| CATEGORII DOCUMENTE |
| Astronomie | Biofizica | Biologie | Botanica | Carti | Chimie | Copii |
| Educatie civica | Fabule ghicitori | Fizica | Gramatica | Joc | Literatura romana | Logica |
| Matematica | Poezii | Psihologie psihiatrie | Sociologie |
METODA DE IDENTIFICARE SI DETERMINARE CANTITATIVA PENTRU a MONO GLICERIL 4-AMINOBENZOAT
1.SCOP SI DOMENIU DE APLICARE
Aceasta metoda reglementeaza identificarea alfa - mono (gliceril 4-aminobenzoatului si identificarea etil 4-aminobenzoatul (benzocaina : INN) ce poate fi prezent ca impuritate.
2.PRINCIPIU
Aceasta identificare este facuta prin cromotografie in strat subtire pe silicagel cu un indicator de fluorescenta si detectia grupului de amine primare libere prin formarea de unui colorant diazo pe placa.
3.REACTIVI
Toti reactivii trebuie sa fie de puritate analitica.
3.1. Amestec solvent: ciclohexan / 2-propanol / diclormetan stabilizat 48 / 64 / 9 (v/v/v).
3.2. Solvent de developare: gazolina (40-60)/benzen/ acetona/solutie hidroxid de amoniu (NH3 minim 25%): 35/35/35/1 (v/v/v/v).
3.3. Solutie de developare:
(a) azotit de sodiu: 1g in 100ml acid clorhidric 1M (preparat imediat inainte de folosire).
(b) 2-naftol: 0,2g in 100ml de hidroxid de potasiu 1M
3.4. Solutii standard:
alfa-monogliceril 4-aminobenzoat: 0,05g in 100ml solvent amestecat 3.1
etil 4-aminobenzoat : 0,05g in 100ml solvent amestecat 3.1
3.5. Placi cu silicagel 60 F254, 0,25 mm grosime, 200 mm x 200 mm
4.APARATURA
4.1. Aparatura uzuala pentru cromatografie in strat subtire
4.2. Vibrator ultrasonic.
4.3. Filtru millipore FH 0,5 mm ori echivalent
5.PROCEDEU
5.1. Prepararea probei
Se cantaresc 1,5g produs de analizat intr-un balon cotat cu dop de 10ml. Se aduce la semn cu solvent (3.1). Se inchide si se lasa pentru o ora la temperatura camerei in intr-un vibrator ultrasonic (4.2). Se filtreaza printr-un filtru Millipore (4,3), si se foloseste filtratul pentru cromotagrafiere.
5.2.Cromatografiere in strat subtire
Se pun cu pipeta pe placa (3.5) cate 10ml solutie proba (5.1) si din fiecare solutie standard (3.4).
Se developeaza cromatograma pana la o inaltime de 150mm intr-un tanc saturat anterior cu solvent 3.2. Se lasa placa sa se usuce la temperatura ambianta.
5.3.Developare
5.3.1. Se observa placa in lumina UV la 254 nm.
5.3.2. Se pulverizeaza placa complet uscata cu solutie 3.3 (a).
Se lasa sa se usuce la temperatura camerei pentru 1 minut si imediat se pulverizeaza cu solutie 3.3. Se usuca placa intr-o etuva la 60C. Spoturile ce apar au o culoare oranj. Alfa-monoglicerii 4-aminobenzoat: Rf 0.07; etil 4-aminobenzonat: Rf 0,5.
B. DETERMINARE
1.SCOP SI DOMENIU DE APLICARE
Aceasta metoda reglementeaza determinarea alfa monogliceril-(4-aminobenzoatul)ului. Se determina de asemenea etil-4-aminobezoatul. nu se poate determina mai mult de 5% (m/m) alfa-mono-gliceril 4-aminobenzoat si mai mult de 1% (m/m) etil- 4-aminobenzoat.
2.DEFINITIE
Continutul de alfa-mono-gliceril 4-aminobenzoat si de etil- 4-aminobenzoat masurat prin aceasta metoda este exprimat in procente masice (% m/m) de produs.
3.PRINCIPIU
Produsul de analizat este suspendat in metanol si dupa un tratament adecvat al probei este determinat prin cromatografie de lichide de inalta performanta (HPLC).
4.REACTIVI
Toti reactivii trebuie sa fie de puritate analitica si trebuie sa fie adecvati pentru HPLC.
4.1.Metanol
4.2.Ortofosfat diacid de potasiu (KH2PO4)
4.3.Diacetat de zinc (Zn (CH3COO)2 2H2O
4.4.Acid acetic (d420 = 1,05)
4.5.Hexacianoferat de tetrapotasiu (K4(Fe(CN)6) 3H2O)
4.6. Etil 4-hidroxibenzoat
Alfa monogliceril 4-aminobenzoat
4.8. Etil 4-aminobenzoat
4.9. Solutie tampon de fosfat (0,02M): se dizolva 2,72 g ortofosfat diacid de potasiu (4.2) intr-un litru de apa
4.10. Eluent: solutie tampon de fosfat (4,9)/metanol (4.1) 61 / 39 (v/v)
Compozitia fazei mobile poate fi schimbata in scopul de a obtine un factor de resolutie R
|
|
unde:
R1 si R2 = timpii de retentie, in minute, ai picurilor
W1 si W2 = latimea picurilor la jumatatea inaltimii, in milimetri
d' = viteza de inregistrare a graficului, in milimetri per minut
4.11.Solutie stoc de alfamonogliceril 4-aminobenzoat: intr-un balon cotat de 100ml se cantaresc cu precizie 40 mg alfamonogliceril 4-aminobenzoat. Se dizolva in 40ml metanol (4.1). Se aduce la semn cu solutie tampon (4.9) si se amesteca.
4.12. Solutie stoc de etil 4-aminobenzoat: intr-un balon cotat de 100ml se cantaresc cu precizie 40mg etil 4-aminobenzoat. Se dizolva in 40ml metanol (4.1). Se aduce la semn cu solutie tampon (4.9) si se amesteca.
4.13. Solute standard intern: intr-un balon cotat de 100ml se cantaresc cu precizie 5mg etil 4-hidroxibenzoat. Se dizolva in 40ml metanol (4.1). Se aduce la semn cu solutie tampon (4.9) si se amesteca.
4.14.Solutii standard: se prepara patru solutii standard prin dizolvarea in 100ml eluent (4.10) conform tabelului de mai jos:
|
Alfa monogliceril 4-aminobenzoat |
Etil 4-aminobenzoat |
Etil 4-hidroxibenzoat |
mg/ml (*) |
ml (4.11) |
mg/ml (*) |
ml (4.12) |
mg/ml (*) |
ml (4.13) |
||||||||||||||||||||||||||||||||||
|
I | ||||||||||||||||||||||||||||||||||||||||||
|
II | ||||||||||||||||||||||||||||||||||||||||||
|
III | ||||||||||||||||||||||||||||||||||||||||||
|
IV | ||||||||||||||||||||||||||||||||||||||||||
|
(!)Aceste valori sunt date ca o indicatie si corespund la mase exacte din 4.11, 4.12 si 4.13. NB: aceste solutii pot fi preparate in moduri diferite. |
Solutie Carrez I: se dizolva 26,5g de hexacianoferat de tetrapotasiu (4.5) in apa si se aduce la volum de 250ml. Solutie Carrez II: se dizolva 54,9g de diacetat de zinc (4.3) si 7,5ml acid acetic (4.4) in apa si se aduce la volum de 250ml. 4.17. Merck Lichrosorb RP-18, sau echivalent, cu dimensiunea medie a particulelor 5 mm. 5.APARATURA 5.1. Echipamenet uzual de laborator. 5.2. Echipament pentru cromatografia de inalta performanta cu detector UV cu lungime de unda variabila si camera termostatata setata la 45C. 5.3.Coloana de otel inoxidabil: lungime 250mm, diametru interior 4,6mm; umplutura lichrosorb RP 5.4. Baie ultrasonica. 6.PROCEDEU Preparare proba 6.1.1. Se cantareste cu precizie 1g de proba intr-un pahar de 100ml si se adauga 10ml metanol (4.1). 6.1.2. Se pune paharul in baia ultrasonica (5.4) timp 20 de minute pentru a se obtine o suspensie. Se transfera cantitativ suspensia astfel obtinuta intr-un balon cotat de 100ml cu nu mai mult de 75ml de eluant (4.10). Se adauga succesiv 1ml solutie carrez I (4.15) si 1ml solutie carrez II (4.16) si se amesteca dupa fiecare adaugare. Se aduce la semn cu eluant (4.10),se reamesteca si se filtreaza printr-o hartie de filtru cutata. 6.1.3.Se transfera cu o pipeta 3,0ml din filtratul obtinut la 6.1.2. si 5,0ml solutie standard intern (4.13) intr-un balon cotat de 50ml. Se aduce la semn cu eluant (4.10) de si se amesteca. Se foloseste solutia astfel obtinuta pentru realizarea analizei cromatografice descrisa la 6.2. 6.2.Cromatografiere 6.2.1. Se regleaza debitul fazei mobile (4.10) la 1,2 ml/min si se seteaza temperatura coloanei la 45C. 6.2.2. Se seteaza detectorul (5.2) la 274nm. 6.2.3.Cu o microseringa se injecteaza cel putin de doua ori 20ml de solutie (6.1.3) in cromatograf si se masoara suprafata picului. 6.3.Curba de etalonare 6.3.1. Se injecteaza 20ml din fiecare dintre solutiile standard (4.1.4) si se masoara suprafata picului. 6.3.2. pentru fiecare concentratie se calculeaza raportul dintre suprafetele picurilor pentru alfamonogliceril 4-aminobenzoat si suprafetele picurilor pentru standardul intern. Se traseaza acest raport pe abscisa iar pe ordonata raportul maselor corespunzatoare. 6.3.3. Se procedeaza in acelasi mod pentru etil 4-hidroxibenzoat. 7.CALCUL 7.1. Din curba de etalonare obtinuta la 6.3. se citesc raporturile de mase (RP1, RP2), corespunzand cu raporturile dintre suprafetele picurilor calculate la 6.2.3, unde: RP1 = masa alfamonogliceril 4-aminobenzoat / masa etil 4-hidroxibenzoatRP2 = masa etil 4-aminobenzoat / masa etil 4-hidroxibenzoat7.2. Din raportul de mase obtinute in acest mod se calculeaza continutul de alfamonogliceril 4-aminobeanzoat si respectiv etil 4-aminobenzoat, ca procente masice (% m/m) cu ajutorul formulelor:
q = cantitatea de etil 4-aminobenzoat (standard intern) cantarita, in miligrame, in 4.12 p = cantitatea de proba, in grame, cantarita in 6.1.1 8. REPETABILITATE (ISO 5725) 8.1 Pentru un continut de 5% (m/m) alfamonogliceril 4-aminobenzoat, diferenta dintre rezultatele a doua determinari efectuate in paralel pe aceeasi proba nu trebuie sa depaseasca 0,25%. Pentru un continut de 1% (m/m) etil 4-aminobenzoat, diferenta dintre rezultatele a doua determinari efectuate in paralel pe aceeasi proba nu trebuie sa depaseasca 0,10%. 9. NOTE 9.1. Inainte de efectuarea unei analize, se verifica daca proba contine substante care ar fi pasibile sa se suprapuna pe cromatograma peste picul standardului intern (etil 4-aminobenzoat). 9.2. Pentru a verifica absenta oricarei interferente, se repeta determinarea prin schimbarea proportiei de metanol in faza mobila cu 10% relativ. ANEXA nr.26 METODA DE DETERMINARE CANTITATIVA PENTRU CLORBUTANOL 1.SCOP SI DOMENIU DE APLICARE Aceasta este metoda
reglementata pentru determinarea clorbutanolului (denumire conform 2.DEFINITIE Continutul de clorbutanol masurat prin aceasta metoda este exprimat in procente masice % (m/m) de produs. 3.PRINCIPIU Dupa un tratament adecvat al produsului de analizat determinarea este realizata prin cromatografie de gaze folosind 2,2,2-tricloretanol ca standard intern. 4.REACTIVI Toti reactivii trebuie sa fie de puritate analitica. 4.1. Clorbutanol (1,1,1-triclor-2-metilpropan-2-ol) 4.2. 2,2,2-tricloretanol 4.3. Etanol absolut 4.4. Solutie standard de clorbutanol: 0,025g in 100ml etanol (4.3) (m/v) 4.5. Solutie standard de 2,2,2-tricloretanol: 4 mg in 100ml etanol (4.3) (m/v) 5.APARATURA 5.1. Echipament uzual de laborator. 5.2. cromatograf de gaze cu detector cu electroni, Ni 63. 6.PROCEDEU 6.1.Preparare proba Se cantareste cu precizie intre 0,1 si 0,3g ('p'g) de proba. Se pune intr-un balon cotat de 100ml. Se dizolva in etanol (4.3), se adauga 1ml solutie standard intern (4.5) si se aduce la semn cu etanol (4.3). 6.2.Conditii pentru cromatografia de gaze 6.2.1.Conditiile de operare trebuie sa realizeze un factor de rezolutie R
unde: R1 si R2 = timpii de retentie, in minute, ai picurilor W1 si W2 = latimea picurilor la jumatatea inaltimii, in milimetri d' = viteza de inregistrare a graficului, in milimetri per minut 6.2.2.Ca exemple, urmatoarele conditii de operare furnizeaza rezolutia necesara:
6.3.Curba standard Folosind 5 baloane cotate de 100ml, se adauga 1ml solutie standard (4.5) si respectiv 0,2, 0,3, 0,4, 0,5 si 0,6ml solutie 4.4, se aduce la semn cu etanol (4.3) si se amesteca. Se injecteaza 1ml din fiecare din aceste solutii in cromatograf in conformitate cu conditiile de operare descrise in 6.2.2. si se construieste o curba de etalonare prin trasarea pe abscisa a raportului dintre masa de clorbutanol si cea de 2,2,2-tricloretanol si pe ordonata raportul suprafetelor picurilor corespondente. 6.4.Se injecteaza 1ml solutie obtinuta la 6.1. si se procedeaza in conformitate cu conditiile descrise la 6.2.2.
7.1Se calculeaza de pe curba standard (6.3) cantitatea 'a' exprimata ca mg de clorbutanol, in solutia 6.1 Continutul de clorbutanol in proba este calculat folosind urmatoarea formula:
Pentru un continut de clorbutanol de 0,5% (m/m) diferenta dintre rezultatele a doua determinari efectuate in paralel pe aceeasi proba nu trebuie sa depaseasca 0,01%. Nota Daca rezultatul este egal cu sau depaseste concentratia maxim admisa este necesara verificarea absentei interferentelor. ANEXA nr.27 METODA DE IDENTIFICARE SI DETERMINARE CANTITATIVA PENTRU CHININA IN PRODUSELE PENTRU INGRIJIREA PARULUI A. IDENTIFICARE 1.SCOP SI DOMENIU DE APLICARE Aceasta este metoda reglementata pentru identificarea si determinarea cantitativa a chininei in sampoane si lotiunile pentru par. 2.PRINCIPIU Identificarea este realizata prin cromatografie in strat subtire pe silicagel. detectia chininei este realizata prin fluorescenta albastra a chininei in conditii acide la 360nm. Pentru confirmare ulterioara, fluorescenta poate fi eliminata prin vapori de brom, si vaporii de amoniac vor determina aparitia unei fluorescente galbui 3.REACTIVI Toti reactivii trebuie sa fie de puritate analitica. 3.1. Placi cu silicagel, fara indicatori fluorescenti, 0,25mm grosime, 200 mm x 200 mm. 3.2. Solvent de developare: toluen / dietileter / diclormetan / dietilamina 20 / 20 / 20 / 8 (v/v/v/v) 3.3. Metanol 3.4. Acid sulfuric (96%; d420 =1,84) 3.5. Dietileter 3.6. Agent de developare: se adauga cu grija 5 de acid sulfuric (3.4) la 95 ml dietileter (3.5) intr-un recipient racit. 3.7. Brom 3.8. Solutie hidroxid de amoniu (28%; d420 = 0,90) 3.9. Chinina, anhidra 3.10. Solutie standard: se cantaresc cu precizie 100,0mg chinina anhidra (3.9) intr-un balon cotat si se dizolva in 100mg metanol (3.3). 4.APARATURA 4.1.Echipament uzual pentru cromotografie in strat subtire. 4.2.Baie ultrasonica. 4.3.Filtru Millipore, FH 0,5mm sau echivalent cu echipament de filtrare adecvat. 5.PROCEDEU 5.1.Preparare proba intr-un balon cotat de 100ml se cantareste cu precizie o cantitate de proba ce poate contine circa 100mg chinina, se dizolva si se aduce la semn cu metanol. Se astupa balonul si se lasa pentru o ora la temperatura camerei intr-un vibrator ultrasonic. Se filtreaza si se foloseste filtratul pentru cromatografie. 5.2.Cromatografiere in strat subtire Se pune cu pipeta 1,0ml solutie standard (3.10) si 1,0ml solutie proba (5.1) pe placa cu silicagel (3.1). se developeaza cromatograma pe o distanta de 150mm folosind solvent 3.2 intr-un tanc saturat anterior cu solvent (3.2). 5.3.Developare 5.3.1. Se usuca placa la temperatura camerei 5.3.2. Se pulverizeaza cu reactivul 3.6. 5.3.3. Se lasa placa la uscat pentru o ora la temperatura camerei. 5.3.4. Se observa in lumina unei lampi UV reglata la o lungime de unda de 360nm. Chinina apare ca un spot fluorescent albastru intens. Ca exemplu tabelul de mai jos indica valorile Rf ale principalilor alcaloizi derivati din chinina cand se developeaza cu solvent 3.2
5.3.5. Pentru confirmarea ulterioara a prezentei chininei, placa este expusa pentru circa o ora la vapori de brom (3.7). Fluorescenta dispare. Cand aceasi placa este expusa la vapori de amoniac (3.8), spoturile reapar cu o culoare maro, si cand placa este examinata din nou sub lumina uv la 360nm se poate observa o fluorescenta galbuie.
1. SCOP SI DOMENIU DE APLICARE Aceasta este metoda reglementata pentru determinarea chininei. poate fi utilizata pentru a determina concentratia maxima admisa de 0,5% (m/m) in sampoane si 0,2 % in lotiunile pentru par. DEFINITIE Continutul de chinina determinat prin aceasta metoda este exprimat in procente masice (% m/m) de produs. PRINCIPIU Dupa un tratament adecvat al produsului de analizat, determinarea este realizata prin cromatografie de lichide de inalta performanta (HPLC) REACTIVI Toti reactivii trebuie sa fie de puritate analitica si adecvati HPLC. Acetonitril Fosfat diacid de potasiu (KH2PO4) Acid ortofosforic (85%; d420 = 1,7) Bromura de tetrametilamoniu Chinina, anhidra Metanol Solutie acid ortofosforic (0,1 M): se cantaresc 11,53g acid ortofosforic (4.3) si se dizolva in 1 000ml apa intr-un balon cotat. Solutie fosfat diacid de potasiu (0,1 M): se cantaresc 13,6g fosfat diacid de potasiu (4.2) si se dizolva in 1 000ml apa intr-un balon cotat. Solutie bromura de tetrametilamoniu: se dizolva 15,40g bromura de tetrametilamoniu in 1 000ml apa intr-un balon cotat. Eluent: acid ortofosforic (4.7)/fosfat diacid de potasiu (4.8)/bromura de tetrametilamoniu (4.9)/ apa / acetonitril (4.1) 10 / 50 / 100 / 340 / 90 (v/v/v/v/v) Compozitia acestei faze mobile poate fi schimbata in scopul de a obtine un factor de rezolutie R
unde: R1 si R2 = timpii de retentie, in minute, ai picurilor W1 si W2 = latimea picului la jumatate inaltimii, in milimetri d' = viteza de inregistrare a graficului, in milimetri per minut silice tratata cu octadecilsilan (10mm) Solutii standard: intr-un set de baloane cotate de 100ml se cantaresc cu precizie cca. 5,0, 10,0, 15,0 si respectiv 20,0mg chinina anhidrida (4.5). Se aduce la semn cu metanol (4.6) si se agita continutul baloanelor pana la dizolvarea chininei. Se filtreaza fiecare proba printr-un filtru de 0,5mm. APARATURA Echipament uzual de laborator. Baie ultrasonica. 5.3. Echipament pentru cromatografie de lichid de inalta performanta, cu detector cu lungime de unda variabila. 5.4. Coloana: lungime 250mm; diametru interior 4,6mm; umplutura silice (4.11). 5.5. Filtru millipore FH 0,5mm, sau echivalent, cu aparatura de filtrare adecvata. PROCEDEU 6.1 Preparare proba intr-un balon cotat de 100 ml se cantareste cu precizie o cantitate suficienta de produs pentru a contine 10,0 mg chinina anhidra, se adauga 20 ml metanol (4.6) si se pune balonul intr-o baie ultrasonica (5.2) pentru 20 minute. Se aduce la semn cu metanol (4.6). Se amesteca solutia si apoi se filtreaza o portiune (5.5). Cromatografiere Debit: 1,0 ml/min. lungime de unda pentru detector (5.3): 332nm. Volum de injectare: 10ml din solutia filtrata (6.1) Masurare: suprafata picului. 6.3. Curba de etalonare Se injecteaza de cel putin 3 ori 10,0ml din fiecare solutie de referinta (4.12), se masoara suprafata picurilor si se calculeaza suprafata medie pentru fiecare concentratie. Se realizeaza curba de etalonare si se verifica ca aceasta sa fie rectilinie. CALCUL 7.1. Din curba de etalonare (6.3) se determina cantitatea in mg de chinina anhidra prezenta in volumul injectat (6.2) 7.2.Concentratia de chinina anhidra in proba, ca procentaj masic (% m/m), este obtinuta utilizand formula urmatoare:
unde: B este cantitatea, in micrograme, de chinina anhidra determinata in cei 10ml de solutie filtrata (6.1) A este masa probei, in grame (6.1) REPETABILITATE (ISO 5725) Pentru un continut de chinina anhidra de 0,5 % (m/m), diferenta dintre rezultatele a doua determinari efectuate in paralel pe aceeasi proba nu trebuie sa depaseasca 0,02%. Pentru un continut de chinina anhidra de 0,2 % (m/m), diferenta dintre rezultatele a doua determinari efectuate in paralel pe aceeasi proba nu trebuie sa depaseasca 0,01%. ANEXA nr.28 METODA DE IDENTIFICARE SI DETERMINARE CANTITATIVA PENTRU SULFITI ANORGANICI SI SULFITI ACIZI SCOP SI DOMENIU DE APLICAREMetoda reglementeaza identificarea si determinarea a sulfitilor anorganici si sulfitilor acizi in produsele cosmetice. este aplicabila numai produselor care au o faza apoasa sau alcoolica si pentru concentratii de pana la 0,2% dioxid de sulf. A. IDENTIFICARE PRINCIPIU Proba se incalzeste in acid clorhidric si dioxidul de sulf care se elibereaza este identificat prin mirosul sau sau prin efectul sau asupra hartiei indicatoare. REACTIVI Toti reactivii trebuie sa fie de puritate analitica. Acid clorhidric (4M). Hartie iod-amidonata (iodat de potasiu) ori echivalent. APARATURA Echipament uzual de laborator. Flacon (25 ml) echipat cu un condensator de reflux scurt. PROCEDEU Se pun circa 2,5g proba in flacon (3.2) cu 10ml de acid clorhdric (2.1) Se amesteca si se incalzeste pana la fierbere. Se testeaza emisia de dioxid de sulf prin miros sau cu hartie indicatoare (2.2). B. DETERMINARE 1.DEFINITIE Continutul de sulfit sau sulfit acid in proba determinat conform acestei metode este exprimat in procente masice de dioxid de sulf. 2.PRINCIPIU Dupa acidifierea probei, dioxidul de sulf eliberat este distilat intr-o solutie de apa oxigenata. Acidul sulfuric format este titrat cu o solutie de hidroxid de sodiu standardizata. 3.REACTIVI Toti reactivii trebuie sa fie de puritate analitica. 3.1. Apa oxigenata 0,2% (m/v). Se prepara zilnic. 3.2. Acid ortofosforic (d425 = 1,75) 3.3. Metanol 3.4. solutie standardizata de hidroxid de sodiu (0,01M) 3.5. Azot 3.6. Indicator: amestec 1:1 (v/v) de rosu de metil (0,03% m/v in etanol) si albastru de metilen (0,05% m/v in etanol). Se filtreaza solutia. 4.APARATURA 4.1. Echipament uzual de laborator 4.2. Aparatura de distilare (vezi figura). 5. PROCEDEU 5.1. in balonul de distilare A (vezi figura) se cantaresc cu precizie circa 2,5g proba. 5.2. Se adauga 60ml apa si 50ml metanol (3.3) si se amesteca. 5.3. Se pun 10ml apa oxigenata (3.1), 60ml apa si cateva picaturi de indicator (3.6) in colectorul de distilare D (vezi figura). Se adauga cateva picaturi de hidroxid de sodiu (3.4) pana la virarea in verde a indicatorului. 5.4. Se repeta 5.3. pentru flaconul de spalare E (vezi figura) 5.5. Se asambleaza aparatura si se regleaza debitul de azot (3.5) la circa 60 bule per minut. 5.6. Se pun 15ml acid ortofosforic (3.2) din palnie in balonul de distilare A. 5.7. Se incalzeste rapid pana la fierbere si apoi se fierbe usor la foc mic pe o perioada totala de 30 minute; 5.8. Se detaseaza colectorul de distilare D. Se clateste tubul si apoi se titreaza solutie de hidroxid de sodiu (3.4) pana la virarea in verde a indicatorului (3.6) 6.CALCUL Se calculeaza continutul de sulfit sau sulfit acid, in procente masice, cu formula:
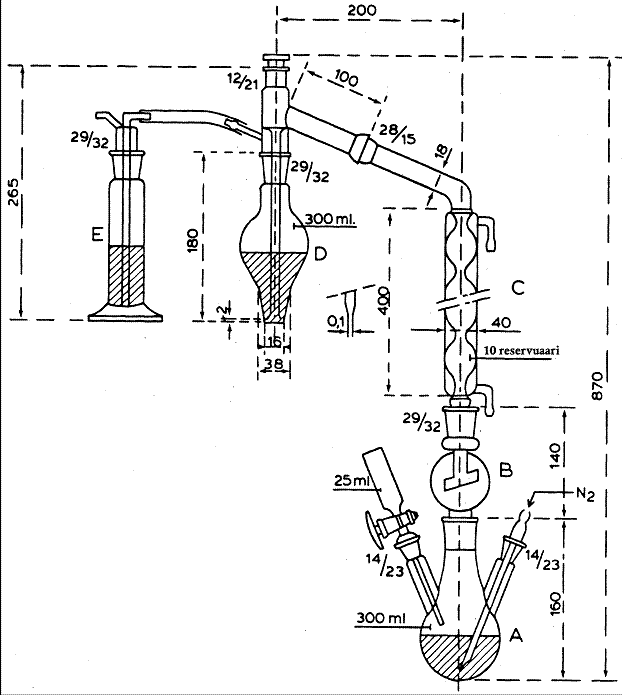
unde: M = concentratie molara a solutiei de hidroxidde sodiuV = volum de hidroxid de sodiu (3.4) necesar pentru titrare (5.8), in mililitrim = masa probei (5.1), in grame. 7. REPETABILITATE (ISO 5725) Pentru un continut de 0,2% m/m dioxid de sulf diferenta dintre doua determinari efectuate in paralel pe aceeasi proba nu trebuie sa fie mai mare de 0,006%. Aparatura de distilare a dioxidului de sulf in conformitate cu Tanner Toate dimensiunile sunt in mm.
ANEXA nr29 METODA DE IDENTIFICARE SI DETERMINARE CANTITATIVA PENTRU CLORATI AI METALELOR ALCALINE SCOP SI DOMENIU DE APLICAREMetoda reglementeaza identificarea si determinarea cloratilor metalelor alcaline in pastele de dinti si alte produse cosmetice A. IDENTIFICARE 1.PRINCIPIU Cloratii sunt separati de alti halogenati prin cromatografie in strat subtire si identificati prin oxidarea iodurii cu formare de iod. 2.REACTIVI Toti reactivii trebuie sa fie de puritate analitica. 2.1.Solutii de referinta: solutii apoase de clorat, bromat si iodat de potasiu (0,2% m/v) proaspat preparate. 2.2.Solvent de developare: solutie amoniacala (28% m/v) / acetona / butanol (60 / 130 / 30 v/v/v) 2.3. Iodura de potasiu, solutie apoasa (5% m/v) 2.4. Solutie de amidon (1 la 5% m/v) 2.5. Acid clorhidric (1M) 2.6. Placi pentru cromatografie in strat subtire cu celuloza, gata preparate (0,25 mm) 3.APARATURA Echipament normal pentru cromatografia in strat subtire. 4.PROCEDEU 4.1.Se extrage circa 1g de proba cu apa, se filtreaza si dilueaza la circa 25 ml. 4.2 Se pun cu pipeta 2ml de solutie (4.1) pe placa (2.6) impreuna cu 2ml portiuni din fiecare din cele trei solutii de referinta (2.1). 4.3.Se pune placa intr-un tanc si se developeaza prin cromatografie ascendenta circa trei sferturi din lungimea placi (2.6) cu solvent 2.2. 4.4. Se scoate din tanc si se lasa sa se evaporeze solventul (Nota Bene: aceasta poate dura pana la 2 ore) 4.5. Se pulverizeaza placa cu iodura de potasiu (2.3) si se lasa la uscat pentru circa 5 minute 4.6. Se pulverizeaza placa cu solutie de amidon (2.4) si se lasa la uscat pentru circa 5 minute 4.7. Se pulverizeaza placa cu acid clorhidric (2.5) 5.EVALUARE Daca cloratul este prezent ,un spot albastru (posibil un spot maro) va aparea dupa o jumatate de ora cu o valoare Rf de aproximativ 0,7 pana la 0,8
Trebuie remarcat faptul ca bromatii si iodatii dau reactie imediata. Trebuie avut grija a nu se confunda spoturile de la bromati si clorati. B. DETERMINARE 1.DEFINITIE Continutul de clorat din proba determinat conform acestei metode este exprimat in procente masice de clorat. 2.PRINCIPIU Cloratul este redus de pulberea de zinc in conditii acide. Clorura formata este masurata prin titrare potentiometrica folosind o solutie de nitrat de argint. O determinare similara inainte reducerii permite determinarea posibilei prezente a halogenurilor. 3.REACTIVI Toti reactivii trebuie sa fie de puritate analitica. 3.1. Acid acetic, 80% (m/m) 3.2. Pulbere de zinc 3.3. Solutie standard de azotat de argint (0,1M) 4.APARATURA 4.1. Echipament uzual de laborator 4.2. Potentiometru echipat cu electrod indicator de argint. 5.PROCEDEU 5.1. Preparare proba intr-o eprubeta pentru centrifuga se cantareste cu precizie o cantitate 'm' de aproximativ 2g. Se adauga circa 15ml acid acetic (3.1) si se amesteca cu grija. Se asteapta 30 minute si se centrifugheaza 15 minute la 2000 rot/min. Se transfera solutia supernatanta intr-un balon cotat de 50 ml. Se repeta centrifugarea de doua ori prin adaugarea a 15ml acid acetic (3.1) la reziduu. Se colecteaza solutia continand clorat in acelasi balon cotat. Se aduce la semn cu acid acetic (3.1). 5.2 Reducere clorat Se iau 20ml solutie 5.1. si se adauga 0,6g pulbere de zinc (3.2). Se aduce la fierbere intr-un balon echipat cu un tub condensator. Dupa 30 minute de fierbere, se raceste si se filtreaza. Se clateste balonul cu apa. Se filtreaza si se reuneste filtratul cu apele de clatire. 5.3. Determinare clorura Se titreaza 20ml solutie 5.2. cu azotat de argint (3.3.) prin folosirea potentiometrului (4.2). Se titreaza in acelasi mod 20ml solutie 5.1. cu azotat de argint (5.3) Nota Bene: daca produsul contine derivati de brom sau iod care pot elibera bromuri sau ioduri dupa reducere, curba de titrare va avea mai multe puncte de inflexiune. In acest caz volumul solutiei titrate (3.3) corespunzand clorurii este diferenta dintre ultimul si penultimul punct de inflexiune. 6.CALCUL Continutul de clorat al probei (% m/m) este calculat cu formula:
unde : V = volumul in mililitri, de solutie de azotat de argint (3.3) folosit la titrarea solutiei 5.2V' = volum in mililitri, de solutie de azotat de argint (3.3) folosit la titrarea a 20ml de solutie 5.1M = molaritatea solutiei standard de azotat de argint (3.3) m = masa probei, in grame. 7. REPETABILITATE (ISO 5725) Pentru un continut de clorat de 3 pana la 5% m/m, diferenta dintre rezultatele a doua determinari efectuate in paralel pe aceeasi proba nu trebuie sa depasieasca 0,07% m/m. ANEXA nr.30 METODA DE IDENTIFICARE SI DETERMINARE CANTITATIVA PENTRU IODAT DE SODIU SCOP SI DOMENIU DE APLICAREMetoda reglementeaza procedeul de identificare si determinare a compozitiei chimice a produselor cosmetice continand iodat de sodiu. A. IDENTIFICARE 1.PRINCIPIU Iodatul de sodiu este separat de alti halogenati prin cromatografie in strat subtire si identificat prin oxidarea iodurii cu formare de iod. 2.REACTIVI Toti reactivii trebuie sa fie de puritate analitica. 2.1 Solutii de referinta: solutii apoase de clorat, bromat si iodat de potasiu (0,1% m/v) proaspat preparate. 2.2. Solvent de developare: solutie amoniacala 28% (m/v) / acetona / butanol (60 / 130 / 30 v/v/v) 2.3. solutie apoasa de iodura de potasiu (5% m/v) 2.4. Solutie de amidon (1 pana la 5% m/v) 2.5. Acid clorhidric (1M) 3.APARATURA 3.1. Placi pentru cromatografie in strat subtire cu celuloza (0,25 mm) gata preparate. 3.2. Echipament normal pentru crtomatografie in strat subtire. 4.PROCEDEU 4.1. Se extrage circa 1g de proba cu apa, se filtreaza si se dilueaza la circa 10ml. 4.2. Se pun cu pipeta 2ml din aceasta solutiei pe linia de baza a placii (3.1) impreuna cu 2ml portiuni din fiecare dintre cele trei solutii de referinta. 4.3. Se pune placa intr-un tanc si se developeaza prin cromatografie ascendenta circa trei sferturi din lungimea placii (2.6) cu solvent (2.2). 4.4. Se indeparteaza placa din tanc si se lasa sa se evapore solventul la temperatura ambianta (Nota Bene: aceasta poate dura pana la doua ore) 4.5. Se pulverizeaza placa cu iodura de potasiu (2.3) si se lasa sa se usuce circa de 5 minute. 4.6. Se pulverizeaza placa cu solutie de amidon (2.4) si se lasa sa se usuce circa de 5 minute 4.7. In final se pulverizeaza cu acid clorhidric (2.5) 5.EVALUARE Daca iodatul este prezent un spot albastru (culoarea poate fi maro sau poate deveni maro daca sta) va aparea imediat cu o valoare Rf de aproximativ 0 pana la 0,2. Trebuie remarcat ca bromatii dau reactii imediate la valori Rf de aproximativ 0,5 pana la 0,6 iar cloratii, dupa circa 30 minute, la valori Rf de 0,7 respectiv 0,8. B. DETERMINARE 1.DEFINITIE Continutul de iodat de sodiu determinat conform acestei metode este exprimat ca procent masic de iodat de sodiu. 2.PRINCIPIU Iodatul de sodiu se dizolva in apa si este determinat prin intermediul cromatografiei de lichid de inalta performanta, folosind o coloana C18 cu faza inversa si o coloana cu schimbator de anioni inseriate. 3.REACTIVI Toti reactivii trebuie sa fie de puritate analitica si in special adecvati cromatografiei de lichide de inalta performanta (HPLC). 3.1. Acid clorhidric (4M) 3.2. Sulfit de sodiu apos, 5% m/v 3.3. solutie stoc iodat de sodiu: se prepara o solutie stoc continand 50mg iodat de sodiu per 100 ml apa. 3.4.ortofosfat diacid de potasiu 3.5.ortofosfat acid de disodiu 2H2O 3.6.faza mobila HPLC: se dizolva 3,88g ortofosfat diacid de potasiu (3.4) si 1,19g ortofosfat acid de disoidiu 2 H2O (3.5) intr-un litru de apa.pH-ul solutiei rezultate este 6,2. 3.7.Hartie indicatoare universala, ph 1-11. 4.APARATURA 4.1.Aparatura de laborator obisnuita. 4.2.Hartie de filtru circulara, diametru 110 mm, Schleicher si Schll nr. 575, sau echivalent. 4.3.Cromatograf de lichid de inalta performanta cu detector cu lungime de unda variabila. 4.4.Coloane: lungime 120 mm; diametru interior: 4,6 mm; numar: doua conectate in serie; prima coloana NecleosilR 5 C18 sau echivalent; a doua colana - VydacTM -301-SB sau echivalent 5.PROCEDEU 5.1.Preparare proba Probe fluide (sampoane) Se cantareste cu precizie o proba pentru testare de aproximativ 1,0g proba intr-un cilindru gradat cu dop de sticla sau un balon cotat. se aduce la semn cu apa si se amesteca. Daca este necesar, se filtreaza solutia. Se determina iodatul din solutie prin intermediul HPLC conform descrierii din sectiune 5.2. Probe solide (sapunuri) Se ia o cota parte din proba si se cantareste cu precizie o proba pentru testare de circa 1,0g intr-un cilindru gradat cu dop de sticla. Se umple pana la 50ml cu apa si se agita puternic timp de un minut. Se centrufugheaza si se filtreaza prin hartie de filtru (4.1) sau se lasa amestecul sa stea pentru cel putin o noapte. Cromatografiere Debit:1 ml/min. lungime de unda a detectorului (4.2): 210 nm Volum de injectare: 10 ml. Masurare : suprafata piculuiEtalonare In baloane cotate de 50 ml se pun cu pipeta 1,0, 2,0, 5,0, 10,0 si 20,0ml solutie stoc de iodat de sodiu (3,3). Se aduce la semn cu apa si se amesteca. solutiile astfel obtinute contin 0,01, 0,02, 0,05, 0,10 si respectiv 0,20mg iodat de sodiu per mililitru. Se injecteaza o portiune de 10ml din fiecare solutie standard de iodat in cromatograful de lichide (4.2) si se obtine o cromatograma. Se determina suprafata picului pentru iodat si se traseaza o curba prin relationarea suprafatei picului cu concentratia de iodat de sodiu. CALCUL Se calculeaza continutul de iodat de sodiu, in procente masice (% m/m), folosind formula:
unde: m este masa, in grame, a probei de testare (5.1) V este volumul total a solutiei proba, in mililitri, obtinuta conform descrierii de la 5.1c este concentratia, in miligrame per milimetru iodat de sodiu, obtinuta din curba de etalonare (5.3). 7. REPETABILIATE (ISO 5725) Pentru un continut de iodat de sodiu de 0,1% (m/m) diferenta dintre rezultatele a doua determinari efectuate in paralel pe aceeasi proba nu trebuie sa depasesca 0,002%. 8. CONFIRMARE 8.1. Principiu Intr-o solutie acidifiata a unui produs cosmetic, iodatul (IO3 ) este redus la iod (I ) cu sulfit si solutia rezultata se investigheaza prin intermediul HPLC. Daca un pic avand un timp de retentie corespunzator timpului de retentie al iodatului dispare dupa tratament cu sulfit, picul original poate fi cel mai probabil atribuit iodatului. 8.2. Procedeu Intr-un balon cotat se pune cu pipeta o portiune de 5ml din solutia proba obtinuta conform descrierii de la punctul 5.1. Se ajusteaza pH-ul solutiei la valoarea 3 ori mai mica cu acid clorhidric (3.1); hartie indicatoare universala (3.7). Se adauga 3 picaturi de solutie de sulfit de sodiu (3.2) si se amesteca. Se injecteaza o portiune de10 ml de solutie in cromatograful de lichid (4.2) Se compara aceasta cromatograma cu cromatograma obtinuta conform descrierii de la punctul 5 pentru aceeasi proba.
DISTRIBUIE DOCUMENTUL
Comenteaza documentul:Te rugam sa te autentifici sau sa iti faci cont pentru a putea comentaCreaza cont nou Termeni si conditii de utilizare | Contact
| |||||||||||||||||||||||||||||||||||||||||