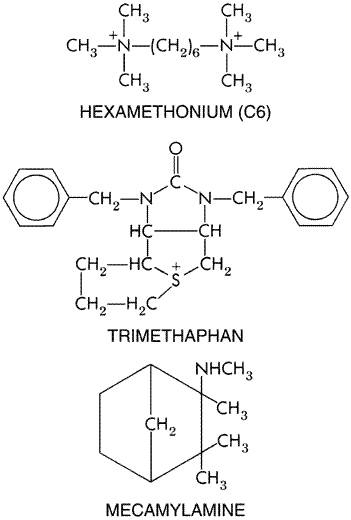
| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Anticholinesterase Agents
Overview
|
This chapter covers agents that prolong the existence of acetylcholine after it is released from cholinergic nerve terminals. These agents inhibit acetylcholinesterase, which is concentrated in synaptic regions and is responsible for the rapid catalysis of the hydrolysis of acetylcholine. Anticholinesterase agents have therapeutic utility in the treatment of glaucoma and other ophthalmologic conditions (see also Chapter 66: Ocular Pharmacology), the facilitation of gastrointestinal and bladder motility, and influencing activity at the neuromuscular junction of skeletal muscle to enhance muscle strength in myasthenia gravis. Anticholinesterase agents that cross the bloodbrain barrier have shown limited efficacy in the treatment of Alzheimer's disease (see also Chapter 22: Treatment of Central Nervous System Degenerative Disorders). Antidotal therapy of the toxic effects of cholinesterase inhibitors used as insecticides and chemical warfare agents is directed to blocking the effects of excessive acetylcholine stimulation and reactivating the phosphorylated, inhibited enzyme. Modification of activity at cholinergic synapses by activation or blockade of muscarinic or nicotinic acetylcholine receptors is discussed in Chapters 7: Muscarinic Receptor Agonists and Antagonists and 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia, respectively. |
Anticholisesterase Agents: Introduction
|
The function of acetylcholinesterase (AChE) in terminating the action of acetylcholine (ACh) at the junctions of the various cholinergic nerve endings with their effector organs or postsynaptic sites is considered in Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems. Drugs that inhibit AChE are called anticholinesterase (anti-ChE) agents. They cause ACh to accumulate in the vicinity of cholinergic nerve terminals and thus are potentially capable of producing effects equivalent to excessive stimulation of cholinergic receptors throughout the central and peripheral nervous systems. In view of the widespread distribution of cholinergic neurons, it is not surprising that the anti-ChE agents as a group have received extensive application as toxic agents, in the form of agricultural insecticides and potential chemical warfare 'nerve gases.' Nevertheless, several members of this class of compounds are widely used as therapeutic agents; others that cross the bloodbrain barrier have been approved or are in clinical trial for the treatment of Alzheimer's disease. Prior to World War II, only the 'reversible' anti-ChE agents were generally known, of which physostigmine is the outstanding example. Shortly before and during World War II, a new class of highly toxic chemicals, the organophosphates, was developed chiefly by Schrader, of I. G. Farbenindustrie, first as agricultural insecticides and later as potential chemical warfare agents. The extreme toxicity of these compounds was found to be due to their 'irreversible' inactivation of AChE, which resulted in long-lasting inhibitory activity. Since the pharmacological actions of both classes of anti-ChE agents are qualitatively similar, they are discussed here as a group. Interactions of anti-ChE agents with other drugs acting at peripheral autonomic synapses and the neuromuscular junction are described in Chapters 7: Muscarinic Receptor Agonists and Antagonists and 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia. History Physostigmine, also called eserine, is an alkaloid obtained from the Calabar
or ordeal bean, the dried ripe seed of Physostigma venenosum, Balfour,
a perennial plant found in tropical The Calabar bean was brought to As a result of the basic research of Stedman (1929a,b) and associates in elucidating the chemical basis of the activity of physostigmine, others began systematic investigations of a series of substituted aromatic esters of alkyl carbamic acids. Neostigmine, a promising member of this series, was introduced into therapeutics in 1931 for its stimulant action on the intestinal tract. It was reported subsequently to be effective in the symptomatic treatment of myasthenia gravis. It is remarkable that the first account of the synthesis of a highly potent organophosphorus anti-ChE, tetraethyl pyrophosphate (TEPP), was published by Clermont in 1854. More remarkable still is the fact that the investigator survived to report on the compound's taste; a few drops should have been lethal. Modern investigations of the organophosphorus compounds date from the 1932 publication of Lange and Krueger on the synthesis of dimethyl and diethyl phosphorofluoridates. The authors' statement that inhalation of these compounds caused a persistent choking sensation and blurred vision apparently was instrumental in leading Schrader to explore this class for insecticidal activity. Upon synthesizing approximately 2000 compounds, Schrader (1952) defined the structural requirements for insecticidal (and, as learned subsequently, for anti-ChE) activity (see below; Gallo and Lawryk, 1991). One compound in this early series, parathion (a phosphorothioate), later became the most widely used insecticide of this class. Malathion, which currently is used extensively, also contains the thionophosphorus bond found in parathion. Prior to and during World War II, the efforts of Schrader's group were directed toward the development of chemical warfare agents. The syntheses of several compounds of much greater toxicity than parathion, such as sarin, soman, and tabun, were kept secret by the German government. Investigators in the Allied countries also followed Lange and Krueger's lead in the search for potentially toxic compounds; diisopropyl phosphorofluoridate (diisopropyl fluorophosphate; DFP), synthesized by McCombie and Saunders (1946), was studied most extensively by British and American scientists. In the 1950s, a series of aromatic carbamates was synthesized and found to have a high degree of selective toxicity against insects and to be potent anti-ChE agents (Ecobichon, 2000). Structure of Acetylcholinesterase AChE exists in two general classes of molecular forms: simple homomeric oligomers of catalytic subunits (i.e., monomers, dimers, and tetramers) and heteromeric associations of catalytic subunits with structural subunits (Massoulie, 2000; Taylor et al., 2000). The homomeric forms are found as soluble species in the cell, presumably destined for export, or associated with the outer membrane of the cell through either an intrinsic hydrophobic amino acid sequence or an attached glycophospholipid. One heterologous form, largely found in neuronal synapses, is a tetramer of catalytic subunits disulfide-linked to a 20,000-dalton lipid-linked subunit. Similar to the glycophospholipid-attached form, it is found in the outer surface of the cell membrane. The other consists of tetramers of catalytic subunits, disulfide linked to each of three strands of a collagen-like structural subunit. This molecular species, whose molecular mass approaches 106 daltons, is associated with the basal lamina of junctional areas of skeletal muscle. Molecular cloning revealed that a single gene encodes vertebrate AChEs (Schumacher et al., 1986; Taylor et al., 2000). However, multiple gene products are found; this diversity arises from alternative processing of the mRNA. The different forms differ only in their carboxyl-termini; the portion of the gene encoding the catalytic core of the enzyme is invariant. Hence, the individual AChE species can be expected to show identical substrate and inhibitor specificities. A separate, structurally related gene encodes butyrylcholinesterase,
which is synthesized in the liver and is primarily found in plasma (Lockridge
et al., 1987). The cholinesterases define a superfamily of proteins
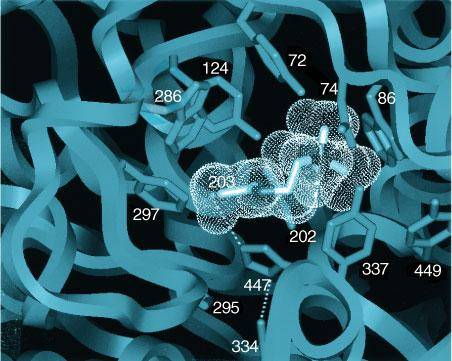
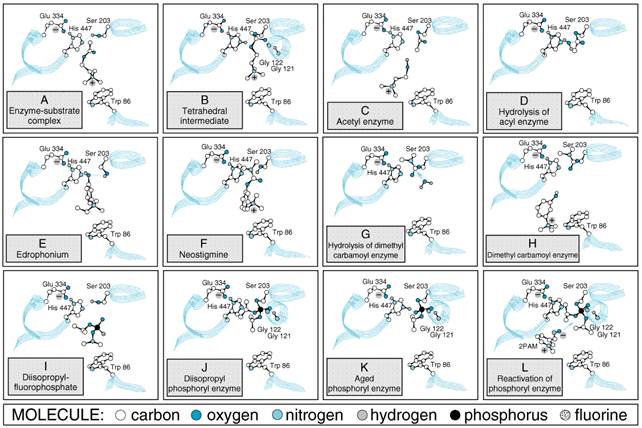
whose structural motif is the The three-dimensional structures of AChEs show the active center to be nearly centrosymmetric to each subunit and reside at the base of a narrow gorge about 20 in depth (Sussman et al., 1991; Bourne et al., 1995). At the base of the gorge lie the residues of the catalytic triad: serine 203, histidine 447, and glutamate 334 (Figure 81). The catalytic mechanism resembles that of other hydrolases, where the serine hydroxyl group is rendered highly nucleophilic through a charge-relay system involving the carboxyl from glutamate, the imidazole on the histidine, and the hydroxyl of the serine (Figure 82A).
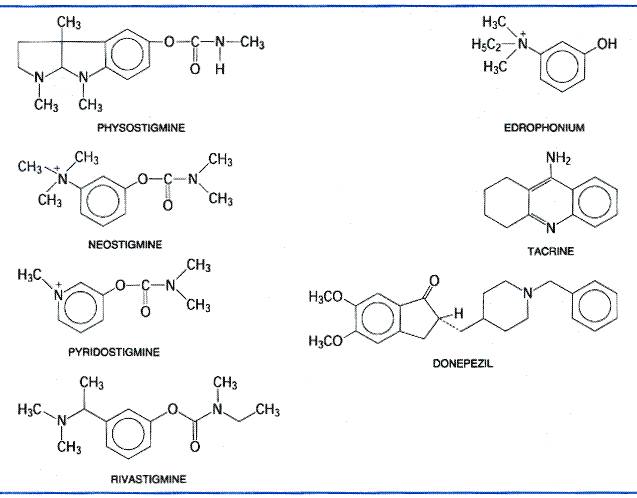
During enzymatic attack of acetylcholine, an ester with trigonal geometry, a tetrahedral intermediate between enzyme and substrate is formed (Figure 82B) that collapses to an acetyl enzyme conjugate with the concomitant release of choline (Figure 82C). The acetyl enzyme is very labile to hydrolysis, which results in the formation of acetate and active enzyme (Figure 82D; see Froede and Wilson, 1971; Rosenberry, 1975). AChE is one of the most efficient enzymes known and has the capacity to hydrolyze 6 x 105 ACh molecules per molecule of enzyme per minute; this yields a turnover time of 150 microseconds. Mechanism of Action of AChE Inhibitors The mechanisms of action of compounds that typify the three classes of anti-ChE agents also are shown in Figure 82E to L. Three distinct domains on AChE constitute binding sites for inhibitory ligands and form the basis for specificity differences between AChE and butyrylcholinesterase: the acyl pocket of the active center, the choline subsite of the active center, and the peripheral anionic site (Taylor and Radić, 1994; Reiner and Radić, 2000). Reversible inhibitors such as edrophonium and tacrine bind to the choline subsite in the vicinity of tryptophan 86 and glutamate 202 (Silman and Sussman, 2000) (Figure 82E). Edrophonium has a brief duration of action owing to its quaternary structure and the reversibility of its binding to the AChE active center. Additional reversible inhibitors, such as donepezil, bind with higher affinity to the active center. Other reversible inhibitors, such as propidium and the peptide toxin fasciculin, bind to the peripheral anionic site on AChE. This site resides at the lip of the gorge and is defined by tryptophan 286 and tyrosines 72 and 124 (Figure 81). Drugs that have a carbamoyl ester linkage, such as physostigmine and neostigmine, are hydrolyzed by AChE, but much more slowly than is ACh. Both the quaternary amine neostigmine and the tertiary amine physostigmine exist as cations at physiological pH. By serving as alternate substrates with a similar binding orientation as acetylcholine (see Figure 82F, G), attack by the active center serine gives rise to the carbamoylated enzyme. The carbamoyl moiety resides in the acyl pocket outlined by phenylalanines 295 and 297. In contrast to the acetyl enzyme, methylcarbamoyl AChE and dimethylcarbamoyl AChE are far more stable (t1/2 for hydrolysis of the dimethylcarbamoyl enzyme is 15 to 30 minutes; see Figure 82H). Sequestration of the enzyme in its carbamoylated form thus precludes the enzyme-catalyzed hydrolysis of ACh for extended periods of time. In vivo, the duration of inhibition by the carbamoylating agents is 3 to 4 hours. The organophosphorus inhibitors, such as diisopropyl fluorophosphate (DFP), serve as true hemisubstrates, since the resultant conjugate with the active center serine phosphorylated or phosphonylated is extremely stable (see Figure 82I, J, K). The organophosphorus inhibitors are tetrahedral in configuration, a configuration that resembles the transition state formed in carboxyl ester hydrolysis. Similar to the carboxyl esters, the phosphoryl oxygen binds within the oxyanion hole of the active center. If the alkyl groups in the phosphorylated enzyme are ethyl or methyl, spontaneous regeneration of active enzyme requires several hours. Secondary (as in DFP) or tertiary alkyl groups further enhance the stability of the phosphorylated enzyme, and significant regeneration of active enzyme usually is not observed. Hence, the return of AChE activity depends on synthesis of new enzyme. The stability of the phosphorylated enzyme is enhanced through 'aging,' which results from the loss of one of the alkyl groups (see Figure 82K; see also Aldridge, 1976). From the foregoing account, it is apparent that the terms reversible and irreversible as applied to the carbamoyl ester and organophosphorus anti-ChE agents, respectively, reflect only quantitative differences in rates of deacylation of the acyl enzyme. Both chemical classes react covalently with the enzyme in essentially the same manner as does ACh. Action at Effector Organs The characteristic pharmacological effects of the anti-ChE agents are due primarily to the prevention of hydrolysis of ACh by AChE at sites of cholinergic transmission. Transmitter thus accumulates, and the response to ACh that is liberated by cholinergic impulses or that is spontaneously released from the nerve ending is enhanced. Virtually all the acute effects of moderate doses of organophosphates are attributable to this action. For example, the characteristic miosis that follows local application of DFP to the eye is not observed after chronic postganglionic denervation of the eye because there is no source from which to release endogenous ACh. The consequences of enhanced concentrations of ACh at motor end-plates are unique to these sites and are discussed below. The tertiary amine and particularly the quaternary ammonium anti-ChE compounds all may have additional direct actions at certain cholinergic receptor sites. For example, the effects of neostigmine on the spinal cord and neuromuscular junction are based on a combination of its anti-ChE activity and direct cholinergic stimulation. Chemistry and StructureActivity Relationships The structureactivity relationships of anti-ChE drugs have been reviewed extensively (see previous editions of this book). Only those agents of general therapeutic or toxicological interest are considered here. Noncovalent Inhibitors While drugs of this class interact by reversible and noncovalent association with the active site in AChE, they differ in their disposition in the body and their affinity for the enzyme. Edrophonium, a quaternary drug whose activity is limited to peripheral nervous system synapses, has a moderate affinity for AChE. Its volume of distribution is limited and renal elimination is rapid, accounting for its short duration of action. By contrast, tacrine and donepezil have higher affinities for AChE, are more hydrophobic, and readily cross the bloodbrain barrier to inhibit AChE in the central nervous system (CNS). Their partitioning into lipid and their higher affinities for AChE account for their longer durations of action. 'Reversible' Carbamate Inhibitors Drugs of this class that are of therapeutic interest are shown in Figure 83. Stedman's early studies (1929a,b) showed that the essential moiety of the physostigmine molecule was the methyl carbamate of a basically substituted simple phenol. The quaternary ammonium derivative neostigmine is a compound of greater stability and equal or greater potency. Pyridostigmine is a close congener that also is employed in the treatment of myasthenia gravis.
An increase in anti-ChE potency and duration of action can result from the linking of two quaternary ammonium moieties. One such example is the miotic agent demecarium, which essentially consists of two neostigmine molecules connected by a series of ten methylene groups. The second quaternary group confers additional stability to the interaction by associating with a negatively charged amino side chain, Asp74, near the lip of the gorge. Carbamoylating inhibitors with high lipid solubilities readily cross the bloodbrain barrier and have longer durations of action. Such agents (rivastigmine) have been approved by the United States Food and Drug Administration (FDA) for the treatment of Alzheimer's disease (Giacobini, 2000; Corey-Bloom et al., 1998; see Chapter 22: Treatment of Central Nervous System Degenerative Disorders). The carbamate insecticides, carbaryl (SEVIN), propoxur (BAYGON), and aldicarb (TEMIK), which are used extensively in garden products, inhibit ChE in a fashion identical with other carbamoylating inhibitors. The symptoms of poisoning closely resemble those of the organophosphates (Baron, 1991; Ecobichon, 2000). Carbaryl has a particularly low toxicity from dermal absorption. It is used topically for control of head lice in some countries. Not all carbamates in garden formulations are cholinesterase inhibitors; the dithiocarbamates are fungicidal. Organophosphorus Compounds The general formula for this class of cholinesterase inhibitors is presented in Table 81. A great variety of substituents is possible: R1 and R2 may be alkyl, alkoxy, aryloxy, amido, mercaptan, or other groups, and X, the leaving group, a conjugate base of a weak acid, is found as a halide, cyanide, thiocyanate, phenoxy, thiophenoxy, phosphate, thiocholine, or carboxylate group. For a compilation of the organophosphorus compounds and their toxicity, see Gallo and Lawryk (1991). DFP produces virtually irreversible inactivation of AChE and other esterases by alkylphosphorylation. Its high lipid solubility, low molecular weight, and volatility facilitate inhalation, transdermal absorption, and penetration into the CNS. The 'nerve gases'tabun, sarin, and somanare among the most potent synthetic toxic agents known; they are lethal to laboratory animals in submilligram doses. Insidious employment of these agents has occurred in warfare and terrorism attacks (Nozaki and Aikawa, 1995). Because of its low volatility and stability in aqueous solution,
parathion (ETILON) became
widely used as an insecticide. Its acute and chronic toxicity has limited its
agricultural use in the Malathion (CHEMATHION MALA-SPRAY) also requires replacement of a
sulfur atom with oxygen in vivo. This insecticide can be detoxified by
hydrolysis of the carboxyl ester linkage by plasma carboxylesterases, and
plasma carboxylesterase activity dictates species resistance to malathion.
The detoxification reaction is much more rapid in mammals and birds than in
insects (see Costa et al., 1987). In recent years, malathion
has been employed in aerial spraying of relatively populous areas for control
of citrus-orchard-destructive Mediterranean fruit flies and mosquitoes that
harbor and transmit viruses harmful to human beings, such as the Among the quaternary ammonium organophosphorus compounds (group E in Table 81), only echothiophate is useful clinically and is limited to ophthalmic administration. Being positively charged, it is not volatile and does not readily penetrate the skin. Metrifonate is a low-molecular-weight organophosphate that is spontaneously converted to the active phosphoryl ester: dimethyl 2,2-dichlorovinyl phosphate (DDVP, dichlorvos). Both metrifonate and DDVP readily cross the bloodbrain barrier to inhibit AChE in the CNS. Metrifonate originally was developed for the treatment of schistosomiasis (see Chapter 42: Drugs Used in the Chemotherapy of Helminthiasis). Its capacity to inhibit AChE in the CNS and its reported low toxicity led to its clinical trial in Alzheimer's disease (Cummings et al., 1999). |
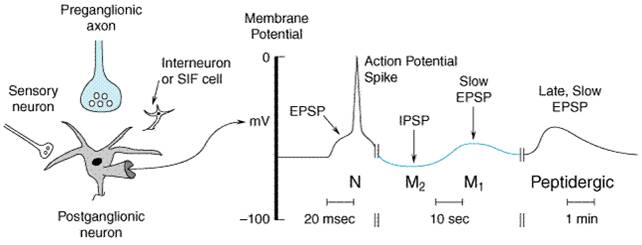
Pharmacological Properties
|
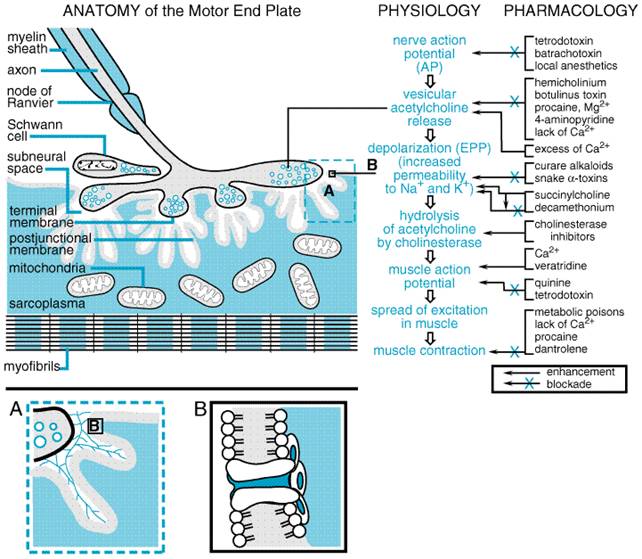
Generally, the pharmacological properties of anti-ChE agents can be predicted by knowing those loci where ACh is released physiologically by nerve impulses, the degree of nerve impulse activity, and the responses of the corresponding effector organs to ACh (see Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems). The anti-ChE agents potentially can produce all the following effects: (1) stimulation of muscarinic receptor responses at autonomic effector organs; (2) stimulation, followed by depression or paralysis, of all autonomic ganglia and skeletal muscle (nicotinic actions); and (3) stimulation, with occasional subsequent depression, of cholinergic receptor sites in the CNS. Following toxic or lethal doses of anti-ChE agents, most of these effects can be noted (see below). However, with smaller doses, particularly those used therapeutically, several modifying factors are significant. In general, compounds containing a quaternary ammonium group do not penetrate cell membranes readily; hence, anti-ChE agents in this category are absorbed poorly from the gastrointestinal tract or across the skin and are excluded from the CNS by the bloodbrain barrier after moderate doses. On the other hand, such compounds act preferentially at the neuromuscular junctions of skeletal muscle, exerting their action both as anti-ChE agents and as direct agonists. They have comparatively less effect at autonomic effector sites and ganglia. In contrast, the more lipid-soluble agents are well absorbed after oral administration, have ubiquitous effects at both peripheral and central cholinergic sites, and may be sequestered in lipids for long periods of time. The lipid-soluble organophosphorus agents also are well absorbed through the skin, and the volatile agents are transferred readily across the alveolar membrane (Storm et al., 2000). The actions of anti-ChE agents on autonomic effector cells and on cortical and subcortical sites in the CNS, where the receptors are largely of the muscarinic type, are blocked by atropine. Likewise, atropine blocks some of the excitatory actions of anti-ChE agents on autonomic ganglia, since both nicotinic and muscarinic receptors are involved in ganglionic neurotransmission (see Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia). The sites of action of anti-ChE agents of therapeutic importance are the CNS, eye, intestine, and the neuromuscular junction of skeletal muscle; other actions are of toxicological consequence. Eye When applied locally to the conjunctiva, anti-ChE agents cause conjunctival hyperemia and constriction of the sphincter pupillae muscle around the pupillary margin of the iris (miosis) and the ciliary muscle (block of accommodation reflex with resultant focusing to near vision). Miosis is apparent in a few minutes and can last several hours to days. Although the pupil may be 'pinpoint' in size, it generally contracts further when exposed to light. The block of accommodation is more transient and generally disappears before termination of the miosis. Intraocular pressure, when elevated, usually falls as the result of facilitation of outflow of the aqueous humor (see Chapter 66: Ocular Pharmacology). Gastrointestinal Tract In human beings, neostigmine enhances gastric contractions and increases the secretion of gastric acid. After bilateral vagotomy, the effects of neostigmine on gastric motility are greatly reduced. The lower portion of the esophagus is stimulated by neostigmine; in patients with marked achalasia and dilation of the esophagus, the drug can cause a salutary increase in tone and peristalsis. Neostigmine augments the motor activity of the small and large bowel; the colon is particularly stimulated. Atony produced by muscarinic-receptor antagonists or prior surgical intervention may be overcome, propulsive waves are increased in amplitude and frequency, and movement of intestinal contents is thus promoted. The total effect of anti-ChE agents on intestinal motility probably represents a combination of actions at the ganglion cells of Auerbach's plexus and at the smooth muscle fibers, as a result of the preservation of ACh released by the cholinergic preganglionic and postganglionic fibers, respectively. Neuromuscular Junction Most of the effects of potent anti-ChE drugs on skeletal muscle can be explained adequately on the basis of their inhibition of AChE at neuromuscular junctions. However, there is good evidence for an accessory direct action of neostigmine and other quaternary ammonium anti-ChE agents on skeletal muscle. For example, the intraarterial injection of neostigmine into chronically denervated muscle, or muscle in which AChE has been inactivated by prior administration of DFP, evokes an immediate contraction, whereas physostigmine does not. Normally, a single nerve impulse in a terminal motor-axon branch
liberates enough ACh to produce a localized depolarization (end-plate
potential) of sufficient magnitude to initiate a propagated muscle action
potential. The ACh released is rapidly hydrolyzed by AChE, such that the
lifetime of free ACh within the synapse ( The anti-ChE agents will reverse the antagonism caused by competitive neuromuscular blocking agents (see Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia). Neostigmine normally is not effective against the skeletal muscle paralysis caused by succinylcholine, since this agent also produces neuromuscular blockade by depolarization. Actions at Other Sites Secretory glands that are innervated by postganglionic cholinergic fibers include the bronchial, lacrimal, sweat, salivary, gastric (antral G cells and parietal cells), intestinal, and pancreatic acinar glands. Low doses of anti-ChE agents augment secretory responses to nerve stimulation, and higher doses actually produce an increase in the resting rate of secretion. Anti-ChE agents increase contraction of smooth muscle fibers of the bronchioles and ureters, and the ureters may show increased peristaltic activity. The cardiovascular actions of anti-ChE agents are complex, since they reflect both ganglionic and postganglionic effects of accumulated ACh on the heart and blood vessels. The predominant effect on the heart from the peripheral action of accumulated ACh is bradycardia, resulting in a fall in cardiac output. Higher doses usually cause a fall in blood pressure, often as a consequence of effects of anti-ChE agents on the medullary vasomotor centers of the CNS. Anti-ChE agents augment vagal influences on the heart. This shortens the effective refractory period of atrial muscle fibers, and increases the refractory period and conduction time at the SA and AV nodes. At the ganglionic level, accumulating ACh initially is excitatory on nicotinic receptors, but at higher concentrations, ganglionic blockade ensues as a result of persistent depolarization of the cell membrane. The excitatory action on the parasympathetic ganglion cells would tend to reinforce the diminished cardiac output, whereas the opposite sequence would result from the action of ACh on sympathetic ganglion cells. Excitation followed by inhibition also is elicited by ACh at the medullary vasomotor and cardiac centers. All of these effects are complicated further by the hypoxemia resulting from the bronchoconstrictor and secretory actions of increased ACh on the respiratory system; hypoxemia, in turn, would reinforce both sympathetic tone and ACh-induced discharge of epinephrine from the adrenal medulla. Hence, it is not surprising that an increase in heart rate is seen with severe cholinesterase inhibitor poisoning. Hypoxemia probably is a major factor in CNS depression that appears after large doses of anti-ChE agents. The CNS-stimulant effects are antagonized by atropine, although not as completely as are the muscarinic effects at peripheral autonomic effector sites. Absorption, Fate, and Excretion Physostigmine is absorbed readily from the gastrointestinal tract, subcutaneous tissues, and mucous membranes. The conjunctival instillation of solutions of the drug may result in systemic effects if measures (e.g., pressure on inner canthus) are not taken to prevent absorption from the nasal mucosa. Physostigmine, administered parenterally, is largely destroyed in the body within 2 hours, mainly by hydrolytic cleavage by plasma esterases; renal excretion plays only a minor role in its elimination. Neostigmine and pyridostigmine are absorbed poorly after oral administration, such that much larger doses are needed than by the parenteral route. Whereas the effective parenteral dose of neostigmine is 0.5 to 2 mg, the equivalent oral dose may be 15 to 30 mg or more. Neostigmine and pyridostigmine are destroyed by plasma esterases, and the quaternary alcohols and parent compounds are excreted in the urine; the half-life of these drugs is only 1 to 2 hours (Cohan et al., 1976). Organophosphorus anti-ChE agents with the highest risk of toxicity are highly lipid-soluble liquids; many have high vapor pressures. The less volatile agents that are commonly used as agricultural insecticides (e.g., parathion, malathion) generally are dispersed as aerosols or as dusts adsorbed to an inert, finely particulate material. Consequently, the compounds are absorbed rapidly through the skin and mucous membranes following contact with moisture, by the lungs after inhalation, and by the gastrointestinal tract after ingestion (Storm et al., 2000). Following their absorption, most organophosphorus compounds are excreted almost entirely as hydrolysis products in the urine. Plasma and liver esterases are responsible for hydrolysis to the corresponding phosphoric and phosphonic acids. However, the cytochrome P450s are responsible for converting the inactive phosphorothioates containing a phosphorus-sulfur (thiono) bond to phosphorates with a phosphorus-oxygen bond, resulting in their activation. These mixed-function oxidases also play a role in deactivation of certain organophosphorus agents. The organophosphorus anti-ChE agents are hydrolyzed in the body by two families of enzymes known as the carboxylesterases and the paraoxonases (A-esterases). These enzymes are found in the plasma and liver and scavenge or hydrolyze a large number of organophosphorus compounds (paraoxon, DFP, TEPP, chlorpyrifos, oxon, tabun, sarin) by cleaving the phosphoester, anhydride, PF, or PCN bonds. The paraoxonases are metalloenzymes not related in structure to the cholinesterases and do not appear to form stable intermediates with organophosphates. They are associated with high-density lipoproteins and may prevent oxidation of endogenous lipids (La Du et al., 1999). A genetic polymorphism (Arg192Gln) that governs organophosphate substrate specificity has been found (Furlong et al., 2000). Wide variations in paraoxonase activity exist among animal species. Young animals are deficient in carboxylesterases and paraoxonases, and this may account for age-related toxicities seen in newborn animals and suspected to be a basis for toxicity in human beings (Padilla et al., 2000). In addition, plasma and hepatic carboxylesterases (aliesterases) and plasma butyrylcholinesterase are inhibited irreversibly by organophosphorus compounds (Lockridge and Masson, 2000); their scavenging capacity for the organophosphates can afford partial protection against inhibition of acetylcholinesterase in the nervous system. The carboxylesterases also catalyze hydrolysis of malathion and other organophosphorus compounds that contain carboxyl-ester linkages, rendering them less active or inactive. Since carboxylesterases are inhibited by organophosphates, toxicity from exposure to two organophosphorus insecticides can be synergistic. |
Toxicology
|
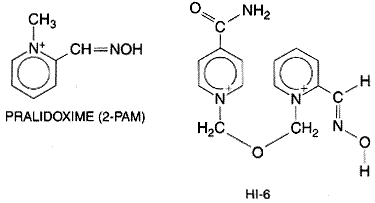
The toxicological aspects of the anti-ChE agents are of practical importance to the physician. In addition to numerous cases of accidental intoxication from the use and manufacture of organophosphorus compounds as agricultural insecticides (over 40 have been approved for use in the United States), these agents have been used frequently for homicidal and suicidal purposes, largely because of their accessibility. Organophosphorus agents account for as much as 80% of pesticide-related hospital admissions. The World Health Organization documents pesticide toxicity as a widespread global problem; most poisonings occur in developing countries (Bardin et al., 1994; Landrigan et al., 2000). Occupational exposure occurs most commonly by the dermal and pulmonary routes, while oral ingestion is most common in cases of nonoccupational poisoning. In the Acute Intoxication The effects of acute intoxication by anti-ChE agents are manifested by muscarinic and nicotinic signs and symptoms and, except for compounds of extremely low lipid solubility, by signs referable to the CNS. Systemic effects appear within minutes after inhalation of vapors or aerosols. In contrast, the onset of symptoms is delayed after gastrointestinal and percutaneous absorption. The duration of effects is determined largely by the properties of the compound: its lipid solubility, whether or not it must be activated to form the oxon, the stability of the organophosphorus-AChE bond, and whether or not 'aging' of the phosphorylated enzyme has occurred. After local exposure to vapors or aerosols or after their inhalation, ocular and respiratory effects generally appear first. Ocular effects include marked miosis, ocular pain, conjunctival congestion, diminished vision, ciliary spasm, and brow ache. With acute systemic absorption, miosis may not be evident due to sympathetic discharge in response to hypotension. In addition to rhinorrhea and hyperemia of the upper respiratory tract, respiratory effects consist of 'tightness' in the chest and wheezing respiration, caused by the combination of bronchoconstriction and increased bronchial secretion. Gastrointestinal symptoms occur earliest after ingestion, and include anorexia, nausea and vomiting, abdominal cramps, and diarrhea. With percutaneous absorption of liquid, localized sweating and muscle fasciculations in the immediate vicinity are generally the earliest manifestations. Severe intoxication is manifested by extreme salivation, involuntary defecation and urination, sweating, lacrimation, penile erection, bradycardia, and hypotension. Nicotinic actions at the neuromuscular junctions of skeletal muscle usually consist of fatigability and generalized weakness, involuntary twitchings, scattered fasciculations, and eventually severe weakness and paralysis. The most serious consequence is paralysis of the respiratory muscles. The broad spectrum of effects on the CNS includes confusion, ataxia, slurred speech, loss of reflexes, CheyneStokes respiration, generalized convulsions, coma, and central respiratory paralysis. Actions on the vasomotor and other cardiovascular centers in the medulla oblongata lead to hypotension. The time of death after a single acute exposure may range from less than 5 minutes to nearly 24 hours, depending upon the dose, route, agent, and other factors. The cause of death primarily is respiratory failure, usually accompanied by a secondary cardiovascular component. Peripheral muscarinic and nicotinic as well as central actions all contribute to respiratory embarrassment; effects include laryngospasm, bronchoconstriction, increased tracheobronchial and salivary secretions, compromised voluntary control of the diaphragm and intercostal muscles, and central respiratory depression. Blood pressure may fall to alarmingly low levels and cardiac irregularities intervene. These effects usually result from hypoxemia; they often are reversed by assisted pulmonary ventilation. Delayed symptoms appearing after one to four days and marked by persistent low blood cholinesterase and severe muscle weakness are termed the intermediate syndrome (Marrs, 1993; DeBleecker et al., 1992, 1995). A delayed neurotoxicity also may be evident after severe intoxication (see below). Diagnosis and Treatment The diagnosis of severe, acute anti-ChE intoxication is made readily from the history of exposure and the characteristic signs and symptoms. In suspected cases of milder acute or chronic intoxication, determination of the ChE activities in erythrocytes and plasma generally will establish the diagnosis (Storm et al., 2000). Although these values vary considerably in the normal population, they usually will be depressed well below the normal range before symptoms are evident. Treatment is both specific and effective. Atropine in sufficient dosage (see below) effectively antagonizes the actions at muscarinic receptor sites, including increased tracheobronchial and salivary secretion, bronchoconstriction, bradycardia, and, to a moderate extent, peripheral ganglionic and central actions. Larger doses are required to get appreciable concentrations of atropine into the CNS. Atropine is virtually without effect against the peripheral neuromuscular compromise. The last-mentioned action of the anti-ChE agents as well as all other peripheral effects can be reversed by pralidoxime (2-PAM), a cholinesterase reactivator. In moderate or severe intoxication with an organophosphorus anti-ChE agent, the recommended adult dose of pralidoxime is 1 to 2 g, infused intravenously within not less than 5 minutes. If weakness is not relieved or if it recurs after 20 to 60 minutes, the dose may be repeated. Early treatment is very important to assure that the oxime reaches the phosphorylated AChE while the latter still can be reactivated. Many of the alkylphosphates are extremely lipid-soluble, and if extensive partitioning into body fat has occurred, toxicity will persist and symptoms may recur after initial treatment. In some cases it has been necessary to continue treatment with atropine and pralidoxime for several weeks. In addition, general supportive measures are important. These include (1) termination of exposure, by removal of the patient or application of a gas mask if the atmosphere remains contaminated, removal and destruction of contaminated clothing, copious washing of contaminated skin or mucous membranes with water, or gastric lavage; (2) maintenance of a patent airway, including endobronchial aspiration; (3) artificial respiration if required; (4) administration of oxygen; (5) alleviation of persistent convulsions with diazepam (5 to 10 mg, intravenously); and (6) treatment of shock (Marrs, 1993; Bardin et al., 1994). Atropine should be given in doses sufficient to cross the bloodbrain barrier. Following an initial injection of 2 to 4 mg, given intravenously if possible, otherwise intramuscularly, 2 mg should be given every 5 to 10 minutes until muscarinic symptoms disappear, if they reappear, or until signs of atropine toxicity appear. More than 200 mg may be required on the first day. A mild degree of atropine block then should be maintained for up to 48 hours or as long as symptoms are evident. Whereas the AChE reactivators can be of great benefit in the therapy of anti-ChE intoxication (see below), their use must be regarded as a supplement to the administration of atropine. Cholinesterase Reactivators Although the phosphorylated esteratic site of AChE undergoes
hydrolytic regeneration at a slow or negligible rate, Several bis-quaternary oximes were shown subsequently to be
even more potent as reactivators for insecticide and nerve gas poisoning (see
below); an example is HI-6, which is used in
The velocity of reactivation of phosphorylated AChE by oximes depends on their accessibility to the active center serine (Wong et al., 2000). Furthermore, certain phosphorylated AChEs can undergo a fairly rapid process of 'aging,' so that within the course of minutes or hours they become completely resistant to the reactivators. 'Aging' probably is due to the loss of one alkoxy group, leaving a much more stable monoalkyl- or monoalkoxy-phosphoryl-AChE (Fleisher and Harris, 1965; see Figure 82K). Organophosphorus compounds containing tertiary alkoxy groups are more prone to 'aging' than are the congeners containing the secondary or primary alkoxy groups (Aldridge, 1976). The oximes are not effective in antagonizing the toxicity of the more rapidly hydrolyzing carbamoyl ester inhibitors, and since pralidoxime itself has weak anti-ChE activity, they are not recommended for the treatment of overdosage with neostigmine or physostigmine and are contraindicated in poisoning with carbamoylating insecticides such as carbaryl. Pharmacology, Toxicology, and Disposition The reactivating action of oximes in vivo is most marked at the skeletal neuromuscular junction. Following a dose of an organophosphorus compound that produces total blockade of transmission, the intravenous injection of an oxime can restore the response to stimulation of the motor nerve within a few minutes. Antidotal effects are less striking at autonomic effector sites, and the quaternary ammonium group restricts entry into the CNS. High doses of pralidoxime and related compounds can in themselves cause neuromuscular blockade and inhibition of AChE; such actions are minimal at the dose rates recommended as an antidote. If pralidoxime is injected intravenously at a rate more rapid than 500 mg per minute, it can cause mild weakness, blurred vision, diplopia, dizziness, headache, nausea, and tachycardia. The oximes as a group are metabolized largely by the liver, and the breakdown products are excreted by the kidney. Delayed Neurotoxicity of Organophosphorus Compounds Certain fluorine-containing alkylorganophosphorus anti-ChE agents (e.g.,
DFP, mipafox) have in common with the triarylphosphates, of which triorthocresylphosphate
(TOCP) is the classical example, the property of inducing delayed
neurotoxicity. This syndrome first received widespread attention following
the demonstration that TOCP, an adulterant of The clinical picture is that of a severe polyneuropathy that begins several days after a single exposure to the toxic compound. It is manifested initially by mild sensory disturbances, ataxia, weakness, muscle fatigue and twitching, reduced tendon reflexes, and tenderness to palpation. In severe cases, the weakness may progress eventually to complete flaccid paralysis, which, over the course of weeks or months, is often succeeded by a spastic paralysis with a concomitant exaggeration of reflexes. During these phases, the muscles show marked wasting. Recovery may require several years and may be incomplete. Because only certain triarylphosphates and fluorine-containing alkylphosphates have the greatest propensity to produce the organophosphate-induced delayed polyneuropathy (OPIDR), toxicity is not dependent upon inhibition of AChE or other cholinesterases. Evidence points to inhibition of a different esterase, termed a neurotoxic esterase, as being linked to the lesions (Johnson, 1993). The enzyme has been isolated and its gene cloned. Its substrate specificity is directed to hydrophobic esters, but its natural substrate and function are unknown (Glynn, 2000). Experimental myopathies that result in generalized necrotic lesions and changes in end-plate cytostructure also are found after long-term exposure to organophosphates (Dettbarn, 1984; DeBleeker et al., 1992). |
Therapeutic Uses
|
Although anti-ChE agents have been recommended for the treatment of a wide variety of conditions involving the peripheral nervous system, their widespread acceptability has been established mainly in four areas: atony of the smooth muscle of the intestinal tract and urinary bladder, glaucoma, myasthenia gravis, and termination of the effects of competitive neuromuscular blocking drugs (see Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia). Long-acting and hydrophobic cholinesterase inhibitors are the only inhibitors with efficacy, albeit limited, in the treatment of dementia symptoms of Alzheimer's disease. Physostigmine, with its shorter duration of action, is useful in the treatment of intoxication by atropine and several drugs with anticholinergic side effects (see below); it also is indicated for the treatment of Friedreich's or other inherited ataxias. Edrophonium can be used for terminating attacks of paroxysmal supraventricular tachycardia. Available Therapeutic Agents The compounds described here are those commonly used as anti-ChE drugs
and cholinesterase reactivators in the Physostigmine salicylate ANTILIRIUM) is available for injection. Physostigmine sulfate ophthalmic ointment and physo-stigmine salicylate ophthalmic solution also are available. Pyridostigmine bromide is available for oral (MESTINON) or parenteral (REGONOL, MESTINON) use. Neostigmine bromide (PROSTIGMIN) is available for oral use. Neostigmine methylsulfate (PROSTIGMIN) is marketed for parenteral injection. Ambenonium chloride (MYTELASE) is available for oral use. Edrophonium chloride (TENSILON, others) is marketed for parenteral injection. Tacrine (COGNEX), donepezil (ARICEPT), rivastigmine (EXELON), and galantamine (REMINYL) have been approved for the treatment of Alzheimer's disease. Pralidoxime chloride PROTOPAM CHLORIDE) is the only AChE reactivator currently available in the Paralytic Ileus and Atony of the Urinary Bladder In the treatment of both these conditions, neostigmine generally is the most satisfactory of the anti-ChE agents. The direct parasympathomimetic agents, discussed in Chapter 7: Muscarinic Receptor Agonists and Antagonists, are employed for the same purposes. Neostigmine is used for the relief of abdominal distension and acute colonic pseudoobstruction from a variety of medical and surgical causes (Ponec et al., 1999). The usual subcutaneous dose of neostigmine methylsulfate for postoperative paralytic ileus is 0.5 mg, given as needed. Peristaltic activity commences 10 to 30 minutes after parenteral administration, whereas 2 to 4 hours are required after oral administration of neostigmine bromide (15 to 30 mg). A rectal tube should be inserted to facilitate expulsion of gas, and it may be necessary to assist evacuation with a small low enema. The drug should not be used when the intestine or urinary bladder is obstructed, when peritonitis is present, when the viability of the bowel is doubtful, or when bowel dysfunction is a consequence of inflammatory disease. When neostigmine is used for the treatment of atony of the detrusor muscle of the urinary bladder, postoperative dysuria is relieved, and the time interval between operation and spontaneous urination is shortened. The drug is used in a similar dose and manner as in the management of paralytic ileus. Glaucoma and Other Ophthalmologic Indications Glaucoma is a disease complex characterized chiefly by an increase in intraocular pressure that, if sufficiently high and persistent, leads to damage to the optic disc at the juncture of the optic nerve and the retina; irreversible blindness can result. Of the three types of glaucomaprimary, secondary, and congenitalanti-ChE agents are of value in the management of the primary as well as of certain categories of the secondary type (e.g., aphakic glaucoma, following cataract extraction); the congenital type rarely responds to any therapy other than surgery. Primary glaucoma is subdivided into narrow-angle (acute congestive) and wide-angle (chronic simple) types, based on the configuration of the angle of the anterior chamber where reabsorption of the aqueous humor occurs. Narrow-angle glaucoma is nearly always a medical emergency in which drugs are essential in controlling the acute attack, but the long-range management is often surgical (e.g., peripheral or complete iridectomy). Wide-angle glaucoma, on the other hand, has a gradual, insidious onset and is not generally amenable to surgical improvement; in this type, control of intraocular pressure usually is dependent upon continuous drug therapy. Since the cholinergic agonists and cholinesterase inhibitors also
block accommodation and induce myopia, these agents produce transient
blurring of far vision and loss of vision at the margin when instilled in the
eye. With long-term administration of the cholinergic agonists and anti-ChE
agents, the compromise of vision diminishes. Nevertheless, other agents
without these side effects, such as Anti-ChE agents have been employed locally in the treatment of a variety of other ophthalmologic conditions, including accommodative esotropia and myasthenia gravis confined to the extraocular and eyelid muscles. Adie (or tonic pupil) syndrome results from dysfunction of the ciliary body, perhaps because of local nerve degeneration. Low concentrations of physostigmine are reported to decrease the blurred vision and pain associated with this condition. In alternation with a mydriatic drug such as atropine, short-acting anti-ChE agents have proven useful for the breaking of adhesions between the iris and the lens or cornea. (For a complete account of the use of anti-ChE agents in ocular therapy, see Chapter 66: Ocular Pharmacology.) Myasthenia Gravis Myasthenia gravis is a neuromuscular disease characterized by weakness
and marked fatigability of skeletal muscle (see Drachman, 1994);
exacerbations and partial remissions occur frequently. Jolly (1895) noted the
similarity between the symptoms of myasthenia gravis and curare poisoning in
animals and suggested that physostigmine, an agent then known to antagonize
curare, might be of therapeutic value. Forty years elapsed before his
suggestion was given systematic trial ( The defect in myasthenia gravis is in synaptic transmission at the neuromuscular junction. When a motor nerve of a normal subject is stimulated at 25 Hz, electrical and mechanical responses are well sustained. A suitable margin of safety exists for maintenance of neuromuscular transmission. Initial responses in the myasthenic patient may be normal, but they diminish rapidly, which explains the difficulty in maintaining voluntary muscle activity for more than brief periods. The relative importance of prejunctional and postjunctional defects in myasthenia gravis was a matter of considerable debate until Patrick and Lindstrom (1973) found that rabbits immunized with the nicotinic receptor purified from electric eels slowly developed muscular weakness and respiratory difficulties that resembled the symptoms of myasthenia gravis. The rabbits also exhibited decremental responses following repetitive nerve stimulation, enhanced sensitivity to curare, and symptomatic and electrophysiological improvement of neuromuscular transmission following administration of anti-ChE agents. Although this experimental allergic myasthenia gravis and the naturally occurring disease differ somewhat, this animal model prompted intense investigation into whether or not the natural disease represented an autoimmune response directed toward the ACh receptor. Antireceptor antibody soon was identified in patients with myasthenia gravis (Almon et al., 1974). Receptor-binding antibodies now are detectable in sera of 90% of patients with the disease, although the clinical status of the patient does not correlate precisely with the antibody titer (Drachman et al., 1982; Drachman, 1994; Lindstrom, 2000). The picture that emerges is that myasthenia gravis is caused by an
autoimmune response primarily to the ACh receptor at the postjunctional
end-plate. Antibodies, which also are present in plasma, reduce the number of
receptors detectable either by snake In a subset of approximately 10% of patients presenting with a myasthenic syndrome, muscle weakness has a congenital rather than an autoimmune basis. Characterization of biochemical and genetic bases of the congenital condition has shown mutations to occur in the acetylcholine receptor which affect ligand-binding and channel-opening kinetics (Engel et al., 1998). Other mutations occur as a deficiency in the form of acetylcholinesterase that contains the collagen-like tail unit (Ohno et al., 2000). As expected, following administration of anti-ChE agents (see below), subjective improvement is not seen in most congenital myasthenic patients. Diagnosis Although the diagnosis of autoimmune myasthenia gravis usually can be made from the history, signs, and symptoms, its differentiation from certain neurasthenic, infectious, endocrine, congenital, neoplastic, and degenerative neuromuscular diseases is challenging. However, myasthenia gravis is the only condition in which the aforementioned deficiencies can be improved dramatically by anti-ChE medication. The edrophonium test for evaluation of possible myasthenia gravis is performed by rapid intravenous injection of 2 mg of edrophonium chloride, followed 45 seconds later by an additional 8 mg if the first dose is without effect; a positive response consists of brief improvement in strength, unaccompanied by lingual fasciculation (which generally occurs in nonmyasthenic patients). An excessive dose of an anti-ChE drug results in a cholinergic crisis. The condition is characterized by weakness resulting from generalized depolarization of the motor end-plate; other features result from overstimulation of muscarinic receptors. The weakness resulting from depolarization block may resemble myasthenic weakness, which is manifest when anti-ChE medication is insufficient. The distinction is of obvious practical importance, since the former is treated by withholding, and the latter by administering, the anti-ChE agent. When the edrophonium test is performed cautiously, limiting the dose to 2 mg and with facilities for respiratory resuscitation immediately available, a further decrease in strength indicates cholinergic crisis, while improvement signifies myasthenic weakness. Atropine sulfate, 0.4 to 0.6 mg or more intravenously, should be given immediately if a severe muscarinic reaction ensues (for complete details, see Osserman et al., 1972; Drachman, 1994). Detection of antireceptor antibodies in muscle biopsies or plasma is now widely employed to confirm the diagnosis. Treatment Pyridostigmine, neostigmine, and ambenonium are the standard anti-ChE drugs used in the symptomatic treatment of myasthenia gravis. All can increase the response of myasthenic muscle to repetitive nerve impulses, primarily by the preservation of endogenous ACh; with equivalent release of ACh, receptors over a greater cross-sectional area of the end-plate then presumably are exposed to concentrations of ACh that are sufficient for channel opening and production of a postsynaptic end-plate potential. When the diagnosis of myasthenia gravis has been established, the optimal single oral dose of an anti-ChE agent can be determined empirically. Baseline recordings are made for grip strength, vital capacity, and a number of signs and symptoms that reflect the strength of various muscle groups. The patient then is given an oral dose of pyridostigmine (30 to 60 mg), neostigmine (7.5 to 15 mg), or ambenonium (2.5 to 5 mg). The improvement in muscle strength and changes in other signs and symptoms are noted at frequent intervals until there is a return to the basal state. After an hour or longer in the basal state, the drug is given again with the dose increased to one and one-half times the initial amount, and the same observations are repeated. This sequence is continued, with increasing increments of one-half the initial dose, until an optimal response is obtained. The duration of action of these drugs is such that the interval between oral doses required to maintain a reasonably even level of strength usually is 2 to 4 hours for neostigmine, 3 to 6 hours for pyridostigmine, or 3 to 8 hours for ambenonium. However, the dose required may vary from day to day, and physical or emotional stress, intercurrent infections, and menstruation usually necessitate an increase in the frequency or size of the dose. In addition, unpredictable exacerbations and remissions of the myasthenic state may require adjustment of the dosage upward or downward. Although all patients with myasthenia gravis should be seen by a physician at regular intervals, most can be taught to modify their dosage regimens according to their changing requirements. Pyridostigmine is available in sustained-release tablets containing a total of 180 mg, of which 60 mg is released immediately and 120 mg over several hours; this preparation is of value in maintaining patients for 6- to 8-hour periods, but should be limited to use at bedtime. Muscarinic cardiovascular and gastrointestinal side effects of anti-ChE agents generally can be controlled by atropine or other anticholinergic drugs (see Chapter 7: Muscarinic Receptor Agonists and Antagonists). However, these anticholinergic drugs mask many side effects of an excessive dose of an anticholinesterase agent. In most patients, tolerance develops eventually to the muscarinic effects, so that anticholinergic medication is not necessary. A number of drugs, including curariform agents and certain antibiotics and general anesthetics, interfere with neuromuscular transmission (see Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia); their administration to patients with myasthenia gravis is hazardous without proper adjustment of anti-ChE dosage and other appropriate precautions. Other therapeutic measures should be considered as essential elements in the management of this disease. Controlled studies reveal that corticosteroids promote clinical improvement in a high percentage of patients. However, when treatment with steroids is continued over prolonged periods, a high incidence of side effects may result (see Chapter 60: Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones). Gradual lowering of maintenance doses and alternate-day regimens of short-acting steroids are used to minimize side effects. Initiation of steroid treatment augments muscle weakness; however, as the patient improves with continued administration of steroids, doses of anti-ChE drugs can be reduced (Drachman, 1994). Other immunosuppressive agents such as azathioprine and cyclosporine also have been beneficial in more advanced cases. Thymectomy should be considered in myasthenia associated with a thymoma or when the disease is not controlled adequately by anti-ChE agents and steroids. The relative risks and benefits of the surgical procedure versus anti-ChE and corticosteroid treatment require careful assessment in each case. Since the thymus contains myoid cells with nicotinic receptors (Schluep et al., 1987) and a predominance of patients have thymic abnormalities, the thymus may be responsible for the initial pathogenesis. It also is the source of autoreactive T helper cells. However, the thymus is not required for perpetuation of the condition. In keeping with the presumed autoimmune etiology of myasthenia gravis, plasmapheresis and immune therapy have produced beneficial results in patients who have remained disabled despite thymectomy and treatment with steroids and anti-ChE agents (Drachman, 1994, 1996). Improvement in muscle strength correlates with the reduction of the titer of antibody directed against the nicotinic cholinergic receptor. Prophylaxis in Cholinesterase Inhibitor Poisoning Studies in experimental animals have shown that pretreatment with pyridostigmine
reduces the incapacitation and mortality associated with 'nerve
agent' poisoning, particularly for agents, such as soman, that show
rapid aging. The first large-scale administration of pyridostigmine to human
beings occurred in 1990 in anticipation of nerve-agent attack in the Intoxication by Anticholinergic Drugs In addition to atropine and other muscarinic agents, many other drugs, such as the phenothiazines, antihistamines, and tricyclic antidepressants, have central as well as peripheral anticholinergic activity. Physostigmine is potentially useful in reversing the central anticholinergic syndrome produced by overdosage or an unusual reaction to these drugs (Nilsson, 1982). The effectiveness of physostigmine in reversing the anticholinergic effects of these agents has been clearly documented. However, other toxic effects of the tricyclic antidepressants and phenothiazines (see Chapters 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders and 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania), such as intraventricular conduction deficits and ventricular arrhythmias, are not reversed by physostigmine. In addition, physostigmine may precipitate seizures; hence, its usually small potential benefit must be weighed against this risk. The initial intravenous or intramuscular dose of physostigmine is 2 mg, with additional doses given as necessary. Physostigmine, a tertiary amine, crosses the bloodbrain barrier, in contrast to the quaternary anti-AChE drugs. The use of anti-ChE agents to reverse the effects of competitive neuromuscular blocking agents is discussed in Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia. Alzheimer's Disease A deficiency of intact cholinergic neurons, particularly those extending from subcortical areas such as the nucleus basalis of Maynert, has been observed in patients with progressive dementia of the Alzheimer's type (Markesbery, 1998). Using a rationale similar to that in other CNS degenerative diseases (see Chapter 22: Treatment of Central Nervous System Degenerative Disorders), therapy for enhancing concentrations of cholinergic neurotransmitters in the central nervous system was investigated (Mayeux and Sano, 1999). In 1993, the FDA approved tacrine (tetrahydroaminoacridine) for use in mild to moderate Alzheimer's disease, but a high incidence of hepatotoxicity and frequent liver function tests limit the efficacy of this drug. About 30% of the patients receiving low doses of tacrine within three months have alanine aminotransferase values of three times normal; upon discontinuing the drug, liver function values return to normal in 90% of the patients. Other side effects are typical of acetylcholinesterase inhibitors. More recently, donepezil was approved for clinical use. There are efficacy data from multiple trials, most involving several hundred patients (Dooley and Lamb, 2000). At 5- and 10-mg daily oral doses, improved cognition and global clinical function were seen in the 21- to 81-week intervals studied. In long-term studies, the drug delayed symptomatic progression of the disease for periods up to 55 weeks. Side effects are largely attributable to excessive cholinergic stimulation, with nausea, diarrhea, and vomiting being most frequently reported. The drug is well tolerated in single daily doses. Usually, 5-mg doses are administered at night for 4 to 6 weeks; if this dose is well tolerated, the dose can be increased to 10 mg daily. Rivastigmine,
a long-acting carbamoylating inhibitor, recently has been approved for use in
the Therapeutic strategies with new compounds are directed at maximizing the ratio of central to peripheral cholinesterase inhibition and the use of cholinesterase inhibitors in conjunction with selective cholinergic agonists and antagonists. Combination therapy with agents that are directed to slowing the progression of the degenerative disease also are being considered. |
Chapter 9. Agents Acting at the Neuromuscular Junction and Autonomic Ganglia
Overview
|
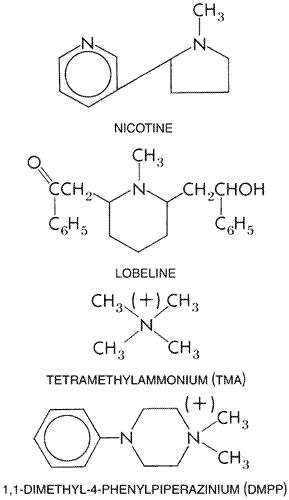
The nicotinic acetylcholine receptor mediates neurotransmission at the neuromuscular junction and peripheral autonomic ganglia; in the central nervous system, it largely controls release of neurotransmitters from presynaptic sites. This chapter focuses on agonists and antagonists at the nicotinic acetylcholine receptor and their clinical utility at the neuromuscular junction or autonomic ganglia. The text begins with an overview of current structural and functional insights regarding the nicotinic acetylcholine receptor and its subtypes. A variety of neuromuscular blocking agents with varying mechanisms of blockade and pharmacokinetic properties are used to produce muscle relaxation during anesthesia (see also Chapter 14: General Anesthetics). Nicotine transiently stimulates nicotinic receptors on ganglia but is best known for its addictive properties arising from its presynaptic actions influencing neurotransmitter release in the brain (see Chapter 24: Drug Addiction and Drug Abuse). The use of ganglionic blocking agents for management of hypertension has been eclipsed by superior agents (see Chapter 33: Antihypertensive Agents and the Drug Therapy of Hypertension), although these agents are sometimes useful alternatives when other agents fail to control blood pressure in life-threatening circumstances (e.g., in the case of an acute dissecting aortic aneurysm) and in surgery where controlled hypotension is indicated. |
Agents Acting at the Neuromuscular Junction and Autonomic Ganglia: Introduction
|
Several drugs have as their major action the interruption or mimicry of transmission of the nerve impulse at the neuromuscular junction of skeletal muscle and/or autonomic ganglia. These agents can be classified together, since they interact with a common family of receptors; these receptors are called nicotinic acetylcholine (also commonly called nicotinic cholinergic) receptors, since they are stimulated by both the neurotransmitter acetylcholine (ACh) and the alkaloid nicotine. Distinct subtypes of nicotinic receptors exist at the neuromuscular junction and the ganglia, and several pharmacological agents that act at these receptors discriminate between them. Neuromuscular blocking agents are distinguished by whether or not they cause depolarization of the motor end plate and, for this reason, are classified either as competitive (stabilizing) agents, of which curare is the classical example, or as depolarizing agents, such as succinylcholine. The competitive and depolarizing agents are used widely to achieve muscle relaxation during anesthesia. Ganglionic agents act by stimulating or blocking nicotinic receptors on the postganglionic neuron. |
The Nicotinic Acetylcholine Receptor
|
The concept of the nicotinic acetylcholine receptor, with which ACh combines to initiate the end-plate potential (EPP) in muscle or an excitatory postsynaptic potential (EPSP) in nerve, is introduced in Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems. Classical studies of the actions of curare and nicotine made this the prototypical pharmacological receptor over a century ago. By taking advantage of specialized structures that have evolved to mediate or block cholinergic neurotransmission, peripheral and then central nicotinic receptors have been isolated and characterized over the last 30 years. These accomplishments represent landmarks in the development of molecular pharmacology. The electric organs from the aquatic species of Electrophorus
and, especially, Torpedo provide rich sources of nicotinic receptor.
The electric organ is derived embryologically from myoid tissue; however, in
contrast to skeletal muscle, a significant fraction (30% to 40%) of the
surface of the membrane is excitable and contains cholinergic receptors. In
vertebrate skeletal muscle, motor end plates occupy 0.1% or less of the cell
surface. The discovery of seemingly irreversible antagonism of neuromuscular
transmission by Purification of the receptor from Torpedo ultimately led to the isolation of complementary DNAs (cDNAs) that encode each of the subunits. These cDNAs, in turn, have permitted the cloning of genes encoding the multiple receptor subunits from mammalian neurons and muscle (Numa et al., 1983). By simultaneously expressing the genes that encode the individual subunits in cellular systems in various permutations and by measuring binding and the electrophysiological events that result from activation by agonists, researchers have been able to correlate functional properties with details of primary structures of the receptor subtypes (Lindstrom, 2000; Karlin and Akabas, 1995; Paterson and Nordberg, 2000). Nicotinic Receptor Structure The nicotinic receptor of the electric organ and vertebrate skeletal
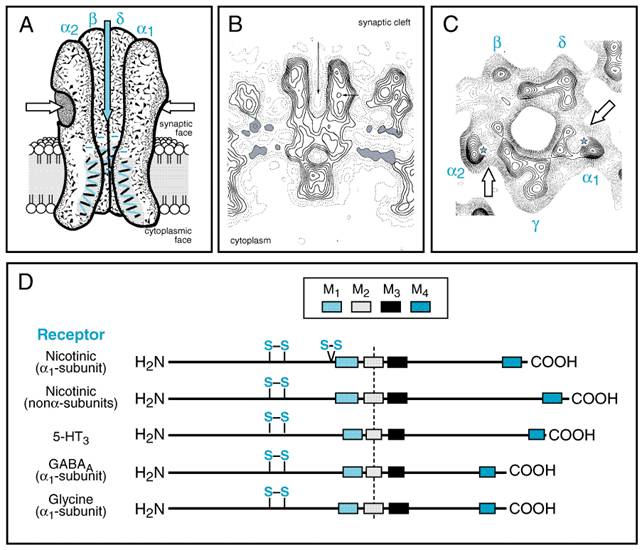
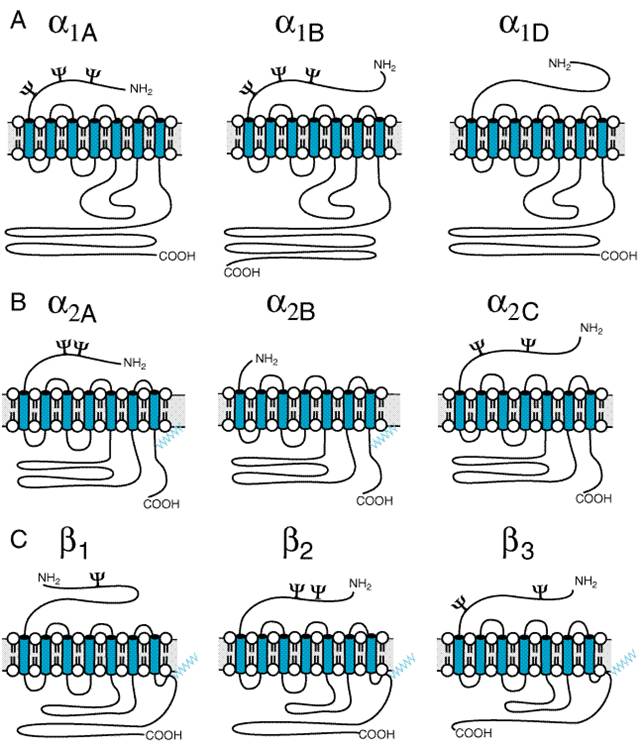
muscle is a pentamer composed of four distinct subunits ( The nicotinic receptor has become the prototype for other ligand-gated ion channels, which include the receptors for the inhibitory amino acids (gamma-aminobutyric acid and glycine) and certain serotonin (5-HT3) receptors. The family of ligand-gated ion channels are pentamers of homologous subunits, each having a molecular mass of 40,000 to 60,000 daltons. The amino-terminal 210 residues constitute virtually all of the extracellular domain. This is followed by four transmembrane-spanning domains, with the region between the third and fourth domain forming most of the cytoplasmic component (Figure 91).
Each of
the subunits within the nicotinic acetylcholine receptor has an extracellular
and an intracellular exposure on the postsynaptic membrane. The five subunits
are arranged to circumscribe an internally located channel in a fashion
similar to petals on a lily (Unwin, 1993; Karlin and Akabas, 1995; Changeux
and Edelstein, 1998). The receptor is an asymmetrical molecule (14 nm x 8 nm) of 250,000 daltons, with
the bulk of the nonmembrane-spanning domain on the extracellular surface. In junctional
areas (i.e., the motor end plate in skeletal muscle and the ventral
surface of the electric organ) the receptor is present at high densities
(10,000/ As is the case for other proteins where cooperativity of both binding
and functional responses is evident, the binding sites are found at the
subunit interfaces, but of the five interfaces, only two in muscle, Measurements of membrane conductances demonstrate that rates of ion translocation are sufficiently rapid (5 x 107 ions per second) to require ion translocation through an open channel, rather than by a rotating carrier of ions. Moreover, agonist-mediated changes in ion permeability (typically an inward movement of primarily Na+ and secondarily Ca2+) occur through a cation channel intrinsic to the receptor structure. The second transmembrane-spanning region on each of the five subunits forms the internal perimeter of the channel. The agonist-binding site is intimately coupled with an ion channel; simultaneous binding of two agonist molecules in muscle results in a rapid conformational change that opens the channel. Details on the kinetics of channel opening have evolved from electrophysiological patchclamp techniques that enable one to distinguish the individual opening and closing events of a single receptor molecule (Sakmann, 1992). Cloning by sequence homology enabled investigators to identify the
genes encoding the nicotinic receptor for higher vertebrates, initially in
muscle and then in neurons. Neuronal nicotinic receptors found in ganglia and
the central nervous system (CNS) also exist as pentamers of subunits composed
of one, two, or more subunits. Although only a single subunit of the type
sequence |
Neuromuscular Blocking Agents
|
History, Sources, and Chemistry Curare is
a generic term for various South American arrow poisons. The drug has a long
and romantic history. It has been used for centuries by the Indians along the
Amazon and Curare was the important tool that Claude Bernard used to demonstrate a locus of drug action at or near the nerve terminations of muscle (Bernard, 1856). The modern clinical use of curare apparently dates from 1932, when West employed highly purified fractions in patients with tetanus and spastic disorders. Research on curare was greatly accelerated by the work of Gill (1940),
who, after prolonged and intimate study of the native methods of preparing
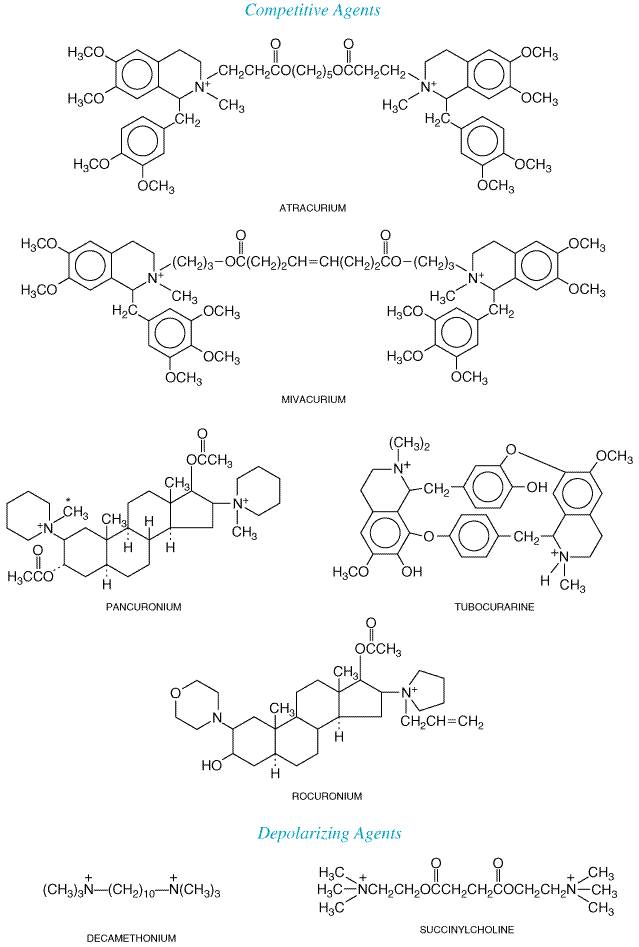
curare, brought to the Details of the fascinating history of curare, its nomenclature, and the chemical identification of the curare alkaloids are presented in McIntyre, 1947, and Bovet, 1972, and previous editions of this textbook. The essential structure of tubocurarine was established by King in 1935 (Figure 92). A synthetic derivative, metocurine (formerly called dimethyl tubocurarine), contains three additional methyl groups, one of which quaternizes the second nitrogen; the other two form methyl ethers at the phenolic hydroxyl groups. This compound possesses two to three times the potency of tubocurarine in human beings.
The
most potent of all curare alkaloids are the toxiferines, obtained from Strychnos
toxifera. A semisynthetic derivative, alcuronium chloride (N,N'-diallylnortoxiferinium
dichloride), was in wide use clinically in Gallamine is one of a series of synthetic substitutes for curare described by Bovet and coworkers in 1949 (see review by Bovet, 1972). Early structureactivity studies led to the development of the polymethylene bis-trimethylammonium series (referred to as the methonium compounds) (Barlow and Ing, 1948; Paton and Zaimis, 1952). The most potent agent at the neuromuscular junction was found when the chain contained ten carbon atoms [decamethonium (C10), see Figure 92]. The member of the series containing six carbon atoms in the chainhexamethonium (C6)was found to be essentially devoid of neuromuscular blocking activity but is particularly effective as a ganglionic blocking agent (see below). In 1949, the curariform action of succinylcholine was described, and its clinical application for relaxation of short duration soon followed (see Dorkins, 1982). Classification and Chemical Properties of Neuromuscular Blocking Agents At present, only a single depolarizing agent, succinylcholine, is in general clinical use, whereas multiple competitive or nondepolarizing agents are available (see Figure 92). Therapeutic selection should be based on achieving a pharmacokinetic profile consistent with the duration of the interventional procedure and minimizing cardiovascular compromise or other side effects (see Table 91). Two general classifications are useful, since they prove helpful in distinguishing side effects and pharmacokinetic behavior. The first relates to the duration of drug action, and these agents are categorized as intermediate-, and short-acting. The persistent blockade and difficulty in complete reversal after surgery with d-tubocurarine, metocurine, pancuronium, and doxacurium led to the development of vecuronium and atracurium, agents of intermediate duration. This was followed by the development of a short-acting agent, mivacurium. Often, the long-acting agents are the more potent, requiring the use of low concentrations. The necessity of administering these agents in low concentrations delays their onset. Rocuronium and rapacuronium are agents of intermediate duration but of rapid onset and lower potency. Their rapid onsets allow them to be used as alternatives to succinylcholine in relaxing the laryngeal and jaw muscles to facilitate tracheal intubation (Bevan, 1994; Savarese et al., 2000). The second classification is derived from the chemical nature of the agents and includes the natural alkaloids or their congeners, the ammonio steroids, and the benzylisoquinolines (Table 91). The natural alkaloid, d-tubocurarine, and semisynthetic alkaloid, alcuronium, while of historical importance, seldom are used. Apart from a shorter duration of action, the newer agents exhibit greatly diminished frequency of side effects, chief of which are ganglionic blockade, block of vagal responses, and histamine release. Metocurine shows diminished histamine release and ganglionic blockade when compared with d-tubocurarine, but it is not devoid of these side effects. The prototype ammonio steroid, pancuronium, shows virtually no histamine release; however, it blocks muscarinic receptors, and this antagonism primarily is manifested in vagal blockade and tachycardia. Tachycardia is eliminated in the newer ammonio steroids: vecuronium, rocuronium, rapacuronium, and pipecuronium. The benzylisoquinolines appear to be devoid of vagolytic and ganglionic blocking actions but still show a slight propensity for release of histamine. The unusual metabolism of the prototype compound, atracurium, and its newer congener mivacurium confers special indications for use of these compounds. For example, atracurium's disappearance from the body depends on hydrolysis of the ester moiety by plasma esterases and by a spontaneous or Hofmann degradation (cleavage of the N-alkyl portion in the benzylisoquinoline). Hence, two routes for degradation are available, both of which remain functional in renal failure. Mivacurium is extremely sensitive to cholinesterase catalysis, therein accounting for its short duration of action. StructureActivity Relationships Several structural features distinguish competitive and depolarizing neuromuscular blocking agents. The competitive agents are relatively bulky, rigid molecules (e.g., tubocurarine, the toxiferines, the benzylisoquinolines, and the ammonio steroids), whereas the depolarizing agents (e.g., decamethonium, succinylcholine) generally have a more flexible structure that enables free bond rotation (see Figure 92; see also Bovet, 1972). While the distance between quaternary groups in the flexible depolarizing agents can vary up to the limit of the maximal bond distance (1.45 nm for decamethonium), the distance for the rigid competitive blockers is typically 1.0 0.1 nm. l-Tubocurarine is considerably less potent than d-tubocurarine. While the two enantiomers have similar internitrogen distances, the d-isomer has all of the hydrophilic groups localized uniquely to one surface. Pharmacological Properties Skeletal Muscle A localized paralytic action of curare was first described by Claude Bernard in the 1850s. That the site of action of d-tubocurarine and other competitive blocking agents was the motor end plate was subsequently established by modern techniques, including fluorescence and electron microscopy, microiontophoretic application of drugs, patchclamp analysis of single channels, and intracellular recording. In brief, competitive antagonists combine with the nicotinic acetylcholine receptor at the postjunctional membrane and thereby competitively block the binding of ACh. When the drug is applied directly to the end plate of a single isolated muscle fiber, the muscle cell becomes insensitive to motor-nerve impulses and to directly applied ACh; however, the end-plate region and the remainder of the muscle fiber membrane retain their normal sensitivity to K+ depolarization, and the muscle fiber still responds to direct electrical stimulation. To analyze the action of antagonists at the neuromuscular junction further, it is first important to consider certain details of receptor activation by acetylcholine. The steps involved in the release of ACh by the nerve action potential, the development of miniature end-plate potentials (MEPPs), their summation to form a postjunctional end-plate potential, the triggering of the muscle action potential, and contraction are described in Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems. Biophysical experimentation has revealed that the fundamental event elicited by acetylcholine or other agonists is an 'all-or-none' opening and closing of the individual receptor channels, which gives rise to a square-wave pulse with an average open-channel conductance of 20 to 30 pS and a duration that is exponentially distributed around a time of about 1 millisecond. The duration of channel opening is far more dependent on the nature of the agonist than is the magnitude of the open-channel conductance (see Sakmann, 1992). The influence of increasing concentrations of the competitive antagonist tubocurarine is to diminish progressively the amplitude of the postjunctional end-plate potential. The amplitude of this postjunctional potential may fall to below 70% of its initial value before it is insufficient to initiate the propagated muscle action potential; this provides a safety factor in neuromuscular transmission. Analysis of the antagonism of tubocurarine on single-channel events shows that, as expected for a competitive antagonist, it reduces the frequency of channel-opening events but does not affect the conductance or duration of opening for a single channel (Katz and Miledi, 1978). At higher concentrations, curare and other competitive antagonists will block the channel directly in a fashion that is noncompetitive with agonists and dependent on membrane potential (Colquhoun et al., 1979). The decay time of the MEPP is of the same duration as the average
lifetime of channel opening (1 to 2 milliseconds). Since the MEPPs are a
consequence of the spontaneous release of one or more quanta of ACh ( If anticholinesterase (anti-ChE) drugs are present, the EPP (or end-plate current) is prolonged up to 25 to 30 milliseconds, which is indicative of the rebinding of transmitter to neighboring receptors before diffusion from the synapse. It is therefore not surprising that anti-ChE agents and tubocurarine act in opposing directions, since increasing the duration of ACh retained in the synapse should favor occupation of the receptor by transmitter and displace tubocurarine. Simultaneous binding by two agonist molecules at the respective The depolarizing agents, such as succinylcholine and decamethonium, act by a different mechanism. Their initial action is to depolarize the membrane by opening channels in the same manner as ACh. However, they persist for longer durations at the neuromuscular junction, primarily because of their resistance to acetylcholinesterase. The depolarization thus is longer lasting, resulting in a brief period of repetitive excitation that may elicit transient muscle fasciculations. The initial phase is followed by block of neuromuscular transmission and flaccid paralysis. This arises because released acetylcholine binds to receptors on an already depolarized end plate. It is the change in end-plate potential elicited by the transient increases in ACh that triggers action potentials. An end plate depolarized from 80 mV to 55 mV by a depolarizing blocking agent is resistant to further depolarization by acetylcholine. In human beings, a sequence of repetitive excitation (fasciculations) followed by block of transmission and neuromuscular paralysis is elicited by depolarizing agents; however, this sequence is influenced by such factors as the anesthetic agent used concurrently, the type of muscle, and the rate of drug administration. The different characteristics of depolarization and competitive blockade are listed in Table 92. In other animal species and occasionally in human beings, decamethonium and succinylcholine produce a blockade that has unique features, some of which combine those of the depolarizing and the competitive agents; Zaimis (1976) has termed this type of action a 'dual' mechanism. In such cases, the depolarizing agents produce initially the characteristic fasciculations and potentiation of the maximal twitch, followed by the rapid onset of neuromuscular block; this block is potentiated by anti-ChE agents. However, following the onset of blockade, there is a poorly sustained response to tetanic stimulation of the motor nerve, intensification of the block by tubocurarine, and usual reversal by anti-ChE agents. The dual action of the depolarizing blocking agents also is seen in intracellular recordings of membrane potential; when agonist is applied continuously, the initial depolarization is followed by a gradual repolarization. The second phase, repolarization, resembles receptor desensitization (Katz and Thesleff, 1957). Under clinical conditions, with increasing concentrations of succinylcholine and in time, the block may convert slowly from a depolarizing to a nondepolarizing type, termed phase I and phase II block (Durant and Katz, 1982). The pattern of neuromuscular blockade produced by depolarizing drugs in anesthetized patients appears to depend, in part, on the anesthetic; fluorinated hydrocarbons may be more apt to predispose the motor end plate to nondepolarization blockade after prolonged use of succinylcholine or decamethonium (see Zaimis, 1976; Fogdall and Miller, 1975). The characteristics of phase I and phase II block are shown in Table 93. During the initial phase of application, depolarizing agents produce channel opening, which can be measured by the statistical analysis of fluctuation of muscle EPPs. The probability of channel opening associated with the binding of drug to the receptor is less with decamethonium than with ACh (Katz and Miledi, 1978). The diminished probability of channel opening would serve to classify decamethonium as a partial agonist at the end plate. Higher concentrations of decamethonium also block the channel directly and thereby interfere with ion permeability (Adams and Sakmann, 1978). Although the observed fasciculations also may result from stimulation of the prejunctional motor-nerve terminal by the depolarizing agent, giving rise to stimulation of the motor unit in an antidromic fashion, the primary site of action of both competitive and depolarizing blocking agents is the postjunctional membrane. Presynaptic actions of the competitive agents may become significant upon repetitive, high-frequency stimulation, since prejunctional nicotinic receptors may be involved in the mobilization of ACh for release from the nerve terminal (Bowman et al., 1990; Van der Kloot and Molgo, 1994). Many drugs and toxins block neuromuscular transmission by other mechanisms, such as interference with the synthesis or release of ACh (see Van der Kloot and Molgo, 1994; see also Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems), but most of these agents are not employed clinically for this purpose. One exception is botulinum toxin, which has been administered locally into muscles of the orbit in the management of ocular blepharospasm and strabismus and has been used to control other muscle spasms and to facilitate facial muscle relaxation (see Chapters 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems and Chapter 66: Ocular Pharmacology). This toxin also has been injected into the lower esophageal sphincter to treat achalasia (see Chapter 38: Prokinetic Agents, Antiemetics, and Agents Used in Irritable Bowel Syndrome). Another exception is dantrolene, which blocks release of Ca2+ from the sarcoplasmic reticulum and is used in the treatment of malignant hyperthermia (see below). The sites of action and interrelationship of several agents that serve as pharmacological tools are shown in Figure 93.
Sequence and Characteristics of Paralysis When an appropriate dose of a competitive blocking agent is injected intravenously in human beings, motor weakness gives way to a total flaccid paralysis. Small, rapidly moving muscles such as those of the eyes, jaw, and larynx relax before those of the limbs and trunk. Ultimately the intercostal muscles and finally the diaphragm are paralyzed, and respiration then ceases. Recovery of muscles usually occurs in the reverse order to that of their paralysis, and thus the diaphragm ordinarily is the first muscle to regain function (see Feldman and Fauvel, 1994; Savarese et al., 2000). After a single intravenous dose of 10 to 30 mg of succinylcholine, muscle fasciculations, particularly over the chest and abdomen, occur briefly; then relaxation occurs within 1 minute, becomes maximal within 2 minutes, and disappears as a rule within 5 minutes. Transient apnea usually occurs at the time of maximal effect. Muscle relaxation of longer duration is achieved by continuous intravenous infusion. After infusion is discontinued, the effects of the drug usually disappear rapidly because of its rapid hydrolysis catalyzed by the butyrylcholinesterase of the plasma and liver. Muscle soreness may follow the administration of succinylcholine. Small prior doses of competitive blocking agents have been employed to minimize fasciculations and muscle pain caused by succinylcholine. However, this procedure is controversial, since it increases the requirement for the depolarizing drug. During prolonged depolarization, muscle cells may lose significant quantities of K+ and gain Na+, Cl, and Ca2+. In patients in whom there has been extensive injury to soft tissues, the efflux of K+ following continued administration of succinylcholine can be life-threatening. The life-threatening complications of succinylcholine-induced hyperkalemia are discussed later in this chapter, but it is important to stress that there are many conditions for which succinylcholine administration is contraindicated or must be undertaken with great caution. The change in the nature of the blockade produced by succinylcholine (from phase I to phase II) presents an additional complication with long-term infusions. Central Nervous System Tubocurarine and other quaternary neuromuscular blocking agents are virtually devoid of central effects following the intravenous administration of ordinary clinical doses because of their inability to penetrate the bloodbrain barrier. The most decisive experiment performed to resolve whether or not curare significantly affects central functions in the dose range used clinically was that of Smith and associates (1947). Smith (an anesthesiologist) permitted himself to receive intravenously two and one-half times the amount of tubocurarine necessary for paralysis of all skeletal muscles. Adequate respiratory exchange was maintained by artificial respiration. At no time was there any evidence of lapse of consciousness, clouding of sensorium, analgesia, or disturbance of special senses. Despite adequate artificially controlled respiration, 'shortness of breath' was experienced, and the accumulation of unswallowed saliva in the pharynx caused the sensation of choking. The experience was decidedly unpleasant. It was concluded that tubocurarine given intravenously even in large doses has no significant central stimulant, depressant, or analgesic effects, and that its sole action in anesthesia is the peripheral paralytic effect on skeletal muscle. Autonomic Ganglia and Muscarinic Sites Neuromuscular blocking agents show variable potencies in producing ganglionic blockade. Just as at the motor end plate, ganglionic blockade by tubocurarine and other stabilizing drugs is reversed or antagonized by anti-ChE agents. At the doses of tubocurarine used clinically, partial blockade probably is produced, both at autonomic ganglia and at the adrenal medulla, which results in a fall in blood pressure and tachycardia. Pancuronium and metocurine show less ganglionic blockade at common clinical doses. Atracurium, vecuronium, doxacurium, pipecuronium, mivacurium, and rocuronium are even more selective (Pollard, 1994; Savarese et al., 2000). The maintenance of cardiovascular reflex responses usually is desired during anesthesia. Pancuronium has a vagolytic action, presumably from blockade of muscarinic receptors. This leads to tachycardia. Of the depolarizing agents, succinylcholine at doses causing neuromuscular relaxation rarely causes effects attributable to ganglionic blockade. However, cardiovascular effects are sometimes observed that are probably due to the successive stimulation of vagal ganglia (manifested by bradycardia) and of sympathetic ganglia (resulting in hypertension and tachycardia). Histamine Release Tubocurarine produces typical histamine-like wheals when injected intracutaneously or intraarterially in human beings, and certain clinical responses to tubocurarine (bronchospasm, hypotension, excessive bronchial and salivary secretion) appear to be caused by the release of histamine. Metocurine, succinylcholine, mivacurium, doxacurium, and atracurium also cause histamine release, but to a lesser extent unless administered rapidly. The ammonio steroids, pancuronium, vecuronium, pipecuronium, and rocuronium, have even less tendency to release histamine after intradermal or systemic injection (Basta, 1992; Watkins, 1994). Histamine release typically is a direct action of the muscle relaxant on the mast cell rather than IgE-mediated anaphylaxis (Watkins, 1994). Actions of Neuromuscular Blocking Agents with Life-Threatening Implications The depolarizing agents can release K+ rapidly from intracellular sites; this may be a factor in production of the prolonged apnea that has been noted in patients who receive these drugs while in electrolyte imbalance (Dripps, 1976). As indicated above, succinylcholine-induced hyperkalemia is a life-threatening complication of the drug. For example, such alterations in the distribution of K+ are of particular concern in patients with congestive heart failure who are receiving digitalis or diuretics. For the same reason, caution should be used or depolarizing blocking agents should be avoided in patients with extensive soft-tissue trauma or burns. A higher dose of a competitive blocking agent often is indicated in these patients. In addition, succinylcholine administration is contraindicated or should be given with great caution in patients with nontraumatic rhabdomyolysis, ocular lacerations, spinal cord injuries with paraplegia or quadriplegia, or with muscular dystrophies. Succinylcholine no longer is indicated for children 8 years old and younger unless emergency intubation or securing an airway is necessary. Hyperkalemia, rhabdomyolysis, and cardiac arrest have been reported. A subclinical dystrophy frequently is associated with these adverse responses (Savarese et al., 2000). Neonates also may have an enhanced sensitivity to competitive neuromuscular blocking agents. Synergisms and Antagonisms The interactions between the competitive and depolarizing neuromuscular blocking agents already have been considered. From a clinical viewpoint, the most important pharmacological interactions of these drugs are with certain general anesthetics, certain antibiotics, Ca2+ channel blockers, and anti-ChE compounds. Since the anti-ChE agents neostigmine, pyridostigmine, and edrophonium preserve endogenous ACh and also act directly on the neuromuscular junction, they can be used in the treatment of overdosage with competitive blocking agents. Similarly, upon completion of the surgical procedure many anesthesiologists employ neostigmine or edrophonium to reverse and decrease the duration of competitive neuromuscular blockade. Succinylcholine should never be administered after reversal of competitive blockade with neostigmine; in this circumstance a prolonged and intense blockade often is achieved. A muscarinic antagonist (atropine or glycopyrrolate) is used concomitantly to prevent stimulation of muscarinic receptors and thereby avoid slowing of the heart rate. The anti-ChE agents, however, are synergistic with the depolarizing blocking agents, particularly in their initial phase of action. Since they will not reverse depolarizing neuromuscular blockade and, in fact, can enhance it, the distinction in the type of neuromuscular blocking agent must be clear. Many inhalational anesthetics (e.g., halothane, isoflurane, and enflurane) exert a stabilizing effect on the postjunctional membrane and therefore act synergistically with the competitive blocking agents. Consequently, when such blocking drugs are used for muscle relaxation as adjuncts to these anesthetics, their doses should be reduced (see Fogdall and Miller, 1975). Aminoglycoside antibiotics produce neuromuscular blockade by inhibiting ACh release from the preganglionic terminal (through competition with Ca2+) and to a lesser extent by noncompetitively blocking the receptor. The blockade is antagonized by calcium salts, but only inconsistently by anti-ChE agents (see Chapter 46: Antimicrobial Agents: The Aminoglycosides). The tetracycline antibiotics also can produce neuromuscular blockade, possibly by chelation of Ca2+. Additional antibiotics that have neuromuscular blocking action, through both presynaptic and postsynaptic actions, include polymyxin B, colistin, clindamycin, and lincomycin (see Pollard, 1994). Ca2+ channel blockers enhance neuromuscular blockade produced by both competitive and depolarizing antagonists. It is not clear whether this is a result of a diminution of Ca2+-dependent release of transmitter from the nerve ending or is a postsynaptic action. When neuromuscular blocking agents are administered to patients receiving these agents, dose adjustments should be considered; if recovery of spontaneous respiration is delayed, Ca2+ salts may facilitate recovery. Miscellaneous drugs that may have significant interactions with either competitive or depolarizing neuromuscular blocking agents include trimethaphan, opioid analgesics, procaine, lidocaine, quinidine, phenelzine, phenytoin, propranolol, magnesium salts, corticosteroids, digitalis glycosides, chloroquine, catecholamines, and diuretics (see Zaimis, 1976; Pollard, 1994; Savarese et al., 2000). Toxicology The important untoward responses of the neuromuscular blocking agents include prolonged apnea, cardiovascular collapse, and those resulting from histamine release. Failure of respiration to become adequate in the postoperative period may not always be due directly to the drug. An obstruction of the airway, decreased arterial carbon dioxide tension secondary to hyperventilation during the operative procedure, or the neuromuscular depressant effect of excessive amounts of neostigmine used to reverse the action of the competitive blocking drugs also may be implicated. Directly related factors may include alterations in body temperature; electrolyte imbalance, particularly of K+ (discussed earlier); low plasma butyrylcholinesterase levels, resulting in a reduction in the rate of destruction of succinylcholine; the presence of latent myasthenia gravis or of malignant disease such as small-cell carcinoma of the bronchus (myasthenic syndrome); reduced blood flow to skeletal muscles, causing delayed removal of the blocking drugs; and decreased elimination of the muscle relaxants secondary to reduced renal function. Great care should be taken when administering these agents to dehydrated or severely ill patients. Malignant Hyperthermia Malignant hyperthermia is a potentially life-threatening event triggered by the administration of certain anesthetics and neuromuscular blocking agents. The clinical features include contracture, rigidity, and heat production from skeletal muscle resulting in severe hyperthermia, accelerated muscle metabolism, metabolic acidosis, and tachycardia. Uncontrolled release of Ca2+ from the sarcoplasmic reticulum of skeletal muscle is the initiating event. Although the halogenated hydrocarbon anesthetics (halothane, isoflurane, and sevoflurane) and succinylcholine alone have been reported to precipitate the response, most of the incidents arise from the combination of depolarizing blocking agent and anesthetic. Susceptibility to malignant hyperthermia, an autosomal dominant trait, is associated with certain congenital myopathies such as central core disease. In the majority of cases, however, no clinical signs are visible in the absence of anesthetic intervention. Determination of susceptibility is made with an in vitro contracture test (IVCT) on a fresh biopsy of skeletal muscle, where contractures in the presence of various concentrations of halothane and caffeine are measured. In over 50% of the families, a linkage is found between the IVCT phenotype and a mutation in the gene (RyR-1) encoding the skeletal muscle ryanodine receptor (RYR-1). Over 20 mutations in a region of the gene that encodes the cytoplasmic face of the receptor have been described. Other loci have been identified on the L-type Ca2+ channel (voltage-gated dihydropyridine receptor) and on other associated proteins or channel subunits. The large size of RyR-1 and the genetic heterogeneity of the condition have precluded the development of a genotypic determination for malignant hyperthermia (Hopkins, 2000; Jurkat-Rott et al., 2000). Current treatment entails an intravenous administration of dantrolene (DANTRIUM), which blocks Ca2+ release and the metabolic sequelae. Dantrolene inhibits Ca2+ release from the sarcoplasmic reticulum of skeletal muscle by limiting the capacity of Ca2+ and calmodulin to activate RYR-1 (Fruen et al., 1997). RYR-1 and the L-type Ca2+ channel are juxtaposed to associate at a triadic junction formed between the T- tubule and sarcoplasmic reticulum. The L-type channel with its T-tubular location serves as the voltage sensor receiving the depolarizing activation signal. The intimate coupling of the two proteins at the triad, along with a host of modulatory proteins in the two organelles and the surrounding cytoplasm, regulate the release of and response to Ca2+ (Lehmann-Horn and Jurkat-Rott, 1999). Rapid cooling, inhalation of 100% oxygen, and control of acidosis should be considered adjunct therapy in malignant hyperthermia. Declining fatality rates for malignant hyperthermia relate to anesthesiologists' awareness of the condition and the efficacy of dantrolene. Patients with central core disease, so named because of the presence
of myofibrillar cores seen upon biopsy of slow-twitch muscle fibers, show
muscle weakness in infancy and delayed motor development. These individuals
have a high susceptibility to malignant hyperthermia with the combination of
an anesthetic and a depolarizing neuromuscular blocker. Central core disease
has five allelic variants of RyR-1 in common with malignant
hyperthermia. Patients with other muscle syndromes or dystonias also have an
increased frequency of contracture and hyperthermia in the anesthesia
setting. Succinylcholine in susceptible individuals also induces masseter
muscle rigidity, which may complicate endotracheal tube insertion and
airway management. This condition has been correlated with a mutation in the
gene encoding the Respiratory Paralysis Treatment of respiratory paralysis arising from an adverse reaction or overdose of a neuromuscular blocking agent should be by positive-pressure artificial respiration with oxygen and maintenance of a patent airway until the recovery of normal respiration is assured. With the competitive blocking agents, this may be hastened by the administration of neostigmine methylsulfate (0.5 to 2 mg, intravenously) or edrophonium (10 mg, intravenously, repeated as required) (Watkins, 1994). Interventional Strategies for Other Toxic Effects Neostigmine antagonizes only the skeletal muscular blocking action of the competitive blocking agents effectively, and it may aggravate such side effects as hypotension or induce bronchospasm. In such circumstances, sympathomimetic amines may be given to support the blood pressure. Atropine or glycopyrrolate is administered to counteract muscarinic stimulation. Antihistamines are definitely beneficial to counteract the responses that follow the release of histamine, particularly when administered before the neuromuscular blocking agent. Absorption, Fate, and Excretion Quaternary ammonium neuromuscular blocking agents are very poorly and irregularly absorbed from the gastrointestinal tract. This fact was well known to the South American Indians, who ate with impunity the flesh of game killed with curare-poisoned arrows. Absorption is quite adequate from intramuscular sites. Rapid onset is achieved with intravenous administration. The more potent agents, of course, must be given in lower concentrations, and diffusional requirements slow their rate of onset. When long-acting, competitive blocking agents, such as d-tubocurarine and pancuronium, are administered, blockade may diminish after 30 minutes owing to redistribution of the drug, yet residual blockade and plasma levels of the drug persist for longer periods. Subsequent doses show diminished redistribution. Long-acting agents may accumulate with multiple doses. The ammonio steroids contain ester groups that are hydrolyzed in the liver. Typically, the metabolites have about one-half of the activity of the parent compound and contribute to the total relaxation profile. Ammonio steroids of intermediate duration of action, such as vecuronium, rocuronium, and rapacuronium (see Table 91), are more rapidly cleared by the liver than are pancuronium and pipecuronium. The more rapid offset of neuromuscular blockade with compounds of intermediate duration argues for sequential dosing of these agents, rather than administering a single dose of a long-duration neuromuscular blocking agent (Savarese et al., 2000). Atracurium is converted to less-active metabolites by plasma esterases and spontaneous degradation. These alternative routes of metabolism are responsible for atracurium not exhibiting an increase in half-life in patients with compromised renal function. Hence, it becomes the agent of choice under these conditions (Hunter, 1994). Mivacurium shows an even greater susceptibility to butyrylcholinesterase catalysis, thus conferring to it the shortest duration among the nondepolarizing blockers. The extremely brief duration of action of succinylcholine also is due largely to its rapid hydrolysis by the butyrylcholinesterase of liver and plasma. Among the occasional patients who exhibit prolonged apnea following the administration of succinylcholine or mivacurium, most have an atypical plasma cholinesterase or a deficiency of the enzyme, due to allelic variations (Pantuck, 1993; Primo-Parmo et al., 1996), hepatic or renal disease, or a nutritional disturbance; however, in some the enzymatic activity in plasma is normal (Whittaker, 1986). Therapeutic Uses The main clinical use of the neuromuscular blocking agents is as an adjuvant in surgical anesthesia to obtain relaxation of skeletal muscle, particularly of the abdominal wall, so that operative manipulations are facilitated. With muscle relaxation no longer dependent upon the depth of general anesthesia, a much lighter level of anesthesia suffices. This situation is of obvious advantage, since the risk of respiratory and cardiovascular depression is minimized. Moreover, the postanesthetic recovery period is shortened. These considerations notwithstanding, neuromuscular blocking agents cannot be used to substitute for inadequate depth of anesthesia in the surgical planes. Otherwise, a risk of reflex responses to painful stimuli and conscious recall may occur. Muscle relaxation is also of value in various orthopedic procedures, such as the correction of dislocations and the alignment of fractures. Neuromuscular blocking agents of short duration often are used to facilitate intubation with an endotracheal tube and have been used to facilitate laryngoscopy, bronchoscopy, and esophagoscopy in combination with a general anesthetic agent. Neuromuscular blocking agents are administered parenterally and nearly always intravenously. As potentially hazardous drugs, they should be administered to patients only by anesthesiologists and other clinicians who have had extensive training in their use and in a setting where facilities for respiratory and cardiovascular resuscitation are immediately at hand. Detailed information on dosage and monitoring the extent of muscle relaxation can be found in anesthesiology textbooks (Pollard, 1994; Savarese et al., 2000). Measurement of Neuromuscular Blockade in Human Beings Assessment of neuromuscular block usually is performed by stimulation of the ulnar nerve. Responses are monitored from compound action potentials or muscle tension developed in the adductor pollicis (thumb) muscle. Responses to repetitive or tetanic stimuli are most useful for evaluation of blockade of transmission, since individual measurements of twitch tension must be related to control values obtained prior to the administration of drugs. Thus, stimulus schedules such as the 'train of four' and the 'double burst' or responses to tetanic stimulation are preferred procedures (Waud and Waud, 1972; Drenck et al., 1989). Rates of onset of blockade and recovery are more rapid in the airway musculature (jaw, larynx, and diaphragm) than in the thumb. Hence, tracheal intubation can be performed before onset of complete block at the adductor pollicis, while partial recovery of function of this muscle allows sufficient recovery of respiration for extubation (Savarese et al., 2000). Differences in rates of onset of blockade, recovery from blockade, and intrinsic sensitivity between the stimulated muscle and those of the larynx, abdomen, and diaphragm should be considered. Use to Prevent Trauma during Electroshock Therapy Electroconvulsive therapy of psychiatric disorders occasionally is complicated by trauma to the patient; the seizures induced may cause dislocations or fractures. Inasmuch as the muscular component of the convulsion is not essential for benefit from the procedure, neuromuscular blocking agents and thiopental are employed. The combination of the blocking drug, the anesthetic agent, and postictal depression usually results in respiratory depression or temporary apnea. An endotracheal tube and oxygen always should be available. An oropharyngeal airway should be inserted as soon as the jaw muscles relax (after the seizure) and provision made to prevent aspiration of mucus and saliva. Succinylcholine or mivacurium is most often used because of the brevity of relaxation. A cuff may be applied to one extremity to prevent the effects of the drug in that limb; evidence of an effective electroshock is provided by contraction of the group of protected muscles. Control of Muscle Spasms Several agents, many of which have rather limited efficacy, have been
used to treat spasticity involving the The anaerobic bacterium Clostridium botulinum produces a family of toxins targeted to presynaptic proteins and that block the release of acetylcholine (ACh) (see Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems). Botulinum toxinA (BOTOX), in blocking ACh release, produces flaccid paralysis of skeletal muscle and diminished activity of parasympathetic and sympathetic cholinergic synapses. Inhibition lasts from several weeks to 3 to 4 months, and restoration of function requires nerve sprouting. Immunoresistance may develop with continued use (Davis and Barnes, 2000). Originally approved for the treatment of the ocular conditions of strabismus and blepharospasm and for hemifacial spasms, botulinum toxin has received wider use in the treatment of spasms and dystonias such as adductor spasmodic dysphonia, oromandibular dystonia, cervical dystonia, and spasms associated with the lower esophageal sphincter and anal fissures. Its dermatological uses include treatment of hyperhidrosis of the palms and axillae that is resistant to topical and iontophoretic remedies and removal of facial lines associated with excessive nerve stimulation and muscle activity. Treatment involves local intramuscular or intradermal injections (Boni et al., 2000). In addition to its use in managing an acute attack of malignant hyperthermia (see above), dantrolene also has been explored in the treatment of spasticity and hyperreflexia. With its peripheral action, it causes a generalized weakness. As such, its use should be reserved to nonambulatory patients with severe spasticity. Hepatotoxicity has been reported with continued use, requiring liver function tests (Kita and Goodkin, 2000). |
Ganglionic Neurotransmission
|
Neurotransmission in autonomic ganglia has
long been recognized to be a far more complex process than that described by
a single neurotransmitterreceptor system; intracellular recordings reveal at
least four different changes in potential that can be elicited by stimulation
of the preganglionic nerve (Eccles and Libet, 1961; Weight et al.,
1979) (Figure 94). The primary event involves a rapid depolarization of
postsynaptic sites by ACh. The receptors are nicotinic, and the pathway is
sensitive to classical blocking agents such as hexamethonium and
trimethaphan. Activation of this primary pathway gives rise to an initial
excitatory postsynaptic potential (EPSP). This rapid depolarization is due
primarily to an inward Na+ and perhaps Ca2+ current
through a neuronal type of nicotinic receptor channel. Multiple nicotinic
receptor subunits or their mRNAs (
An action potential is generated in the postganglionic neuron when the initial EPSP attains a critical amplitude. In mammalian sympathetic ganglia in vivo, it may be necessary for multiple synapses to be activated before transmission is effective. Discrete end plates with focal localization of receptors do not exist in ganglia; rather, the dendrites and nerve cell bodies contain the receptors. Iontophoretic application of ACh to the ganglion results in a depolarization with a latency of less than 1 millisecond; this decays over a period of 10 to 50 milliseconds (Ascher et al., 1979). Measurements of single-channel conductances indicate that the characteristics of nicotinic receptor channels of the ganglia and the neuromuscular junction are quite similar. The secondary events that follow the initial depolarization are insensitive to hexamethonium or other nicotinic antagonists. They include the slow EPSP, the late slow EPSP, and an inhibitory postsynaptic potential (IPSP). The slow EPSP is generated by ACh acting on muscarinic receptors, and it is blocked by atropine or antagonists that are selective for M1 muscarinic receptors (see Chapter 7: Muscarinic Receptor Agonists and Antagonists). The slow EPSP has a longer latency and a duration of 30 to 60 seconds. In contrast, the late slow EPSP lasts for several minutes and is initiated by the action of peptides released from presynaptic nerve endings or interneurons in specific ganglia (Dun, 1983). The peptides and ACh may be released from the same nerve ending, but the enhanced stability of the peptide in the ganglion extends its sphere of influence to postsynaptic sites beyond those in immediate proximity to the nerve ending. The slow EPSPs result from decreased K+ conductance (Weight et al., 1979). The K+ conductance has been called an M current, and it regulates the sensitivity of the cell to repetitive fast-depolarizing events (Adams et al., 1982). Like the slow EPSP, the IPSP is unaffected by the classical
nicotinic-receptor blocking agents. Substantial electrophysiological and
morphological evidence has accumulated to suggest that catecholamines
participate in the generation of the IPSP. Dopamine and norepinephrine cause
hyperpolarization of ganglia, and both the IPSP and the catecholamine-induced
hyperpolarization are blocked by The relative importance of the secondary pathways and even the nature
of the modulating transmitters appear to differ among individual ganglia and
between parasympathetic and sympathetic ganglia. A variety of peptides,
including luteinizing hormonereleasing hormone, substance P, angiotensin, calcitonin
gene-related peptide, vasoactive intestinal polypeptide, neuropeptide Y, and
enkephalins, have been identified in ganglia by immunofluorescence. They
appear localized to particular cell bodies, nerve fibers, or SIF cells; are
released upon nerve stimulation; and are presumed to mediate the late slow
EPSP (Dun, 1983; Elfvin et al., 1993). Other neurotransmitter
substances, such as 5-hydroxytryptamine and gamma-aminobutyric acid, are
known to modify ganglionic transmission. Precise details of their modulatory
actions are not understood, but they appear to be most closely associated
with the late slow EPSP and inhibition of the M current in various ganglia.
It should be emphasized that the secondary synaptic events only modulate the
initial EPSP. Conventional ganglionic blocking agents can inhibit ganglionic
transmission completely; the same cannot be said for muscarinic antagonists
or Drugs that stimulate cholinergic receptor sites on autonomic ganglia can be grouped into two major categories. The first group consists of drugs with nicotinic specificity, including nicotine itself. Their excitatory effects on ganglia are rapid in onset, are blocked by ganglionic nicotinic-receptor antagonists, and mimic the initial EPSP. The second group is composed of agents such as muscarine, McN-A-343, and methacholine. Their excitatory effects on ganglia are delayed in onset, blocked by atropine-like drugs, and mimic the slow EPSP. Ganglionic blocking agents acting on the nicotinic receptor may be classified into two groups. The first group includes those drugs that initially stimulate the ganglia by an ACh-like action and then block them because of a persistent depolarization (e.g., nicotine); prolonged application of nicotine results in desensitization of the cholinergic receptor site and continued blockade. (See review by Volle, 1980.) The blockade of autonomic ganglia produced by the second group of blocking drugs, of which hexamethonium and trimethaphan can be regarded as prototypes, does not involve prior ganglionic stimulation or changes in ganglionic potentials. These agents impair transmission either by competing with ACh for ganglionic nicotinic receptor sites or by blocking the channel when it is open. Trimethaphan acts by competition with ACh, analogous to the mechanism of action of curare at the neuromuscular junction. Hexamethonium appears to block the channel after it opens. This action shortens the duration of current flow, since the open channel either becomes occluded or closes (Gurney and Rang, 1984). Regardless of the mechanism, the initial EPSP is blocked and ganglionic transmission is inhibited. Ganglionic Stimulating Drugs History Two natural alkaloids, nicotine and lobeline, exhibit peripheral actions by stimulating autonomic ganglia. Nicotine (see Figure 95) was first isolated from leaves of tobacco, Nicotiana tabacum, by Posselt and Reiman in 1828, and Orfila initiated the first pharmacological studies of the alkaloid in 1843. Langley and Dickinson (1889) painted the superior cervical ganglion of rabbits with nicotine and demonstrated that its site of action was the ganglion, rather than the preganglionic or postganglionic nerve fiber. Lobeline, from Lobelia inflata, has many of the same actions as nicotine but is less potent.
A number of synthetic compounds also have prominent actions at ganglionic receptor sites. The actions of the 'onium' compounds, of which tetramethylammonium (TMA) is the simplest prototype, were explored in considerable detail in the last half of the nineteenth century and in the early twentieth century. Nicotine Nicotine is of considerable medical significance because of its toxicity, presence in tobacco, and propensity for conferring a dependence on its users. The chronic effects of nicotine and the untoward effects of the chronic use of tobacco are considered in Chapter 24: Drug Addiction and Drug Abuse. Nicotine is one of the few natural liquid alkaloids. It is a colorless, volatile base (pKa= 8.5) that turns brown and acquires the odor of tobacco on exposure to air. Pharmacological Actions The complex and often unpredictable changes that occur in the body after administration of nicotine are due not only to its actions on a variety of neuroeffector and chemosensitive sites but also to the fact that the alkaloid can stimulate and desensitize receptors. The ultimate response of any one system represents the summation of stimulatory and inhibitory effects of nicotine. For example, the drug can increase heart rate by excitation of sympathetic or paralysis of parasympathetic cardiac ganglia, and it can slow heart rate by paralysis of sympathetic or stimulation of parasympathetic cardiac ganglia. In addition, the effects of the drug on the chemoreceptors of the carotid and aortic bodies and on brain centers influence heart rate, as do also the cardiovascular compensatory reflexes resulting from changes in blood pressure caused by nicotine. Finally, nicotine elicits a discharge of epinephrine from the adrenal medulla, and this hormone accelerates cardiac rate and raises blood pressure. Peripheral Nervous System The major action of nicotine consists initially of transient stimulation and subsequently of a more persistent depression of all autonomic ganglia. Small doses of nicotine stimulate the ganglion cells directly and may facilitate the transmission of impulses. When larger doses of the drug are applied, the initial stimulation is followed very quickly by a blockade of transmission. Whereas stimulation of the ganglion cells coincides with their depolarization, depression of transmission by adequate doses of nicotine occurs both during the depolarization and after it has subsided. Nicotine also possesses a biphasic action on the adrenal medulla; small doses evoke the discharge of catecholamines, and larger doses prevent their release in response to splanchnic nerve stimulation. The effects of nicotine on the neuromuscular junction are similar to those on ganglia. However, with the exception of avian and denervated mammalian muscle, the stimulant phase is largely obscured by the rapidly developing paralysis. In the latter stage, nicotine also produces neuromuscular blockade by receptor desensitization. Nicotine, like ACh, is known to stimulate a number of sensory receptors. These include mechanoreceptors that respond to stretch or pressure of the skin, mesentery, tongue, lung, and stomach; chemoreceptors of the carotid body; thermal receptors of the skin and tongue; and pain receptors. Prior administration of hexamethonium prevents the stimulation of the sensory receptors by nicotine but has little, if any, effect on the activation of the sensory receptors by physiological stimuli. Central Nervous System Nicotine markedly stimulates the CNS. Low doses produce weak analgesia; with higher doses, tremors leading to convulsions at toxic doses are evident. The excitation of respiration is a prominent action of nicotine; although large doses act directly on the medulla oblongata, smaller doses augment respiration reflexly by excitation of the chemoreceptors of the carotid and aortic bodies. Stimulation of the CNS with large doses is followed by depression, and death results from failure of respiration due to both central paralysis and peripheral blockade of muscles of respiration. Nicotine induces vomiting by both central and peripheral actions. The central component of the vomiting response is due to stimulation of the emetic chemoreceptor trigger zone in the area postrema of the medulla oblongata. In addition, nicotine activates vagal and spinal afferent nerves that form the sensory input of the reflex pathways involved in the act of vomiting. Studies in isolated higher centers of the brain and spinal cord reveal that the primary sites of action of nicotine in the CNS are prejunctional, causing the release of other transmitters. Accordingly, the stimulatory and pleasure-reward actions of nicotine appear to result from release of excitatory amino acids, dopamine, and other biogenic amines from various CNS centers (MacDermott et al., 1999). Chronic exposure to nicotine in several systems causes an increase in the density or number of nicotinic receptors (see Di Chiara et al., 2000; Stitzel et al., 2000). While the details of the mechanism are not yet understood, the response may be compensatory to the desensitization of receptor function by nicotine. Cardiovascular System When administered intravenously to dogs, nicotine characteristically produces an increase in heart rate and blood pressure. The latter is usually a more sustained response. In general, the cardiovascular responses to nicotine are due to stimulation of sympathetic ganglia and the adrenal medulla, together with the discharge of catecholamines from sympathetic nerve endings. Also contributing to the sympathomimetic response to nicotine is the activation of chemoreceptors of the aortic and carotid bodies, which reflexly results in vasoconstriction, tachycardia, and elevated blood pressure. Gastrointestinal Tract The combined activation of parasympathetic ganglia and cholinergic nerve endings by nicotine results in increased tone and motor activity of the bowel. Nausea, vomiting, and occasionally diarrhea are observed following systemic absorption of nicotine in an individual who has not been exposed to nicotine previously. Exocrine Glands Nicotine causes an initial stimulation of salivary and bronchial secretions that is followed by inhibition. Absorption, Fate, and Excretion Nicotine is readily absorbed from the respiratory tract, buccal
membranes, and skin. Severe poisoning has resulted from percutaneous
absorption. Being a relatively strong base, its absorption from the stomach
is limited, and intestinal absorption is far more efficient. Nicotine in
chewing tobacco, because it is more slowly absorbed than inhaled nicotine,
has a longer duration of effect. The average cigarette contains 6 to 11 mg of
nicotine and delivers about 1 to 3 mg of nicotine systemically to the smoker;
bioavailability can increase as much as threefold with intensity of puffing
and technique of the smoker (Henningfield, 1995; Benowitz, 1998). Nicotine is
available in several dosage forms to help achieve abstinence from tobacco
use. Efficacy primarily results from preventing a withdrawal or abstinence
syndrome. Nicotine may be administered orally as a gum (nicotine polacrilex; NICORETTE), transdermal patch (NICODERM,
HABITROL, others), a
nasal spray ( Approximately 80% to 90% of nicotine is altered in the body, mainly in the liver but also in the kidney and lung. Cotinine is the major metabolite, with nicotine-1'-N-oxide and 3-hydroxycotinine and conjugated metabolites found in lesser quantities (Benowitz, 1998). The profile of metabolites and the rate of metabolism appear to be similar in the smoker and nonsmoker. The half-life of nicotine following inhalation or parenteral administration is about 2 hours. Both nicotine and its metabolites are eliminated rapidly by the kidney. The rate of urinary excretion of nicotine is dependent upon the pH of the urine; excretion diminishes when the urine is alkaline. Nicotine also is excreted in the milk of lactating women who smoke; the milk of heavy smokers may contain 0.5 mg per liter. Acute Nicotine Poisoning Poisoning from nicotine may occur from accidental ingestion of nicotine-containing insecticide sprays or in children from ingestion of tobacco products. The acutely fatal dose of nicotine for an adult is probably about 60 mg of the base. Smoking tobacco usually contains 1% to 2% nicotine. Apparently, the gastric absorption of nicotine from tobacco taken by mouth is delayed because of slowed gastric emptying, so that vomiting caused by the central effect of the initially absorbed fraction may remove much of the tobacco remaining in the gastrointestinal tract. The onset of symptoms of acute, severe nicotine poisoning is rapid; they include nausea, salivation, abdominal pain, vomiting, diarrhea, cold sweat, headache, dizziness, disturbed hearing and vision, mental confusion, and marked weakness. Faintness and prostration ensue; the blood pressure falls; breathing is difficult; the pulse is weak, rapid, and irregular; and collapse may be followed by terminal convulsions. Death may result within a few minutes from respiratory failure. Therapy Vomiting should be induced with syrup of ipecac or gastric lavage should be performed. Alkaline solutions should be avoided. A slurry of activated charcoal is then passed through the tube and left in the stomach. Respiratory assistance and treatment of shock may be necessary. Other Ganglionic Stimulants Stimulation of ganglia by tetramethylammonium (TMA) or 1,1-dimethyl-4-phenylpiperazinium iodide (DMPP) differs from that produced by nicotine in that the initial stimulation is not followed by a dominant blocking action. DMPP is about three times more potent and slightly more ganglion-selective than nicotine. Although parasympathomimetic drugs stimulate ganglia, their effects usually are obscured by stimulation of other neuroeffector sites. McN-A-343 represents an exception to this; in certain tissues its primary action appears to occur at muscarinic M1 receptors in ganglia. Ganglionic Blocking Drugs The chemical diversity of compounds that block autonomic ganglia without causing prior stimulation is shown in Figure 96.
History and StructureActivity Relationship Although Triethylsulfoniums, like the quaternary and bis-quaternary ammonium ions, possess ganglionic blocking actions. This knowledge led to the development of sulfonium ganglionic blocking agents such as trimethaphan (see Figure 96). Mecamylamine, a secondary amine, was introduced into therapy for hypertension in the mid-1950s. Pharmacological Properties Nearly all of the physiological alterations observed after the administration of ganglionic blocking agents can be anticipated with reasonable accuracy by a careful inspection of Figure 61 and by knowing which division of the autonomic nervous system exercises dominant control of various organs (Table 94). For example, blockade of sympathetic ganglia interrupts adrenergic control of arterioles and results in vasodilation, improved peripheral blood flow in some vascular beds, and a fall in blood pressure. Generalized ganglionic blockade may result also in atony of the bladder and gastrointestinal tract, cycloplegia, xerostomia, diminished perspiration, and, by abolishing circulatory reflex pathways, postural hypotension. These changes represent the generally undesirable features of ganglionic blockade, which severely limit the therapeutic efficacy of ganglionic blocking agents. Cardiovascular System The importance of existing sympathetic tone in determining the degree to which blood pressure is lowered by ganglionic blockade is illustrated by the fact that blood pressure may be decreased only minimally in recumbent normotensive subjects but may fall markedly in sitting or standing subjects. Postural hypotension is a major problem in ambulatory patients receiving ganglionic blocking drugs; it is relieved to some extent by muscular activity and completely by recumbency. Changes in cardiac rate following ganglionic blockade depend largely on existing vagal tone. In human beings, mild tachycardia usually accompanies the hypotension, a sign that indicates fairly complete ganglionic blockade. However, a decrease may occur if the heart rate is initially high. Cardiac output often is reduced by ganglionic blocking drugs in patients with normal cardiac function as a consequence of diminished venous return resulting from venous dilation and peripheral pooling of blood. In patients with cardiac failure, ganglionic blockade frequently results in increased cardiac output due to a reduction in peripheral resistance. In hypertensive subjects, cardiac output, stroke volume, and left ventricular work are diminished. Although total systemic vascular resistance is decreased in patients who receive ganglionic blocking agents, changes in blood flow and vascular resistance of individual vascular beds are variable. Reduction of cerebral blood flow is small unless mean systemic blood pressure falls below 50 to 60 mm Hg. Skeletal muscle blood flow is unaltered, but splanchnic and renal blood flow decrease following the administration of a ganglionic blocking agent. Absorption, Fate, and Excretion The absorption of quaternary ammonium and sulfonium compounds from the enteric tract is incomplete and unpredictable. This is due both to the limited ability of these ionized substances to penetrate cell membranes and to the depression of propulsive movements of the small intestine and gastric emptying. Although the absorption of mecamylamine is less erratic, a danger exists of reduced bowel activity leading to frank paralytic ileus. After absorption, the quaternary ammonium and sulfonium blocking agents are confined primarily to the extracellular space and are excreted mostly unchanged by the kidney. Mecamylamine concentrates in the liver and kidney and is slowly excreted in an unchanged form. Untoward Responses and Severe Reactions Among the milder untoward responses observed are visual disturbances, dry mouth, conjunctival suffusion, urinary hesitancy, decreased potentia, subjective chilliness, moderate constipation, occasional diarrhea, abdominal discomfort, anorexia, heartburn, nausea, eructation and bitter taste, and the signs and symptoms of syncope caused by postural hypotension. More severe reactions include marked hypotension, constipation, syncope, paralytic ileus, urinary retention, and cycloplegia. Therapeutic Uses Of the ganglionic blocking agents that have appeared on the
therapeutic scene, only mecamylamine (INVERSINE) and trimethaphan (ARFONAD) currently are utilized in the Ganglionic blocking agents have been supplanted by superior agents for
the treatment of chronic hypertension (see Chapter 33:
Antihypertensive Agents and the Drug Therapy of Hypertension). Alternative
agents also are available for management of acute hypertensive crises (Murphy,
1995; see Chapter 33: Antihypertensive Agents and the Drug Therapy of
Hypertension). A remaining use of ganglionic blockers in a hypertensive
crisis is for the initial control of blood pressure in patients with acute
dissecting aortic aneurysm, particularly when preexisting conditions make An additional therapeutic use of the ganglionic blocking agents is in the production of controlled hypotension; a reduction in blood pressure during surgery may be sought deliberately to minimize hemorrhage in the operative field, to reduce blood loss in various orthopedic procedures, and to facilitate surgery on blood vessels (Fukusaki et al., 1999). Trimethaphan, as an infusion, may be used as an alternative to or in combination with sodium nitroprusside, since some patients are resistant to the latter drug. Trimethaphan blunts the sympathoadrenal stimulation caused by nitroprusside and reduces the required dosage (Fahmy, 1985). Trimethaphan can be used in the management of autonomic hyperreflexia or reflex sympathetic dystrophy. This syndrome typically is seen in patients with injuries of the upper spinal cord and results from a massive sympathetic discharge. Since normal central inhibition of the reflex is lacking in such patients, the spinal reflex is dominant. |
Ganglionic Neurotransmission
|
Neurotransmission
in autonomic ganglia has long been recognized to be a far more complex
process than that described by a single neurotransmitterreceptor system;
intracellular recordings reveal at least four different changes in potential
that can be elicited by stimulation of the preganglionic nerve (Eccles and
Libet, 1961; Weight et al., 1979) (Figure 94). The primary event
involves a rapid depolarization of postsynaptic sites by ACh. The receptors
are nicotinic, and the pathway is sensitive to classical blocking agents such
as hexamethonium and trimethaphan. Activation of this primary pathway gives
rise to an initial excitatory postsynaptic potential (EPSP). This rapid
depolarization is due primarily to an inward Na+ and perhaps Ca2+
current through a neuronal type of nicotinic receptor channel. Multiple
nicotinic receptor subunits or their mRNAs (
An action potential is generated in the postganglionic neuron when the initial EPSP attains a critical amplitude. In mammalian sympathetic ganglia in vivo, it may be necessary for multiple synapses to be activated before transmission is effective. Discrete end plates with focal localization of receptors do not exist in ganglia; rather, the dendrites and nerve cell bodies contain the receptors. Iontophoretic application of ACh to the ganglion results in a depolarization with a latency of less than 1 millisecond; this decays over a period of 10 to 50 milliseconds (Ascher et al., 1979). Measurements of single-channel conductances indicate that the characteristics of nicotinic receptor channels of the ganglia and the neuromuscular junction are quite similar. The secondary events that follow the initial depolarization are insensitive to hexamethonium or other nicotinic antagonists. They include the slow EPSP, the late slow EPSP, and an inhibitory postsynaptic potential (IPSP). The slow EPSP is generated by ACh acting on muscarinic receptors, and it is blocked by atropine or antagonists that are selective for M1 muscarinic receptors (see Chapter 7: Muscarinic Receptor Agonists and Antagonists). The slow EPSP has a longer latency and a duration of 30 to 60 seconds. In contrast, the late slow EPSP lasts for several minutes and is initiated by the action of peptides released from presynaptic nerve endings or interneurons in specific ganglia (Dun, 1983). The peptides and ACh may be released from the same nerve ending, but the enhanced stability of the peptide in the ganglion extends its sphere of influence to postsynaptic sites beyond those in immediate proximity to the nerve ending. The slow EPSPs result from decreased K+ conductance (Weight et al., 1979). The K+ conductance has been called an M current, and it regulates the sensitivity of the cell to repetitive fast-depolarizing events (Adams et al., 1982). Like
the slow EPSP, the IPSP is unaffected by the classical nicotinic-receptor
blocking agents. Substantial electrophysiological and morphological evidence
has accumulated to suggest that catecholamines participate in the generation
of the IPSP. Dopamine and norepinephrine cause hyperpolarization of ganglia,
and both the IPSP and the catecholamine-induced hyperpolarization are blocked
by The
relative importance of the secondary pathways and even the nature of the
modulating transmitters appear to differ among individual ganglia and between
parasympathetic and sympathetic ganglia. A variety of peptides, including
luteinizing hormonereleasing hormone, substance P, angiotensin, calcitonin
gene-related peptide, vasoactive intestinal polypeptide, neuropeptide Y, and
enkephalins, have been identified in ganglia by immunofluorescence. They
appear localized to particular cell bodies, nerve fibers, or SIF cells; are
released upon nerve stimulation; and are presumed to mediate the late slow
EPSP (Dun, 1983; Elfvin et al., 1993). Other neurotransmitter
substances, such as 5-hydroxytryptamine and gamma-aminobutyric acid, are
known to modify ganglionic transmission. Precise details of their modulatory
actions are not understood, but they appear to be most closely associated
with the late slow EPSP and inhibition of the M current in various ganglia.
It should be emphasized that the secondary synaptic events only modulate the
initial EPSP. Conventional ganglionic blocking agents can inhibit ganglionic
transmission completely; the same cannot be said for muscarinic antagonists
or Drugs that stimulate cholinergic receptor sites on autonomic ganglia can be grouped into two major categories. The first group consists of drugs with nicotinic specificity, including nicotine itself. Their excitatory effects on ganglia are rapid in onset, are blocked by ganglionic nicotinic-receptor antagonists, and mimic the initial EPSP. The second group is composed of agents such as muscarine, McN-A-343, and methacholine. Their excitatory effects on ganglia are delayed in onset, blocked by atropine-like drugs, and mimic the slow EPSP. Ganglionic blocking agents acting on the nicotinic receptor may be classified into two groups. The first group includes those drugs that initially stimulate the ganglia by an ACh-like action and then block them because of a persistent depolarization (e.g., nicotine); prolonged application of nicotine results in desensitization of the cholinergic receptor site and continued blockade. (See review by Volle, 1980.) The blockade of autonomic ganglia produced by the second group of blocking drugs, of which hexamethonium and trimethaphan can be regarded as prototypes, does not involve prior ganglionic stimulation or changes in ganglionic potentials. These agents impair transmission either by competing with ACh for ganglionic nicotinic receptor sites or by blocking the channel when it is open. Trimethaphan acts by competition with ACh, analogous to the mechanism of action of curare at the neuromuscular junction. Hexamethonium appears to block the channel after it opens. This action shortens the duration of current flow, since the open channel either becomes occluded or closes (Gurney and Rang, 1984). Regardless of the mechanism, the initial EPSP is blocked and ganglionic transmission is inhibited. Ganglionic Stimulating Drugs History Two natural alkaloids, nicotine and lobeline, exhibit peripheral actions by stimulating autonomic ganglia. Nicotine (see Figure 95) was first isolated from leaves of tobacco, Nicotiana tabacum, by Posselt and Reiman in 1828, and Orfila initiated the first pharmacological studies of the alkaloid in 1843. Langley and Dickinson (1889) painted the superior cervical ganglion of rabbits with nicotine and demonstrated that its site of action was the ganglion, rather than the preganglionic or postganglionic nerve fiber. Lobeline, from Lobelia inflata, has many of the same actions as nicotine but is less potent.
A number of synthetic compounds also have prominent actions at ganglionic receptor sites. The actions of the 'onium' compounds, of which tetramethylammonium (TMA) is the simplest prototype, were explored in considerable detail in the last half of the nineteenth century and in the early twentieth century. Nicotine Nicotine is of considerable medical significance because of its toxicity, presence in tobacco, and propensity for conferring a dependence on its users. The chronic effects of nicotine and the untoward effects of the chronic use of tobacco are considered in Chapter 24: Drug Addiction and Drug Abuse. Nicotine is one of the few natural liquid alkaloids. It is a colorless, volatile base (pKa= 8.5) that turns brown and acquires the odor of tobacco on exposure to air. Pharmacological Actions The complex and often unpredictable changes that occur in the body after administration of nicotine are due not only to its actions on a variety of neuroeffector and chemosensitive sites but also to the fact that the alkaloid can stimulate and desensitize receptors. The ultimate response of any one system represents the summation of stimulatory and inhibitory effects of nicotine. For example, the drug can increase heart rate by excitation of sympathetic or paralysis of parasympathetic cardiac ganglia, and it can slow heart rate by paralysis of sympathetic or stimulation of parasympathetic cardiac ganglia. In addition, the effects of the drug on the chemoreceptors of the carotid and aortic bodies and on brain centers influence heart rate, as do also the cardiovascular compensatory reflexes resulting from changes in blood pressure caused by nicotine. Finally, nicotine elicits a discharge of epinephrine from the adrenal medulla, and this hormone accelerates cardiac rate and raises blood pressure. Peripheral Nervous System The major action of nicotine consists initially of transient stimulation and subsequently of a more persistent depression of all autonomic ganglia. Small doses of nicotine stimulate the ganglion cells directly and may facilitate the transmission of impulses. When larger doses of the drug are applied, the initial stimulation is followed very quickly by a blockade of transmission. Whereas stimulation of the ganglion cells coincides with their depolarization, depression of transmission by adequate doses of nicotine occurs both during the depolarization and after it has subsided. Nicotine also possesses a biphasic action on the adrenal medulla; small doses evoke the discharge of catecholamines, and larger doses prevent their release in response to splanchnic nerve stimulation. The effects of nicotine on the neuromuscular junction are similar to those on ganglia. However, with the exception of avian and denervated mammalian muscle, the stimulant phase is largely obscured by the rapidly developing paralysis. In the latter stage, nicotine also produces neuromuscular blockade by receptor desensitization. Nicotine, like ACh, is known to stimulate a number of sensory receptors. These include mechanoreceptors that respond to stretch or pressure of the skin, mesentery, tongue, lung, and stomach; chemoreceptors of the carotid body; thermal receptors of the skin and tongue; and pain receptors. Prior administration of hexamethonium prevents the stimulation of the sensory receptors by nicotine but has little, if any, effect on the activation of the sensory receptors by physiological stimuli. Central Nervous System Nicotine markedly stimulates the CNS. Low doses produce weak analgesia; with higher doses, tremors leading to convulsions at toxic doses are evident. The excitation of respiration is a prominent action of nicotine; although large doses act directly on the medulla oblongata, smaller doses augment respiration reflexly by excitation of the chemoreceptors of the carotid and aortic bodies. Stimulation of the CNS with large doses is followed by depression, and death results from failure of respiration due to both central paralysis and peripheral blockade of muscles of respiration. Nicotine induces vomiting by both central and peripheral actions. The central component of the vomiting response is due to stimulation of the emetic chemoreceptor trigger zone in the area postrema of the medulla oblongata. In addition, nicotine activates vagal and spinal afferent nerves that form the sensory input of the reflex pathways involved in the act of vomiting. Studies in isolated higher centers of the brain and spinal cord reveal that the primary sites of action of nicotine in the CNS are prejunctional, causing the release of other transmitters. Accordingly, the stimulatory and pleasure-reward actions of nicotine appear to result from release of excitatory amino acids, dopamine, and other biogenic amines from various CNS centers (MacDermott et al., 1999). Chronic exposure to nicotine in several systems causes an increase in the density or number of nicotinic receptors (see Di Chiara et al., 2000; Stitzel et al., 2000). While the details of the mechanism are not yet understood, the response may be compensatory to the desensitization of receptor function by nicotine. Cardiovascular System When administered intravenously to dogs, nicotine characteristically produces an increase in heart rate and blood pressure. The latter is usually a more sustained response. In general, the cardiovascular responses to nicotine are due to stimulation of sympathetic ganglia and the adrenal medulla, together with the discharge of catecholamines from sympathetic nerve endings. Also contributing to the sympathomimetic response to nicotine is the activation of chemoreceptors of the aortic and carotid bodies, which reflexly results in vasoconstriction, tachycardia, and elevated blood pressure. Gastrointestinal Tract The combined activation of parasympathetic ganglia and cholinergic nerve endings by nicotine results in increased tone and motor activity of the bowel. Nausea, vomiting, and occasionally diarrhea are observed following systemic absorption of nicotine in an individual who has not been exposed to nicotine previously. Exocrine Glands Nicotine causes an initial stimulation of salivary and bronchial secretions that is followed by inhibition. Absorption, Fate, and Excretion Nicotine
is readily absorbed from the respiratory tract, buccal membranes, and skin.
Severe poisoning has resulted from percutaneous absorption. Being a
relatively strong base, its absorption from the stomach is limited, and
intestinal absorption is far more efficient. Nicotine in chewing tobacco,
because it is more slowly absorbed than inhaled nicotine, has a longer
duration of effect. The average cigarette contains 6 to 11 mg of nicotine and
delivers about 1 to 3 mg of nicotine systemically to the smoker;
bioavailability can increase as much as threefold with intensity of puffing
and technique of the smoker (Henningfield, 1995; Benowitz, 1998). Nicotine is
available in several dosage forms to help achieve abstinence from tobacco
use. Efficacy primarily results from preventing a withdrawal or abstinence
syndrome. Nicotine may be administered orally as a gum (nicotine polacrilex; NICORETTE), transdermal patch (NICODERM,
HABITROL, others), a
nasal spray ( Approximately 80% to 90% of nicotine is altered in the body, mainly in the liver but also in the kidney and lung. Cotinine is the major metabolite, with nicotine-1'-N-oxide and 3-hydroxycotinine and conjugated metabolites found in lesser quantities (Benowitz, 1998). The profile of metabolites and the rate of metabolism appear to be similar in the smoker and nonsmoker. The half-life of nicotine following inhalation or parenteral administration is about 2 hours. Both nicotine and its metabolites are eliminated rapidly by the kidney. The rate of urinary excretion of nicotine is dependent upon the pH of the urine; excretion diminishes when the urine is alkaline. Nicotine also is excreted in the milk of lactating women who smoke; the milk of heavy smokers may contain 0.5 mg per liter. Acute Nicotine Poisoning Poisoning from nicotine may occur from accidental ingestion of nicotine-containing insecticide sprays or in children from ingestion of tobacco products. The acutely fatal dose of nicotine for an adult is probably about 60 mg of the base. Smoking tobacco usually contains 1% to 2% nicotine. Apparently, the gastric absorption of nicotine from tobacco taken by mouth is delayed because of slowed gastric emptying, so that vomiting caused by the central effect of the initially absorbed fraction may remove much of the tobacco remaining in the gastrointestinal tract. The onset of symptoms of acute, severe nicotine poisoning is rapid; they include nausea, salivation, abdominal pain, vomiting, diarrhea, cold sweat, headache, dizziness, disturbed hearing and vision, mental confusion, and marked weakness. Faintness and prostration ensue; the blood pressure falls; breathing is difficult; the pulse is weak, rapid, and irregular; and collapse may be followed by terminal convulsions. Death may result within a few minutes from respiratory failure. Therapy Vomiting should be induced with syrup of ipecac or gastric lavage should be performed. Alkaline solutions should be avoided. A slurry of activated charcoal is then passed through the tube and left in the stomach. Respiratory assistance and treatment of shock may be necessary. Other Ganglionic Stimulants Stimulation of ganglia by tetramethylammonium (TMA) or 1,1-dimethyl-4-phenylpiperazinium iodide (DMPP) differs from that produced by nicotine in that the initial stimulation is not followed by a dominant blocking action. DMPP is about three times more potent and slightly more ganglion-selective than nicotine. Although parasympathomimetic drugs stimulate ganglia, their effects usually are obscured by stimulation of other neuroeffector sites. McN-A-343 represents an exception to this; in certain tissues its primary action appears to occur at muscarinic M1 receptors in ganglia. Ganglionic Blocking Drugs The chemical diversity of compounds that block autonomic ganglia without causing prior stimulation is shown in Figure 96.
History and StructureActivity Relationship Although
Triethylsulfoniums, like the quaternary and bis-quaternary ammonium ions, possess ganglionic blocking actions. This knowledge led to the development of sulfonium ganglionic blocking agents such as trimethaphan (see Figure 96). Mecamylamine, a secondary amine, was introduced into therapy for hypertension in the mid-1950s. Pharmacological Properties Nearly all of the physiological alterations observed after the administration of ganglionic blocking agents can be anticipated with reasonable accuracy by a careful inspection of Figure 61 and by knowing which division of the autonomic nervous system exercises dominant control of various organs (Table 94). For example, blockade of sympathetic ganglia interrupts adrenergic control of arterioles and results in vasodilation, improved peripheral blood flow in some vascular beds, and a fall in blood pressure. Generalized ganglionic blockade may result also in atony of the bladder and gastrointestinal tract, cycloplegia, xerostomia, diminished perspiration, and, by abolishing circulatory reflex pathways, postural hypotension. These changes represent the generally undesirable features of ganglionic blockade, which severely limit the therapeutic efficacy of ganglionic blocking agents. Cardiovascular System The importance of existing sympathetic tone in determining the degree to which blood pressure is lowered by ganglionic blockade is illustrated by the fact that blood pressure may be decreased only minimally in recumbent normotensive subjects but may fall markedly in sitting or standing subjects. Postural hypotension is a major problem in ambulatory patients receiving ganglionic blocking drugs; it is relieved to some extent by muscular activity and completely by recumbency. Changes in cardiac rate following ganglionic blockade depend largely on existing vagal tone. In human beings, mild tachycardia usually accompanies the hypotension, a sign that indicates fairly complete ganglionic blockade. However, a decrease may occur if the heart rate is initially high. Cardiac output often is reduced by ganglionic blocking drugs in patients with normal cardiac function as a consequence of diminished venous return resulting from venous dilation and peripheral pooling of blood. In patients with cardiac failure, ganglionic blockade frequently results in increased cardiac output due to a reduction in peripheral resistance. In hypertensive subjects, cardiac output, stroke volume, and left ventricular work are diminished. Although total systemic vascular resistance is decreased in patients who receive ganglionic blocking agents, changes in blood flow and vascular resistance of individual vascular beds are variable. Reduction of cerebral blood flow is small unless mean systemic blood pressure falls below 50 to 60 mm Hg. Skeletal muscle blood flow is unaltered, but splanchnic and renal blood flow decrease following the administration of a ganglionic blocking agent. Absorption, Fate, and Excretion The absorption of quaternary ammonium and sulfonium compounds from the enteric tract is incomplete and unpredictable. This is due both to the limited ability of these ionized substances to penetrate cell membranes and to the depression of propulsive movements of the small intestine and gastric emptying. Although the absorption of mecamylamine is less erratic, a danger exists of reduced bowel activity leading to frank paralytic ileus. After absorption, the quaternary ammonium and sulfonium blocking agents are confined primarily to the extracellular space and are excreted mostly unchanged by the kidney. Mecamylamine concentrates in the liver and kidney and is slowly excreted in an unchanged form. Untoward Responses and Severe Reactions Among the milder untoward responses observed are visual disturbances, dry mouth, conjunctival suffusion, urinary hesitancy, decreased potentia, subjective chilliness, moderate constipation, occasional diarrhea, abdominal discomfort, anorexia, heartburn, nausea, eructation and bitter taste, and the signs and symptoms of syncope caused by postural hypotension. More severe reactions include marked hypotension, constipation, syncope, paralytic ileus, urinary retention, and cycloplegia. Therapeutic Uses Of the
ganglionic blocking agents that have appeared on the therapeutic scene, only mecamylamine
(INVERSINE) and trimethaphan (ARFONAD) currently are utilized in the Ganglionic
blocking agents have been supplanted by superior agents for the treatment of
chronic hypertension (see Chapter 33: Antihypertensive Agents and the
Drug Therapy of Hypertension). Alternative agents also are available for
management of acute hypertensive crises (Murphy, 1995; see Chapter 33:
Antihypertensive Agents and the Drug Therapy of Hypertension). A remaining
use of ganglionic blockers in a hypertensive crisis is for the initial
control of blood pressure in patients with acute dissecting aortic aneurysm,
particularly when preexisting conditions make An additional therapeutic use of the ganglionic blocking agents is in the production of controlled hypotension; a reduction in blood pressure during surgery may be sought deliberately to minimize hemorrhage in the operative field, to reduce blood loss in various orthopedic procedures, and to facilitate surgery on blood vessels (Fukusaki et al., 1999). Trimethaphan, as an infusion, may be used as an alternative to or in combination with sodium nitroprusside, since some patients are resistant to the latter drug. Trimethaphan blunts the sympathoadrenal stimulation caused by nitroprusside and reduces the required dosage (Fahmy, 1985). Trimethaphan can be used in the management of autonomic hyperreflexia or reflex sympathetic dystrophy. This syndrome typically is seen in patients with injuries of the upper spinal cord and results from a massive sympathetic discharge. Since normal central inhibition of the reflex is lacking in such patients, the spinal reflex is dominant. |
Chapter 10. Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists
Overview
|
Catecholamines released by the sympathetic nervous system and adrenal medulla are involved in regulating a host of physiological functions, particularly in integrating responses to a range of stresses that would otherwise threaten homeostatic mechanisms. Norepinephrine is the major neurotransmitter in the peripheral sympathetic nervous system, whereas epinephrine is the primary hormone secreted by the adrenal medulla in mammals. Activation of the sympathetic nervous system occurs in response to diverse stimuli, including physical activity, psychological stress, blood loss, and many other normal or disease-related provocations. Because the functions mediated or modified by the sympathetic nervous system are diverse, drugs that mimic, alter, or antagonize its activity are useful in the treatment of many clinical disorders, including hypertension, cardiovascular shock, arrhythmias, asthma, and anaphylactic reactions. Some of these indications are discussed elsewhere ( see Chapters 28: Drugs Used in the Treatment of Asthma, 32: Drugs Used for the Treatment of Myocardial Ischemia, 33: Antihypertensive Agents and the Drug Therapy of Hypertension, 34: Pharmacological Treatment of Heart Failure, and 35: Antiarrhythmic Drugs). The physiological and metabolic responses that follow stimulation of sympathetic nerves in mammals usually are mediated by the neurotransmitter norepinephrine, although cotransmitters such as peptides potentially may contribute to sympathetic effects. As part of the response to stress, the adrenal medulla also is stimulated, resulting in elevation of the concentration of epinephrine in the circulation; epinephrine functions as a hormone, acting at distant sites in the circulation. The actions of these two catecholamines are very similar at some sites but differ significantly at others. For example, both compounds stimulate the myocardium; however, epinephrine dilates blood vessels to skeletal muscle, whereas norepinephrine causes constriction of blood vessels in skin, mucosa, and kidney. Dopamine is a third naturally occurring catecholamine. Although it is found predominantly in the basal ganglia of the central nervous system (CNS), dopaminergic nerve endings and specific receptors for this catecholamine have been identified elsewhere in the CNS and in the periphery. The role of catecholamines in the CNS is detailed in Chapter 12: Neurotransmission and the Central Nervous System and elsewhere. As might be expected, sympathomimetic aminesnaturally occurring catecholamines and drugs that mimic their actionsand adrenergic receptor antagonistsdrugs that block the effects of sympathetic stimulationconstitute two of the more extensively studied groups of pharmacological agents. Many of the actions of agonists or antagonists that activate or inhibit adrenergic receptors are understandable in terms of the known physiological effects of catecholamines. Whereas endogenous catecholamines such as epinephrine are sometimes used as drugs, most of the available agonists are structural analogs of epinephrine or norepinephrine. These synthetic compounds have a variety of advantages as therapeutic agentssuch as oral bioavailability, prolonged duration of action, and specificity for particular subtypes of adrenergic receptorswhich serve to enhance their therapeutic actions and to diminish potential adverse effects. The structure, cellular function, and physiological effects of adrenergic agonists and antagonists are outlined in this chapter. |
Catecholamines and Sympathomimetic Drugs
|
Most of the actions of catecholamines and sympathomimetic agents can be classified into seven broad types: (1) a peripheral excitatory action on certain types of smooth muscle, such as those in blood vessels supplying skin, kidney, and mucous membranes, and on gland cells, such as those in salivary and sweat glands; (2) a peripheral inhibitory action on certain other types of smooth muscle, such as those in the wall of the gut, in the bronchial tree, and in blood vessels supplying skeletal muscle; (3) a cardiac excitatory action, responsible for an increase in heart rate and force of contraction; (4) metabolic actions, such as an increase in rate of glycogenolysis in liver and muscle and liberation of free fatty acids from adipose tissue; (5) endocrine actions, such as modulation (increasing or decreasing) of the secretion of insulin, renin, and pituitary hormones; (6) central nervous system (CNS) actions, such as respiratory stimulation and, with some of the drugs, an increase in wakefulness and psychomotor activity and a reduction in appetite; and (7) presynaptic actions that result in either inhibition or facilitation of the release of neurotransmitters such as norepinephrine and acetylcholine; physiologically, the inhibitory action is more important than the excitatory action. Many of these actions and the receptors that mediate them are summarized in Tables 61 and 63. Not all sympathomimetic drugs show each of the above types of action to the same degree. However, many of the differences in their effects are only quantitative, and descriptions of the effects of each compound would be unnecessarily repetitive. Therefore, the pharmacological properties of these drugs as a class are described in detail for the prototypical agent, epinephrine. Appreciation of the pharmacological properties of the drugs that are
described in this chapter is critically dependent on understanding the
classification, distribution, and mechanism of action of the various subtypes
of adrenergic receptors (
History The pressor effect of adrenal extracts was first shown by Oliver and Schfer in 1895. The active principle was named epinephrine by Abel in 1899 and synthesized independently by Stolz and Dakin (seeHartung, 1931). The development of our knowledge of epinephrine and norepinephrine as neurohumoral transmitters is outlined in Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems. Barger and Dale (1910) studied the pharmacological activity of a large series of synthetic amines related to epinephrine and termed their action sympathomimetic. This important study determined the basic structural requirements for activity. When it was later found that cocaine or chronic denervation of effector organs reduced the responses to ephedrine and tyramine but enhanced the effects of epinephrine, it became clear that the differences between sympathomimetic amines were not simply quantitative. It was suggested that epinephrine acted directly on the effector cell, whereas ephedrine and tyramine had an indirect effect by acting on the nerve endings. The discovery that reserpine depletes tissues of norepinephrine (Bertler et al., 1956) was followed by evidence that tyramine and certain other sympathomimetic amines do not act on tissues from animals that have been treated with reserpine; this, too, indicated that they act by releasing endogenous norepinephrine (Burn and Rand, 1958). Chemistry and Structure-Activity Relationship of Sympathomimetic Amines
Many directly acting sympathomimetic drugs influence both Separation of Aromatic Ring and Amino Group By far the greatest sympathomimetic activity occurs when two carbon atoms separate the ring from the amino group. This rule applies with few exceptions to all types of action. Substitution on the Amino Group The effects of amino substitution are most readily seen in the actions
of catecholamines on Substitution on the Aromatic Nucleus Maximal Hydroxyl groups in positions 3 and 5 confer Since substitution of polar groups on the phenylethylamine structure makes the resultant compounds less lipophilic, unsubstituted or alkyl-substituted compounds cross the bloodbrain barrier more readily and have more central activity. Thus, ephedrine, amphetamine, and methamphetamine exhibit considerable CNS activity. In addition, as noted above, the absence of polar hydroxyl groups results in a loss of direct sympathomimetic activity. Catecholamines have only a brief duration of action and are ineffective when administered orally, because they are rapidly inactivated in the intestinal mucosa and in the liver before reaching the systemic circulation (seeChapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems). Compounds without one or both hydroxyl substituents are not acted upon by catechol-O-methyltransferase (COMT), and their oral effectiveness and duration of action are enhanced. Groups other than hydroxyls have been substituted on the aromatic
ring. In general, potency at
Substitution on the This substitution blocks oxidation by monoamine oxidase (MAO), greatly
prolonging the duration of action of noncatecholamines because their degradation
depends largely on the action of MAO. The duration of action of drugs such as
ephedrine or amphetamine is thus measured in hours rather than in minutes.
Similarly, compounds with an Substitution on the Substitution of a hydroxyl group on the Optical Isomerism Substitution on either Physiological Basis of Adrenergic Receptor Function An important factor in the response of any cell or organ to
sympathomimetic amines is the density and proportion of The ultimate response of a target organ to sympathomimetic amines is
dictated not only by the direct effects of the agents but also by the reflex
homeostatic adjustments of the organism. One of the most striking effects of
many sympathomimetic amines is a rise in arterial blood pressure caused by
stimulation of vascular Indirectly Acting Sympathomimetic Drugs For many years, it was presumed that sympathomimetic amines produced their effects by acting directly on adrenergic receptors. However, this notion was dispelled by the finding that the effects of tyramine and many other noncatecholamines were reduced or abolished after chronic postganglionic denervation or treatment with cocaine or reserpine. Under these circumstances, the effects of exogenously administered epinephrine, and especially norepinephrine, often were enhanced. These observations led to the proposal that tyramine and related amines act indirectly, following uptake into the adrenergic nerve terminal, by displacing norepinephrine from storage sites in the synaptic vesicles or from extravesicular binding sites. Norepinephrine could then exit from the adrenergic nerve terminal and interact with receptors to produce sympathomimetic effects. The depletion of tissue stores of catecholamines that follows treatment with reserpine or degeneration of adrenergic nerve terminals would explain the lack of effect of tyramine under these conditions. In the presence of cocaine, the high-affinity neuronal transport system for catecholamines and certain congeners is inhibited, and tyramine and related amines are unable to enter the adrenergic nerve terminal. In this manner, cocaine inhibits the actions of indirectly acting sympathomimetic amines while potentiating the effects of directly acting agents that are normally removed from the synaptic cleft by this transport system (seeChapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems). In assessing the proportion of direct and indirect actions of a sympathomimetic amine, the most common experimental procedure is to compare the dose-response curves for the agent on a particular target tissue before and after treatment with reserpine (Trendelenburg, 1972). Those drugs whose actions are essentially unaltered after treatment with reserpine are classified as directly acting sympathomimetic amines (e.g., norepinephrine, phenylephrine), whereas those whose actions are abolished are termed indirectly acting (e.g., tyramine). Many agents exhibit some degree of residual sympathomimetic activity after the administration of reserpine, but higher doses of these amines are required to produce comparable effects. These are classified as mixed-acting sympathomimetic amines; that is, they have both direct and indirect actions. The proportion of direct and indirect actions can vary considerably among different tissues and species. In some cases, relatively little is known about these properties in human beings. Since the actions of norepinephrine are more marked on False-Transmitter Concept As indicated above, indirectly acting amines are taken up into
adrenergic nerve terminals and storage vesicles, where they replace norepinephrine
in the storage complex. Phenylethylamines that lack a This hypothesis, known as the false-transmitter concept, is a
possible explanation for some of the effects of inhibitors of MAO.
Phenylethylamines normally are synthesized in the gastrointestinal tract as a
result of the action of bacterial tyrosine decarboxylase. The tyramine formed
in this fashion usually is oxidatively deaminated in the gastrointestinal
tract and the liver, and the amine does not reach the systemic circulation in
significant concentrations. However, when a MAO inhibitor is administered,
tyramine may be absorbed systemically and is transported into adrenergic
nerve terminals, where its catabolism is again prevented because of the
inhibition of MAO at this site; it is then Despite such functional impairment, patients who have received MAO inhibitors may experience severe hypertensive crises if they ingest cheese, beer, or red wine. These and related foods, which are produced by fermentation, contain a large quantity of tyramine and, to a lesser degree, other phenylethylamines. When gastrointestinal and hepatic MAO are inhibited, the large quantity of tyramine that is ingested is absorbed rapidly and reaches the systemic circulation in high concentration. A massive and precipitous release of norepinephrine can result, with consequent hypertension that can be severe enough to cause myocardial infarction or a stroke. The properties of various MAO inhibitors (reversible or irreversible; selective or nonselective at MAO-A and MAO-B) are discussed in Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders. Endogenous Catecholamines Epinephrine Epinephrine
(adrenaline) is a potent stimulant of both Blood Pressure Epinephrine is one of the most potent vasopressor drugs known. If a pharmacological dose is given rapidly by an intravenous route, it evokes a characteristic effect on blood pressure, which rises rapidly to a peak that is proportional to the dose. The increase in systolic pressure is greater than the increase in diastolic pressure, so that the pulse pressure increases. As the response wanes, the mean pressure may fall below normal before returning to control levels. The mechanism of the rise in blood pressure due to epinephrine is
threefold: (1) a direct myocardial stimulation that increases the strength of
ventricular contraction (positive inotropic action); (2) an increased heart
rate (positive chronotropic action); and (3) vasoconstriction in many
vascular bedsespecially in the precapillary resistance vessels of skin,
mucosa, and kidneyalong with marked constriction of the veins. The pulse
rate, at first accelerated, may be slowed markedly at the height of the rise
of blood pressure by compensatory vagal discharge. Small doses of epinephrine
(0.1 The effects are somewhat different when the drug is given by slow
intravenous infusion or by subcutaneous injection. Absorption of epinephrine
after subcutaneous injection is slow due to local vasoconstrictor action; the
effects of doses as large as 0.5 to 1.5 mg can be duplicated by intravenous
infusion at a rate of 10 to 30
Vascular Effects The chief vascular action of epinephrine is exerted on the smaller arterioles and precapillary sphincters, although veins and large arteries also respond to the drug. Various vascular beds react differently, which results in a substantial redistribution of blood flow. Injected epinephrine markedly decreases cutaneous blood flow,
constricting precapillary vessels and small venules. Cutaneous
vasoconstriction accounts for a marked decrease in blood flow in the hands
and feet. The 'after congestion' of mucosae following the
vasoconstriction from locally applied epinephrine probably is due to changes
in vascular reactivity as a result of tissue hypoxia rather than to Blood flow to skeletal muscles is increased by therapeutic doses in
human beings. This is due in part to a powerful The effect of epinephrine on cerebral circulation is related to systemic blood pressure. In usual therapeutic doses, the drug has relatively little constrictor action on cerebral arterioles. It is physiologically advantageous that the cerebral circulation does not constrict in response to activation of the sympathetic nervous system by stressful stimuli. Indeed, autoregulatory mechanisms tend to limit the increase in cerebral blood flow caused by increased blood pressure. Doses of epinephrine that have little effect on mean arterial pressure
consistently increase renal vascular resistance and reduce renal blood flow
by as much as 40%. All segments of the renal vascular bed contribute to the
increased resistance. Since the glomerular filtration rate is only slightly
and variably altered, the filtration fraction is consistently increased.
Excretion of Na+, K+, and Cl is decreased;
urine volume may be increased, decreased, or unchanged. Maximal tubular
reabsorptive and excretory capacities are unchanged. The secretion of renin
is increased as a consequence of a direct action of epinephrine on Arterial and venous pulmonary pressures are raised. Although direct pulmonary vasoconstriction occurs, redistribution of blood from the systemic to the pulmonary circulation, due to constriction of the more powerful musculature in the systemic great veins, doubtless plays an important part in the increase in pulmonary pressure. Very high concentrations of epinephrine may cause pulmonary edema precipitated by elevated pulmonary capillary filtration pressure and possibly by 'leaky' capillaries. Coronary blood flow is enhanced by epinephrine or by cardiac
sympathetic stimulation under physiological conditions. The increased flow
occurs even with doses that do not increase the aortic blood pressure and is
the result of two factors. The first is the increased relative duration of
diastole at higher heart rates (see below); this is partially offset
by decreased blood flow during systole because of more forceful contraction
of the surrounding myocardium and an increase in mechanical compression of
the coronary vessels. The increased flow during diastole is further enhanced
if aortic blood pressure is elevated by epinephrine, and, as a consequence,
total coronary flow may be increased. The second factor is a metabolic
dilator effect that results from the increased strength of contraction and
myocardial oxygen consumption due to the direct effects of epinephrine on
cardiac myocytes. This vasodilation is mediated in part by adenosine released
from cardiac myocytes, which tends to override a direct vasoconstrictor
effect of epinephrine that results from activation of Cardiac Effects Epinephrine is a powerful cardiac stimulant. It acts directly on the
predominant In accelerating the heart, epinephrine preferentially shortens systole
so that the duration of diastole usually is not reduced. Indeed, activation
of Some effects of epinephrine on cardiac tissues are largely secondary to the increase in heart rate, and are small or inconsistent when the heart rate is kept constant. For example, the effect of epinephrine on repolarization of atrial muscle, Purkinje fibers, or ventricular muscle is small if the heart rate is unchanged. When the heart rate is increased, the duration of the action potential is consistently shortened, and the refractory period is correspondingly decreased. Conduction through the Purkinje system depends on the level of membrane potential at the time of excitation. Excessive reduction of this potential results in conduction disturbances, ranging from slowed conduction to complete block. Epinephrine often increases the membrane potential and improves conduction in Purkinje fibers that have been excessively depolarized. Epinephrine normally shortens the refractory period of the human
atrioventricular (AV) node by direct effects on the heart, although doses of
epinephrine that slow the heart through reflex vagal discharge may indirectly
tend to prolong it. Epinephrine also decreases the grade of AV block that
occurs as a result of disease, drugs, or vagal stimulation. Supraventricular
arrhythmias are apt to occur from the combination of epinephrine and
cholinergic stimulation. Depression of sinus rate and AV conduction by vagal
discharge probably plays a part in epinephrine-induced ventricular
arrhythmias, since various drugs that block the vagal effect confer some
protection. The action of epinephrine in enhancing cardiac automaticity and
its action in causing arrhythmias are effectively antagonized by Cardiac arrhythmias have been seen in patients after inadvertent intravenous administration of conventional subcutaneous doses of epinephrine. Ventricular premature systoles can appear, which may be followed by multifocal ventricular tachycardia or ventricular fibrillation. Pulmonary edema also may occur. Epinephrine decreases the amplitude of the T wave of the electrocardiogram (ECG) in normal persons. In animals given relatively larger doses, additional effects are seen on the T wave and the ST segment. After decreasing in amplitude, the T wave may become biphasic, and the ST segment can deviate either above or below the isoelectric line. Such ST-segment changes are similar to those seen in patients with angina pectoris during spontaneous or epinephrine-induced attacks of pain. These electrical changes therefore have been attributed to myocardial ischemia. Also, epinephrine as well as other catecholamines may cause myocardial cell death, particularly after intravenous infusions. Acute toxicity is associated with contraction band necrosis and other pathological changes. Recent interest has focused on the possiblilty that prolonged sympathetic stimulation of the heart, such as in congestive cardiomyopathy, may promote apoptosis of cardiomyocytes. Effects on Smooth Muscles The effects of epinephrine on the smooth muscles of different organs
and systems depend on the type of adrenergic receptor in the muscle (seeTable
61). The effects on vascular smooth muscle are of major physiological
importance, whereas those on gastrointestinal smooth muscle are relatively
minor. Gastrointestinal smooth muscle is, in general, relaxed by epinephrine.
This effect is due to activation of both The responses of uterine muscle to epinephrine vary with species,
phase of the sexual cycle, state of gestation, and dose given. Epinephrine
contracts strips of pregnant or nonpregnant human uterus in vitro by
interaction with Epinephrine relaxes the detrusor muscle of the bladder as a result of
activation of Respiratory Effects Epinephrine affects respiration primarily by relaxing bronchial muscle. It has a powerful bronchodilator action, most evident when bronchial muscle is contracted because of disease, as in bronchial asthma, or in response to drugs or various autacoids. In such situations, epinephrine has a striking therapeutic effect as a physiological antagonist to substances that cause bronchoconstriction. The beneficial effects of epinephrine in asthma also may arise from
inhibition of antigen-induced release of inflammatory mediators from mast
cells, and to a lesser extent from diminution of bronchial secretions and
congestion within the mucosa. Inhibition of mast cell secretion is mediated
by Effects on the Central Nervous System Because of the inability of this rather polar compound to enter the CNS, epinephrine in conventional therapeutic doses is not a powerful CNS stimulant. While the drug may cause restlessness, apprehension, headache, and tremor in many persons, these effects in part may be secondary to the effects of epinephrine on the cardiovascular system, skeletal muscles, and intermediary metabolism; that is, they may be the result of somatic manifestations of anxiety. Some other sympathomimetic drugs more readily cross the bloodbrain barrier. Metabolic Effects Epinephrine has a number of important influences on metabolic
processes. It elevates the concentrations of glucose and lactate in blood by
mechanisms described in Chapter 6: Neurotransmission: The Autonomic and
Somatic Motor Nervous Systems. Insulin secretion is inhibited through an
interaction with Epinephrine raises the concentration of free fatty acids in blood by
stimulating Miscellaneous Effects Epinephrine reduces circulating plasma volume by loss of protein-free
fluid to the extracellular space, thereby increasing erythrocyte and plasma
protein concentrations. However, conventional doses of epinephrine do not
significantly alter plasma volume or packed red-cell volume under normal
conditions, although such doses are reported to have variable effects in the
presence of shock, hemorrhage, hypotension, and anesthesia. Epinephrine
rapidly increases the number of circulating polymorphonuclear leukocytes,
likely due to The effects of epinephrine on secretory glands are not marked; in most
glands secretion usually is inhibited, partly owing to the reduced blood flow
caused by vasoconstriction. Epinephrine stimulates lacrimation and a scanty
mucous secretion from salivary glands. Sweating and pilomotor activity are
minimal after systemic administration of epinephrine, but occur after
intradermal injection of very dilute solutions of either epinephrine or norepinephrine.
Such effects are inhibited by Mydriasis is readily seen during physiological sympathetic stimulation but not when epinephrine is instilled into the conjunctival sac of normal eyes. However, epinephrine usually lowers intraocular pressure from normal levels and in wide-angle glaucoma; the mechanism of this effect is not clear, but both reduced production of aqueous humor due to vasoconstriction and enhanced outflow probably occur (seeChapter 66: Ocular Pharmacology). Although epinephrine does not directly excite skeletal muscle, it
facilitates neuromuscular transmission, particularly that following prolonged
rapid stimulation of motor nerves. In apparent contrast to the effects of Epinephrine promotes a fall in plasma K+ largely due to
stimulation of K+ uptake into cells, particularly skeletal muscle,
due to activation of Large or repeated doses of epinephrine or other sympathomimetic amines
given to experimental animals lead to damage to arterial walls and
myocardium, which is so severe as to cause the appearance of necrotic areas
in the heart indistinguishable from myocardial infarcts. The mechanism of
this injury is not yet clear, but Absorption, Fate, and Excretion As indicated above, epinephrine is not effective after oral administration, because it is rapidly conjugated and oxidized in the gastrointestinal mucosa and liver. Absorption from subcutaneous tissues occurs relatively slowly because of local vasoconstriction and the rate may be further decreased by systemic hypotension, for example in a patient with shock. Absorption is more rapid after intramuscular injection. In emergencies, it may be necessary in some cases to administer epinephrine intravenously. When relatively concentrated solutions (1%) are nebulized and inhaled, the actions of the drug largely are restricted to the respiratory tract; however, systemic reactions such as arrhythmias may occur, particularly if larger amounts are used. Epinephrine is rapidly inactivated in the body. The liver, which is rich in both of the enzymes responsible for destroying circulating epinephrine (COMT and MAO), is particularly important in this regard (seeFigure 65). Although only small amounts appear in the urine of normal persons, the urine of patients with pheochromocytoma may contain relatively large amounts of epinephrine, norepinephrine, and their metabolites. Epinephrine is available in a variety of formulations geared for different clinical indications and routes of administration, such as by injection (usually subcutaneously but sometimes intravenously), by inhalation, or topically. Several practical points are worth noting. First, epinephrine is unstable in alkaline solution; when exposed to air or light, it turns pink from oxidation to adrenochrome and then brown from formation of polymers. Epinephrine injection is available in 1:1,000, 1:10,000, and 1:100,000 solutions. The usual adult dose given subcutaneously ranges from 0.3 mg to 0.5 mg. The intravenous route is used cautiously if an immediate and reliable effect is mandatory. If the solution is given by vein, it must be adequately diluted and injected very slowly. The dose is seldom as much as 0.25 mg, except for cardiac arrest, when larger doses may be required. Epinephrine suspensions (e.g., SUS-PHRINE) are used to slow subcutaneous absorption and must never be injected intravenously. Also, a 1% (1:100) formulation is available for administration via inhalation; every precaution must be taken not to confuse this 1:100 solution with the 1:1000 solution designed for parenteral administration, because inadvertent injection of the 1:100 solution can be fatal. Toxicity, Adverse Effects, and Contraindications Epinephrine may cause disturbing reactions, such as restlessness, throbbing headache, tremor, and palpitations. The effects rapidly subside with rest, quiet, recumbency, and reassurance. More serious reactions include cerebral hemorrhage and cardiac arrhythmias. The use of large doses or the accidental, rapid intravenous injection of epinephrine may result in cerebral hemorrhage from the sharp rise in blood pressure. Ventricular arrhythmias may follow the administration of epinephrine. Angina may be induced by epinephrine in patients with coronary artery disease. The use of epinephrine generally is contraindicated in patients who
are receiving nonselective Therapeutic Uses Epinephrine has limited clinical uses. In general, these are based on
the actions of the drug on blood vessels, heart, and bronchial muscle. In the
past, the most common use of epinephrine was to relieve respiratory distress
due to bronchospasm; however, Norepinephrine Norepinephrine (levarterenol, l-noradrenaline, l- Pharmacological Properties The pharmacological actions of norepinephrine and epinephrine have
been extensively compared in vivo and in vitro (seeTable
102). Both drugs are direct agonists on effector cells, and their actions
differ mainly in the ratio of their effectiveness in stimulating Cardiovascular Effects The cardiovascular effects of an intravenous infusion of 10 Unlike epinephrine, small doses of norepinephrine do not cause
vasodilation or lower blood pressure, since the blood vessels of skeletal
muscle constrict rather than dilate; Other Effects Other responses to norepinephrine are not prominent in human beings. The drug causes hyperglycemia and other metabolic effects similar to those produced by epinephrine, but these are observed only when large doses are given; that is, norepinephrine is not as effective a 'hormone' as epinephrine. Intradermal injection of suitable doses causes sweating that is not blocked by atropine. Absorption, Fate, and Excretion Norepinephrine, like epinephrine, is ineffective when given orally and is absorbed poorly from sites of subcutaneous injection. It is rapidly inactivated in the body by the same enzymes that methylate and oxidatively deaminate epinephrine (see above). Small amounts normally are found in the urine. The excretion rate may be greatly increased in patients with pheochromocytoma. Toxicity, Adverse Effects, and Precautions The untoward effects of norepinephrine are similar to those of epinephrine, although there is typically greater elevation of blood pressure with norepinephrine. Excessive doses can cause severe hypertension, so careful blood pressure monitoring generally is indicated during systemic administration of this agent. Care must be taken that necrosis and sloughing do not occur at the
site of intravenous injection owing to extravasation of the drug. The
infusion should be made high in the limb, preferably through a long plastic
cannula extending centrally. Impaired circulation at injection sites, with or
without extravasation of norepinephrine, may be relieved by infiltrating the
area with phentolamine, an Therapeutic Uses and Status Norepinephrine (norepinephrine bitartrate, LEVOPHED) has only limited therapeutic value. The use of it and other sympathomimetic amines in shock is discussed later in this chapter. In the treatment of low blood pressure, the dose is titrated to the desired pressor response. Dopamine Dopamine (3,4-dihydroxyphenylethylamine) (seeTable 101) is the immediate metabolic precursor of norepinephrine and epinephrine; it is a central neurotransmitter particularly important in the regulation of movement (Chapters 12: Neurotransmission and the Central Nervous System, 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania, and 22: Treatment of Central Nervous System Degenerative Disorders) and possesses important intrinsic pharmacological properties. Dopamine is a substrate for both MAO and COMT and thus is ineffective when administered orally. Classification of dopamine receptors is described in Chapter 22: Treatment of Central Nervous System Degenerative Disorders. Pharmacological Properties Cardiovascular Effects The cardiovascular effects of dopamine are mediated by several distinct types of receptors that vary in their affinity for dopamine (Goldberg and Rajfer, 1985). At low concentrations, the primary interaction of dopamine is with vascular D1-dopaminergic receptors, especially in the renal, mesenteric, and coronary beds. By activating adenylyl cyclase and raising intracellular concentrations of cyclic AMP, D1-receptor stimulation leads to vasodilation (Missale et al., 1998). Infusion of low doses of dopamine causes an increase in glomerular filtration rate, renal blood flow, and Na+ excretion. As a consequence, dopamine has pharmacologically appropriate effects in the management of states of low cardiac output associated with compromised renal function, such as severe congestive heart failure. At somewhat higher concentrations, dopamine exerts a positive
inotropic effect on the myocardium, acting on Other Effects Although there are specific dopamine receptors in the CNS, injected dopamine usually has no central effects because it does not readily cross the bloodbrain barrier. Precautions, Adverse Reactions, and Contraindications Before dopamine is administered to patients in shock, hypovolemia should be corrected by transfusion of whole blood, plasma, or other appropriate fluid. Untoward effects due to overdosage generally are attributable to excessive sympathomimetic activity (although this also may be the response to worsening shock). Nausea, vomiting, tachycardia, anginal pain, arrhythmias, headache, hypertension, and peripheral vasoconstriction may be encountered during an infusion of dopamine. Extravasation of large amounts of dopamine during infusion may cause ischemic necrosis and sloughing. Rarely, gangrene of the fingers or toes has followed the prolonged infusion of the drug. Dopamine should be avoided or used at a much reduced dosage (one-tenth or less) if the patient has received a MAO inhibitor. Careful adjustment of dosage also is necessary for the patient who is taking tricyclic antidepressants, as responses may be particularly variable. Therapeutic Uses Dopamine (dopamine hydrochloride;INTROPIN) is used in the treatment of severe congestive failure, particularly in patients with oliguria and with low or normal peripheral vascular resistance. The drug also may improve physiological parameters in the treatment of cardiogenic and septic shock. While dopamine may acutely improve cardiac and renal function in severely ill patients with chronic heart disease or renal failure, there is relatively little evidence supporting long-term changes in clinical outcome (Marik and Iglesias, 1999). The management of shock is discussed later in this chapter. Dopamine hydrochloride is used only intravenously. The drug is
initially administered at a rate of 2 to 5 Related drugs include fenoldopam and dopexamine.
Fenoldopam (CORLOPAM) is a
D1-receptor-selective agonist, which lowers blood pressure in
severe hypertension (Elliott et al., 1990; Nichols et al., 1990).
Fenoldopam does not appear to activate
Epinephrine was first used as a bronchodilator at the beginning of this
century, and ephedrine was introduced into western medicine in 1924, although
it had been used in
Isoproterenol Isoproterenol (isopropylarterenol, isopropylnorepinephrine, isoprenaline,
isopropylnoradrenaline, d,l- Pharmacological Actions The major cardiovascular effects of isoproterenol (compared with epinephrine and norepinephrine) are illustrated in Figure 102. Intravenous infusion of isoproterenol lowers peripheral vascular resistance, primarily in skeletal muscle but also in renal and mesenteric vascular beds. Diastolic pressure falls. Systolic blood pressure may remain unchanged or rise, although mean arterial pressure typically falls. Cardiac output is increased because of the positive inotropic and chronotropic effects of the drug in the face of diminished peripheral vascular resistance. The cardiac effects of isoproterenol may lead to palpitations, sinus tachycardia, and more serious arrhythmias; large doses of isoproterenol may cause myocardial necrosis in animals. Isoproterenol relaxes almost all varieties of smooth muscle when the
tone is high, but this action is most pronounced on bronchial and
gastrointestinal smooth muscle. It prevents or relieves bronchoconstriction.
Its effect in asthma may be due in part to an additional action to inhibit
antigen-induced release of histamine and other mediators of inflammation;
this action is shared by Absorption, Fate, and Excretion Isoproterenol is readily absorbed when given parenterally or as an aerosol. It is metabolized primarily in the liver and other tissues by COMT. Isoproterenol is a relatively poor substrate for MAO and is not taken up by sympathetic neurons to the same extent as are epinephrine and norepinephrine. The duration of action of isoproterenol therefore may be longer than that of epinephrine, but it still is brief. Toxicity and Adverse Effects Palpitations, tachycardia, headache, and flushed skin are common. Cardiac ischemia and arrhythmias may occur, particularly in patients with underlying coronary artery disease. Therapeutic Uses Isoproterenol (isoproterenol hydrochloride;ISUPREL) may be used in emergencies to stimulate heart rate in patients with bradycardia or heart block, particularly in anticipation of inserting an artificial cardiac pacemaker or in patients with the ventricular arrhythmia torsades de pointes. In disorders such as asthma and shock, isoproterenol largely has been replaced by other sympathomimetic drugs (see below and Chapter 28: Drugs Used in the Treatment of Asthma). Dobutamine Dobutamine
resembles dopamine structurally but possesses a bulky aromatic substituent on
the amino group (seeTable 101). The pharmacological effects of
dobutamine are due to direct interactions with Cardiovascular Effects The cardiovascular effects of racemic dobutamine represent a composite
of the distinct pharmacological properties of the () and (+) stereoisomers.
Dobutamine has relatively more prominent inotropic than chronotropic effects
on the heart compared to isoproterenol. The explanation for this useful
selectivity is not clear. It may be due in part to the fact that peripheral
resistance is relatively unchanged. Alternatively, cardiac In animals, administration of dobutamine at a rate of 2.5 to 15 Adverse Effects In some patients, blood pressure and heart rate may increase significantly during administration of dobutamine; this may require reduction of the rate of infusion. Patients with a history of hypertension may be at greater risk of developing an exaggerated pressor response. Since dobutamine facilitates atrioventricular conduction, patients with atrial fibrillation are at risk of marked increases in ventricular response rates; digoxin or other measures may be required to prevent this from occurring. Some patients may develop ventricular ectopic activity. As with any inotropic agent, dobutamine potentially may increase the size of a myocardial infarct by increasing myocardial oxygen demand. This risk must be balanced against the patient's overall clinical status. The efficacy of dobutamine over a period of more than a few days is uncertain; there is evidence for the development of tolerance (Unverferth et al., 1980). Therapeutic Uses Dobutamine (dobutamine hydrochloride;DOBUTREX) is indicated for the short-term treatment of cardiac decompensation that may occur after cardiac surgery or in patients with congestive heart failure or acute myocardial infarction. Dobutamine increases cardiac output and stroke volume in such patients, usually without a marked increase in heart rate. Alterations in blood pressure or peripheral resistance usually are minor, although some patients may have marked increases in blood pressure or heart rate. Clinical evidence of longer-term efficacy remains uncertain. Interestingly, an infusion of dobutamine in combination with echocardiography is useful in the noninvasive assessment of patients with coronary artery disease (Madu et al., 1994). Stressing of the heart with dobutamine may reveal cardiac abnormalities in carefully selected patients. Dobutamine has a half-life of about 2 minutes; the major metabolites
are conjugates of dobutamine and 3-O-methyldobutamine. The onset of
effect is rapid. Consequently, a loading dose is not required, and
steady-state concentrations generally are achieved within 10 minutes of
initiation of an infusion. The rate of infusion required to increase cardiac
output is typically between 2.5 and 10
Some of the major adverse effects of A second strategy that has increased the usefulness of several A final strategy to enhance preferential activation of pulmonary Administration of In the treatment of asthma, Metaproterenol Metaproterenol (called orciprenaline in Terbutaline Terbutaline
is a Albuterol Albuterol
(salbutamol; VENTOLIN, PROVENTIL, others) is a selective Isoetharine Isoetharine
was the first drug with Pirbuterol Pirbuterol
is a relatively selective Bitolterol Bitolterol (bitolterol mesylate;TORNALATE) is a novel Fenoterol Fenoterol BEROTEC) is a Formoterol Formoterol FORADIL) is a long-acting Procaterol Procaterol MASCACIN, others) is a Salmeterol Salmeterol SEREVENT) is a Ritodrine Ritodrine
is a selective Therapeutic Uses Ritodrine may be administered intravenously to selected patients to
arrest premature labor. Ritodrine and related drugs can prolong pregnancy (King
et al., 1988). However, Adverse Effects of The major adverse effects of Skeletal muscle tremor is a relatively common adverse effect of the Tachycardia is a common adverse effect of systemically administered Arterial oxygen tension may fall when treatment of patients with an acute exacerbation of asthma is begun; this may be due to drug-induced pulmonary vascular dilation, which leads to increased mismatching of ventilation and perfusion. This effect usually is small and transient. Supplemental oxygen should be given if necessary. Severe pulmonary edema has been reported in women receiving ritodrine or terbutaline for premature labor. The results of a number of epidemiologic studies have suggested a
possible adverse connection between prolonged use of There is some evidence suggesting that regular use of Large doses of
The major clinical effects of a number of sympathomimetic drugs are
due to activation of Methoxamine Methoxamine (methoxamine hydrochloride;VASOXYL;seeTable 101) is a relatively specific Phenylephrine Phenylephrine is an Mephentermine Mephentermine (seeTable 101) is a sympathomimetic drug that acts both directly and indirectly; it has many similarities to ephedrine (see below). After an intramuscular injection, the onset of action is prompt (within 5 to 15 minutes), and effects may last for several hours. Since the drug releases norepinephrine, cardiac contraction is enhanced, and cardiac output and systolic and diastolic pressures usually are increased. The change in heart rate is variable, depending on the degree of vagal tone. Adverse effects are related to CNS stimulation, excessive rises in blood pressure, and arrhythmias. Mephentermine (mephentermine sulfate;WYAMINE SULFATE) is used to prevent hypotension, which frequently accompanies spinal anesthesia. Metaraminol Metaraminol (metaraminol bitartrate;ARAMINE) (seeTable 101) is a
sympathomimetic drug with prominent direct effects on vascular Midodrine Midodrine PROAMATINE) is an orally effective,
Clonidine Clonidine,
an imidazoline, was synthesized in the early 1960s and found to produce
vasoconstriction that was mediated by
Pharmacological Effects The major pharmacological effects of clonidine involve changes in
blood pressure and heart rate, although the drug has a variety of other
important actions. Intravenous infusion of clonidine causes an acute rise in
blood pressure, apparently because of activation of postsynaptic Data obtained using [3H]clonidine as a radioligand in
receptor-binding assays suggest that noradrenergic imidazoline-preferring
binding sites exist in the brain. These sites, however, do not bind
catecholamines and thus cannot mediate the centrally mediated hypotensive
effects of norepinephrine. There is increasing evidence that these
imidazoline-preferring sites may represent a new family of receptors through
which clonidine and other imidazolines may elicit hypotensive effects (van
Zwieten, 1999). However, the lack of an antihypertensive effect of clonidine
and other imidazoline-site ligands in genetically engineered mice lacking
functional Clonidine decreases discharges in sympathetic preganglionic fibers in
the splanchnic nerve as well as in postganglionic fibers of cardiac nerves (Langer
et al., 1980). These effects are blocked by Absorption, Fate, and Excretion Clonidine is well absorbed after oral administration, and bioavailability is nearly 100%. The peak concentration in plasma and the maximal hypotensive effect are observed 1 to 3 hours after an oral dose. The elimination half-life of the drug ranges from 6 to 24 hours, with a mean of about 12 hours (Lowenthal et al., 1988). About half of an administered dose can be recovered unchanged in the urine, and the half-life of the drug may increase with renal failure. There is good correlation between plasma concentrations of clonidine and its pharmacological effects. A transdermal delivery patch permits continuous administration of clonidine as an alternative to oral therapy. The drug is released at an approximately constant rate for a week; 3 or 4 days are required to reach steady-state concentrations in plasma. When the patch is removed, plasma concentrations remain stable for about 8 hours and then decline gradually over a period of several days; this decrease is associated with a rise in blood pressure (Langley and Heel, 1988; Lowenthal et al., 1988). Adverse Effects The major adverse effects of clonidine are dry mouth and sedation. These responses occur in at least 50% of patients and may require discontinuation of drug administration. However, they may diminish in intensity after several weeks of therapy. Sexual dysfunction also may occur. Marked bradycardia is observed in some patients. These and some of the other adverse effects of clonidine are frequently related to dose, and their incidence may be lower with transdermal administration of clonidine, since antihypertensive efficacy may be achieved while avoiding the relatively high peak concentrations that occur after oral administration of the drug; however, this possibility requires further evaluation (Langley and Heel, 1988). About 15% to 20% of patients develop contact dermatitis when using clonidine in the transdermal system. Withdrawal reactions follow abrupt discontinuation of long-term therapy with clonidine in some hypertensive patients (Parker and Atkinson, 1982; see also Chapter 33: Antihypertensive Agents and the Drug Therapy of Hypertension). Therapeutic Uses The major therapeutic use of clonidine (clonidine hydrochloride;CATAPRES) is in the treatment of
hypertension (seeChapter 33: Antihypertensive Agents and the Drug
Therapy of Hypertension). Clonidine also has apparent efficacy in the
treatment of a range of other disorders. Stimulation of Acute administration of clonidine has been used in the differential
diagnosis of patients with hypertension and suspected pheochromocytoma. In
patients with primary hypertension, plasma concentrations of norepinephrine
are markedly suppressed after a single dose of clonidine; this response is
not observed in many patients with pheochromocytoma (Bravo et al.,
1981). The capacity of clonidine to activate postsynaptic Apraclonidine Apraclonidine IOPIDINE) is a
relatively selective Guanfacine Guanfacine is a phenylacetylguanidine derivative. Its structural formula is as follows:
Guanfacine (guanfacine hydrochloride;TENEX) is an Guanabenz Guanabenz and guanfacine are closely related chemically and pharmacologically. The structural formula of guanabenz is as follows:
Guanabenz (guanabenz acetate;WYTENSIN) is a centrally acting Methyldopa Methyldopa Tizanidine Tizanidine ZANAFLEX) is a muscle relaxant drug used
for the treatment of spasticity associated with cerebral and spinal
disorders. It also is an Brimonidine Brimonidine tartrate ALPHAGAN) is
an Miscellaneous Adrenergic Agonists Amphetamine Amphetamine,
racemic Cardiovascular Responses Amphetamine given orally raises both systolic and diastolic blood pressure. Heart rate is often reflexly slowed; with large doses, cardiac arrhythmias may occur. Cardiac output is not enhanced by therapeutic doses, and cerebral blood flow does not change much. The l isomer is slightly more potent than the d isomer in its cardiovascular actions. Other Smooth Muscles In general, smooth muscles respond to amphetamine as they do to other sympathomimetic amines. The contractile effect on the sphincter of the urinary bladder is particularly marked, and for this reason amphetamine has been used in treating enuresis and incontinence. Pain and difficulty in micturition occasionally occur. The gastrointestinal effects of amphetamine are unpredictable. If enteric activity is pronounced, amphetamine may cause relaxation and delay the movement of intestinal contents; if the gut already is relaxed, the opposite effect may occur. The response of the human uterus varies, but there usually is an increase in tone. Central Nervous System Amphetamine is one of the most potent sympathomimetic amines in stimulating the CNS. It stimulates the medullary respiratory center, lessens the degree of central depression caused by various drugs, and produces other signs of stimulation of the CNS. These effects are thought to be due to cortical stimulation and possibly to stimulation of the reticular activating system. In contrast, the drug can obtund the maximal electroshock seizure discharge and prolong the ensuing period of depression. In elicitation of CNS excitatory effects, the d isomer (dextroamphetamine) is three to four times as potent as the l isomer. The psychic effects depend on the dose and the mental state and personality of the individual. The main results of an oral dose of 10 to 30 mg include wakefulness, alertness, and a decreased sense of fatigue; elevation of mood, with increased initiative, self-confidence, and ability to concentrate; often, elation and euphoria; and increase in motor and speech activities. Performance of simple mental tasks is improved, but, although more work may be accomplished, the number of errors may increase. Physical performancein athletes, for exampleis improved, and the drug often is abused for this purpose. These effects are not invariable, and may be reversed by overdosage or repeated usage. Prolonged use or large doses are nearly always followed by depression and fatigue. Many individuals given amphetamine experience headache, palpitation, dizziness, vasomotor disturbances, agitation, confusion, dysphoria, apprehension, delirium, or fatigue (seeChapter 24: Drug Addiction and Drug Abuse). Fatigue and Sleep Prevention and reversal of fatigue by amphetamine have been studied extensively in the laboratory, in military field studies, and in athletics. In general, the duration of adequate performance is prolonged before fatigue appears, and the effects of fatigue are at least partly reversed. The most striking improvement appears to occur when performance has been reduced by fatigue and lack of sleep. Such improvement may be partly due to alteration of unfavorable attitudes toward the task. However, amphetamine reduces the frequency of attention lapses that impair performance after prolonged sleep deprivation and thus improves execution of tasks requiring sustained attention. The need for sleep may be postponed, but it cannot be avoided indefinitely. When the drug is discontinued after long use, the pattern of sleep may take as long as two months to return to normal. Analgesia Amphetamine and some other sympathomimetic amines have a small analgesic effect, but it is not sufficiently pronounced to be therapeutically useful. However, amphetamine can enhance the analgesia produced by morphine-like drugs. Respiration Amphetamine stimulates the respiratory center, increasing the rate and depth of respiration. In normal human beings, usual doses of the drug do not appreciably increase respiratory rate or minute volume. Nevertheless, when respiration is depressed by centrally acting drugs, amphetamine may stimulate respiration. Depression of Appetite Amphetamine and similar drugs have been used for the treatment of obesity, although the wisdom of this use is at best questionable. Weight loss in obese human beings treated with amphetamine is almost entirely due to reduced food intake and only in small measure to increased metabolism. The site of action is probably in the lateral hypothalamic feeding center; injection of amphetamine into this area, but not into the ventromedial satiety center, suppresses food intake. Neurochemical mechanisms of action are unclear but may involve increased release of norepinephrine and/or dopamine (Samanin and Garattini, 1993). In human beings, tolerance to the appetite suppression develops rapidly. Hence, continuous weight reduction usually is not observed in obese individuals without dietary restriction (Silverstone, 1992; Bray, 1993). Mechanisms of Action in the CNS Amphetamine appears to exert most or all of its effects in the CNS by
releasing biogenic amines from their storage sites in nerve terminals. The
alerting effect of amphetamine, its anorectic effect, and at least a
component of its locomotor-stimulating action are presumably mediated by
release of norepinephrine from central noradrenergic neurons. These effects
can be prevented in experimental animals by treatment with Toxicity and Adverse Effects The acute toxic effects of amphetamine usually are extensions of its therapeutic actions and, as a rule, result from overdosage. The central effects commonly include restlessness, dizziness, tremor, hyperactive reflexes, talkativeness, tenseness, irritability, weakness, insomnia, fever, and sometimes euphoria. Confusion, aggressiveness, changes in libido, anxiety, delirium, paranoid hallucinations, panic states, and suicidal or homicidal tendencies occur, especially in mentally ill patients. However, these psychotic effects can be elicited in any individual if sufficient quantities of amphetamine are ingested for a prolonged period. Fatigue and depression usually follow central stimulation. Cardiovascular effects are common and include headache, chilliness, pallor or flushing, palpitation, cardiac arrhythmias, anginal pain, hypertension or hypotension, and circulatory collapse. Excessive sweating occurs. Symptoms referable to the gastrointestinal system include dry mouth, metallic taste, anorexia, nausea, vomiting, diarrhea, and abdominal cramps. Fatal poisoning usually terminates in convulsions and coma, and cerebral hemorrhages are the main pathological findings. The toxic dose of amphetamine varies widely. Toxic manifestations occasionally occur as an idiosyncrasy after as little as 2 mg, but are rare with doses of less than 15 mg. Severe reactions have occurred with 30 mg, yet doses of 400 to 500 mg are not uniformly fatal. Larger doses can be tolerated after chronic use of the drug. Treatment of acute amphetamine intoxication may include acidification
of the urine by administration of ammonium chloride; this enhances the rate
of elimination. Sedatives may be required for the CNS symptoms. Severe
hypertension may require administration of sodium nitroprusside or an Chronic intoxication with amphetamine causes symptoms similar to those of acute overdosage, but abnormal mental conditions are more common. Weight loss may be marked. A psychotic reaction with vivid hallucinations and paranoid delusions, often mistaken for schizophrenia, is the most common serious effect. Recovery usually is rapid after withdrawal of the drug, but occasionally the condition becomes chronic. In these persons, amphetamine may act as a precipitating factor hastening the onset of an incipient schizophrenia. The abuse of amphetamine as a means of overcoming sleepiness and of increasing energy and alertness should be discouraged. The drug should be used only under medical supervision. The amphetamines are schedule II drugs under federal regulations (seeAppendix I). The additional contraindications and precautions for the use of amphetamine generally are similar to those described above for epinephrine. Its use is inadvisable in patients with anorexia, insomnia, asthenia, psychopathic personality, or a history of homicidal or suicidal tendencies. Dependence and Tolerance Psychological dependence often occurs when amphetamine or dextroamphetamine is used chronically, as discussed in Chapter 24: Drug Addiction and Drug Abuse. Tolerance almost invariably develops to the anorexigenic effect of amphetamines, and is often seen also in the need for increasing doses to maintain improvement of mood in psychiatric patients. Tolerance is striking in individuals who are dependent on the drug, and a daily intake of 1.7 g without apparent ill effects has been reported. Development of tolerance is not invariable, and cases of narcolepsy have been treated for years without requiring an increase in the initially effective dose. Therapeutic Uses Amphetamine and dextroamphetamine are used chiefly for their CNS effects. Dextroamphetamine (dextroamphetamine sulfate;DEXEDRINE), with greater CNS action and less peripheral action, generally is preferred to amphetamine; it is used in obesity, narcolepsy, and attention-deficit hyperactivity disorder. These uses are discussed later in this chapter. Methamphetamine Methamphetamine (methamphetamine hydrochloride;DESOXYN) is closely related chemically to amphetamine and ephedrine (seeTable 101). Small doses have prominent central stimulant effects without significant peripheral actions; somewhat larger doses produce a sustained rise in systolic and diastolic blood pressures, due mainly to cardiac stimulation. Cardiac output is increased, although the heart rate may be reflexly slowed. Venous constriction causes peripheral venous pressure to increase. These factors tend to increase the venous return and, therefore, the cardiac output. Pulmonary arterial pressure is raised, probably owing to increased cardiac output. Methamphetamine is a schedule II drug under federal regulations and has high potential for abuse (seeChapter 24: Drug Addiction and Drug Abuse and Appendix I). It is used principally for its central effects, which are more pronounced than those of amphetamine and are accompanied by less prominent peripheral actions. These uses are discussed below in the section of this chapter on therapeutic uses. Methylphenidate Methylphenidate is a piperidine derivative that is structurally related to amphetamine and has the following formula:
Methylphenidate (methylphenidate hydrochloride;RITALIN) is a mild CNS stimulant with more prominent effects on mental than on motor activities. However, large doses produce signs of generalized CNS stimulation that may lead to convulsions. Its pharmacological properties are essentially the same as those of the amphetamines. Methylphenidate also shares the abuse potential of the amphetamines. Methylphenidate is effective in the treatment of narcolepsy and attention-deficit hyperactivity disorder, as described below. Methylphenidate is readily absorbed after oral administration and reaches peak concentrations in plasma in about 2 hours. Methylphenidate is a racemate; its more potent (+) enantiomer has a half-life of about 6 hours, and the less potent () enantiomer has a half-life of about 4 hours. Concentrations in the brain exceed those in plasma. The main urinary metabolite is a deesterified product, ritalinic acid, which accounts for 80% of the dose. The use of methylphenidate is contraindicated in patients with glaucoma. Pemoline Pemoline CYLERT) is structurally dissimilar to methylphenidate but elicits similar changes in CNS function with minimal effects on the cardiovascular system. It is employed in treating attention-deficit hyperactivity disorder and can be given once daily because of its long half-life. Clinical improvement may require treatment for 3 to 4 weeks. Its use has been associated with severe hepatic failure. Ephedrine Ephedrine (ephedrine sulfate) is both an Pharmacological Actions Ephedrine does not contain a catechol moiety, and it is effective
after oral administration. The drug stimulates heart rate and cardiac output
and variably increases peripheral resistance; as a result, ephedrine usually
increases blood pressure. Stimulation of the Therapeutic Uses and Toxicity In the past, ephedrine was used to treat Stokes-Adams attacks with
complete heart block and as a CNS stimulant in narcolepsy and depressive
states. It has been replaced by alternative modes of treatment in each of
these disorders. In addition, its use as a bronchodilator in patients with
asthma has become much less extensive with the development of Untoward effects of ephedrine include the risk of hypertension, particularly after parenteral administration or with higher than recommended oral dosing. Insomnia is a common CNS adverse effect. Tachyphylaxis may occur with repetitive dosing. Concerns have been raised about the safety of ephedrine. Usual or higher than recommended doses may cause important adverse effects in susceptible individuals and be especially of concern in patients with underlying cardiovascular disease that might be unrecognized. Of potentially greater cause for concern, large amounts of herbal preparations containing ephedrine (ma huang, Ephedra) are utilized around the world. There can be considerable variability in the content of ephedrine in these preparations, which may lead to inadvertent consumption of higher than usual does of ephedrine and its isomers. Other Sympathomimetic Agents Several sympathomimetic drugs are used primarily as vasoconstrictors
for local application to the nasal mucous membrane or the eye. The structures
of propylhexedrine (BENZEDREX), naphazoline hydrochloride (PRIVINE, NAPHCON, others), tetrahydrozoline
hydrochloride (TYZINE, VISINE ORIGINAL, others), oxymetazoline hydrochloride
(AFRIN,
OCUCLEAR, others),
and xylometazoline hydrochloride (OTRIVIN) are depicted in Table 101 and Figure
103. Ethylnorepinephrine hydrochloride (BRONKEPHRINE) (seeTable 101) is a
Phenylephrine (see above), pseudoephedrine (SUDAFED, others) (a stereoisomer of ephedrine), and phenylpropanolamine (PROPAGEST, others) are the sympathomimetic drugs that have been used most commonly in oral preparations for the relief of nasal congestion. Pseudoephedrine hydrochloride is available without a prescription in a variety of solid and liquid dosage forms. Phenylpropanolamine hydrochloride shares the pharmacological properties of ephedrine and is approximately equal in potency except that it causes less CNS stimulation. The drug has been available without prescription in tablets and capsules. In addition, numerous proprietary mixtures marketed for the oral treatment of nasal and sinus congestion contain one of these sympathomimetic amines, usually in combination with an H1-histamine receptor antagonist. Also, phenylpropanolamine suppresses appetite by mechanisms possibly different from those of amphetamines (Wellman, 1992). Concern about the possibility that phenylpropanolamine increases the risk of hemorrhagic stroke in young women led the United States Food and Drug Administration (FDA) recently to consider banning the sale of the drug. The FDA has issued a public warning about the risk and has asked manufacturers of over-the-counter products containing phenylpropanolamine to stop marketing them; several manufacturers have complied with the request. Therapeutic Uses of Sympathomimetic Drugs The success that has attended efforts to develop therapeutic agents that can influence adrenergic receptors selectively and the variety of vital functions that are regulated by the sympathetic nervous system have resulted in a class of drugs with a large number of important therapeutic uses. Shock Shock is a clinical syndrome characterized by inadequate perfusion of tissues; it usually is associated with hypotension and ultimately with the failure of organ systems (Hollenberg et al., 1999). Shock is an immediately lifethreatening impairment of delivery of oxygen and nutrients to the organs of the body. Causes of shock include hypovolemia (due to dehydration or blood loss), cardiac failure (extensive myocardial infarction, severe arrhythmia, or cardiac mechanical defects such as ventricular septal defect), obstruction to cardiac output (due to pulmonary embolism, pericardial tamponade, or aortic dissection), and peripheral circulatory dysfunction (sepsis or anaphylaxis). The treatment of shock consists of specific efforts to reverse the underlying pathogenesis as well as nonspecific measures aimed at correcting hemodynamic abnormalities. Regardless of the etiology, the accompanying fall in blood pressure generally leads to marked activation of the sympathetic nervous system. This, in turn, causes peripheral vasoconstriction and an increase in the rate and force of cardiac contraction. In the initial stages of shock these mechanisms may maintain blood pressure and cerebral blood flow, although blood flow to the kidneys, skin, and other organs may be decreased, leading to impaired production of urine and metabolic acidosis (Ruffolo, 1992). The initial therapy of shock involves basic life-support measures. It
is essential to maintain blood volume, which often requires monitoring of
hemodynamic parameters. Specific therapy (e.g., antibiotics for
patients in septic shock) should be initiated immediately. If these measures
do not lead to an adequate therapeutic response, it may be necessary to use
vasoactive drugs in an effort to improve abnormalities in blood pressure and
flow. This therapy is generally empirically based on response to hemodynamic
measurements. Many of these pharmacological approaches, while apparently
clinically reasonable, are of uncertain efficacy. Adrenergic agonists may be
used in an attempt to increase myocardial contractility or to modify
peripheral vascular resistance. In general terms, Cardiogenic shock due to myocardial infarction has a poor prognosis;
therapy is aimed at improving peripheral blood flow. Definitive therapy, such
as emergency cardiac catheterization following surgical revascularization or
angioplasty, may be very important. Mechanical left ventricular assist
devices also may be important in maintaining cardiac output and coronary
perfusion in critically ill patients. In the setting of severely impaired
cardiac output, falling blood pressure leads to intense sympathetic outflow
and vasoconstriction. This may further decrease cardiac output as the damaged
heart pumps against a higher peripheral resistance. Medical intervention is
designed to optimize cardiac filling pressure (preload), myocardial
contractility, and peripheral resistance (afterload). Preload may be
increased by administration of intravenous fluids or reduced with drugs such
as diuretics and nitrates. A number of sympathomimetic amines have been used
to increase the force of contraction of the heart. Some of these drugs have
disadvantages: isoproterenol is a powerful chronotropic agent and can greatly
increase myocardial oxygen demand; norepinephrine intensifies peripheral
vasoconstriction; and epinephrine increases heart rate and may predispose the
heart to dangerous arrhythmias. Dopamine is an effective inotropic agent that
causes less increase in heart rate than does isoproterenol. It also promotes
renal arterial dilation; this may be useful in preserving renal function.
When given in high doses (greater than 10 to 20 In some patients in shock, hypotension is so severe that
vasoconstricting drugs are required to maintain a blood pressure that is
adequate for perfusion of the CNS (Kulka and Tryba, 1993). The hemodynamic abnormalities in septic shock are complex and are not well understood. Most patients with septic shock initially have low or barely normal peripheral vascular resistance, possibly owing to excessive effects of endogenously produced nitric oxide as well as normal or increased cardiac output. If the syndrome progresses, myocardial depression, increased peripheral resistance, and impaired tissue oxygenation occur. The primary treatment of septic shock is antibiotics. Data on the comparative value of various adrenergic agents in the treatment of septic shock are limited (Chernow and Roth, 1986). Therapy with drugs such as dopamine or dobutamine is guided by hemodynamic monitoring, with individualization of therapy depending on the patient's overall clinical condition. Hypotension Drugs with predominantly Patients with orthostatic hypotension (excessive fall in blood
pressure with standing) represent a pharmacological challenge in many cases.
There are diverse causes for this disorder, including the ShyDrager syndrome
and idiopathic autonomic failure. There are several therapeutic approaches
including physical maneuvers and a variety of drugs (fludrocortisone,
prostaglandin synthesis inhibitors, somatostatin analogs, caffeine, vasopressin
analogs, and dopamine antagonists). A number of sympathomimetic drugs have
been used in treating this disorder. The ideal agents would enhance venous
constriction prominently and produce relatively little arterial constriction
so as to avoid supine hypertension. No such agent currently is available.
Drugs used in this disorder to activate Hypertension Centrally acting Cardiac Arrhythmias Cardiopulmonary resuscitation in patients with cardiac arrest due to
ventricular fibrillation, electromechanical dissociation, or asystole may be
facilitated by drug treatment. Epinephrine is an important therapeutic agent
in patients with cardiac arrest; epinephrine and other In patients with paroxysmal supraventricular tachycardias,
particularly those associated with mild hypotension, careful infusion of an Congestive Heart Failure Sympathetic stimulation of Local Vascular Effects of Epinephrine is used in many surgical procedures in the nose, throat,
and larynx to shrink the mucosa and improve visualization by limiting
hemorrhage. Simultaneous injection of epinephrine with local anesthetics
retards the absorption of the anesthetic and increases the duration of
anesthesia (seeChapter 15: Local Anesthetics). Injection of Nasal Decongestion
For decongestion, Asthma Use of adrenergic agents in the treatment of asthma is discussed in Chapter 28: Drugs Used in the Treatment of Asthma. Allergic Reactions Epinephrine is the drug of choice to reverse the manifestations of
serious, acute hypersensitivity reactions (e.g., from a food, bee
sting, or drug allergy). A subcutaneous injection of epinephrine rapidly
relieves itching, hives, and swelling of lips, eyelids, and tongue. In some
patients, careful intravenous infusion of epinephrine may be required to
ensure prompt pharmacological effects. This treatment may be lifesaving when
edema of the glottis threatens patency of the airway or when there is
hypotension or shock in patients with anaphylaxis. In addition to its
cardiovascular effects, epinephrine is thought to activate Ophthalmic Uses Application of various sympathomimetic amines for diagnostic and therapeutic ophthalmic use is discussed in Chapter 66: Ocular Pharmacology. Narcolepsy Narcolepsy is characterized by hypersomnia, including attacks of sleep
that may occur suddenly under conditions that are not normally conducive to
sleep. Some patients respond to treatment with tricyclic antidepressants or
MAO inhibitors. Alternatively, CNS stimulants such as amphetamine, dextroamphetamine,
or methamphetamine may be useful (Mitler et al., 1993). Modafinil
(PROVIGIL), a CNS stimulant, may have
benefit in narcolepsy (Fry, 1998). In the Weight Reduction Obesity arises as a consequence of positive caloric balance. Optimally, weight loss is achieved by a gradual increase in energy expenditure from exercise combined with dieting to decrease the caloric intake. However, this obvious approach has a relatively low success rate. Consequently, alternative forms of treatment, including surgery or medications, have been developed in an effort to increase the likelihood of achieving and maintaining weight loss. Amphetamine was found to produce weight loss in early studies of patients with narcolepsy and was subsequently used in the treatment of obesity (Silverstone, 1986). The drug promotes weight loss by suppressing appetite rather than by increasing energy expenditure. Other anorexiant drugs include methamphetamine, dextroamphetamine, phentermine, benzphetamine, phendimetrazine, phenmetrazine, diethylpropion, mazindol, and phenylpropanolamine. In short-term (up to 20 weeks), double-blind, controlled studies, amphetamine-like drugs have been shown to be more effective than placebo in promoting weight loss; the rate of weight loss is typically increased by about 0.5 pound per week with these drugs. There is little to choose among these drugs in terms of efficacy. However, long-term weight loss has not been demonstrated unless these drugs are taken continuously (Bray, 1993). In addition, other important issues have not yet been resolved; these include the selection of patients who might be benefited by these drugs, whether the drugs should be administered continuously or intermittently, and the duration of treatment (Silverstone, 1986). Adverse effects of treatment include the potential for drug abuse and habituation, serious worsening of hypertension (although in some patients blood pressure may actually fall, presumably as a consequence of weight loss), sleep disturbances, palpitations, and dry mouth. These agents may be effective as adjuncts in the treatment of obese patients. However, available evidence does not support the isolated use of these drugs in the absence of a more comprehensive program that stresses exercise and modification of diet. Attention-Deficit Hyperactivity Disorder (ADHD) This syndrome, usually first evident in childhood, is characterized by excessive motor activity, difficulty in sustaining attention, and impulsiveness. Children with this disorder frequently are troubled by difficulties in school, impaired interpersonal relationships, and excitability. Academic underachievement is an important characteristic. A substantial number of children with this syndrome have characteristics that persist into adulthood, although in modified form (American Psychiatric Association, 1987). Behavioral therapy may be helpful in some patients. Catecholamines may be involved in the control of attention at the level of the cerebral cortex. A variety of stimulant drugs have been utilized in the treatment of ADHD, and they are particularly indicated in moderate-to-severe cases. Dextroamphetamine has been demonstrated to be more effective than placebo (Klein et al., 1980); methylphenidate also is effective in children with ADHD, although information about the long-term efficacy of both drugs is limited. Treatment may start with a dose of 5 mg of methylphenidate in the morning and at lunch; the dose is increased gradually over a period of weeks depending on the response as judged by parents, teachers, and the physician. The total daily dose generally should not exceed 60 mg; because of its short duration of action, most children require two or three doses of methylphenidate each day. The timing of doses is adjusted individually in accordance with rapidity of onset of effect and duration of action. Some children may not respond, and the drug should be discontinued after one month of dosage adjustment. Methylphenidate and dextroamphetamine probably have similar efficacy in ADHD and are the preferred drugs for this disorder (Elia et al., 1999). Pemoline appears to be less effective, although it may be used once daily in some children (Klein et al., 1980). Potential adverse effects of these medications in children include insomnia, abdominal pain, anorexia, and weight loss that may be associated with suppression of growth. Minor symptoms may be transient or may respond to adjustment of dosage or administration of the drug with meals. Other drugs that have been utilized include tricyclic antidepressants, antipsychotic agents, and clonidine (Fox and Rieder, 1993). There is evidence that stimulant medications are effective in adults with similar disorders (Chiarello and Cole, 1987). |
Adrenergic Receptor Antagonists
|
Many types of drugs interfere with the function of the sympathetic nervous system and thus have profound effects on the physiology of sympathetically innervated organs. Several of these drugs are important in clinical medicine, particularly for the treatment of cardiovascular diseases. Drugs that decrease the amount of norepinephrine released as a consequence of sympathetic nerve stimulation as well as drugs that inhibit sympathetic nervous activity by suppressing sympathetic outflow from the brain are discussed in Chapter 33: Antihypertensive Agents and the Drug Therapy of Hypertension. The remainder of this chapter focuses on drugs termed adrenergic
receptor antagonists, which inhibit the interaction of norepinephrine, epinephrine,
and other sympathomimetic drugs with adrenergic receptors. Almost all of
these agents are competitive antagonists in their interactions with either
Chemistry The structural formulas of a number of
Pharmacological Properties Cardiovascular System The most important effects of
Blockade of Blockade of
Although certain vascular beds contain Other Actions of
Phenoxybenzamine and Related Haloalkylamines Phenoxybenzamine is a haloalkylamine that blocks Chemistry The haloalkylamine adrenergic blocking drugs are closely related
chemically to the nitrogen mustards; as in the latter, the tertiary amine
cyclizes with the loss of chlorine to form a reactive ethyleniminium or
aziridinium ion (seeChapter 52: Antineoplastic Agents). The molecular
configuration directly responsible for blockade is probably a highly reactive
carbonium ion formed upon cleavage of the three-membered ring. It is presumed
that the arylalkyl amine moiety of the molecule is responsible for the
relative specificity of action of these agents, since the reactive
intermediate probably reacts with sulfhydryl, amino, and carboxyl groups in
many proteins. Because of these chemical reactions, phenoxybenzamine is
covalently conjugated with Pharmacological Properties The major effects of phenoxybenzamine result from blockade of Phenoxybenzamine inhibits the uptake of catecholamines into both
adrenergic nerve terminals and extraneuronal tissues. In addition to blockade
of The pharmacokinetic properties of phenoxybenzamine are not well
understood. The half-life of phenoxybenzamine is probably less than 24 hours.
However, since the drug inactivates Therapeutic Uses A major use of phenoxybenzamine (phenoxybenzamine hydrochloride;DIBENZYLINE) is in the treatment of
pheochromocytoma. Pheochromocytomas are tumors of the adrenal medulla and
sympathetic neurons that secrete enormous quantities of catecholamines into
the circulation. The usual result is hypertension, which may be episodic and
severe. The vast majority of pheochromocytomas are treated surgically;
however, phenoxybenzamine is frequently used to treat the patient in
preparation for surgery. The drug controls episodes of severe hypertension
and minimizes other adverse effects of catecholamines, such as contraction of
plasma volume and injury of the myocardium. A conservative approach is to
initiate treatment with phenoxybenzamine (at a dosage of 10 mg twice daily) 1
to 3 weeks before the operation. The dose is increased every other day until
the desired effect on blood pressure is achieved. Therapy may be limited by
postural hypotension; nasal stuffiness is another frequent adverse effect.
The usual total daily dose of phenoxybenzamine in patients with
pheochromocytoma is 40 to 120 mg given in two or three divided portions. Some
physicians do not use phenoxylenzamine preoperatively in patients with
pheochromocytoma (Boutros et al., 1990). Prolonged treatment with
phenoxybenzamine may be necessary in patients with inoperable or malignant
pheochromocytoma. In some patients, particularly those with malignant
disease, administration of metyrosine may be a useful adjuvant (Brogden et
al., 1981; Perry et al., 1990). Metyrosine is a competitive
inhibitor of tyrosine hydroxylase, the rate-limiting enzyme in the synthesis
of catecholamines (seeChapter 6: Neurotransmission: The Autonomic and
Somatic Motor Nervous Systems). Phenoxybenzamine was the first Toxicity and Adverse Effects The major adverse effect of phenoxybenzamine is postural hypotension. This is often accompanied by reflex tachycardia and other arrhythmias. Hypotension can be particularly severe in hypovolemic patients or under conditions that promote vasodilation (administration of vasodilator drugs, exercise, ingestion of alcohol or large quantities of food). Reversible inhibition of ejaculation and aspermia after orgasm may occur because of impaired smooth muscle contraction in the vas deferens and ejaculatory ducts. Phenoxybenzamine has mutagenic activity in the Ames test, and repeated administration of this drug to experimental animals causes peritoneal sarcomas and lung tumors (IARC, 1980). The clinical significance of these findings is not known. Phentolamine and Tolazoline Phentolamine, an imidazoline, is a competitive The pharmacokinetic properties of phentolamine are not known, although the drug is extensively metabolized. Tolazoline is well absorbed after oral administration and is excreted in the urine. Therapeutic Uses Phentolamine (phentolamine mesylate;REGITINE) can be used in the short term to
control hypertension in patients with pheochromocytoma. Rapid infusions of
phentolamine may cause severe hypotension, and the drug should be
administered cautiously. Phentolamine also may be useful to relieve
pseudoobstruction of the bowel in patients with pheochromocytoma; this
condition may result from the inhibitory effects of catecholamines on intestinal
smooth muscle. Phentolamine has been used locally to prevent dermal necrosis
after the inadvertent extravasation of an Tolazoline (tolazoline hydrochloride;PRISCOLINE) has been used in the treatment of persistent pulmonary hypertension of the newborn and as an aid in visualizing distal peripheral vessels during arteriography (Gouyon and Francoise, 1992; Wilms et al., 1993). The use of tolazoline in the newborn may be replaced by the use of prostaglandins or nitric oxide (Gouyon and Francoise, 1992). Toxicity and Adverse Effects Hypotension is the major adverse effect of phentolamine. In addition, reflex cardiac stimulation may cause alarming tachycardia, cardiac arrhythmias, and ischemic cardiac events, including myocardial infarction. Gastrointestinal stimulation may result in abdominal pain, nausea, and exacerbation of peptic ulcer. Phentolamine should be used with particular caution in patients with coronary artery disease or a history of peptic ulcer. Prazosin and Related Drugs Prazosin,
the prototype of a family of agents that contain a piperazinyl quinazoline
nucleus, is a very potent and selective Pharmacological Properties Prazosin The major effects of prazosin are a result of its blockade of Prazosin (prazosin hydrochloride;MINIPRESS) is well absorbed after oral
administration, and bioavailability is about 50% to 70%. Peak concentrations
of prazosin in plasma are generally reached 1 to 3 hours after an oral dose.
The drug is tightly bound to plasma proteins (primarily The initial dose should be 1 mg, usually given at bedtime, so that the
patient will remain recumbent for at least several hours to reduce the risk
of syncopal reactions that may follow the first dose of prazosin. Therapy is
begun with 1 mg given two or three times daily, and the dose is titrated
upward depending on the blood pressure. A maximal effect generally is
observed with a total daily dose of 20 mg in patients with hypertension. In
the treatment of benign prostatic hyperplasia (BPH), doses from 1 to 5 mg
twice daily typically are used. The twice-daily dosing requirement for
prazosin is a disadvantage compared with newer Terazosin Terazosin (terazosin hydrochloride;HYTRIN) is a close structural analog of prazosin
(Kyncl, 1993; Wilde et al., 1993). It is less potent than prazosin but
retains high specificity for Doxazosin Doxazosin CARDURA) is another structural analog of prazosin.
It, too, is a highly selective antagonist at Alfuzosin Alfuzosin
is a quinozoline-based Tamsulosin Tamsulosin FLOMAX), a benzenesulfonamide, is an Adverse Effects A major potential adverse effect of prazosin and its congeners is the so-called first-dose phenomenon; marked postural hypotension and syncope are sometimes seen 30 to 90 minutes after a patient takes an initial dose. Occasionally, syncopal episodes also have occurred with a rapid increase in dosage or with the addition of a second antihypertensive drug to the regimen of a patient who is already taking a large dose of prazosin. The mechanisms responsible for such exaggerated hypotensive responses or for the development of tolerance to these effects are not clear. An action in the CNS to reduce sympathetic outflow may contribute (see above). The risk of the first-dose phenomenon is minimized by limiting the initial dose to 1 mg at bedtime, by increasing the dosage slowly, and by introducing additional antihypertensive drugs cautiously. Since orthostatic hypotension may be a problem during long-term treatment with prazosin or its congeners, it is essential to check standing as well as recumbent blood pressure. Nonspecific adverse effects such as headache, 'dizziness,' and asthenia do not often limit treatment with prazosin. The nonspecific complaint of 'dizziness' is generally not due to orthostatic hypotension. Although not extensively documented, the adverse effects of the structural analogs of prazosin appear to be similar to those of the parent compound. For tamsulosin, at a dose of 0.4 mg daily, effects on blood pressure are not expected, although impaired ejaculation may occur. Therapeutic Uses Prazosin and its congeners have been used successfully in the
treatment of primary systemic hypertension (seeChapter 33:
Antihypertensive Agents and the Drug Therapy of Hypertension). The most
important distinction among these drugs relates to their duration of action
and thus the required dosing interval. Considerable recent interest has
focused on the use of Congestive Heart Failure
Benign Prostatic Hyperplasia
Other Disorders Although anecdotal evidence suggested that prazosin might be useful in the treatment of patients with variant angina (Prinzmetal's angina) due to coronary vasospasm, several small controlled trials have failed to demonstrate a clear benefit (Robertson et al., 1983b; Winniford et al., 1983). Some studies have indicated that prazosin can decrease the incidence of digital vasospasm in patients with Raynaud's disease; however, its relative efficacy as compared with other vasodilators (e.g., Ca2+ channel blockers) is not known (Surwit et al., 1984; Wollersheim et al., 1986). Prazosin may have some benefit in patients with other vasospastic disorders (Spittell and Spittell, 1992). Prazosin decreases ventricular arrhythmias induced by coronary artery ligation or reperfusion in laboratory animals; the therapeutic potential for this use in human beings is not known (Davey, 1986). Prazosin also might be useful for the treatment of patients with mitral or aortic valvular insufficiency, presumably because of reduction of afterload; additional data are needed (Jebavy et al., 1983; Stanaszek et al., 1983). Ergot Alkaloids The ergot alkaloids were the first adrenergic blocking agents to be
discovered, and most aspects of their general pharmacology were disclosed in
the classic studies of Dale (1906). Ergot alkaloids exhibit a complex variety
of pharmacological properties. To varying degrees, these agents act as
partial agonists or antagonists at Chemistry Details of the chemistry of the ergot alkaloids are presented in Chapter
11: 5-Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists. In
general, compounds of the ergonovine type, which lack a peptide side chain,
have no adrenergic blocking activity. Of the natural ergot preparations,
'ergotoxine' has the greatest Pharmacological Properties Both the natural and the dihydrogenated peptide alkaloids produce The most important effects of the ergot alkaloids are due to actions on the CNS and direct stimulation of smooth muscle. The latter occurs in many different organs (seeChapter 11: 5-Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists), and even dihydroergotoxine (ergoloid mesylate) has been observed to produce spastic contractions of the intestine. The peptide ergot alkaloids can reverse the pressor response to epinephrine to a depressor action. However, all the natural ergot alkaloids cause a significant rise in blood pressure as a result of peripheral vasoconstriction, which is more pronounced in postcapillary than in precapillary vessels. Although hydrogenation reduces this action, dihydroergotamine still is an effective vasoconstrictor; a residual constrictor action of dihydroergotoxine also is demonstrable. Ergotamine, ergonovine, and other ergot alkaloids can produce coronary vasoconstriction, often with associated ischemic changes and anginal pain in patients with coronary artery disease. The ergot alkaloids usually induce bradycardia even when the blood pressure is not increased. This is predominantly due to increased vagal activity, but a central reduction in sympathetic tone and direct myocardial depression also may be involved. Toxicity and Adverse Effects The dose of dihydroergotoxine in human beings is limited by the occurrence of nausea and vomiting. Prolonged or excessive administration of any of the natural peptide ergot alkaloids can cause vascular insufficiency, including myocardial ischemia and gangrene of the extremities due to marked arterial constriction (Galer et al., 1991). This is particularly likely to occur in the presence of preexisting vascular pathological processes. In severe cases, prompt vasodilation is essential. There have been no comparative studies on the treatment of this sporadic condition, but a directly acting drug such as nitroprusside appears to be most effective (Carliner et al., 1974). Toxic effects of the ergot alkaloids are described in more detail in Chapter 11: 5-Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists. Therapeutic Uses The primary uses of ergot alkaloids are to stimulate contraction of the uterus postpartum and to relieve the pain of migraine (Mitchell and Elbourne, 1993; Saxena and De Deyn, 1992; seeChapter 11: 5-Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists). However, newer alternatives, such as sumatriptan and other 5-HT1-receptor agonists, may have better efficacy and safety in migraine (Dechant and Clissold, 1992; see also Chapter 11: 5-Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists). Ergonovine and methylergonovine are useful in preventing and treating postpartum hemorrhage due to uterine atonia, probably by stimulating uterine contraction, which compresses bleeding blood vessels. Synthetic preparations of the posterior pituitary hormone oxytocin also are used to enhance uterine contractions (seeChapter 56: Pituitary Hormones and Their Hypothalamic Releasing Factors); this may have the benefit not only of preventing or treating uterine hemorrhage but also of inducing or augmenting labor. Dinoprostone (prostaglandin E2) also inhibits postpartum bleeding and may be efficacious if there is an inadequate response to ergot alkaloids or oxytocin (Winkler and Rath, 1999). Ergot alkaloids have been used clinically in many settings: diagnostically to stimulate coronary artery contraction; as putative cognition enhancers (Wadworth and Chrisp, 1992); and in the management of orthostatic hypotension (Stumpf and Mitrzyk, 1994). The effect of bromocriptine on the secretion of prolactin is described in Chapter 56: Pituitary Hormones and Their Hypothalamic Releasing Factors. Additional Indoramin Indoramin
is a selective, competitive The bioavailability of indoramin is generally less than 30% (with
considerable variability), and it undergoes extensive first-pass metabolism (Holmes
and Sorkin, 1986; Pierce, 1990). Little unchanged drug is excreted in the
urine, and some of the metabolites may be biologically active. The
elimination half-life is about 5 hours. Some of the adverse effects of
indoramin include sedation, dry mouth, and failure of ejaculation. Although
indoramin is an effective antihypertensive agent, it has complex
pharmacokinetics and lacks a well-defined place in current therapy. Indoramin
currently is not available in the Labetalol Labetalol,
a potent Ketanserin Although developed as a 5-HT-receptor antagonist, ketanserin
also blocks Urapidil Urapidil
is a novel, selective Bunazosin Bunazosin
is an Yohimbine Yohimbine YOCON) is a competitive antagonist that
is selective for Neuroleptic Agents Natural and synthetic compounds of several other chemical classes
developed primarily because they are antagonists of D2dopamine
receptors also exhibit
History Ahlquist's hypothesis that the effects of catecholamines were mediated
by activation of distinct Propranolol has equal affinity for Chemistry The structural formulas of some
Pharmacological Properties As in the case of Cardiovascular System The major therapeutic effects of Since catecholamines have positive chronotropic and inotropic actions,
Although high concentrations of many The cardiovascular effects of Coronary arterial blood flow increases during exercise or stress to
meet the metabolic demands of the heart. By increasing heart rate,
contractility, and systolic pressure, catecholamines increase myocardial
oxygen demand. However, in patients with coronary artery disease, fixed
narrowing of these vessels attenuates the expected increase in flow, leading
to myocardial ischemia. Activity As Antihypertensive Agents
Presynaptic As indicated above, some Propranolol and other nonselective Pulmonary System Nonselective Metabolic Effects
Other Effects
Non-Subtype-Selective Propranolol In view of the extensive experience with propranolol (propranolol
hydrochloride;INDERAL), it
is useful as a prototype (seeTable 103). Propranolol interacts with Absorption, Fate, and Excretion Propranolol is highly lipophilic and is almost completely absorbed after oral administration. However, much of the drug is metabolized by the liver during its first passage through the portal circulation; on average, only about 25% reaches the systemic circulation. In addition, there is great interindividual variation in the presystemic clearance of propranolol by the liver; this contributes to enormous variability in plasma concentrations (approximately 20-fold) after oral administration of the drug and contributes to the wide range of doses in terms of clinical efficacy. In other words, a clinical disadvantage of propranolol is that multiple, increasing steps in drug dose may be required over time. The degree of hepatic extraction of propranolol declines as the dose is increased. The bioavailability of propranolol may be increased by the concomitant ingestion of food and during long-term administration of the drug. Propranolol has a large volume of distribution (4 liters/kg) and
readily enters the CNS. Approximately 90% of the drug in the circulation is
bound to plasma proteins. It is extensively metabolized, with most
metabolites appearing in the urine. One product of hepatic metabolism is
4-hydroxypropranolol, which has some Analysis of the distribution of propranolol, its clearance by the
liver, and its activity is complicated by the stereospecificity of these
processes (Walle et al., 1988). The ()-enantiomers of propranolol and
other A sustained-release formulation of propranolol (INDERAL LA) has been developed to maintain therapeutic concentrations of propranolol in plasma throughout a 24-hour period (Nace and Wood, 1987). Suppression of exercise-induced tachycardia is maintained throughout the dosing interval, and patient compliance may be improved. Therapeutic Uses For the treatment of hypertension and angina, the initial oral dose of
propranolol is generally 40 to 80 mg per day. The dose may then be titrated
upward until the optimal response is obtained. For the treatment of angina,
the dose may be increased at intervals of less than one week, as indicated
clinically. In hypertension, the full blood-pressure response may not develop
for several weeks. Typically, doses are less than 320 mg per day. If
propranolol is taken twice daily for hypertension, blood pressure should be
measured just prior to a dose to ensure that the duration of effect is
sufficiently prolonged. Adequacy of Nadolol Nadolol CORGARD) is a long-acting antagonist with
equal affinity for Absorption, Fate, and Excretion Nadolol is very soluble in water and is incompletely absorbed from the
gut; its bioavailability is about 35% (Frishman, 1981). Interindividual
variability is less than with propranolol. The low solubility of nadolol in
fat may result in lower concentrations of the drug in the brain as compared
with more lipid-soluble Timolol Timolol (timolol maleate;BLOCADREN) is a potent,
non-subtype-selective Absorption, Fate, and Excretion Timolol is well absorbed from the gastrointestinal tract and is subject to moderate first-pass metabolism. It is metabolized extensively by the liver, and only a small amount of unchanged drug appears in the urine. The half-life in plasma is about 4 hours. Interestingly, the ocular formulation of timolol (TIMOPTIC), used for the treatment of glaucoma, may be extensively absorbed systemically (seeChapter 66: Ocular Pharmacology); adverse effects can occur in susceptible patients, such as those with asthma or congestive heart failure. Pindolol Pindolol VISKEN) is a non-subtype-selective Although only limited data are available, Absorption, Fate, and Excretion Pindolol is almost completely absorbed after oral administration and has moderately high bioavailability. These properties tend to minimize interindividual variation in the plasma concentrations of the drug that are achieved after its oral administration. Approximately 50% of pindolol ultimately is metabolized in the liver. The principal metabolites are hydroxylated derivatives that subsequently are conjugated with either glucuronide or sulfate before renal excretion. The remainder of the drug is excreted unchanged in the urine. The plasma half-life of pindolol is about 4 hours; clearance is reduced in patients with renal failure. Labetalol Labetalol (labetalol hydrochloride;NORMODYNE, TRANDATE) is representative of a class of
drugs that act as competitive antagonists at both The pharmacological effects of labetalol have become clearer since the
four isomers were separated and tested individually. The R,R isomer is
about four times more potent as a The actions of labetalol on both Labetalol is available in oral form for therapy of chronic hypertension and as an intravenous formulation for use in hypertensive emergencies. Labetalol has been associated with hepatic injury in a limited number of patients (Clark et al., 1990). Absorption, Fate, and Excretion Although labetalol is completely absorbed from the gut, there is extensive first-pass clearance; bioavailability is only about 20% to 40% and is highly variable (McNeil and Louis, 1984). Bioavailability may be increased by food intake. The drug is rapidly and extensively metabolized in the liver by oxidative biotransformation and glucuronidation; very little unchanged drug is found in the urine. The rate of metabolism of labetalol is sensitive to changes in hepatic blood flow. The elimination half-life of the drug is about 8 hours. The half-life of the R,R isomer of labetalol (dilevalol) is about 15 hours. Labetalol provides an interesting and challenging example of pharmacokinetic-pharmacodynamic modeling applied to a drug that is a racemic mixture of isomers with different kinetics and pharmacological actions (Donnelly and Macphee, 1991). Carvedilol Carvedilol COREG) is a non-subtype-selective Absorption, Fate, and Excretion Carvedilol has a bioavailability of about 25% to 35% because of extensive first-pass metabolism. Carvedilol is eliminated by hepatic metabolism and has a terminal half-life of 7 to 10 hours, but most of the drug is eliminated with a half-life of about 2 hours. Therapeutic Uses In the treatment of hypertension, the usual starting dose is 6.25 mg twice daily; if an adequate therapeutic response is not achieved, the dose may be increased progressively over time, typically to a maximum of 25 mg twice daily. In the treatment of congestive heart failure, dosing is much more cautious due to possibility of acutely worsening heart failure. Dosing often starts at 3.125 mg twice per day with cautious increases over time.
Metoprolol Metoprolol (metoprolol tartrate;LOPRESSOR) is a Absorption, Fate, and Excretion Metoprolol is almost completely absorbed after oral administration, but bioavailability is relatively low (about 40%) because of first-pass metabolism. Plasma concentrations of the drug vary widely (up to 17-fold), perhaps because of genetically determined differences in the rate of metabolism (Benfield et al., 1986). Metoprolol is extensively metabolized by the hepatic monooxygenase system, and only 10% of the administered drug is recovered unchanged in the urine. The half-life of metoprolol is 3 to 4 hours. An extended-release formulation (TOPROL XL) is available for once-daily administration (Plosker and Clissold, 1992). Therapeutic Uses For the treatment of hypertension, the usual initial dose is 100 mg per day. The drug is sometimes effective when given once daily, although it is frequently used in two divided doses. Dosage may be increased at weekly intervals until optimal reduction of blood pressure is achieved. If the drug is taken only once daily, it is important to confirm that blood pressure is controlled for the entire 24-hour period. Metoprolol is generally used in two divided doses for the treatment of stable angina. Conventional dosage forms of metoprolol have been extensively established for indications in hypertension and ischemic heart disease. The extended-release formulation, which provides relatively constant rates of drug delivery over 24-hour periods, may be given once daily. For the initial treatment of patients with acute myocardial infarction, an intravenous formulation of metoprolol tartrate is available. Oral dosing is initiated as soon as the clinical situation permits. Metoprolol generally is contraindicated for the treatment of acute myocardial infarction in patients with heart rates of less than 45 beats per minute, heart block greater than first degree (PR interval greater than or equal to 0.24 second), systolic blood pressure less than 100 mm Hg, or moderate-to-severe heart failure. Atenolol Atenolol TENORMIN) is a Absorption, Fate, and Excretion Atenolol is incompletely absorbed (about 50%), but most of the absorbed dose reaches the systemic circulation. There is relatively little interindividual variation in the plasma concentrations of atenolol; peak concentrations in different patients vary over only a fourfold range (Cruickshank, 1980). The drug is excreted largely unchanged in the urine, and the elimination half-life is about 5 to 8 hours. The drug accumulates in patients with renal failure, and dosage should be adjusted for patients whose creatinine clearance is less than 35 ml/minute. Therapeutic Uses The initial dose of atenolol for the treatment of hypertension usually is 50 mg per day, given once daily. If an adequate therapeutic response is not evident within several weeks, the daily dose may be increased to 100 mg; higher doses are unlikely to provide any greater antihypertensive effect. Atenolol has been shown to be efficacious, in combination with a diuretic, in elderly patients with isolated systolic hypertension. Esmolol Esmolol (esmolol hydrochloride;BREVIBLOC) is a Absorption, Fate, and Excretion Esmolol has a half-life of about 8 minutes and an apparent volume of
distribution of approximately 2 liters/kg. The drug contains an ester
linkage, and it is hydrolyzed rapidly by esterases in erythrocytes. The
half-life of the carboxylic acid metabolite of esmolol is far longer (4
hours), and it accumulates during prolonged infusion of esmolol (seeBenfield
and Sorkin, 1987). However, this metabolite has very low potency as a The onset and cessation of Since esmolol is used in urgent settings where immediate onset of Acebutolol Acebutolol (acebutolol hydrochloride;SECTRAL) is a selective Absorption, Fate, and Excretion Acebutolol is well absorbed, but it is extensively metabolized to an active metabolite, diacetolol, which accounts for most of the drug's activity (Singh et al., 1985). The elimination half-life of acebutolol is typically about 3 hours, but the half-life of diacetolol is 8 to 12 hours; it is excreted in the urine. Therapeutic Uses The initial dose of acebutolol in hypertension is usually 400 mg per day; it may be given as a single dose, but two divided doses may be required for adequate control of blood pressure. Optimal responses usually occur with doses of 400 to 800 mg per day (range 200 to 1200 mg). For treatment of ventricular arrhythmias, the drug should be given twice daily. Other A plethora of other Adverse Effects and Precautions The most common adverse effects of Cardiovascular System
Bradycardia is a normal response to Some patients complain of cold extremities while taking Abrupt discontinuation of Pulmonary Function A major adverse effect of Central Nervous System The adverse effects of Metabolism As described above, Miscellaneous The incidence of sexual dysfunction in men with hypertension who are
treated with Overdosage The manifestations of poisoning with Drug Interactions Both pharmacokinetic and pharmacodynamic interactions have been noted
between Other drug interactions have pharmacodynamic explanations. For
example, Therapeutic Uses Cardiovascular Diseases
Myocardial Infarction A great deal of interest has focused on the use of Congestive Heart Failure It is a common clinical observation that acute administration of Alterations in cardiac responsiveness to catecholamines have been
found in heart failure. A consistent observation is that sympathetic nervous
system activity is increased in patients with congestive heart failure (Bristow,
1993). Infusions of It is of potential interest that The mechanism(s) utilized by A number of mechanisms have been proposed to play a role in the
beneficial effects of Studies involving numerous patients have demonstrated that certain Because of the real possibility of acutely worsening cardiac function
in patients with congestive heart failure, particular caution and the
involvement of an experienced physician are required in initiating therapy
with a Other Cardiovascular Diseases
Other Uses Many of the signs and symptoms of hyperthyroidism are reminiscent of
the manifestations of increased sympathetic nervous system activity. Indeed,
excess thyroid hormone increases the expression of Propranolol, timolol, and metoprolol are effective for the prophylaxis of migraine (Tfelt-Hansen, 1986); the mechanism of this effect is not known, and these drugs are not useful for treatment of acute attacks of migraine. Propranolol and other
Selection of a The various |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 7086
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved