| CATEGORII DOCUMENTE |
| Astronomie | Biofizica | Biologie | Botanica | Carti | Chimie | Copii |
| Educatie civica | Fabule ghicitori | Fizica | Gramatica | Joc | Literatura romana | Logica |
| Matematica | Poezii | Psihologie psihiatrie | Sociologie |
IONII IN SOLUTIILE DE ELECTROLIT
1. Originea ionilor in solutie
Ionii pot fi produsi in solutiile de elctrolit pe cale fizica sau pe cale chimica.
Pe cale fizica pot fi produsi ioni din saruri ionice intrinsece (cristale ionice NaCl, KCl, etc) prin: a) topire cand se obtin topituri ionice (lichide ionice pure); b) dizolvare in solventi cand se obtin solutii de electrolit. Acesti electroliti se numesc dupa Conway si Fuoss electroliti adevarati sau ionofori (purtatori de ioni).
Pe cale chimica ionii se produc in urma unei reactii de solvoliza, iar gradul de ionizare depinde de termodinaimca reactiei formatoare de ioni.
Pentru substantele neionice ce reactioneaza cu solventul formand electroliti s-a propus denumirea de electroliti potentiali sau ionogeni (generatori de ioni).
Cateva din grupele de reactii formatoare de ioni in solutie, sunt prezentate mai jos:
a) Reactii de disociere a acizilor HA in solventi acceptori de protoni S (Brnsted)
in care S este solventul (apa, cetona, alcool, etc), iar Ka este constanta de disociere.
b) Reactii de protoliza
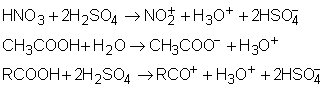
c) In solventii protici apar reactii de autoprotoliza de tipul:
![]()
dintre care mentionam:
![]()
d) Reactiile interfaciale pot fi producatoare de ioni:
![]() reactie
anodica
reactie
anodica
e) Ionii mai pot proveni si din diferite procese heterogene, ca de exemplu:
![]() produs
Grignard
produs
Grignard
![]()
Subliniem la acest paragraf privitor la originea ionilor in solutie, distinctia ce se face intre electroliti "adevarati" (intrinseci) si electroliti "potentiali". Aceasta impartire este preferabila celei clasice de electroliti "tari" si "slabi", care se refera la gradul de disociere, deoarece gradul de disociere depinde pentru un electrolit dat de solventul in care acesta se dizolva.
Teoria clasica a disocierii electrolitice
In teoriile vechi asupra solutiilor de electrolit nu a fost recunoscuta existenta independenta a ionilor in solutie, cu toate ca inca incepand cu Grotthus in 1806 au aparut unele idei despre existenta unor specii incarcate in solutie. Chiar Faraday care mentiona termenul de ion in corespondenta sa cu Whewel (inclusiv termenii de de anion si cation) nu folosea conceptul de ion liber in teoria sa asupra electrolizei si conductiei solutiilor de electrolit.
Abia mai tarziu, in 1857 Clausius afirma pentru prima data faptul ca particulele incarcate conducatoare de electricitate sunt produse direct ca rezultat al dizolvarii unui electrolit in apa.
Aceasta conceptie a fost dezvoltata de catre Arrhenius in 1883 si 1887, iar prin lucrarile lui Ostwald si Kohlrausch (1888) teoria clasica a solutiilor de electroliti capata forma definitiva care se pastreaza si astazi.
In continuare se descrie succint bazele teoriei clasice ale disociatiei solutiilor electrolit.
Teoria lui Arrhenius (1887). Lucrarile efectuate in acea vreme au aratat ca solutiile bune conducatoare de lectricitate fac exceptie de la legile solutiilor diluate. Astfel, presiunea osmotica, scaderea tensiunii de vapori, cresterea punctului de fierbere, scaderea punctului de congelare, au valori de 2 - 3 ori mai mari decat cele normale. De exemplu, o solutie de NaCl (electrolit binar) produce un efect osmotic dublu fata de cel masurat la o solutie de neelectrolit (de exemplu zahar). Aceasta comportare a solutiilor de electrolit fata de cea a solutiilor de neelectrolit are o cauza comuna care consta in aceea ca ionii independenti proveniti din disocierea electrolitului (de exemplu din punctul de vedere al presiunii osmotice), se comporta ca moleculele de neelectrolit.
Van'T Hoff a reprezentat aceste efecte anormale prin factorul "i" introdus in ecuatia pentru presiunea osmotica:
p = i CRT (1)
in care C = concentratia speciei active in solutie; iar R si T au semnificatia cunoscuta (constanta gazelor si respectiv temperatura absoluta.
Van'T Hoff a definit si a interpretat acest factor prin relatia:
![]()
Pentru a explica aceasta comportare privitoare la presiunea osmotica si la celelalte marimi enumerate mai sus, Arrhenius a sugerat ideea ca solutiile bune conducatoare de electricitate sunt in mare masura disociate in ioni (anioni si cationi). Astfel, in conceptia lui Arrhenius factorul i este privit ca o masura a cresterii numarului total de particule in solutie ca urmare a disocierii. In acest fel i devine :
Tot in legatura cu disocierea solutiilor de electrolit Arrhenius a definit gradul de disociere a ca reprezentand fractiunea de molecule care se disociaza in ioni, din numarul total de molecule de electrolit prezente in solutie. Este evident ca a este un numar cuprins intre 0 si 1.
Pornind de la definitia gradului de disociere a se poate calcula concentratia moleculelor nedisociate si a speciilor disociate. Astfel, daca C este concentratia electrolitului, numarul total de molecule de electrolit nedisociat este: C(1 - a), iar concentratia speciilor disociate este data de relatia nCa in care n reprezinta numarul ionilor in care se disociaza molecula de electrolit. Tinand seama de aceste valori ale concentratiei electrolitului nedisociat si a electrolitului disociat se poate exprima intr-o forma cantitativa factorul van'T Hoff (i):
![]()
care prin rearanjare devine:
i = 1 + a n
Introducand valoarea lui i in ecuatia (1) se obtine:
p a n - 1)]CRT (4)
Ecuatia (4) este expresia aplicarii teoriei disociatiei electrolitice a electrolitilor pentru explicarea abaterii acestor solutii de la legile solutiilor diluate.
Arrhenius a aratat ca gradul de disociere a creste odata cu diluarea solutiei. Aceasta crestere este prezentata pentru unii electroliti in tabelul 1.
La acizii polibazici si la bazele poliacide, precum si la sarurile metalelor polivalente disocierea se face in trepte dupa cum urmeaza:
![]() I
I
![]() II
II
Variatia gradului de disociere cu concentratia electrolitului in apa
|
molaritate |
|||
|
electrolit | |||
|
a |
a |
a |
|
|
HCl | |||
|
CH3COOH | |||
|
NaOH | |||
Prima treapta de ionizare reprezinta ionizarea primara, treapta a doua ionizarea secundara, treapta a treia tertiara s.a.m.d. Ionizarea primara este mult mai importanta decat cea secundara sau tertiara. Deoarece disocierea este o reactie exoterma gradul de disociere va scadea odata cu cresterea temperaturii. Astfel in cazul HCl: a = 92 la 18 C si a = 89,7 la 100 C. Apa face exceptie de la aceasta regula gradul ei de disociere crescand cu temperatura.
Tinand seama de a in teoria clasica a disocierii, electrolitii se impart in urmatoarele categorii:
a. Electrolitii tari. Sunt electrolitii care se dizolva in solventi in proportie mai mare de 50% pentru care valoarea a > 0,5. Pentru acesti electroliti, a nu variaza mult cu concentratia (vezi si tabelul 1) valoarea sa ramanand totdeauna apropiata de 1.
b. Electroliti slabi. Sunt electrolitii care se dizolva greu in solventi (sub 1%) avand valoarea lui a £ 0,01. In cazul acestor electroliti a variaza mult cu concentratia, ea tinzand spre zero la concentratii mari.
In legatura cu nomenclatura electrolitilor mentionam ca electrolitii care disociaza in doi ioni (KCl, SO4Mg etc.) se numesc electroliti binari. Cei care disociaza in trei ioni se numesc ternari (BaCl2), iar cei care formeaza 4 ioni se numesc cuaternari Al(Cl)3.
Electrolitii binari pot fi uniunivalenti (KCl) si bibivalenti (CuSO4), electrolitii ternari pot fi unibivalenti (K2SO4), iar cei cuaternari terunivalenti (FeCl3).
3. Constanta de disociere. Legea dilutiei a lui Oswald
Conform teoriei lui Arrhenius in solutia unui electrolit, in afara de ionii rezultati din disocierea moleculelor mai exista si molecule ramase nedisociate, deoarece disocierea completa are loc numai la dilutie infinita (unde a
Deci se poate considera ca disocierea este o reactie reversibila, o parte din molecule disociaza, iar o parte din ioni se recombina, stabilindu-se urmatorul echilibru de reactie:
Daca se aplica acestui proces de echilibru legea maselor se obtine relatia cunoscuta:
![]()
unde K este constanta de echilibru, iar in acest caz particular poarta denumirea de constanta de disociere sau constanta de ionizare. Constanta de disociere este independenta de concentratie.
Intr-o solutie, ce contine un mol de substanta AB al carei grad de disociere este a dizolvata in V litri de apa, concentratiile diferitelor specii componente vor fi:
a) moli de AB nedisociat
a ioni gram de A+ si de B-
Tinand seama de aceste relatii intre a a), [A+] si [AB], concentratiile speciilor A+, B- si AB vor putea fi exprimate de relatiile:
![]()
![]()
![]()
inlocuind aceste valori in ecuatia 6 se obtine:
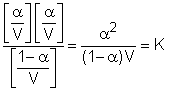
Deoarece concentratia este inversul dilutiei adica C = 1/V relatia (7) se poate scrie:
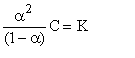
Aceasta expresie poarta denumirea de legea dilutiei a lui Oswald sau legea disociatiei electrolitice. Ea permite calcularea constantei de disociere K cu ajutorul marimii a. Aceasta marime la randul ei, poate fi obtinuta experimental prin masuratori de conductanta electrica.
In cazul electrolitilor slabi a 0 si deci expresia (1 - a In aceasta situatie legea dilutiei devine:
![]() sau
sau ![]()
sau atunci cand V =
1, a ![]()
Legea dilutiei a lui Oswald nu se aplica decat electrolitilor slabi. Ea este astfel considerata o lege limita deoarece pentru electrolitii tari se poate aplica numai in cazul unor dilutii foarte mari (pentru concentratii mai mici decat 10-3 mol/1).
Pentru electrolitii tari in locul legii dilutiei a lui Oswald, Kraus a propus o relatie care are forma:
![]()
in care m = n - 2, iar n = constanta ce variaza putin de la valoarea 1,5 in functie natura electrolitului.
4. Solvatarea ionilor
Fenomenul de solvatare se datoreaza faptului ca ionul care apare printre moleculele de solvent, schimba insusirile si ordinea distribuirii acestora in solutie. Daca moleculele de solvent au moment de dipol, atunci ele interactioneaza cu ionii formand invelisuri de solvatare. Totodata interactiunea electrostatica nu este singura cauza a solvatarii ionilor. Solvatarea poate sa apara si datorita fortelor chimice. Multe saruri formeaza hidrati nu numai in solutii, dar si in stare solida. De altfel este cunoscut ca spre o astfel de structura complexa tind aproape toate sarurile. De exemplu, formarea hidratilor sarurilor de cupru este un proces tip de formare a unei structuri complexe. In astfel de combinatii, legatura dintre ioni si moleculele de apa este pur chimica, fiind conditionata de valenta de coordinatie respectiva. Mobilitatea ionilor depinde de vascozitatea mediului si raza lor si se determina pe baza legii lui Stockes.In legatura cu solvatarea ionilor este interesant de comparat marimea mobilitatii si marimea razelor lor cristalografice.
Conform ecuatiei mobilitatii este de asteptat ca razei maxime sa-i corespunda o mobilitate minima.
In realitate, intre razele ionice cristalografice si razele obtinute din mobilitatile ionilor dupa ecuatia Stokes, nu este nici o corespondenta. Aceasta nepotrivire este rezultatul faptului ca in solutie se afla ioni solvatati care constau din ionul propriu zis si invelisul lui de solvatare format din molecule de solvent, care se misca impreuna cu el.
Compararea razelor Stokes ale ionilor solvatati ai metalelor alcaline arata ca raza ionica cea mai mare o au in solutie ionii de litiu si cea mai mica ionii de cesiu. Invers, raza cristalografica cea mai mica este a litiului si cea mai mare a cesiului.
Totodata mentionam ca razele ionice cristalografice se deosebesc in masura mult mai mare decat mobilitatea ionilor. Astfel de raporturi se pot explica prin aceea ca ionilor cu raze mici le corespund la una si aceeasi sarcina o mare intensitate a campului electric din jurul lor. Ca rezultat, ionii cu raza mica au o mare putere de atractie a moleculei solventului care, astfel, se ataseaza ionilor formand un invelis puternic legat de acesta, ceea ce duce la o mobilitate mai mica a ionilor mici in comparatie cu cei mari. Corespunzator, ionii cu raze mai mici se caracterizeaza printr-o energie de solvatare mai mare.
Pentru a explica marea discrepanta intre datele furnizate de diferitele metode de masurare a hidratarii, Bockris a definit doua tipuri de hidratare pe care el le-a denumit "primara" si "secundara". Hidratarea primara se refera la moleculele de apa imobilizate de ion cu care acesta se misca formand o entitate distincta pe cand hidratarea secundara se refera la interactiile dintre ion si solventul asezat dincolo de stratul primar de hidratare. Samoilov foloseste pentru hidratarea primara denumirea de hidratare apropiata, iar pentru hidratarea secundara denumirea de hidratare indepartata. In sfarsit J.E.Desnoyers si C. Jolicoeur pentru aceleasi notiuni folosesc denumirea de hidratare hidrofilica si respectiv hidratare de polarizare.
4.1. Numarul de solvatare
Au fost efectuate numeroase incercari de a stabili conditiile critice pentru care o molecula de apa poate fi considerata ca facand parte din invelisul de solvatare, numarul total de molecule de apa (solvent) cuprins in invelisul de hidratare denumindu-se numar de hidratare (solvatare). Exemple ale unor criterii propuse in acest scop sunt: energia cinetica, pierderea proprietatilor de translatie si compresibilitate a moleculelor de apa. Practic insa, diferitele metode experimentale de estimare a gradului de hidratare (determinarea numarului de hidratare) masoara diferitele proprietati ale moleculei de apa si deoarece ionul cu moleculele de apa din invelisul de hidratare nu formeaza o entitate stabila nu se poate astepta de la aceste metode sa furnizeze date similare. In continuare se vor descrie cateva metode experimentale de determinare a numarului de hidratare.
Metoda Hittorf
Unul din procedeele clasice ale determinarii numarului moleculelor de solvent care se leaga cu ionul, este calculul pe baza determinarii numerelor de transport prin metoda Hittorf.
Acest procedeu de determinare al numerelor de hidratare este insuficient de exact deoarece, odata cu marirea concentratiei, numerele de transport se schimba cu variatia cantitatii apei transportata de ioni, dar si cu schimbarea mobilitatii, ca rezultat al interactiunii dintre ioni. In afara de aceasta, pe aceasta cale se poate determina numai diferenta in numerele de hidratare a ionilor.
Metodele bazate pe legea lui Stoks si pe viteza de difuzie
Una din metodele determinarii numarului de hidratare pentru ionii singulari este bazata pe determinarea razei ionului direct din ecuatia mobilitatii, adica plecand de la ecuatia lui Stokes:
![]()
in care: r = raza ionului; h = vascozitatea; u = mobilitatea ionului; z = valenta; e0 = sarcina electronului.
Dupa
aceasta ecuatie din mobilitatea si vascozitatea mediului, se
determina marimea r, iar pe baza lui r - volumul ionului ![]() . Volumul
ionului solvatat se compara cu volumul ionului nesolvatat bazandu-se pe
raza cristalografica a ionului
. Volumul
ionului solvatat se compara cu volumul ionului nesolvatat bazandu-se pe
raza cristalografica a ionului ![]() . Diferenta intre
aceste volume este volumul invelisului de solvatare:
. Diferenta intre
aceste volume este volumul invelisului de solvatare:
![]()
Daca acest volum se imparte la volumul moleculei de solvent, aflam numarul de solvatare al ionului nh, adica:
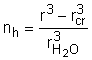
Pe baza unor astfel de date s-a aratat ca invelisul apos din jurul ionului consta din cateva straturi de molecule de apa.
Apropiata de metoda prezentata mai sus este si metoda difuziei. Dupa aceasta metoda, plecand de la vitezele de difuzie, se determina constanta de difuzie D:
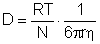
Pornind de la coeficientul de difuzie se determina raza r si volumul ionului solvatat v. Scotand din volumul obtinut v, volumul propriu al ionului, se afla volumul invelisului de solvatare si se determina numarul de solvatare.
Metoda compresibilitatii
Una din metodele moderne pentru determinarea numarului de solvatare este metoda ultrasonica care se bazeaza pe faptul ca viteza undelor ultrasonice depinde de compresibilitatea solutiei permitand astfel determinarea coeficientilor de compresibilitate b
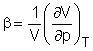
Numarul de hidratare se obtine din valorile coeficientilor de compresibilitate al solventului pur (apa) si al solutiei de electrolit obtinut din masuratorilor ultrasonice, folosind relatia:
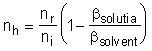
in care na = numarul de moli de apa; ni = numarul de moli de ioni.
Explicarea acestui mod de determinare a numarului de hidratare consta in aceea ca, datorita efectului de electrostrictiune, moleculele de solvent din stratul primar de solvatare au un inalt grad de comprimare, deci solutiile de ioni au un grad de comprimare mai mare decat solventii puri, astfel incat coeficientul de compresibilitate al solutiei, b va fi mai mic decat coeficientul de compresibilitate al solventului b
Adaugam ca electrostrictiunea se datoreste comprimarii solventului in jurul ionului sub influenta intensitatii mari a campului electric din jurul ionului solvatat.
Toate metodele de determinare a numarului primar de solvatare au un anumit grad de incertitudine care poate merge pana la 25%. Valorile obtinute pentru diferiti ioni monovalenti si pentru diferite metode sunt prezentate in tabelul
|
Ionul |
Metoda compresibilitatii |
Metoda mobilitatii |
Metoda teoretica |
Valoarea integrala |
|
Li+ | ||||
|
Na+ | ||||
|
K+ | ||||
|
F- | ||||
|
Cl- | ||||
|
Br- |
|
|||
|
I- |
In concluzie se poate aprecia ca in cursul anilor s-au depus eforturi pentru a decide daca un anumit numar de molecule de apa sau solvent (numar de solvatare sau hidratare) este asociat cu un anumit ion dand nastere unei specii moleculare tipice (ionul solvatat sau hidratat).
Examinarea datelor experimentale mai vechi privitoare la numerele de solvatare obtinute prin diverse metode (mobilitatea ionilor, compresibilitatii, constantei dielectrice, densitati, studii de dializa) conduce la aprecierea ca prin unele metode se obtin valori exagerate, de exemplu s-a obtinut pentru Na+ un numar de hidratare egal cu 700. Aceasta este o dovada ca unele metode masoara efecte care implica mai multe strate de solvent din vecinatatea ionului si deasemeni este o dovada ca metodele detecteaza influenta ionului la distante diferite.
Deasemenea o examinare a datelor furnizate de aceste metode arata o mare variabilitate in rezultate depinzand de metoda folosita.
De la aceasta variatie a aparut conceptul de sfera de coordinare interna (strat de solvatare primara) si externa (strat de solvatare secundara) in functie de distanta de la ion.
Studii mai
noi efectuate cu ajutorul razelor X si cu ajutorul izotopilor radioactivi, au
aratat ca numarule de hidratare pentru Cr+3
este 6, deci ionul hidratat de Cr+3 este de forma ![]()
Studii de
rezonanta magnetica nucleara folosind O17 in H2O
(H2O17) s-au indreptat mai ales asupra vitezei de schimb
a apei intre ionul hidratat si solvent. Aceste lucrari au aratat
ca viata medie pentru speciile hidratate sunt: Fe3+ cca 10-4
sec; Mn++ cca 10-7 sec; Co++ cca 10-6
sec; Ni++ cca 10-4 sec pentru apa ecuatoriala si
cca 10-8 sec pentru apa axiala. Aceste vieti medii se
aplica speciilor octaedrice ![]()
4. Determinarea experimentala a energiei de hidratare si solvatare
Dupa determinarea numarului de solvatare urmeaza sa se stabileasca schimbarile energetice care au loc in timpul interactiei dintre ion si moleculele de solvent, aceasta fiind una din problemele centrale ale intregii teorii a solutiilor de electrolit. Pentru a se produce dizolvarea sarii, trebuie sa se invinga interactiunea dintre ioni, adica sa se depaseasca valoarea energiei retelei cristaline. Energia care se degaja la dizolvarea sarii (Udis) este egala cu diferenta dintre totalul energiei de solvatare (hidratare) a ionilor (Uhidr) si energia retelei cristaline Ucr.
![]()
Daca diferenta dintre energia de hidratare si energia retelei cristaline este pozitiva, atunci se va produce dizolvarea sarii, iar daca este negativa, dizolvarea nu se produce.
Deoarece diferenta dintre entalpie si potentialul izobar pentru sistemele condensate nu este mare atunci caldura de hidratare se poate determina experimental din diferenta dintre caldura de retea si caldura de dizolvare:
![]()
Energia retelei cristaline
Energia retelei cristaline Ucr poate fi calculata aproximativ din reprezentarile electrostatice. In cazul simplu al unei molecule biatomice energia de atractie dintre ionii zi si zj se calculeaza conform legii lui Columb
![]()
Aceasta energie potentiala coulombiana U este egala cu lucrul efectuat pentru a aduce un ion de la infinit la distanta r de celalalt ion. Astfel zi si zj fiind de semn contrar lucrul este negativ adica exoterm si in consecinta energia potentiala a sistemului descreste. Daca r este suficient de mic incep sa fie efective si fortele de repulsie ceea ce inseamna ca, daca dorim sa apropiem in continuare ionii, va fi necesar sa se dea un lucru sistemului. Energia repulsiva creste foarte rapid deoarece fortele repulsive au un domeniu efectiv mult mai scurt decat fortele de atractie. In acord cu Born energia de repulsie este data de relatia:
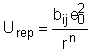
unde bij este o constanta specifica substantei, iar n = 9,
Astfel, energia potentiala U a 2 ioni in functie de distanta r, este data de relatia:
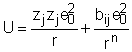
Suprapunand potentialele de atractie si de repulsie se obtine curba continua din fig.1.
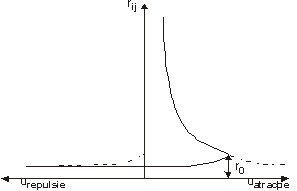
Fig. 1
Distanta de echilibru:
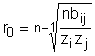
reprezinta separatia interionica intr-o molecula stabila.
Diferenta intre energiile potentiale la separatie r0 si r¥ este egala cu lucrul efectuat de sistem cand ionii se apropie unul de altul de la o distanta infinita pentru a forma molecula. Evident aceasta va fi egala cu energia de disociere Udis a moleculei. Eliminand pe bij din ecuatiile (21), (22) se va obtine urmatoarea relatie pentru energia de disociere:
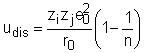
Ecuatiile obtinute pana acum se refera la molecule ionice individuale in stare de vapori. Pentru a extinde validitatea lor la cristalele ionice consideram energia de retea Ucr (care inlocuieste pe Udis) si care se defineste ca energia consumata in construirea retelei din ionii liberi. Deoarece fortele implicate in acest proces sunt culombiene in marea majoritate, putem porni de la o ecuatie similara cu ecuatia 21. Ea trebuie sa fie imbunatatita numai cu un factor care sa tie seama de tipul retelei cristaline si care poate fi calculat:
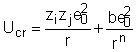
Distanta de echilibru r0 este obtinuta din ecuatia:
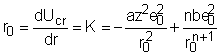
iar coeficientul de repulsie b va fi dat de expresia:
![]()
in care a este constanta lui Madelung care se poate calcula pentru fiecare retea.
Substituind valoarea ec.(24) se obtine energia potentiala a unei perechi de ioni din retea in conditii de echilibru:
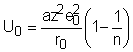
Pentru a obtine energia totala de retea pe mol de sare trebuie sa se considere si numarul de coordinare. Molecula contine 2 N ioni dar nu se iau in consideratie decat N pentru a nu se lua fiecare legatura de 2 ori. Astfel energia de retea per mol devine:
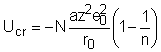
Energia de retea Ucr se defineste drept caldura molara de formare (DHretea) a cristalului din ioni liberi gazosi la T = 0.
In prezent energiile retelelor cristaline sunt bine studiate si determinate cu exactitate suficienta pentru unele substante.
In tabelul 3 prezentam valori ale energiilor de retea pentru cateva substante.
Energiile de retea D Hretea, caldurile de dizolvare D Hdis si
caldurile de hidratare D Hhidr in Kcal/mol la 250C
|
Sarea |
DH0retea |
DH0dizolv |
DH0hidr |
|
LiF | |||
|
NaF | |||
|
KF | |||
|
NaCl | |||
|
KCl | |||
|
NaBr | |||
|
KBr | |||
|
KI |
Caldurile de dizolvare si solvatare
Din tabelul 3 se vede ca energiile de retea au o valoare mare chiar pentru saruri ce contin ioni monovalenti. Este deci de asteptat sa fie necesare deasemenea valori mari de energie pentru a elibera ionii din reteaua cristalina. In consecinta, dizolvarea usoara (energia libera de dizolvare mica DG0dis) a multor substante ionofore sau ionogene in solventi potriviti (proces care in esential consta din ruperea legaturilor dintre componentii substantei respective si crearea ionilor liberi) poate fi explicata numai daca se presupune ca in timpul dizolvarii cristalului au loc anumite procese care produc o cantitate de energie de un ordin de marime similar cu acela al energiei de retea. Acest lucru se poate observa din ciclurile Haber-Born pentru
a) electroliti ionofori
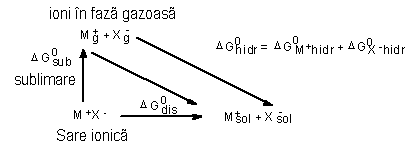 |
b) electroliti ionogeni
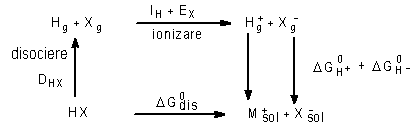 |
In aceste cicluri termenii au urmatoarea semnificatie:
![]() =
energie libera de sublimare
=
energie libera de sublimare
![]() = energie
libera de hidratare (solvatare)
= energie
libera de hidratare (solvatare)
![]() = energie
libera de dizolvare
= energie
libera de dizolvare
DHX = energie de disociere
IH = energie de ionizare
EX = afinitate electronica
Din ciclul Haber-Born se observa ca factorul major care determina solubilitatea generala a unui electrolit este DG0hidr.
4.3. Evaluarea marimilor termodinamice de hidratare pentru ioni individuali
De un mare interes stiintific si practic sunt marimile termodinamice ale ionilor individuali. Obtinerea lor pe cale experimentala este foarte dificila deoarece nu se cunosc metode experimentale pentru determinarea marimilor termodinamice ale ionilor ci numai pentru marimile termodinamice ale electrolitilor ca atare, care sunt ansambluri de ioni pozitivi si negativi. Din acest motiv, conventional, s-a stabilit ca functiile termodinamice ale ionului H+ in stare standard sa fie considerate egale cu zero.
Pe de alta parte, aproape toate teoriile solvatarii se bazeaza pe calcule electrostatice referitoare la ioni singulari. Astfel se iveste necesitatea ca marimile calculate teoretic prin metodele electrostatice pentru ioni, sa fie in mod necesar comparate cu marimile termodinamice absolute ale ionilor si nu cu cele conventionale ale electrolitilor.
Cele mai multe metode de acest fel au in vedere impartirea la doi a functiilor termodinamice pentru perechi selectionate de anion-cation folosind anumite proceduri de extrapolare care implica raza ionului.
Metoda a
fost utilizata prima oara de catre Bernal si Fowler care
au presupus ca deoarece razele K+ si F sunt
practic egale in scara Pauling este justificat sa se divizeze energia
totala de hidratare a KF aproape in parti egale pentru fiecare
ion. Aceasta procedura a condus la obtinerea valorii: ![]() = -94
Kcal/mol pentru K+ si -97 Kcal/mol pentru F
= -94
Kcal/mol pentru K+ si -97 Kcal/mol pentru F
Deoarece apa nu este un solvent fara dimensiuni si fara structura, anionii si cationii de marime egala nu pot fi in mod necesar aceiasi. Din acest motiv unii autori au adaugat diferite incremente la raza (cristalina) a anionului sau a cationului, considerand orientarea diferita a apei in jurul lor.
Astfel pentru determinarea functiilor termodinamice ale ionilor singulari devine critica mai ales alegerea unei scari corecte ale razelor ionice.
In afara acestei incertitudini cu privire la marirea ionului pot aparea complicatii suplimentare datorita marimii finite a moleculei de apa.
In adevar, in solutiile apoase marimea moleculelor de solvent este comparabila cu cea a ionilor, astfel ca exista un spatiu liber suficient de mare intre ion si solvent asa cum se observa in figura
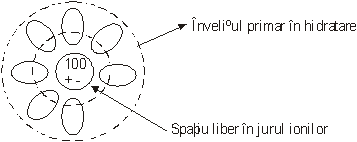 |
Fig.2
Ca o consecinta a acestui spatiu liber apar dificultati la efectuarea calculelor pe baza unei simetrii sferice pana la suprafata ionului. Din cele prezentate mai sus reiese ca sunt necesare metode de calcul pentru functiile termodinamice ale ionilor singulari care sa fie independente de valoarea razelor ionice.
In acest sens, se pare ca singura tehnica dezvoltata pentru evaluarea marimilor termodinamice absolute ale ionilor singulari fara implicarea razelor ionice este masurarea potentialului Volta intre o solutie de electrolit si o alta suprafata de solutie prin vid sau prin gaz inert.
Randles a aplicat metoda masurarii potentialului Volta* pentru determinarea energiei absolute de hidratare pentru ionul K+.
Metoda experimentala a lui Randles consta in principal dintr-un tub de sticla prin centrul caruia curge un mic jet de mercur, iar solutia de KCl curge in lungul suprafetei interioare a tubului de sticla. Solutia de KCl si jetul de Hg sunt izolati unul de altul de un strat de azot.
Daca se ia in considerare celula:
Hg jet/N2/KCl[a = 1]/Hg2Cl2/Hg
Tensiunea electromotoare a acestei celule da diferenta de potential Volta a celor doua lichide (FDY
Considerand si celula:
K/Hg/Hg2Cl2/KCl[a
= 1]/K ![]() )
)
impreuna cu
limita fotoelectrica a mercurului (W) si energiile de sublimare si
ionizare a potasiului solid![]() , se obtine energie de
solvatare a ionului K+ folosind urmatoarea secventa de
ecuatii:
, se obtine energie de
solvatare a ionului K+ folosind urmatoarea secventa de
ecuatii:
1)
![]() FDy
FDy
2)
![]() W
= hn = 4,51 l.v
W
= hn = 4,51 l.v
3)
![]()
![]()
4)
![]()
![]()
![]()
![]()
Folosind
aceasta metoda Randles a gasit valoarea de -80.6 0.5 Kcal/ion g
pentru ![]() . Cu
aceasta valoare absoluta a lui
. Cu
aceasta valoare absoluta a lui ![]() gasita
de Randles se poate elabora o scara a marimilor termodinamice
absolute asa cum se observa in tabelul 4.
gasita
de Randles se poate elabora o scara a marimilor termodinamice
absolute asa cum se observa in tabelul 4.
Valorile absolute ale functiilor termodinamice ale hidratarii la 250C.
|
Ion |
|
S0 gbs/ion gr |
|
|
|
H+ | ||||
|
Li+ | ||||
|
Na+ | ||||
|
K+ | ||||
|
Rb+ | ||||
|
Cs+ | ||||
|
F | ||||
|
Cl | ||||
|
Br | ||||
|
I | ||||
|
OH |
Este
interesant de notat ca valoarea absoluta a lui ![]() obtinuta
de Blandamer si Symons este de 260.7 adica foarte apropiata de valoarea
lui
obtinuta
de Blandamer si Symons este de 260.7 adica foarte apropiata de valoarea
lui ![]() din tabelul 4. Ei
au obtinut aceasta valoare a lui
din tabelul 4. Ei
au obtinut aceasta valoare a lui ![]() pornind de la
separarea valorii energiei de hidratare a RbCl in doua parti
egale deoarece ionii Rb+ si Cl au in scara
Gourary-Adrian raze egale. Astfel se poate conchide ca daca
metoda Randles pentru obtinerea energiilor libere de hidratare a ionilor
singulari este corecta, iar metoda experimentala a lui Gourary si
Adrian pentru determinarea razelor ionice in solutie este cea mai
corecta si deasemeni ea arata ca orientarea moleculelor de
apa nu este semnificativa pentru determinarea energiei de
hidratare.
pornind de la
separarea valorii energiei de hidratare a RbCl in doua parti
egale deoarece ionii Rb+ si Cl au in scara
Gourary-Adrian raze egale. Astfel se poate conchide ca daca
metoda Randles pentru obtinerea energiilor libere de hidratare a ionilor
singulari este corecta, iar metoda experimentala a lui Gourary si
Adrian pentru determinarea razelor ionice in solutie este cea mai
corecta si deasemeni ea arata ca orientarea moleculelor de
apa nu este semnificativa pentru determinarea energiei de
hidratare.
5. Interactia dintre ioni - apa si neelectroliti
Hidratarea
este un proces care poate influenta puternic dizolvarea neelectrolitilor in apa,
deoarece o parte din cantitatea de apa, folosita la dizolvarea
neelectrolitului va fi folosita in procesul de solvatare a ionilor
atunci cand in solutia de neelectroliti se introduce si un electrolit. In
adevar daca un litru de apa contine: ![]() moli de
apa, atunci cand se dizolva un electrolit (de exemplu NaCl in
concentratie de 10-2 mol) un numar de molecule de apa
vor fi imobilizate in invelisul de hidratare a ionului.
moli de
apa, atunci cand se dizolva un electrolit (de exemplu NaCl in
concentratie de 10-2 mol) un numar de molecule de apa
vor fi imobilizate in invelisul de hidratare a ionului.
Astfel daca ne referim tot la solutia de NaCl 0.02 molar tinand seama ca numarul de hidratare a acestei sari este 7, inseamna ca un numar de 0,07 moli/l apa vor fi folositi la hidratare si vor fi deci scosi din cei 55.5 mol/l de apa. Cu alte cuvinte apa libera neimplicata in solvatare va fi in concentratie de 55.45 moli. In continuare daca se considera o solutie 1N de NaCl se vor imobiliza prin solvatare 7 moli l apa, iar apa libera va fi de 48.5 mol/l, iar pentru o solutie de 5N NaCl este evident ca aproape jumatate din apa va fi blocata in invelisul de solvatare.
Din cele aratate mai sus reiese clar ca atunci cand intr-o solutie apoasa de neelectrolit se introduce un electrolit, din cauza solvatarii ionilor electrolitului, o parte de neelectrolit va precipita. Acest proces are o larga utilizare pentru separarea neelectrolitilor din solutii apoase in special in domeniul chimiei organice.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3985
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved