| CATEGORII DOCUMENTE |
| Alimentatie nutritie | Asistenta sociala | Cosmetica frumusete | Logopedie | Retete culinare | Sport |
UNIVERSITATEA "OVIDIUS" CONSTANTA
FACULTATEA DE FARMACIE
SPECIALIZAREA FARMACIE
EVALUAREA IN VITRO A COMPRIMATELOR CU ASPIRINA TAMPONATA DE PE PIATA FARMACEUTICA DIN ROMANIA
CAPITOLUL I
I. 1. Comprimate. Istoric. Definitie
Prima informatie despre o forma asemanatoare cu comprimatele se intalneste in literatura medicala araba din secolul X, in care este descris procesul de obtinere al comprimatului din particule de substanta presate intre capetele a doua bete de ebonita, aplicand o forta cu ajutorul unui ciocan. Detalii despre procesul de comprimare au fost publicate in anul 1843 de catre englezul William Brockedon, ce inventeaza si o masina de comprimat, pentru care obtine brevetul 9977: "Manufacturing pills and medicinal lozengez by causing materials when in a state of granulation, dust powder to be made into form and solidified by pressure in dies". In acest caz, forta aplicata era tot un ciocan.
Prima substanta farmaceutica ce a fost tratata astfel a fost bicarbonatul de potasiu. Utilizarea pilulelor comprimate a crescut rapid, acestea au fost denumite cu termenul de tableta (engl. Tablet), in SUA, in 1870, si apoi ciocanul a fost inlocuit cu prese ce actionau cu putere. Alte brevete care au avut ca subiect maisinile de comprimat au fost: J.A. Mc.Frran, 1874, J.P. Remington, 1875, J.Dunton, 1876. Deja in 1874, existau ambele prese: masina cu excentric si masina rotativa, care operau in mod asemanator cu masinile utilizate azi. Nu dupa mult timp, au fost concepute masinile care permiteau acoperirea comprimatelor cu un strat protector. Primele lucrari stiintifice, privind introducerea comprimatelor in terapeutica, se datoreaza lui J. Dunton, in 1874, in SUA si lui Rosenthal, in Germania.
Catre sfarsitul secolului trecut, comprimatele erau larg raspandite, mai ales in tarile anglo-saxone, dar introducerea lor in farmacopei a fost efectuata mai tarziu. Prima monografie: tabletele de nitroglicerina a fost introdusa in Farmacopeea Britanica , in 1885, dar nici o alta monografie de tablete nu apare pana in 1945, de cand utilizarea acestei forme creste vertiginos; de exemplu, Ed. B. Ph. Din 2000 contine 320 de monografii de tablete.
Prima farmacopee care contine tablete in SUA este UPS IX din 1916, care includea o singura monografie.
In Europa, aceasta noua forma farmaceutica, care a inlocuit vechile pilule, este introdusa in secolul XX, cand pentru prima data este inscrisa in Formularul Oficial ,in 1906, iar in Farmacopeea Franceza, este introdusa ca monografie in anul 1937, sub numele de: Comprimes, lat. Compressi (care se refera la operatia farmaceutica). Comprimatele au o vechime mai mare de 160 de ani, dar ele reprezinta 50-60% din formele farmaceutice administrate si 75% din formele dozate solide.
Cu toate ca fabricarea comprimatelor a fost cunoscuta de o perioada lunga de timp, studiile stiintifice privind fizica comprimarii au fost initiate in SUA si Europa in 1960, mai intai pe masinile de comprimat care s-au modernizat si automatizat si mai tarziu pe simulatori, procesul de presare fiind astazi bine cunoscut.
Pentru prima data, comprimatele sunt mentionate in FR V, in 1943, cu denumirea: Comprimate, Tablete (lat. Tablettae, Compressa), care continea patru monografii; in editiile urmatoare, numarul exemplarelor creste: VII-16, VIII-21, IX-46, X-33 monografii. Suplimentul 2004 al FR X le inscrie cu denumirea: Comprimate, lat. Compressi.
Comprimatele sunt preparate farmaceutice solide, care contin doze unitare din una sau mai multe substante active. Se obtin, in general, prin comprimarea unui volum constant de particule. Comprimatele sunt destinate administrarii orale. Anumite comprimate sunt inghitite sau mestecate, unele sunt dizolvate sau dispersate in apa inaintea administrarii, iar altele sunt mentinute in cavitatea bucala pentru a elibera substanta activa (Comprimate, Compressi, FR X, suplimentul 2004; Tabletes, Compressi, Ph Eur 4 th 2002, suplimentul 4.1, 2003)
Din punct de vedere fizic, comprimatele sunt incluse in categoria sistemelor macroeterogene, de tip gaz/solid (G / S).
Particulele sunt constituite din una sau mai multe substante active, asociate sau nu cu excipienti: diluanti, lianti, dezagreganti, agenti de curgere, lubrifianti, compusi care pot influenta comportamentul preparatului in tubul digestiv, coloranti si aromatizanti autorizati. Cerintele acestei monografii nu trebuie sa fie aplicate preparatelor prezentate sub forma de comprimate destinate utilizarii pe alte cai de administrare decat calea orala.
Pentru aceste preparate, cerintele specifice sunt descrise in alte monografii, de exemplu: preparate rectale, preparate vaginale si preparate buco-faringiene. De asemenea, prevederile acestei monografii nu se aplica pastilelor, liofilizatelor orale, granulelor orale, gumelor orale, care au monografii separate si se fabrica prin alte metode si nu prin comprimare. Prevederile acestei monografii generale pot sa nu se aplice comprimatelor pentru uz veterinar in anumite cazuri.
Denumirea acestor forme farmaceutice provine de la verbul latin comprimo, comprimere (a presa). Deseori, este intalnita si denumirea de tablete care deriva de la tabula - tabla, care sugereaza forma preparatelor.
In general, comprimatele au forma de cilindru, ale caror extremitati pot fi plane sau taiate oblic. Pot sa prezinte santuri de subdivizare, o sigla sau alte inscriptionari.
Dimensiunile si masa comprimatelor variaza in anumite limite, in functie de cantitatea de substanta medicamentoasa (doza) si modul de utilizare. Astfel, comprimatele au, in general, un diametru de 5-17 mm si greutatea intre 0,1 si 1 g.
I. 2. Clasificarea formelor farmaceutice cu cedare conventionala
Comprimatele se clasifica in functie de diferite criterii, dintre acestea cele mai importante sunt:
Numarul de substante medicamentoase, se deosebesc comprimate simple si compuse;
Solubilitatea in apa si permeabilitatea substantelor active, pot fi:
-comprimate cu substante active din clasa I (sistemul de clasificare biofarmaceutica, abrev. SCB): cu solubilitate mare si permeabilitate mare: teofilina, ketoprofen, verapamil, metoprolol tartrat;
-comprimate cu substante active din clasa II (SCB): cu solubilitate mica si permeabilitate mare : piroxicam , griseofulvina;
-comprimate cu substante active din clasa III (SCB): cu solubilitate mare si permeabilitate mica: ranitidina, cimetidina, captopril, atenolol;
-comprimate cu substante active din clasa IV (SCB): cu solubilitate mica si permeabilitate mica: furosemid, hidroclorotiazida;
In functie de forma, se deosebesc comprimate: rotunde, ovale, oblonge, mai rar: patrate, hexagonale, triunghiulare sau dreptrunghiulare. Comprimatele de forma circulara (discuri) si cele ovale pot fi : cu margini intacte, suprafata plana sau convexa:
-comprimate plate cu margini;
-comprimate plate cu bordura;
-comprimate bombate (convexe);
-comprimate biconvexe.
Comprimatele convexe (bombate) se pot clasifica in functie de raportul dintre raza de curbura si diametru.
In functie de inscriptionare sau santurile de pe suprfata acestea pot fi:
-comprimate inscriptionate: semn, marca, dozaj
-comprimate fractionabile: cu santuri: -cu sant in V
-cu sant in U
-cu sant in forma de cruce
In functie de masa, diametrul si grosime sunt redate in tabelul I:
Tabel 1. Dimensiunea comprimatelor recomandate in functie de masa lor [Dobre E. 1996]
|
Masa comprimatului ( mg ) |
Diametrul (mm ) |
Grosimea (mm ) |
In mod obisnuit, diametrul comprimatelor de forma circulara este cuprins intre 6 si 17 mm, iar masa variaza intre 0.1 si 0.8 g.
In functie de modul de formulare:
-comprimate neacoperite: -cu strat mic
-cu straturi multiple;
-comprimate acoperite: cu unul sau mai multe straturi, formate din amestecuri de substante diverse: rasini, gume, gelatine, zaharuri, plastifianti, ceruri, polioli, coloranti si aromatizanti si uneori substante active: - cu zahar ( drajeuri )
- cu pelicule
In functie de procedeul de fabricare, comprimatele neacoperite pot fi:
cu strat unic
cu straturi multiple paralele
cu straturi multiple concentrice
In functie de viteza de eliberare a substantei active comprimatele cu eliberare conventionala se clasifica in :
-comprimate pentru administrare perorala;
-comprimate pentru administrare pe mucoase;
-comprimate pentru solutii injectabile si oftalmice;
in functie de calea de administrare:
a). Comprimate administrate pe calea perorala (gastrointestinala):
-comprimate care se inghit;
-comprimate care se transforma in solutie sau suspensie;
-comprimate efervescente;
-comprimate orodispersabile;
b). Comprimate administrate pe mucoasa:
1. Comprimate bucofaringiene (bucale, oromucozale):
-comprimate de mestecat;
-comprimate bucale;
-comprimate sublinguale;
-comprimate de supt (pastile);
-comprimate alveolare (conuri alvolare);
-comprimate mucoadezive bucale;
2. Comprimate vaginale:
-se introduc in vagin, unde se dizolva sau se dezagrega;
-comprimatele vaginale destinate a fi dizolvate pentru irigatii vaginale;
3. Comprimate oftalmice (oculare):
-inserte, mici discuri sterile, solubile (se dizolva rapid dupa aplicare)
-comprimate destinate prepararii de solutii si suspensii oftalmice, sterile;
4. Comprimate pentru inhalatii
c).Comprimate sterile administrate pe caile parenterale:
-comprimate sterile destinate prepararii de solutii si suspensii injectabile;
1. 2. 1. Preformularea
Dintre formele farmaceutice in care substanta medicamentoasa poate fi formulata si preparata, cele care constituie ponderea cea mai mare in productia industriala o reprezinta comprimatele (40-50%). In aceasta situatie se gasesc si noile substante medicamentoase active pe cale orala, care primesc autorizatia de introducere pe piata.
Inainte ca aceste forme sa fie formulate si preparate, este necesara stabilirea unor proprietati fundamentale, fizice si chimice ale moleculei substantei medicamentoase, precum si unele proprietati care deriva din acestea pentru particulele substantei medicamentoase. Aceste informatii vor determina etapele consecutive si caile posibile de abordare a formularii prepararii produsului, sau informatia care sa justifice modificari in structura moleculara (1-10). Aceasta prima faza de studiu despre substanta medicamentoasa este cunoscuta sub numele de preformulare. Ea se realizeaza de obicei cu ocazia studiilor legate de introducerea unui nou compus bioactiv in terapeutica.
Caracterizarea unei substante medicamentoase in faza preformularii include:
Proprietati fundamentale ale substantei medicamentoase:
Spectroscopie U.V.
Solubilitate
-in apa (solubilitatea intrinseca, influenta pH-ului)
-pKa (controlul solubilitatii, formarea de saruri)
-saruri (solubilitate, higroscopie, stabilitate)
-solventi (vehicule, extractie)
-coeficient de partitie k (lipofilie, relatie, structura-activitate)
-dizolvare (biofarmacie)
3. Punct de topire ( ATD- polimorfism, hidrati, solvati)
4. Metoda de dozare (U.V., CSS, HPLC)
5. Stabilitatea in solutie si in stare solida (termica, hidroliza, oxidare, fotoliza, ioni metalici, pH)
Proprietati derivate din structura chimica
6. Microscopie (marimea particulelor, morfologie)
7. Curgerea pulberii
-densitatea patului (formularea comprimatelor)
-unghiul de repaus (formularea comprimatelor)
8. Proprietati de comprimare: compresibilitate, compactibilitate (ajuta la alegerea excipientilor)
9. Compatibilitatea excipientilor (trierea preliminara prin ATD, confirmare CSS)
Evaluarea acestor proprietati se face in ordinea enumerata, in functie de existenta unei cantitati de substanta medicamentoasa, care in aceasta faza preliminara a cercetarii este redusa (miligrame-grame).
Atentia se va concentra asupra a doua proprietati esentiale ale noului compus bioactiv: solubilitatea intrinseca (Co) si constanta de disociere (pKa). Ele vor indica imediat necesitatea si posibilitatea de a prepara mai multe saruri solubile ale substantei medicamentoase pentru a inlatura biodisponibilitatea redusa datorita solubilitatii reduse, mai ales in cazul formelor farmaceutice solide.
In afara determinarii mentionate in lista prezentata, se va tine seama de proprietatile analitice pe care chimistii le-au determinat dupa sinteza noului compus bioactiv:
-identificare (RMN, I.R, CSS, ATD, rotatie optica unde este cazul)
-dozare (titrare, U.V., HPLC)
-calitate (aspect, miros, culoare, pH, p.t)
Pana in momentul in care substanta medicamentoasa poate fi furnizata in cantitate mai mare, pentru aprecierea caracteristicilor patului (masei) de pulbere, cercetatorii se vor orienta asupra urmatoarelor proprietati:
1. Solubilitate: -din Co si pKa (in apa)
-Tm ( punctul de topire)
-log P (apa si solventi)
2. Dizolvare: - din Co si pKa
3. Alegerea sarii: -din structura, pKa, log P si Tm
4. pka (in serie omoloaga): -din valorile Hammett
5. Stabilitate: -din valorile Hammett si echilibrul ionic (solubilitate)
6. Higroscopie: -din solubilitate si alegerea sarii (log P)
7. log P (comportamentul de repartitie): -din valorile si f
8. Puritatea: -din valoarea Tm
9. Stabilirea dozarii (HPLC): -din solubilitate si log P
Cu aceste date, de evaluare preliminara si folosind optimizarea moleculara, poate incepe preformularea pe o cantitate mai mare de substanta medicamentoasa, sau caracterizarea patului pulberii.
Evaluarea preliminara si optimizarea moleculara
A. Spectroscopia
Primul lucru in preformulare il constituie stabilirea unei metode analitice simple de determinare cantitativa. Majoritatea substantelor medicamentoase absorb in U.V (190-390). Caracterul acid sau bazic se poate deduce din grupele functionale. Acestea vor indica solventii in care substanta va fi ionizata sau neionizata (starea ionica poate modifica spectrul U.V., prin cresterea absorbtiei sau prin modificari batocrome (rosu) sau hipsocrome (albastru) ale maximului sau minimului de absorbtie sau ambelor). Se poate alege lungimea de unda maxima la care sa se faca dozarea in solutie. Cantitatea de lumina absorbita de solutia unei substante medicamentoase este proportionala cu concentratia si distanta strabatuta, conform ecuatiei Lambert-Beer in care este coeficientul de absorbtie molara:
Absorbanta (A)=log(I0/I)= .C.l , [relatia 1]
Unde I0 si I sunt intensitatea luminii incidente, respectiv a celei transmise. Se obisnuieste sa se foloseasca coeficientul de absorbtie specifica E1% 1cm sau extinctia, cand lungimea parcursa este 1 cm, iar concentratia solutiei este 1% (g/v).
B. Solubilitatea
Determinarile pe cantitatile mici de substanta nou sintetizata incep cu solubilitatea si pKa. pKa ofera informatii despre pH-ul de realizat pentru a mentine solubilitatea si pentru alegerea sarii care sa dea o biodisponibilitate din starea solida si sa asigure stabilitatea. O solutie in apa sub 1% (10mg/ml) in domeniul de pH de la 1 la 7 la 30C va crea probleme de biodisponibilitate necorespunzatoare. Daca viteza de dizolvare intrinseca este sub 0,1 mg/cm2/min ea va fi limitanta de viteza de absorbtie.
O solubilitate mai mica de 1 mg/ml necesita folosirea de saruri solubile daca formularea se face pentru comprimate. Cresterea solubilitatii in mediul acid sugereaza o baza slaba, iar in mediul bazic, un acid slab. O crestere a solubilitatii in mediul acid si bazic denota un caracter amfoteric, iar absenta modificarii solubilitatii in functie de pH arata o molecula neionizabila la care nu se poate masura pKa. Daca ea nu se poate manipula in acest fel se vor cauta mijloace de crestere a solubilitatii prin dizolvare in alt solvent.
B. 1. Solubilitatea intrinseca Co
Solubilitatea substantei pure in mediul acid sau bazic este solubilitatea intrinseca Co. Ea se masoara la 40C pentru a asigura stabilitatea solutiei si la 370 C pentru studiile biofarmaceutice. In cazul cand puritatea este nesigura, se face o diagrama a solubilitatii de faza in care se masoara solubilitatea (mg/ml) in functie de diferite raporturi in greutate intre substanta si solvent. O deviere de la orizontala arata in cazul cresterii solubilitatii, o solubilizare prin asociere, complexare sau solubilizare datorita impuritatilor, iar in cazul scaderii solubilitatii, o salefiere.
B. 2. Valoarea pKa din solubilitate
Un procent de 75% din substantele medicamentoase sunt baze slabe, 20% sunt acizi slabi si 5% sunt neionice. Ecuatia Henderson-Hasselbalch arata ca:
Pentru baze slabe:
pH=pKa+ log10 ([B]/[BH+]) [ relatia 2 ]
iar pentru acizi slabi:
pH=Pka + log10 ([A-]/[HA]) [ relatia 2' ] ;
aceste ecuatii se folosesc pentru a determina pKa dupa modificari in solubilitate; de a prevedea solubilitatea la orice pH daca se cunoaste solubilitatea intrinseca Co si pKa; si de a usura alegerea sarii convenabile si a-l prevedea solubilitatile in functie de pH.
B. 3. Saruri
O imbunatatire a solubilitatii se poate face prin alegerea sarii. Sarurile cu acizi sau baze tari sunt usor solubile dar pot fi higroscopice ( instabile). Se prefera sarurile cu acizi sau baze slabe, care vor avea un pH mai tolerabil fiziologic, o stabilitate mai buna in ambalaje si un gust mai acceptabil .
In vivo solubilitatea lor depinde in primul rand de solubilitatea in stratul de difuzie din jurul particulelor care se dizolva (care este egal cu cel al unei solutii saturate). Sarurile acizilor slabi au in stomac un pH mai mare decat al sucului gastric (1,5) ceea ce le grabeste dizolvarea. in intestin sarea nu scade pH-ul, valoarea sa din stratul de difuzie favorizeaza dizolvarea. Sarurile bazelor slabe se dizolva rapid in stomac, dar absorbtia nu are loc fiind ionizate, pana in intestin unde dizolvarea are loc rapid pentru sarea datorita pH-ului stratului de difuzie care compenseaza pe cel al mediului, nefavorabil dizolvarii bazei slabe.
Formarea de saruri modifica proprietatile fizico-chimice ale substantei medicamentoase. Schimbari in solubilitate si viteza de dizolvare influenteaza viteza si marimea absorbtiei. De aceea orice noua sare trebuie examinata separat printr-un screening al preformularii.
B. 4. Solventii
Formularea unei solutii apoase a substantei medicamentoase este necesara pentru studiile preclinice. Daca solubilitatea in apa nu este suficienta, sau substanta este instabila in apa, se formuleaza solutii in cosolventi sau in alti solventi. Dintre solventii neaposi miscibili cu apa mai acceptabili sunt glicerolul, alcoolul, propilenglicolul. Solventii utili sunt cercetati si pentru a usura extractia si separarea (pentru cromografie).
B. 5. Coeficientul de partitie
Coeficientul de partitie este fractia de substanta distribuita intre un solvent si apa. Aplicatiile sale in preformulare pot fi pentru:
-solubilitate (amestecuri de solventi)
-absorbtie in vivo (in serii omoloage pentru reltia structura-activitate (SAR)
-cromatografia de partitie (alegerea coloanei HPLC, alegerea fazei mobile)
O importanta practica deosebita o are coeficientul de distributie lipide: apa Ku a care permite o clasificare a polaritatii sau lipofiliei unei substante medicamentoase.
S-a constatat o relatie liniara a solubilitatii intr-un solvent ) log1(Cs) si coeficientul de repartitie:
(log Ku a =log P):
log Cs=logCo + f(log + 0,89logP + 0,03), [relatia 3]
unde este puterea relativa a solventului din amestecul cu lichid nemiscibil ( exemplu, pentru glicerol, sau propilenglicol sau PEG 400, etanol si dimetoxisulfoxid, valorile sunt respectiv de 0,5; 1;2 si 4).
Coeficientul de partitie lipide:apa este o masura a lipofiliei relative a substantei de obicei in forma neionizata, masurata prin distributia intre o faza apoasa (solutie tampon) si un solvent nemiscibil lipofil sau un ulei. Frecvent utilizat ca faza lipofila a fost octanolul. Metoda agitarii flaconului s-a folosit frecvent pentru determinare. Substanta medicamentoasa dizolvata intr-una din faze s-a agitat 30 minute in faza apoasa, Ca. Stiind concentratia in faza apoasa inainte de experiment, C, se poate calcula coeficientul de repartitie ulei:apa:
log Ku a = ( C-Ca) / Ca; [relatia 4]
cand coeficientul de repartitie este mai mare se reduce volumul fazei uleioase de la 1:1 la 1:4 sau 1:9.
B. 6. Relatii structura-activitate
Exista cercetari de pionierat in aprecierea relatiei structura-activitate biologica, (SAR) (Meyer, 1899; Overton,1901). Aceasta relatie este folosita in chimia de sinteza de astazi pentru a facilita o abordare stiintifica in sinteza de noi compusi biologic activi, analogi, mai activi. Aplicarea SAR depinde de cunoasterea proprietatilor fizico-chimice ale fiecarei substante din clasa terapeutica investigata. Se considera (Collander, 1951) ca:
log Ku a = a - log Koctanol apa + b [relatia 5]
relatia este valabila pentru solventii polari sau semipolari dar nu se respecta pentru solventii nepolari (hexan, heptan, ciclohexan) si depinde de solubilitatea substantei in apa. De aceea octanolul trebuie saturat cu apa, iar apa cu octanolul, inaintea determinarii.
Masurarea logP se face nu in apa ci in solutii cu diferite valori de pH, folosind solutii tampon. Ȋn functie de gradul de disociere a acizilor si bazelor slabe, se va obtine un coeficient de repartitie aparent si nu unul absolut.
Speciile ionizate au o solubilitate mai mare in apa si o lipofilie mai mica, deci valoarea coeficientului de repartitie va fi inevitabil mai mica. Datorita importantei log P pentru SAR valorile se determina si la pH fiziologic de 7,4.
Relatia cantitativa structura-activitate (QSAR) se bazeaza pe premisa ca absorbtia substantelor medicamentoase este un proces prin membranele celulare si depinde de lipofilia substantei, iar viteza de patrundere este proportionala cu coeficientul de repartitie in vitro.
Modelulu fizico-chimic pentru activitatea biologica presupune ca activitatea unui compus este corelata cu urmatorii factori dependenti de structura moleculara: - electronic (sarcina)
-steric (marimea spatiala)
- efecte hidrofobe (partiala)
Activitatea biologica = f[( electronic) + (steric) + (hidrofob) + (structural/teoretic)]
Parametrul electronic este cuantificat de constanta de substituent sigma σ Hammett (1940) si reflecta reactivitatea chimica intr-o serie omoloaga. Constanta de substituent este pozitiva pentru grupele atragatoare de electroni ( acizi), si negativa pentru gruparile donatoare de electroni (baze). Se poate folosi la prevederea pKa.
Efectele sterice se manifesta atunci cand exista o interactiune directa intre substituent si nucleul parinte si depinde si de marimea substituentului. Valorile pozitive pentru parametrul efectului seric Es indica efecte sterice semnificative cu impedanta intra si intermoleculara pentru o reactie sau legatura la un loc activ.
Componentul hidrofob este masurat prin distributia compusului intre faza apoasa si un lichid nemiscibil. S-a demonstrat o relatie intre coeficientul de partitie intr-o serie, folosind o constanta de substituent aditiva ( ) (Hansch si Fujita,1964). Aceasta constanta este legata aproape in intregime de efectul unui substituient particular si mai putin de compusul parinte, iar aceasta permite prevederea coeficientului de partitie log P, a unui nou derivat, cu o buna precizie, deoarece este un nou derivat, cu o buna precizie.
Constanta de substituient poate fi legata si de efectul biologic deoarece este un component aditiv al coeficientului de partitie. Aceasta a dus la o aplicare larga a SAR luand in considerare efectele densitatii electronice a nucleului aromatic si efectele sterice, in studiile cantitative SAR. Studiile QSAR au evoluat, dar log P ramane unul din cei mai utili parametrii fizici de la care se pot obtine interpretari si corelari valoroase.
B. 7. Dizolvarea
Dizolvarea este importanta din punct de vedere biofarmaceutic atunci cand viteza sa este limitanta a procesului de absorbtie. In cazul dizolvarii solidelor se foloseste ecuatia modificata de Nernst si Noyes-Whitney.
B. 8. Viteza intrinseca de dizolvare
Daca dizolvarea este controlata numai de difuzie (controlulu transportului) viteza de difuzie este direct proportionala cu concentratia de saturatie a substantei in solutie (solubilitatea). Ȋn acest caz constanta de viteza K1 este:
K1 = 0,62.D2/3 x x [relatia 6]
Unde este vascozitatea cinetica, este viteza angulara a discului care se roteste a substantei medicamentoase. Mentinanad constanta suprafata solidului care se dizolva (un disc compactat din substanta pura expusa cu o singura fata a mediului de dizolvare) se poate calcula viteza de dizolvare dm/dt, iar in cazul in care Cs >>> C (conditii indepartate de solubilitatea de saturatie sau conditii sink):
dC / dt= (A/V) x K1 x Cs ; [relatia 7]
iar viteza intrinseca de dizolvare = K1 x Cs (mg x cm2/min)
Aceasta constanta de viteza nu este identica cu cea obtinuta la dizolvarea comprimatelor la care marimea suprafetei se schimba in timpul dizolvarii. Viteza intrinseca de dizolvare depinde de proprietatile intrinseci ale substantei medicamentoase si nu depinde de formulare.
Ȋn cazul in care dizolvarea are loc in mediul acid sau bazic pentru substante medicamentoase acizi sau baze slabe, se tine seama de ecuatia Henderson-Hasselbalch:
Viteza intrinseca de dizolvare= k'(Co(1+ antilog (pKa - pH),
adica viteza de dizolvare a substantei depinde de solubilitatea intrinseca Co, de constanta de disociere (pKa) si de pH, fie in mediul de dizolvare fie in stratul de difuzie din jurul particulei sarii care se dizolva.
Cunoasterea valorii constantei intrinseci de dizolvare are importanta pentru comportamentul in vivo al sarii fata de substanta originara. Ȋn cazul ca este mai mare, exista un avantaj al utilizarii sarii fata de compusul parinte.
Influenta ionului comun asupra solubilitatii are o consecinta insemnata. Adaugarea unui ion comun reduce solubilitatea unui electrolit putin solubil (salefiere). Ȋn schimb, ionii mai mari (benzoat, salicilat), pot modifica structura apei, au un efect hidrotrop, crescand solubilitatea compusilor putin solubili in apa.
C. Punctul de topire
Punctul de topire se poate determina prin tehnici diferite:
-topirea in capilara
-microscopie cu placa incalzita
-analiza termica prin calometrie diferentiala
Metoda topirii in capilara se face prin observarea topirii substantei dintr-o capilara in contact cu un material incalzit. Microscopul cu placa incalzita se foloseste tot in observarea topirii, dar viteza de incalzire este controlata si inregistrarea este mai precisa. Analiza termica diferentiala (ATD) sau calorimetrica de scanare diferentiala (DSC) este mult mai versatila, iar marimea probelor este de 2-5 mg.
Analiza termica diferentiala masoara diferenta de temperatura intre proba si o referinta in functie de temperatura sau timpul in care incalzirea este constanta. Analiza calorimetrica de scanare diferentiala este asemanatoare cu analiza termica diferentiala cu deosebirea ca aparatul masoara cantitatea de energie necesara pentru a tine proba la aceeasi temperatura ca si martorul, adica masoara entalpia de tranzitie.
Cand in proba nu exista schimbare fizica sau chimica, nu exista nici schimbare de temperatura, nici necesar de energie pentru a mentine o izoterma. Dar cand au loc schimbari de faza, caldura latenta suprima cresterea de temperatura si schimbarea de temperatura sau energia izoterma necesara se inregistreaza pe un inregistrator ca un rezultat al unui semnal electric generat de termocupluri. Astfel se pot cuantifica tranzitii cristaline, evaporarea si sublimarea.
Obiectivele studiului in preformulare sunt polimorfismult, masurarea p.t. si alte tranzitii de faza. Confirmarea se face cu date ale difractiei de raze X si cu spectroscopia I.R.
D. Polimorfismul
Un polimorf este un material solid cu cel putin doua aranjamente moleculare diferite fiecare producand specii cristaline diferite. Aceste diferente dispar in stare lichida sau de vapori. Importanta decelarii formelor polimorfe este pentru solubilitate si viteza de dizolvare. Speciile cu p.t. mai coborat sunt in general mai stabile, iar alti polimorfi sunt instabili sau metastabili si sufera conversia in forma stabila. Polimorfii nu sunt entitati chimice diferite. Dar unele proprietati fizice difera: p.t., densitatea, forma cristalina, proprietatile optice si electrice, presiunea de vapori, solubilitatea. Polimorfismult se intalneste la 2/3 din substantele medicamentoase organice. Formele polimorfe se noteaza cu cifre romane.
In preformulare intereseaza: numarul de forme polimorfe; stabilitatea formelor metastabile; existenta starii amorfe sticloase; posibilitatea de stabilizare a formelor metastabile; solubilitatea fiecarei forme; existenta posibilitatii ca forma solubila sa reziste prelucrarilor tehnologice si perioadei de stocare. Este necesar sa se cunoasca si formele pseudopolimorfe deoarece multi polimorfi se pot obtine prin schimbarea solventului de recristalizare. Recristalizarea cu molecule de apa sau de solvent (hidrat sau solvati) poate fi confundata cu adevaratii polimorfi si de aceea se numesc pseudopolimorfi. Forma de cristalizare se schimba. Diferentierea intre ei se face observand p.t. al lor dupa dispersare in ulei de silicoane. Pseudopolimorfii evolueaza spre gaz, provocand bule in ulei, polimorfii adevarati se topesc producand o a doua faza.
Polimorfismul adevarat se obtine prin manipularea solventului, sau prin sublimare si recristalizarea topiturii. Supraracirea topiturii ajuta la descoperirea modificarilor instabile. Identificarea acestor forme necesita observarea atenta a temperaturii, una prea rapida va masca tranzitia endoterma, iar una prea lenta va favoriza degradarea. Se lucreaza cu doua viteze, de 2 si 200C pe minut.
Diferenta intre p.t. intre polimorfi este o masura a stabilitatii polimorfilor. Cand diferenta este <10C nu are semnificatie. Daca diferenta p.t. este de 25-500C atunci specia cu p.t mai mic cristalizeaza dificil in topitura si se va transforma rapid. Daca diferenta este mica (1-250C) forma instabila se poate obtine usor inainte ca sa se manifeste transformarea solid-solid.
In cazul in care se identifica polimorfii, trebuie efectuate studii pentru a determina proprietatile lor.
E. Puritatea cristalului
Analiza termica se folosette pentru determinarea puritatii. Prezenta impuritatilor modifica unele proprietati fizice ale substantei.
F. Solubilitatea
Unul din cele mai importante consecinte ale determinarii p.t. este pentru solubilitatea substantei datorita legaturii intre p.t. si solubilitate prin caldura de topire. Caldura latenta de topire este cantitatea de caldura produsa in timpul topirii. Este energia necesara cresterii distantelor interatomice sau intermoleculare pentru a usura dezordinea spre a distruge reteaua cristalina si a incepe topirea. Un cristal cu legaturi slabe se va topi la temperatura joasa si are o caldura de topire mica. Solubilitatea este influentata si ea de ruperea structurii cristaline pentru a permite dispersia moleculara in solvent si ea depinde de asemenea de fortele intermoleculare.
Polimorfii difera in privinta solubilitatii. De aceea folosirea unor polimorfi diferiti in formulari diferite, poate conduce la diferente in proprietatile biofarmaceutice, viteza de dizolvare sau viteza de absorbtie.
G. Metoda de dozare
Dozarea substantei medicamentoase este necesara pentru a determina apoi stabilitatea substantei in solutie si in stare solida. in unele cazuri se foloseste spectrofotometria U>V> separarea substantei de produsii de degradare si excipienti se face cu ajutorul cromatografiei. CSS ajuta la identificarea numarului produsilor de degradare si la separari. Cromatografia de lichide cu inalta presiune (CLIP), sau de inalta performanta (HPLC) este una din metodele cele mai corespunzatoare pentru dozare.
H. Stabilitatea substantei medicamentoase
Produsele farmaceutice ar trebui sa aiba o perioada de valabilitate de 5 ani. Ȋn acest timp concentratia nu trebuie sa scada sub 90%, iar produsii de degradare sa nu fie toxici. Ȋn scopul formularii unui astfel de preparat este necesar ca in primul rand substanta medicamentoasa sa fie stabila, iar adaosul de excipienti si tehnologia de preparare sa nu-i scada stabilitatea.
Degradarea substantei medicamentoase poate avea loc prin diferite mecanisme: hidroliza, oxidare, fotoliza, cataliza prin urme de ioni metalici. Hidroliza si oxidarea sunt caile cele mai obisnuite, iar lumina si ionii metalici catalizeaza procesele oxidative ulterioare.
Conditiile de stres in perioada de stocare a studiului de preformulare sunt redate astfet pentru substanta solid: -caldura (0C): 4;20;30;37 la 50% si 75% U.R.
-absorbtia umiditatii: 30;45;60 si 90% u.r la 250C
-factori fizici: pulverizarea in moara cu bile.
I. Temperatura
Poate influenta toate cele patru mecanisme de degradare, accelerandu-le. O crestere a temperaturii cu 100C produce o crestere a descompunerii de 2-5 ori.
J. Hidroliza
Hidroliza este cea mai frecventa cauza a instabilitatii substantelor medicamentoase. Reactia hidrolitica implica atacul nucleofil al apei la legaturile labile (lactamica > ester > amida > imida ) ale substantei medicamentoase in solutie si este de ordinul intai. Daca acest atac este efectuat de un alt solvent decat apa, se numeste solvoliza. Hidroliza poate fi catalizata de: prezenta ionilor oxidril, prezenta ionilor de hidrogen, prezenta ionilor metalelor divalente, caldura, lumina, polaritatea solutiei si taria ionica, concentratii medicamentoase mai tari; hidroliza ionica este mai rapida decat cea moleculara.
K. Influenta pH
Hidroliza celor mai multe substante medicamentoase are loc in conditii extreme de pH, adica este catalizata de concentratii mari de ( H3O+) si (OH-). Multe substante medicamentoase sunt mai slabe la pH intre 4 si 8. Ȋn cazul solutiilor injectabile ajustarea pH-ului trebuie facuta cu sisteme tampon de capacitate mica, spre a se putea ajunge usor la valoarea pH-ului fiziologic de 7,4. Acizii slabi si bazele slabe sunt mai solubile in forma ionizata, deci instabilitatea este frecventa la speciile incarcate electric. Aceasta constituie o problema, deoarece cresterea concentratiei solutiei pana la valoarea dorita se face prin ionizare. O micsorare a degradarii se poate realiza prin asocierea la apa a unor solventi miscibili, care scad ionizarea, inlatura necesitatea unor valori extreme de pH, contribuie la dizolvarea substantei medicamentoase si reduc activitatea apei micsorand polaritatea solventului.
Reactiile de hidroliza in mediul apos sunt catalizate de pH, iar acest lucru se determina prin masurarea vitezei de degradare in functie de pH, mentinand constante temperatura, taria ionica si concentratia solventului. Sistemele tampon folosite sunt: acetat, lactat, citrat, fosfat si ascorbat.
L. Oxidarea
Oxidarea este puternic influentata de factorii de mediu: oxigen, lumina, urme de metale. Procesul de oxidare trebuie compensat cu unul de reducere. Oxidarea este o pierdere de electroni, iar agentul oxidant trebuie sa fie capabil sa preia, sa castige electroni. Oxidarea este sinonima si cu dehidrogenarea (pierderea de hidrogen). Cei mai multi antioxidanti cedeaza electroni sau ioni de hidrogen care sunt acceptati de un radical liber spre a termina reactia in lant. Antioxidantul spre a fi eficient trebuie sa se oxideze mai repede, mai usor, decat substanta medicamentoasa pe care numai astfel o poate proteja de oxidare.
Agentii chelanti
Agentii de chelare sut eficienti in blocarea urmelor de ioni metalici (care sunt catalizatori de oxidare) care sunt capabili sa formeze mai multe legaturi.
M. Fotoliza
Oxidarea si intr-o masura si hidroliza sunt catalizate de lumina. Energia produsa de radiatie este cu atat mai mare cu cat lungimea de unda scade (U.V. > vizibil > I.R.) si este independenta de temperatura. Cand molecula substantei medicamentoase absoarbe radiatie electromagnetica (lumina, fotoni) la lungimile de unda caracteristice, acesta produce o crestere a starii energetice a compusului. Aceasta energie produce descompunerea, poate fi retinuta sau transferata, poate fi transformata in caldura, sau poate produce emisie de lumina la o alta lungime de unda (fluorescenta, fosforescenta). Lumina naturala este in domeniul lungimilor de unda 290-780 nm din care numai cea U.V. produce fotodegradarea.
De aceea fotoliza trebuie evitata prin ambalare corespunzatoare, sticle colorate, cutii de carton, folii de aluminiu. Sticla incolora absoarbe radiatiile (80% in domeniul 290-380) in timp ce ambalajele din plastic absorb numai jumatate din aceasta cantitate de radiatie.
N. Stabilitatea in stare solida
Stabilitatea substantei medicamentoase in stare solida difera de cea discutata anterior, care are loc in solutie. Ȋn produsele solide exista totusi umiditate. Aceasta apa actioneaza ca vector pentru reactii chimice intre substanta medicamentoasa si excipienti, ca solutii saturate.
O. Higroscopia
O substanta care absoarbe suficienta apa din atmosfera ajungand sa se dizolve, este delicvescentaa. O substanta care pierde apa si formeaza un hidrat cu mai putine molecule de apa sau devine anhidra, se numeste eflorescenta. Aceste situatii sunt extreme. Substantele medicamentoase se pot comporta indiferent din punctul de vedere mentionat, ori castiga ori pierd apa din atmosfera in functie de umiditatea relativa. Materialele care ajung in echilibru cu umiditatea din atmosfera se numesc higroscopice. De aceea astfel de materiale, se vor prelucra in aer conditionat cu U.R sub 50%, iar cele foarte higroscopice la sub 40% U.R. Unele materiale hidrofile care se degradeaza usor, daca au aceasta calitate, trebuie sa fie ferite de umiditate spre a evita degradarea chimica si se vor conditiona in ambalaje ermetice sau in prezenta unui agent deshidratant.
P. Microscopia
Microscopia se foloseste in timpul formularii in cristalografie, pentru a determina morfologia cristalelor (structura, forma), polimorfismul si solvatii, precum si pentru a determina marimea particulelor.
I. 2. 2. Formularea
Pe parcursul ultimelor patru decenii, industria farmaceutica a investit numeroase fonduri si mult timp pentru studiul compactarii comprimatelor. Aceste cheltuieli sunt rezonabile, luand in considerare valoarea comprimatelor ca forma farmaceutica. Deoarece formele farmaceutice pot fi auto-administrate de catre pacient, acestea sunt vadit mai profitabile ca produs, decat formele medicamentoase parenterale, care trebuie administrate, in majoritatea cazurilor de catre personal instruit. Aceasta se reflecta si prin faptul ca peste 80% din medicamentele care sunt comercializate ca produse farmaceutice, formulate pentru a produce efecte sistemice, sunt forme pentru uz intern.
Comparativ cu alte forme medicamentoase orale, comprimatele sunt forma aleasa de catre producatori, pentru costurile joase de productie, ambalaj si transport, stabilitate inalta si rezistenta.
Ca sisteme de eliberare a substantei active, comprimatele pot varia de la formulari cu eliberare conventionala, relativ simpla, la formulari complexe de forme cu eliberare prelungita sau modificata.
Cel mai important rol al sistemului de eliberare consta in cedarea substantei active la locul de actiune, in concentratie suficienta si cu viteza adecvata, dorita.
Astfel, formularea comprimatelor trebuie sa realizeze un compromis intre:
-obtinerea unor produse cu proprietatile dorite privind dezagregarea si dizolvarea rapida sau controlata a particulelor primare constituente;
-si procedeul de fabricare al unui produs solid compact, estetic, dar cu rezistenta mecanica mare la manipulare, ambalare, depozitare si transport.
Formularea urmareste, in principal, realizarea de comprimare care trebuie sa indeplineasca urmatoarele criterii:
exactitatea si uniformitatea continutului de substanta activa pe fiecare unitate de dozare si de la un lot de fabricare la altul;
dizolvarea optima a substatei active, deci disponibilitate din forma farmaceutica pentru absorbtie, compatibila cu calea de utilizare destinata (eliberare conventionala sau prelungita);
stabilitate fizico-chimica, ce include stabilitatea substantei active, dezagregarea, viteza si timpul de dizolvare a substantei active din comprimate pentru o perioada lunga de timp;
acceptabilitatea pacientului: pe cat posibil, produsul final sa aiba un aspect atractiv, forma, marimea adecvata, culoare, gust, in scopul de a mari complianta cu regimul de dozare prescris;
posibilitati de fabricare avantajoasa: formularea trebuie sa permita o productie eficienta, practica, cu randament bun si la un pret de cost mic.
Factorii care influenteaza formularea comprimatelor
In etapa de formulare a comprimatelor se vor avea in vedere:
1. tipul, marimea si forma comprimatelor;
2. calea de administrare a comprimatelor;
3. proprietatile materiilor prime;
Tipul, marimea si forma comprimatelor
Cea mai raspandita forma farmaceutica orala, utilizata in prezent o constituie comprimatele, care pot fi definite ca produse solide de medicamente prin compactare. Majoritatea constau din amestecuri de pulberi care sunt presate in matrita (spatiu limitat) pentru a produce un singur corp rigid.
Desi se cunosc o varietate de tipuri de comprimate, care se administreaza pe cai diferite, toate se fabrica prin procedeul de comprimare:
-cele mai raspandite tipuri de comprimate sunt acelea destinate a fi inghitite, care apoi se dezagrega si elibereaza medicamentul in tractul gastrointestinal;
-un tip de comprimate, mai putin raspandit, care isi castiga popularitatea foarte rapid este formulat pentru a permite dizolvarea sau dispersarea in apa iainte de administrare. Ideal pentru acest tip de comprimate ar fi ca toate ingredientele sa fie solubile, dar frecvent poate fi acceptata o suspensie fina;
-unele comprimate sunt destinate sa fie mestecate (masticabile). Acest tip de comprimate este utilizat cand se doreste o absorbtie din cavitatea bucala sau pentru a mari dispersia inainte de inghitire;
-alternativ, comprimatele denumite pastile (engl. Lozenges) pot fi destinate unei dizolvari lente in cavitatea bucala, pentru o actiune locala;
Un alt factor important in formulare il constituie selectarea marimii si formei de comprimat adecvata. Acestea sunt determinate de substanta activa. Substantele active continute in cantitati foarte mici in formularea comprimatelor, de ordinul microgramelor (acid folic, digoxin, rezerpina, dexametazon) necesita folosirea unor cantitati mari de diluanti si alte substante auxiliare, pentru a obtine un amestec care sa permita obtinerea unui comprimat de forma si marimea acceptata de pacient.
O marime convenabila si comoda a comprimatelor cu substante active in doze mici este de aproximativ 6,35 mm diametru si cu o forma rotunda sau alta forma.
Comprimatele mici sunt dificil de utilizat de catre pacienti; in general acestea au o masa de 150 mg sau mai mult, in functie de densitatea excipientilor folositi in formulare.
Daca substanta activa se afla in cantitate mai mare intre 100-200 mg, masa comprimatului poate fi cuprinsa intre 150-300 mg; se pot folosi matrite rotunde cu diametrul cuprins intre 6.35 si 11.1 mm, in functie de densitatea si compresibilitatea amestecului de pulberi folosit.
Cu toate ca in majoritatea cazurilor, diametrul comprimatelor este constant, grosimea lor difera, din considerente de formulare, datorate flexibilitatii compozitiei.
1. 2. 3.Excipienti.Generalitati.
Comprimatele pot avea destinatii diferite de utilizare, iar modul lor de preparare difera. Acest fapt duce la formulari ce prezinta anumite particularitati. O categorie de comprimate este cea a celor cu cedare conventionala. Comprimatele conventionale, contin cateva categorii de excipienti cu ajutorul carora se pot formula si prepara: diluanti, lianti, dezagreganti, lubrifianti. Acestora li se adauga si alte cateva categorii, uneori puternic diferentiate dependent de destinatia speciala de utilizare a comprimatului respectiv.
Diluanti
Diluantii se asociaza intr-un comprimat atunci cand cantitatea de substanta medicamentoasa se foloseste intr-o doza foarte mica, spre a putea prepara un comprimat de marime convenabila. Diluantii trebuie sa fie excipienti inerti chimic si fiziologic, netoxici si cu proprietati bune de compresibilitate si compactibilitate. Uneori rolul de diluant se combina si cu o alta proprietate, cum ar fi cea de dezagregant.
Excipientul diluant cel mai folosit este -lactoza. Se poate folosi ca lactoza anhidra sau hidratata, precum si un sortiment aglomerat obtinut din lactoza monohidratata prin aerosolizare-uscare (atomizare, spray dried) in care este asociata cu 10% material amorf. Lactoza este solubila in apa, dar lent. Este stabila si nereactiva, si poate da o reactie de culoare bruna cu substante cu grupari aminice (reactia Maillard), in special forma hidratata. Lactoza anhidra are proprietati corespunzatoare de coenzima, dar curgerea este mai dificila. Lactoza obtinuta prin aerosolizare (atomatizata) are proprietati bune de curgere, dar necesita lianti. Un sortiment obtinut din lactoza cristalizata si amorfa, Fast Flo, poseda bune proprietati de compresibilitate si dizolvare. Lactoza confera duritate ridicata comprimatelor in care se asociaza in proportii suficient de mari.
Amidonurile ( Amylum)
Amidonul este un excipient de natura vegetala.
Se disting: amidonuri native si amidonuri modificate.
Amidonurile native sunt extrase din vegetale si se pot imparti in amidonuri propriu-zise si feculele.
a) Amidonurile propriu-zise sunt extrase in general din grauntele de graminee sau sau de leguminoase, care contin rezerva de hidrati de carbon. Gasim in aceasta grupa amidonurile de porumb, grau si orez.
b). Feculele sunt amidonurile extrase din tuberculi (exemple: amidonul de cartof sau de manioc).
Sub aceasta denumire (Amylum) se recunosc mai multe sorturi de amidon, in functie de provenienta, de cracteristicile macro si microscopice pe care le detin.
Astfel, in F.R. X figureaza o monografie individuala, intitulata Amylum, care reuneste date despre trei sorturi de amidon (de grau, de porumb si cartof ).
Amidonul se prezinta ca o pulbere fina, alba, fara miros si fara gust, cu urmatoarele caracteristici microscopice in functie de provenienta, redate in tabelul 2.
Tabel 2. Caracteristicile sorturilor de amidon (F.R. X)
|
Nr. Crt. |
Tipul de amidon |
Forma granulelor |
Diametrul granulelor |
|
grau |
granule simple, mici, rotunde granule simple, mari, sferice sau lenticulare |
2-10 μm 10-45 μm |
|
|
porumb |
granule simple, poliedrice granule simple rotunde |
2-23 μm 25-32 μm |
|
|
cartof |
granule simple neregulat-ovoidale, cu o extremitate mai ingusta decat cealalta Granule simple rotunde |
30-100 μm 10-35 μm |
Sorturile de amidon difera de asemenea prin proportia intre amiloza si amilopectina pe care o detin, astfel amidonul de porumb contine 27% amiloza, iar cel de cartof 22%.
Conform FR X amidonul se pastreaza in recipiente bine inchise etans, pe eticheta specificandu-se natura amidonului (Amylum tritici, Amylum maydis sau Amylum solani).
Indiferent de natura sa amidonul nu trebuie sa contina Echerichia coli si Salmonella sp. Aceste diferente modifica proprietatile fizice pe care le au, ceea ce nu permite inlocuirea reciproca a acestor sorturi in formularile farmaceutice.
Farmacopeea Europeana, editia a sasea prezinta monografii individuale pentru fiecare sort de amidon utilizat in practica farmaceutica. Astfel, pentru sorturile de amidon de grau, porumb si cartof caracteristicile macroscopice si microscopice coincid cu cele din Farmacopeea Romana in vigoare, in plus se prevad limite pentru continutul de SO2 (maximum 50 ppm), iar calitatea microbiologica a acestora presupune un continut de maximum 103 bacterii si 102 fungi per gram (numarul total de germeni aerobi viabili) si absenta E.coli.
Ȋn formularea pudrelor cel mai utilizat este amidonul de orez, care are partcicule foarte mici. Se mai pot folosi si amidonurile de grau sau de porumb. Cel mai putin recomandat este amidonul de cartof.
Avantajele pe care le confera pudrei sunt:
capacitate buna de absorbtie a apei si a substantelor grase;
confera pudrei o buna aderenta;
prezinta o actiune racoritoare si calmanta;
are o capacitate de curgere corespunzatoare;
este inert din punct de vedere fiziologic;
in prezenta umiditatii fermenteaza si este usor invadat de catre microorganisme (chiar in caz de transpiratie si la temperatura corpului) ;
se sterilizeaza usor.
Farmacopeea Europeana, editia a 6 a prezinta monografii individuale pentru fiecare sort de amidon utilizat in practica farmaceutica. Astfel, pentru sorturile de amidon de grau, porumb si cartof, cracteristicile macroscopice si microscopice coincid cu cele din Farmacopeea Romana in vigoare, in plus se prevad limite pentru continutul in SO2 (maximum 50 ppm), iar calitatea microiologica a acestora presupune un continut de maximun 103 bacterii si 102 fungi per gram (numarul total de germeni aerobi viabili) si absenta E.coli.
Amidonul poate fi folosit ca excipient diluant in obtinerea comprimatelor in proportie de 30%; o proportie mai mare da comprimate moi si friabile sau ca aglutinant (mucilag din amidon in concentratie de 5-15%, optim 10%) si dezagregant in concentratie de 5-10%.
Trebuie cunoscut foarte bine continutul sau in umiditate, pentru a nu obtine o masa prea moale.
Amidonurile modificate au fost preparate initial in industria alimentara, mai mult pentru a imbunatati proprietatile de gel si vascozitatea in solutie, si mai putin pentru a ameliora proprietatile mecanice ale granulelor de amidon intregi.
Cel mai utilizat in industria farmaceutica este amidonul pregelatinizat inscris in Farmacopeele americane si englezesti. Sunt obtinute in general prin coacerea pe rulouri incalzite a laptelui de amidon. Produsul obtinut uscat este apoi cules sub forma unor solzi. Dupa dispersarea acestuia in apa, acest produs prezinta mai rar cateva granule de amidon mai mult sau mai putin explodate.
Amidonurile pregelatinizate sunt cunoscute sub diverse denumiri comerciale (Lycatab PGS, Starch 1500), sunt folosite in farmacie mai ales ca agenti de granulare (figura nr.1)

Fig. 1. Amidon pregelatinizat Lycatab
Sorturile de amidon pregelatinizat prezinta proprietati de curgere si compresibilitate crescute, ceea ce permite utilizarea acestora ca agenti lianti utilizati in procesele de comprimare directa (tabelul 3 ).
Tabel 3. Proprietatile de curgere si compresibilitate ale amidonului pregelatinizat
|
Nr. Crt. |
Concentratie % |
Utilizare |
|
diluant (capsule gelatinoase tari) |
||
|
aglutinant (comprimare directa) |
||
|
algutinant (granulare umeda) |
||
|
dezagregant (comprimate) |
Farmacopeea Europeana editia a 6 a specifica ca obtinerea amidonului pregelatinizat se realizeaza din amidonul de porumb, cartof si de orez care se incadreaza in limitele referitoare la limitele de SO2 (maximum 50 ppm) si ale calitatii microbiologice caracteristice de amidon oficinale.
Manitolul a dobandit o utilizare extinsa in formulari orale, comprimate pentru supt sau comprimate masticabile. Are o putere de indulcire de 72% din cea a zaharului. Este necalorigen. Caldura de dizolvare este negativa lasand o senzatie de racorire. Solubilitatea in apa este 1:5,5. Este nehigroscopic. Proprietatile de curgere sunt slabe si necesita asocierea cu lubrefianti in cantitati mari.
Sorbitolul este un izomer optic al manitolului. Spre deosebire de acesta este higroscopic.
Zaharul se foloseste ca diluant, in cantitati moderate. Este usor higroscopic. Are proprietati adezive de matrita si ponsoane. Dezagregarea este prelungita in prezenta sa. Ȋn unele sortimente se asociaza cu alti excipienti: Sugartab (90-93% zaharoza si 7-10% zahar invertit); Di-Pac (97% zaharoza si 3% dextrine modificate); Nu-Tab ( 95% zaharoza, 4% zahar invertit si 0,1-0,2% amidon de porumb si stearat de magneziu). Aceste varietati se folosesc la comprimarea directa pentru comprimate bucale sau perorale.
Celuloza microcristalina reprezinta celuloza purificata si depolimerizata partial prin tratarea cu acizi minerali diluati ai α- celulozei sub forma microcristalina, prezinta o structura fibroasa. Este prezenta in diverse forme comerciale, care difera prin forma particulei, diametrul, continutul in umiditate, in functie de care prezinta proprietati si utilizari diferite (figura 2).

Fig. 2. Celuloza microcristalina SEM
Una din varietatile comerciale este cunoscuta sub numele de Avicel. Aceasta se prezinta ca o pulbere alba, cristalina, fara miros si gust, inslolubila in apa, cu densitatea aparenta cuprinsa intre 0,2 - 0,5 g/cm3.
Produsul este extrem de compresibil, permite obtinerea de comprimate de duritate mare, nu se foloseste singur, se asociaza cu lactoza, amidon si fosfat de calciu.
Se cunosc mai multe sorturi, Avicel 101 (pulbere cu particule de 50 μm) si Avicel 102 (cristale granulate cu diametrul particulelor de 90 μm).
Este preferata utilizarea sortului 102 fata de 101, deoarece are o capacitate de curgere si de lubrifiere mai buna imprimata de dimensiunea particulelor (care este mai mare), de forma acestora (granulara) si de o compresabilitate mai scazuta.
Derivatii de celuloza (metilceluloza si carboximetilceluloza) se folosesc ca lianti sub forma de mucilagii, sau ca atare ca pulberi umectate, dar au capacitate de legare mica.
Unele saruri anorganice se folosesc ca diluanti: carbonatul de calciu, fosfatul dicalcic, fosfatul tricalcic, carbonatul de magneziu, clorura de sodiu. O utilizare mai frecventa o are fosfatul dicalcic dihidratat, insolubil, cu o curgere si stabilitate buna, dar cu caracteristici slabe de comprimare. Este usor alcalin. In unele sortimente exista asocieri: Emcompress A (fosfat dicalcic 80-95%, asociat cu amidon, stearat de magneziu, celuloza microcristalina si Primojel-amindonglicolat de sodiu) pentru a imbunatati comprimarea si dezagregarea.
Excipientii solubili precum si triturarile unor substante medicamentoase cu acestia ( digoxina cu lactoza) cresc viteza de dizolvare a substantei medicamentoase greu solubile.
Lianti
Excipientii lianti sau aglutinanti se folosesc pentru a mari coeziunea particulelor pulberii in vederea formularii granulelor si pentru a creste coeziunea particulelor in timpul comprimarii. Alegerea liantului se face in functie de compatibilitatea cu componentii formulei, capacitatea de legare, fara a intarzia prea mult dezagregarea. In granularea umeda se folosesc sub forma dispersiilor lor in apa (mucilagii).
Guma arabica in solutii apoase de 10-25% este bun liant, dar confera rezistenta mecanica crescuta comprimatelor si poate prelungi dezagregarea dupa pastrarea comprimatelor in timp.
Tragacanta se poate asocia sub forma de pulbere, iar umectarea se face cu apa.
Zaharul se foloseste ca sirop in concentratie de 50-65%. Comprimatele au rezistenta mecanica buna, dar sunt friabile.
Gelatina este un liant corespunzator. Se foloseste sub forma de solutii apoase calde spre a preveni gelificarea la rece. Prepararea mucilagiului se poate face usor, iar rezistenta mecanica pe care o confera comprimatelor este buna, fara a intarzia dezagregarea in timp. Ȋn concentratii mari, creste rezistenta mecanica si timpul de dezagregare. Se pot folosi si solutii (alcoolice de gelatina cu pH=2 sau pH=11 dar procedeul de preparare este mai complicat.
Coca de amidon are capacitate coeziva redusa, ceea ce face ca dezagregarea comprimatelor sa fie rapida. Se poate folosi amidonul sub forma de pulbere, legarea fiind asigurata de adaugarea de apa calda sau vapori de apa, dar formarea granulelor este mai dificila in acest mod. Asocierea mucilagiului de gelatina la coca de amidon creste capacitatea de coeziune a particulelor si rezistenta mecanica a comprimatelor.
Derivatii de celuloza : metilceluloza, carboximetilceluloza sodica si mai ales hidroxipropilmetilceluloza (HPLC) sunt polimeri hidrosolubili care au o buna capacitate de legare a pulberilor in granule. Sunt preferabili derivatii lor naturali. Este necesara cunoasterea tipului, a gradului de polimerizare, deoarece vascozitatea sortimentelor difera. Hidroxipropilmetilceluloza se dizolva in clorura de metilen sau amestecuri hidroalcoolice. Etilceluloza se foloseste ca solutie alcoolica 0,5-2%.
Polivinilpirolidona este utilizata atat ca solutie apoasa cat si ca solutie alcoolica. Solutiile alcoolice se folosesc la granularea substantelor sensibile la umiditate. Granularea se face usor, uscarea este rapida iar comprimarea usoara. Rezistenta mecanica a comprimatelor este insa destul de mica.
Dezagreganti
Dezagregantii au rolul de a invinge forta de coeziune a materialului conferita prin comprimare si prin prezenta excipientilor lianti, in scopul eliberarii continutului medicamentos in stomac dupa inghitirea comprimatului, sau in apa inaintea administrarii. Dezagregantii se adauga amestecului de pulberi inainte de granulare (intragranular) sau granulelor inainte de comprimare (intergranular) sau in ambele faze. Dupa mecanismul prin care actioneaza se disting cateva categorii de dezagreganti: accentuarea fortelor capilare de incorporare a apei in comprimat, umflare in contact cu apa, eliberare de gaze, topire la temperatura corpului, actiunea unor enzime.
Dezagregantii care actioneaza prin favorizarea efectelor capilare dizolva puntile agentului liant din reteaua capilara dintre granulele comprimatului si reduc tensiunea superficiala. In acest fel favorizeaza patrunderea apei in structura poroasa a comprimatului, prin forte capilare, care determina dezagregarea acestuia. Cei mai utilizati dezagreganti de acest tip sunt amidonurile. Adaugarea amidonului se face in proportie de 5-20% din masa comprimatului. Este avantajos pentru grabirea dezagregarii, ca din cantitatea totala sa se adauge 25-30% intragranular.
Amidonul intragranular asigura dezagregarea comprimatului, iar cel intragranular asigura dezagregarea granulelor eliberand particulele substantei medicamentoase. Imbibarea amidonului la temperatura corpului are un rol minor in dezagregare, intrucat umflarea amidonului incepe la temperaturi peste 40oC. Amidonul formeaza cu apa legaturi de hidrogen, ceea ce favorizeaza patrunderea in comprimat.
Se pare ca actiunea de dezagregare optima se realizeaza atunci cand granululele de amidon formeaza un film in jurul aglomerarilor de particule ce constituie granulatul. Concentratii mai mari de amidon pot reduce timpul de dezagregare, dar scade rezistenta mecanica a comprimatului. De aceea se cauta o concentratie optima in formulare.
Unii derivati de amidon au proprietati dezagregante superioare, fiind numiti si superdezagreganti. Asa sunt sunt sortimentele de Primojel ( aminoglicolat de sodiu); Explotab (carboximetil amidon); Crosspovidone (polivinilpirolidona reticulara). Imbibarea comprimatelor care contin amidon se face cu o crestere de volum de 20-25% in timp ce aceste amidonuri modificate isi modifica volumul cu apa pana la 200-300%.
Dezagregantii care se imbiba si isi maresc volumul in prezenta apei se folosesc in concentratii reduse pentru a nu intarzia dezagregarea din cauza vascozitatii prea mari care se realizeaza in jurul comprimatului si in interiorul hidratant al acestuia. Din aceasta categorie se folosesc carboximetilceluloza sodica si s-au folosit in mai mica masura si agarul, tragacanta, alginatul de sodiu.
Dezagregantii generatori de gaz se folosesc pentru dezagregarea rapida a comprimatelor cand alti dezagreganti nu au dat rezultate satisfacatoare, sau au in formularea comprimatelor solubile (comprimate efervescente). Utilizarea lor este conditionata de o umiditate redusa in timpul prepararii (atmosfera cu umiditate controlata, sub 20% u.r.) si conservarii comprimatului. Cei mai obisnuiti sunt amestecurile de acid citric cu bicarbonat de sodiu sau alti carbonati, in raporturi stoechiometrice intre ei si proportii diferite in comprimate.
Unii dezagreganti actioneaza prin simpla dizolvare in apa, sau prin topire la temperatura corpului. Ȋn anumite cazuri se asociaza enzime care rup liantul: amilaze pentru amidon, celulaze pentru derivatii de celuloza, hemicelulaze pentru gume, proteaze pentru gelatina, invertaza pentru zaharoza, dar utilizarea acestora este redusa.
Lubrefianti
Denumirea de lubrefianti se da excipientilor care micsoreaza adeziunea dintre comprimat pe o parte si matrita si ponsoanele pe de alta parte, precum si intre granulele care alimenteaza matrita, micsorand frecarea dintre acestea. De fapt in toate cazurile are loc o micsorare a fortelor de frecare fapt pentru care unii ii denumesc agenti antifrictionali. O impartire a lubrefiantilor se poate face in :
Lubrefianti propriu-zisi, care micsoreaza frecarea suprafetei laterale a comprimatului de matrita si favorizeaza eliminarea comprimatului;
Antiaderenti, care micsoreaza lipirea materialului de comprimat de ponsoane si matrita;
Glisanti, care imbunatatesc curgerea granulatului in matrita.
Lubrefiantii (stearatul de magneziu, uleiurile minerale) actioneaza printr-un mecanism diferit. Uleiul mineral (ulei de vaselina) actioneaza prin separarea suprafetelor care vin in contact intre care se manifesta adeziunea, printr-un film lubrifiant lichid.
Stearatul de magneziu actioneaza printr-o lubrefiere marginala, manifestata prin adeziunea gruparilor polare ale moleculei de peretii matritei, iar cu lanturile hidrocarbonate spre material. Eficienta lor este mai mare decat a lubrifiantilor lichizi, deoarece adeziunea gruparii polare fata de matrita metalica este mai puternica.
Lubrefiantii tind sa egalizeze distributia presiunii de comprimare in masa comprimatului. Eficienta lor este determinata de un grad de dispersie avansat al particulelor. Intrucat sunt higrofobi, prelungesc timpul de dezagregare al comprimatelor si timpul de dizolvare al substantei medicamentoase. De aceea nu se recomanda sa se depaseasca anumite proportii in comprimat. Acidul stearic, stearatul de magneziu, stearatul de calciu se folosesc in concentratii de 0,1-0,25% si nu va depasi concentratia de 1%. Parafina lichida cu vascozitate mica si uleiurile hidrogenate se pot folosi ca lubrefianti, fata de cei solizi, necesitand in plus dispersarea prin intermediul unui solvent organic volatil.
Talcul si dioxidul de siliciu coloidal (Aerosoli) si un silicoaluminat de sodiu hidratat (Cab-O-Sil) sunt folositi ca agenti glisanti pentru usurarea curgerii granulatului din palnia de umplere in matrita. Sortimentele de talc cu particule mari sunt abrazive. Concentratiile lui nu vor depasi 3%.
Se folosesc si amestecuri de excipienti cu rol lubrifiant si glisant: talc + stearat de magneziu (9:1); talc + aerosil + stearat de magneziu ( 8:1:1); talc + alcool etilic (9:1).
Lubrefiantii solubili se folosesc in cazul comprimatelor solubile, cum sunt comprimatele efervescente. Laurilsulfatul de sodiu sau cel de magneziu au bune proprietati de curgere, favorizeaza dezagregarea, nu influenteaza negativ rezistenta mecanica. Ei se adauga si la celelalte comprimate la care se folosesc lubrifianti insolubili, pentru a contracara efectul hidrofobizant al stearatului de magneziu.
Polietilenglicolii (PEG 400, PEG 600) se folosesc sub forma de pulbere fina ca lubrefianti solubili; ei au o putere lubrefianta mai redusa decat a unor concentratii egale de stearil. Se mai poi folosi benzoatul de sodiu si esteri ca triacetatul de gliceril.
I. 2. 4. Procedee tehnologice de obtinere a formelor farmaceutice cu cedare conventionala
Descrierea procesului tehnologic si a echipamentului folosit:
Procesul tehnologic al realizarii comprimatelor cuprinde urmatoarele operatii:
Divizarea (pulverizarea) substantelor la gradul de diviziune necesar unor amestecuri omogene cat si in vederea obtinerii efectului terapeutic;
Clasarea operatia de sortare a particulelor cu dimensiuni apropiate;
Cantarirea
Amestecarea - operatia prin care se asigura omogenitatea amestecului de pulberi cu consecinte in realizarea unor comprimate dozate uniform;
Granularea umeda - este o operatie frecvent intrebuintata in metoda comprimarii indirecte;
Uscarea
Divizarea (calibrare)
Pudrarea
Comprimarea - este operatia in care amestecul de pulberi sau granulate sunt supuse la doua forte opuse;
Ambalarea primara
Ambalarea secundara
1. Divizarea- pulverizarea - este operatia de maruntire a materialelor solide pana la anumite dimensiuni.
Principiul metodei: infrangerea coeziunii particulelor care formeaza corpul solid si crearea unor suprafete noi.
Scopul: accelerarea operatiilor fizice (amestecare, uscare) si a reactiilor chimice prin cresterea suprafatei de contact dintre fazele care participa la proces. Functie de natura fortelor cu care se invinge coeziunea, se impart in divizare prin: strivire, lovire, frecare si despicare. Prin pulverizare are loc o crestere a gradului de dispersie prin reducerea marimii particulelor. Marimea suprafetei pe unitatea de greutate, cunoscuta ca suprafata specifica, creste prin micsorarea marimii particulelor.
In operatia de micsorare a marimii particulelor se lucreaza in mai multe etape. Se incepe cu o maruntire grosiera ( concasare ), in raportul de reducere a diametrului D1/D2 este intre 3 si 7. Se continua cu pulverizarea sau macinarea, in care gradul de reducere a marimii particulelor poate merge pana la 100.
Pulverizarea are loc prin impact (lovire, percutare) cu carcasa, precum si cu ciocanele.
Vom face o diviziune coloidala, cu dimensiunea particulelor de 0,175mm, cu ajutorul morii coloidale (figura 3), astfel:
Mod de functionare: discul se roteste cu viteza foarte mare, el are pe partile laterale inele concentrice care se rotesc intre sicane, fixate pe peretii carcasei. Materialul de macinat e introdus sub forma de suspensie lichida prin racord. Viteza discului trebuie sa fie foarte mare pentru a realiza socuri foarte mari, comparativ cu loviturile de ciocan.

Fig. 3. Moara coloidala
Clasarea este etapa necesara pentru a separa particulele care au granulatia dorita unui amestec cu diferite granulatii.
Urmeaza cantarirea substantelor folosite pentru fabricarea comprimatului:
Acid acetilsalicilic 0,500
Carbonat de calciu 0,250
Celuloza microcristalina 101 0,020
Amidon porumb 0,0265
Talc 0,0015
Dioxid de siliciu coloidal 0,002
Apa purificata 0,080
Urmatoarea etapa este amestecarea materialelor. Aceasta etapa are ca obiectiv accelerarea reactiilor chimice si omogenizarea amestecului de materiale. Amestecatorul cu pat fluidizat actioneaza prin intermediul unui flux de aer asupra pulberilor. Dispozitivul se foloseste si la uscare si granulare.
Se introduc: Substanta active: acidul acetilsalicilic
Agentul de tamponare: carbonatul de caliciu
..Agent de diluare: celuloza microcristalina
Dezagregant, aglutinant, agent de diluare: amidonul de porumb
..Agent de lubrefiere: talcul; dioxidul de siliciu
Dupa uscare, se poate face granularea. Granularea este procesul prin care se mareste dimensiunea particulelor prin aglomerare si aderarea particulelor mai mici in agregate permanente mai mari, denumite granule. In ele pot fi indentificate particulele initiale.
In urma granularii,
-se imbunatatesc proprietatile de curgere ale amestecului (posibile datorita coeziunii mari)
-imbunatatirea proprietatilor de comprimare,
-e prevenita separarea componentelor unui amestec de pulberi datorita densitatilor diferite.
- granulele fiind mai dense ocupa un volum mai mic, deci sunt mai usor de pastrat si manipulat.
Granularea cu pat fluidizant foloseste acelasi principiu ca la uscare. Fluidizarea pulberii intr-un curent de aer ascendent, granularea cu solutia de liant aerosolizata in aparat, si uscarea granulelor obtinute in aceeasi incinta. Lichidul de granulare este pompat printr-o duza, in particule mici, aerosolizate. El va determina aderarea particulelor umezite in momentul ciocnirii lor. Granulele uscate cad pe la peretii vasului. Particulele mai fine sunt retinute in sacii de filtrare ai aerului.

Fig. 4. Granulator in pat fluidizat
Uscarea este o etapa importanta pentru ca asigura conservarea materialelor, iar materialele uscate permit granularea. Uscarea in pat fluidizat are loc prin contactul aerului cald cu particulele solide supuse uscarii. Mecanismul este prin convectie si este dinamic. Patul pulberii se fluidizeaza cu ajutorul curentului de aer cu viteza mare, care este suficienta sa antreneze particulele solide si sa produca transportul lor.
Avantajele uscarii in pat fluidizat:
Transfer eficient de caldura si masa, viteza mare de uscare;
Uscarea se face asupra particulelor si nu a intregului pat, deci viteza de uscare este constanta;
Temperatura este uniforma si precis controlata;
Materialul uscat are o curgere libera;
Particulele au forma sferica.
Divizarea se realizeaza in acelasi aparat ca si granularea si uscarea.
Se pudreaza granulele obtinute cu Lubrifiantul solid: talcul si dioxidul de siliciu
Dezagregantul: amidonul de porumb
Comprimarea
Se realizeaza prin presarea substantelor medicamentoase sub forma de pulbere sau granule in stare uscata cu sau fara adaos de substante ajutatoare, inerte din punct de vedere terapeutic.
Etape: - rearanjarea particulelor
- deformarea in puncte de contact
- fragmentarea
- legarea
Vom comprima cu ajutorul masinii de comprimat cu excentric. Are palnie mobila si matrita fixa, conform figurii 5:
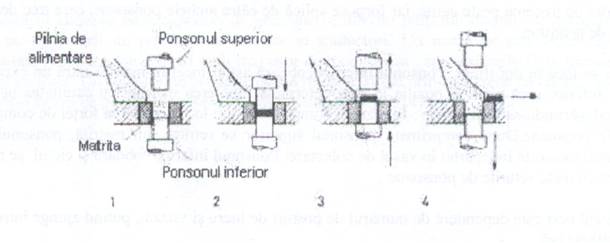
Fig. 5. Masina de comprimat cu excentric
Se umple matrita cu materia, cu ajutorul pistoanelor se comprima materialul. Prin ridicarea pistonului se evacueaza comprimatul de matrita, apoi din matina si opertiunea se repeta.
Faza nr.1 - ponsonul superior si inferior cat si palnia de umplere se gasesc in pozitie initiala-spatiul matritei este umplut cu material de comprimat.
Faza nr.2- ponsonul superior preseaza materialul in matrita formand comprimatul.
Faza nr.3- ponsonul superior se retrage in pozitia initiala si cel inferior se ridica la suprafata impingand comprimatul la marginea matritei.
Faza nr.4- palnia de umplere aluneca inainte, impingand comprimatul pe banda de evacuare, in acelasi timp ponsonul inferior ajunge la baza matritei si masa de comprimat va umple din nou matrita.
I. 2. 5. Controlul calitatii formelor farmaceutice cu cedare conventionala
Comprimatele trebuie sa fie formulate, fabricate si conditionate, in asa fel incat sa asigure calitatea in relatie cu biodisponibilitatea, stabilitatea fizico-chimica si absenta contaminarii microbiene.
Calitatea comprimatelor cu cedare conventionala este asigurata prin respectarea regulilor Bonne Partique de Fabrication (BPF) si a farmacopeilor si trebuie sa se pastreze neschimbata pe toata perioada de valabilitate.
In acest scop, controalele se vor efectua incepand cu materiile prime, pe fazele procesului tehnologic (controlul in cursul fabricarii) si asupra produsului finit.
Controlul materiilor prime
Acest control este efectuat pe materiile achizitionate si aflate in depozitul fabricii, inainte ca ele sa intre in procesul de productie.
Fiecare container cu materia prima (substante active si substante auxiliare) este insotit de un buletin de analiza, care atesta identitatea si puritatea sa.
Ȋn cadrul etapelor de preformulare si formulare, se verifica o serie de proprietati fizico-mecanice, care sunt importante pentru procesul comprimarii: forma cristalina, marimea particulelor, curgerea, compresibilitatea si compactibilitatea.
Dintre substantele auxiliare, diluantii sunt componentii in proportia cea mai mare. Pentru diluanti, sunt importante proprietatile fizice: culoarea, gustul, mirosul, cat si puritatea chimica.
In cazul excipientilor lianti, se verifica vascozitatea, proprietate importanta pentru capacitatea de aglutinare.
Eficacitatea lubrifiantilor depinde de marimea particulelor care se determina prin anumite procedee. De asemenea, aromatizantii, edulcorantii si colorantii se controleaza prin metode specifice pentru identificare si puritate.
Controlul in cursul fabricarii
Acest control este de asemenea, obligatoriu pentru a obtine comprimate cu o calitate corespunzatoare. In aceasta etapa, se efectueaza diferite controale, in functie de fazele procesului tehnologic ales:
-palnia de alimentare, care trebuie sa contina o cantitate de pulbere sau granule corespunzatoare;
-uniformitatea masei, prin controlul periodic al greutatii comprimatului, la intervale de 5-30 de minute;
-duritatea comprimatelor;
-eventual, dezagregarea comprimatelor;
Controlul produsului finit
Acest control cuprinde o serie de verificari impuse de farmacopei sau de normele de fabricare a comprimatelor. FR X si suplimentele prevad determinarea urmatorilor parametrii: aspect, miros, gust, culoare, uniformitatea masei, uniformitatea continutului, testul de dezagregare si testul de dizolvare.
In plus, se efectueaza si alte determinari specifice comprimatelor: rezistenta mecanica la rupere (duritateta), friabilitatea, modul de sectionare a comprimatului, pH-ul, studii de stabilitate in timp, absorbtia apei.
Determinari fizico-chimice
Examenul caracterelor macroscopice (organoleptic)
Acest control se refera la urmarirea caracterelor vizuale: aspect, culoare, gust, miros si tactile: la atingere sau de adeziune.
a) Caractere vizuale
-aspect: comprimatele neacoperite trebuie sa prezinte forme de discuri sau alte forme, aspect uniform, margini intacte, suprafata plana sau convexa; o culoare uniforma fara pete, asperitati sau crapaturi.
Eventualele inscriptii, santuri trebuie sa fie vizibile si foarte netede; acestea pot fi prezente pe una sau pe ambele fete ale comprimatului.
Prezenta unor striuri verticale pe marginea comprimatelor este cauzata de dificultatea de alunecare in matrita; aceasta denota o cantitate de lubrifiant insuficienta sau un lubrefiant necorespunzator.
Schimbarea aspectului comprimatelor indica eventuale modificari aparute in timpul depozitarii.
-omogenitatea: se observa cu ochiul liber sau cu lupa.
Prezenta petelor releva o dispersare neuniforma a materiilor prime, a amestecului de pulberi sau a granulelor dar si a unor interactiuni dintre componente.
Omogenitatea se observa si pe un comprimat rupt in doua bucati; se examineaza suprafata interna care trebuie sa fie asemanatoare cu cea externa.
-culoare: in general, comprimatele sunt albe. Comprimatele colorate trebuie sa prezinte o coloratie constanta, uniforma pentru productia unei sarje, cat si pe timpul depozitarii; aceasta denota pastrarea proprietatilor comprimatelor si deci eficacitatea.
b).Caractere olfactive
-gustul si mirosul: sunt caracteristice componentelor asociate. Trebuie sa se detecteze un miros specific al substantelor medicamentoase sau a substantelor auxiliare: mentol, acizi, arome, parfumuri, etc.
c).Caractere tactile
-la atingerea cu mana, sa prezinte o suprafata neteda lucioasa, fara asperitati sau crapaturi.
Determinarea caracterelor dimensionale
Pentru evaluarea calitatii comprimatelor, sunt foarte importanti si parametrii ca: masa, inaltimea, diametrul comprimatelor. Determinarea diametrului si a grosimii comprimatelor sunt necesare chiar in timpul procesului tehnologic de fabricare.
La acelasi diametru si greutate, un comprimat mai subtire este un comprimat mai puternic decat unul gros. Daca masina de comprimat este reglata bine, diferentele de grosime ale comprimatelor nu trec de 1-1,5%.
Uniformitatea dimensiunilor este necesara pentru exactitatea dozarii, dar si pentru a nu se produce dificultati la ambalarea in tuburi sau in folii.
Daca unele comprimate sunt mai mari, nu intra in tuburi, iar daca sunt mai mici, se vor sfarama. Grosimea marginilor comprimatelor are importanta si in procesul de acoperire a acestora.
Comprimatele cu marginea mai subtire se pot acoperi mai usor, dar astfel de samburi sunt mai fragili la manevrele din turbina de drajfiere.
Dimensiunea comprimatului se determina clasic cu micrometrul standard cu surub (MOORE si WRIGHT).
Suprafata unui compimat se calculeaza dupa formula:
A = 2πr(r+h) ; [relatia 8]
A= aria suprafetei, in mm2
r = raza comprimatului, in mm
h = inaltimea comprimatului, in mm
Astazi se utilizeaza aparate moderne, care determina mai multi parametrii: masa, inaltimea, diametrul comprimatelor si rezistenta mecanica.
Determinarea pH-ului
Acest parametru este foarte important, pentru a nu modifica pH-ul fiziologic care difera dupa calea de administrare:
-calea oro-faringiana: pH = 6.7-7
-calea vaginala:pH = 4-4.5
-calea oculara:pH = 7.1-7.6
-calea gastro-intestinala:
-stomac: pH = 1-2 pe nemancate si 3 in timpul meselor;
-duoden: pH = 4.6
-jejun: pH= 7-8
pH-ul se determina potentiometric, conform FR: un comprimat se disperseaza in 30 ml apa distilata, la 370C .
Determinari farmacotehnice
Uniformitatea masei
Este un parametru important care caracterizeaza comprimatele si se determina in cursul fabricarii. Evaluarea statistica a masei medii si a comprimatelor verifica operarea regulata a masinii de comprimat si asigura o productie regulata si economica; ea se efetueaza continuu. Uniformitatea masei asigura si o uniformiate a dozarii si este necesara pentru conditionarea automata. Farmacopeile prevad ca proba sa se efectueze pe 20 de comprimate neacoperite prelevate la intamplare, care se cantaresc individual si li se calculeaza masa medie si abaterea relativa standard, de la masa comprimatelor.
Fata de masa medie calculata, masa individuala poate sa prezinte urmatoarele abateri procentuale redate in tabelul 4:
Tabel 4. Uniformitatea masei comprimatelor-limite admise de FR X suplimentul 2004
|
Forma farmaceutica |
Masa medie |
Deviatie % |
|
Comprimate neacoperite |
80 mg sau mai putin de 80 mg Peste 80 mg si mai putin de 250 mg 250 mg si peste 250 mg |
Atunci cand masa medie este mai mica sau egala cu 40 mg, nu se determina uniformitatea masei, ci uniformitatea continutului preparatelor unidoza.
Uniformitatea continutului
Este un test pentru stabilirea omogenitatii seriei. Se efectueaza dozarea substantei active pe 10 comprimate individual, apoi se determina media aritmetica si se calculeaza abaterea relativa standard. Criteriile corespun daca uniformitatea continutului se incadreaza in limitele de 85-115% din continutul declarat pe eticheta si nu este mai mare de 6%. In farmacopei, pentru testarea suplimentara, este inclusa o clauza in caz ca una sau mai multe unitati nu corespund cerintelor standard.
Determinarea se repeta pe inca 20 de comprimate, daca pentru cel mult un comprimat, continutul in substanta activa este in afara limitelor de 85% si 115% fata de valoarea medie, dar pentru un comprimat, continutul nu este in afara limitelor de 75% si 125%.
Testul se aplica in functie de tinutul de substanta activa/comprimat.
Sunt controlate prin acest test comprimatele cu pana la 2 mg substanta activa si cele al caror continut in substanta activa reprezinta 50% din masa comprimatului.
Pentru aceasta din urma, nu mai este necesara determinarea uniformitatii masei, daca se determina uniformitatea continutului.
Rezistenta la rupere
Este definita ca rezistenta pe care o opune comprimatul unor forte de solicitare externe, care actioneaza pe partile laterale, diametral opuse, pana la rupere.
In majoritatea cazurilor, comprimatul este plasat deasupra unei nicovale fixe, la care este transmisa forta prin intermediul unui piston mobil. O serie de dispozitive de acest tip sunt accesibile comercial, inclusiv Stokes (sau Monsanto), Strong-Cobb.
Pistonul poate fi miscat atat manual, cat si electronic. Comparand diferitele tipuri de dispozitive pentru determinarea rezistentei mecanice, s-a dovedit ca rezultatele obtinute la aparatele electronice sunt mai reproductibile decat cele obtinute cu dispozitivele manuale. Cu dispozitivele electronice, in cea mai mare parte, se obtin rezultate constante.
Forta aplicata asupra grosimii comprimatului este numita rezistenta la flexiune notata cu δτ.
David S.T. si Augsburger L.L. au constatat ca rezistenta la flexiune este aceeasi, indiferent de metoda utilizata daca se folosesc calcule adecvate.
Pot fi prezentate doua tipuri de erori: una asociata cu instalarea incorecta a punctului zero sau folosirea unei scari care nu indica exact forta aplicata.
Aparatele utilizate pentru determinarea rezistentei la rupere sunt de mai multe feluri: cu greutate, cu resort sau cu presiune pneumatica. Se determina greutatea la rupere, exprimata in kg.
O greutate cu cursor, prin intermediul unui piston, preseaza comprimatul plasat vertical intr-un lacas. Cresterea presiunii are loc lent si uniform. Se noteaza greutatea care produce ruperea comprimatului.
Farmacopeea Europeana prevede o metoda care utilizeaza o forta de presare.
Aparatul consta intr-un dispozitiv cu doi clesti care are suprafetele orizontale perpendiculare pe directia de miscare. Suprafetele de rupere ale clestilor sunt orizontale si mai mari decat zona de contact cu comprimatul. Aparatul este calibrat, utilizand un sistem cu o precizie de 1 N.
Determinarea se efectueaza la 10 comprimate prin plasarea comprimatelor intre clesti, in acelasi sens cu directia de aplicare a fortei.
Se calculeaza media valorilor minime si maxime ale fortelor masurate, exprimate in newtoni. Acest procedeu este in intregime automatizat.
Friabilitatea (abraziunea)
Testul de friabilitate sau rezistenta la abraziune (radere) este destinat sa determine in conditiile definite, abraziunea comprimatelor, fenomen prin care suprafetele comprimatelor sunt deteriorate si/sau evidentiaza laminare sau rupere, cand sunt supuse la socul mecanic de frecare.
Masurarea rezistentei la abraziune, notata cu B este exprimata ca pierderea procentuala in masa. Friabilitatea poate fi raportata in functie de timp sau de numarul de rotatii.
In literatura, sunt descrise diferite teste care evolueaza friabilitatea: Roche, Burlinson, Shafer.
Majoritatea testelor masoara pierderile care survin dupa amestecarea comprimatelor peste un anumit interval de timp. Mai specific un numar de comprimate cantarite (w0), sunt supuse unei operatii de amestecat intr-un container inchis geometric-fixat pentru un timp specific. Apoi comprimatele sunt recantarite (w).
Determinarea friabilitatii se efectueaza in conditii bine stabilite, cu aparate numite friabilatoare.
In mare, aparatul este alcatuit dintr-o toba rotativa transparenta, din material plastic, cu proprietati antistatice, cu suprafata interna polizata.
In interiorul tobei, este fixata o lama recurenta care ridica comprimatele pana la jumatate din inaltimea tobei si apoi se lasa sa cada. La fiecare rotatie, comprimatele sunt ridicate si coborate.
Toba are diametrul interior intre 283 si 291 mm si o adancime intre 36 si 40 mm.
O parte a tobei este mobila; ea este atasata la o axa orizontala a unui dispozitiv, care o roteste cu 25 rpm.
Comprimatele sunt rostogolite, la fiecare intoarcere a tobei, cu o curba de protectie, cu raza interna intre 75.5 si 85.5 mm, care se extinde in mijlocul tobei spre pereti.
Determinarea se efectueaza pe o cantitate mica de 6 g (20 comprimate); daca comprimatele sunt mai mari de 0.65 g fiecare, se utilizeaza 10 comprimate.
Comprimatele se plaseaza in sita numarul 1000, se elimina praful cu aer sub presiune.
Se scot de pe sita, se cantaresc exact si se introduc in toba, care se roteste de 100 ori (4 minute); se elimina orice praf anterior. Daca tabletele nu sunt rupte, crapate sau sparte se cantaresc.
In general testul se efectueaza o singura data.
Uzura se exprima in procente fata de masa comprimatelor. Limita maxima de friabilitate admisa este de 0.8-1%. Daca rezultatele sunt indoielnice, sau daca masa pierduta este peste 1%, se repeta testul de doua ori si se deremina media celor trei teste.
Dezagregarea
Dezagregarea este o constanta fizica prin care se poate aprecia daca substanta activa incorporata in comprimat se va elibera si va fi resorbita in organism.
Prin capacitatea de dezagregare a unui comprimat se intelege dizolvarea totala (comprimate solubile) sau dezagregarea in particule componente (granule, particule).
Timpul de dezagregare este diferit in functie de tipul comprimatului.
Farmacopeea Romana X prevede dezagregarea sau dizolvarea comprimatelor in apa in cel mult 15 minute.
Farmacopeea verifica capacitatea de dezagregare introducand 6 comprimate intr-un vas conic de 500 ml cu 300 ml apa mentinuta la temperatura de 35-390C. In timpul determinarii vasul se agita prin usoara rotire de 2 ori pe minut. Dupa trecerea timpului prevazut, continutul din vas se filtreaza printr-o sita cu latura ochiurilor de 0,8 mm, vasul se spala cu 300 ml apa la 37 20 C, care se aduce peste particulele ramase de la dezagregare. Se admit sa ramana pe sita particule care la usoara apasare cu o bagheta aplatizata la unul din capete trec prin sita. Daca rezultatul determinarii este negativ, proba se repeta de doua ori cu cate 6 comprimate, stabilindu-se durata dezagregarii pentru ambele determinari. Produsul este considerat corespunzator daca media acestor doua determinari nu depaseste timpul prevazut cu mai mult de 20%.
Prin testul de dezagregare se determina timpul necesar transformarii comprimatelor in particule fine sau in timpul necesar eliberarii continutului din invelisul capsulelor, atunci cand acestea sunt introduse intr-un mediu lichid in conditii de lucru specifice.
Tehnica de lucru
Se introduce in fiecare dintre cele sase tuburi ale dispozitivului cate un comprimat si se aplica cate un disc pe fiecare tub, daca nu se prevede altfel. Se introduce dispozitivul cu cele sase tuburi in vasul cilindric care contine mediul lichid prevazut si aparatul se pune in functiune. Dupa trecerea timpului prevazut in monografia respectiva dispozitivul se scoate din lichid.
Testul de dezagregare este considerat corespunzator daca nu mai exista nici un reziduu pe sita metalica a dispozitivului sau daca reziduul ramas este constituit dintr-o masa moale, fara nucleul palpabil sau din fragmente formate din invelisul comprimatelor.
Daca rezultatul este necorespunzator datorita aderentei comprimatelor pe discuri, testul se repeta pe inca sase comprimate fara a folosi discurile.
Dizolvarea
Testul de dizolvare permite o apreciere mai completa a comprimatelor, privind bio-disponibilitatea substantei active din comprimate. Viteza de dezagregare a comprimatelor nu poate constitui un indiciu si asupra dizolvarii substantei active din comprimat. Din acest motiv testul dizolvarii se asociaza determinarii vitezei de dezagregare, mai ales cand substanta medicamentoasa are solubilitate redusa in apa. Timpul de dizolvare al substantei active din comprimat trebuie sa se incadreze in limita timpului impus in determinarea dezagregarii. Prin testul de dizolvare prevazut in Farmacopee se stabileste cantitatea de substanta activa dizolvata intr-o forma farmaceutica solida cu administrare orala, intr-un anumit timp. Ȋn monografia individuala se prevede: tipul de aparat, mediul de dizolvare, numarul pe rotatii pe minut, metoda de dozare a substantei dizolvate, cantitatea minima de substanta activa (in procene fata de continutul declarat), care trebuie sa se dizolve in timpul test.
Aparatul nr.1 (agitator cu cosulet rotativ): Este format dintr-un agitator, un vas cilindric cu fundul emisferic si o baie termostatata.

Fig. 9. Aparat de dizolvare
Agitatorul este constituit dintr-o tija metalica verticala prevazuta la partea inferioara cu un cosulet de forma cilindrica, confectionat din doua parti din otel inoxidabil: partea superioara formata dintr-o placa prevazuta cu un orificiu de 2 mm, sudata de tija agitatorului si partea inferioara, de forma cilindrica, constituita dintr-o sita metalica din sarma de otel inoxidabil, cu dimensiunea ochiurilor de 420 micrometri, daca nu se prevede altfel. Tija se plaseaza in centrul vasului, astfel incat distanta dintre cosulet si fundul vasului sa fie de 25 2 mm. Partea superioara a tijei se cupleaza la partea mecanica a aparatului care este prevazuta cu un regulator de viteza. Miscarea de rotatie a agitatorului trebuie sa fie uniforma si fara oscilatii vizibile.
Vasul cilindric, cu fundul emisferic si capacitatea de 1000 ml, este confectionat din sticla sau din alt material transparent adecvat si este prevazut cu un capac care prezinta un orificiu central, permitand trecerea tijei agitatorului, precum si cu alte orificii colaterale care permit introducerea unui termometru si a dispozitivelor de prelevare a probei.
Baia termostatata, in care se introduce vasul cilindric, care trebuie sa asigure mentinerea temperaturii mediului de dizolvare in timpul determinarii la 37 0,50C.
Tehnica de lucru
Se introduce in vasul cilindric al aparatului volumul prevazut din mediul de dizolvare in prealabil incalzit la 37 0,50 C. Proba de analizat se introduce in cosuletul uscat, iar in cazul aparatului nr 2, proba de analizat se aseaza pe fundul vasului cilindric, ca atare, sau prin intermediul unui dispozitiv, daca proba prezinta tendinta de flotare. Se indeparteaza bulele de aer de la suprafata probei de analizat, se pune in functiune aparatul si se regleaza viteza de rotatie, conform prevederilor din monografia respectiva.
Se preleveaza probe la timpul sau la intervalele de timp prevazute, de la jumatatea distantei dintre suprafata mediului de dizolvare si baza cosuletului sau a paletei si la cel putin 10 mm de peretele vasului. Probele prelevate se filtreaza la 370 C printr-un fitru, astfel ales incat marimea porilor sa fie de aproximativ 1 micrometru, sa nu retina substanta activa continuta in solutie si sa nu contina substante extractile in mediul de dizolvare, care ar putea interfera dozarea.
Se determina concentratia substantei active dizolvate conform prevederilor din monografia respectiva.
Ȋn cazul prevederilor multiple se aduce in vasul cilindric un volum din mediul de dizolvare folosit, egal cu volumul prelevat.
CAPITOLUL II
II. 1. Acid acetilsalicilic-Caracterizare fizico-chimica.
Denumire chimica:acid 2-acetoxibenzoic
Denumire comerciala: Aspirina
Formula chimica: 2- CH3COOC6H4COOH
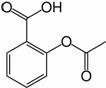
Acidul acetilsalicilic este un produs de sinteza obtinut prin:
Acetilarea acidului salicilic prin actiunea anhidridei acetice in diferite conditii de temperatura si in prezenta de deshidratanti precum acidul sulfuric, acetat de sodiu anhidru:
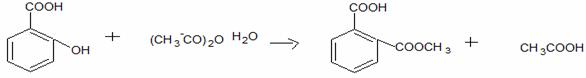
Acetilarea acidului salicilic cu clorura de acetil la cald in solutie piridinica:
Acidul acetilsalicilic se prezinta sub forma de cristale aciculare incolore sau pulbere cristalina alba, fara miros sau cu miros foarte slab de acid acetic, gust acru, punct de topire cuprins intre 134-1380C.
Foarte putin solubila in apa rece (1:300), solubila ȋn 1:5 parti alcool etilic, 1:20 parti eter,1: 25 parti cloroform; se dizolva in solutii de hidroxizi sau carbonati alcalini cu formare de saruri. Hidrolizeaza partial in aer umed, la fierbere, in apa sau la dizolvare in hidroxizi sau in carbonati alcalini. Solutiile apoase au reactie acida.
Punctul de fierbere: 1400C
Punctul de topire 1330C
Datorita functiei sale acide poate da saruri cu oxizii sau hidroxizii metalici si esteri cu alcooli si fenoli (R):
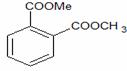
Hidroxizii alcalini dizolva aspirina prin neutralizarea gruparii carboxil cu formare de saruri. La cald hidroxizii saponifica imediat molecula aspirinei cu formare de salicilat de sodiu si acetat de sodiu, substante solubile:
Prin acidulare cu acizi minerali precipita acidul salicilic care poate fi separat si identificat. Ȋn solutie ramane acid acetic. Se dozeaza prin neutralizarea aspirinei in solutie alcoolica la rece cu hidroxid de sodiu sau potasiu titrat, in prezenta de fenolftaleina:
Controlul dozarii se face adaugand peste aspirina neutralizata o cantitate dubla de hidroxid titrat si se incalzette la reflux 15 minute, se produce saponificarea. Se titreaza excesul de hidroxid. Continutul in acid salicilic obtinut la ultima titrare poate sa difere mult cu cel mult 1% fata de continutul obtinut la prima titrare.
Metode de analiza a substantei active
Se prepara o solutie A astfel: 1,5g aspirina se agita cu 30 ml apa timp de 5 minute si se filtreaza. Solutia filtrata se completeaza la 30 ml prin spalarea filtratului cu apa.
Identificare
Prin hidroliza alcalina, acidul acetilsalicilic formeaza acid salicilic si acid acetic. Acidul salicilic poate fi identificat prin reactia cu clorura de fer (III) sau pe baza punctului de topire, iar acidul acetic prin reactia de formare a acetatului de etil.
-0,5g substanta se dizolva in 5 ml solutie 100g/l hidroxid de sodium, se fierbe 3 minute si , dupa racire, se aciduleaza cu solutie de 200 g/l acid sulfuric. Se percepe miros de acid acetic si rezulta un precipitat cristalin, alb, care dupa separare , spalare cu apa si uscare are p.t = 157-1610C.
Daca se dizolva o cantitate mica din precipitat in 2 ml apa la cald si se adauga dupa racire o picatura solutie de 30g/l clorura fer (III), rezulta o coloratie albastra violet (acidul salicilic).
Peste 2-3 ml filtrat de la separarea precipitatului se adauga 1 ml alcool etilic si 2 ml acid sulfuric, concentrate, se formeaza acetatul de etil cu miros caracteristic de mere verzi (acidul acetic).
Cu azotitul de sodiu si in prezenta acidului sulfuric, acidul acetilsalicilic formeaza un colorant cu structura indofenolica, formarea acestuia manifestandu-se printr-o gama de coloratii.
-0,005g substanta se trateaza cu 2 ml acid sulfuric concentrat si se agita. Dupa racire, se adauga cateva picaturi de solutie 100 g/l nitrit de sodium. Initial se obtine o coloratie galbena, care vireaza in galben portocaliu, rosu portocaliu si, in final in rosu inchis.
Conditii de puritate
Aspectul solutiei: solutia obtinuta prin dizolvarea a 1 g substanta in 10 ml alcool etilic trebuie sa fie limpede si incolora.
Arsen: se admit cel mult 0,0002%.
2,50 g substanta se trateaza conform modului descris la "Controlul limitei de arsen- Procedeul II".
Cloruri: se admit cel mult 0,004%. 10 ml solutie A se compara cu 2 ml solutie etalon completata cu apa la 10 ml (0,02 ion clorura) dupa adaugarea in ambele solutii reactivii pentru caracterizarea ionului clor.
Metale grele: se admit cel mult 0,001% . 10 ml solutie extractiva, obtinuta prin tratarea reziduului de la calcinare dupa metoda generala, se compara cu 5ml solutie etalon completata cu apa la 10 ml ( 0,005 mg ion plumb), dupa care s-au adaugat in ambele solutii reactivii pentru caracterizarea ionului plumb.
Sulfati: se admit cel mult 0,02% . 10 ml solutie A se compara cu 10 ml solutie etalon (0,1 mg ion sulfat) dupa adaugarea in ambele solutii a reactivilor de identificare a ionului sulfat.
Acid salicilic liber: se admite cel mult 0,1%. Acidul salicilic, avand grupa hidroxil fenolica acetilata, nu formeaza cu ionii Fe3+ combinatii complexe colorate; acidul salicilic, prin gruparea fenolica libera, formeaza o asemenea combinatie de culoare albastra- violet. 0,10 g substanta se dizolva in 2 ml alcool etilic si solutia se dilueaza cu apa la 50 ml. Separat, se prepara o solutie etalon din 2 ml alcool etilic, 1 ml solutie 0,001g/l acid salicilic si apa la 50 ml. La ambele solutii se adauga 1 ml solutie acida 2 g/l sulfat de amoniu fer (III) si 1 ml solutie 0,1 M acid clorhidric. Coloratia solutiei proba, la 30 secunde, nu trebuie sa fie mai intensa decat a etanolului.
II. 2. Acidul acetilsalicilic. Profil farmacologic
Farmacocinetica
Absorbtie per os relativ buna, majoritar la nivelul stomacului si duodenului superior.
Un prim pasaj intestinal si hepatic redus, cu o disponibilitate peste medie (cca. 68%)
Biodisponibilitatea per os este in functie de forma fizica si farmaceutica:
Forme polimorfe: forma I (cristalizata in etanol) realizeaza concentratii plasmatice duble fata de forma II (cristalizata din n-hexan);
Comprimatele efervescente dau concentratie plasmatica dubla fata de comprimatele obisnuite;
Formele enterosolubile realizeaza o absorbtie intarziata, concentratie plasmatica mai scazuta si concentratie plasmatica maxima de aproximativ 6 ore (fata de cea de o ora in cazul formelor obisnuite gastrosolubile);
Alimentele intarzie absorbtia gastrointestinala.
Biotransformarea prin hidroliza functiei ester, la acid acetic si acid salicilic, este catalizata de catre esterazele intestinale, hepatice si sanguine. In timpul primului pasaj intestinal si hepatic, acidul acetilsalicilic este hidrolizat mai putin. In sange este repede hidrolizat. T ½ al acidului acetilsalicilic la salicilat este foarte scurt (T 1/1 aproximativ 15 min).
Salicilatul este in continuare biotransformat in ficat, pe trei cai:
Prioritar, prin glicinoconjugare, la acid salicikuric;
Glucuronoconjugare la fenolglucuronida si acilglucuronida;
In mica masura, hidroxilarea la acid gentizic (acid 2,5 dihidroxibenzoic), metabolitul activ.
T ½ al salicilatului este relativ scurt (T1/2= 2-4 ore), la doze de acid acetilsalicilic mici si medii.
Cinetica de biotransformare a salicilatului este de ordinal 2, de tip Michaelis-Menten, realizandu-se cinetica de saturare a sistemelor enzimatice, chiar la dozele medii, analgezicele - antipireticele, de acid acetilsalicilic. Aceasta deoarece glicinoconjugatele si fenolglucuronoconjugazele au capacitate enzimatica limitata.
Capacitatea maxima de biotransformare corespunde unei concentratii plasmatice de salicilat (concentratia plasmatica cca. 50 mcg/ml) inferioare celei realizate de dozele analgezice-antipiretice de acid acetilsalicilic (concentratia plasmatica cca. 60 mcg/ml). Consecinta consta intr-o corelatie nu directa, ci inverse, intre dozele de acid acetilsalicilic si vitezele de biotransformare, respective clearance-urile salicilatului, cu un alt salt anormal al timpului de injumatatire al salicilatului (T1/2 = 15-30 ore) la dozele mari antiinflamatoare (tabel 5).
Tabel 5: Corelatia dozelor de acid acetilsalicilic cu timpul de injumatatire
|
Doze (g/) |
T ½ (h) |
|
0,3-0,6 |
2-4 |
|
3-4 |
Din punct de vedere clinic, gravitatea rezulta din faptul ca, la dozele terapeutice mari, o crestere relativ mica a dozelor de acid acetilsalicilic poate antrena o crestere neproportionala masiva a cresterilor plasmatice de salicilat, cu riscul atingerii zonelor de concentratii plasmatice toxice de salicilat, fara o reala supradozare absoluta. In consecinta, la tratament cu doze mari de acid acetilsalicilic, monitorizarea pe criteriul farmacocinetic poate fi de utilitate reala.
Transportul in sange se face sub forma libera si in forma legata de albumine, in proportii relativ egale.
Metabolitul salicilat este legat de albuminele plasmatice intr-un procent inalt (80-90%), la doze terapeutice de acid acetilsalicilic, inclusiv la dozele mari antiinflamatorii.
La concentratiile toxice de salicilat (peste 400 mcg/ml), procentul de legare al salicilatului de catre albuminele plasmatice se reduce mult (la cca 50%). In aceste conditii, forma libera de salicilat se ridica la circa jumatate din concentratia plasmatica de salicilat, depasind cu mult capacitatea de saturatie a sintetazelor si fiind disponibila pentru difuziune in tesuturi, cu efecte toxice grave.
Procentul de legare al salicilatului de albuminele plasmatice este redus semnificativ si in alte situatii: hipoalbuminemie, hiperbilirubinemie, precum si la asocierea cu alte medicamente care circula legate de albuminele plasmatice in procent ridicat (alte AINS).
Difuziunea si distributia are loc la nivelul tuturor tesuturilor, dar inegal. Difuziunea prin bariera hematoencefalica este semnificativa, dar lenta. Difuziunea prin placenta este rapida, cu reactii adverse la fat.
In intoxicatia acuta, in perioada secundara, de acidoza metabolica, este favorizata difuziunea in tesuturi, inclusiv in creier (datorita cresterii la pH acid a proportiei de forma neionizata), cu accentuarea fenomenelor toxice, spre deosebire de perioada primara de alcaloza respiratorie, cand e favorizata eliminarea.
Eliminarea se face pe cale renala (filtrare glomerulara si secretie tubulara active), sub forma de metabolit, predominant acid saliciluric (respective 70% acid saliciluric, 20% glucuronizi si 3% salicilat, pentru 1g acid acetilsalicilic si la pH=5)
Procentul de salicilat nebiotransformat si eliminat urinar creste cu doza de acid acetilsalicilic, datorita saturarii sistemelor enzimatice precum si cu pH-ul urinii) (tabelul).
Tabel 6: Corelatia intre doza de acid acetilsalicilic administrate si eliminarea sa pe cale renala
|
Doza de acid acetilsalicilic (g) |
pH urinar |
Salicilat in urina (%) |
|
1 |
5 |
3 |
|
1 |
7 |
25 |
|
5 |
50 |
|
|
8 |
80 |
Farmacodinamie
Actiuni farmacodinamice
Analgezic moderat (mecanism central talamic si periferic de inhibare a biosintezei de PGE1, ce contribuie la durerea din inflamatie prin sensibilizarea terminatiilor nervoase aferente, la actiunea algogena a histaminei si bradikininei);
Antipiretic moderat (mecanism hipotalamic)
Antiinflamator, antireumatic puternic (mecanism de inhibitie de PG inflamatoare, prin acetilarea ireversibila a ciclooxigenazei inductibile tip COX-2);
Antiagregant plachetar la doze mici subanalgezice (mecanism de inhibare a biosintezei plachetare de tromboxon 2 proagregant, prin acetilarea ireversibila a ciclooxigenazei constitutive tip COX-1);
Uricozuric, prin inhibarea reabsorbtiei tubulare active la acid uric (la doze de cca 2g/zi); hipocolesterolemiant, hipoglicemiant.
Farmacotoxicologie
a) Efecte secundare
Aparat digestiv
Efect ulcerigen, prin hipersecretie gastrica acida si scaderea cantitatii de mucus protector (mecanism de inhibare a biosintezei de prostaglandine I2 si respectiv a PGE2 citoprotectoare, prin acetilarea inversa a COX-1) cu gastralgie si reactivarea ulcerelor gastrice la ulcerosi
Microhemoragii gastrice (la 1-3 g/zi)
Sange
Hipocoagulare (antiagregant plachetar la doze mici si hipoprotrombinizant la doze mari), cu favorizarea microhemoragiilor si anemiei;
Hiperagregare plachetara, la dozele foarte mari antiinflamatoare (inhibarea biosintezei de PGI2, antiagregante prin acetilarea ireversibila a COX-1 din endoteliu vascular), cu favorizarea accidentelor trombotice.
Aparat respirator
Bronhoconstrictie, cu agravarea astmului bronsic (mecanism de inhibare a biosintezei de PGE2 bronhodilatatoare, prin acetilarea ireversibila a COX-1);
Aparat renal
Reducerea filtrarii glomerulare (mecanism de inhibare a biosintezei de PGE2 vasodilatatoare, prin inhibarea ireversibila a COX-1) si retentie hidrosalina;
SNC si analizatori
Euforie
Acufene, tulburari de echilibru si auditive
b) Reactii alergice mai frecvent pe teren alergic:
Eruptii cutanate (eritem polimorf, eritem nodos, eritem pigmentat fix), purpura, porfirie;
Edem angioneurotic si laringian
Soc anafilactic.
Sensibilizare incrucisata in grupa AINS.
b) Sindrom Reye (hepatita fulminanta si edem cerebral), frecvent fatal, este precipitat la copii sub 4 ani, tratati cu acid acetilsalicilic ca antipiretic in infectiile virale (gripa, varicela, hepatita).
Intoxicatia acuta
Initial, alcaloza respiratorie ( prin stimularea centrului respirator cu hiperventilatie);
Ulterior, acidoza metabolica ( prin paralizia centrului, cu acumulare de CO2);
Convulsii, delir;
Deces la copil (la 10g acid acetilsalicilic).
Farmacoepidemiologie
Contraindicatii
Ulcer gastro-duodenal, astm bronsic;
Diateza hemoragica;
Sarcina (intarzie travaliul; sangerare postpartum);
Alergie la salicilati;
Contraindicata inainte de o interventie chirurgicala ( cu minim 1 saptamana).
Farmacoterapie si farmacografie
Indicatii
Algii moderate ( nevralgii, mialgii, atralgii, cefalee)
Febra de etiologie diversa ( procese inflamatorii, infectii microbiene si virale acute, fiind excluse infectiile virale la copiii sub 4 ani)
Afectiuni reumatismale inflamatorii (poliartrita reumatoida)
Afectiuni tromboembolice (tromboze arteriale, profilaxia infarctului de miocard)
Timpul optim de administrare raportat la mese. Administrarea acidului acetilsalicilic in functie de forma farmaceutica administrata:
intre mese in administrare ocazionala;
Dupa mese, la administrari repetate sau daca apar efecte de irigatie gastrica (pirozis, epigastralgie).
Administrarea in formele enterosolubile se face intre mese, netriturate si nemestecate.
Posologie , per os:
Ca analgezic si antipiretic, la adult, 0,5g (0,350 - 0,650g) de 4-6 ori/zi, iar la copii 65mg/kg/zi, fractionat la 4-6 ore;
Ca antiinflamator, la adult, 3-5 g (2,5-5,5g)/zi, in 3-5 prize, iar la copii, 100 mg (90-130mg) kg/zi fractionat la 4-6 ore;
Ca antiagregant plachetar, la adult, 0,1-0,3g o data/zi sau 0,3-0,5g la 2-3 zile
in profilaxia infarctului de miocard, 0,3g
in reumatism poliarticular acut, la copil doza de atac de 0,1g/kg/zi, fractionat la 4-6 ore, timp de 20-30 zile, apoi 2/3 din doza de atac, timp de 10-20 zile si 1/2 - 1/3 timp de 30-40 zile
Contraindicat: ca antipiretic, la copii cu viroze febrile ( gripa, varicela), deoarece poate induce sindromul Reye.
Contraindicata automedicatia continua, mai mult de 10 zile la adult si mai mult de 5 zile la copil.
Monitorizarea reactiilor adverse:
Tinitus (zgomote in urechi)
Melena (scaun negru, moale, lucios)
Sangerari (echimoze, gingiovoragii)
Interactiuni
Bauturile alcoolice potenteaza tendinta la microhemoragii;
AINS potenteaza efectele secundare consecutive inhibitiei COX-1 (gastrite, respiratorii, renale); asocierea acidului acetilsalicilic cu AINS este contraindicata.
Nu are nici o influenta asupra capacitatii de a conduce vehicule sau de a folosi utilaje
Influentarea testelor de laborator: pH, colesterol, acid uric, etc
Sarciana si alaptarea
Acidul acetilsalicilic s-a dovedit teratogen in studii experimentale, la animalele de laborator.
Studii epidemiologice la femeia insarcinata nu au evidentiat efecte teratogene si fetotoxice in conditiile administrarii in primele 2 trimestre de sarcina, dar experienta sa este limitata pentru utilizarea cronica de doze mari.
Folosirea in ultimult trimesru de sarcina a fost asociata cu toxicitate cardio-pulmonara si renala la fat, inchiderea premature a canalului arterial, intarzierea si prelungirea travaliului si cresterea frecventei accidentelor hemoragice (inclusiv pentru dozele mici).
In primele doua trimestre de sarcina se poate administra acid acetilsalicilic, dar numai la indicatia medicului; se recomanda evitarea tratamentului cronic cu doze mai mari de 150 mg/zi. in ultimul trimestru de sarcina acidul acetil salicilic este contraindicat (cu exceptia utilizarii punctuale pentru anumite indicatii cardiologice si obstetricale foarte limitate).
Deoarece acidul acetilsalicilic se excreta in laptele matern, folositea in timpul alaptarii trebuie evitata, sau alaptarea se intrerupe, in functie de raportul risc terapeutic/beneficiul potential la sugar.
II. 3. Forme farmaceutice cu cedare conventionala cu acid acetilsalicilic tamponat
Acid acetilsalicilic 500 mg, gluconat de calciu 150 mg, amidon de porumb, polietilenglicol 600, aerosil, stearat de magneziu, talc;
Acid acetilsalicilic 500 mg, gluconat de calciu 150 mg, amidon de porumb, macrogol 6000, dioxid de siliciu coloidal anhidru, talc, amidon de cartofi, stearat de magneziu;
Acid acetilsalicilic 500 mg, carbonat de calciu 250 mg, amidon pregelinizat, celuloza microcristalina pH 103, hipromeloza 4000, dioxid de siliciu coloidal anhidru, talc, acid stearic;
Acid acetilsalicilic 500 mg, carbonat de calciu, amidon de porumb, talc, stearat de magneziu, oxid de siliciu coloidal anhidru;
Acid acetilsalicilic 500mg, carbonat de calciu 250mg, celuloza microcristalina, amidon de porumb, talc, dioxid de siliciu coloidal si apa purificata.
acetilsalicilic 500 mg si excipienti: carbonat de calciu, amidon de porumb, stearat de magneziu, dioxid de siliciu coloidal anhidru, talc.
II. 4. Excipientii utilizati la obtinerea comprimatelor cu acid acetilsalicilic tamponat
Un comprimat de acid acetilsalicilic tamponat este alcatuit din: acid acetilsalicilic 500mg, carbonat de calciu 250mg, celuloza microcristalina, amidon de porumb, talc, dioxid de siliciu coloidal si apa purificata.
Talc
Talcul este un excipient anorganic, un silicat de magneziu hidratat, purificat si pulverizat, o pulbere alba, foarte fina, onctuoasa la pipait, aderenta, fara miros, fara gust, lipsita de granulatii nisipoase.
Se utilizeaza singur sau in asociere cu alti excipienti pentru actiunea de catifelare a pielii, prevenirea si reducerea secretiilor si transpiratiilor. Conform F.R. X talcul nu trebuie sa contina mai mult de 500 microorganisme aerobe pe gram.
Poate fi sterilizat in prealabil (1700C), o ora, prezinta o granulometrie specifica gradului de finete imprimat de sita VIII si este folosit la pudrarea manusilor chirurgicale si in cazul pudrelor care se plica pe pielea sugarilor.Nu se aplica pe plagi deschise deoarece formeaza talcoame.
Se poate folosi si in formularea comprimatelor ca agent lubrifiant, are un bun efect antiaderent, usureza curgerea granulatului in palnie, iar F.R X limiteaza adaosul de talc la 3%.
Este lubrifiantul cel mai folosit deoarece are un bun efect antiaderent si, in mare parte, urmeaza curgerea granulatului in palnie. Pentru a obtine rezultate bune este necesara o proportie de 3% .
Calitatea de lubrifiant a talcului este o urmare a caracteristicilor cristalelor care, datorita onctuozitatii aluneca intre ele. Cu toate ca talcul este un bun lubrifiant, datorita faptului ca nu este complet independent din punct de vedere fiziologic putand provoca aparitia unor granuloame de talc, s-a propus inlocuirea lui cu alte substante. Se utilizeaza si amestecuri de talc, aerosil si stearat de magneziu.
Solubilitate. Practic insolubil in apa si in acizi, partial solubil in solutii de hidroxizi alcalini.
Celuloza microcristalina
Celuloza microcristalina reprezinta celuloza purificata si depolimerizata partial prin tratarea cu acizi minerali diluati a α- celulozei sub forma microcristalina, prezinta o structura fibroasa. Este prezenta in diverse forme comerciale, care difera prin forma particulei, diametrul, continutul in umiditate, in functie de care prezinta proprietati si utilizari diferite (figura 10).
Fig.10. Celuloza microcristalina
Una din varietatile comerciale este cunoscuta sub numele de Avicel. Aceasta se prezinta ca o pulbere alba, cristalina, fara miros si gust, inslolubila in apa, cu densitatea aparenta cuprinsa intre 0,2 - 0,5 g/cm3. Produsul este extrem de compresibil, permite obtinerea de comprimate de duritate mare, nu se foloseste singur, se asociaza cu lactoza, amidon si fosfat de calciu.
Se cunosc mai multe sorturi, Avicel 101 (pulbere cu particule de 50 μm) si Avicel 102 (cristale granulate cu diametrul particulelor de 90 μm).
Este preferata utilizarea sortului 102 fata de 101, deoarece are o capacitate de curgere si de lubrifiere mai buna imprimata de dimensiunea particulelor (care este mai mare), de forma acestora (granulara) si de o compresabilitate mai scazuta.
Sinonime
Avicel PH; Celex; celuloza gel, E 460, Fibrogel, Tabuloso, Vivapur, Emcocel, Ethisheres.
Denumire chimica: Celuloza [9004-34-6]
Formularea empirica si greutatea moleculara
(C6H10O5)n n
Formula structurala
Categorii functionale
Absorbant, agent de suspendare, diluant al comprimatelor si al capsulelor, dezagregant.
Aplicatii in formularea sau tehnica farmaceutica
Celuloza microcristalina este folosita in mare masura in industria farmaceutica (conform tabelului 7), in primul rand ca diluant al comprimatelor orale sau capsulelor, fiind folosita in procesul de granulare umeda si procesul comprimarii directe. Celuloza microcristalina este utilizata si in industria cosmetica si cea alimentara.
Tabel 7. Utilizarile farmaceutice ale celulozei microcristaline
|
Utilizare |
Concentratie (%) |
|
Adsorbant | |
|
Antiaderent | |
|
Liant/diluant al capsulelor | |
|
Dezintegrant al comprimatelor | |
|
Liant/diluant al comprimatelor |
Descriere
Celuloza microcristalina reprezinta celuloza purificata, partial depolarizata, care se prezinta ca o pudra cristalina alba, fara miros si gust, compusa din particule poroase. Comercial este utilizata in diferite dimensiuni ale pariculelor cu diferite grade de hidratare, ce au diferite proprietati si aplicatii.
Proprietati
Densitatea aparenta : 0.337 g/cm3;
Densitatea reala: 1.512-1.668 g/cm3
Capacitate de curgere : 1.41 g/s pentru Emcocel 90M
Punt de topire 260 -270C
Continutul de umiditate: in general mai putin de 5%. Cu toate acestea, diferite sortimente pot avea diferite cantitati de apa. Celuloza microcristalina este higroscopica.
Solubilitate: usor solubila in solutie hidroxid de sodiu 5%; practic insolubila in apa, acizi diluati si majoritatea solventilor organici.
Stabilitate si conditii de conditionare: celuloza microcristalina desi este higroscopica este stabila. Aceasta trebuie conditionata in recipiente bine inchise, tinute la rece, intr-un loc lipsit de umiditate.
Incompatibilitati: celuloza microcristalina este incompatibila cu agenti oxidanti puternici.
Metode de fabricare: celuloza microcristalina este fabricata prin hidroliza controlata cu solutii de acizi minerali ai -celulozei, obtinuta sub forma de pulpa din planta fibroasa. In urma hidrolizei este obtinuta hidroceluloza, care este purificata prin filtrare, iar suspensia apoasa este uscata.
Celuloza microcristalina este folosita cu intelepciune in formularea formelor farmaceutice, dar si in industria alimentara. Studiile efectuate au aratat ca este netoxica si neiritanta pentru organism. Celuloza microcristalina nu este absorbita sistemic, astfel are un mic potential toxic. Consumul in cantitati mari de celuloza poate avea un efect laxativ, desi este prea putin probabil sa existe aceasta problema cand celuloza este folosita ca excipient intr-o forma farmaceutica.
Amidonul de porumb
Amidonul de porumb (Amylum maydis) este un excipient de natura vegetala, fiind extras din cariopsele de Zea Mays.
Amidonul de porumb se prezinta ca o pulbere fina, alba, fara miros si fara gust, prezinta granule simple sau poliedrice cu diametrul cuprins intre 2 - 23 μm, si granule simple rotunde cu diametrul cuprins intre 25 - 32 μm, cu urmatoarele caracteristici microscopice redate in figura 11:

Fig. 11. Amidonul de porumb
Amidonul de porumb difera de celelalte sorturi de amidon prin diferenta intre amiloza si amilopectina pe care o detine de 27% amiloza. Conform F.R. X amidonul de porumb se pastreaza in recipiente inchise etans, pe eticheta specificandu-se denumirea acestuia.
Amidonul de porumb poate fi folosit ca excipient diluant in obtinerea comprimatelor in proportie de 30%; o proportie mai mare da comprimate moi si friabile sau ca aglutinant (mucilag din amidon in concentratie de 5-15%, optim 10%) si dezagregant in concentratie de 5-10%. Trebuie cunoscut foarte bine continutul sau in umiditate, pentru a nu obtine o masa prea moale.
Carbonatul de calciu este utilizat ca agent de tamponare. Carbonatul de calciu se prezinta sub forma unei pulberi albe, inodora si incolora, stabila la temperatura camerei, ce trebuie depozitata in containere bine inchise, la loc rece si uscat.Este incompatibila cu acizii si sarurile de amoniu. Carbonatul de calciu mai este regasit si sub urmatoarele denumiri: Destab, MagGran CC, Micromite.
Denumirea chimica: acid carbonic sau sare de calciu (1:1).
Formula empirica si greutatea moleculara: CaCO3 - 100.09
Formula structurala: CaCO3
Carbonatul de calciu poate fi folosit ca agent de tamponare, agent de acoperire, diluant al comprimatelor, agent terapeutic.
Carbonatul de calciu mai este folosit ca o baza pentru preparatele dentare, ca aditiv in diferite preparate alimentare sau antiacid si in anumite suplimente cu calciu.
Carbonatul de calciu este netoxic, dar administrat oral in cantitati mai mari poate produce constipatie sau flatulente. Consumul de cantitati mari (4-60 g/zi) poate provoca afectiuni renale sau hiperglicemie. Ca antiacid sunt folosite doze de 1,5 g, iar in tratamentul hiperfosfatemiei in cazul pacientilor cu insuficienta renala cronica se administreaza doze cuprinse intre 2,5-17 g. Carbonatul de calciu poate limita absorbtia altor substante daca sunt administrate concomitent.
Cand carbonatul de calciu este folosit in comprimate impreuna cu acidul acetilsalicilic sau substante inrudite, urmele de fier pot cauza decolorarea. Acest neajuns poate fi corectat prin adaugarea unui agent chelator corespunzator.
Dioxidul de siliciu coloidal
Dioxidul de siliciu coloidal (Aerosil) este folosit ca agent glisant pentru usurarea curgerii granulatului din palnia de umplere in matrita. Se mai foloseste ca aglutinant numai in asociere cu alti lianti, cel mai frecvent, cu amidonul, in mod special, cu amidonul hidrolizat. Este un bun agent aglutinant, mai ales in comprimarea directa si la comprimarea substantelor hidrofile. Se recomanda amestecul compus din aerosil si amidon hidrolizat (85:15) in care amidonul hidrolizat actioneaza ca aglutinant, iar aerosilul hidrofilizeaza substantele lipofile. O alta compozitie (Complex FTS) constituita din aerosil, talc, stearat de magneziu (1:8:1) se propune in cantitate de 3%, rezultand astfel o actiune tripla: de aglutinare, de lubrifiere si o curgere buna.
Apa purificata
Apa este cel mai utilizat solvent, un solvent ideal pentru substantele solubile in apa, pentru cele care nu hidrolizeaza in mediu apos sau nu sufera alte procese de degradare fizico-chimica. Suplimentul 2006 al Farmacopeei Romane editia a X a prevede mai multe monografii individuale pentru apa folosita la obtinerea formelor farmaceutice dozate nesterile (Apa purificata vrac, apa purificata conditionata in recipiente).
CAPITOLUL III
PARTEA EXPERIMENTALA
EVALUAREA IN VITRO A COMPRIMATELOR CU ASPIRINA TAMPONATA DE PE PIATA FARMACEUTICA DIN ROMANIA
In partea experimentala a lucrarii ne-am axat pe evaluarea in vitro a 3 categorii de comprimate cu acid acetilsalicilic tamponat, marci inregistrate , prezente pe piata farmaceutica romaneasca sub denumirile de : ACID ACETILSALICILIC MCC TAMPONAT 500 mg, ACID ACETILSALICILIC - RICHTER 500 mg si OZOPIRIN T.
Studiul experimental a vizat evaluarea in vitro a:
v caracterelor organoleptice (aspect, omogenitate, culoare, miros),
v caracteristicilor dimensionale (masa, grosime, inaltime),
v parametrilor farmacotehnici (uniformitatea masei, dezagregarea, friabilitatea),
v capacitatii de neutralizare a acidului clorhidric.
In acest scop s-au folosit comprimatele cu aspirina tamponata cu urmatoarea compozitie redate in tabelul 8.
Tabel 8. Compozitia comprimatelor cu aspirina tamponata, marci inregistrate in Romania
|
COMPOZITIE |
ACID ACETILSALICILIC MCC TAMPONAT 500 mg |
acid acetilsalicilic - richter 500 mg |
Ozopirin t |
|
Acid acetilsalicilic |
500 mg |
500 mg |
500 mg |
|
Carbonat de calciu |
250 mg |
Nu este mentionata |
|
|
Gluconat de calciu |
150 mg | ||
|
Excipienti |
Amidon pregelinizat |
Amidon de porumb |
Amidon de porumb |
|
Avicel PH 103 |
PEG 6000 |
Talc |
|
|
HPMC 4000 |
Talc |
Dioxid de siliciu coloidal anhidru |
|
|
Dioxid de siliciu coloidal anhidru |
Dioxid de siliciu coloidal anhidru |
Stearat de magneziu |
|
|
Talc |
Amidon de cartofi | ||
|
Acid stearic |
Stearat de magneziu |
III. I. 1. Controlul organoleptic
Aspectul examinarea omogenitatii suprafetei comprimatelor ofera indicatii asupra uniformitatii dispersarii. Suprafata comprimatului trebuie sa aiba o culoare uniforma, fara pete, asperitati sau crapaturi. Prezenta de puncte sau pete de intensitate diferita a coloratiei este un indiciu al unei dispersari neuniforme a comprimatelor sau a unor incompatibilitati dintre componente.
Schimbarea aspectului poate da indicatii asupra unor eventuale modificari petrecute in timpul stocarii. Se recomanda si examinarea comprimatelor dupa ce au fost fracturate, deoarece examinarea suprafetei rupturii, comparativ cu suprafata comprimatului, poate da indicatii asupra eventualelor degradari ale comprimatelor. Cercetarea marimii diametrului ca si a grosimii este pretioasa chiar in timpul productiei. La acelasi diametru si greutate, un comprimat mai subtire este comprimat mai puternic decat unul mai gros sau are o masa diferita. Gustul, mirosul si culoarea sunt imprimate de caracteristicile substantelor folosite. Aceste caracteristici sunt redate in tabelul nr. 9.
Tabel 9. Caracteristicile organoleptice ale comprimatelor de aspirina tamponata marci inregistrate in Romania
|
Caracteristici |
ACID ACETILSALICILIC MCC TAMPONAT mg (seria 15789) |
acid acetilsalicilic - richter 500 mg seria 03030309) |
Ozopirin t seria 157) |
|
Aspect |
Comprimate neacoperite, oblonge, cu un sant median pe ambele fete, cu o structura compacta si amorfa, cu margini intregi. |
Comprimate neacoperite, forma rotunda, plate, prevazute cu un sant median pe o fata, cu o structura compacta si amorfa, cu margini intregi. |
Comprimate neacoperite, neinscriptionate, plate, cu o forma rotunda, cu margini rotunde. Pe suprafata acestora sunt prezente cristale fine. |
|
Miros |
un miros slab de acid acetic |
un miros slab de acid acetic |
miros persistent de acid acetic |
|
Culoare |
alba |
alba |
alba, marmorata |
Din datele prezente in tabelul 9 se observa ca in cazul comprimatelor cu acid acetilsalicilic tamponat produs de Ozone prezinta pe suprafata lor cristale foarte fine, sunt marmorate si au un miros persistent de acid acetic.
IV. I. 2. Determinarea caracteristicilor dimensionale ale comprimatelor
Pentru evaluarea calitatii comprimatelor, sunt foarte importanti si parametrii ca: masa, inaltimea, diametrul comprimatelor. Determinarea diametrului si a grosimii comprimatelor sunt necesare chiar in timpul procesului tehnologic de fabricare. La acelasi diametru si greutate, un comprimat mai subtire este un comprimat mai puternic decat unul gros. Daca masina de comprimat este reglata bine, diferentele de grosime ale comprimatelor nu trec de 1 - 1,5%. Uniformitatea dimensiunilor este necesara pentru exactitatea dozarii, dar si pentru a nu se produce dificultati la ambalarea in tuburi sau in folii.
Grosimea comprimatului ofera informatii valoroase corelat cu celelalte teste mecanice. Poate indica neregularitati in compozitia materialului comprimabil, sau in procesul de comprimare. Pentru determinarea rezistentei mecanice s-a folosit aparatul Dr. Schleuniger Pharmatron ablet Tester 8 M (figura nr.9). Acest aparat permite evaluarea simultana a grosimii si a diametrului comprimatelor. In acest caz, rezultatele se exprima (mm) ca valoare medie a 10 determinari D.S (deviatia standard) si sunt redate in tabelele 10 - 12.
Tabel 10. Caractere dimensionale pentru ACID ACETILSALICILIC MCC TAMPONAT 500 mg
|
Nr. crt. |
Grosimea (mm) |
Diametrul (mm) |
|
Valoare medie D.S. |
Tabel 11. Caractere dimensionale pentru ACID ACETILSALICILIC
- RICHTER 500 mg
|
Nr. crt. |
Grosimea (mm) |
Diametrul (mm) |
|
Valoare medie D.S |
Tablel 12. Caractere dimensionale pentru OZOPIRIN T
|
Nr. crt. |
Grosimea (mm) |
Diametrul (mm) |
|
|
Valoare Medie D.S |
12,235 0,0479 |
||
Valorile obtinute pentru acesti parametrii dimensionali se incadreaza in diferentele de grosime de 1 - 1,5%.
IV. I. 3. Determinari farmacotehnice
Uniformitatea masei preparatelor unidoza
Se determina conform F.R. X, Suplimentul 2004; proba se efectueaza pe 20 de comprimate neacoperite prelevate la intamplare, care se cantaresc individual si carora li se calculeaza masa medie si abaterea relativa standard, de la masa comprimatelor (tabel 13).
Fata de masa medie calculata, masa individuala a cel mult 2 din cele 20 de comprimate poate sa prezinte o abatere procentuala mai mare de 5%. Pentru efectuarea operatiei de cantarire s-a folosit o balanta analitica cu patru zecimale.
% deviatia standard = ![]()
Evaluarea statistica a masei medii a comprimatelor verifica operarea regulata a masinii de comprimat si uniformitatea dozarii.
Tabel 13. Uniformitatea masei comprimatelor-limite admise de FR X suplimentul 2004
|
Forma farmaceutica |
Masa medie |
Deviatie % |
|
Comprimate neacoperite |
80 mg sau mai putin de 80 mg Peste 80 mg si mai putin de 250 mg 250 mg si peste 250 mg |
Rezultatele determinarilor se regasesc in tabelele 14 - 16.
ACID ACETILSALICILIC
MCC TAMPONAT 500 mg
Masa totala (g) = 23,3625 g
Masa medie g/comprimat = 1,1688 g
Tabel 14. Uniformitatea masei preparatelor unidoza - ACID ACETILSALICILIC
MCC TAMPONAT 500 mg
|
Nr. crt. |
Masa individuala comprimat (g |
Deviatia standard (%) |
|
Masa medie = 1,1688 g |
||
ACID ACETILSALICILIC- RICHTER 500 mg
Masa totala (g) = 17,002 g
Masa medie g/comprimat = 0,8503 g
Tabel 15. Uniformitatea masei preparatelor unidoza - ACID ACETILSALICILIC- RICHTER 500 mg
|
Nr. crt |
Masa individuala comprimat (g) |
Devia ia standard |
|
| ||
|
Masa medie = 0.8503 g |
||
OZOPIRIN T
Masa totala (g) = 15.4140 g
Masa medie g/comprimat = 0.7707g
Tabel 16. Uniformitatea masei comprimatelor OZOPIRIN T
|
Nr. crt |
Masa individuala comprimat (g) |
Deviatia standard (%) |
|
Masa medie = 0.7707 g |
||
Din analiza rezultatelor obtinute la aceasta determinare, se constata ca toate comprimatele cu aspirina tamponata se incadreaza in limitele admise de F.R. X, supliment 2004, pentru aceasta determinare.
IV. I. 4. Determinarea rezistentei mecanice (FR X, Suplimentul 2001)
Rezistenta mecanica asigura mentinerea integra a formei comprimatelor, la manipulare, in cursul conditionarii, ambalarii, transportului si depozitarii pana la administrare. Totusi ea nu poate depasi anumite limite, deoarece o rezistenta mecanica prea mare influenteaza negativ dezagregarea, implicit eliberarea substantei active si eficienta terapeutica.
Este de dorit obtinerea unui comprimat suficient de dur, cu o valoare de peste 7 kP. Testul de rezistenta mecanica poate indica si neuniformitati in procesul de comprimare, precum si inceputul capping-ului.
Aceasta metoda se utilizeaza pentru a determina, in conditii bine definite, rezistenta comprimatelor la rupere, masurata prin forta necesara pentru a provoca ruperea comprimatelor prin strivire. Mod de lucru: se aseaza comprimatul intre brate tinand cont, daca este cazul de forma, de santul median si de agravarea acestuia in acelasi fel fata de directia de aplicare a fortei. Se efectueaza masurarea pe 10 comprimate, avand grija sa se indeparteze orice fragment de comprimat inainte de fiecare determinare.
Pentru determinarea rezistentei mecanice s-a folosit aparatul Dr. Schleuniger Pharmatron ablet Tester 8 M (figura nr. 9).
Rezultatele se exprima indicand valoarea medie, valorile minime si maxime ale fortelor masurate, toate exprimate in Newtoni. Datele experimentale obtinute sunt redate in tabelul 17.

Fig. nr. 9. Aparat de masurare a rezistentei la rupere
Tabel 17. Rezistenta mecanica a comprimatelor cu aspirina tamponata, marci inregistrate in Romania
|
Nr. cpr. |
Rezistenta mecanica (N) |
||
|
ACID ACETILSALICILIC MCC TAMPONAT 500 mg |
ACID ACETILSALICILIC- RICHTER 500 mg |
OZOPIRIN T |
|
|
31 | |||
|
Valoare minima (N) | |||
|
Valoare maxima (N) | |||
|
Valoare medie (N) | |||
Pentru ACIDACETILSALICILIC- RICHTER 500 mg si OZOPIRIN T se remarca valori mici ale rezistentei mecanice, corelate cu neuniformitati in procesul de comprimare, cu afectarea procesului de dezagregare.
IV. I. 5. Testul de friabilitate (F.R. X, Suplimentul 2001)
Testul de friabilitate sau rezistenta la abraziune (radere) este destinat sa determine in conditiile definite, abraziunea comprimatelor, fenomen prin care suprafetele comprimatelor sunt deteriorate si/sau evidentiaza laminarea sau ruperea, cand sunt supuse la socul mecanic de frecare. Determinarea se efectueaza pe 20 comprimate; daca comprimatele sunt mai mari de 0.65 g fiecare, se utilizeaza 10 comprimate.
Mod de lucru
Comprimatele se plaseaza in sita numarul 1000 si se indeparteaza eventuala pulbere cu un jet de aer comprimat sau cu o perie moale. Se cantaresc comprimatele, se introduc an tambur si se indeparteaza eventuala pulbere la fel ca cea de mai sus. Daca nici un comprimat nu este crapat, fisurat sau spart, se cantaresc comprimatele cu precizie de 1 mg.
Ca regula generala, determinarea se efectueaza o singura data. Totusi, daca rezultatele sunt ambigue sau daca masa pierduta este mai mare de 1% se repeta determinarea de doua ori si se calculeaza media celor trei rezultate. Pierderea maxima de masa, considerata acceptabila pentru majoritatea produselor este de 1% din masa comprimatelor supuse determinarii.
In cazul comprimatelor cu diametrul mai mare sau egal cu 13 mm pot sa apara probleme de reproductibilitate legate de neregularitatea miscarii. In acest caz se ajusteaza pozitia tamburului astfel incat sa se evite alipirea comprimatelor in pozitia de repaus, ceea ce le impiedica sa cada liber. In general aceste rezultate se pot obtine inclinand tamburul astfel incat axul sa formeze un unghi de 10 grade. Pentru efectuarea testul am folosit un friabilator Electrolab (figura nr. 10).

Fig. nr. 10. Friabilatorul Electrolab
Exprimarea rezultatelor
Fiabilitatea se exprima in termeni de masa pierduta si se calculeaza in procente din masa initiala, cu ajutorul relatiei de mai jos:
Friabilitate% =![]()
unde:
mi = masa initiala a comprimatelor (mg),
mf= masa finala a comprimatelor dupa ce au fost supuse testului.
Valorile experimentale ale friabilitatii pentru cele 3 categorii de comprimate cu aspirina tamponata, marci inregistrate, prezente pe piata farmaceutica romaneasca figureaza in tabelul 18.
Tabel 18. Friabilitatea comprimatelor cu aspirina tamponata (marci inregistrate) in Romania
|
Marca inregistrata |
Friabilitate % |
Rezultat |
|
ACID ACETILSALICILIC MCC TAMPONAT 500 mg |
| |
|
ACID ACETILSALICILIC- RICHTER 500 mg |
F = | |
|
OZOPIRIN T |
F = F = F = |
Media =9,6766 |
Rezultatele obtinute la aceasta determinare denota ca in cazul comprimatelor cu aspirina tamponata produsa de Ozone si Gedeon Richter, testul nu este valid, corelat cu valori mici ale rezistentei mecanice ale comprimatelor.
IV. I. 6. Dezagregarea comprimatelor
Dezagregarea este o constanta fizica prin care se poate aprecia daca substanta activa incorporata in comprimat se va elibera si va fi resorbita in organism.
Prin capacitatea de dezagregare a unui comprimat se intelege dizolvarea totala (comprimate solubile) sau dezagregarea in particule componente (granule, particule).
Timpul de dezagregare este diferit in functie de tipul comprimatului.
Farmacopeea Romana X prevede dezagregarea sau dizolvarea comprimatelor in apa in cel mult 15 minute.
Farmacopeea verifica capacitatea de dezagregare introducand 6 comprimate intr-un vas conic de 500 ml cu 300 ml apa mentinuta la temperatura de 35-390C. In timpul determinarii vasul se agita prin usoara rotire de 2 ori pe minut. Dupa trecerea timpului prevazut, continutul din vas se filtreaza printr-o sita cu latura ochiurilor de 0,8 mm, vasul se spala cu 300 ml apa la 37 20 C, care se aduce peste particulele ramase de la dezagregare.
Se admit sa ramana pe sita particule care la usoara apasare cu o bagheta aplatizata la unul din capete trec prin sita. Daca rezultatul determinarii este negativ, proba se repeta de doua ori cu cate 6 comprimate, stabilindu-se durata dezagregarii pentru ambele determinari. Produsul este considerat corespunzator daca media acestor doua determinari nu depaseste timpul prevazut cu mai mult de 20%.
Determinare se efectueaza conform prevederilor F.R. X, Suplimentul 2004; s-a folosit un aparat Pharma test (figura nr. 11). Se introduce cate un comprimat in fiecare din cele sase tuburi si se apilica cate un disc pe fiecare tub. Se introduce dispozitivul cu cele sase tuburi in vasul cilindric care contine apa incalzita la 370C 20C si aparatul se pune in functiune. Dupa trecerea timpului prevazut (15 min.) dispozitivul se scoate din apa.
Proba analizata este corespunzatoare daca cele sase comprimate se dezagrega in timpul prevazut. Daca rezultatul este necorespunzator datorita aderentei comprimatelor pe discuri, testul se repeta inca pe sase comprimate, fara a folosi discurile.

Fig. nr. 11. Tester pentru dezagregarea comprimatelor
Rezultate experimentale ale testului de dezagregare se regasesc in tabelul 19 si sunt exprimate ca valori medii a 3 determinari pentru fiecare dintre exemplele luate in studiu.
Tabel 19. Valorile timpului de dezagregare pentru comprimatele cu aspirina (marci inregistrate)
|
Marca inregistrata |
valori individuale Timp de dezagregare (secunde) |
Timp de dezagregare (Valoare medie), secunde |
|
ACID ACETILSALICILIC MCC TAMPONAT 500 mg |
t1=42 | |
|
t2 =32 |
||
|
t3 = 39 |
||
|
ACID ACETILSALICILIC- RICHTER 500 mg |
t1 =17 | |
|
t2=19 |
||
|
t3=18 |
||
|
OZOPIRIN T |
t1 = 16 | |
|
t2 =19 |
||
|
t3 =17 |
Valorile obtinute se incadreaza in limita admisa de 15 minute pentru acest test, dar observam ca in cazul Ozopirin T si ACID ACETILSALICILIC- RICHTER 500 mg valori mici ale timpului de dezagregare, corelate cu rezistenta mecanica redusa.
De asemenea dezagregarea rapida s-ar putea datora concentratiei mari de dezagregant din formula si unei forte de comprimare insuficienta la obtinerea comprimatului.
IV. I. 7. Capacitatea de neutralizare a acidului clorhidric (U.S.P. 30, 2007 )
Conform monografiei individuale din U.S.P, nu mai putin de 1.9 mEq de acid sunt consumati pentru fiecare 325 mg de aspirina dintr-un comprimat.
Reactivi si aparatura utilizatii
-HCl 1.0 N,
-NaOH 0.5 N
-pH-metru,
-agitator mecanic (300 30 rpm).
Tehnica de lucru
Determinarea se face la 37oC 3 oC incepand din momentul agitarii mecanice.
Se cantaresc 20 de comprimate si se determina masa medie, se tritureaza comprimatele pana la obtinerea unei pulberi fine omogene. Pulberea obtinuta din comprimatele corespunzatoare la 325 mg acid acetilsalicilic se transfera cantitativ intr-un balon Erlenmayer de 250 ml si se adauga 5 ml alcool (neutralizat in prealabil la pH=3.5) pentru umectare.
Se adauga apoi 70 ml apa si se agita mecanic la 300 rpm timp de 1 minut. Sub agitare continua se adauga 30 ml HCl 1,0 N si se continua agitarea timp de 15 minute. Se titreaza (sub agitare mecanica) excesul de HCl 1,0 N cu NaOH 0,5 N pana la obtinerea solutiei cu un pH= 3,5 in maxim 5 minute (pH-ul trebuie sa ramana constant 10-15 secunde). 1 ml HCl 1.0 N corespunde la 1 mEq acid consumat.
In aceleasi conditii de lucru se efectueaza si o proba martor.
Se calculeaza numarul de mEq de HCl consumat cu ajutorul formulei:
Totalul mEq = ( 30 x NHCI) - (VNaOH x NNaOH)
in care NHCI si NNaOH reprezinta concentratiile normale ale acidului clorhidric, respectiv ale hidroxidului de sodiu, iar VNaOH reprezinta volumul de hidroxid de sodiu folosit in timpul titrarii. Se calculeaza rezultatul in mEq de acid consumat pe gram de substanta folosita.
Formula din USP a fost modificata, raportandu-ne la masa medie a comprimatelor luate in lucru, masa cantarita corespunzatoare unui continut de 325 mg aspirina.
Astfel vom face urmatoarele notatii:
VNaOH martor = volumul NaOH 0,5 N folosit la titrarea probei martor, (ml),
VNaOH proba = volumul NaOH 0,5 N folosit la titrarea probei, (ml),
masa medie (20 cp), masa probei luata in lucru (g).
Valorile capacitatii de neutralizare a comprimatelor cu aspirina, marci inregistrate de pe piata romaneasca sunt redate in tabelul 20.
Tabel 20. Capacitatea de neutralizare a comprimatelor cu aspirina tamponata (marci inregistrate pe piata romaneasca)
|
Marca inregistrata |
Date experimentale |
mEq acid consumat mg acid acetilsalicilic |
|
ACID ACETILSALICILIC MCC TAMPONAT 500 mg |
VNaOH martor = 58,9 ml, VNaOH proba = 54,2 ml, masa medie (20 cp) =1.1688 g masa probei luata in lucru=0,781g. | |
|
ACID ACETILSALICILIC- RICHTER 500 mg |
VNaOH martor = 58,9 ml, VNaOH proba = 60,05ml, masa medie (20 cp) =0,8503 masa probei luata in lucru =0,553g | |
|
OZOPIRIN T |
VNaOH martor = 58,9 ml, VNaOH proba = 54,95 ml, masa medie (20 cp) =0,770 g masa probei luata in lucru =0,500g |
Aceasta forma de acid acetil salicilic(ACID ACETILSALICILIC- RICHTER 500 mg) nu se incadreaza in limitele de neutralizare ale acidului clorhidric.
Diferentele se pot datora si utilizarii unui agent de tamponare diferit, gluconat de calciu pentru formula necorespunzatoare la acest parametru si carbonat de calciu pentru celelalte 2 formule.
CAPITOLUL IV
CONCLUZII
Evaluarea in vitro a comprimatelor cu aspirina tamponata marci inregistrate de pe piata farmaceutica din Romania, a evidentiat:
OZOPIRIN T
v examen organoleptic necorespunzator, comprimate marmorate cu miros persistent de acid acetic,
v prezenta unor cristele fine pe suprafata comprimatelor),
v rezistenta mecanica scazuta corelata cu o dezagregare rapida,
v friabilitate peste limita admisa de farmacopee (9,667%),
v capaciatea de neutralizare a acidului clorhidrica in limitele admise de U. S. P. 30,
ACID ACETILSALICILIC- RICHTER 500 mg
v examen organoleptic corespunzator,
v rezistenta mecanica scazuta corelata cu o dezagregare rapida,
v friabilitate peste limita admisa de farmacopee (2,04%),
v capaciatea de neutralizare a acidului clorhidrica nu se incadreaza in limitele admise de U.S.P. 30 ,
ACID ACETILSALICILIC MCC TAMPONAT 500 mg
v examen organolepticcorespunzator,
v rezistenta mecanica crescuta corelata cu un timp de dezagregare mai mare comparativ cu celelalte marci inregistrate,
v friabilitate in limitele admise (0,082%),
v capaciatea de neutralizare a acidului clorhidrica se incadreaza in limitele admise de U.S.P. 30 .
BIBLIOGRAFIE
1. Cristea A. N., Tratat de farmacologie, editia I, Editura Medicala, Bucuresti, 2006.
2. Ghiran D., Muresan A., Pitea M., Medicamente Antiinflamatoare Nesteroidiene, Editura Dacia, Cluj-Napoca; 2004.
3. Hatieganu E., Dumitrescu D., Stecoza C., Morusciag L., Chimie Terapeutica Volumul I, Editura Medicala, Bucuresti, 2006.
4. Leucuta S.E., Biofarmacie si Farmacocinetica, Editura Dacia, Cluj-Napoca, 2002.
5. Leucuta S.E., Tehnologie Farmaceutica Industriala, Editia a II-a, Editura Dacia, Cluj-Napoca, 2007.
6. Lupuleasa D., Popovici I., Tehnologie Farmaceutica vol. 2, Editura Polirom, Bucuresti, 2008.
7. Lupuleasa D., Popovici I., Tehnologie Farmaceutica vol. 3, Editura Polirom, Bucuresti, 2009.
8. Otsuka M, Ohtani H, Otsuka K, Kaneniwa N. Effect of humidity on solid-state isomerization of various kinds of lactose during grinding. J Pharm Pharmacol 1993; 45: 2-5.
9. Paronen P. Behaviour of some direct compression adjuvants during the tabletting process. STP Pharma 1986; 2(19): 682-688.
10. Rajabi-Siahboomi A.R., Tiwari S.B., Modulation of drug release from hydrophilic matrices, Springerlink, 2008.
11. Zuurman K, Riepma KA, Bolhuis GK, et al. The relationship between bulk density and compactibility of lactose granulations. Int J Pharm 1994; 102: 1-9.
12. *** Farmacopeea Romana editia a X-a, Editura Medicala, Bucuresti, 2008.
13. *** Farmacopeea Romana editia a X-a, Suplimentul 2001, Editura Medicala, Bucuresti, 2001.
14. *** Farmacopeea Romana editia a X-a, Suplimentul 2004, Editura Medicala, Bucuresti, 2004.
15. *** Farmacopeea Romana editia a X-a, Suplimentul 2006, Editura Medicala, Bucuresti, 2006.
16. *** The United States Pharmacopeia XXX edition, The National Formulary XXVII, Unites States Pharmacopoeial Convention, Inc., Rockville, M. D., 2007.
17. ***European Pharmacopoeia, 6.0., Council of Europe, Strassbourg, Cedex, France, 2008.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 7919
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved
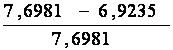 x100
x100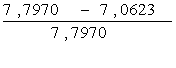 x100
x100