| CATEGORII DOCUMENTE |
| Alimentatie nutritie | Asistenta sociala | Cosmetica frumusete | Logopedie | Retete culinare | Sport |
FIZIOPATOLOGIA HEMOSTAZEI SI FIBRINOLIZEI
Echilibrul fluido-coagulant se realizeaza prin functionarea normala a mecanismelor componente ale acestuia, hemostaza primara, hemostaza secundara si fibrinoliza (hemostaza tertiara)
In conditii patologice, studiul echilibrului fluido-coagulant poate decela:
- sindroame hemoragice
- vasculopatii;
- trombocitopenii;
- trombopatii;
- coagulopatii;
- boala tromboembolica;
- sindrom fibrinolitic primar sau secundar (CID).
Hemostaza, reactia inflamatoare acuta si reactia imuna, reprezinta reactii tisulare de aparare locala. Hemostaza fiziologica este declansata de aparitia unei leziuni vasculare si are rol in oprirea sangerarii la nivelul vaselor de calibru mic sau mijlociu (realizata in principal prin formarea unui dop fibrinoplachetar). In conditiile lezarii peretelui uniu vas de calibru mare, hemostaza este declansata si se poate desfasura normal dar este ineficienta.
Schema clasica de derulare a evenimentelor hemostazei este valabila in vitro (fenomenele studiate sunt influentate de conditiile de laborator in care acestea se desfasoara). In vivo, etapele hemostazei nu au loc intr-o stricta succesiune.
Timpul vasculo-plachetar (hemostaza primara)
Hemostaza primara realizeaza conditiile locale reologice (dependente de fluxul sanguin si de caracteristicile vasului) necesare formarii unui dop plachetar (trombocitar) care opreste temporar sangerarea. Dopul strict plachetar (dop plachetar alb), insuficient ancorat la nivelul peretelui vascular si relativ permeabil, nu reprezinta o solutie definitiva pentru oprirea sangerarii.
Hemostaza primara presupune parcurgerea celor 3 evenimente principale
a) modificarile hemodinamice si reologice locale;
b) aderarea plachetara la nivelul peretelui vascular lezat;
c agregarea trombocitelor aderate
In vivo, leziunea vasculara induce:
- vasoconstrictie locala;
- expunerea sangelui extravazat la:
- factori tisulari (colagenul vascular ce induce aderarea plachetara);
- factori vasculari cu efect procoagulant (tromboplastina tisulara ce declanseaza calea extrinseca a coagularii, cu aparitia unor cantitati reduse de trombina).
Trombina astfel formata, care nu genereaza retea de fibrina, stimuleaza:
agregarea plachetara;
stimularea coagularii pe calea intrinseca (activeaza FXI, FVIII, FV).
a) Modificarile hemodinamice si reologice locale
Modificarile hemodinamice locale constau in vasoconstrictia activa a vasului lezat realizata prin reflex local de axon (este declansat de factorul mecanic inductor al leziunii vasculare si are durata scurta). Vasoconstrictia (de intensitate si durata variabile, in functie de particularitatile reactive ale vasului lezat) este mentinuta in timp prin actiunea mediatorilor eliberati din trombocitele activate metabolic (serotonina, endotelina, tromboxan A2, catecolamine etc.).
In conditii de vasoconstrictie, fluxul sanguin aferent vasului lezat este deviat in colaterale neafectate din tesuturi invecinate, datorita vasodilatatiei (efect al eliberarii locale de histamina, prostaciclina etc.).
In aceasta faza au loc si modificari reologice locale: aplatizarea pasiva a vasului lezat, cresterea vascozitatii sangelui (datorita plasmexodiei locale), scaderii presiunii hidrostatice intravasculare (prin pierderea de sange), precum si aglomerarea eritrocitelor la nivelul leziunii vasculare.
Vasoconstrictia contribuie la scaderea marcata sau chiar oprirea fluxului de sange din vasul lezat si favorizeaza aderarea si agregarea plachetara, fiind o conditie obligatorie pentru un efect hemostatic eficient al acestor mecanisme trombocitare (legaturile dintre vas si trombocit, prin FvW, dar si cele dintre trombocite aderate, prin molecule de fibrinogen, sunt slabe).
Toate aceste modificari hemodinamice au loc doar in conditiile in care, anterior momentului aparitiei unei leziuni parietale, vasele interesate sunt normale din punct de vedere structural si functional.
b) Aderarea plachetara la nivelul peretelui vascular lezat
Aderarea plachetara reprezinta procesul de atasare (legare) a trombocitelor la fibrele de colagen din structura peretelui vascular. In conditii normale, fibrele de colagen sunt structuri subendoteliale si nu vin in contact direct cu fluxul sanguin. In conditiile unei leziuni endoteliale, acestea sunt expuse intravascular. Aderarea plachetara este un proces pasiv (nu necesita consum de energie, activare metabolica plachetara sau modificari conformationale ale membranei plachetare).
Aderarea plachetara se realizeaza prin contact direct, sub forma unor punti intre membrana plachetara si fibrele de colagen. La formarea acestor punti de aderare participa:
- factorul von Willebrand (componenta a factorului plasmatic VIII
- receptori ai colagenului specifici pentru factorul von Willebrand;
- receptorul trombocitar specific pentru FvW, glicoproteina Ib-IX (GPIb-IX).
# Factorul von Willebrand (FvW)
FvW realizeaza punti intre membrana trombocitara si fibrele de colagen. Aceste legaturi sunt mediate prin structuri receptoare (receptori ai colagenului si receptorul trombocitar, respectiv, GPIb-IX).
FvW este o molecula proteica ce se ataseaza cu unul dintre capete la nivelul receptorilor colagenici fata de care are o mare afinitate (fenomenul este pasiv). Dupa atasarea de fibrele de colagen, FvW sufera modificari conformationale ce induc aparitia unei afinitati pentru receptorii trombocitari specifici. Aceasta modificare conformationala permite legarea FvW, cu celalalt capat, la nivelul receptorului trombocitar (GPIb-IX), rezultatul fiind aparitia puntilor de aderare.
# Receptorii colagenului specifici pentru FvW
Structurile receptoare ale colagenului, specifice pentru FvW, au un continut bogat in hidroxilizina si hidroxiprolina
# Receptorul trombocitar specific pentru FvW (GPIb-IX)
GPIb-IX este un complex de glicoproteine (heterotrimer) care formeaza doua componente diferite din punct de vedere functional: GPIb si GPIX.
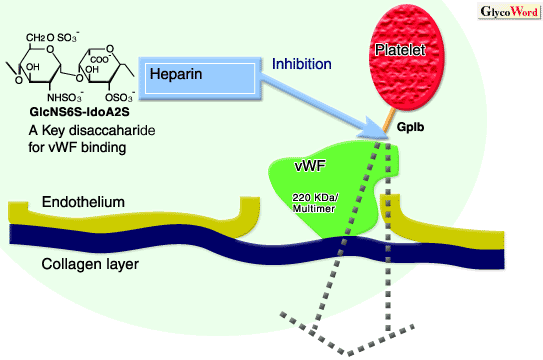
* GPIb (glicocalicina) este un heterodimer transmembranar.
- Lantul alfa, strict implicat in aderarea plachetara, este format din 3 segmente:
1. segmentul extramembranar contine spre capatul extern (NH2-terminal) situs-ul de legare pentru FvW; acesta este un domeniu format din 7 secvente repetitive (SR, short repeats), cu un continut crescut de leucina;
2. segmentul intramembranar;
3. segmentul intracitoplasmatic (in contact cu enzime plachetare).
- Lantul beta nu participa la aderare. Acest lant polipeptidic este legat (prin legaturi S-S) de lantul alfa pe care il leaga de GPIX ; lantul beta are rol structural.
* GPIX este receptorul trombocitar pentru FXI plasmatic (Rosenthal) al coagularii. GPIX este un cofactor. Aceasta componenta nu participa la fenomenul de aderare plachetara.
GPIb angajeaza legaturi cu FvW (eliberat de celulele endoteliale) doar in conditiile in care acesta s-a fixat anterior pe receptorii colagenici specifici. In contact cu receptorii colagenici, FvW sufera modificari conformationale care favorizeaza recunoasterea FvW de catre GPIb si GPIX (in acest fel, GPIX se activeaza si devine apta sa fixeze si sa activeze FXI plasmatic; prin activarea FXI, este declansata coagularea pe calea intrinseca).
In concluzie, aderarea plachetara presupune derularea a doua evenimente:
atasarea FvW la nivelul receptorilor colagenici, cu aparitia modificarilor conformationale ale FvW;
legarea FvW la nivelul receptorilor trombocitari (GPIb), cu aparitia puntilor de aderare plachetara.
In paralel, are loc declansarea coagularii pe calea extrinseca (CE) din care rezulta cantitati reduse de trombina. Trombina rezultata din activarea CE nu are capacitatea de a transforma fibrinogenul in fibrina dar indeplineste alte functii:
activeaza factorii plasmatici XI, VIII, V;
este implicata in activarea metabolica plachetara, prin stimularea receptorilor membranari de tip PAR (receptori activati de proteaze), respectiv PAR-1 si PAR-4; acesti receptori sunt stimulati si de FXa, precum si de complexul factor tisular-FVIIa.
In conditii normale, mecanismele coagularii (hemostaza secundara) au loc concomitent cu cele ale hemostazei primare si se desfasoara doar la suprafata membranei trombocitare.
In conditii patologice, scaderea aderarii plachetare la fibrele de colagen poate avea drept cauza:
- afectarea plachetara cantitativa (trombocitopenie) sau calitativa (trombopatie, respectiv deficit de GP Ib-IX, in sindromul numit distrofie trombocitara hemoragica);
- afectarea plasmatica (deficit de FvW, in boala von Willebrand);
- alterarea sintezei fibrelor de colagen din punct de vedere cantitativ sau calitativ (in vasculopatii).
c) Agregarea plachetara a trombocitelor aderate
Agregarea plachetara reprezinta stabilirea de legaturi multiple si punctiforme intre trombocite care au aderat la fibrele de colagen. Legaturile sunt realizate prin intermediul pseudopodelor (emise de trombocitele aderate si activate metabolic) si a moleculelor de fibrinogen care se leaga la nivelul receptorilor trombocitari specifici (GPIIb-IIIa). Agregarea plachetara este un proces activ care necesita consum de energie (rezultata din activarea metabolica a trombocitelor aderate).
Inductorii fenomenului de activare si agregare plachetara sunt:
trombina (rezultat al activarii coagularii pe CE);
ADP-ul (eliberat din celulele endoteliale lezate);
tromboxanul A2 (TxA2) eliberat din trombocitele activate metabolic.
Trombina si ADP-ul activeaza doar plachetele aderate, nu si plachetele circulante. In conditiile in care acesti inductori sunt diluati intravascular, iar trombina este neutralizata de anticoagulanti circulanti (antitrombina III), este limitata posibilitatea trombozarii vaselor lezate.
Fixarea trombinei la nivelul receptorilor membranari de tip PAR determina efecte controlate prin semnale transmembranare. In urma legarii trombinei de PAR apar modificari conformationale ale proteinei G care se activeaza si, prin intermediul unor mesageri, exercita efecte:
activarea fosfolipazei C (PLC);
activarea MLCK (miozinkinaza lanturilor usoare);
activarea fosfolipazei A2 (PLA2).
Fosfolipaza C activata are urmatoarele efecte:
a) mobilizeaza Ca din depozitele intraplachetare;
b) actioneaza asupra fosfolipidelor membranare si activeaza cascada fosfatidil-inozitolilor, cu formarea de IP3 (inozitol-trifosfat) si DAG (diacilglicerol) care au rol esential in metabolismul plachetar;
-IP3 este o substanta hidrosolubila (difuzibila in citoplasma)
- IP3 se fixeaza la nivelul granulelor dense (care prezinta receptori pentru IP3) si determina mobilizarea Ca de la acest nivel, cu cresterea concentratiei intracitoplasmatice a Ca;
- IP3 stimuleaza fosforilarea lanturilor usoare de miozina care interactioneaza cu actina favorizand, astfel, deplasarea granulelor si modificarea formei plachetare;
- DAG activeaza proteinkinaza C (PKC) care
- fosforileaza MLCK si stimuleaza filamentele de actina;
activeaza (prin fosforilare) receptorii trombocitari (GPIIb-IIIa) implicati in agregare (segmentul intracitoplasmatic al acestui tip de receptori se afla in contact cu PKC).
MLCK activata fosforileaza miozina implicata in reactia de eliberare plachetara (release reaction). Expulzia granulatiilor plachetare este insotit de exprimarea membranara a receptorului GPIIb-IIIa .
* Fosfolipaza A2 activata desprinde din fosfolipidele membranei trombocitare acidul arahidonic din care, pe calea ciclooxigenazei, rezulta endoperoxizi ciclici intermediari (PGG2 si PGH2), iar pe calea tromboxan-sintetazei se formeaza TxA2. TxA2 exercita urmatoarele efecte:
TxA2 eliberat extracelular stimuleaza miocitele si mentine in timp vasoconstrictia locala;
TxA2 stimuleaza activitatea plachetara (efect autocrin); fixarea TxA2 pe receptori membranari specifici (R-TxA2), aflati in contact cu proteina G, determina activarea suplimentara a acesteia, cu intensificarea reactiei de eliberare trombocitara si cresterea gradului de exprimare a GPIIb-IIIa pe suprafata membranei plachetare.
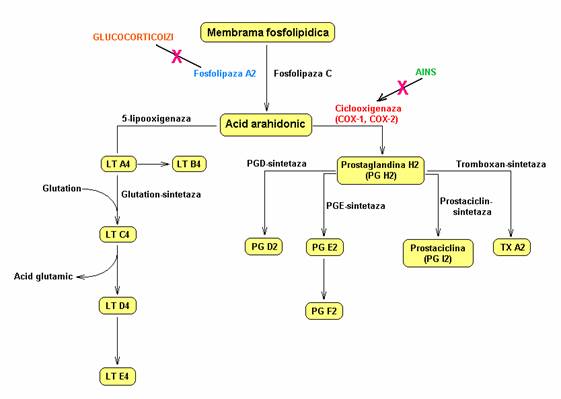
Metabolismul acidului arahidonic
Receptorii specifici agregarii plachetare
GPIIb-IIIa sunt receptori exprimati pe membrana plachetara, aflati in contact cu PKC si formati din doua componente.
* GPIIb, fosforilata in segmentul intracitoplasmatic de PKCsrc(sarcoma cells), sufera modificari conformationale ce se transmit componentei GPIIIa.
* GPIIIa de la nivelul unui pseudopod devine, astfel, apta sa fixeze lantul gamma al unei molecule de fibrinogen circulant. Se formeaza punti de fibrinogen intre pseudopode vecine.
PKCsrc intensifica fosforilarile, cu efecte:
fosforilarea PLC determina activarea suplimentara a cascadei fosfatidilinozitolilor ;
fosforilarea unor proteine membranare induce exprimarea pe suprafata plachetara a unor fosfolipide electronegative (fosfatidilcolina, fosfatidilserina); prin translocarea fosfolipidelor, membrana plachetara devine intens electronegativa si apta de a fixa proteine plasmatice (factori ai coagularii) electropozitive;
fosforilarea segmentului intracitoplasmatic al receptorilor tip PAR induce internalizarea si distrugerea intralizozomala a acestora, cu inhibarea procesului de aderare plachetara.
In cursul derularii fenomenelor hemostazei primare, se constata ca aderarea plachetara stimuleaza procesul de agregare, iar agregarea plachetara induce inhibarea aderarii.
In concluzie, formarea dopului plachetar alb, ca rezultat al hemostazei primare, parcurge 3 etape :
activarea cascadei inozitolfosfatilor si activarea MLCK;
formarea TxA2, cu efecte care persista si dupa epuizarea mecanismelor hemostazei primare;
activarea PKCsrc, cu sistarea procesului de aderare plachetara.
Initial, dopul plachetar alb (format exclusiv din plachete) este insuficient hemostatic, deoarece:
- puntile de fibrinogen realizate intre trombocite sunt friabile;
- intre pseudopodele trombocitare legate prin punti de fibrinogen exista spatii prin care pot trece elemente figurate sanguine cu plasticitate crescuta (eritrocite).
Metamorfoza vascoasa a trombocitelor agregate
Inductorul fenomenului de metamorfoza vascoasa este trombina. Trombina rezulta din activarea CE a coagularii de catre tromboplastina tisulara (eliberata din celulele endoteliale lezate).
Trombina se fixeaza pe receptori trombocitari specifici (GP-V) si exercita urmatoarele efecte:
- transforma fibrinogenul in fibrina, cu aparitia unor punti mai solide intre trombocite;
- creste permeabilitatea membranei trombocitare pentru Na; patrunderea Na in trombocit, insotita de patrunderea apei (efect osmotic), determina fenomenul de hiperhidratare celulara prin care se desfiinteaza spatiile dintre pseudopode si dopul plachetar devine mai hemostatic.
Acest dop plachetar rezultat din hemostaza primara opreste sangerarea doar pe perioada fenomenului de vasoconstrictie. Cand vasoconstrictia inceteaza, dopul plachetar poate fi antrenat odata cu reluarea fluxului sanguin (in lipsa retelei de fibrina, dopul plachetar nu este suficient de solid ancorat la peretele vascular). Ancorarea dopului plachetar este consolidata odata cu aparitia retelei de fibrina rezultata din derularea procesului de coagulare. Colagenul vascular are receptori pentru fibrina.
Fiziopatologia hemostazei primare
- Vasculopatii
- Anomalii plachetare
- calitative (trombopatii)
- cantitative (trombocitopenii)
- Boala von Willebrand
Tulburarea hemostazei primare determina aparitia (uneori spontan) de sindroame hemoragipare la nivel cutanat si al mucoaselor (acestea contin vase de calibru mic in care trombocitele sunt eficiente in oprirea sangerarii de la nivelul leziunii parietale)
- purpura, petesii, echimoze la nivel cutanat;
- epistaxis, gingivoragii, hemoptizii, hematemeze, melene, hematurie, sangerari genitale (menometroragii).
Purpura anafilactica (purpura reumatoida Henoch-Schnlein)
Purpura anafilactica apare mai frecvent la copii sau tineri in contextul unor infectii cu streptococ beta-hemolitic (angina acuta, reumatism articular acut etc.) si se exprima clinic prin:
- febra;
- eruptie cutanata purpurica localizata in special in jumatatea inferioara a corpului;
- dureri articulare;
- dureri abdominale cu caracter colicativ;
- insuficiente de organ (insuficienta renala acuta in forme severe de boala).
Mecanismul patogen al bolii este de tip imun (reactie de hipersensibilitate de tip III) si consta in aparitia de complexe imune circulante (CIC) ce determina leziuni multiple capilare si parenchimatoase.
CIC sunt formate din antigen streptococic si anticorpi de tip IgG, in prezenta unui exces de antigen (suprastimulare antigenica). CIC au dimensiuni mici si pot trece printre jonctiunile interendoteliale de la nivelul peretelui capilar, in anumite zone preferentiale. In aceste zone (tegument, perete intestinal, rinichi, membrane sinoviale etc.), exista conditii hemodinamice particulare: flux sanguin turbulent (vase cu bifurcatii sau curburi), regim de filtrare, presiune hidrostatica mare, toate acestea favorizand extravazarea si depozitarea CIC perivascular. Prezenta CIC in spatiul perivascular determina declansarea reactiei inflamatoare. Inflamatia explica simptomatologia dureroasa si febra la bolnavii cu purpura anafilactica.
CIC activeaza local complementul, cu aparitia anafilatoxinelor (C3a si C5a) care au ca efecte:
- degranularea mastocitelor situate perivascular, cu eliberare de histamina care creste permeabilitatea vasculara, produce vasodilatatie si favorizeaza constituirea edemului
- stimularea chemotactismului, cu atragerea in focar a celulelor proinflamatoare; acestea se activeaza metabolic si elibereaza enzime proteolitice (hidrolaze, proteaze etc.) si radicali liberi de oxigen cu efect distructiv atat asupra endoteliilor, cu aparitia de microfocare hemoragice purpura), cat si asupra celulelor parenchimatoase, cu aparitia insuficientelor de organ.
In cazuri de boala cu evolutie severa, inflamatia importanta la nivel glomerular poate determina scaderea capacitatii de filtrare, cu aparitia insuficientei renale acute. Leziunile endoteliale intinse pot explica aparitia microtrombilor plachetari ce pot realiza obstructii, pana la aparitia de micronecroze (insuficienta de organ).
Scorbutul (avitaminoza C)
Vitamina C intervine in procesul de sinteza a colagenului vascular. Deficitul de vitamina C tulbura acest proces, peretele vascular devine friabil, cu microfisuri ce explica manifestarile hemoragipare.
Purpura mecanica apare, la persoane care au o structura vasculara fragila, dupa ortostatism prelungit. Cresterea presiunii hidrostatice intravasculare (datorita stazei) la nivelul membrelor inferioare suprasolicita structura peretelui vascular.
La varste inaintate, procesele degnerative cu caracter generalizat afecteaza si structura colagenica a peretelui vascular, cu posibilitatea aparitiei sindromului purpuric.
Hipercorticismul si hipertiroidismul altereaza structura proteica normala a peretelui vascular si pot duce la aparitia unui sindrom purpuric.
In contextul unor boli cu patogenie autoimuna (lupus eritematos sistemic, sclerodermie, dermatomiozita etc.) apar in circulatie imunoglobuline G anormale care se comporta ca Ac fata de Ig normale. Complexele Ag-Ac se depun la nivelul peretilor vasculari, precum si perivascular, determinand aparitia angeitei. Fenomenele inflamatoare fragilizeaza vasele si explica aparitia fenomenelor hemoragice.
Sindroamele Ehlers-Danlos sunt boli ereditare, cu transmitere autosomal dominanta, caracterizate prin tulburarea sintezei fibrelor de colagen, din punct de vedere cantitativ sau calitativ. Unele dintre cele 11 tipuri de sindroame Ehlers-Danlos (tipul 4 si tipul 6) evolueaza cu vasculopatie.
Din punct de vedere clinic, aceste sindroame se caracterizeaza prin:
- hipermobilitate articulara datorata unei hiperlaxitati ligamentare;
- hiperelasticitate tegumentara, cu pliuri cutanate de dimensiuni mari;
- manifestari hemoragice de o mare gravitate (exista anomalii structurale ale vaselor de calibru mare).
Tipul 4 se caracterizeaza printr-un deficit major in sinteza colagenului de tip III care intra in structura peretelui arcului aortic si a peretelui intestinal. In aceste conditii, pot aparea:
- anevrisme aortice care se pot rupe si pot determina hemoragii de o gravitate extrema;
- rupturi intestinale, cu hemoragii intestinale si peritonite.
Tipul 6 se caracterizeaza prin absenta congenitala a lizilhidroxilazei (rol in formarea hidroxilizinei din structura receptorilor colagenici pentru FvW). Apare un defect de aderare a trombocitelor la fibrele de colagen, cu posibilitatea manifestarilor hemoragice.
Telangiectazia ereditara hemoragica (boala Rendu-Ősler)
Telangiectazia ereditara hemoragica este o boala ereditara, cu transmitere autosomal dominanta, rezultat al unui defect de angiogeneza a vaselor mici (mai ales venule), cu caracter generalizat, in care vasele mici prezinta leziuni segmentare.
Defectul vascular se caracterizeaza prin absenta din structura peretelui a tunicii musculare si conjunctive (a fibrelor de colagen), la nivelul anumitor zone care devin surse de hemoragii. Peretii vasculari sunt subtiri, formati aproape exclusiv din endoteliu si prezinta ectazii (dilatatii) ce se pot rupe usor, la eforturi mici.
La persoanele care prezinta acest tip de anomalie a structurii peretelui vascular, hemostaza primara este afectata prin:
- defect de aderare trombocitara datorat lipsei colagenului vascular;
- lipsa vasoconstrictiei reflexe (peretii vasculari sunt areactivi) datorata absentei fibrelor musculare netede.
Caracteristici clinice si paraclinice ale trombopatiilor:
- numar de trombocite variabil (normal, scazut sau crescut);
- trombocite alterate din punct de vedere functional
- deficite ale receptorilor trombocitari (deficit de GPIb-IX, GPIIb-IIIa);
- deficite ale cailor de activare metabolica implicate in reactia de eliberare plachetara (deficit de PLA2, ciclooxigenaza, tromboxan-sintetaza); trombocitele nu au capacitate de agregare in prezenta inductorilor;
- deficite ale granulatiilor plachetare; in sindromul placutelor cenusii, absenta granulelor α (contin FvW, GPIIb-IIIa) si a granulelor β (contin Ca, ADP) exista deficit de aderare si agregare plachetara;
- sindroamele hemoragipare caracteristice tulburarilor hemostazei primare pot aparea spontan (trombocitele intervin in procesul fiziologic de mentinere a integritatii peretelui vascular).
Trombopatii ereditare
Distrofia trombocitara hemoragica (Sindromul Bernard-Soulier)
Distrofia trombocitara hemoragica se transmite autosomal recesiv (defect al cromozomului 9, respectiv al genelor care codifica sinteza celor 3 lanturi ale GPIb-IX). Trombocitele sunt anormale din punct de vedere functional (trombopatie).
Din punct de vedere clinic, gravitatea bolii difera:
-in forma homozigota hemoragiile sunt severe;
- in forma heterozigota hemoragiile sunt moderate sau chiar lipsesc.
# Anomalii functionale trombocitare
1. Deficitul de GPIb (receptor trombocitar specific pentru FvW) determina defectul de aderare plachetara (incapacitatea plachetelor de a adera la fibrele de colagen ) care poate fi explicat prin:
- defecte ale domeniului extramembranar al lantului α al GPIb (scaderea continutului de leucina) care explica deficitul de legare cu FvW;
- deficit de sinteza a lantului β al GPIb, cu afectarea formarii complexului receptorial GPIb-IX;
- deficit de glicoziltransferaza, cu afectarea glicozilarii lanturilor GPIb-IX si a exprimarii acestui receptor pe suprafata membranei trombocitare.
2. Deficitul de GPIX (receptor trombocitar specific pentru FXI) determina tulburarea procesului de coagulare pe calea intrinseca (deficitul tromboplastinoformarii).
# Anomalii morfologice trombocitare
Trombocitele au dimensiuni mari si au un numar crescut de granulatii (dense si clare), bine dezvoltate si situate central in celula (aspect "limfocitoid").
Trombastenia Glanzman este o trombopatie cu transmitere autosomal recesiva (defect al cromozomului 17, respectiv al genelor care codifica sinteza lanturilor componente ale GPIIb-IIIa), in care exista anomalii functionale trombocitare ce explica deficitul agregarii plachetare
# Anomalii functionale trombocitare
1. Deficitul de sinteza a GPIIb-IIIa (receptor plachetar pentru fibrinogen) induce deficitul de agregare plachetara, cu lipsa formarii dopului plachetar alb.
2. Deficite enzimatice:
- deficit de PTKsrc, cu scaderea fosforilarilor diverselor substrate plachetare;
- deficitul enzimelor implicate in glicoliza trombocitara (gliceraldehid-3-fosfatdehidrogenaza, piruvatkinaza) explica scaderea productiei de ATP, cu tulburarea proceselor energodependente (reactia de eliberare trombocitara, emiterea de pseudopode, retractia cheagului).
3. Deficitul de actomiozina plachetara (trombostenina sau factor plachetar 7) determina afectarea reactiei de eliberare, cu scaderea expresiei membranare a GPIIb-IIIa (este accentuat deficitul de GPIIb-IIIa).
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 4055
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved