| CATEGORII DOCUMENTE |
| Alimentatie nutritie | Asistenta sociala | Cosmetica frumusete | Logopedie | Retete culinare | Sport |
DEFINITIE. CLASIFICARE
Aceste anomalii cromozomiale pot fi intilnite intr-o varietate de situatii clinice: 50 % sunt recunoscute sau diagnosticate in perioada graviditatii in urma avorturilor (jumatate din aceste avorturi sunt datorate unor mutatii cromozomiale). Mai tarziu, in perioada gestatiei, 6 % dintre copiii nou nascuti si tot atatia n.n. morti prezinta anomalii cromozomiale. Fetusul purtator al unei mutatii cromozomiale este predispus la malformatii, tulburari de dezvoltare sau hidrops. La fel, copiii distrofici sau dismorfici pot fi purtatorii unei anomalii cromozomiale.
Anomalii mostenite cromozomiale pot fi suspectate in cuplurile sterile sau cu avorturi spontane repetate, sau care au copii care prezinta dismorfisme. Cand diagnosticul anomaliei cromozomiale este facut prenatal prognosticul copiiilor este greu de stabilit. Cele mai valoroase informatii referitoare la consecintele clinice ale anomaliilor cromozomiale sunt date de observarea copiilor diagnosticati din cauza problemelor care apar dupa nastere.
Tipuri de anomalii cromozomiale
Anomaliile numerice pot fi reprezentate de aneuploidii si poliploidii
a. aneuploidia este data de aditia sau pierderea unuia sau rareori a doi cromozomi dintr-o pereche
b. poliploidia este aditia unuia sau a mai multor seturi complete haploide de cromozomi
Anomalii de structura sunt rearanjamente ale materialului genetic in cadrul aceluiasi cromozom sau intre doi cromozomi diferiti.
Aceste rearanjamente pot fi echilibrate cand nu se produc modificari esentiale in interiorul materialului genetic, sau neechilibrate cand rezulta un castig sau o pierdere in segmentele esentiale ale cromozomului.
Frecventa anomaliilor cromozomiale
50 % din avorturile spontane si mai mult de 5 % din toate sarcinile cunoscute sunt purtatoare de anomalii cromozomiale
aproximativ 0,5 % (1/200) din nou-nascuti au anomalii cromozomiale
In tabelul 2 sunt prezentate succint diferitele tipuri de mutatii cromozomiale si diagnosticul lor.
a. aneuploidiile cromozomilor sexuali sunt responsabile pentru 33 % din mutatiile cromozomiale
b. aneuploidiile autosomale determina 25 % din anomaliile cromozomiale
c. rearanjamentele cromozomiale de structura echilibrate raspund pentru 33 % din anomaliile cromozomiale
d. rearanjamentele cromozomiale structurale neechilibrate cauzeaza 8 % din anomaliile cromozomiale
doar anomaliile structurale echilibrate si mutatiile cromozomilor sexuali pot fi compatibile uneori cu un fenotip normal.
Tabloul clinic al anomaliilor cromozomiale
Exista mai multe situatii clinice cand putem suspecta o anomalie cromozomiala:
Cuplurile sterile
2 - 4 % din cuplurile sterile pot prezenta un rearanjament autosomal; uneori unul dintre membrii cuplului poate avea un numar modificat de gonosomi
anomaliile cromozomiale pot aparea in deficiente ale gametogenezei sau defecte de nidatie sau implantare a zigotului
Avorturile spontane si nou nascutii morti
Aceste avorturi spontane se pot asocia cu tulburari de crestere si dezvoltare embrionare ale sacului gestational sau cu embrioni si fetusi malformati (pierduti dupa primul trimestru de sarcina)
Aneuploidia se poate oberva la 50 % sau mai mult dintre avorturile spontane si poate aparea si la nou nascutul mort care prezinta frecvent malformatii
Nou nascutii vii cu anomalii:
Au deficiente clinice comune cum ar fi malformatiile, tulburarile de dezvoltare si crestere intarziata , afectarea aparatului de reproducere
Sunt rezultatul unor aneuploidii sau rearanjamente structurale neechilibrate aparute intamplator la genitor si purtate de gametii care se fecundeaza.
|
Anomalia cromozomiala |
Rezultatul aberatiei |
Aparitia |
|
Triploidie |
Trei cromozomi in fiecare pereche ( 3 n) |
La avortoni |
Monosomie- Autosomala - Cromozomul x |
Un singur cromozom in pereche (2 n-1) Un singur cromozom sexual |
Letala in primele saptamani de sarcina Este frecvent letala in prima parte a sarcinii, dar poate fi descoperita numai in prima copilarie sau in adolescenta |
Trisomie- Autosomala - Pe hetero-cromozomi |
3 cromozomi intr-o pereche (2 n +1) Un cromozom sexual in plus |
Este letala in sarcina pentru toti autosomii cu exceptia cr. 13,18,21 De obicei dgs se pune la pubertate sau uneori la adult |
Deletii-Autosomale-pe cromozomul X |
Monosomie partiala pe un singur autosom. Monosomie partiala a unui cr. X |
De obicei prezenta la nastere sau in timpul primei copilarii Poate fi detectata de la nastere pana la varsta adulta |
DuplicatiiAutosomale |
Trisomie partiala |
De la nastere pana in prima copilarie |
TABEL nr.2 TIPURI DE ANOMALII CROMOZOMIALE
MUTATII CROMOZOMIALE DE NUMAR
Trisomia - consta in prezenta a 3 cromozomi intr-o pereche in care de obicei gasim doar 2 cromozomi omologi. Au fost observate trisomii pentru fiecare dintre autosomi cu exceptia cromozomului 1
cele mai multe din trisomii duc la moartea embrionului, de aceea sunt cele mai frecvente mutatii cromozomiale care sunt depistate la embrionii avortati spontan
viabilitatea embrionilor care poarta o trisomie este data de marimea si continutul materialului genetic purtat de cel de-al treilea cromozom. Cu exceptii foarte rare doar trisomiile 13,18,21 supravietuiesc postnatal si pot fi diagnosticate in populatie
trisomiile cromoxomilor sexuali (XXX, XXY) au ca efect deficiente in dezvoltare; uneori sunt depistate la avortoni sau fetusi morti dar mai frecvent sunt diagnosticate la nou nascutii morti
* Mecanismele de producere si factorii asociati
Nondisjunctia consta intr-o alterare a segregarii cromozo-miale sau cromatidiene care afecteaza diviziunea celulara in timpul meiozei sau mitozei.
Nondisjunctia din prima diviziune meiotica (fig. 11. - meioza I) consta in lipsa de separare (segregare) a cromozomilor omologi.
Nondisjunctia din a doua diviziune meiotica (meioza II) rezulta din lipsa de separare a cromatidelor surori din cromozomii bicromatidici.
Ambele mecanisme pot duce la formarea gametilor anormali, disomici sau nulosomici, pentru cromozomul respectiv. In urma fecundarii acestor gameti rezulta zigoti purtatori de aneuploidie (fie trisomie, fie monosomie).
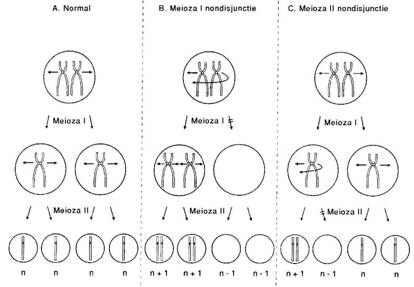
Fig. 11. Meioza
Nondisjunctia mitotica in celulele somatice este asemanatoare cu nondisjunctia din meioza II, cand nu se realizeaza disjunctia cromatidelor in anafaza; vor rezulta celule trisomice si monosomice.
Nondisjunctia apare cu o frecevnta mai mare in ovogeneza decat in spermatogeneza (studiile citogenetice arata ca in 90 % din cazurile de sindrom Down extracromozomul este de origine materna, in urma nondisjunctiei meiotice primare). Nu s-au putut atribui erorile mecanismului de diviziune celulara unor factori din mediul extern cum ar fi radiatiile, administrarea de hormoni sau consumul de alcool.
Nu a fost stabilita etiologia genetica a nondisjunctiei primare la om. Singura influenta sigura este varsta inaintata a mamei. Varsta materna avansata favorizeaza avorturile spontane si aparitia trisomiilor la n.n. Cu toate eforturile considerabile care au fost depuse nu s-a putut depista natura factorilor care influenteaza desfasurarea procesului de segregare cromozomiala. In celulele trisomice poate aparea nondisjunctia secundara in timpul diviziunii celulare. Astfel femeile cu sindrom Down pot sa produca la randul lor gameti disomici sau monosomici pentru cromozomul 21. Riscul recurentei pentru trisomii 13,18 sau 21 este 1-2 % pentru mamele al caror prim copil trisomic s-a nascut cand ea avea mai putin de 30 de ani. Peste 30 de ani riscul creste dupa o curba exponentiala
*Sindroame clinice
Sindromul Down sau trisomia 21 apare cu incidenta de 1/680 n.n. Frecventa este dubla la embrionii de 10-12 saptamani, trisomia 21 fiind o cauza extrem de frecventa a avorturilor spontane.
prenatal: ecografia poate pune in evidenta hidropsul fetal, stenoza sau atrezia duodenala, femurul scurtat. Nivelul crescut al a fetoproteinei materne sporeste sansa diagnosticarii unui sindrom Down la fetus
nou-nascutul are un facies caracteristic rotund cu radacina nasului larga, epicantus, pete Brushfield la nivelul irisului, limba mare proeminenta, urechi mici si occipital turtit. Investigatiile paraclinice pot pune in evidenta hiperbilirubinemie, rare leziuni cardiace (defecte ale canalului atrioventricular, septului atrial si ventricular)
in copilarie sau la adultul tanar se constata retardul mental care de obicei este moderat. Alte semne clinice pot fi: hipotrofia staturala, boli autoimune, surditate. Femeile au o fertilitate redusa si un risc de 50 % de a avea o sarcina purtatoare de trisomie 21. Adultii in varsta sunt predispusi la dementa presenila tip Alzheimer. Odata ce individul supravietuieste primului an de viata poate sa ajunga la varsta inaintata de 50-60 ani.
Trisomia 13 are o incidenta de 1/5000 n.n.
Prenatal: majoritatea embrionilor cu trisomie 13 sunt avortati in primul trimestru al sarcinii. In perioada celui de-al doilea trimestru, anomaliile care pot fi observate prin examinarea ultrasonografica includ o intarziere in crestere si dezvoltare, leziuni congenitale cardiace, dezvoltarea redusa a encefalului, leziuni faciale (holoprosencephal) si omphalocel.
Nou-nascutul prezinta anomalii la nivelul extremitatii cefalice: dismorfism craniofacial cu gura de lup, buza de iepure, anencefalie, microcefalie, ciclopie. Aproape toti nou-nascutii afectati au anomalii cardiace letale si un retard mental profund - greu de decelat la aceasta varsta, dar sigur prezent.
Trisomia 18 are o incidenta de 1/3500 n.n.
Prenatal: cele mai multe din trisomiile 18 sunt diagnosticate la avortoni sau la n.n. morti. Anomaliile sunt severe si sunt date de intarzierea in dezvoltare, leziuni cardiace congenitale, hernii diafragmatice (detectate prin ultrasonografie)
La nastere nou nascutul cu trisomie 18 are un facies mic cu ocipital proeminent, urechi mici si ascutite, pliuri cutanate, piele incretita. Majoritatea prezinta malformatii cardiace si la nivelul altor organe interne. Este mai frecventa la nou nascutul de sex feminin care sunt profund handicapati
Sindromul Klinefelter (47,XXY) apare cu o incidenta 1/800 n.n.. Semnele clinice caracteristice acestui sindrom include :
Baiat tanar slab cu purtare imatura
Baiat la pubertate cu testicule mici
Barbat adult cu un aspect eunucoid, ginecomastie si musculatura slab dezvoltata
Barbat aparent normal cu sterilitate si azoospermie.
Acesti indivizi au un risc crescut pentru cancer de san si schizofrenie
Productia de testosteron este deficitara datorita hialinizarii si fibrozarii progresive a tubilor seminiferi, majoritatea acestor barbati sunt sterili Este necesara suplimentarea de durata cu testosteron,.
Sindromul 47, XYY apare cu aceeasi frecventa ca si sindromul 47, XXY
Barbatii cu acest sindrom nu prezinta dismorfisme
Diagnosticul este pus cel mai adesea accidental in urma unor screninguri prenatale, sau la nou-nascuti
Semnele clinice includ statura inalta si uneori problemele sociale pe care acesti indivizi le prezinta; in prezent este negata agresivitatea extrema si criminalitatea care a fost atribuita la inceput indivizilor cu acest sindrom si care este enumerata in foarte multe publicatii de specialitate.
Femeile cu sindrom 47, XXX nu au, in majoritatea cazurilor, manifestari clinice. Ele sunt fertile si pot da nastere la urmasi normali. La un numar mic dintre ele poate fi prezenta sinostoza radioulnara, oligomenoreea, amenoreea secundara si menopauza prematura. Ele prezinta un risc ridicat pentru boli psihice, in special pentru schizofrenie.
Monosomiile autosomale sunt letale asupra produsului de conceptie si deci nu au putut fi puse in evidenta la nou-nascutii vii
Monosomnia pentru cromozomul X (45,X) este o forma comuna de mutatie cromozomiala. Se diagnosticheaza atat la avortoni dar si la paciente in viata cu sindrom Turner.
Semne clinice in sindrom Turner (incidenta 1/2500 n.n de sex feminim)
In primul trimestru al sarcinii 10 % din avorturile spontane sunt cauzate de tulburarile majore de embriogeneza pe care le induce monosomia X
In al doilea trimestru al sarcinii examinarea cu ultrasunete poate pune in evidenta hidropsul fetal, coarctatia aortei si malformatiile rinichiului. Majoritatea acestor fetusi se vor naste morti.
la nou nascutul cu sindrom Turner aspectul poate fi normal sau putem inalni semne caracteristice: gatul palmat, toracele in scut, coarctatia aortei, edeme la maini si picioare
in copilarie trasaturile caracteristice sunt statura mica si suflul cardiac. Fetele la pubertate prezinta amenoree primara sau secundara si lipsa de dezvoltare a caracterelor sexuale secundare; majoritatea sunt sterile (ovarele se transforma in bandelete fibroase). Dezvoltarea mentala este normala dar pot aparea anomalii de perceptie spatiala.
la femeia adulta care nu a primit un tratament hormonal de suplinire apare osteoporoza si boli cardiovasculare cu debut precoce
citogenetica in sindromul Turner poate pune in evidenta nu numai monosomia X(45,X) dar si mozaicismul (46,XX/45,X/47,XXX), sau prezenta unui izocromozom 46,XXqi, sau o monosomie partiala (46,XXp-)
Monozomia x poate rezulta prin nondisjunctie dar si prin anafaza intarziata. Riscul recurentei in sindromul Turner nu creste odata cu varsta avansata a mamei
Mozaicismul in aneuploidii
Mozaicismul consta in prezenta a doua sau mai multe linii celulare cu diferite cariotipuri la acelasi individ. O linie celulara normala coexista cu o linie celulara anormala care poate prezenta o mutatie cromozomiala de numar sau structura. O anumita linie celulara poate sa apara in toate tesuturile, dar poate sa fie gasita si numai intr-unul singur sau o parte din tesuturi.
Aspectul clinic pentru fiecare mozaicism depinde de continutul cromozomului implicat dar si de proportia liniilor celulare normale sau patologice.
Printre mozaicismele mai frecvent intalnite putem mentiona: trisomia 8 (copil cu dismorfism facial, retardare mentala, intarziere in crestere, contractura fexorilor degetelor de la maini si picioare), mozaicisme pentru trisomiile 13,18,21, sau sindrom Turner cand femeile pot fi fertile, etc.
Mozaicismele rezulta prin nondisjunctie cromozomiala in perioada embriogenezei, deci dupa prima diviziune a celulei ou.
Disomia uniparentala
Este prezenta intr-o linie celulara diploida cand amandoi cromozomii unei perechi provin de la un singur parinte, sau in cazul unei celule trisomice cand 2 cromozomi provin de la acelasi parinte (unul este cromozomul "lenes" din anafaza intarziata). In acest al doilea caz liniile celulare diploide rezultate prin diviziunea celulei trisomice pot prezenta disomie uniparentala.
Consecintele posibile ale disomiei uniparentale pot fi:
Amprentarea (imprintingul) unei gene sau regiuni cromozomiale in functie de originea parentala (materna sau paterna) a cromozomului implicat, se traduce prin nivele anormale ale proteinei sintetizate. Ex: sindromul Prader-Willi pentru disomia 15 materna, si sindrom Angelman pentru disomia uniparentala 15 (paterna)
Poliploidia
Triploidia (3 n) se caracterizeaza prin 69 cromozomi cu sexul genetic XXX, XXY sau XYY
Tetraploidia (4 n) = 92 cromozomi si sex genetic XXXX, XXYY
Reprezinta 20 % din anomaliile cromozomiale care induc avort spontan in primul trimestru; placenta prezinta hiperplazie trofoblastica si poate aparea mola hidatiforma partiala: in al II-lea si al III-lea trimestru sarcina se caracterizeaza prin retard in dezvoltare; se dezvolta un oligohidroamios progresiv, placenta este mica iar fatul are macrocefalee, leziuni cardiace si sidactilie, nou-nascutii vii sunt foarte rari si supravietuiesc o perioada mica de timp.
In cazul triploidiilor avortul se produce frecvent in primul trimestru, iar in cazurile rare cand sarcina evolueaza fetusul are malformatii cardiace majore, microcefalie, retard in dezvoltare.
ANOMALII CROMOZOMIALE DE STRUCTURA
(rearanjamente cromozomiale)
Rezulta in urma rupturii si schimburilor segmentelor cromozomiale la diferite nivele. Multe din structurile noi , cum ar fi fragmentele acentrice cromozomiale si cromozomii dicentrici, sunt foarte instabile in timpul diviziunii celulare, ceea ce duce la pierderea materialului cromozomial si moartea celulei. Se pot produce de asemenea o varietate de alterari stabile ale cromozomului uman.
1. Deletia reprezinta o pierdere de material genetic la nivelul bratului unui cromozom.
Clasificare:
Deletia terminala intereseaza unul din capetele telomerice ale cromozomului si consta intr-o ruptura a bratului cromozomial. Fragmentul acentric rezultat este pierdut in timpul diviziunii celulare.
Deletia interstitiala rezulta dupa realizarea a doua linii de ruptura si pierderea fragmentului interstitial rezultat
Cromozomul in inel rezulta prin doua rupturi ale capetelor terminale ale cromozomului de fiecare parte a centromerului si fuzionarea la nivelul punctelor de ruptura, dupa incurbarea prealabila a bratelor legate de centromer. Segmentele distale rezultate in urma rupturilor sunt pierdute si astfel indivizii cu cromozom in inel au deletii atat pe bratul lung (q) cat si pe cel scurt (p) al cromozomului implicat
Deletiile pot sa apara ca rezultat al segregarii unor cromozomi cu inversii sau translocatii pe cromozomii urmasilor din parinti care au suferit aceste rearanjamente structurale echilibrate in timpul vietii.
Sindroamele clinice care apar in urma deletiilor pe bratele cromozomiale se caracterizeaza printr-un fenotip anormal. Semnele clinice caracteristice sunt secundare unei pierderi de material genetic de la nivelul unei benzi sau subbenzi a bratului cromozomial, material genetic care contine o anumita informatie, cu o anumita functie genetica. Sunt trei sindroame caracteristice mai frecvente:
a. Sindromul "cri du chat"(cat cry) - "mieunatul pisicii" (46,XX,5p- ; 46,XY,5p-)
Nou nascutul are faciesul rotund si un tipat asemanator cu mieunatul pisicii care dispare cu timpul, de asemenea prezinta malformatii cardiace si un retard mental sever.
De obicei este vorba de o deletie terminala "de novo" pe bratul scurt al cromozomului 5: zona critica este 5p15
b. Sindromul Wolf-Hirschhorn (4 p -)
Nou-nascutii au dismorfism cranio-facial, frunte proeminenta, retard in crestere, trunchi lung, membre subtiri, malformatii cardiace, renale, ale SNC si retard mental. 10 - 50 % din aceste deletii sunt asociate cu translocatii familiale, regiunea implicata este 4p16
c. Cromozomul 14 in inel (14 r)
Punctele de ruptura observate mai frecvent sunt 14p11 si 14q32. Nou-nascutul prezinta dismorfism facial cu sau fara retard mental.
Sindroamele date de microdeletii rezulta din deletii mici care intereseaza benzile cu rezolutie inalta evidentiate prin diagnosticul citogenetic.
* sindromul Prader-Willi: in acest sindrom semnele clinice evolueaza dupa nastere:
La n.n. apare o hipotonie profunda care impune un diagnostic diferential cu bolile metabolice sau miopatiile
In prima copilarie , intre al 2-lea si al 3-lea an de viata, copilul cu sindrom Prader - Willi dezvolta apetit foarte crescut si obezitate tronculara. Mainile si picioarele sunt mici, copii sunt hipotonici, cu retard mental
In perioada pubertara se accentueaza obezitatea si apare gonadismul daca nu se instituie o terapie corespunzatoare
*Sindromul Angelman : copiii prezinta convulsii, ataxie, retard mental; sunt veseli si hiperactivi
2. Duplicatiile constau intr-un castig de material genetic si se traduc prin trisomii partiale pentru anumite segmente cromozomiale.
Duplicatiile segmentelor cromozomiale pot rezulta din rearanjamente structurale familiale cum ar fi translocatia sau inversia
Riscul recurentei nu este mare in afara de cazul in care duplicatia este un rearanjament cromozomial familial
Izocromozomul: unul din bratele cromozomului (p sau q) este duplicat si tot materialul genetic al celuilalt brat este pierdut. Cele 2 brate ale cromozomului, aflate de o parte si de alta a centromului sunt imagini in oglinda ale aceleiasi informatii genetice.
Pentru majoritatea autosomilor izocromozomii induc un rearanjament neechilibrat care este letal. Cu toate acestea exista si cazuri viabile de izocromozomi pentru bratele lungi ale unor cromozomi acrocentrici, cazuri care prezinta un fenotip normal
Izocromozomul pentru bratul lung al cromozomului 21 [45,XX, i(21 q) duce la lipsa bratelor scurte ale celor doi cromozomi 21, pe cand bratele lungi sunt prezente de doua ori intr-un cromozom. Este o mutatie cromozomiala similara translocatiei robertsoniene in care pierderea materialului bratului scurt al cromozomului acrocentric implicat nu se traduce prin efecte fenotipice deosebite.
Daca un cromozom sau regiune cromozomiala sufera o amprentare genetica, disomia uniparentala creata de un izocromozom are consecinte clinice. De exemplu izocromozomul 15 q duce la aparitia sindromului Prader- Willi
In cazul cromozomilor de sex, izocromozomul Xq se intalneste la aproximativ 10 - 15 % din femeile cu s. Turner. Aceste femei au un cromozom X normal, celalalt fiind un izocromozom pentru bratul lung. Functional Xq i se traduce printr-o monosomie Xp. Aspectul clinic este identic cu un sindrom Turner 45,X.
Mecanismul de producere: separarea celor doua brate cromozomiale in timpul diviziunii celulare nu se realizeaza in plan longitudinal ci in plan transversal, cu pierderea bratelor scurte in cazul izocromozomului Xq, sau a bratelor lungi in cazul Xpi. Un mecanism si mai complex duce la formarea izocromozomului dicentric.
Riscul de recurenta: daca parintii au un izocromozom pe un autosom (21 qi) riscul de aparitie pentru o trisomie la urmasi este de 100 %. Deoarece izocromozomul merge intr-unul din gameti in timpul meiozei, pot rezulta gameti nulosomici sau disomici. Fecundarea unui gamete nulosomic da o monosomie letala pentru celula ou. Fecundarea gametelui disomic duce la trisomie, caz frecvent intalnit in trisomia 21.
Inversiile se produc prin modificarea ordinei materialului genetic de pe un cromozom ; segmentul dintre doua linii de ruptura se rasuceste cu 1800 .
De cele mai multe ori cromozomul rezultat chiar daca nu pierde sau castiga material genetic isi modifica morfologia. Pot sa apara "de novo" sau pot fi prezente intr-o familie pe parcursul a mai multe generatii
* Tipuri:
a) inversia pericentrica este rezultatul a doua rupturi si rearanjamentului materialului genetic de ambele parti ale centromerului
b)inversia paracentrica este rezultatul unor rupturi si a unui rearanjament de aceeasi parte a centromerului. Cromozomul rezultat va avea avelasi aspect morfologic dar va prezenta o ordine modificata a benzilor din structura.
Inversia poate sa se produca in timpul diviziunii meiotice
a) inversia pericentrica: deoarece purtatorii unei inversii au in mod obisnuit un cromozom normal si unul care poarta aceasta inversie, perechea de cromozomi omologi va suferi o recombinare in profaza meiozei primare, printr-un crossing over in bucla; rezulta cromozomi cu duplicatii sau deletii pentru segmentele aflate in afara punctelor de ruptura care determina inversia.
Cromozomii neimplicati in recombinare sunt similari cu cromozomii parentali(unul normal, unul inversat)
Embrionii care primesc un cromozom recombinat au un cariotip neechilibrat. Supravietuirea lor depinde atat de marirea materialului duplicat sau deletat cat cat si de cromozomul afectat.
b). Inversiile paracentrice
Ca si in cazul reversiei pericentrice se poate forma in timpul meiozei o bucla intre punctele de jonctiune ale segmentului inversat.
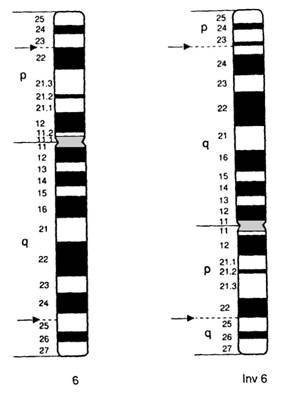
Fig. 12. Inversie
o singura recombinare im interiorul buclei produce cromozomii recombinati instabili (fie dicentrici, fie acentrici) care in mod obisnuit sunt letali si rareori pot fi depistati la nou-nascutii vii
Daca nu se produce crossingoverul in bucla cromozomii rezultati vor fi identici cu cromozomii parentali
Semnele clinice
purtatorii inversiilor paracentrici si pericentrici au un fenotip normal
diagnosticul se poate pune intamplator prin testare pentru diagnosticul prenatal, la avorturile spontane (cand rearanjamentul neechilibrat prezinta macroduplicatii sau deletii) si la nou-nascutii. morti sau vii cu duplicatii sau deletii mici rezultate in urma inversiei
Translocatiile de fuziune centrica rezulta prin fuzionarea bratelor lungi intre doi cromozomi acrocentrici; se intalneste mai frecvent intre cromozomii 13-15, 21 si 22
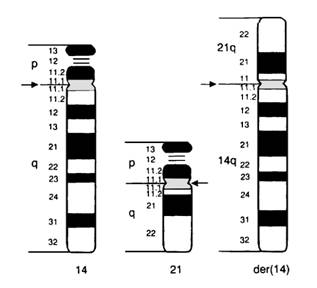
Fig. 13. Translocatie de fuziune centrica
Punctele de ruptura de la care pornesc aceste rearanjamente sunt la nivelul sau in aproprierea centromerului la amandoi cromozomii implicati.
Bratele lungi ale cromozomilor fuzioneaza iar bratele scurte se pierd astfel ca heterozigotii purtatori ai acestui rearanjament prezinta doar 45 cromozomi.
Purtatorii translocatiei robertsoniene sunt normali si translocatia este echilibrata pentru ca bratele scurte ale cromozomilor acrocentrici sunt bogate in material genetic heterocromatinizat si gene care codifica r-ARN, aceasta informatie se gaseste in copii multiple si pe alti cromozomi acrocentrici.
Cea mai comuna translocatie robertsoniana este 45,XX sau XY, t(13q14q) si se intalneste cu o frecventa de 1/1500 de indivizi. De asemenea translocatia 14q21q este comuna si este frecvent o cauza de sindrom Down.
Consecintele meiozei la purtatorii cu translocatie robertsoniana: gametii care rezulta in urma segregarii vor prezenta rearanjamentele echilibrate si neechilibrate; in acest tip de translocatie 3 cromozomi sunt de obicei implicati pentru ca bratele scurte s-au pierdut.
Cromozomii parentali purtatori ai unei translocatii robertsoniene (14q21q )+ sunt reprezentati in fig. 14. Complexul reprezinta 3 structuri cu segmente omoloage pentru cromozomii interesati. B. Sunt posibile 3 tipuri de segregare. In tipul 1, cromozomul translocat (der.14) segrega de cromozomii 14 si 21. Fecundarea gametilor rezultati va produce un cariotip cu translocatie echilibrata sau unul normal in ambele situatii fenotipul fiind normal. In segregarile tip 2 si 3 toti gametii rezultati vor fi anormali si va rezulta o trisomie sau monosomie pentru bratul lung al cromozomului q atat pentru cromozomul 21 cat si pentru cromozomul 14. Numai unul din gametii tipului 2 (der. 14 si 21) vor permite dupa fecundare realizarea unui zigot viabil. Acesti copii vor prezenta s. Down(trisomie 21 q)].
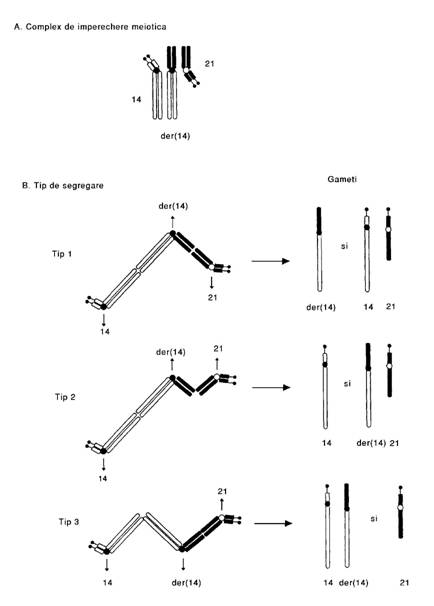
Fig. 14. Segregarea cromozomilor cu translocatie Robertsoniana
Semne clinice in t (14 q 21 q): purtatorii acestei translocatii sunt clinic normali.
In urma fecundarii rearanjamentul devine neechilibrat si zigotul poate prezenta trisomie 21 cand sarcina poate ajunge la termen, trisomie 14 (avort spontan precoce) sau monosomie atat pentru cromozomul 14 cat si 21 (avort spontan precoce).
Urmasii cu rearanjamente echilibrate pot avea cromozomii normali, dar ca si parintii ei vor fi purtatori ai translocatiei
Familiile care au aceasta translocatie pot avea numerosi membrii cu sindrom Down sau cu avorturi repetate. De aceea este important sa se faca cariotipul cuplurilor care au avorturi.
5. Translocatia reciproca se produce prin ruptura si schimbul unor segmente intre cromozomii neomologi. Punctele de ruptura la nivelul carora se face schimbul pot avea orice localizare pe cromozom.
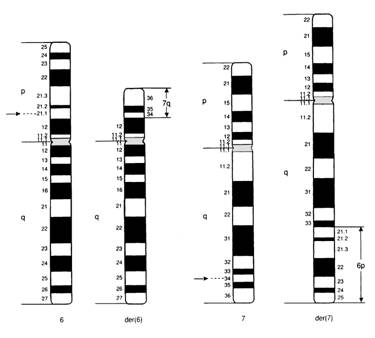
Fig. 15. Translocatia reciproca
Este un rearanjament cromozomial echilibrat la parinti.
* Consecintele meiozei la purtatorii de translocatii reciproce:
segmentele omoloage ale celor doua perechi de cromozomi implicati in translocatie si cromozomii translocati (der 6, der 7) vor forma un complex in cruce (cu 4 structuri)
segregarea cromozomilor pereche va duce la formarea gametilor care pot fi cu cromozomi normali, cu translocatie echilibrata sau neechilibrata
disjunctia complexului "in cruce" in anafaza distribuie in mod normal doi cromozomi unei celule si alti doi cromozomi altei celule (2:2) dar uneori este posibila si o distributie 3:1.
A. Segmentele omoloage ale perechilor de cromozomi implicati se aseaza in cruce. B. Segregarea se poate face in 3 tipuri dupa cum centromerii perechilor de cromozomi vor fi alternanti sau adiacenti fusului de diviziune in anafaza. Gametii rezultati vor purta o translocatie echilibrata sau vor fi normali cand segregarea este alternativa. In segregarile adiacente 1 si 2 se produc gameti anormali fie cu duplicatii fie cu deficiente de segmente cromozomiale.
Pentru majoritatea translocatiilor semnele clinice anormale rezulta dupa segregarea adiacenta 1 la urmasi viabili anormali sau la avortoni.
In segregarea adiacenta II gametii rezultati vor prezenta duplicatii si deletii majore ceea ce duce dupa fecundare la eliminarea zigotului inainte de nidarea in mucoasa uterina.
gametii cu translocatii neechilibrate rezultati dintr-o disjunctie 2:2 au amandoi duplicatii si deletii pentru segmentele implicate in translocatii; cei rezultati din disjunctie 3:1 pot avea duplicatii sau deficiente pentru segmentele implicate ale celor 2 cromozomi sau pot fi nulosomici sau disomici pentru fiecare dintre ei.
(Riscul de recurenta pentru purtatori 20 % - 30 .
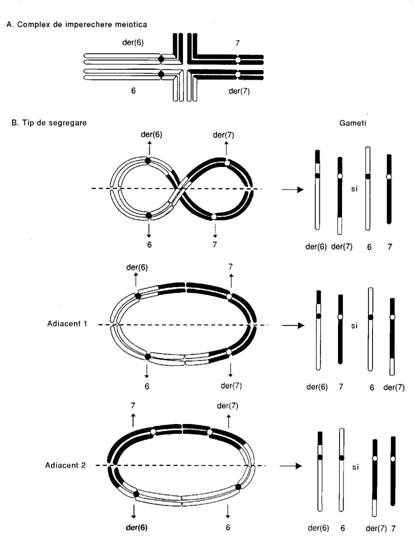
Fig. 16. Meioza la un purtator de translocatii t(6.7)
Situsurile fragile ale cromozomilor
Sunt puncte care pot fi vizualizate daca culturile celulare sunt tratate in anumite conditii. Sunt transmise genetic si au localizari specifice pe cromozomi.
Sunt peste 100 de situsuri fragile raportate; se stie ca grupul cu sensibilitate la folat este localizat in vecinatatea tripletelor repetitive.
Acest grup a fost identificat pe autosomi si pe cromozomul X. Numai doua din situsurile de pe cromozomii X sunt asociate cu fenotipuri clinice, cel mai comun este retardarea mentala cu X fragil (Xq 27.3).
Cromozomii markeri
Deriva din rearanjamente structurale si sunt gasiti in mod obisnuit sub forma extracromozomilor
Cromozomii marker se formeaza prin rearanjament structural si in mod obisnuit sunt gasiti ca piese extracromozomiale.
1. Mecanismul formarii si cromozomii de origine sunt necunoscuti. Marea majoritate a cromozomilor care se prezinta ca si markeri constitutionali sunt mai mici decat cromozomul 22. Pot sa fie cromozomi in inel, sau cromozomi bisatelitati, sau pot fi metacentrici.
2. Markerii mici pot manifesta instabilitate mitotica si pot avea un mozaic de forme. Ei pot prezenta variatii ale heterocromatinei si eucromatinei.
3.Numerosi markeri sunt compatibili cu o dezvoltare normala si inteligenta normala. Ei pot fi transmisi ereditar fara a avea consecinte fenotipice. Alterori, ca in cazul unei duplicatii a cromozomului 15, pot induce anomalii fenotipice.
4.Identificarea markerilor se poate face folosind hibridizarea in situ cu fluorescenta.
5.Implicatiile clinice depind de originea si continutul markerului.
Ruperea cromozomilor: cromozomii rupti rezulta in urma unor leziuni ale cromozomilor in metafaza, care pot duce la deletii sau translocatii
*inducerea leziunilor rezulta din surse interne (procese de sinteza sau reparatorii ale ADN-ului) sau externe ( radiatii, substante chimice)
In acest grup de boli sunt incluse afectiuni hematologice, malformatii si chiar afectiuni cu un risc crescut de malignizare.
Anemia Fanconi (pancitopenica)
Copiii pot prezenta o varietate de malformatii interne si externe. Malformatia caracteristica este la nivelul radiusului si policelui, apar de asemenea pete pigmentare pe trunchi, boli cardiace congenitale si malformatii renale. Prin hipoplazia medulara a oaselor acesti copii dezvolta in timp pancitopenie, si in stadiile finale leucemie, necesitand un transplant de maduva.
Citogenetica evidentiaza un numar mare de rupturi cromozomiale in culturile de limfocite, testarea este mai fidela daca in tratarea culturilor se foloseste un agent alchilant (DEB)= diepoxybutanul.
Sindromul Bloom
Copiii afectati prezinta retard in dezvoltare, teleangiectazii la nivelul fetei. Au risc crescut de a dezvolta procese maligne.
Teleangiectazia
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 13746
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved