| CATEGORII DOCUMENTE |
| Alimentatie nutritie | Asistenta sociala | Cosmetica frumusete | Logopedie | Retete culinare | Sport |
BOLI MONOGENICE
Transmiterea ereditara
Transmiterea mendeliana intereseaza acele situatii in care o singura gena mutanta are un efect semnificativ asupra starii de sanatate a omului prin transmiterea ereditara a genei, respectand anumite principii. Mendel a postulat legile transmiterii autosomale si heterosomale, dominante si recesive din 1866, genele fiind caracterizate prin caracterele fizice si semnele chimice pe care le determina. Analiza pedigreului este esentiala pentru determinarea modelului transmiterii caracterelor.
Definitie = modelul transmiterii caracterelor monogenice este dat de fenotipul realizat de cuplul heterozigot (dominant) de gene sau numai de cuplul homozigot ( recesiv).
Gena = factorul ereditar care prin interactiunea cu mediul inconjurator determina un caracter.
Alele = forme alternative ale unei gene pentru un locus dat; pot fi alele multiple, normale sau anormale pentru aceeasi gena.
Locus = localizarea fizica specifica a genei pe cromozom; pentru ca organismul uman este diploid, individul prezinta doua alele pe fiecare locus (exceptie genele hemizigote din perechea XY)
Genotipul = defineste constitutia genetica a unui individ care este data de perechea de gene alele implicata in realizarea unui caracter
Fenotipul = cuprinde totalitatea caracterelor realizate prin exprimarea genelor si actiunea factorilor de mediu.
Homozigot = cuplu de gene alele identice (izomorfe) localizate pe acelasi locus geni pe cromozomi omologi.
Heterozigot = alelele de pe acelasi locus geni sunt heteromorfe (ex: una dominanta/una recesiva, una normala/una mutanta)
Dominanta = gena se manifesta fenotipic si in cuplu heterozigot
Recesivitate = caracterul se manifesta fenotipic numai daca gena responsabila se gaseste in cuplu homozigot (dominanta si recesivitatea se refera la expresivitatea clinica a caracterelor si nu la gene propriuzise).
Autosomal = cromozomii autosomi sunt reprezentati de 22 de perechi de cromozomi nonsexuali.
X sau Y lincat = gene localizate pe cromozomul X sau Y (sex-lincat = X-lincat)
Heterozigotul dublu = este un individ cu 2 alele mutante, fiecare pentru un locus diferit, dar interesand acelasi caracter.
Modelul de transmitere ereditara pentru caracterele monogenice se poate determina prin examinarea istoricului familial. Dupa realizarea anchetei familiale, datele culese sunt reprezentate grafic in detalii. Indivizii sunt reprezentati corespunzator sexului si generatiei careia apartin si relatiilor biologice dintre ei (realizarea arborelui genealogic).
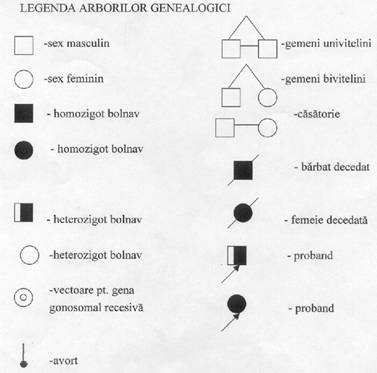
Fig. 8. Legenda arbore genealogic
Mai mult de 1/2 din bolile descrise sunt transmise dupa modelul autosomal dominant, aproximativ 1/3 sunt recesive si 1/10 sunt transmise X-lincat.
Dominanta implica prezenta alelei mutante cel putin intr-o singura copie (heterozigot) pentru realizarea unui fenotip.
*Criteriile transmiterii autosomal dominante
Transmiterea bolii se face din generatie in generatie; in pedigreeul cu transmitere autosomal dominanta pot exista mai multi indivizi bolnavi in fiecare generatie
Cu exceptia mutatiilor "de novo" fiecare copil afectat va avea cel putin un parinte afectat
Din incrucisarea unui heterozigot bolnav cu un homozigot sanatos (situatia cea mai frecventa), fiecare copil are 50 % sansa de a mosteni alela anormala si sa prezinte boala, iar 50 % din ei au sansa de a mosteni alela normala.
Transmiterea genei mutante nu este influentata de sexul parintilor bolnavi sau al copiilor (mai putin atunci cand este vorba de o alela mutanta care limiteaza dezvoltarea unei structuri specifice unui anumit sex- ca de exemplu uterul)
* Caracteristici: In transmiterea autosomal dominanta intervin un numar important de aspecte particulare care trebuiesc luate in considerare pentru realizarea unui sfat genetic corect:
mutatiile noi: un caz nou cu o boala autosomal dominanta poate fi rezultatul unei mutatii "de novo"; trebuie luate in considerare si alte cauze: heterogenitatea genetica sau o expresivitate foarte slaba a genei in fenotipul parintilor
mutatiile "de novo" apar mai frecvent in cazul bolilor care sunt atat de severe incat persoanele afectate au o capacitate de reproducere mult diminuata (ex. acondroplazia)
cresterea varstei parentale este asociata cu o crestere a numarului mutatiilor dominante pentru anumiti loci. Se considera ca aceasta crestere poate fi legata de acumularile diviziunilor celulare suferite de ovocite in timp
trebuie sa se tina seama de implicarea unor mutatii noi in inducerea bolilor dominante in momentul realizarii sfatului genetic
Penetranta redusa: penetranta se refera la exprimarea fenotipului mutant si se supune legii "totul sau nimic". Este reprezentata de procentul indivizilor care au gena mutanta si care sunt afectati fenotipic. Penetranta redusa apare atunci cand frecventa exprimarii unui genotip este mai mica de 100 %. Nonpenetranta se refera la situatiile in care alela mutanta este transmisa dar nu are exprimare fenotipica.
Pentru a stablili penetranta intr-o boala anume, se imparte numarul total de indivizi care prezinta boala la numarul total de indivizi care au mostenit alela mutanta (ex.: pentru polidactilie penetranta este 65 %)
Poate fi influentata de
factori cum ar fi varsta (ex: boala
Expresivitate variabila. Expresivitatea se refera la gradul si modul de exprimare fenotipica a unei alele la un individ.
Expresivitatea variabila este des intilnita in bolile comune, ceea ce insemneaza ca toti membrii familiei trebuiesc examinati cu atentie tinand cont de intregul tablou simptomatic al bolii.
Indivizii afectati care sunt capabili de reproducere sunt in general mai putin sever afectati, o expresivitate mai severa poate sa apara la copiii lor care pot sa prezinte intregul spectru al sindromului
Varsta de debut variabila: boala rinichiului polichistic (APKD) este o boala autosomal dominanta dar chisturile renale apar in timp, de aceea diagnosticul (fara test molecular) la o persoana tanara de foarte multe ori scapa. In unele cazuri chiar heterozigotii pot sa moara, din alte cauze, inainte ca problemele renale sa se manifeste.
Mozaicismul daca un parinte prezinta un mozaic de linii celulare, datorita unei mutatii care a survenit dupa prima diviziune a celulei ou, acesta poate sa fie clinic neafectat. Daca mozaicismul intereseaza gonadele (implicit celulele sexuale) creste riscul de recurenta pentru mutatia "de novo". De exemplu osteogeneza imperfecta tip II este datorata unei mutatii dominante. Riscul de recurenta observat in caz de mozaicism este de 6 %, diferit fata de cel calculat in conditii normale. Prezenta mozaicismului in liniile celulare sexuale ale tatalui, a fost observata si la doi dintre copiii sai afectati.
Heterogenitatea genetica. Mutatiile diferite pe acelasi locus sau pe loci diferiti pot sa fie responsabile pentru acelasi tablou clinic. Acest lucru face mult mai dificila consilierea unui cuplu in care partenerii au aceeasi boala si nu sunt inruditi (un barbat si o femeie care au amandoi surditate si vor sa aiba copii). In acest caz, ca si in cazul retinitei pigmentare boala se poate transmite autosomal dominant, autosomal recesiv sau X-lincat.
Fenocopia. Este o manifestare a unui fenotip produs de obicei de un genotip specific, dar care poate fi cauzata si de factorii de mediu care afecteaza primordiul embrionar fara sa produca mutatii la nivelul canalului genetic.
Simptomatologia clinica diferita influentata de
sex. Severitatea unei boli dominante poate depinde de sexul parintelui
afectat. In boala
* Corectitudinea observatiilor clinice si paraclinice sta la baza realizarii unui sfat genetic corect (la acordarea sfatului nu trebuie sa ne bazam pe diagnosticul altor medici sau cel enuntat de familie).
Exemple clinice:
Boala Huntington este o boala autosomal dominanta, cu debut la adulti interesand selectiv moartea celulelor din nucleul caudat. Tabloul clinic include miscari coreice, pierderea progresiva a activitatii mentale; moartea survine dupa 15 ani de la debut, debut care are loc in a patra decada de viata (daca tatal este parintele afectat boala poate debuta mai devreme la copilul sau). Gena este localizata pe 4 p, mutatia cea mai comuna fiind un trinucleotid repetat instabil.
Sindromul Marfan: este o boala a tesutului conjunctiv cu expresivitate variabila interesand scheletul (proportii anormale, scolioza, arahnodactilie) si sistemul cardiovascular (prolapsul valvei mitrale, dilatatia aortei cu risc de ruptura si leziuni oculare). Diagnosticul se bazeaza pe semnele clinice care intereseaza cel putin doua sisteme. Mutatia genei (fibrilina) este localizata pe 15q
TRANSMITEREA AUTOSOMAL RECESIVA
Fenotipul se poate observa numai la indivizii homozigoti si pedigreeul tipic demonstreaza ca rudele, parintii sau copiii indivizilor bolnavi indiferent de sex, pot fi sanatosi. Acest tip de transmitere apare mai frecvent in familiile consangvine. De asemenea se poate suspecta acest tip de transmitere in enzimopatii in care nivelul unei enzime este redus sau absent la indivizii afectati, in timp ce la parinti valorile sunt normale.
daca boala este rara parintii si rudele (altele decat fratii) sunt de obicei normali.
din incrucisarea a doi indivizi heterozigoti raportul de segregare pentru fiecare sarcina este 3:4 indivizi sanatosi 1:4 homozigoti afectati
daca genele recesive sunt totdeauna alele, cand amandoi parintii sunt afectati toti copiii lor vor fi afectati.
ambele sexe sunt afectate in proportie egala
daca boala este rara, probabilitatea de consangvinitatea parentala este crescuta
Frecventa heterozigotilor: indivizii afectati sunt aproape intotdeauna urmasii unor heterozigoti normali. Relatia dintre frecventa genelor din diferite genotipuri localizate pe acelasi locus este data de legea Hardy Weinberg (2 pq pentru heterozigoti). Legea demonstreaza ca frecventa heterozigotilor sanatosi este mult mai mare decat cea a homozigotilor (bolnavi). Calcularea frecventei homozigotilor este importanta pentru conturarea programelor de screening
Statusul de vector (purtator): indivizii sunt de obicei neafectati dar unii pot avea nivele scazute enzimatice sau semne clinice minime. Majoritatea purtatorilor nu prezinta manifestari fenotipice ale bolii.
Consangvinitatea: de exemplu in alcaptonurie 60 % dintre copiii afectati au parinti inruditi (veri) . Indivizii consanguini au un numar mare de gene comune: verii primari au 1/8 din gene comune iar urmasii lor au din genele lor comune. Consangvinitatea este crescuta in izolate (religioase, geografice).
Fenotipurile autosomal recesive in populatiile locale. Este important de cunoscut grupul populational studiat pentru ca in unele situatii exista circumstante (grupuri rasiale) in care alela mutanta este mai frecventa datorita efectului fondator sau avantajului heterozigotilor. Ex: anemia cu celule in secera (la negrii), boala Tay Sachs la evreii Ashkenazi, fibroza chistica la europenii nordici
Heterogenitatea: alelele mutante sunt localizate pe diferiti loci dar dau manifestari clinice similare (albinism, surditatea neurosenzoriala, hipotroidismul congenital). Daca parintii surzi au loci diferiti implicati, copiii lor vor avea auzul normal (acestia vor fi dublu heterozigoti)
Galactozemia. Copilul afectat poate prezenta hepatomegalie sau cataracta si poate sa moara prin acidoza metabolica. Absenta galactozo-1-fosfat-uridiltransferazei duce la imposibilitatea metabolizarii lactozei. Pentru tratament se instituie o dieta care elimina produsele ce contin lactoza (in primul rand laptele). Copiii netratati care supravietuiesc vor avea retard mental. Au fost descrise mai multe alele mutante implicate in producerea bolii.
Homocistinuria. Pentru ca acesti indivizi prezinta o atitudine corporala anormala pot fi confundati cu bolnavii cu sindrom Marfan. Doua treimi din acesti bolnavi au retard mental sau probleme psihice. Deficienta cistin-β sintetazei creste nivelul homocistinei si metioninei. Trebuie instituita o dieta redusa in metionina. Poate surveni moartea subita prin accidente tromboembolice.
Fibroza chistica. Aproximativ 1:5000 copii din Europa nordica prezinta aceasta boala manifestata prin infectii cronice respiratorii si malabsorbtie. Este afectat mirosul, nivelul clorului si sodiului este ridicat in secretii (saliva, sudoare); 90 % din pacienti mor prin complicatii ale afectiunilor pulmonare care debuta deja la varsta de 1 an. Gena mutanta este localizata pe cromozomul 7q; tratamentul este paleativ: fizioterapie, antibiotice.
TRANSMITEREA X- LINCATA
Poate fi atat dominanta cat si recesiva si afectiunile datorate genelor X-lincate se manifesta fenotipic in mod obisuit la barbati. Aceasta transmitere poate fi suspectata daca sunt afectate mai multe rude de sex masculin pe linie materna. Barbatii sunt hemizigoti pentru alela mutanta (X*Y), stare care confera exprimare fenotipica chiar si pentru o alela recesiva.
* Criterii
Boala este rara in familie, parintii si rudele lor (exceptand barbatii pe linie materna) sunt de obicei normali.
Barbatii hemizigoti afectati transmit gena doar la fetele lor si nu la baieti (fetele vor fi heterozigote, purtatoare ale alelei mutante). Nu exista transmitere de la barbat la barbat.
Femeile heterozigote purtatoare sunt clinic normale, dar vor transmite gena la baieti (50 % din ei vor fi bolnavi) si la fetele lor care pot fi (50 %) purtatoare sanatoase.
Fetele bolnave rezulta din barbati bolnavi si mame purtatoare
Cu exceptia cazurilor cu mutatii noi, fiecare baiat afectat se naste dintr-o femeie heterozigota.
Daca gena mutanta este dominanta toti descendentii de sex feminin ai unui barbat afectat vor fi bolnavi. In aceeasi situatie 50 % din descendentii de sex feminin sau masculin ai unei femei heterozigote vor prezenta boala. Nici in aceasta situatie nu exista transmitere de la barbat la barbat
Cazurile sporadice. Daca numai un mascul este afectat, fara ca alti membrii ai familiei sa fie afectati, se pune intrebarea daca boala este rezultatul unei noi mutatii sau mama este heterozigota.
Femeile heterozigote pot fi depistate uneori prin semne clinice minore, chiar nesesizabile, sau nivele enzimatice intermediare. In aproximativ 2/3 din cazurile cu boli x-lincate recesive, care sunt frecvent letale in copilarie si in care apare un singur individ de sex masculin afectat in familie, mama este heterozigota
Heterogenitatea: albinismul se poate transmite prin gena autosomal recesiva (albinismul oculo cutanat) sau prin gena X-lincata (albinismul ocular)
Femeile bolnave. Compensarea dozajului genelor de pe perechea XX se face prin inactivarea (lyonizarea) intamplatoare a unui cromozom X la nivelul blastocistului. Rezulta un status de hemizigotie pentru majoritatea cromozomilor X. Femeile care sunt heterozigote pentru alele mutante X-linkate prezinta clone, linii celulare in care una sau cealalta alela este inactivata.
Femeile pot prezenta boli X- linkate in urmatoarele situatii:
inactivarea intamplatoare este mai frecventa pe liniile cu X normal (femeia prezinta o hemizigotie pentru alela mutanta)
descendentul de sex feminin rezulta din casatoria mamei heterozigote si a tatalui afectat (risc de 25 %)
sindrom Turner 45,X in care cromozomul X poarta gena detrimentala
un barbat afectat poate sa transmita boala la fiica sa, daca aceasta primeste si pe X-ul matern aceeasi alela mutanta, rezultata la mama printr-o mutatie noua.
o femeie heterozigota poate da nastere la o fata bolnava daca aceasta mosteneste un X cu aceeasi noua mutatie de la tatal sau.
inactivarea neintamplatoare a cromozomului X in timpul formariii izocromozomilor
bolile X lincate letale - daca boala este letala la barbati orice nou-nascut de sex masculin moare la nastere sau inainte. Femeile heterozigote sunt normale dar din descendentii lor de sex masculin vor muri. In final se altereaza raportul intre sexe (sex ratio) pentru afectiunea respectiva (doua femei afectate la fiecare barbat)
Aspecte clinice: - incidenta 1:3600 de baieti prezinta aceasta boala cu debut in primul an de viata. Semnele caracteristice includ slabiciune musculara progresiva, hipertrofia muschilor gambei si moarte, prin afectiuni cardiace si respiratorii intre 10 si 20 ani.
Aspecte genetice: gena mutanta este localizata pe Xp21. Produsul normal de sinteza al acestei gene este distrofina care actioneaza probabil ca un protector al miofibrilei. Aproximativ 60 % dintre barbatii afectati prezinta deletii detectabile prin tehnici moleculare de analiza, iar restul familiei necesita analize lincate.
Sindromul X fragil
Aspecte clinice:- retardul mental este mai frecvent la barbati, sugerand existenta unei mutatii X- lincate. Cea mai comuna cauza a retardarii mentale ereditare este acest sindrom care apare cu o frecventa de 1 : 500 barbati. Masculii afectati vor avea urechi mari, barbie proeminenta, testicule mari dupa pubertate si retard mental moderat. Copiii afectati pot avea tulburari de limbaj si de auz. Probabilitatea instalarii retardului mental intr-o familie este amplificata de numarul generatiilor peste care trece mutatia (paradoxul Sherman). Si femeile pot fi afectate intr-o proportie de 1/3 din totalul cazurilor
Aspecte genetice: diagnosticul s-a facut initial prin culturi cromozomiale (cu folat) si au identificat un situs fragil pe Xq27.3 Recent s-a identificat ca este o mutatie dinamica la nivelul unui triplet instabil de nucleotide (CGG) interesand gena FMR-1 pe Xq27. Cresterea numarului secventelor CGG intre 50 si 200 face ca aceasta secventa sa devina instabila, stare numita premutatie. Barbatii care poarta premutatia o vor transmite fiicelor lor, care vor avea inteligenta normala, dar baietii acestora au un risc mare ca prin expansiunea tripletului (de la 200 la 3000 de repetari) premutatia sa devina mutatie. Transmiterea ereditara a sindromului X fragil poate fi considerata o transmitere atipica legata de cromozomul X.
EREDITATE MITOCONDRIALA
Celulele umane au sute de mitocondrii dispersate in citoplasma, fiecare continand molecule de ADN circular. Mutatiile ADN-ului mitocondrial sunt acum cunoscute si sunt raspunzatoare pentru un numar mic de boli genetice.
* Criterii
Fiecare mitocondrie contine un numar de copii ale genomului circular. Enzimele mitocondriale sunt de obicei codate de gene nucleare dar unele dintre citocromoxidaze sunt codate numai de ADN-mt
Aproape tot ADN-ul mitocondrial este mostenit pe linie materna. Pedigreeurile cu ereditate mitocondriala pot arata ca toti copiii unei mame afectate sunt bolnavi si toti copiii unei mame neafectate sunt sanatosi.
Proportia mitocondriilor este specifica pentru diferitele tesuturi. Sunt bogate in mitocondrii celulele muschilor striati si cardiac, renale si ale SNC. De aceea bolile mitocondriale sunt de obicei miopatii, cardiomiopatii si afectiuni ale SNC.
Exemple clinice:
Sindromul Kearns-Sayre este o boala sporadica cu debut dupa varsta de 20 ani. Se caracterizeaza prin oftalmoplegie, anomalii ale pigmentului melanic, retinita pigmentara, ataxie cerebeloasa. Indivizii prezinta deficiente musculare datorate ADN-ului mitocondrial.
Neuropatia optica Leber. Se caracterizeaza prin atrofie optica, miscari dezordonate, modificari ECG care se transmit exclusiv de la femeie la copilul sau de ambe sexe. Boala este secundara mutatiilor unei gene care codeaza o subunitate din complexul I al caii transportatoare de electroni.
MECANISMELE MUTATIEI
Dezvoltarea tehnologiei ADN a fost urmata de descrierea mutatiilor la nivelul secventei de ADN implicata. In genomul uman se intalnesc tipuri si mecanisme diferite de mutatii incluzand rearanjamente majore genice si mutatii punctiforme. Aceste mutatii pot afecta transcriptia si/sau translatia si pot altera structura sau functia unei proteine.
Rearanjamente genice majore
Deletiile sunt detectate folosind tehnologia ADN-ului recombinat si sunt indicate de absenta sau modificarea dimensiunii unui fragment de ADN.
a talasemia este datorata deletiei genei cluster a globina, care are patru secvente inalt inrudite una in vecinatatea celeilalte:
- din pricina similitudinii dintre aceste gene poate aparea imperecherea gresita a cromozomilor precursori meiozei
- dupa crossing-over un cromozom va contine o deficienta de material genetic
- crossing-overul inegal in regiunea genei cluster a globin are ca rezultat un cromozom lipsit de genele functionale pentru a globina (acesti cromozomi sunt comuni in populatiile din Asia de S-E).
- fetusul cu deficienta genelor a globin moare cu edeme generalizate pe parcursul sarcinii sau la scurt timp dupa nastere.
Deficienta hormonului de crestere este o alta afectiune comuna a carei cauza sunt deletiile :
gena cluster pentru hormonul de crestere este formata din secvente similare de ADN asezate una in vecinatatea celeilalte
imperecherea anormala si crossing over inegal in meioza cauzeaza deletii la nivelul acestei gene, mutatii care duc la aparitia piticismului (nanismului) la homozigoti
Hipercolesterolemia familiala (FM): rezulta din defecte ale genei receptorului LDL (lipoproteine cu densitate mica); mai poate fi cauzata de deletii datorate unui crossing over inegal interesand secventa inalt repetitica alu.
2. Duplicatiile secventelor de ADN sunt comune in evolutie si se pot datora imperecherii gresite intre secventele ADN homologe din vecinatate apropiata, cu duplicarea materialului genetic care este continut in interiorul genei. Mecanismul este asemanator cu duplicatia prezentata la a talasemie. Duplicatiile pot afecta citirea matritei de sinteza. FH si DMD sunt exemple de boli genetice care pot fi cauzate de duplicatii.
3. Insertiile sunt cauze rare ale mutatiei in genomul uman. Cu toate acestea, transpozitia de ADN nu este rar intalnita in genomul uman, desi in mod obisnuit nu implica secventa codanta. Cand transpozitia afecteaza gena poate sa apara manifestarea clinica.
Secventele lungi repetitive (LINE) si scurte alu (sau SINE) sunt exemple de secvente care se pot insera in interiorul genelor.
Hemofilia este o boala care rezulta prin inserarea unei secvente LINE in gena pentru factorul VIII, afectandu-i functia. La fel neurofibromatoza poate rezulta prin insertia unei secvente alu
in gena implicata.
Mutatiile punctiforme
Substitutia unui singur nucleotid, similara aceleia intalnita la bacterii si virusuri, sunt cele mai comune cauze ale mutagenezei in genomul uman. Daca codonii sunt afectati de o singura substitutie aceasta mutatie se numeste mutatie missens. Bolile care se datoresc unor mutatii in mai multe puncte diferite sunt β talasemia, fibroza chistica, fenilcetonuria (PKU), boala Tay-Sachs
B talasemia a fost prima boala la care s-au identificat mutatiile punctiforme ca fiind cauza aparitiei unei hemoglobine anormale prin afectarea transcriptiei, matisarii ARN-mesager si translatiei.
Aspecte clinice - copiii homozigoti nu au la nastere anemie datorita nivelului crescut al hemoglobinei fetale. Cu toate acestea boala se manifesta dupa primul an de viata cu anemie, paloare si slabiciune. Apare hepatosplenomegalia datorata hematopoezei extramedulare. Apare o poliferare a maduvei osoase in bosele frontale. Dezvoltarea fizica si sexuala este intarziata. Tratamentul se face cu transfuzii sanguine si transplante medulare.
*Fenilcetonuria (PKU) se transmite printr-o gena autosomal recesiva care determina diferite mutatii ale fenilalanin-hidroxilazei care metabolizeaza fenilalanina in tirozina. La acesti bolnavi fenilalanina nu este metabolizata si se acumuleaza ceea ce duce la afectarea SNC si retard mental.
Screeningul neonatal pentru PKU poate depista precoce acesti copii bolnavi; dieta instituita precoce poate preveni retardul mental.
Mutatiile missens pot duce la alterarea unui codon care codeaza o proteina structurala (anemia cu celule in secera rezulta pentru ca pe catena α a hemoglobinei in pozitia 6 acidul glutamic este inlocuit de valina), sau pot afecta functia proteinei cand mutatia are implicatii asupra unui lant metabolic, sau modifica structura tridimensionala a proteinei compromitandu-i functia.
Mutatiile dinamice
Se datoresc expansiunii secventelor repetate de trinucleotide in gene noi. Aceste trinucleotide pot fi prezente in capatul 5' al genei, in regiunea codanta a genelor si la capatul 3' al genei
Expansiunea tripletului CGG in capatul 5' de peste 200 de ori induce situsuri fragile (ex. sindromul X fragil)
Expansiunea tripletului in capatul 3' a genei duce la distrofie miotonica
O alta categorie de mutatii dinamice se realizeaza prin expansiunea mai mica (intre 35 si 150 de ori) a trinucleotidului CAG in regiunea codanta a genei: rezulta afectiuni neurodegenerative inclusiv boala Huntington, ataxie spinocerebeloasa, etc.
Anticipatia este aparitia unei boli genetice cu severitate mai mare sau la o varsta mai tanara in generatiile tinere ale familiei. Se poate datora extinderii regiunii de trinucleotide repetate daca mutatia dinamica trece prin meioza si primele faze ale dezvoltarii embrionare.
In bolile care sunt cauzate de mutatii dinamice (sindrom X fragil, boala Huntington, distrofie miotonica) exista o corelatie stransa intre dimensiunea extinderii trinucleotidelor repetate si severitatea bolii sau varsta de debut.
In unele situatii, daca numai una din alelele unui locus functioneaza normal, organismul nu reuseste sa sintetizeze suficient produs de sinteza (ex: hipercolesterolemia familiala). Astfel se considera ca mutatia determina un efect dominant.
Alteori in urma acestor mutatii rezulta un produs anormal care interfereaza cu functiile produsului normal de sinteza al alelei interesate (ex: in deficientele de crestere in osteogeneza imperfecta)
Exista o distributie etnica specifica a mutatiilor. Acest lucru face ca anumite boli genetice sa apara cu o frecventa mai mare in anumite populatii: anemia falciforma este mai frecventa la negrii, hipercolesterolemia familiala este mai frecventa la canadienii francezi.
Frecventa mai crescuta a unor genopatii poate fi explicata prin avantajul selectiei acestor indivizi (persoanele care au hematii in secera sunt protejate in fata malariei) sau prin efectul de cuplu fondator intr-o populatie.
BAZELE BIOCHIMICE ALE BOLILOR GENETICE
Exista situatii in care dezvoltarea normala sau aparitia unei stari de boala se produce prin interactiunea factorului genetic ("intrinsec") si nongenetic ("extrinsec"). Chiar si atunci cind se iau in considerare numai factorii genetici e important sa se aminteasca ca genele mutante nu actioneaza izolat, ele influentand actiunea multor altor gene.
Erorile innascute de metabolism sunt determinate de mutatii genetice largi care pot fi cauzate de o singura gena cu transmitere mendeliana (boala Tay-Sachs) sau un proces mult mai complex (boala arterelor coronariene). Multe boli pot fi tratate sau ameliorate prin dieta sau alte modalitati nongenetice, dar stabilirea unor strategii necesita o cunoastere a fiziopatologiei sau proceselor fundamentale.
Relatia gene-proteine
Fiecare dintre genele structurale specifica secventa de aminoacizi a proteinei sau controleaza rata de sinteza a unei proteine specifice. Consecinta imediata a unei mutatii genice este schimbarea calitatii sau cantitatii proteinei specifice. Astazi, peste 500 modificari biochimice sunt partial / complet cunoscute.
Medicii trebuie sa fie familiarizati cu modul in care variatele tipuri de mutatii influenteaza proteina finala si determina patologia clinica.
Conceptul "o gena - o enzima" este acum numit "un cistron - un polipeptid" pentru ca produsul genei nu este totdeauna o enzima si pentru ca polipeptidele pot fi subunitati in interiorul enzimelor. Principiile care sustin acest concept sunt urmatoarele:
Toate procesele biochimice sunt controlate genetic.
Fiecare cale biochimica parcurge etape, fiecare dintre acestea fiind controlate de diferite enzime.
Fiecare enzima e codificata de una sau mai multe gene.
O singura mutatie genetica poate cauza o alterare a unei reactii biochimice. Toate perturbarile unei singure gene au ca rezultat o molecula proteica anormala sau un control anormal al unor molecule.
Functiile proteinelor si modificarile functiei proteinelor
Proteinele functionale (enzime) sunt implicate in metabolism si proteinele plastice in structura organismului. Mutatia poate cauza o tulburare genetica specifica pentru fiecare functie a proteinei
1. Efectele directe biologice Multe mutatii actioneaza direct in producerea bolii:
Osteogeneza imperfecta cuprinde un grup de tulburari care implica defecte in producerea colagenului tip I. Colagenul de tip I este o proteina majora din structura osului. Are o structura helicoidala compusa din 2 lanturi de procolagen pro a 1 si pro a 2. Copiii cu acest tip sever de osteogeneza imperfecta au fost descoperiti ca prezinta o mutatie punctiforma ce implica o substitutie a Glicinei , care produce instabilitatea colagenului, afecteaza producerea acestuia sau asamblarea fibrilelor de colagen in matricea extracelulara. Copiii afectati sunt heterozigoti pentru mutatie si frecvent mor cu fracturi severe .
Beta-talasemia: este consecinta unor mutatii pe gena - globinei ceea ce afecteaza sinteza lanturilor . Functia normala a moleculei de hemoglobina este transportul oxigenului de la plamani la tesuturile periferice. Mutatiile altereaza transportul oxigenului si stimuleaza distrugerea hematiilor, inducand o anemie severa. Homozigotii cu anemie severa necesita repetate transfuzii sanguine.
Sindromul Kartagener (sindromul cililor imobili)
Acest sindrom autosomal recesiv combina bronsiectazia, dextrocardia si sterilitatea masculina. La microscopul electronic sunt absente cozile spermatozoizilor cauzand imobilitatea lor. Tot electronomicroscopic se observa ca miscarea cililor mucoasei bronsice e absenta ducand la o lipsa de curatire a secretiilor pulmonare Deoarece cilii necesita multe proteine structurale e mai mult ca sigur ca mutatiile sunt heterogene.
2. Proteine care afecteaza metabolismul altor molecule
Multe defecte biochimice provoaca efecte secundare. Daca sinteza sau transportul lor este afectat, ele pot cauza fiecare o deficienta a produsului sau acumularea daunatoare a precursorului. Daca membrana de transport este implicata poate exista o malabsorbtie sau o crestere a pierderii renale. Adesea, mecanismul exact de producere al simptomatologiei bolii este neclar.
Fenilcetonuria. Fenilalanin-hidroxilaza hepatica transforma aminoacidul fenilalanina in tirozina. Deficitul de fenilalanin-hidroxilaza va determina cresterea nivelului de fenilalanina si al metabolitilor sai indolici, care vor produce retardare mentala si paloare la copiii afectati. Tratamentul implica o restrictie severa orala de fenilalanina.
Galactozemia. Cea mai comuna enzima deficitara implicata este galactozo-1-fosfat-uridil transferaza. Copiii prezinta hepatomegalie, icter si hipotonie. Moartea se produce prin acidoza. Deficienta enzimei produce acumularea galactozei care nu poate fi transformata in glucoza. Tratamentul impune eliminarea lactozei din dieta.
Boala Tay-Sachs. Hexozaminidaza A este implicata in metabolismul lipidelor. Aceasta muta zaharuri dintr-o ramificatie particulara a lantului lung al lipidelor (ceramide), care actioneaza ca o suprafata membranara receptoare in creier. La pacientii afectati sinteza anormala a enzimei conduce la acumularea masiva de lipide in celulele nervoase. Afecteaza progresiv copiii prin pierderea indemanarii si vederii. Moartea survine la varste tinere. Nu exista tratament.
Mucopolizaharidoze(MPZ). Nivelele deficitare de α1 iduronidaza apar in 3 ipostaze fenotipice clinice, care fac parte din MPZ :
a. Sindromul Hurler: copiii afectati prezinta anchiloze articulare, hepatosplenomegalie, cataracta si retardare mintala progresiva. Copiii mor prin infectii respiratorii sau afectiuni cardiace.
b. Sindromul Scheie: este asemanator (anchiloze articulare, cataracta) cu sindromul Hurler, dar efectele sunt mai atenuate si copiii nu dezvolta retard mintal. Diagnosticul nu poate fi stabilit inainte de varsta adulta.
c. Sindromul Hunter-Scheie: este un fenotip intermediar cu atingere articulara si cataracta, si cu retardare mintala redusa.
Toate cele trei stari clinice sunt asociate cu o productie anormala de mucopolizaharide si nivele scazute de α1 iduronidaza.
Fibroza chistica Gena implicata a fost gasita responsabila de codificarea unei proteine reglatoare a conductantei transmembranare care e implicata in transportul clorului prin membrana celulara la nivelul pancreasului, plamanului si glandelor sudoripare. Este indus un defect in permeabilitatea clorului cu aparitia unei boli obstructive cronice pulmonare si insuficienta pancreatica. Tratamentul este inca numai simptomatic.
Deficienta hormonilor tiroidieni Exista numeroase defecte ale hormonogenezei tiroidiene (deficit de tiroid, peroxidaza, substanta caraus anormala, receptor anormal pentru iod) fiecare fiind mostenite ca trasaturi autosomal recesive.
Unele deficite ale hormonului tiroidian, cum ar fi deficitul peroxidazei tiroidiene ce implica conversia necorespunzatoare, sunt produse de un numar anormal de alele care raspund de severitatea clinica. Rezultatul final este hipotiroidismul congenital care induce la copil trasaturi grosolane, intarziere in crestere si eventual retard mintal. Tratamentul consta in administrarea exogena de hormoni tiroidieni.
3. Tipuri de alterari proteice:
Sinteza proteica e rezultatul transcriptiei ADN in mARN si in proteine la nivelul ribozomilor. Transcriptia este realizata de la nucleotid la nucleotid prin complementaritate, in timp ce translatia se face de la nucleotid inspre secventa de aminoacizi.
Reglarea transcriptiei si translatiei determina procesul normal de dezvoltare. Procesul de sinteza a proteinelor poate fi intrerupt in urmatoarele etape:
Transcriptia si splicing-ul ARN-ului mesager
Alterarea in totalitate a mARN-ului transcris se observa cand deletiile/mutatiile implica aranjarea sau lipirea situsurilor, sau introducere unui codon stop sinteza. Aceste modificari se traduc prin incapacitatea de citire a unei secvente complete de ADN si imposibilitateade transcrierii ei in mARN(talasemiile rezulta prin reducerea sau absenta totala a sintezei globinei).
Translatia. Daca o mutatie se produce prin modificarea structurii ADN transcriptia poate avea loc, insa translatia informatiei in polipeptid poate fi oprita de un codon stop sinteza aparut in urma mutatiei si pozitionat la inceput .
Structura secundara si tertiara a polipeptidelor. Mutatiile care afecteaza structura secundara sau tertiara a proteinelor se reflecta in productia proteinei. Plicaturarea anormala poate duce la o molecula instabila.
Mutatiile colagenului pot conduce la o legare incrucisata anormala avand ca rezultat alterarea proteinei structurale. Un exemplu de alterare a proteinelor structurale datorata mutatiilor colagenului este observata in sindromul Ehlers-Danlos cand pielea are mare elasticitate si hiperextensibilitate.
Localizarea tridimensionala. O proteina completa poate ocupa o pozitie specifica in structura celulei. Modificarea secventelor de aminoacizi poate interfera cu capacitatea proteinei de a fi localizata la locul potrivit. Multe proteine necesita o localizare normala pentru o functie normala.
a. La unii pacienti cu metilmalonil-acidemie, enzimei ii lipseste secventa lider, ceea ce impiedica patrunderea enzimei in mitocondrie. Enzima nu functioneaza normal decat in interiorul mitocondriei . Copiii prezinta manifestari ca voma si stare de rau.
Diagnosticul enzimopatiilor
Implica identificarea acumularii sau pierderii metabolitilor, masurarea nivelurilor specifice enzimatice sau identificarea variantelor de proteine.
Suspectarea bolii biochimice. Desi au fost caracterizate multe dintre mutatiile responsabile de bolile metabolice, clinicianul si biochimistul noteaza in primul rand simptomele pacientului sau profilul sau biochimic. Pana cand defectul primar este clarificat, tot ceea ce clinicianul observa este consecinta varietatii tipurilor de mutatii.
* primele suspiciuni le ridica unele modificari biochimice care sunt diagnosticate ca urmare a testelor screening la nastere inainte de aparitia oricaror simptome clinice. Semnele clinice pot deasemenea sugera modificari ale unui sistem. De exemplu: sangerarea excesiva poate fi prima manifestare a hemofiliei; slabiciunea musculara poate fi semnalul distrofiei musculare Duchenne; ambiguitatea genitala poate indica lipsa sensibilitatii la androgeni.
Analiza directa a proteinelor sau enzimelor Cand calea biochimica este cunoscuta, o apreciere directa a enzimei sau produsului enzimatic este posibila. Poate fi utilizat sange, culturi de fibroblasti din piele sau urina. De exemplu un copil poate fi suspectat ca avand boala Tay - Sachs daca prezinta o deteriorare progresiva fizica si mentala, chiar daca a parut normal in primele luni de viata. Pentru confirmarea diagnosticului masurarea activitatii hexozaminidazei A in sange poate fi utila.
Studiul culturilor celulare. Orice fibroblasti din piele sau limfoblasti transformati pot fi folositi pentru o examinare biochimica completa. Incorporarea precursorilor radioactivi in cresterea celulelor, folosirea unor studii performante si compararea rezultatelor de la un numar de indivizi cu aceeasi boala reprezinta avantajele unei culturi pe termen lung.
Tehnicile moleculare. Analiza linkage-ului si a mutatiei directe sunt utile pentru un numar mare de modificari biochimice. Acestea fac posibil diagnosticul precoce la fel de bine ca identificarea heterozigotilor.
Relatia genotip - fenotip
O intrebare la care trebuie sa se raspunda este daca aspectul clinic poate fi prevazut prin cunoasterea genei implicate. Cunoasterea actuala a mutatiilor specifice sugereaza urmatoarele:
Mutatiile alelice pot aparea in diferite fenotipuri clinice. Mutatiile genei distrofinei pot conduce la distrofie musculara Duchenne sau distrofie musculara Becker, depinzand in totalitate de functionalitatea distrofinei. La pacientul cu talasemie deficienta poate prezenta variatii mari, depinzand de tipul si localizarea mutatiilor alelice.
Aspectul clinic nu poate fi prevazut prin cunoasterea mutatiilor specifice. Desi defectul primar poate fi stiut evolutia ulterioara a bolii poate fi neclara. De exemplu, schimbarile identificate in situsul X fragil nu pot explica intarzierea vorbiri sau alte manifestari ale sindromului X fragil.
Mostenirea caracterelor nu este intotdeauna recesiva Majoritatea starilor de deficienta enzimatica sunt manifeste doar la homozigoti, desi heterozigotii normali pot avea nivele intermediare ale enzimei implicate. Oricum, multe boli biochimice (hipercolesterolemia familiala, osteogeneza imperfecta tip I) care implica proteine structurale sau de transport se manifesta la heterozigoti. Homozigotii sunt rar diagnosticati, doar in anumite conditii.
Consideratii clinice dupa diagnostic. Odata ce modificarea metabolica a fost diagnosticata precis, trebuie actionat in urmatoarele directii:
1. Sfatul genetic al individului afectat sau al familiei.
2 Identificarea heterozigotilor
3. Diagnosticul prenatal.
4. Tratamentul imediat al individului afectat.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 10484
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved