| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
DOCUMENTE SIMILARE |
|
TERMENI importanti pentru acest document |
|
Reakční médium
Solvatace
Diskuse o reaktivitě a mechanismu jsou většinou založeny na chování izolovaných molekul a rozpouštědlo je obvykle uvažováno jako inertní složka, která se sice občas zapojuje do reakce, ale pouze značně pasivně. Realita bývá ovšem značně vzdálena od této představy. Interakce mezi rozpouštědlem a rozpuštěnou molekulou mohou mít výrazný, někdy dominantní vliv na průběh reakce. Je ovšem pravdou, že naše současné poznatky
o charakteru tohoto vlivu jsou dost omezené. Souvisí to se skutečností, že velmi málo je prozkoumána struktura kapalného stavu, byť i čistých médií. Samozřejmě ještě komplikovanější je studium změn v těsné blízkosti rozpuštěné molekuly. Je samozřejmě možné vytvořit řadu modelů popisujících toto chování, ale dosud chybí ucelená teorie o vlivu rozpouštědla na rychlost a rovnováhu reakcí.
Mezimolekulární síly
V případě nepolárních molekul jsou jedinými přitažlivými silami slabé, ale vždy přítomné Londonovské či disperzní síly. Jejich původ je v pohybu elektronů v molekule za vzniku přechodných dipolových momentů, které mohou polarizovat okolní molekuly a držet je pohromadě. Síla těchto přitažlivých sil závisí na druhé mocnině molekulové polarizability a. Ta dále závisí na počtu elektronů v molekule a na jejich pohyblivosti. Pentan je tedy polarizovatelnější než butan jen proto, že má víc elektronů. Butadien je ovšem polarizovatelnější než butan díky přítomnosti pohyblivějších p elektronů, i když má o 4 elektrony méně. Charakteristikou polarizability molekul je index lomu (n), poněvadž odráží schopnost elektronů přizpůsobovat svůj pohyb oscilujícímu elektrickému poli světla. Jsou svázány vztahem:
a = (3Vm/4pL)(n2 - 1)/(n2 + 2)
kde Vm je molární objem a L je Avogadrova konstanta.
Molekuly, které samy mají permanentní dipolový moment, mohou přitahovat okolní molekuly indukcí, tj. polarizací sousední molekuly přímou dipól-dipolovou interakcí. Tyto interakce jsou omezeny na relativně velmi blízkou oblast a dipól-dipolové interakce jsou i omezeny nutností vhodné vzájemné orientace, což zřejmě nemůže být splněno současně pro všechny molekuly. Disperzní síly ovšem působí ve všech orientacích interagujících molekul a v celém objemu kapaliny. Zřejmě tedy převládají intermolekulární přitažlivé síly i v polárních rozpouštědlech, s výjimkou těch, kde jsou molekuly malé a výrazně dipolární, anebo kde jsou vázány vodíkovými můstky.
Vodíkové můstky hrají speciální roli v rozpouštědlových interakcích, zvláště v rozpouštědlech obsahujících OH skupiny. Především proto, že vodíkový atom na kladném konci OH dipólu je obvykle orientován vně molekuly a tedy dobře přístupný pro záporně nabité okolní částice. U kladně nabitých částic tomu tak nebývá. Centrum kladného náboje obvykle bývá uvnitř molekuly a jeho přístupnost je obvykle malá. Vodíkové vazby také mohou být mimořádně silné. To je důsledkem jednak malé sterické náročnosti, ale také charakteru této vazby: oba vazebné elektrony OH vazby, ale i volný elektronový pár vodíkové vazby, jsou delokalizovány přes všechny tři účastníky. Vzdálenost kyslík - kyslík v O-H..O vazbě je obvykle podstatně kratší než součet van der Waalsových poloměrů - zřejmě tedy atraktivní interakce převládají nad repulzí.
Solvatace iontů
Silné elektrolyty mohou být rozpuštěny pouze tehdy, jestliže interakce rozpouštědla s ionty překonají jejich přitažlivé síly. To zřejmě vyžaduje působení rozpouštědel s vysokou dielektrickou konstantou, poněvadž velikosti interakcí mezi náboji jsou úměrné převrácené hodnotě dielektrické konstanty rozpouštědla. Rozpouštědlo musí mít také schopnost solvatovat vzniklé ionty. V případě malých iontů jsou síly solvatace především elektrostatické - každý iont přitahuje bipolární molekuly rozpouštědla. Voda je typickým rozpouštědlem - má vysokou dielektrickou konstantu, anionty jsou solvatovány pomocí vodíkových vazeb
a kationty jsou přitahovány volnými el. páry kyslíku. Ty molekuly vody, které jsou v přímém kontaktu s ionty, jsou potom polarizovány a mohou přitahovat další molekuly vody. Tímto způsobem se vytváří solvatační obal až z několika vrstev molekul vody. V koncentrovanějších roztocích a zvláště v přítomnosti vícenásobně nabitých iontů nejsou síly solvatace schopny zabránit jejich částečné agregaci. Začínají se vytvářet iontové páry a vyšší asociáty. Zůstávají sice obklopeny solvatující sférou, ale ionty patří do většího agregátu, který se v rozpouštědle chová jako samostatná „supermolekula“. V nevodných roztocích většinou dochází k asociaci iontů při nižších koncentracích než ve vodě. Jen velmi málo rozpouštědel (mimo vodu) kombinuje vysokou dielektrickou konstantu se schopností solvatovat kationty i anionty. V případě velkých iontů, nebo těch, které mají velkou uhlíkatou molekulu, nebo velmi polarizovatelných anorganických iontů je situace ještě komplikovanější. Jen ve velmi řídkých případech jsou nepolární rozpouštědla s nízkou dielektrickou konstantou schopna rozpustit elektrolyty.
Solvatace elektricky neutrálních molekul
Nepolární látky se většinou dobře rozpouštějí v nepolárních rozpouštědlech - disperzní síly jsou téměř stejné jako v obou původních komponentách. Lze jen velmi obtížně hovořit o nějaké specifické interakci látka - rozpouštědlo a hovořit tedy o solvataci je sotva možné. Situace u polárnějších látek je podobná situaci u velkých iontů - disperzní síly působí, ale míra jejich efektivity je závislá na polarizabilitě obou partnerů. Tvorba vodíkových vazeb a dipól-dipólové interakce jsou silně ovlivněny rozsahem nábojové delokalizace rozpouštěné látky a geometrií interakce. Rozsah solvatace potom závisí na míře kompenzace mezi energií interakce látka - rozpouštědlo a energií porušení intermolekulárních interakcí čisté látky i rozpouštědla.
Termodynamika rozpouštění
Rozpouštění musí být stabilizující interakcí - kdyby tomu tak nebylo, tak prostě neexistuje. Musí tedy být vždy doprovázeno poklesem Gibbsovy energie, která je ovšem ovlivněna jak enthalpickými, tak entropickými příspěvky. Solvatace vzniká působením přitažlivých sil mezi látkou a rozpouštědlem - enthalpie rozpouštění tedy musí být záporná. Avšak jak vzniká solvatace, tak část molekul rozpouštědla má omezenou pohyblivost a dochází tedy ke zmenšení jejich entropie. Entropie solvatace je tedy také záporná - čím silnější, efektivnější solvatace, tím větší entropický efekt.
To je nejlépe vidět na hydrataci protonu. Ve vodných roztocích je hydratovaný proton většinou uvažován jako H3O+. Experimentální poznatky ovšem ukazují na existenci čtyř molekul vody vázaných k protonu a dalších molekul volněji vázaných ve vnějších solvatačních vrstvách.
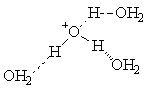
Toto chování se odráží v enormně velkých hodnotách enthalpie i entropie hydratace protonu (-1129 kJ/mol a -131 J/molK) a je i příčinou extrakce značného množství vody při extrakci silně kyselého roztoku do organického rozpouštědla.
Rovnováha mezi entropickým a enthalpickým příspěvkem je často schována v celkové Gibbsově energii. Ukažme si příklad tohoto chování. Kyselina trichloroctová je ve vodě silnější kyselinou než kyselina octová v souladu s vyšší elektronegativitou chloru. Z měření ovšem vychází, že enthalpie ionizace kys. octové ji favorizuje. Ve skutečnosti tedy entropický člen způsobuje posun rovnováhy ve prospěch ionizace trichloroctové kyseliny.
|
(při 25oC) |
DG0 / kJ mol -1 |
DH0 / kJ mol -1 |
DS0 / J K-1mol -1 |
|
CH3COOH | |||
|
CCl3COOH |
+ 2.9 |
- 9 |
Vysvětlení je zcela pochopitelné, uvážíme-li hydrataci obou aniontů. Acetátový anion je malou částicí, jejíž náboj je koncentrován na obou kyslících. Ve vodných roztocích je silně vázán vodíkovými vazbami a přispívá tedy k záporné enthalpii ionizace. Na druhé straně ovšem přispívá k tvorbě velmi silné solvatace dobře organizovanými molekulami vody a tím vede k velkému zápornému entropickému příspěvku. Trichloracetátový anion má naopak mnohem delokalizovanější záporný náboj, je méně efektivně hydratován a enthalpie
a speciálně entropie ionizace jsou mnohem méně záporné.
Celková termodynamika ionizace je ovšem výsledkem příspěvku všech pochodů - jak samotné ionizace, tak i hydratace všech přítomných částic. Moderní metody hmotové spektrometrie umožnily separaci některých těchto procesů. Uvažujme např. protonaci aminů:
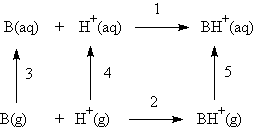
V tomto schématu termodynamické parametry rovnice 1 odpovídají normální protonační rovnováze ve vodě, reakce 2 v plynné fázi a reakce 3 odpovídá hydrataci neutrální báze. Tyto údaje jsou přístupné přímým měřením. Termodynamika rovnice 4 nemůže být přímo měřena, ale je ji možno s velkou mírou pravděpodobnosti odhadnout. Parametry ostatních rovnic lze vypočítat z uvedených čtyř pomocí termodynamických cyklů. Výsledky pro methylamin jsou tyto :
|
Reakce |
DG0 kJ/mol |
DH0 kJ/mol |
DS0 J/Kmol |
Výsledky ukazují na zcela určující vliv solvatace. Gibbsovy energie -DG0 hydratace a protonace pro soubor alifatických aminů jsou uvedeny v další tabulce :
|
Reakce |
amoniak |
methylamin |
dimethylamin |
trimethylamin |
Nepravidelné pořadí bazicit (řádek 1) primárních, sekundárních a terciálních aminů bylo dlouho předmětem spekulací. Nyní je zřejmé, že je způsobeno součtem jednotlivých efektů - vlastní bazicity izolovaných molekul (2) a solvatace kationtů (5).
Vliv solvatace na reakční rychlost
Porovnání rychlostí v plynné a kapalné fázi ukazuje na velmi výrazný vliv solvatace. Tento efekt může být doslova dramatický. Např. u nukleofilní substituce za pokojové teploty (OH- a brommethan) ve vodném roztoku je rychlost 1.4 x 10-4 l/mol s, v plynné fázi potom 6 x 1011 l/mol s. V této studii bylo možné studovat kinetiku přechodu z plynné fáze do roztoku, poněvadž bylo možné sledovat jednotlivé specificky hydratované částice.
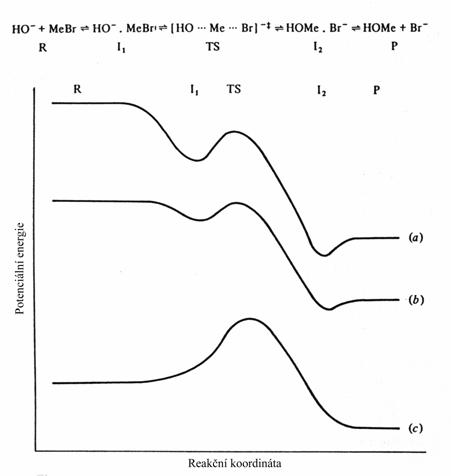
Kvalitativní zobrazení změn energie v průběhu reakce OH iontu s brommethanem: a) v plynné fázi s nesolvatovaným OH-, b) v plynné fázi s OH- .(H2O)2, c) ve vodném roztoku
Výsledky mohou být interpretovány takto :
v plynné fázi reagující částice nejprve vytvoří bezbariérově stabilní adukt, ve kterém je záporný náboj mnohem více delokalizován než v izolovaném OH aniontu. Potom dochází k reorganizaci přes klasický SN2 tranzitní stav. Ve vodném roztoku je OH iont silně stabilizován solvatací a nemůže bez dodání energie vytvořit komplex s brommethanem (musí být odštěpeny některé molekuly vody).
Většina našich znalostí o efektu rozpouštědla má původ v kvalitativních úvahách o způsobu ovlivnění reakční rychlosti a bylo vytvořeno několik empirických škál kvantitativně popisujících tento efekt. Detailnější analýza rozdílného efektu solvatace může být učiněna jen na základě analýzy jednotlivých příspěvků k celkovému efektu. Abychom překonali těžkosti s určením DS, zaveďme veličinu G, popisující změnu standardní Gibbsovy energie při přechodu látky z jednoho rozpouštědla do druhého. Pro neutrální molekuly může být G stanoveno na základě distribučních koeficientů za předpokladu, že obě rozpouštědla jsou nemísitelná, nebo jestliže jsou mísitelná, z relativních rozpustností nebo distribučních koeficientů vzhledem k třetímu rozpouštědlu, se kterým jsou obě nemísitelná. Pro iont jsou ovšem takovéto výpočty neproveditelné. Není totiž možné rozpustit samotný kationt bez patřičného aniontu. Jak ale ohodnotit jejich příspěvky? Termodynamika sama o sobě nemůže tuto otázku řešit bez dalších předpokladů. Nejběžnější z nich je, že náboj hraje zanedbatelnou roli v solvataci, jestliže je lokalizován uvnitř velkého uhlovodíkového zbytku velkého polarizovatelného organického iontu. Tedy např. pro soli Ph4As+ Ph4B- je pozorovaná hodnota G rozdělena rovnoměrně mezi oba ionty. Takto je možné získat G hodnotu pro jakýkoliv iont X- jednoduše odečtením hodnoty pro Ph4As+ od změřené hodnoty Ph4As+X-. Touto cestou byly stanoveny hodnoty G přechodu z jednoho rozpouštědla do druhého pro řadu jednoduchých iontů. Konečně, jestliže měření provedeme v každém rozpouštědle, můžeme získat DG kompletací termodynamického cyklu uvedeného na následujícím obrázku.
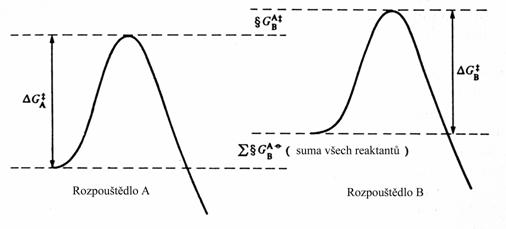
Výpočet Gibbsovy energie přechodu tranzitního stavu z rozpouštědla A do rozpouštědla B.
Aplikace této techniky je uvedena pro klasické SN2 reakce MeI + Cl-, MeI + SCN- a Me3S+ + SCN- v tab.
|
Reaktant |
MeI |
Me3S+ |
Cl- |
SCN- |
|
G kJ/mol | ||||
|
Reakce |
MeI + Cl- |
MeI + SCN- |
Me3S+ + SCN- | |
|
kDMF/kMeOH | ||||
|
DG DMF DG MeOH | ||||
|
G |
Sulfoniový iont a v malém rozsahu i methyljodid jsou stabilizovány přechodem do DMF, důležitá je polarizabilita, resp. schopnost amidového kyslíku solvatovat kationt. Na druhé straně jsou anionty destabilizovány v DMF v důsledku nemožnosti tvorby vodíkových vazeb. Toto je zřejmě příčinou enormního rozdílu v rychlostech prvních dvou reakcí (stabilita tranzitních stavů není solvatací téměř ovlivněna). Oba tranzitní stavy jsou záporně nabity, ale oba jsou také snadno polarizovatelné a pravděpodobně vodíkové vazby a van der Waalsovské interakce jsou v rovnováze. Oproti tomu ve třetí reakci se přechod do jiného rozpouštědla téměř neprojeví na solvataci reaktantů, relativně malé rozdíly v reakčních rychlostech jsou zřejmě způsobeny rozdílnou solvatací tranzitního stavu. Ten je prakticky elektricky neutrální (NCS Me SMe2 ), velmi málo citlivý na vodíkové vazby a stabilizovaný více DMF než methanolem. Ideální by bylo rozdělit G na komponenty H a S . H hodnoty je možno získat jako rozdíl mezi rozpouštěcími teply ve dvou rozpouštědlech a S je potom roven ( H - G)/T. Naneštěstí řada publikovaných rozpouštěcích tepel elektrolytů je zřejmě nereálná a jejich rozdělení na enthalpie jednotlivých iontů je založeno na řadě různých předpokladů, takže přímé porovnání s G je velmi nejisté. V případech, kde se to zdá být oprávněné, výsledky mohou být překvapující. Např. azidový iont je značně destabilizován přechodem z methanolu do DMSO ( G = 17.6 kJ/mol), ale přechod je ve skutečnosti exotermní H = -4.2 kJ/mol. V DMSO musí být azid silněji vázán než v methanolu bez ohledu na absenci vodíkové vazby, ale vzniklý solvát je vysoce orientovaný a S = -73 J/K mol. K úplné termodynamické analýze nám chybí hodnoty H a §S které mohou být získány způsobem zobrazeným na minulém obrázku z měření reakčních rychlostí pro každé rozpouštědlo pro řadu teplot.
Empirické korelace rozpouštědlových efektů
Rigorózní analýza uvedená v předcházejícím odstavci je poměrně mladého data a mnohem širší uplatnění nalezly empirické kvantitativní korelace. Jestliže nepolární reaktant vytváří nepolární tranzitní stav, nebude mít solvatace zřejmě výrazný vliv na reakční rychlost. Např. rychlost Diels - Alderovy dimerace cyklopentadienu je za stejné teploty prakticky stejná v plynné fázi, v substanci i v roztoku rozpouštědel tak různých, jako jsou parafín a kys. octová. Na druhé straně, jestliže v průběhu reakce dochází k výrazné změně nábojové distribuce, potom rozpouštědlo bude pravděpodobně hrát značnou roli. Z kvalitativního pohledu platí - jestliže tranzitní stav je polárnější než reaktanty, bude stabilizován polárními rozpouštědly a reakce bude tedy urychlována a naopak. Klasicky byly tyto efekty studovány na příkladě nukleofilních substitučních reakcí, kde může vzniknout celá řada situací s různou nábojovou distribucí. Reakce, které vycházejí z relativně nepolárních reaktantů ( reakce
1-4 ), vedou k tranzitním stavům s významnou separací náboje. V reakcích 2, 3 a 6 je jeden z reaktantů iont, jehož náboj je delokalizován v tranzitním stavu. V reakci 5 mezi opačně nabitými ionty dochází k značné redukci nábojové separace a tranzitní stav, ačkoliv je dipolární, je elektroneutrální.
|
Typ reakce |
Náboje v zákl. stavu |
Náboje v tranzitním stavu |
Zvýšení polarity rozp. |
|
SN1 1 |
R - Z |
Rd .Z d |
značné urychlení |
|
2 |
R - Z+ |
Rd . Z d |
malé zpomalení |
|
SN2 3 |
X- + RZ |
Xd . R Z d |
malé zpomalení |
|
4 |
X + RZ |
Xd . R Z d |
značné urychlení |
|
5 |
X- + RZ+ |
Xd . R Z d |
značné zpomalení |
|
6 |
X + RZ+ |
Xd . R Z d |
malé zpomalení |
Bylo nalezeno mnoho konkrétních příkladů v souladu s tímto obecným modelem. SN2 hydrolýza methyl- nebo ethyljodidu OH iontem ve vodném ethanolu (typ 3) je zpomalována vzrůstající koncentrací vody, ale SN1 solvolýza terc.butylchloridu (typ 1) je urychlována. V případě, že odcházející skupina je kladně nabitá, je realita jiná, než je předvídána. Jak hydrolýza Me3S+ hydroxidovým iontem (typ 5), tak solvolýza t-butylSMe2+ (typ 2) ve vodném ethanolu jsou zpomalovány přídavkem vody. Menschutkinova reakce (Et3N + EtI, typ 4) je urychlována rostoucí polaritou rozpouštědla v pořadí hexan < benzen < aceton < ethanol < methanol, ale reakce Me3N s Me3S+ (typ 6) je zpomalována polárními rozpouštědly: voda < methanol < ethanol < nitromethan.
Podobných experimentálních poznatků byla nalezena celá řada a byla více či méně exaktně analyzována. Cílem bylo nalézt pokud možno univerzální popis důsledků změny rozpouštědla. Vznikla řada empirických rovnic od nejjednodušších lineárních až po užití multiregresních metod. I přes to, že bylo dosaženo některých povzbudivých výsledků, není cílového stavu dosud dosaženo. Získané vztahy sice naznačují některá zobecnění, ale ucelená teorie dosud neexistuje.
Efekt izotopově značeného rozpouštědla
Rychlosti reakcí prováděných v těžké vodě nebo v deuterovaném alkoholu jsou často velmi odlišné od rychlostí reakcí prováděných v normálních rozpouštědlech. To může být důsledkem primárního izotopového efektu, jestliže proton z rozpouštědla nebo jiné částice, která jej s rozpouštědlem vyměňuje, je účastníkem reakčního kroku, který určuje rychlost. Velikost tohoto efektu může být většinou vypočtena s rozumnou přesností s pomocí infračervených vazebných vibrací a z odhadu rozsahu, v jakém je izotop přenesen v tranzitním stavu (např. pomocí Brønstedových koeficientů).
Sekundární izotopový efekt může být také velmi důležitý. Může mít dva zdroje: a) izotopově značená částice vstupuje do reakce, byť nedochází k přenosu izotopu. Např. nukleofilita HO- nemusí být stejná jako DO-. b) druhou příčinou je rozdílná solvatace. Např. D2O je více uspořádaná než H2O.
Velikost sekundárního izotopového efektu lze měřit na základě rovnovážné konstanty rovnice:
![]()
kde ROH je rozpouštědlo. Rovnovážná konstanta této reakce (nazývaná také izotopový frakční faktor) je mírou rozsahu, v jakém sekundární izotopový efekt upřednostňuje tvorbu XD vůči XH v závislosti na poměru ROD/ROH.
![]()
Bylo nalezeno, že f závisí především na druhu a náboji atomu, na který je mobilní vodík vázán. Např.:
|
fO = 1.0 |
fO |
fO |
|
fN = 0.9 |
fN |
fO |
Tyto faktory mohou být užity k odhadu vlivu izotopového efektu rozpouštědla na polohu rovnováh. Pro rovnováhu dvou systémů XH a YH platí :
![]()
KH = [YH]/[XH] KD = [YD]/[XD]
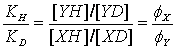
Tento poměr je samozřejmě přesně aplikovatelný jen na měření ve směsi ROH/ROD, ale je použitelný i pro porovnání rovnováh měřených v čisté H2O a D2O. Pro rovnováhy zahrnující několik izotopově odlišitelných poloh se f faktory násobí. Uvažujme disociace XOH kyseliny:
![]()
V produktu jsou tři H-O+ vazby, v reaktantech tři H-O vazby. Jestliže všechny protony jsou zaměněny za deuterium, platí:
KH/KD = (fO fO
To ukazuje, že kyseliny jsou přibližně 3x silnější ve vodě než v těžké vodě. Jinak řečeno D3O+ je 3x silnější kyselinou než H3O+. Podobně DO- je silnější než HO-.
![]()
KH/KD = (fO fO . fO
Analýza kinetického izotopového rozpouštědlového efektu vyžaduje odhad vlivu tohoto efektu na tranzitní stav. Jednoduchý přístup předpokládá, že sekundární efekt se mění pravidelně se zánikem a vznikem vazby, tedy že kinetický efekt bude průměrem vypočteného efektu pro systémy na obou stranách rychlost determinujícího kroku. Např. pro kysele katalyzované otevírání ethylenoxidu můžeme uvažovat SN1 i SN2 mechanismus:
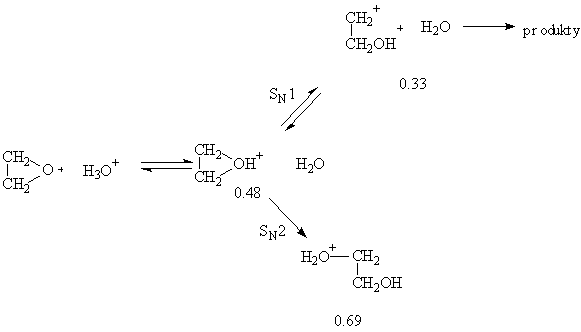 Hodnoty
udávají poměr KH/KD pro různé stupně reakce. Pro SN1
reakci by podle Hammondova postulátu měl tranzitní stav ležet blíže
k acyklickému karbokationtu a KH/KD by měl být cca
0.33. Pro SN2 mechanismus bude tranzitní stav blíže oxoniovému iontu
a KH/KD by měl být kolem 0.48. Pozorovaná hodnota 0.45
není tedy úplně přesná, přesto zcela jasně ukazuje na převahu SN2
mechanismu.
Hodnoty
udávají poměr KH/KD pro různé stupně reakce. Pro SN1
reakci by podle Hammondova postulátu měl tranzitní stav ležet blíže
k acyklickému karbokationtu a KH/KD by měl být cca
0.33. Pro SN2 mechanismus bude tranzitní stav blíže oxoniovému iontu
a KH/KD by měl být kolem 0.48. Pozorovaná hodnota 0.45
není tedy úplně přesná, přesto zcela jasně ukazuje na převahu SN2
mechanismu.
Kinetický efekt elektrolytů
Rychlost řady reakcí, zvláště těch, které zahrnují ionty, je velmi ovlivňována přídavkem elektrolytů, i když ty se vlastní reakce nezúčastní. Příčina tohoto chování, nazývaného efekt solí, tkví v elektrostatické interakci mezi ionty, což způsobuje odchylky od ideálního chování i při velmi nízkých koncentracích. Tyto odchylky závisí na všech iontech a platí pro všechny ionty přítomné v roztoku. Změna koncentrací nereagujících iontů ovlivňuje aktivity reagujících iontů. Uvažujme reakci, v níž dvě výchozí látky tvoří tranzitní stav a posléze produkty:
![]()
![]() A + B AB produkty
A + B AB produkty
rychlost = n AB
kde n je rychlost rozpadu tranzitního stavu. Za neideálních podmínek musí být tato pseudorovnovážná konstanta vyjádřena v aktivitách:
K = aAB /aAaB = y‡ AB ]/yAyB[A][B]
rychlost = k[A][B] = n K yAyB/ y‡)[A][B]
k = n K yAyB/ y‡
Použijeme-li nyní Debye-Hückelovu teorii k vyjádření aktivitních koeficientů iontů
log y = -Dz2I1/2
kde D je konstanta pro dané rozpouštědlo a danou teplotu, z je náboj iontu a I je iontová síla roztoku definovaná jako:
I = 0.5Scizi, kde ci je koncentrace iontu.
Náboj v tranzitním stavu tedy musí být z‡ = zA + zB. Kombinací předešlých rovnic získáváme:
log k = log n K - D I1/2 = log n K + 2D zAzB I1/2
Pro extrémní případ, kdy I = 0, se rovnice zjednoduší na
log (k/k0) = 2D zAzB I1/2, kde k0 je rychlostní konstanta při nekonečném zředění. Ve vodě při 25oC je 2D = 1
a rovnice má tvar:
log (k/k0) = zAzB I1/2
Ve zředěných roztocích platí tento vztah prakticky kvantitativně. Vzrůst iontové síly roztoku zvyšuje jeho polaritu. Reakce mezi ionty s kvalitativně stejným nábojem je potom urychlována přídavkem elektrolytů a mezi ionty opačného znaménka je zpomalována. Odchylky od této rovnice se vyskytují většinou v případě reakcí zahrnujících neutrální molekuly, které přesto často vykazují efekt elektrolytu. Mnoho reakcí mezi neutrálními, relativně nepolárními reaktanty probíhá přes tranzitní stav, který, byť je nenabitý, je dipolární. Může být tedy stabilizován iontovou interakcí a vykazuje veliký kladný efekt elektrolytu. Na druhé straně i reakce mezi ionty a neutrálními molekulami často vykazuje malý záporný efekt elektrolytů, poněvadž náboj v tranzitním stavu je více delokalizován než v iontovém reaktantu. Existence elektrolytického efektu má zvláštní význam v provádění kinetických experimentů, protože není vždy zcela jasné, jestli změna v reakční rychlosti je důsledkem skutečné účasti přidaných iontů v reakci nebo jen efektem elektrolytu. Je vhodné provést kontrolní experiment za užití inertního elektrolytu pro srovnání s chováním potenciálně reaktivní částice. Experiment také může být proveden v přítomnosti velkého přebytku inertního elektrolytu, takže iontová síla zůstává prakticky konstantní. Popsaný efekt se nazývá primárním kinetickým elektrolytickým efektem, protože přímo ovlivňuje rychlost determinující krok. Ionty mohou také ovlivňovat reakční rychlost změnou polohy rovnováhy předcházejícího kroku a tento jev se potom se nazývá sekundární kinetický elektrolytický efekt. Často se vyskytuje u reakcí kysele katalyzovaných, kde rychlost je úměrná [H3O+]. Jestliže katalyzátor HA je relativně slabou kyselinou, potom [H3O+] může být ovlivněna působením elektrolytu na disociaci. Úpravou této rovnice a aplikací Debye-H ckelovy teorie dostáváme:
log [H3O ] = log Ka - log [A [HA] + I1/2
To ukazuje, že vzrůstající iontová síla urychluje reakce posunutím protonační rovnováhy.
Elektrolyty ovlivňují podobným způsobem i bazicky katalyzované reakce - katalýza relativně slabou neutrální bází, jako je amoniak, je zvyšována rostoucí iontovou silou, protože ta zvyšuje [OH-].
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1284
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved