| CATEGORII DOCUMENTE |
| Alimentatie nutritie | Asistenta sociala | Cosmetica frumusete | Logopedie | Retete culinare | Sport |
Diabetul - factor de risk major pentru ateroscleroza
Multe studii experimentale "in vitro" demonstreaza ca ateroscleroza este o boala inflamatoare caracterizata de infiltrarea celulelor inflamatoare si de expresia citokinelor [Libby 2001]. Ateroscleroza este o conditie inflamatorie cronica care implica interactii complexe intre celulele inflamatorii (neutrofile, limfocite, monocite/macrofage) si celulele vasculare (CE, CMN). CE primesc semnale de la factori umorali, mediatori inflamatori si forte fizice din circulatie si tesut. Unii din activatorii capabili de inducerea raspunsurilor celulare proinflamatoare si protrombotice au fost identificati, acestia incluzand: lipoproteine modificate, citokine proinflamatoare, chemokine, peptide vasoactive, neuropeptide, hiperglicemia si produsii AGE, fumatul, stresul oxidativ si altii [Ross 1999].
Printre evenimentele initiale ale aterogenezei se numara disfunctia endoteliala si depunerea si retentia particulelor lipoproteice aterogenice bogate in apolipoproteina B in spatiul subendotelial [Simionescu et al., 1986; Williams & Tabas, 1995]. Aceasta depunere promoveaza inflamatia locala, caracterizata de eliberarea factorilor de semnalizare solubili, incluzand chemokine [Gerszten et al., 1999] si de expresia moleculelor de adeziune celulara endoteliala, ca VCAM-1, ICAM-1 si P-slectina. Aceste molecule recruteaza leucocitelor mononucleare, in special monocite, in peretele arterial. Avand acces la spatiul subendotelial, monocitele recrutate se diferentiaza in macrofage, proces controlat de interactiile monocitelor cu matricea extracelulara si citokinele din subendoteliu.
Rolul CE si CMN este de a integra semnale generate de la diferitii agonisti pentru a regla efectiv raspunsul imuno-inflamator, prin expresia moleculelor de adeziune, citokine, chemokine, metaloproteaze matriceale (MMP) si factori de crestere. Producerea acestor mediatori inflamatori duce la migrare celulara, proliferare, producere de matrice extracelulara si dezvoltarea placii [Libby & Hansoon 1991, Tedgui & Mallat 2001]. In timpul inflamatiei, multiple citokine sunt prezente la situsurile inflamate, fiecare din acestea putand influenta natura raspunsului inflamator [Stllberger & Finsterer 2002]. Citokinele proinflamatoare pot fi implicate in destabilizarea si ruperea placii aterosclerotice. Prezenta unui proces inflamator caracterizeaza locul de ruptura sau eroziune a placii, iar citokinele proinflamatoare cresc expresia MMP, care se stie ca sunt implicate in remodelarea vasculaturii si ruperea placii [Blake & Ridker 2001]. Nivelele crescute ale citokinelor proinflamatoare sunt markeri ai bolilor cardiovasculare.
Diabetul mielitus a fost recunoscut ca factor de risc cardiovascular major, independent, inca de la aparitia primelor investigatii epidemiologice la scara larga din 1970s [Kannel &McGee 1979]. In lume exista peste 100 milioane de persoane cu diabet (5-8% din populatia totala) si se asteapta ca acest numar sa creasca semnificativ in viitor. Corespunzator cu datele din studiile clinice, majoritatea persoanelor diabetice mor din cauza bolilor cardiovasculare, iar ateroscleroza este cauza a 8 din 10 din cei care mor din cauza diabetului [Gu et al., 1998]. Diabetul mielitus, in absenta unei boli cardiovasculare anterioare, confera un risc de evenimente cardiovasculare similar cu al persoanelor nondiabetice care au avut deja un infarct miocardic anterior, reprezentand astfel "risc coronarian echivalent". Astfel, pacientii diabetici trebuiesc inclusi in programe de preventie secundara, in functie de istoria lor cardiovasculara [Haffner et al., 1998; Marso 2002]. In studiile clinice si experimentale recente s-a aratat ca pacientii diabetici care constituie intre 15 si 20% din pacientii cu sindrom coronarian acut prezinta un risc considerabil crescut de morbiditate si mortalitate, comparativ cu pacientii nondiabetici, riscul de evenimente adverse la diabetici fiind de doua ori mai mare decat la cei nondiabetici.
Diabetul si bolile metabolice inrudite, ca hiperinsulinemie, rezistenta la insulina si obezitate sunt recunoscute ca contributori majori la morbiditatea si mortalitatea cardiovasculara.
Dovezi recente au aratat rolul semnificativ si independent al inflamatiei sistemice si coronariene in initierea, progresia si agravarea aterosclerozei, suprapunandu-se cu factorii traditionali de risc [Ross 1999]. Data fiind cresterea propagarii diabetului si sindromului dismetabolic, raspandirea aterosclerozei la diabetici, posibilele sinergii ale diabetului si inflamatiei in ateroscleroza, precum si potentialul de preventie si beneficii terapeutice in modularea inflamatiei la diabetici sunt subiecte actuale care asteapta rezolvarea.
1. Infalmatie, diabet, ateroscleroza
Procesul patofiziologic al aterosclerozei a fost sistematic sumarizat in cativa pasi, fiecare din ei fiind cararcterizat de implicarea semnificativa a elementelor umorale si inflamatoare celulare [Ross 1999]. Atat diabetul cat si ateroscleroza sunt conditii multifactoriale care par sa aiba o baza inflamatoare comuna [Pickup & Crook 1998; Fernandez-Real & Ricart 1999 ]. Intr-adevar, interactia dintre inflamatie, diabet si ateroscleroza par sa sustina teoria conform careia sursele vasculare si non-vasculare ale inflamatiei sistemice pot fi importante in patologia ambelor boli. Studii patologice, angiografice si alte studii in vivo au aratat ca diabetul favorizeaza progresia accelerata a aterosclerozei [Dortimer et al., 1978]. Placile diabetice sunt de asemenea mai complicate si reprezinta un risc mai mare pentru complicatiile ulterioare. In plus, persoanele diabetice care mor brusc au un numar crescut de placi aterosclerotice fisurate, comparativ cu nondiabeticii [Davies 1989]. De fapt, rezultatele examinarii angioscopice arata ca pacientii diabetici cu angina instabila au o incidenta mai mare de ulcerare a placii si de formare a trombusilor intracoronarieni decat pacientii nondiabetici, si de regula, placile diabetice au un miez lipidic mai mare si componenta inflamatoare mai bogata, fiind de regula mai complicate datorita trombozei suprapuse [Moreno et al., 2000].
Concentratiile crescute de glucoza duc la disfunctia endoteliala care este asociata cu scaderea nivelelor de NO derivat de la endoteliu, cresterea permeabilitatii vasculare, crestrea moleculelor de adeziune si ca urmare a adezivitatii endoteliale si ingrosarea membranei bazale a vaselor de sange [Stehouwer et al., 1997]. De asemenea, formarea AGE de catre glucoza crescuta induce stres oxidativ, inflamatie si disfunctie vasculara [Brownlee et al.,1988; Schmidt et al., 1994; Schmidt et al., 1995; Wautier et al., 1996]. Scaderea productiei de NO este foarte importanta, fiind asociata cu inflamatia indusa de glucoza. In afara de proprietatile vasodilatatoare binecunoscute, oxidul nitric are functii multiple in vasculatura. Astfel, studiile au arat ca NO inhiba procesul inflamator prin reducerea interactiilor leucocite-endoteliu la nivel celular si molecular [Kubes et al., 1991; De Caterina et al., 1995]. NO inhiba selectiv expresia VCAM-1 indusa de IL-1 pe CE, ceea ce se reflecta in reducerea procesului de adeziune a monocitelor la monostratul endotelial. De asemenea, NO scade expresia endoteliala a altor molecule de adeziune celulara (E-selectina si mai putin ICAM-1) si a citokinelor secretabile (IL-6 si IL-8). Tehnicile moleculare arata ca NO reduce transcriptia genica a VCAM-1, partial prin inhibarea NF-kB [De Caterina et al., 1995]. Tot prin inhibarea NF-kB in celulele endoteliale, NO reduce expresia factorului stimulator de colonii de macrofage (MCSF), un factor important care regleaza formarea celulelor spumoase in leziunile aterosclerotice [Peng et al., 1995]. Mecanismele exacte ale inhibitiei NF-kB mediate de NO sunt probabil legate de inducerea si stabilizarea moleculei IkBa care are rolul de a inhiba translocarea (si deci activarea) NF-kB in nucleu.
Interactiile dintre leucocite si endoteliu sunt un aspect important al procesului inflamator. Endoteliul vascular directioneaza leucocitele catre locurile cu leziuni vasculare ca raspuns la stimuli inflamatori, pentru a preveni infectia. In timp ce acest proces inflamator este necesar pentru a preveni bolile infectioase, o initiere anormala a raspunsului inflamator in timpul conditiilor patologice ca hiperglicemia si diabetul, pot cauza leziuni ale vaselor si ale tesutului inconjurator datorita producerii de mediatori toxici (specii reactive de oxigen si citokine) de catre endoteliul disfunctional sau de leucocitele activate [Weiss 1989]. In acest sens, s-a demonstrat aparitia leziunilor vasculare in microcirculatia din retina diabetica, datorita sechestrarii leucocitelor [Miyamoto et al., 1998]. Mai mult, traficul patologic al leucocitelor poate avea un rol important in dezvoltarea bolilor macrovasculare, traficul crescut al leucocitelor in microcirculatia din vasa vasorum contribuind la patogeneza aterosclerozei [Libby 2001].
Desi este plauzibil sa acceptam diabetul ca declansator/inductor al inflamatiei vasculare, procesul invers (inflamatia sa induca diabetul) este de asemenea adevarat, dovezi substantiale aratand ca inflamatia (de grad scazut) este un determinant patogenic important al diabetului de tip 2. Intr-un studiu de preventie coronariana din Scotia de Vest s-a aratat ca nivelele de CRP cresc riscul dezvoltarii in viitor a diabetului de tip 2 si ca acest risc este independent de greutatea corpului, de nivelele crescute de trigliceride sau glucoza, sau tratamentul cu statine [Freeman et al., 2002]. In plus, numarul crescut de leucocite a fost un predictor independent al unei actiuni inrautatite a insulinei si pentru dezvoltarea diabetului de tip 2 la indienii Pima [Vozarova et al., 2002] si la femeile adulte din U.S. [Ford 2002].
Mai recent, dovezi clinice solide au confirmat rolul patogenic al inflamatiei in aparitia diabetului, aratand ca agentii antiinflamatori ca statine [Freeman et al., 2001], agonistii de PPAR (peroxisome proliferator- activated receptors) [Buchanan et al., 2002] si alte medicamente, incluzand inhibitori ai enzimei de conversie a angiotensinei [Haffner et al., 1998], pot preveni aparitia diabetului la subiectii cu risc crescut.
Aceste rezultate epidemiologice sunt intarite de studii experimentale care demonstreaza efectele hiperglicemiei asupra unor citokine pro-inflamatoare, incluzand IL-6 si TNFα, derivate in special din tesutul adipos. In modelele de rozatoare diabetice, IL-6 modifica eliberarea insulinei stimulata de glucoza din celulele β pancreatice [Sandleret al., 1990] si diminueaza sinteza glicogenului stimulata de insulina in hepatocitele in cultura [Kanemaki et al., 1998]. La oameni, administrarea de IL-6 exogen induce hiperglicemie proportional cu doza folosita, si cresterea corespunzatoare a nivelelor de glucagon [Tsigos et al., 1997]. TNFα poate induce rezistenta la insulina printr-o varietate de mecanisme, incluzand efecte inhibitorii directe asupra proteinei transportor de glucoza, GLUT4, asupra receptorului insulinei si substratelor receptorului insulinei [Hotamisligil & Spiegelman 1994].
In ciuda acestor rezultate, datele de perspectiva care evalueaza relatia dintre inflamatia cronica si incidenta diabetului de tip 2 sunt rare. Intr-un studiu care a urmarit riscul de aparitie al aterosclerozei, markeri ai inflamatiei ca numarul limfocitelor, fibrinogenul si albumina serica scazuta dar si variabilele hemostazei asociate diabetului ca factorul VII si factorul von Willebrand au fost asociate cu riscul diabetului de tip 2 [Duncan et al., 1999]. Totusi, aceasta asociere a fost abolita dupa ajustarea la obezitate.
Daca moleculele de semnalizare adipocitare actioneaza in acord cu mediatori ai inflamatiei implicand un rol esential pentru inflamatie in diabetogeneza, sau daca markeri inflamatori sunt pur si simplu co-eliberati in paralel cu alte substante patogene, ramane o problema care necesita studii suplimentare.
De asemenea, este posibil ca disfunctia endoteliala microvasculara sa fie implicata in dezvoltarea rezistentei la insulina, furizand probabil, o legatura in plus intre diabetul de tip 2 si ateroscleroza [Pinkney et al., 1997]. Endoteliul arteriolar si capilar joaca un rol important in reglarea fluxului de sange in tesuturile sensibile la insulina, ca ale ficatului si muschilor scheletali, functionand de asemena ca bariera pentru transportul insulinei la compartimentele interstitiale. Esecul transportului insulinei la tesuturile active metabolic in starile dinamice, de-a lungul perioadei hiperglicemice poate fi un pas care limiteaza masura de determinare a eficacitatii insulinei. In plus, microalbuminuria, un rezultat al disfunctiei endoteliale renovasculare este puternic predictiva pentru bolile cardiovasculare atat la indivizii diabetici, cat si la cei ne-diabetici si este prezenta uzual in cazul rezistentei la insulina si a diabetului de tip 2. Mai mult, studii experimentale au aratat ca in vivo, vasodilatarea dependenta de endoteliu este afectata de administratrea exogena a citokinelor proinflamatoare [Hingorani et al., 2000]. In ce masura expunerea cronica la nivele scazute de inflamatie induce sau exacerbeaza rezistenta la insulina prin acest proces, sau alternativ, prin afectarea permeabilitatii vasculare nu este pe deplin inteles.
2. Anormalitati dislipidemice in diabet
Metabolismul anormal al lipoproteinelor bogate in trigliceride este crucial in patofiziologia dislipidemiei aterogenice in diabet [Simionescu et al., 1996]. Astfel, rezistenta la insulina si diabetul de tip 2 sunt asociate cu o acumulare/concentrare de lipide plasmatice si anormalitati ale lipoproteinelor, care include scaderea colesterolului HDL, preponderenta de particule de LDL dense si mici, si nivele crescute de trigliceride [Haffner et al., 1990, 2000]. Fiecare din aceste aspecte sunt asociate cu un risc crescut pentru bolile cardiovasculare. Numeroase studii au demonstrat o asociere intre marimea si densitatea LDL si bolile arterelor coronare [Austin et al., 1988; Campos et al., 1992; Coresh et al., 1993; Gardner et al., 1996; Lamarche et al., 1997]. Mai mult, studii recente au aratat ca concentratiile particulelor de LDL, si in special nivelele de LDL mai dense si mai mici, sunt predictive pentru evenimentele coronariene, si ca acest lucru este independent de alti factori de risc ai bolilor arterelor coronare [Blake et al., 2002; Rosenson et al., 2002; St-Pierre et al., 2003]. Cel mai adesea diabetul este asociat cu particule LDL mai mici si mai dense si astfel mai susceptibile la oxidare, avand grad mai mare de aterogenicitate [Ross 1999]. In plus, glicarea LDL si a altor lipoproteine este un proces destul de intalnit in diabet, si face ca lipoproteinele pacientilor diabetici sa fie mai susceptibile la oxidare si mai aterogenice.
Cresterea secretiei hepatice de VLDL mari, bogate in trigliceride si diminuarea preluarii VLDL pare sa aiba o importanta centrala in patofiziologia acestei dislipidemii. Tipic, nivelele plasmatice reduse de HDL s-au dovedit a fi datorate reducerii subspeciei HDL2b si cresterii relative sau absolute ale HDL3b si HDL3c, care sunt mai mici si mai dense [Krauss 2004]. Reducerea HDL asociata cu diabetul de tip 2 este un proces multifactorial, insa, factorul major pare sa fie transferul crescut de colesterol de la HDL la lipoproteinele bogate in trigliceride, cu transfer reciproc de trigliceride la HDL. Particulele de HDL bogate in trigliceride sunt hidrolizate de lipaza hepatica si apoi sunt catabolizate si preluate rapid din plasma [Hopkins & Barter 1986].
Unele studii clinice au aratat beneficiile strategiilor de scadere a lipidelor la diabetici, chiar si in cazurile de crestere modesta sau nivele normale ale colesterolului, aceste beneficii fiind adesea mai importante decat la pacientii nondiabetici [The Heart Protection Study 2002].
3. Rolul citokinelor in ateroscleroza indusa de diabet
Observatiile experimentale au demonstrat rolul central al unor retele de citokine si chemokine in procesul aterosclerotic [Ross 1999]. In aceasta privinta, diabetul, rezistenta la insulina, intoleranta la glucoza par sa aiba toate efectele modulatoare importante, cu activitate pro-aterosclerotica globala. Cel putin partial, ca o consecinta a hiperglicemiei si imbalantei in homeostazia insulinei, diabetul induce anormalitati in expresia si activitatea diferitelor citokine si peptide vasoactive ca TNFa, IL-6, angiotensina II, endotelina-1 [Kroder et al 1996]. Nivelele serice crescute ale TNFa, IL-6 si a unor molecule de adeziune celulara sunt predictori importanti ai evenimentelor cardiovasculare ulterioare la subiectii sanatosi precum si la cei diabetici. La pacientii diabetici, nivelele crescute de VCAM-1 sunt asociate cu mortalitate cardiovasculara [Jager et al., 2000]. IL-1 si TNFa sunt produse de celulele inflamatoare in placa aterosclerotica si induc la randul lor productia de IL-6 de celulele din placa, inclusiv de catre celulele musculare netede. In plus, IL-6 circulant stimuleaza axa hipotalamica-pituitara-adrenala, activare care e asociata cu obezitatea, hipertensiunea si rezistenta la insulina [Yudkin et al., 2000]. Pacientii hiperglicemici au o productie crescuta de produsi finali ai glicarii avansate (AGE), care sunt recunoscuti specific de celulele inflamatoare din placa aterosclerotica si induc eliberarea citokinelor inflamatoare. Acesti mediatori stimuleaza proliferarea endoteliala si a celulelor musculare netede, activarea endoteliala si supraproductia de colagen, contribuind astfel la cresterea si progresia placii. Proteina C reactiva (CRP) - un marker nespecific al inflamatiei, care are de asemenea o activitate inflamatoare directa in ateroscleroza, a fost asociata cu efecte cardiovasculare adverse la pacientii cu boli arteriale coronariene, precum si la subiectii sanatosi [Liuzzo et al., 1994; Ridker et al., 1997]. Interesant, nivelele de CRP sunt mai mari la pacientii diabetici decat la cei fara diabet. Desi nivelele crescute de CRP la diabetici pot depinde de activarea sau disfunctia adipocitelor, o ipoteza suplimentara este ca nivelul crescut de CRP poate fi asociat cu un proces inflamator scazut, datorat patogenilor inflamatori, infectiile fiind uzuale in diabet [Danesh et al., 1997].
4. Anormalitati genetice
Cateva anormalitati genetice ale metabolismului glicemic au fost asociate cu dezvoltarea diabetului, complicatiile diabetice, sau cu aspecte ale sindromului dismetabolic. Toate aceste modificari pot fi de asemenea implicate in ateroscleroza.
Factorii de transcriptie ca PPAR a si g sunt acum studiati intensiv. In timp ce PPARa stimuleaza in primul rand degradarea oxidativa beta a acizilor grasi, PPARg promoveaza stocarea lipidica prin regularea diferentierii adipocitelor. PPARa este tinta moleculara a medicamentelor ce scad concentratia lipidica si inflamatia, iar PPARg interactioneaza cu medicamentele sensibile la insulina ca tiazolidinedione, si pot modula semnificativ ateroginicitatea si inflamatia. In particular, recent s-a aratat ca mutatii ale genei PPARg sunt asociate cu diabetul, intoleranta la glucoza si hipertensiune [Barroso et al., 1999]. Rolul factorilor de transcriptie in homeostazie si boli este complex si inca neclar datorita heterogenitatii, specificitatii celulare, si multitudinii de efecte ale acestora. Totusi, dovezi substantiale au aratat ca PPAR interactionaeaza intracelular si inhiba activarea unui alt factor de transcriptie (NF-kB) ale carui activitati sunt in principal proinflamatoare si proliferative. Soarecii deficienti la PPARa au un raspuns sistemic in faza acuta prelungit si reactie inflamatoare imuna si inascuta locala la stimuli daunatori obisnuiti, in timp ce activarea PPARa pare sa scada expresia VCAM-1 in celulele endoteliale activate [Marx et al., 1999]. In plus, PPARa scade selectiv activitatile secretoare si proliferative ale celulelor musculare netede [Staels et al., 1998]. In particular, liganzii acestui factor de transcriptie inhiba productia de IL-6 si prostaglandina si expresia COX-2 induse de IL-1 in celulele endoteliale. La pacientii hiperlipidemici, activarea PPARa dependenta de fenofibrat scade nivelul de IL-6, fibrinogen si proteina C reactiva (CRP) [Staels et al., 1998]. PPARg activati de tiazolidinedione inhiba proliferarea si migrarea celulelor musculare netetede vasculare. In celulele endoteliale, agonistul de PPARg, troglitazona, scade semnificativ expresia VCAM-1 si ICAM-1 indusa de TNFa, in timp ce soarecii apoE deficienti tratati cu troglitazona au numar redus de monocite/macrofage in placile aterosclerotice [Pasceri et al., 2000]. In final, activarea combinata a PPARa si g pare sa scada productia MCP-1 indusa in celulele endoteliale de CRP, efect similar cu al simvastatinului. In plus, pacientii cu diabet de tip 2 tratati cu troglitazona au nivele crescute ale IkB (proteina inhibitoare a factorului de transcriptie NF-kB), implicit activitate NF-kB redusa, precum si nivele sistemice ale speciilor reactive de oxigen, si ale CRP [Aljada et al., 2001].
Expunerea prelungita la hiperglicemie este cunoscuta ca factor primar in patogeneza complicatiilor diabetice [Laakso 1999; Grundy et al., 1999]. Hiperglicemia induce un numar mare de alterari in tesutul vascular care promoveaza ateroscleroza accelerata. In prezent au fost descrise trei mecanisme majore care sunt implicate in modificarile patologice observate in vasculatura persoanelor diabetice si a animalelor diabetice: 1) glicarea ne-enzimatica, 2) activarea protein kinazei C (PKC) si 3) stresul oxidativ. Important este ca aceste mecanisme nu sunt independente. De exemplu, stresul oxidativ indus de hiperglicemie promoveaza formarea produsilor finali de glicare avansata (AGE) si acivarea PKC [Nishikawa et al., 2000]
1) AGE
Efectele hiperglicemiei sunt adesea ireversibile si duc la disfunctie celulara progresiva [Fioretto et al., 1998]. De exemplu, la pacientii diabetici cu transplant pancreatic functional, patologia renal continua sa progreseze pentru cel putin 5 ani dupa ce diabetul a fost tratat [Fioretto et al., 1998]. Mecanismul implicat in acest proces nu este clar, dar sugereaza ca perturbarile celulare pot persista in ciuda normalizarii concentratiei de glucoza (asa-numitul "efect-memorie"). Astfel, mai degraba persistente decat tranziente, modificarile metabolice acute sunt de importanta majora in patogeneza complicatiilor diabetului.
Unele dintre mecanismele importante, responsabile de ateroscleroza accelerata in diabet este reactia ne-enzimatica dintre glucoza si proteinele sau lipoproteinele din peretele arterial. Glucoza formeaza produsi de glicare timpurii, reversibili cu gruparile amino reactive ale proteinelor circulante sau din peretele vascular (baze Schiff). Unii din produsii de glicare timpurii continua sa sufere o serie complexa de rearanjamente chimice si formeaza produsi finali de glicare avansata (AGE) [Brownlee et al., 1988]. Odata formati, acesti produsi sunt stabili si virtual ireversibili. AGE se acumuleaza continuu pe proteinele peretelui vascular in imbatranire si la o rata accelerata in diabet [Brownlee et al., 1988]. Gradul de glicare ne-enzimatica este determinat in principal de concentratia de glucoza si de timpul de expunere. Totusi, un alt factor critic pentru formarea AGE este potentialul redox al micromediului tesutului. Astfel, in situatii in care potentialul redox local a fost modificat in stres oxidativ, formarea AGE a cescut substantial [Nishikawa et al., 2000; Giardino et al., 1996; Schmidt et al., 1999].
La randul lor, AGE pot accelera procesul de ateroscleroza prin diverse mecanisme, care pot fi clasificate ca a) independente de receptor, sau b) mediate de receptor.
a) mecanisme independente de receptor
Glicozilarea proteinelor si lipoproteinelor poate interfera cu functia lor normala prin distrugerea conformatiei moleculare, modificarea activitatii enzimatice, capacitate de degradare redusa si interferenta cu recunoasterea receptorului. Astfel, modificari in fiziologia normala a proteinelor care sunt relevante pentru ateroscleroza pot promova ateroscleroza la indivizii diabetici.
Poate cel mai studiat exemplu este interferenta cu fiziologia normala a particulelor de lipoproteine de densitate scazuta (LDL). Procesul de glicozilare are loc atat pe apolipoproteina B (apoB) [Bucala et al., 1995], cat si pe componentele fosfolipidice [Bucala et al., 1993] ale LDL, ducand la modificari functionale in preluarea LDL si cresterea susceptibilitatii la modificari oxidative.
Studiile clinice au aratat un nivel crescut al AGE la LDL obtinut de la pacientii diabetici, comparativ cu indivizii normali [Bucala et al., 1993; 1994]. Glicozilarea apoB (proteina de la suprafata LDL) a LDL are loc in pincipal la un reziduu de lizina incarcat pozitiv, din domeniul de legare a receptorului LDL, care este esential pentru recunoasterea specifica a LDL-ului de catre receptorul acestuia. Glicozilarea LDL creste proportional cu nivelele de glucoza, nivelele AGE-apoB fiind de 4 ori mai mari la pacientii diabetici [Bucala et al., 1993]. Glicozilarea apoB duce la o diminuare semnificativa a preluarii LDL mediate de receptor, scazand preluarea in vivo a LDL glicozilat, comparativ cu LDL nativ [Steinbrecher & Witztum 1984]. In plus, macrofagele umane derivate din monocite recunosc LDL glicozilat intr-o masura mult mai mare decat LDL nativ [Klein et al., 1995]. Preluarea LDL glicat de catre macrofage nu este mediata de receptor LDL specific, ci de receptori nespecifici (scavenger) prezenti pe macrofagele umane. Pentru ca glicozilarea LDL creste procesul de preluare a acestuia de celulele intimale aortice [Sobenin et al., 1993] si de macrofagele derivate din monocite [Klein et al., 1995], cu stimularea ulterioara a formarii celulelor spumoase, se crede ca procesul de recunoastere a LDL glicozilat de catre receptorii scavenger promoveaza acumularea intracelulara a esterilor de colesterol si astfel, ateroscleroza.
Un alt efect aterogen al glicarii este ca induce cresterea susceptibilitatii LDL la modificari oxidative. Reactiile de oxidare care au loc normal in timpul glicarii pot oxida componentele fosfolipidice ce contin amine ale LDL [Bucala et al., 1993]. Glicozilarea avansata a acestor componente este insotita de modificarea oxidativa progresiva a reziduurilor acizilor grasi nesaturati [Steinbrecher & Witztum 1984]. Oxidarea LDL ce urmeaza formarii AGE-LDL au loc direct proportional cu concentratia de glucoza si poate fi inhibata de inhibitori ai formarii AGE ca aminoguanidina [Bucala et al., 1993]. Astfel, glicarea confera susceptibilitate crescuta a LDL la modificari oxidative [Steinbrecher & Witztum 1984], proces considerat un pas critic al aterogenicitatii LDL.
Glicozilarea componentelor matricei, ca colagen IV, laminina si vitronectina reduc legarea heparan sulfatului anionic, ducand la un turnover imens al heparan sulfatului. Absenta acestuia din matrice duce la producere excesiva a altor componente matriceale si alterarea separarii factorilor care regleaza cresterea intre proteoglicanii legati de matrice si celule [Brownlee et al., 1988].
b) mecanisme dependente de receptor
Interactiile celulare ale AGE sunt mediate printr-un receptor specific pentru AGE (RAGE), de pe suprafata celulei. Prezenta RAGE, un membru al superfamiliei de receptori imunoglobulinici [Neeper et al., 1992], a fost demonstrat in toate celulele relevante pentru procesul aterosclerotic incluzand macrofagele derivate din monocite, celule endoteliale si celule musculare netede [Schmidt et al., 1999; Bucala et al., 1995].
La animalele mature, RAGE de pe celulele endoteliale este prezent la nivel scazut [Brett et al., 1993]. Totusi, in anumite circumstante patologice are loc cresterea excesiva, sustinuta a RAGE. In leziunile patologice, abundenta celulelor ce exprima RAGE este asociata de regula cu locurile unde sunt acumulati liganzii RAGE. In vasculatura diabetica, celulele care exprina nivele crescute de RAGE sunt adesea langa ariile in care AGE este abundent [Schmidt et al., 1999; Ritthaler et al., 1995].
Interactia AGE cu RAGE de pe celulele endoteliale duce la inducerea stresului oxidativ si in consecinta, a factorului de transcriptie NF-kB [Yan et al., 1994; Wautier et al., 1994] si VCAM-1 (figura 9) [Schmidt et al., 1995].
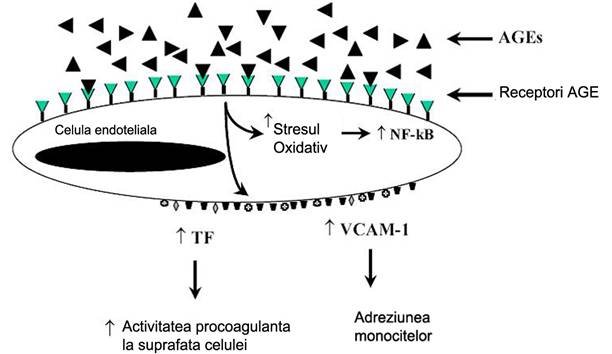
Figura 9. Consecintele interactiei AGE cu receptorii (RAGE) de pe celula endoteliala. (Modificata dupa Aronson D& Rayfield EJ, 2002)
In plus, legarea AGE cu receptorii lor specifici duce la slabirea functiei de bariera a endoteliului [Schmidt et al., 1995; Wautier et al., 1996], datorita cresterii permeabilitatii monostratului de celule endoteliale [Wautier et al., 1996; Esposito e al., 1989]. Astfel, interactia dintre AGE cu celulele ce exprima RAGE poate media evenimentele initiale din ateroscleroza. De exemplu, cresterea permeabilitatii endoteliale poate duce la cresterea intrarii lipidelor in subendoteliu (figura 10). Cresterea interactiilor adezive ale monocitelor cu suprafta endoteliala poate ulterior sa duca la migrarea transendoteliala.
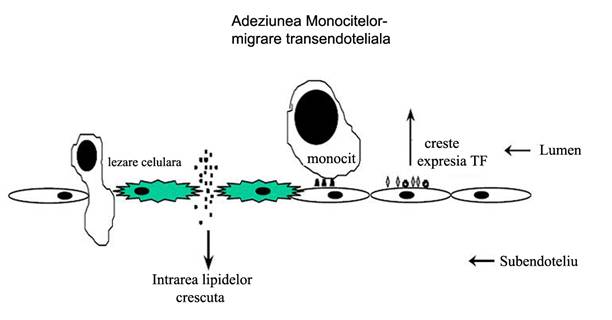
Figura 10. Mecanisme prin care AGE induce disfunctia endoteliala si promoveaza ateroscleroza (Modificata dupa Aronson D& Rayfield EJ, 2002)
Legarea AGE solubili la monocite (ce au RAGE) induce chemotaxia acestora [Schmidt et al., 1993], urmata de infiltrarea mononucleara prin monostratul endotelial intact [Kirstein et al., 1990]. Studiile patologice ale placilor aterosclerotice umane au evidentiat infiltrarea celulelor care exprima RAGE in intima largita [Brett, 1993]. De asemenea, intereactia dintre monocite/macrofage cu AGE are ca rezultat productia diferitilor mediatori ca interleukina-1, TNFα si factorul de crestere insulinic-1 [Kirstein et al., 1990, 992], care au roluri importante in patogeneza aterosclerozei [Ross, 1999]. In celulele musculare netede, legarea proteinelor modificate-AGE la RAGE este asociata cu cresterea prolferarii celulare [Vlassara et al., 1994]. Desi mecanismul exact al acestui raspuns nu este cunoscut, efectele care promoveaza cresterea mediata de RAGE are loc, probabil prn intermediul citokinelor sau factorilor de crestere. Astfel, in conditiile in care depunerea AGE in tesut este crescuta, interactia mediata de receptor a proteinelor-AGE cu celulele peretelui vascular faciliteaza migrarea celulelor inflamatoare in leziune cu eliberarea ulterioara a citokinelor care promoveaza cresterea.
2) Protein kinza C
Consecintele metabolice ale hiperglicemiei pot fi exprimate in celulele in care transportul glucozei este independent de insulina. Hiperglicemia intracelulara rezultata a fost implicata in patogeneza complicatiilor diabetice prin activarea protein kinazei C (PKC) [Feener & King, 1997; Koya & King, 1998]. Concentratiile crescute ale glucozei activeaza PKC prin intermediul diacilglicerolului (DAG), cofactorul celular endogen major pentru activarea PKC.
PKC este o familie de cel putin 12 izoforme de serin si treonin kinaze. Desi unele izoforme ale PKC sunt exprimate in tesutul vascular, in modelul de sobolan diabetic exista o activare preferentiala a PKC β2 in aorta, inima si retina si PKC β in glomeruli [Inoguchi et al., 1992; Koya et al., 1997].
Sistemul PKC este ubicuitar distribuit in celule si este implicat in transcriptia unor factori de crestere si in transducerea semnalului in raspuns la factorii de crestere [Inoguchi et al., 1992; Koya et al., 2000; Park et al., 2000]. Activarea PKC creste expresia factorului de crestere transformator β (TGF β), care este unul dintre cei mai importanti factori ce rgleaza producerea matricei extrcelulare, prin activarea expresiei genice a proteoglicanilor si colagenului si scaderea sintezei enzimelor proteolitice care degradeaza proteinele matriceale [Nabel et al., 1993]. Se crede ca expresia crescuta a TGF β duce la ingrosarea membranei bazale a capilarelor - una din anormalitatile structurale initiale observate in aproape toate tesuturile diabetice. Inhibitorul specific de PKC β (LY333531) reduce expresia glomerulara a TGF β si a proteinelor matricei extracelulare ca fibronectina si colagenul IV [Koya et al., 1997; 2000].
3) Stresul oxidativ
Stresul oxidativ este acceptat ca fiind un mecanism patogenic pentru ateroscleroza. Printre sechelele induse de hiperglicemie, s-a sugerat ca stresul oxidativ este un potential mecanism pentru ateroscleroza accelerata [Nishikawa et al., 2000; Baynes & Thorpe, 1999].
Ce este stresul oxidativ?
Stresul oxidativ este definit in general ca formarea excesiva si/sau indepartarea insuficienta a moleculelor extrem de reactive, ca speciile reactive de oxigen (ROS) sau speciile reactive de nitrogen (RNS). ROS includ radicali liberi ca superoxid (O2-), hidroxil (OH), peroxil (RO2), hidroperoxil (HRO2), precum si specii ne-radical ca peroxid de hidrogen (H2O2) si acid hidrocloros (HOCl). RNS includ radicali liberi ca oxidul nitric (NO) si dioxidul de nitrogen (NO2), precum si specii ne-radiacal ca peroxinitriti (ONOO-), oxid nitros (HNO2) si peroxinitrati alchil (RONOO). Dintre aceste molecule reactive, O2-, NO si ONOO- sunt cele mai studiate specii si joaca roluri importante in complicatiile cardiovasculare.
Exista multiple surse de stres oxidativ in diabet incluzand caile ne-enzimatice, enzimatice si mitocondriala.
Sursele ne-enzimatice ale stresului oxidativ sunt originare din biochimia oxidativa a glucozei. Hiperglicemia poate produce direct nivele crescute de ROS. Glucoza poate suferi auto -oxidare si genereaza radicali OH [Turko et al., 2001]. In plus, asa cum am discutat mai sus, glucoza reactioneaza ne-enzimatic cu proteinele ducand la dezvoltarea produsilor Amadori, urmata de formarea AGE, eliberand ROS de-a lungul intregului proces. In conditii hiperglicemice metabolismul glucozei este de asemenea crescut prin calea poliol (cresterea productiei de sorbitol), care genereaza productie crescuta de O2-.
Sursel enzimatice ale generarii crescute de specii reactive in diabet includ sintaza oxidului nitric (NOS), NADPH oxidaza si xantin oxidaza [Guzik et al., 2000; 2002; Aliciguzel et al., 2003]. Toate izoformele ale NOS necesita cinci cofactori ca dinucleotide flavin-adenine, mononucleotide flavine, hemuri, BH4 si Ca2+- calmodulina. Daca NOS isi pierde substratul, L-arginina sau unul din acesti 5 cofactori, NOS poate produce O2- in loc de NO [Maritim et al., 2003; Guzik et al., 2000; 2002; Aliciguzel et al., 2003]. NADPH oxidaza este o enzima care contine cinci subunitati si este sursa majora de producere a O2- [Guzik et al., 2000; 2002; Kitada et al., 2003; Etoh et al., 2003]. Guzik si colegii au investigat nivelele de O2- in vasculatura pacientiilor diabetici si au testat sursele de O2- folosind inhibitori de NOS, NADPH oxidaza, xantin oxidaza si ai lantului de transport de electroni mitocondrial [Guzik et al., 2002]. Acest studiu a demonstrat ca diabetul creste productia de O2- in principal prin activarea NADPH oxidazei. De asemenea, studiile in vitro recente, au aratat ca NADPH oxidaza este sursa majora de producere a anionilor superoxizi in celulele cardiovasculare [Green et al., 2004]. NADPH oxidaza este o enzima asociata membranei care catalizeaza reducerea unui electron la oxigen folosind NADPH sau NADH ca donori de electroni. Complexul NADPH oxidazei este prezent in neutrofile, monocite/macrophage, celule endoteliale, CMN, miocite cardiace, pericite si fibroblasti adventiciali [Manea et al., 2005]. Acesta consta din cinci subunitati: un citocrom b558 asociat membranei ce contine gp91phox si p22phox si un complex citosolic alcatuit din p40phox, p47phox, p67phox. Cand componentele membranare sunt separte de cele citosolice, enzima este inactiva. Diferiti stimuli, ca glucoza crescuta sau citokinele, activeaza protein kinaza C sau A care fosforileaza componentele citosolice ale enzimei si intregul complex citosolic migreaza la membrana (Figura 11).
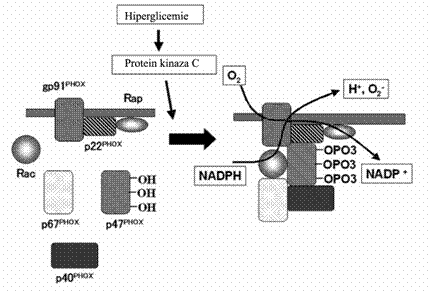
Figura 11. Enzima NADPH oxidaza cuprinde cinci componente: p40phox (phox de la OXidaze PHagocite), p47phox, p67 phox, p22phox si gp91phox : in celulele neactivate, trei din componentele enzimei si anume, p40phox, p47 phox si p67phox, sunt in citoplasma, formand un complex. Celelalte doua componente, p22phox si gp91phox, sunt localizate in membrana. Cand enzima este stimulata de hiperglicemie, prin calea PKC, componentele citosolice devin fosforilate si intregul complex citosolic migreaza catre membrana. (dupa Nakagami et al., 2005 )
Pentru activarea enzimei mai sunt necesare inca doua proteine de masa moleculara mica Rac si Rap. In celulele neactivate, Rac este localizata in citoplasma intr-un complex dimeric cu Rho-GDI (inhibitorul disocieri guanidinei), iar Rap este localizata in membrana. In timpul activarii, Rac se leaga la GTP (guanozin - trifosfat) si migreaza la membrana alaturi de complexul citosolic al NADPH oxidazei. Glucoza crescuta activeaza NADPH oxidaza in celulele vasculare [Dragomir et al., 2004; Manea et al., 2005].
Lantul respirator mitocondrial este o alta sursa de generare ne-enzimatica a speciilor reactive. In timpul procesului de fosforilare oxidativa electronii sunt transferati de la purtatorii de electroni NADH si FADH2, prin patru complexe succesive, in interiorul membranei mitocondriale, la oxigen, generand ATP [Green et al., 2004]. In conditii normale, O2- este eliminat imediat prin mecanismul natural de aparare. Un studiu recent demonstreaza ca generarea O2- indusa de hiperglicemie la nivel mitocondrial este pasul initial care declanseaza cercul vicios al stresului oxidativ in diabet [Nishikawa et al., 2000; Brownlee, 2001]. Cand celulele endoteliale sunt expuse la hiperglicemie, la nivele relevante diabetului clinic, exista o generare crescuta de ROS, in special de O2-, care precede activarea cailor majore implicate in dezvoltarea complicatiilor diadetului. Important este ca blocarea radicalilor O2- prin trei metode diferite previne activarea NF-kB precum si a caii poliol, formarea AGE si activarea PKC. Bazandu-se pe aceasta informatie, s-a postulat de mai multe grupuri ca O2- mitocondrial este bulgarele de zapada care transforma stresul oxidativ intr-o avalansa in diabet prin stimularea producerii mai mari de ROS si de RNS, prin activarea NF-kB, PKC si NADPH oxidaza (Figura 12).
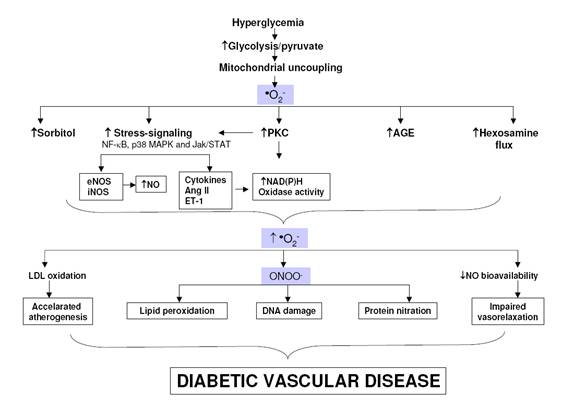
Figura 12. Modelul actual de generare a speciilor reactive si tintele acestora in diabet. Generarea excesiva a ROS mitocondriale, datorita hiperglicemiei, initiaza un cerc vicios prin activarea cailor sensibile la stres ca NF-κB, p38 MAPK si Jak/STAT, caile poliol/sorbitol si hexosaminei, PKC si AGE. Productia crescuta de AGE, sorbitol si citokine proinflamatoare exercita un feedback pozitiv asupra sintezei de ROS si RNS crescand disfunctia vasculara mediata de PKC, prin alterarea expresiei genice dar si a functiei si structurii vasculare. (Modificata dupa Johansen et al., 2005)
Astfel, inhibarea formarii radicalilor liberi intracelulari va furniza o strategie terapeutica pentru preventia stresului oxidativ si a complicatiilor vasculare asociate diabetului.
Efectele celor trei mecanisme majore implicate in modificarile patologice observate in diabet afecteaza in primul rand functia celulelor vasculare. Este bine cunoscut da nivele crescute de glucoza induc disfunctia celulelor endoteliale dar si a celulelor musculare netede.
a) Disfunctia celulei endoteliale indusa de hiperglicemie
Un singur strat de celule endoteliale acopera suprafata interioara a tuturor vaselor de sange, furnizand o interfata activa metabolic intre sange si tesut care moduleaza fluxul de sange, transportul nutrientilor, coagularea si tromboza, si diapedeza leucocitelor [Cines et al., 1998]. Celulele endoteliale sintetizeaza substante bioactive importante, incluzand NO si ROS, prostaglandine, endotelina-1 si angiotensina II, care rgleaza functia si structura vaselor de sange. NO dilateaza vasele si mediaza controlul endoteliului (relaxarea vasculara) [Verma & Anderson, 2001]. In plus, NO inhiba activarea plachetara, limiteaza inflamatia prin reducerea adeziunii leucocitelor la endoteliu si migrarea in peretele vaselor, si diminueaza proliferarea si migrarea celulelor musculare netede. Impreuna, aceste proprietati inhiba aterogeneza si protejeaza vasele de sange.
S-a presupus ca distrugerea sau disfuctia celulelor endoteliale, care duce la diminuarea productiei NO, joaca un rol esential in progresia si/sau dezvoltarea bolilor vasculare in diabet. Diabetul afecteaza vasodilaterea mediata de endoteliu (mediata de NO), inainte de formarea ateromului [Williams et al., 1996]. Un numar de mecanisme fundamentale contribuie la biodisponibilitatea scazuta a NO derivat de endoteliu, in diabet. Hiperglicemia inhiba productia de NO prin blocarea activarii sintazei eNOS si cresterea ROS, in special anionii superoxizi in CE si CMN [De Vriese et al., 2000]. Anionii superoxizi anihileaza direct NO cu formare de ioni peroxinitriti toxici [Milstien & katusic 1999]. Exista si alte mecanisme anormale in diabetul de tip 2 care scad de asemenea NO derivat din endoteliu. Rezistenta la insulina duce la eliberarea excesiva a acizilor grasi liberi din tesutul adipos [Hennes et al., 1996] care activeaza enzima semnalizatoare PKC, inhiba fosfatidilinozitol 3 kinaza (PI-3) si creste productia de ROS - mecanisme care diminueaza producerea de NO sau ii scad biodisponibilitatea [Inoguchi et al., 2000]. Producerea de peroxinitriti scade sinteza de substante vasodilatatoare si antiplachetare [Zou et al., 1999].
Pe langa reducerea NO, diabetul induce expresia vasoconstrictorilor, dintre care cel mai important, endotelina-1 (ET-1), care activeaza receptorul ET-A de pe celulele musculare netede si induce vasoconstrictia. In afara de modularea tonusului vascular, ET-1 creste salinitatea renala si retentia apei, stimuleaza sistemul renina-angiotensina si induce hipertrofia celeulelor musculare netede [Hopfner & Gopalakrishnan, 1999]. Diabetul induce si alte substante vasoactive derivate din endoteliu, ca prostanoizi vasoconstrictori si angiotensina II, investigarea relevantei lor patofiziologice in diabet fiind in curs de cercetare.
Migrarea limfocitelor si monocitelor in intima participa integral la ateroscleroza. Celulele T secreta citokine care moduleaza formarea leziunii [Libby & Aikawa, 1998]. Dupa ce ajung in spatiul subendotelial, monocitele preiau LDL oxidat prin receptori scavenger si devin celule spumoase. Acumularea localizata a celulelor spumoase duce la formarea pliurilor lipidice, trasatura leziunilor initiale ale aterosclerozei [Libby 2001]. Diabetul accelereaza aceste procese patologice. Prin scaderea NO, crsterea stresului oxidativ si a receptorilor pentru AGE, hiperglicemia induce activarea factorilor de transcriptie nucleari NF-kB si AP-1. Acesti factori regleaza expresia genelor ce codifica diferiti mediatori ai aterosclerozei ca molecule de adeziune, chemokine, IL-1, TNFa [Schmidt & Stern, 2000; Rosen et al., 2001]. Anormalitatile lipidice care se gasesc de regula in diabet, ca crestesterea VLDL si eliberarea excesiva a acizilor grasi si liberi, activeaza de asemenea factorul de transcriptie NF-kB, inducand ulterior la expresia moleculelor de adeziune celulara si a citokinelor [Dichtl et al., 1999].
Pe langa accelerarea procesului initial al aterosclerozei, diabetul promoveaza instabilitatea placii. Celulele endoteliale diabetice elaboreaza citokine si scade sinteza de novo a colagenului in celulele musculare netede vasculare [Hussain et al., 1996]. Diabetul creste producerea de metaloproteaze matriceale care duce la distrugerea colagenului [Uemura et al., 2001]. Colagenul confera stabilitate mecanica capului fibros al placii. Cand distrugerea colagenului creste si sinteza scade, placile sunt mai susceptibile la rupturi, ceea ce declanseaza formarea trombusului. In final, un modulator important al severitatii rupturii placii este extinderea ocluziei vasculare prin formarea trombusilor. Productia de factor tisular, procoagulantul major gasit in placa aterosclerotica este crescuta de CE in diabet, alaturi de alterarea factorilor fibrinolitici [Kario et al., 1995].
b) Disfunctia celulelor musculare netede indusa de hiperglicemie
Asa cum am amintit, arterele afectate de diabet si ateroscleroza au functia vasomotoare afectata. In particular, pacientii cu diabet de tip 2 au vasodilatarea dependenta de NO afectata, reflectand o anormalitate a functiei sau a transducerii semnalului in CMN.
Dupa cum stim, CMN sunt contributori importanti la dezvoltarea aterosclerozei, iar diabetul induce activitate aterogenica CMN vasculare. Hiperglicemia activeaza protein kinaza C, receptorii pentru produsii finali ai glicarii avansate si factorul de transcriptie kB in CMN vasculare, ca si in CE. Activarea acestor sisteme duce la cresterea productiei de O2-, contribuind la stresul oxidativ [Inoguchi et al., 2000] care promoveaza procesul aterosclerotic. In plus, odata formate pliurile lipidice bogate in macrofage, CMN vasculare din media si adventicia arterelor migreaza in leziunea intimala care ia nastere, prolifereaza si depun o matrice extracelulara complexa, pasi importanti in progresia catre placa aterosclerotica avansata. CMN vasculare cultivate, obtinute de la pacientii cu diabet de tip 2 prezinta un proces crescut de proliferare comparativ cu CMN provenite de la pacienti fara diabet [Suzuki et al., 1998]. Ca sursa de colagen, CMN vasculare intaresc ateromul facandu-l mai rezistent la ruptura si la tromboza. Intr-adevar, leziunile care s-au rupt din cauza trombozei tind sa aiba mai putine CMN [Libby 2001]. Leziunile aterosclerotice avansate de la pacientii diabetici au mai putine CMN vasculare comparativ cu cei fara diabet [Fukumoto et al., 1998]. Modificarile LDL-ului induse de hiperglicemie pot sa controleze, in parte, migrarea crescuta si apoi apoptoza CMN vasculare in leziunile aterosclerotice. LDL care a suferit glicare non-enzimatica induce migrarea CMN vasculare in vitro, in timp ce LDL glicat oxidat poate induce apoptoza CMN vasculare [Taguchi et al., 2000]. Astfel, diabetul afecteaza functia CMN vasculare, promovand formarea leziunilor aterosclerotice, instabilitatea placii si evenimente clinice.
Astfel, conceptele despre ateroscleroza au evoluat de la ideea degenerarii inevitabile la un scenariu mult mai bine definit de evenimente celulare si moleculare. Pe masura ce cunostintele despre mecanismele fundamentale ale acestei boli au crescut, ateroscleroza a inceput sa fie privita, mai degraba ca un proces modificabil decat unul inevitabil. Ateroscleroza este definita ca un proces ce implica multi factori, reprezentat de o serie de mecanisme celulare si moleculare si de interactii multiple intre metabolismul lipidic, activarea monocitelor, celulele endoteliale, citokine si/sau alte cai metabolice intracelulare. Controlul aterosclerozei ar putea fi realizat prin interventii terapeutice la diferite etape ale procesului inflamator. Tintele terapeutice pot include caile citokinelor, factorii de crestere, factorii de transcriptie, genele modificate si multiple cai metabolice intracelulare.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1877
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved