| CATEGORII DOCUMENTE |
| Astronomie | Biofizica | Biologie | Botanica | Carti | Chimie | Copii |
| Educatie civica | Fabule ghicitori | Fizica | Gramatica | Joc | Literatura romana | Logica |
| Matematica | Poezii | Psihologie psihiatrie | Sociologie |
Studiul solutiilor de electroliti constituie o preocupare centrala din domeniul cercetarilor termodinamicii, ca urmare a numarului mare de procese care cuprind ioni in solutie.
Geneza domeniului este situata inainte de inceputul secolului XX, iar contributiile de pionierat la termodinamica solutiilor binare diluate, au fost concluzionate mai tarziu in cateva legi simple, la care s-au adaugat si unele consideratii suplimentare, specifice numai solutiilor de electroliti.
Dupa fundamentarea unui material faptic primar, obtinut prin experimente simple asupra limitelor de aplicare ale legilor solutiilor perfecte, cercetarile s-au putut extinde si asupra sistemelor neideale, in urma introducerii unor functii termodinamice corective ale concentratiilor, stabilite de Lewis si Randall. La interval de peste un deceniu acestea au fost completate de Raymond si Scatchard, prin alt tip de formulare, cu substrat diferit.
Implicarea solutiilor de electroliti in procesele chimice si biochimice precum si in operatii industriale, din care industria medicamentelor constituie o parte foarte importanta, a constituit un factor stimulativ in dezvoltarea cercetarilor de termodinamica fenomenologica a sistemelor reale, ca suport necesar in interpretarea si orientarea unor procese tehnologice.
In afara metodelor consacrate obtinerii functiilor traditionale de abatere de la idealitate (activitati termodinamice, coeficientii de activitate, functiile termodinamice de exces), s-au aplicat si alte modalitati de evidentiere a neidealitatilor prin prelucrarea unor marimi neconventionale, corelabile cu cele uzuale.
In literatura domeniului se remarca o lipsa acuta de date experimentale, de calitate, necesare caracterizarii termodinamice a solutiilor neideale. Chiar si
in solutiile electrolitilor singulari si cu atat mai mult in sistemele multi-componente, situatia devine critica, in deosebi la concentratii care depasesc valorile de 1,5 3 molal.
Rezultatele aplicarii premizelor modelului lui K. S. Pitzer si a colaboratorilor sai, (transpuse in forma operationala pentru calculul principalelor marimi de abatere de la idealitate, au confirmat valorile coeficientilor de activitate ionici medii si ai celor ionici individuali, a coeficientilor osmotici proprii solventului din solutie precum si a functiilor termodinamice de exces, stabiliti din date experimentale. Volumul imens de calcule se poate realiza prin programe de calcul adaptate particularitatilor fiecarei categorii de functii termodinamice.
In ultimii 10-15 ani s-a accentuat tendinta de amplificare a caracterizarii solutiilor apoase de electroliti printr-o varietate de modele noi, prelucrate statistic.
Aceasta orientare este atestata in publicatiile unor congrese internationale organizate de institutii de prestigiu, ca A.I.Ch.E, (Institutul American de Inginerie Chimica), N.S.F. (Fundatia Nationala de Stiinta), N.B.S. (Biroul National de Standarde). Colectiile de lucrari, ale unor asemenea manifestari stiintifice reprezinta documente fundamentale relevand nu numai liniile directoare ale cercetarilor din acest domeniu dar si perspectivele lor de dezvoltare.
2 Marimi si ecuatii termodinamice implicate in valorificarea datelor experimentale
Starea ionica a solutiilor de electroliti are implicatii bogate in caracterizarea sistemelor ce contin ioni in solutii. Datele primare, fundamentale, descoperite de Arrhenius, asupra disociatiei electrolitice si confirmate de lucrarile reprezentative ale epocii, ca cele ale lui van't Hoff, Brnstedt, van Laar, s.a., au demonstrat experimental, existenta unor abateri ale solutiilor de electroliti de la legile simple, cu caracter coligativ, stabilite pentru solutiile ideale. Prelucrarea cantitativa a acestor abateri, a fost realizata, in decursul timpului, de contributiile lui G. N. Lewis si M. Randall, autorii notiunilor de fugacitate si activitate termodinamica, urmati de cercetatori de prestigiu care le-au continuat opera.
Tratarea unitara si riguroasa a solutiilor de electroliti s-a realizat prin teorii statistice confirmate prin rezultatele experimentelor, valorificate prin diferite sisteme.
2.1 Functii termodinamice in solutii de electroliti
Complexitatea fenomenelor dintr-o solutie de electroliti se poate evidentia prin contributiile efectelor - de natura diferita - ale ionilor din solutie, regasite global, in formularea si valorificarea marimilor termodinamice ale sistemelor.
Din punct de vedere termodinamic, solutiile de electroliti sunt privite ca amestecuri nesimetrice in raport cu componentii sai: electrolitul (electrolitii) dizolvat (dizolvati) si solventul. De exemplu, in cazul energiei libere Gibbs a unei solutii, concura urmatoarele efecte reprezentate de:
* fortele interionice de tip Debye - Hckel ( DH ), Gel ;
* interactiile ion - solvent, Gis si
* interactiile ionice de ordin superior celor de tip DH, GES.
Suma acestor energii libere, responsabile de neidealitatea solutiilor de electroliti, corecteaza marimea ipotetica Gid. Energia libera ideala Gid are un corespondent valabil numai in absenta oricaror interactii ionice in solutie (in cazul unor dilutii extreme ale solutiilor de electroliti).
Urmeaza astfel:
![]() (1)
(1)
Avand in vedere ca interdependentele ion-ion si ion-solvent pot fi cuprinse in GES , rezulta:
![]() (2)
(2)
Cum in general G = Gid + GE, reiese din ultimele doua relatii:
![]() (3)
(3)
In cazul solutilor binare de electroliti tari, Glueckaauf prevede corectia la Gid ca suma dintre Gel (energia electrostatica) si un termen entropic:
![]() (4)
(4)
Pentru un sistem binar (format din
n1 moli de solvent si n2 moli de electrolit) potentialele chimice ale
solventului, ![]() si al dizolvatului,
si al dizolvatului, ![]() ,
au forma cunoscuta:
,
au forma cunoscuta:
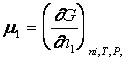 ;
;  (4a,b)
(4a,b)
Forma explicita a potentialelor chimice, in functie de un potential standard si o concentratie corectata (activitatea termodinamica a componentului in cauza) este stabilita cu rigoare in expunerile de termodinamica chimica.
Exprimarea corecta a acestor marimi impune alegerea potentialului chimic standard, adaptat alegerii concentratiei de lucru.
Adesea apar confuzii in stabilirea standardului sau tratari necon-cludente si chiar ambigue, ceea ce justifica o scurta expunere asupra starii standard conventionale.
Stari standard conventionale
Potentialele chimice ale unui
component din orice sistem real sunt definite pana la o constanta aditiva,
arbitrara. In calculele numerice o astfel de constanta se exprima prin potentialul standard, ca: ![]() ,
de tip activitate termodinamica sau
,
de tip activitate termodinamica sau ![]() , de tip fugacitate. Ambele notatii releva standardizari ale compozitiei, in primul caz, prin
, de tip fugacitate. Ambele notatii releva standardizari ale compozitiei, in primul caz, prin ![]() ,
iar in cazul al doilea o standardizare a compozitiei si a presiunii, prin notatia
,
iar in cazul al doilea o standardizare a compozitiei si a presiunii, prin notatia ![]() .
.
In solutii se opereaza cu potentiale standard de tip activitate, care difera intre ele in functie de modalitatile de exprimare ale concentratiilor. Potentialul chimic al unui anumit corp ( fie "i" ) dintr-o solutie neideala data este acelasi la T, P stabilite, dar daca compozitiile se exprima diferit se modifica scala de compozitii si in compesatie se modifica si potentialele standard respective.
In general,
![]() (5)
(5)
Pentru a mentine aceeasi valoare a
potentialului chimic, ![]() - pentru acelasi component in acelasi sistem -
chiar daca se schimba modul de exprimare al compozitiei,
- pentru acelasi component in acelasi sistem -
chiar daca se schimba modul de exprimare al compozitiei, ![]() este termenul care se adapteaza
schimbarii. Conventional, pentru
compozitia in fractii molare,
este termenul care se adapteaza
schimbarii. Conventional, pentru
compozitia in fractii molare, ![]() se noteaza
se noteaza ![]() ; in
molalitati,
; in
molalitati, ![]() , iar in concentratii molare,
, iar in concentratii molare, ![]() . Semnificatia standardului este conditionata
si de conventiile pentru coeficientii de activitate, dictate de natura
componentilor din solutii. Conventia
simetrica convine solutiilor formate din corpuri miscibile in orice proportie
(de exemplu amestecuri lichide) care se trateaza la fel. Oricare ar fi
componentul din solutie, (fie i ) daca
. Semnificatia standardului este conditionata
si de conventiile pentru coeficientii de activitate, dictate de natura
componentilor din solutii. Conventia
simetrica convine solutiilor formate din corpuri miscibile in orice proportie
(de exemplu amestecuri lichide) care se trateaza la fel. Oricare ar fi
componentul din solutie, (fie i ) daca ![]() si
si ![]() , iar
, iar ![]() . Normarea coeficientului de activitate, (
. Normarea coeficientului de activitate, (![]() ),
duce la identitatea
),
duce la identitatea ![]() cand se releva ca sistem de referinta solutia perfecta.
cand se releva ca sistem de referinta solutia perfecta.
Cazul solutiilor de electroliti nu
se poate trata decat printr-o conventie nesimetrica, determinata de
diferenta neta dintre particularitatile
componentilor sai: solventul in cantitate predominanta si electrolitii cu
solubilitate limitata. Ca urmare in stabilirea standardelor se are in vedere ca fractia molara a solventului poate lua valori chiar
pana la ![]() ,
dar fractiile molare ale dizolvatilor se mentin intr-un domeniu restrans.
,
dar fractiile molare ale dizolvatilor se mentin intr-un domeniu restrans.
Pentru solvent,
![]() (6)
(6)
cand se
reproduce cazul relevat prin conventia 1 pentru starea standard (![]() si
si ![]() ),
iar pentru dizolvati (i ¹
),
iar pentru dizolvati (i ¹
![]() (6a)
(6a)
standardizarea
este consecinta diminuarii fractiei molare a dizolvatului i la va-loarea minima ![]() ,
cu normarea
,
cu normarea ![]() ,
cand
,
cand ![]() .
.
Se evidentiaza astfel, prin standardizarile diferite ale dizolvatilor fata de solvent, sistemul de referinta nesimetric este reprezentat de solutia ideala diluata.
Pentru solutiile de electroliti vom opera in mod necesar cu conventia nesimetrica si preferential pe scala molalitatilor.
Pentru electrolitii din solutie (i ¹ 1) potentialul chimic ia forma:
![]() (7)
(7)
sau in functie de activitatea termodinamica:
![]() (7a)
(7a)
unde :
![]() (T,P) reprezinta potentialul chimic standard, cand
(T,P) reprezinta potentialul chimic standard, cand ![]() si
si
![]() , referinta fiind astfel o solutie ipotetica ideala diluata.
, referinta fiind astfel o solutie ipotetica ideala diluata.
Marimi molale partiale in corelatie cu coeficientii de activitate ai componentilor din solutii apoase de electroliti
Marimile molale partiale ale unui electrolit dintr- o solutie se obtin prin metode de calcul fundamentate in termodinamica solutiilor neideale ale solutiilor de neelectroliti, cu adaptarile impuse de structura ionica a solutiilor .
Lucrand consecvent pe scala molalitatilor, cu coeficienti de activitate molali medii, g , si contand pe cunoscutele corelatii dintre potentialul chimic al unui component dintr-o solutie de electrolit - vezi ecuatia (7) - cu alte marimi termodinamice, se obtin urmatoarele ecuatii pentru cele mai uzuale marimi molale partiale, pe scala molalitatilor:
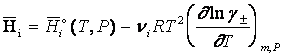 (8)
(8)
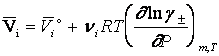 (9)
(9)
 (10)
(10)
![]() (11)
(11)
precum si ![]() , adaptat acestor formulari:
, adaptat acestor formulari:
![]() (12)
(12)
La dilutie infinita, marimile molare partiale devin identice cu marimile standard corespunzatoare. Solutiile de electroliti sunt neutre electric; daca electrolitul disociaza in n cationi cu valenta z+ si n anioni cu valenta z - , se scrie in acest caz:
![]() (13)
(13)
Coeficientul de activitate mediu ionic al unui electrolit din solutie, este:
![]() (14)
(14)
iar activitatea medie ionica, a este:
![]() (14a)
(14a)
care poate fi scris si in forma:
![]() (14b)
(14b)
unde:
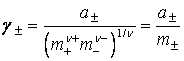 (15)
(15)
In consecinta, potentialul chimic se formuleaza in functie de activitatea ionica medie, pe scala molalitatilor, dupa cum urmeaza:
![]() (16)
(16)
Relatiile dintre molalitatea medie ionica, activitate si coeficientul de activitate mediu ionic pentru solutiile de electroliti de diferite tipuri de valenta sunt prezentate in tabelul 1.
Tabelul 1 Relatii intre marimi moleculare ionice pentru diferite tipuri de electroliti
|
Tipul de electrolit |
|
|
|
|
1:1; 2:2; 3:3 |
|
M |
|
|
2:1 |
|
41/3 m |
|
|
1:2 |
|
41/3 m |
|
|
3:1 |
|
271/4 m |
|
|
1:3 |
|
271/4 m |
|
|
4:1 |
|
2561/5 m |
|
|
1:4 |
|
2561/5 m |
|
|
3:2 |
|
1081/5 m |
|
In acord cu ecuatia (7), nu vom apela la relatii de conversie intre coeficientii de activitate formulati pe scala fractiilor molare sau a concentratiilor molare, lucrand consecvent cu marimile exprimate in functie de molalitate.
Marimile termodinamice caracteristice solutiilor ternare de electroliti se pot obtine din datele unor masuratori foarte precise ebuliometrice si criometrice. Prin specificul lor aceste metode impun tratarea amestecurilor de electroliti ca sisteme bine definite din punct de vedere termodinamic.
2.4 Functii de abatere de la idealitate si
corelarea lor cu diferite marimi termodinamice pentru sisteme ternare de electroliti
In cazul unui sistem ternar constituit din doi electroliti - simbolizati prin indicii (2) si (3), iar solventul cu indicele (1) - gradul de abatere de la idealitate al solutiei de electroliti se poate exprima printr-o functie de abatere, D, formulata in raport cu energiile libere G2 si G3 ale electrolitilor dizolvati in solutiile lor binare si cu energia libera G23 asociata ternarului format prin amestecarea solutiilor binare:
![]()
(17)
![]()
Aceasta ecuatie se prelucreaza formuland fiecare energie libera Gibbs din expresie in functie de potentialele chimice constituente, exprimate pe scala molalitatilor in forma cunoscuta. Prin aceasta prelucrare, avandu-se totodata in vedere si compensarea potentialelor chimice standard din relatia obtinuta precum si a celor din solutiile considerate, se obtine:
![]()
in care: m1 este numarul de moli de solvent; (g )2 este coeficientul de activitate al solventului din solutia binara a electrolitului 2, cu semnificatii analoge pentru (g )3 pentru solutia binara a electrolitului 3; (g )23 este coeficientul de activitate al solventului continut in solutia ternara; g este coeficientul de activitate al electrolitului 2 din solutia binara si analog g pentru electrolitul 3 din solutia binara; iar g coeficientul de activitate al electrolitilor 2 si 3 din solutia apoasa ternara.
Ecuatia (18) scrisa prescurtat este:
![]()
![]()
(19)
Prima paranteza patrata din ecuatia (18) reprezinta functia lgd care este privita ca masura globala a modificarii interactiilor ion-solvent la trecerea de la solutiile binare la amestecul ternar. Functia lgd releva modificarile interactiilor dintre ioni, (ion-ion) in solutia ternara, fata de binarele din care provin si este reprezentata de a doua paranteza patrata din ecuatia (18). Cele doua functii d si d se pot combina cu ecuatii care contin, in forma directa sau implicita, coeficientii de activitate din sisteme, conducand la corelari importante intre functii de abatere in binare si ternare.
Coeficientul de activitate al amestecului de electroliti, g , din solutia ternara, reprezinta media geometrica a coeficientilor de activitate (g )23 si
g )23 , din solutia ternara:
![]() (20)
(20)
sau in forma logaritmica:
![]()
(20a)
Cu ajutorul functiilor de abatere d si d , pot fi evaluate noi marimi de abatere care sa reflecte comportarea solutiilor de electroliti.
Marimile (g )23 si (g )23 includ corectii de interactii, fiind cantitati care caracterizeaza un electrolit in prezenta celuilalt electrolit. O situatie similara este evidentiata de legile lui Harned, cu caracter semiempiric, in formularea data pe cai indirecte. Pentru electrolitii uni-univalenti expresia legilor Harned este:
![]()
(21)
![]()
(21 a)
unde : a b, sunt coeficientii Harned. In majoritatea tratarilor se retin in aceste ecuatii numai coeficientii a si a , ceilalti fiind neglijabili ca valoare.
Ecuatiile (21) si (21a) evidentiaza dependenta dintre coeficientul de activitate al unui electrolit din solutia amestecului ternar si coeficientul de activitate continut in sistem binar, precum si de molalitatea celuilalt electrolit, la molalitate totala constanta. Mai explicit, din relatiile lui Harned rezulta ca intr-un amestec ternar coeficientul de activitate al unui electrolit in amestec este egal cu coeficientul de activitate al aceluiasi electrolit aflat intr-un sistem binar, plus un supliment de interactie. Coeficientii Harned depind de molalitatea totala a ternarului, m si nu de molalitatile individuale, m2 si m3 . In amestecurile de electroliti cu valente superioare celor 1-1 valente, se lucreaza la tarie ionica ( I ) totala constanta, iar legile Harned iau forma:
![]()
(22)
![]() (22a)
(22a)
Legatura acestor coeficienti s-a redat cu rigoare printr-un calcul termodinamic, rezultand:
![]() (23)
(23)
unde: F este coeficientul osmotic al apei din solutia cu electrolitul (2), F este coeficientul osmotic al apei din solutia cu electrolitul (3).
In forma data (21) si (21a), legile Harned nu pot fi valorificate din date experimentale, avand ca necunoscute coeficientii Harned. Combinand ecuatiile (18) cu ecuatiile Harned, se constata ca:
![]() (24)
(24)
Din combinarea ecuatiilor (23) cu (24) rezulta coeficientii Harned:
![]()
(25)
![]()
(26)
a si a se pot valorifica din date criometrice prin intermediul coeficientilor osmotici usor accesibili. Din suma coeficientilor Harned astfel exprimati, se regaseste ecuatia (24).
Punand in ecuatiile Harned aceiasi parametrii, se obtine direct:
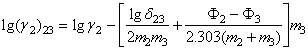 (27)
(27)
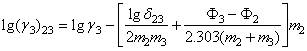 (27a)
(27a)
Valorile unor asemenea marimi sunt greu accesibile prin alte metode, dar nu excluse. S-au realizat, de exemplu, valorificari prin teorii statistice ale solutiilor de electroliti pentru obtinerea unor marimi de exces (de exemplu prin teoria Debye-Hckel extinsa). Datele de referinta raman insa tot cele fundamentate experimental.
In continuarea ecuatiilor statistice, observam ca in sistemele ternare, marimile d a si a sunt corelate cu functiile de exces la amestecarea solutiilor binare de electroliti, de exemplu:
![]() (28)
(28)
![]() (29)
(29)
Transpunandu-se intr-o alta formulare, aceste ecuatii devin:
![]() (30)
(30)
![]() (31)
(31)
Asemenea ecuatii sunt consacrate valorificarii marimilor de exces la amestecare prin date criometrice, dar si prin date ebuliometrice, la temperaturile corespunzatoare masuratorilor.
3 Teorii statistice ale solutiilor de electroliti
3.1 Elemente introductive
Dupa stabilirea suportului primar al configuratiilor ionice din solutiile de electroliti, s-a manifestat tendinta de a se corela, pe bazele mecanicii statistice, marimile macroscopice termodinamice masurabile, cu particularitatile microscopice ale ionilor din solutie.
La initierea unei teorii statistice, a solutiilor de electroliti se au in vedere, in general, cateva repere pentru alegerea modelului fizic cu care se asimileaza sistemul, cum ar fi:
echilibrul dinamic dintre miscarea termica a ionilor si interactiile electrostatice de atractie si repulsie;
natura electrolitului si rolul solventului care mijloceste obtinerea ionilor incarcati electric;
stabilirea functiei de distributie a sarcinilor electrice ale ionilor din solutie;
expresia potentialului electrostatic de interactie (atractiv si repulsiv) s. a.
Un exemplu bine cunoscut de specialisti este prima teorie moderna a solutiilor de electroliti, Debye-Hckel, care va fi expusa in continuare. Incepand de la finele deceniului al doilea al secolului XX si ajungandu-se pana la sfarsit de secol, teoria a constituit un model statistic permanent perfectionat, dar si sursa de inspiratie pentru creatorii de noi teorii .
Exista putine teorii statistice complet originale, independente intre ele, atat prin ideile fizice cat si prin modalitatile de prelucrare matematica, cu toate ca literatura domeniului abunda in asemenea contributii. Majoritatea sunt insa variante de imbunatatire, care decurg din modele preexistente, dar nu lipsesc nici contributii ale unor nume consacrate din literatura domeniului, care au dat consistenta, prin prelucrari noi, acestor teorii.
O alta teorie de referinta, consacrata , este teoria lui Mayer, elaborata dupa anul 1950 si completata pentru solutii de electroliti multicomponente, de Friedman. Teoria este bazata pe premisele modelului clusterelor celulare si prelucrata din elementele teoriei coeficientilor viriali ai gazelor reale. Rigoarea teoriei lui Mayer se datoreza, in mare masura, posibilitatii de evaluare exacta a integralei configurationale din evolutia calculelor desfasurate.
Este remarcabila concordanta obtinuta intre valorile unor functii termodinamice calculate si valorile lor experimentale, in teoria extinsa de Friedman. Pe de alta parte, pentru concentratii relativ scazute, s-au constatat valori foarte apropiate intre marimile termodinamice calculate dupa teoria Mayer si teoria Debye-Hckel, conferindu-se deplina credibilitate acestor teorii, desi ele se declara si sunt de proveniente diferite .
Nu este in intentia noastra de a dezvolta teorii cunoscute, ca cele amintite, si nici sa enumeram numarul mare de contributii noi. Cu toate acestea cerinta unui numar de date calculate ca valori de comparatie pentru marimile termodinamice determinate experimental, se impune. In consecinta am ales pentru prezentare una din cele mai recente teorii a solutiilor de electroliti, elaborata de K.S.Pitzer si colaboratorii sai si consolidata dupa anul 1980, alaturi de teoria limita Debye-Hckel.
3.2 Teoria statistica Debye-Hckel
Teoria Debye-Huckel se bazeaza pe un model simplificat al solutiilor de electroliti, sustinut de un grup de ipoteze:
solventul este reprezentat de un dielectric ideal continuu, in care sunt imersati ionii; propritatile sale sunt independente de taria campului electric;
electrolitul este complet disociat la orice concentratie;
ionii sunt asimilati cu sfere rigide incarcate electric, nepolarizabile,
care creeaza in jurul lor un camp electric de semn opus cu simetrie
sferica;
intre ioni se exercita forte de natura coulombiana care determina o repartitie uniforma a ionilor in solutie, caracterizata de prezenta unei atmosfere ionice, cu o anumita densitate electrica;
fortele coulombiene sunt mai puternice decat fortele inter-moleculare; ele se atenueaza relativ lent cu distanta inter-ionica, facand imposibila separarea in solutia electrolitica a unor clustere ionice bine conturate;
energia de atractie inter-ionica este neglijaila fata de energia agitatiei termice.
In dezvoltarea teoriei, se concentreza observatiile asupra unui singur
ion, i, situat in originea unui sistem de coordonate, inconjurat de o atmosfera ionica. Se considera ionul central pozitiv si un element de volum dV, situat la o distanta r=10-5-10-7 de ionul i. Probabilitatea ca elementul de volum sa fie incarcat negativ este, in virtutea teoremei Maxwell-Boltzman, superioara probabilitatii de a fi incarcat pozitiv. Ionul central este astfel inconjurat de un nor de ioni cu o anumita densitate elecrica cu o sarcina medie in timp egala si de semn contrar cu cea a ionului central.
![]()
Daca r(r) reprezinta
densitatea de sarcina electrica la distanta r de ionul central i si y(r) potentialul
electrostatic corespunzator configuratiei date, functiile Y (r) si r(r) sunt corelate prin ecuatia
Poisson:
![]()
in care D
este constanta dielectrica a solutiei, asimilata cu cea a solventului pur, in
teoria Debye-Hckel. Relatia (33) devine:
![]()
In absenta
interactiilor dintre ioni, probabilitatea ca un ion j sa se gaseasca la
un moment dat in elementul de volum dV este:
Deoarece elementul de volum se gaseste la distanta r de ionul central i
este caracterizat de potentialul electrostatic y(r), rezulta:
![]()
unde eij reprezinta energia potentiala medie a ionului j aflat la distanta r de
ionul i, echivalenta cu lucrul necesar pentru aducerea ionului j de la infinit la
![]()
distanta r de ionul i. Tinand seama de toate configuratiile
posibile, avem:
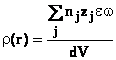
(36)
Densitatea medie de
sarcina este
![]()
Pe baza ipotezei neglijarii energiei de atractie interionica, comparativ cu
energia agitatiei termice, se admite
Ceea ce permite dezvoltarea in serie a exponentialei
![]()
(39)
Din dezvoltare se retin primii doi termeni, iar ceilalti se
neglijeaza, avand in vedere relatia de mai sus. Intrucat solutia este neutra electric, ecuatia
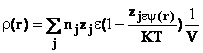
(40) , densitatea de sarcina r(r) decrisa de ecuatia (41), ia forma ecuatiei
![]()
![]()
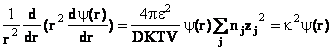
Ecuatia
de mai sus respecta teorema suprapunerii liniare a campurilor, indicand o proportionalitate
directa intre potential si densitatea de sarcini. Se obtine:
(43)
![]()
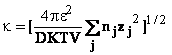
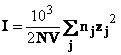
unde
valoarea lui k este
definita de ecuatia (44). Taria ionica I este
definita de relatia (45):
(pentru un electrolit uni-univalent taria ionica este egala cu concentratia in moli litru) si la 00C (cand DH2O=88,23), se obtine k=0,324x108 I1/2cm-1 sau 1/k=3,08/I1/2Å. Pentru I=1,1/k Å.
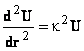
![]()
Substituind in U y(r)r, se obtine ecuatia (46) si solutia generala a acestei ecuatii diferentiale, este ecuatia
unde A si B sunt constante de integrare determinate din conditii la limita.
![]()
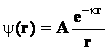
Pentru ca potentialul y(r) sa ramana
finit pentru valori mari ale lui r, trebuie ca B=0 si prin urmare, expresia potentialului chimic este dat de ecuatia (48) si respectiv (49)
(49)
![]()
![]()
Evaluarea
constantei A conduce al expresia (50) si astfel potentialul y(r) este dat de relatia (51):
(51)
![]()
Ecuatia de mai sus, obtinuta de Debye si Hckel, defineste
potentialul mediu in timp intr-un punct situat la o distanta r de un ion cu
valenta zi, intr-o solutie de electrolit, in absenta fortelor exterioare.
Considerand ionul i ca o sarcina punctiforma, (zie , el creeaza
la distanta r, intr-un mediu cu
constanta dielectrica D, potentialul:
![]()
![]()
Conform
principiului suprapunerii liniare a
campurilor, potentialul total y(r) la distanta r, este suma
dintre potentialul ionului central (y )
si potentialul cu care atmosfera sa ionica
actioneaza asupra lui (yi conform relatiei (53). Se obtine astfel expresia fundamentala a teoriei
Debye-Hckel data de relatia (54):
(54)
In solutii diluate (1 k este mult mai mare decat a. Astfel, pentru o solutie 0,01 molala a unui electrolit uni-univalent, raza atmosferei ionice calculata este de 30Å, iar pentru o solutie 1 molala, 1 k=3Å, ceea ce reprezinta mai putin decat distanta normala de maxima apropiere dintre doi ioni. Neaplicabilitatea teoriei Debye-Hckel la solutii concentrate de electroliti, in asemenea cazuri, se explica tocmai prin valoarea 1/ k, care devine mai mica decat raza ionica si presupune astfel atmosfera ionica in interiorul ionului, ceea ce nu este real.
q Functii termodinamice calculate prin teoria Debye-H ckel
Daca G reprezinta energia libea Gibbs a unei solutii de electrolit si Gid este energia libera a unei solutii cu aceeasi concentratie, dar in absenta sarcinilor, atunci (G-Gid) reprezinta energia libera de exces a solutiei de electrolit GE. In domeniul de aplicabilitate al teoriei Debye-Hckel, cand interactiile ionice de ordin superior celor Debye-Hckel sunt nule, GES si deci GE=Gel. Energia libera de exces este o masura a interactiilor reciproce dintre ioni. Intre energia libera de exces si valenta ionilor exista relatia
![]()
(55)
in care yi reprezinta potentialul electric al atmosferei ionului i.
Guggenheim calculeaza energia libera de exces a unei solutii de electrolit transpunand sistemul in conditiile unei distributii invariabile, sarcinile ionilor fiind marite treptat de la zero pana la valoarea lor reala.
q Marimi termodinamice caracteristice solventului
q Coeficientul osmotic si coeficientul de activitate
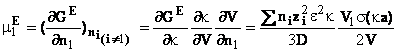
Prima
etapa in calculul marimilor caracteristice solventului consta in stabilirea
expresiei potentialului chimic de exces, cand
(56)
![]()
Deoarece
potentialul chimic de exces este conform ecuatiei (57a), el se poate exprima in
functie de coeficientul osmotic rational, cu ecuatia (57b):
(57a)
(57b):
![]()
Din relatiile de mai sus pentru coeficientul osmotic rational, se obtine
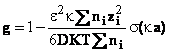
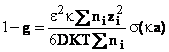
In virtutea egalitatii (60), urmeaza ca activitatea solventului este dat de ecuatia (61)
![]()
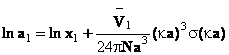
(61)
unde ![]() reprezinta volumul molar partial al solventului.
reprezinta volumul molar partial al solventului.
Folosind definitia data pentru coeficientul osmotic practic se obtine
![]()
Marimi termodinamice caracteristice electrolitilor dizolvati
Expresia potentialului chimic de exces al unui electrolit dizolvat poate fi stabilita pe doua cai
a)

In raport cu sensul fizic al potentialului chimic de
exces, care reprezinta energia electrica
a unui ion i cu sarcina zie , inconjurat de o atmosfera ionica, cand se gaseste
(63)
![]()
b) Potentialul chimic de exces al unui ion poate fi
calculat cu cand se obtine
![]()
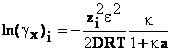
unde Vi reprezinta volumul molar al
ionilor i. Din expresia potentialului chimic de exces se poate calcula
coeficientul de activitate al ionului i
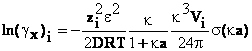
(66)
![]()
![]()
Pe intregul domeniu de concentratii in care este
valabila teoria Debye-Hckel produsul k Vi<<1 si termenul al
doilea din ultima relatie este neglijabil comparativ cu primul. Calculele arata
ca intr-o solutie 4 molala a unui
electrolit valent, termenul al doilea
reprezinta mai putin de 1% din valoarea
coeficientului de activitate. Exprimand coeficientul de activitate in functie
de taria ionica si de parametrul k, se obtine
(69)
unde:
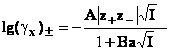
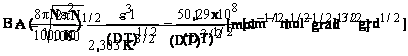
![]()
pentru solventul apa, in solutiile electrolitilor uni-valenti, la 298K, coeficientul A , iar B=0,3291x108.
In
solutiile foarte diluate termenul Ba![]() este foarte
mic, iar coeficientul de activitate mediu se exprima prin legea limita Debye-Hckel
este foarte
mic, iar coeficientul de activitate mediu se exprima prin legea limita Debye-Hckel
![]()
Se regaseste astfel legea empirica Lewis-Randall care arata ca la concentratii mici, specificitatea ionilor dispare, proprietatile lor fiind determinate doar de sarcina si concentratia lor.
Ecuatia de mai sus, completata cu un termen liniar in concentratie, devine identica cu relatia dedusa empiric de Brnsted:
![]()
Adaugand acelasi termen suplimentar ecuatiei se obtine ecuatia Debye-Hckel imbunatatita
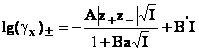
in care B si B sunt parametri ajustabili.
Expresiile stabilite pentru coeficientii de
activitate dupa teoria Debye Hckel nu pot
explica aparitia minimelor pe curbele de variatie ale coeficientilor de
activitate cu concentratia. Fenomenul s-a explicat prin influenta hidratarii ionilor. Neconcordanta dintre valorile
experimentale ale diferitelor marimi termodinamice si cele calculate pe baza
rezultatelor teoriei Debye-Hckel a fost
explicata de diferiti autori in diferite moduri. Astfel, Onsager arata ca
folosirea ecuatiei Poisson-Boltzman impune pentru doi ioni, i si j,
la distanta r unul de altul, respectarea egalitatii
Relatia de mai sus este insa valabila doar daca densitatea de sarcina medie in apropierea perechii de ioni, i si j este mereu suma densitatilor induse de cei doi ioni separati. Acest lucru este posibil numai in cazul solutiilor cu concentratii mici, continand ioni cu diametre ionice mari si sarcini electrice mici. Teoria Debye-Hckel este o lege limita a carei precizie creste cu dilutia. Ca prima teorie statistica a solutiilor de electroliti, teoria Debye-Hckel a avut un succes remarcabil, servind totodata ca baza pentru teoria lui Onsager, a conductivitatii electrice si pentru teoria lui Kirkwood, a difuziei termice in solutii ionice La concentratii mici teoria Debye-Hckel este o formulare limita a rezultatelor teoriilor care i-au urmat, valabile si la concentratii moderate. Dintre acestea amintim teoria lui Bogoliubov si teoria lui Mayer, cu extinderea facuta de Friedman, teoria lui Kirkwood si Poirier.
3.3 Modelul lui Pitzer si al colaboratorilor sai
Modelul propus de Pitzer si a colaboratorilor sai, dintre care amintim: G. Mayorga, J.J. Kim, L.F. Silvester, R.N. Roy, J.R. Peterson, J.C. Piper, are ca suport principal teoria lui Debye-Hckel (DH) imbunatatita, la care s-au adus cateva contributii proprii, unele de natura empirica. In afara exploatarii modelului (DH), autorii au prelucrat si parti din contributiile celor mai reprezentativi cercetatori ai domeniului, precum: Kirkwood, Scatchard, Flower si Rushbrooke, Guggenheim, la care se adauga si alte nume celebre ca, van Rysselberghe si Einsenberg, Ramanathan si Friedman, Rasaiah si Friedman, Scatchard, Rush si Jonson, Vilcu si Sirbu, Wood si Robinson, Rasaiah si Valleau si altii.
Modelul Pitzer (P) include ideile majore ale teoriei Debye-Hckel completate cu elemente noi si numeroase prelucrari, impuse de un formalism matematic incarcat. Introducerea unor marimi carora li se atribuie un sens fizic, dar care nu sunt accesibile practic, a impus suplinirea unor necunoscute cu parametrii ajustabili. In mod necesar numarul parametrilor empirici se mareste odata cu cresterea complexitatii sistemelor caracterizate.
Pentru a discerne contributiile principale ale teoriei Pitzer la studiul solutiilor de electroliti, fata de bazele teoriei (DH), vom prezenta comparativ, esenta acestor teorii, aflate in stransa conexiune.
In ambele teorii marimea fundamentala calculata este potentialul de interactie ionica (Y), care rezulta prin transpunerea in calcule a premizelor lor fizice principale.
Postulatul major al teoriei primare (DH) are in vedere disocierea totala a ionilor in solutie. Ionii, considerati sarcini electrice punctiforme, sunt distribuiti in jurul unui 'ion central' de semn opus 'atmosferei ionice' creeate in jurul ionului; atmosfera ionica are o simetrie sferica cu o anumita densitate electrica, j. Fortele coulombiene exercitate intre ioni sunt superioare energiei agitatiilor termice, neglijabila, daterminand o repartitie uniforma a ionilor in solutie. Functia de distributie a ionilor este calculata dupa legea Maxwell-Boltzmann.
Fata de premisele principale ale teoriei (DH) prezentate, modelul (P) are in vedere simetria sferica a atmosferei ionice, precizand existenta ei 'in conditii de stationaritate' a sistemelor de electroliti si in absenta "fortelor externe'. Fata de teoria (DH), modelul (P) accepta, in plus, existenta unor molecule nedisociate si implicit, existenta unui parametru suplimentar: potentialul intermolecular Uij inaccesibil practic, dar evidentiat in calcule printr-un parametru arbitrar. Functia de distributie radiala a ionilor este calculata tot dupa legea de distributie Maxwell-Boltzmann.
In formularea primara a teoriei (DH) ionii sunt considerati ca sarcini punctiforme, dar prima corectie adusa acestei teorii elimina aceasta premisa simplificatoare introducand in expresia potentialului Y un parametru dimensional, A, reprezentand distanta de minima apropiere dintre ioni (suma razelor ionice). Acest parametru se regaseste si in teoria (P). Suplimentar, Pitzer introduce un parametru denumit 'hard core' (miez rigid) care ar parea echivalent cu parametrul A, dar caruia i se atribuie particularitati speciale fiind exploatat prin consecintele sale, de natura cinetica, asupra unor proprietati ale solutiilor. Se constata, ca urmare, ca presiunea osmotica a solutiilor, p, este continuta in formularea sa clasica, in expresia potentialului de interactie dintre ioni, Y
In modelul matematic (P) fortele de interactie ionica sunt exprimate printr-o dezvoltare in coeficienti viriali, din care se retin doi termeni: primul, lij semnificand interactia dintre ionii i si j si urmatorul, lijk redand interactia ionilor i, j si k. Fortele de mica distanta sunt reprezentate de primul termen, iar al doilea cuprinde interactii la distanta mare. Coeficientii viriali apar ca functii de taria ionica, I.
In ambele modele constanta dielectrica a solutiei se asimileaza cu cea a solventului, (D Do). Remarcam insa ca in teoria (DH), corectata in conditiile aplicarii sale la concentratii ridicate ale solutiilor de electroliti, se lucreaza in virtutea realitatilor fizice cu constanta dielectrica reala a solutiei respective, accesibila practic direct, considerindu-se Do ¹ D. Pe langa corectivele semnalate pentru modelul (DH), avem in vedere si corectia datorata hidratarii ionilor fara a mai pune in discutie reluarile ulterioare de calcule rafinate prin contributiile lui La Mer, Gronwall, Sandved, Glueckaauf, Scatchard, Poirier si alti specialisti.
q Solutii apoase cu un singur electrolit
In teoria lui Pitzer, proprietatile solutiilor de electroliti pot fi exprimate printr-un termen "electrostatic" plus o serie de coeficienti viriali in care coeficientii viriali sunt dependenti de taria ionica a solutiei. Se sugereaza in mod particular ca cel de-al doilea coeficient virial variaza cu taria ionica. De asemenea, s-a propus o forma alternativa imbunatatita pentru termenul "electrostatic", fata de teoria D-H.
Dependenta celui de-al doilea coeficient virial de taria ionica apare implicit si in teoria mai complexa a lui Mayer.
In forma generala, sistemul de ecuatii pentru proprietatile
termodinamice ale electrolitilor puri sau ale amestecurilor acestora, se poate
scrie pornind de la expresia energiei libere Gibbs de exces:
![]() (75)
(75)
In aceste conditii coeficientul osmotic si coeficientul mediu ionic de activitate capata formele:
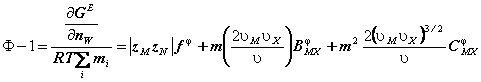 (76)
(76)
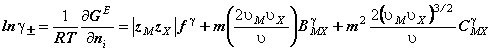
unde: ![]() si
si ![]() sunt numerele ionilor M si X, iar zM si zX sunt sarcinile electrice
respective, n nM nX , iar nW este numarul de Kg solvent
si m este molalitatea.
sunt numerele ionilor M si X, iar zM si zX sunt sarcinile electrice
respective, n nM nX , iar nW este numarul de Kg solvent
si m este molalitatea.
In ecuatiile (75), (76) si (77) s-au considerat trei functii pentru termenul electrostatic de tip f:
![]() (78)
(78)
![]() (79)
(79)
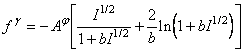
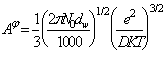 (81)
(81)
unde: ![]() este taria ionica,
este taria ionica, ![]() este constanta D-H pentru coe-ficientul
osmotic si toate cele trei forme se reduc la legea limita D-H la valori mici
ale tariei ionice (I). Valoarea sa la 25 oC este 0,392 pentru
apa, D este constanta dielectrica a
apei, K constanta lui Boltzmann, e este sarcina electrica, No
numarul lui Avogadro iar dW
densitatea apei. Pentru a mentine ecuatii simple pentru solutii de
amestecuri de electroliti, b din ecuatiile (78) trebuie sa ramana acelasi pentru toate
solutiile, iar valoarea selectata a lui I
este constanta D-H pentru coe-ficientul
osmotic si toate cele trei forme se reduc la legea limita D-H la valori mici
ale tariei ionice (I). Valoarea sa la 25 oC este 0,392 pentru
apa, D este constanta dielectrica a
apei, K constanta lui Boltzmann, e este sarcina electrica, No
numarul lui Avogadro iar dW
densitatea apei. Pentru a mentine ecuatii simple pentru solutii de
amestecuri de electroliti, b din ecuatiile (78) trebuie sa ramana acelasi pentru toate
solutiile, iar valoarea selectata a lui I
este de 1,2. De asemenea valoarea a = 2,0 a fost gasita satisfacatoare pentru toate solutiile considerate.
Pentru cel de-al doilea coeficient virial, desemnat prin parametrii b, s-au definit trei forme:
![]() (82)
(82) ![]() (83)
(83)
![]()
unde
parametrii ![]() si
si ![]() dau seama de interactiile pe distanta mica dintre perechile de ioni.
dau seama de interactiile pe distanta mica dintre perechile de ioni.
Pentru parametrii C au fost stabilite de catre Pitzer si Mayorga, urmatoarele forme:
![]() ;
(85)
;
(85) ![]() (86)
(86)
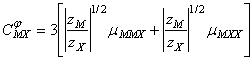 (87)
(87)
Parametrul ![]() defineste
al treilea coeficient virial care, in mod uzual, este mic si uneori complet
neglijabil. Acesta da seama de interactiile triple: MMX, respectiv MXX.
defineste
al treilea coeficient virial care, in mod uzual, este mic si uneori complet
neglijabil. Acesta da seama de interactiile triple: MMX, respectiv MXX.
Pentru obtinerea parametrilor, in cele mai multe cazuri aceste ecuatii au fost fixate dupa datele de coeficienti osmotici recomandate de Robinson si Stockes sau dupa datele proprii ale lui Pitzer.
Evaluarea parametrilor a fost discutata de asemenea in functie de structura solventului si de fortele inter-ionice. Valorile cele mai bune pentru coeficientii viriali au fost obtinute prin metoda celor mai mici patrate. Din analiza facuta trebuie retinut ca al doilea coeficient virial poate lua fie valori pozitive fie negative, in functie de fortele predominante pe distanta mica, atractive sau repulsive.
In literatura de specialitate se gasesc parametrii Pitzer de interactie inter-ionica pentru 304 solutii apoase de electroliti singulari la concentratii mari, pana la molalitati apropiate de saturatie, la calculul acestora contribuind diversi colaboratori, ca: J. J. Kim, Jr., Harvie, Moller si Weare.
q Solutii multicomponente de electroliti
Tratarea termodinamica a solutiilor multicomponente de electroliti a fost simplificata considerabil prin folosirea ecuatiilor care disting clar efectele de amestecare de cele ale componentilor puri.
Scatchard si Guggenheim au evidentiat ca aceasta maniera de tratare este facilitata de posibilitatile de utilizare a ecuatiilor analitice ce reprezinta cu acuratete proprietatile termodinamice ale electrolitilor puri.
Pitzer si colaboratorii sai au stabilit pentru solutii de electroliti micsti un sistem de ecuatii care reprezinta o crestere "viriala" a energiei libere Gibbs de exces, GE, in functie de molalitatea ionilor, incluzand unele expresii provenite din consideratii teoretice si empirice stabilite pentru solutii cu un singur electrolit.
Forma ecuatiei initiale folosite de K. S. Pitzer si J. J. Kim este:
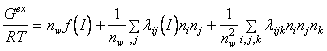 (88)
(88)
unde:
nw este numarul de kg. solvent, ni , nj , nk , sunt numerele de moli de specii ionice i, j si k. Functia f depinde doar de taria ionica I si reprezinta, ca si in modelul D-H, efectele interactiilor coulombiene pe distanta mare.
lij si ![]() constituie al doilea si respectiv al treilea
coeficient virial care reprezinta efectele fortelor pe distanta mica dintre
ioni, considerati cate doi, respectiv cate trei in acelasi timp.
constituie al doilea si respectiv al treilea
coeficient virial care reprezinta efectele fortelor pe distanta mica dintre
ioni, considerati cate doi, respectiv cate trei in acelasi timp.
Functia "f" are forma matematica apropiata de functia conventionala de distributie D-H, conducand la aceeasi lege limita D-H, dar cu un efect mai mic al dimensiunii ionice. Dorind sa utilizeze o singura functie "f" independent de dimensiunea ionilor, autorii au acceptat o expresie aproximativa.
In cazul coeficientilor viriali, pe baza proprietatilor electrolitilor puri, au fost determinate sume adecvate. Proprietatile amestecurilor implica adaugarea la aceste sume a altor termeni, care dau seama de diferenta intre interactiile ionilor diferiti de acelasi semn si interactiile ionilor identici de acelasi semn. Daca s-ar respecta principiul lui Brnsted al interactiilor specifice, toti acesti termeni de diferenta ar fi zero. Pitzer si colaboratorii sai au renuntat la principiul lui Brnsted si au aratat ca acesti termeni, desi mici ca marime si uneori neglijabili, sunt in general mai mari ca zero.
Principalele caracteristici folosite de autori in stabilirea sistemului de ecuatii pentru solutii multicomponente au fost:
- considerarea electrolitilor sub forma ionizata si nu sub forma neutra, moleculara;
- dependenta coeficientului virial lij de taria ionica I, ceea ce face mai usoara adoptarea functiei "f" universale si obtinerea unei convergente mai rapide in seria viriala.
Astfel au stabilit ecuatii pentru coeficientul osmotic al amestecului de electroliti si pentru coeficientul de activitate al fiecarui electrolit neutru:
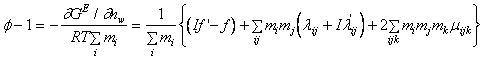
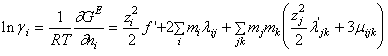 (90)
(90)
S-a notat ![]() si
si ![]() iar mi
= ni/nw
iar mi
= ni/nw
Pentru coeficientul de activitate al unui electrolit neutru se obtine:
 (91)
(91)
Pentru stabilirea setului de ecuatii (89), (90) si (91) s-au folosit marimile:
![]() (91a)
(91a)
cu reluarea ecuatiei (75) si respectarea acelorasi semnificatii pentru marimi;
 (91b)
(91b)
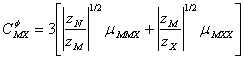 (91c)
(91c)
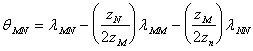
![]() (91d)
(91d)
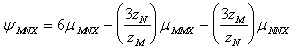 (91e)
(91e)
unde:
![]() este coeficientul Debye-Hckel ;
este coeficientul Debye-Hckel ; ![]() si are valoarea de 0.392 la 25 0C;
D este constanta dielectrica a apei, K constanta Boltzmann si e sarcina
electrica. Parametrul b in ff este un parametru empiric egal cu 1,2 la 25 0C.Valoarea lui a
si are valoarea de 0.392 la 25 0C;
D este constanta dielectrica a apei, K constanta Boltzmann si e sarcina
electrica. Parametrul b in ff este un parametru empiric egal cu 1,2 la 25 0C.Valoarea lui a
Pentru solutii multicomponente cu
ion comun de tip MX - NX sau cu valente superioare este necesar sa se
adauge un termen la parametrul ![]() , care ia forma:
, care ia forma:
![]() (92)
(92)
Pentru electroliti 2-2 valenti a = 1,4 si a = 12,0. Pe baza acestei noi adaugiri, tinand seama ca c si c' acopera toti cationii iar a si a' acopera toti anionii, se poate scrie ecuatia pentru coeficientul osmotic al solutiei sub forma:
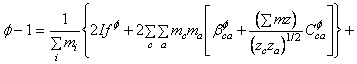
![]()
unde: ![]()
In ecuatia (92) se regasesc termeni care reprezinta suma dubla pentru molalitati precum si pentru al doilea si al treilea coeficient virial in cazul unui electrolit pur. Acesti termeni pot fi evaluati din informatii asupra electrolitilor puri si au fost determinati pentru un numar mare de electroliti.
Pentru coeficientii de activitate
medii ionici ai solutiilor multicomponente de electroliti 2-2 valenti sau cu
valente superioare se adauga la ![]() si
si ![]() parametrii
parametrii ![]() si a cu aceeasi forma ca in termenul
si a cu aceeasi forma ca in termenul![]() . Se obtine:
. Se obtine:
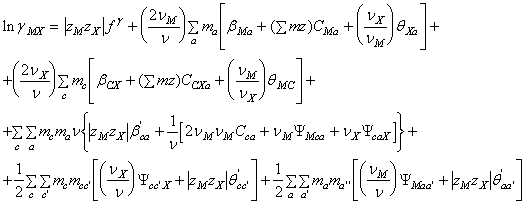
(94)
unde:
![]()
Ecuatia (94) capata forme simplificate in cazul in care toti dizolvatii sunt de acelasi tip de valenta, cand exista un anion sau un cation comun sau cand sunt doar doi dizolvati.
In cazul unui amestec de doi electroliti simetrici cu sarcina z si cu anion comun expresia coeficientului de activitate devine:
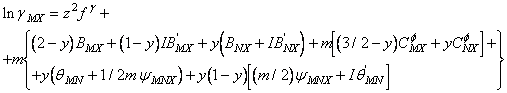 (95)
(95)
unde parametrii "f", b, C q si Y au semnificatiile date de ecuatiile de mai sus.
Pe baza setului de ecuatii stabilit de Pitzer si colaboratorii sai, Harvie, Moller si Weare au stabilit un nou set de ecuatii in anul 1984, mult simplificat. Ei au considerat ca in solutie electrolitul se regaseste sub forma de ioni, fiind astfel necesara tratarea termodinamica a ionilor individuali.
Coeficientul osmotic si coeficientii de activitate individuali devin:
 (96)
(96)
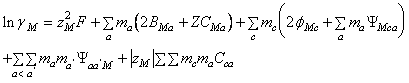 (97)
(97)
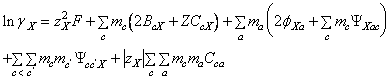 (98)
(98)
unde:
- m i este molalitatea (moli/Kg.solvent), M, c, c' indica cationii, X, a, a' indica anionii, c<c' si a<a' indica perechile deosebite de anioni si cationi, iar celelalte marimi au semnificatiile cunoscute.
AF si F sunt termeni definiti prin expresiile matematice indicate mai jos:
![]()
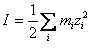 (99)
(99)
identica cu ecuatia (81).
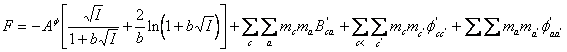
(100)
Parametrii de interactie b b b (2) si CF definesc termenii B si C si au forme diferite in functie de numarul de electroliti din solutie si de valentele ionilor. Astfel, pentru solutii ce contin o sare cu ioni monovalenti acesti parametrii au forma:
![]() ;
; ![]() ;
;
![]()
(101)
unde a=2 , iar g(x) si g'(x) sunt functii de forma:
![]() ;
; ![]() ; (102)
; (102)
![]() ;
; ![]() ;
; ![]() ;
;
Pentru electroliti 2-2 valenti si de valente de tip superior sunt definiti urmatorii parametrii:
![]() ;
; ![]()
![]() (103)
(103)
pentru
electroliti 2-2 valenti sunt valabili: ![]() ;
;
![]() ,
iar pentru electroliti de tip 3-2 si 4-2 valenti :
,
iar pentru electroliti de tip 3-2 si 4-2 valenti :![]() ;
; ![]()
Harvie,
Moller si Weare considera ca pentru o solutie ce contine o singura sare
proprietatile termodinamice depind numai de parametrii de interactie ![]() ,
,
![]() ,
,
![]() si
si
![]() .
Parametrii f si y sunt
utilizati numai pentru amestecuri de minimum doua saruri. Parametrul f raspunde de
interactiile cation-cation si anion-anion, in timp ce parametrul y este definit
pentru interactiile cation-cation-anion si anion-anion-cation. Valorile fij sunt date
de relatiile:
.
Parametrii f si y sunt
utilizati numai pentru amestecuri de minimum doua saruri. Parametrul f raspunde de
interactiile cation-cation si anion-anion, in timp ce parametrul y este definit
pentru interactiile cation-cation-anion si anion-anion-cation. Valorile fij sunt date
de relatiile:
![]() ;
; ![]() ;
; ![]()
unde: qij este doar un parametru ajustabil si este definit pentru fiecare pereche de cationi si fiecare pereche de anioni.
Termenii ![]() si
si ![]() sunt
responsabili de efectele de amestecare electrostatice ale perechilor
nesimetrice cation-cation si anion-anion definite de Pitzer. Pentru ecuatii cu
termeni electrostatici de ordin superior acestia sunt calculati de fiecare
data, utilizand aproximatia Chebishev pentru perechi nesimetrice de anioni si
cationi in integralele de configuratie Pitzer. Valorile
sunt
responsabili de efectele de amestecare electrostatice ale perechilor
nesimetrice cation-cation si anion-anion definite de Pitzer. Pentru ecuatii cu
termeni electrostatici de ordin superior acestia sunt calculati de fiecare
data, utilizand aproximatia Chebishev pentru perechi nesimetrice de anioni si
cationi in integralele de configuratie Pitzer. Valorile ![]() si
si ![]() depind de sarcina si taria ionica si sunt zero
cand perechile de anioni si de cationi au aceeasi sarcina.
depind de sarcina si taria ionica si sunt zero
cand perechile de anioni si de cationi au aceeasi sarcina.
Coeficientul mediu ionic g poate fi calculat dupa ecuatia clasica:
![]() (105)
(105)
Rezultatele satisfacatoare obtinute de autori au permis extinderea utilizarii acestui set de ecuatii in caracterizarea termodinamica a apelor naturale.
Desi complexa, teoria Pitzer a fost adoptata ca urmare a performantelor sale: o concordanta foarte buna intre datele calculate si cele obtinute experimental (uneori aproape pana la coincidenta lor).
Succesul modelului Pitzer, contrar aparentelor de rigoare absoluta si grija de a nu omite nici un parametru fizic din tratare, se explica insa tocmai prin suplinirea unor marimi inaccesibile direct prin parametrii ajustabili, empirici. Ca exemple edificatoare putem alege coeficientii viriali, deja amintiti, atribuiti interactiilor ionice duble si triple. Ei au fost determinati indirect pe baza datelor de coeficienti osmotici, recomandate de Robinson si Stockes sau dupa datele proprii ale lui Pitzer si ale colaboratorii sai. Asemenea parametrii au fost calculati pentru numeroase solutii apoase binare de electroliti, la concentratii mari. Expresii aproximate de autori au fost acceptate si pentru functiile care intervin in marimile termodinamice macroscopice (GE).
In tratarea sistemelor multicomponente s-au avut in vedere marimile pentru componentii puri si efectele de amestecare asociate constituirii lor. In asemenea sisteme creste numarul parametrilor ajustabili.
S-au aplicat aproximari si pentru ecuatii cu termeni electrostatici de ordin superior, utilizandu-se consecvent pentru perechi nesimetrice de anioni si cationi aproximatia Chebishev.
Lucrand in acest mod autorii au stabilit un sistem de calcule de ajustare, contand pe valori experimentale verificate, pentru cazurile examinate, cu care s-au putut corela marimile ajustabile.
In acest mod modelul Pitzer poate acoperi o sfera larga de sisteme, cu rezultate foarte bune.
4 Verificarea cunostintelor
Descrieti functiile termodinamice caracteristice solutiilor de
electroliti.
2. Explicati starile standard conventionale ale solutiilor de
electroliti.
Explicati corelatia marimilor molare partiale cu coeficientii de
activitate ai componentilor din solutiile de electroliti.
Descrieti functiile de abatere de la idealitate si corelatia lor cu
diferite marimi.
5. Explicati teoria limita Debye-Hckel.
6. Explicati teoria lui Pitzer si a colaboratorilor sai.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3284
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved