, bolile prionice au o evolutie clinica cu agravare progresiva de-a lungul mai multor luni sau ani, care duce invariabil la moarte. Experientele pe oi si capre, prin inoculare intracerebrala, orala, subcutanata, intramusculara sau intravenoasa, au aratat existenta gradelor diferite de sensibilitate a diferitelor rase de animale, mai ales dupa inocularea subcutanata, ceea ce sugereaza ca fondul genetic influenteaza decisiv sensibilitatea la infectia prionica. Adeseori, multe dintre animalele inoculate, nu dezvolta maladia.
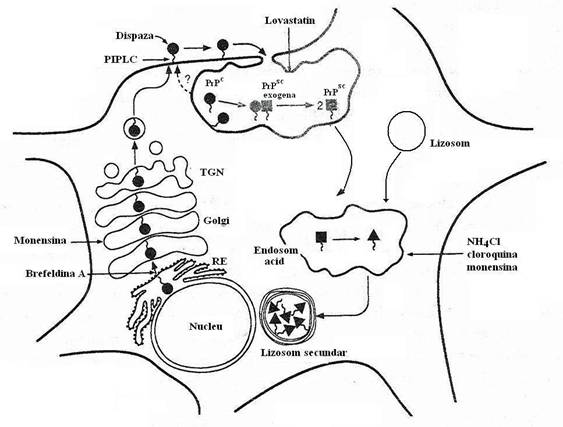
Fig 107. Caile sintezei si degradarii proteinei prionice in celulele cultivate. Patratele simbolizeaza PrPsc (proteina scrapie), iar cercurile semnifica PrPc(proteina prionica). PrPc, precurorul PrPsc, este sensibila la digestia cu dispaza sau la PIPLC adaugate in mediu. PrPc este endocitata, in conditii normale fiind recirculata si ulterior degradata de proteaze, ca parte a metabolismului celular. Intr-un proces infectios, are loc interactiunea PrPc cu proteina PrPsc exogena (infectioasa), in cursul careia PrPc este convertita la proteina patologica, fara implicarea reticulului endoplasmic si a cisternelor Golgi. In celulele cultivate, dar nu in creier, capatul N-terminal al PrPsc este clivat ulterior, in endosomul acid, cu formarea PrP 27-30 (triunghi) si acumularea acesteia in lizosomul secundar
(dupa Prussiner, 1996).
Patogenitate. Maladiile prionice pot fi: infectioase, familiale si sporadice.
Bolile prionice sunt limitate la SNC si evolueaza cu leziuni degenerative tipice pentru encefalitele spongiforme. Ele pot sa apara spontan in populatie, fara nici o cauza aparenta (formele sporadice) sau au caracter ereditar-familial. In ambele tipuri de maladii, prionii sunt infectiosi si pot fi transmisi prin inoculare la organisme ale aceleiasi specii sau ale unei specii inrudite.
Prionii produc o varietate de boli neurologice degenerative, care afecteaza ovinele si caprinele (boala scrapie), bovinele (encefalopatia spongiforma sau boala "vacii nebune"), felinele, cervidele, nurcile etc., precum si omul.
Leziunile produse de prioni sunt limitate la nivelul sistemului nervos central si includ vacuolizarea si necroza neuronala, degenerarea spongiforma a tesutului cerebral, reactii astrogliale cu hipertrofia astrocitelor si proliferarea lor in cortex. Datorita focarelor multiple de liza neuronala, apar "gauri" care confera creierului un aspect spongios. La unele organisme se observa depunerea unor placi de amiloid (colorabile cu rosu de Congo), in sistemul nervos central. Ele ar putea fi formate din proteine prionice sau din agregate de prioni.
Maladia scrapie (tramblanta)apare in populatii de ovine si caprine, cu predispozitie genetica, dar faptul ca se poate propaga prin inoculare, respinge ideia caracterului pur genetic. Boala se instaleaza dupa 2-5 ani de la contaminare, cu tulburari neurologice: prurit intens, dificultate in mers (prin hipoplazia cerebelului), cu agravare progresiva, pana la imobilitate totala, cu sfarsit letal. Principalele leziuni apar insa in creier, care degenereaza lent.
Infectia naturala se face pe cale digestiva cu proteine prionice din ingredientele de origine animala si initial evolueaza la nivelul sistemului limfoid, de unde se disemineaza in organism, pe calea nervilor, a fibrelor din nevrax si pe cale sanguina. Aparitia maladiei trebuie urmata de sacrificarea intregului efectiv de animale.
Maladiile prionice umane sunt exogene (natura infectioasa) sau endogene, adica sunt mostenite prin predispozitie genetica, iar altele au caracter sporadic (apar fara cauze cunoscute). Formele sporadice nu au etiologie infectioasa sau genetica si se pot atribui conversiei spontane a PrPc la starea PrPsc sau existentei unei mutatii somatice nedetectate in proteina, ce favorizeaza conversia la PrPsc.
Formele infectioase ale maladiei prionice sunt rezultatul transmiterii orizontale a prionilor infectiosi. Cele mai cunoscute sunt boala "kuru" si boala Creutzfeldt-Jacoob.
Boala "kuru"* afecteaza exclusiv populatia tribului Fore din Papua-Noua Guinee si este caracterizata prin lipsa coordonarii motorii, accentuata progresiv, astfel incat bolnavii necesita sprijin atat asezati, cat si in pozitie verticala. Maladia se transmite pe cale digestiva, datorita canibalismului ritual (cinstirea inaintasilor prin consumul creierului cadavrelor).
*Termenul "kuru" reflecta principalele simptome ale bolii si este tradus prin "a tremura de frica, " a se infiora" sau "moartea zambind".
Boala Creutzfeldt-Jacob (CJD) are o frecventa de l la un milion si apare in jurul varstei de 60 de ani, cu miscari anormale, rigiditate, tulburari psihice de tip demential. Poate aparea sporadic (caracter ereditar-familial) sau prin infectie exogena, dupa unele practici medicale (transplant de cornee sau de duramater, injectii de hormoni hipofizari, in special a celor de crestere, obtinuti din hipofiza umana recoltata de la cadavre, dupa implantarea electrozilor contaminati pentru EEG sau prin operatii chirurgicale cu instrumente contaminate).
Sindromul Gerstmann-Straussler-Scheinker (GSS) este o varietate naturala a bolii Creutzfeldt-Jacob, caracterizata prin instalarea precoce a incapacitatii de coordonare a miscarilor, prin leziuni cerebeloase si dementa.
Insomnia familiala letala (IFL) debuteaza cu tulburari ale somnului si ale sistemului nevos vegetativ, urmate de dementa.
Descoperirea mutatiilor in genele PrnP la bolnavii cu CJD, GSS si IFL, a dus la concluzia ca bolile prionice sunt atat genetice cat si infectioase.
Semnificatie biologica. Existenta prionilor (proteine infectioase, replicabile pe o cale neobisnuita), a sugerat posibilitatea transmiterii unor maladii infectioase in absenta unui transfer de material genetic codificator al sintezei proteice, ceea ce este in contradictie cu dogmele biologiei moleculare, ale virologiei si microbiologiei.
Ideea ca o proteina este capabila de autoreplicare printr-o interactiune proteina-proteina, in absenta informatiei genetice si a circulatiei ei in celula, dupa modelul clasic este greu de acceptat.
Chesebro si Fields (l996) considera ca este posibil ca PrP (proteina rezistenta la proteaze) sa aiba un rol important in patogeneza maladiei, fara sa fie agentul cauzal. Ea ar functiona ca un receptor sau ca un cofactor necesar pentru agentul cauzal sau pentru un virus ubicvitar, cu mare rezistenta la inactivare.
Modificarea structurala a PrPc la PrPsc ar putea fi rezultatul actiunii unei proteine necunoscute, cu rol de chaperonina (Guillemet si col, l996). Chaperonii sunt proteine care faciliteaza plierea polipeptidelor in timpul biosintezei si transportului in organite si impiedica agregarea proteinelor in conditiile stressului celular (ca de exemplu, socul termic). Chaperonii actioneaza prin legarea de substratul lor, uneori dependenta de ATP si impiedica formarea intermediarilor pliati, nefunctionali. Formarea PrPsc implica modificarea plierii si agregarea moleculelor, procese in care chaperonii au rol important.
Este posibil ca prezenta prionilor sa fie mult mai larga in natura, si oricum, nelimitata strict la mamifere.
Dupa Prusiner (l995), este posibil ca mai multe boli nervoase cronice (dementa presenila umana de tip Alzheimer, maladia Parkinson, scleroza multipla, scleroza laterala amiotrofica etc.), cu etiologie necunoscuta, sa fie produse sau influentate in evolutie, de interventia unor agenti patogeni subvirali, de tipul prionilor.
Bolile prionice exemplifica un mecanism de patogenitate bazata pe schimbarea conformatiei unei proteine care se autopropaga. Probabil ca transmiterea informatiei biologice pe calea conformatiei proteinei este un fenomen mai general in natura. La levuri si la fungii filamentosi s-au identificat proteine care se comporta ca prioni.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1966
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved