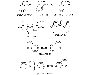| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Analgesic-Antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout
Overview
|
This chapter describes drugs used to treat the symptoms and signs of inflammation and drugs used for gout. Most currently available nonsteroidal antiinflammatory drugs (NSAIDs) inhibit both cyclooxygenase-1 (COX-1; constitutive) and cyclooxygenase-2 (COX-2; induced in settings of inflammation) activities, and thereby synthesis of prostaglandins and thromboxane. The inhibition of COX-2 is thought to mediate, at least in part, the antipyretic, analgesic, and antiinflammatory action of NSAIDs, but the simultaneous inhibition of COX-1 results in unwanted side effects, particularly those leading to gastric ulcers, that result from decreased prostaglandin formation. The potential therapeutic advantage of selective COX-2 inhibitors is discussed. NSAIDs include aspirin, which irreversibly acetylates cyclooxygenase, and several other classes of organic acids, including propionic acid derivatives (ibuprofen, naproxen, etc.), acetic acid derivatives (e.g., indomethacin and others), and enolic acids (e.g., piroxicam), all of which compete with arachidonic acid at the active site of cyclooxygenase. Acetaminophen is a very weak antiinflammatory drug but is effective as an antipyretic and analgesic agent and lacks certain side effects of NSAIDs, such as gastric ulceration and blockade of platelet aggregation. Gold salts, primarily used as second-line drugs to treat patients with unremitting and chronic forms of rheumatoid arthritis, also are discussed. Also covered in this chapter are agents used in prophylaxis of acute gout (e.g., colchicine) or treatment of chronic gout ( allopurinol, uricosuric agents), a disorder caused by deposition of crystals of sodium urate in joints and other sites. Some agents used to treat inflammation are discussed elsewhere in this textbook, including glucocorticoids (see Chapter 60: Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones ) and immunosuppressants (see Chapter 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants). |
NSAIDs: Nonsteroidal Antiinflammatory Drugs
|
The antiinflammatory, analgesic, and antipyretic drugs are a heterogeneous group of compounds, often chemically unrelated (although most of them are organic acids), which nevertheless share certain therapeutic actions and side effects. The prototype is aspirin; hence these compounds are often referred to as aspirin-like drugs; they also are frequently called nonsteroidal antiinflammatory drugs, or NSAIDs, an abbreviation that is used throughout this chapter to refer to these agents. There has been substantial progress in elucidating the mechanism of action of NSAIDs. Inhibition of cyclooxygenase (COX), the enzyme responsible for the biosynthesis of the prostaglandins and certain related autacoids, generally is thought to be a major facet of the mechanism of NSAIDs. Some of the shared properties of NSAIDs are considered first; then the more important drugs are discussed in some detail. History The medicinal effect of the bark of willow and certain other plants
has been known to several cultures for centuries. In The active ingredient in the willow bark was a bitter glycoside called salicin, first isolated in a pure form in 1829 by Leroux, who also demonstrated its antipyretic effect. On hydrolysis, salicin yields glucose and salicylic alcohol. The latter can be converted into salicylic acid, either in vivo or by chemical manipulation. Sodium salicylate was first used for the treatment of rheumatic fever and as an antipyretic in 1875, and the discovery of its uricosuric effects and of its usefulness in the treatment of gout soon followed. The enormous success of this drug prompted Hoffman, a chemist employed by Bayer, to prepare acetylsalicylic acid based on the earlier, but forgotten, work of Gerhardt in 1853. After demonstration of its antiinflammatory effects, this compound was introduced into medicine in 1899 by Dreser under the name of aspirin. The name is said to have been derived from Spiraea, the plant species from which salicylic acid was once prepared. The synthetic salicylates soon displaced the more expensive compounds obtained from natural sources. By the early years of this century, the chief therapeutic benefits of aspirin were known. Toward the end of the nineteenth century, other drugs were discovered that shared some or all of these actions; among these, only derivatives of para-aminophenol (e.g., acetaminophen) are used today. Beginning with the introduction of indomethacin for the treatment of rheumatoid arthritis in 1963, a host of other agents with similar actions have been introduced over the years, culminating in the recent development of selective inhibitors of COX-2 (see below). Mechanism of Action of NSAIDs Although NSAIDs had been known to inhibit a wide variety of reactions in vitro, no convincing relationship could be established with their known antiinflammatory, antipyretic, and analgesic effects until 1971, when Vane and associates and Smith and Willis demonstrated that low concentrations of aspirin and indomethacin inhibited the enzymatic production of prostaglandins (see Chapter 26: Lipid-Derived Autacoids: Eicosanoids and Platelet-Activating Factor). There was, at that time, some evidence that prostaglandins participated in the pathogenesis of inflammation and fever, and this reinforced the hypothesis that inhibition of the biosynthesis of these autacoids could explain a number of the clinical actions of the drugs (see Higgs et al., in Symposium, 1983a). Numerous subsequent observations have reinforced this point of view, including the observations that prostaglandins are released whenever cells are damaged, they appear in inflammatory exudates, and NSAIDs inhibit the biosynthesis of prostaglandins in all cells tested. However, NSAIDs generally do not inhibit the formation of eicosanoids such as the leukotrienes, which also contribute to inflammation, nor do they affect the synthesis of numerous other inflammatory mediators. There are differences of opinion as to whether or not NSAIDs may have other actions that contribute to their therapeutic effects (see below; Abramson and Weissman, 1989; Vane, 1994). Inflammation The inflammatory process involves a series of events that can be elicited by numerous stimuli (e.g., infectious agents, ischemia, antigenantibody interactions, and thermal or other physical injury). Each type of stimulus provokes a characteristic pattern of response that represents a relatively minor variation on a theme. At a macroscopic level, the response usually is accompanied by the familiar clinical signs of erythema, edema, tenderness (hyperalgesia), and pain. Inflammatory responses occur in three distinct phases, each apparently mediated by different mechanisms: (1) an acute transient phase, characterized by local vasodilation and increased capillary permeability; (2) a delayed, subacute phase, most prominently characterized by infiltration of leukocytes and phagocytic cells; and (3) a chronic proliferative phase, in which tissue degeneration and fibrosis occur. Many different mechanisms are involved in the inflammatory process (Gallin et al., 1992; Kelly et al., 1993). The ability to mount an inflammatory response is essential for survival in the face of environmental pathogens and injury, although in some situations and diseases the inflammatory response may be exaggerated and sustained for no apparent beneficial reason. Several classes of leukocytes play an essential role in inflammation. Although earlier ideas emphasized the promotion of migration of cells out of the microvasculature, recent studies have examined the role of the endothelial cell and of cell adhesion molecules, including E-, P-, and L-selectins, intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and leukocyte integrins in the adhesion of leukocytes, platelets, and endothelium at sites of inflammation (see Kishimoto and Anderson, Lasky and Rosen in Gallin et al., 1992; Bevilacqua and Nelson, 1993; and Cronstein and Weissmann, 1993). Activated endothelial cells play a key role in 'targeting' circulating cells to inflammatory sites. Expression of the various adhesion molecules varies among different cell types involved in the inflammatory response. For example, expression of E-selectin is restricted primarily to endothelial cells and is enhanced at sites of inflammation. P-selectin is expressed predominantly on platelets and on endothelial cells and is enhanced by cytokines. L-selectin, in contrast, is a receptor for P-selectin, and L-selectin is expressed on leukocytes and is shed when these cells are activated. Cell adhesion appears to occur by recognition of cell-surface glycoprotein and carbohydrates on circulating cells by the adhesion molecules whose expression has been enhanced on resident cells. Thus, endothelial activation results in adhesion of leukocytes by their interaction with newly expressed L-selectin and P-selectin, whereas endothelial-expressed E-selectin interacts with sialylated Lewis X and other glycoproteins on the leukocyte surface; endothelial ICAM-1 interacts with leukocyte integrins. NSAIDs may inhibit expression or activity of certain of these cell adhesion molecules. Such effects have been described for some NSAIDs and not others, suggesting that interference with action of cell adhesion molecules is not a common mechanism of action of all NSAIDs (see Diaz-Gonzalez and Sanchez-Madrid, 1998). Nonetheless, effects on adhesion molecules may contribute in part to the antiinflammatory actions of some NSAIDs. Novel classes of antiinflammatory drugs directed against cell adhesion molecules are under active development (see, for example, Kavanaugh et al., 1994; Rao et al., 1994; Endemann et al., 1997). The recruitment of inflammatory cells to sites of injury involves the concerted interactions of several types of soluble mediators in addition to the cell adhesion molecules outlined above. These include the complement factor C5a, platelet activating factor, and leukotriene B4. All can act as chemotactic agonists. Several different cytokines also appear to play an essential role in orchestrating the inflammatory process, especially interleukin 1 (IL-1) and tumor necrosis factor (TNF; see Dinarello, 1992). Both IL-1 and TNF are derived from mononuclear cells and macrophages (as well as other cell types) and induce expression of numerous genes to promote the synthesis of a variety of proteins that contribute to inflammatory events. IL-1 and TNF are considered principal mediators of biological responses to bacterial lipopolysaccharides (endotoxins) and many other infectious stimuli. IL-1 and TNF appear to work in concert with each other and with growth factors (such as granulocyte/macrophage colony stimulating factor, GM-CSF) and other cytokines, such as IL-8 and related chemotactic cytokines (chemokines), which can promote neutrophil infiltration and activation. IL-1 comprises two distinct polypeptides (IL-1 TNF, originally termed 'cachectin' because of its ability to
produce a wasting syndrome, is composed of two closely related proteins:
mature TNF (TNF IL-1 and TNF produce many of the same proinflammatory responses, which
include induction of fever, sleep, and anorexia; mobilization and activation
of polymorphonuclear leukocytes; induction of cyclooxygenase and lipoxygenase
enzymes; increase in adhesion molecule expression; activation of B cells, T
cells, and natural killer cells; and stimulation of production of other
cytokines. Other actions of these agents likely contribute to the fibrosis
and tissue degeneration of the chronic proliferative phase of inflammation:
stimulation of fibroblast proliferation, induction of collagenase, and
activation of osteoblasts and osteoclasts. Both IL-1 and TNF increase
expression of many types of genes, probably in part via the activation
of transcription factors, such as NF A naturally occurring IL-1 receptor antagonist (IL-1ra), a 17-kDa protein, competes with IL-1 for receptor binding, blocks IL-1 activity in vitro and in vivo, and can prevent death in animals induced by administration of bacteria or bacterial lipopolysaccharide (Arend, 1993). IL-1ra often appears to achieve high levels in patients with various infections or inflammatory conditions. Thus, the balance between IL-1 and IL-1ra may contribute to the extent of an inflammatory response. Studies are in progress to assess whether IL-1ra or other IL-1 antagonists are beneficial as novel types of antiinflammatory agents. Other cytokines and growth factors (e.g., IL-2, IL-6, IL-8, and
GM-CSF) contribute to manifestations of the inflammatory response. The
concentrations of many of these factors are increased in the synovia of
patients with arthritides, such as rheumatoid arthritis. The concentration of
peptides, such as substance P, which promotes firing of pain fibers, also is
increased at such sites. To counter the effects of proinflammatory mediators,
other cytokines and growth factors have been implicated as having
antiinflammatory activity. These include transforming growth factor- Histamine was one of the earliest mediators of the inflammatory process identified. Although several H1 histamine-receptor antagonists are available, they are useful only for the treatment of vascular events in the early transient phase of inflammation (see Chapter 25: Histamine, Bradykinin, and Their Antagonists). Bradykinin and 5-hydroxytryptamine (serotonin, 5-HT) also may play a role in mediating inflammation, but their antagonists ameliorate only certain types of inflammatory responses (see Chapter 25: Histamine, Bradykinin, and Their Antagonists). Specific inhibitors of leukotriene synthesis, (zileuton, a 5-lipoxygenase inhibitor) and cysteinyl leukotriene-receptor antagonists (montelukast and zafirlukast) exert antiinflammatory actions and have been approved for the treatment of asthma (see Chapter 28: Drugs Used in the Treatment of Asthma). Another lipid autacoid, platelet-activating factor (PAF), has been implicated as an important mediator of inflammation, and inhibitors of its synthesis and action are under study (see Chapter 26: Lipid-Derived Autacoids: Eicosanoids and Platelet-Activating Factor). The effects produced by intradermal, intravenous, or intraarterial injections of small amounts of prostaglandins are strongly reminiscent of inflammation. Prostaglandin E2 (PGE2) and prostacyclin (PGI2) cause erythema and an increase in local blood flow. With PGE2, such effects may persist for up to 10 hours, and they include the capacity to counteract the vasoconstrictor effects of substances such as norepinephrine and angiotensin. These properties are not generally shared by other inflammatory mediators. In contrast to their long-lasting effects on cutaneous vessels and superficial veins, prostaglandin-induced vasodilation in other vascular beds vanishes within a few minutes. Although PGE1 and PGE2 (but not PGF2 Rheumatoid Arthritis Although the pathogenesis of rheumatoid arthritis is largely unknown, it appears to be an autoimmune disease driven primarily by activated T cells, giving rise to T cellderived cytokines, such as IL-1 and TNF. Although activation of B cells and the humoral response also are evident, most of the antibodies generated are IgG of unknown specificity, apparently elicited by polyclonal activation of B cells rather than from a response to a specific antigen. Many cytokines, including IL-1 and TNF, have been found in the rheumatoid synovium. Of the available antiinflammatory drugs, only the adrenocorticosteroids are known to interfere with the synthesis and/or actions of cytokines such as IL-1 or TNF (see Chapter 60: Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones). Although some of the actions of these cytokines are accompanied by the release of prostaglandins and/or thromboxane A2, only their pyrogenic effects are blocked by inhibitors of cyclooxygenase (see below). In addition, many of the actions of the prostaglandins are inhibitory to the immune response, including suppression of the function of helper T cells and B cells and inhibition of the production of IL-1. Thus, it is difficult to ascribe the antirheumatoid effects of aspirin-like drugs solely to inhibition of prostaglandin synthesis. It has been proposed that salicylate and certain other NSAIDs can directly inhibit the activation and function of neutrophils, perhaps by inhibition of membrane-associated processes, independent of their ability to inhibit prostaglandin synthesis (see Abramson and Weissmann, 1989). Furthermore, as mentioned previously, some NSAIDs can inhibit leukocyte adhesion by a mechanism that seems to be independent of their ability to inhibit prostaglandin biosynthesis. Pain NSAIDs usually are classified as mild analgesics, but this classification is not altogether correct. A consideration of the type of pain as well as its intensity is important in the assessment of analgesic efficacy. In some forms of postoperative pain, for example, the NSAIDs can be superior to the opioid analgesics. Moreover, they are particularly effective in settings in which inflammation has caused sensitization of pain receptors to normally painless mechanical or chemical stimuli. Pain that accompanies inflammation and tissue injury probably results from local stimulation of pain fibers and enhanced pain sensitivity (hyperalgesia), in part a consequence of increased excitability of central neurons in the spinal cord ('central sensitization'; see Konttinen et al., 1994). Bradykinin, released from plasma kininogen, and cytokines, such as TNF Large doses of PGE2 or PGF2 Fever Regulation of body temperature requires a delicate balance between the production and loss of heat; the hypothalamus regulates the set point at which body temperature is maintained (see Saper and Breder, 1994). In fever, this set point is elevated, and NSAIDs promote its return to normal. These drugs do not influence body temperature when it is elevated by such factors as exercise or increases in the ambient temperature. Fever may be a result of infection or one of the sequelae of tissue
damage, inflammation, graft rejection, malignancy, or other disease states. A
common feature of these conditions is the enhanced formation of cytokines
such as IL-1 Inhibition of Prostaglandin Biosynthesis by NSAIDs Since the principal therapeutic effects of NSAIDs derive from their ability to inhibit prostaglandin production, the enzymatic activities involved in prostaglandin synthesis are described here briefly (see also Chapter 26: Lipid-Derived Autacoids: Eicosanoids and Platelet-Activating Factor). The mechanisms by which varying NSAIDs interfere with prostaglandin synthesis then are outlined. The first enzyme in the prostaglandin synthetic pathway is prostaglandin endoperoxide synthase, or fatty acid cyclooxygenase. This enzyme converts arachidonic acid to the unstable intermediates PGG2 and PGH2. It is now appreciated that there are two forms of cyclooxygenase, termed cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) (see Vane et al., 1998). COX-1 is a constitutive isoform found in most normal cells and tissues, while COX-2 is induced in settings of inflammation by cytokines and inflammatory mediators (Seibert et al., 1997). However, COX-2 also is constitutively expressed in certain areas of kidney and brain (Breder et al., 1995; Harris et al., 1994). Importantly, COX-1, but not COX-2, is constitutively expressed in the stomach. This accounts for the markedly reduced occurrence of gastric toxicity with the use of selective inhibitors of COX-2 (see below). The fate of PGG2/PGH2 cyclooxygenase products differs from tissue to tissue, depending on the particular PGG2/PGH2-metabolizing enzymatic activities present (see Figure 261). Arachidonic acid also can be converted, via the 5-lipoxygenase pathway, to a variety of leukotrienes. Aspirin and NSAIDs inhibit the cyclooxygenase enzyme and prostaglandin production; they do not inhibit lipoxygenase pathways and, hence, do not suppress leukotriene formation. Glucocorticoids suppress the expression of COX-2 and thus COX-2-mediated prostaglandin production (Masferrer et al., 1994a). This effect may contribute in part to the antiinflammatory actions of glucocorticoids. Table 271 provides a classification of NSAIDs and other analgesic and antipyretic agents based on chemical categories. Structures of these agents are given in subsequent sections describing their therapeutic effects. Individual agents inhibit cyclooxygenase by differing mechanisms. Aspirin covalently modifies both COX-1 and COX-2, thus resulting in an irreversible inhibition of cyclooxygenase activity. This is an important distinction for aspirin, as the duration of the effects of aspirin is related to the turnover rate of cyclooxygenases in different target tissues. In the structure of COX-1, aspirin acetylates serine 530, preventing the binding of arachidonic acid to the active site of the enzyme and thus the ability of the enzyme to make prostaglandins. In COX-2, aspirin acetylates a homologous serine at position 516. Although covalent modification of COX-2 by aspirin also blocks the cyclooxygenase activity of this isoform, an interesting property of COX-2, not shared by COX-1, is that acetylated COX-2 now synthesizes 15(R)-hydroxyeicosatetraenoic acid (15(R)-HETE) (Lecomte et al., 1994; O'Neill et al., 1994). Interestingly, this aspirin-induced product can undergo transcellular metabolism by the 5-lipoxygenase enzyme to yield 15-epilipoxin A4, which exerts potent antiinflammatory actions and therefore may potentiate the antiinflammatory action of aspirin (Claria and Serhan, 1995; Serhan et al., 1999). Platelets are especially susceptible to prolonged, aspirin-mediated,
irreversible inactivation of cyclooxygenase because they have little or no
capacity for protein biosynthesis and thus cannot regenerate the
cyclooxygenase enzyme. In practical terms, this means that a single dose of
aspirin will inhibit the platelet cyclooxygenase for the life of the platelet
(8 to 11 days); in human beings, a daily dose of aspirin as small as 40 mg is
sufficient to produce this effect. The ability of platelets to be inhibited
by such low doses of aspirin is related to the presystemic inhibition of the
cyclooxygenase in the portal circulation before the aspirin is deacetylated
to salicylate in the liver. In contrast to aspirin, salicylic acid has no
acetylating capacity. Nevertheless, it, like aspirin, reduces the synthesis
of prostaglandins in vivo, but whether this is due to a direct effect
on cyclooxygenase and/or an indirect effect due to an ability of salicylate
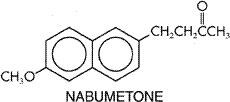
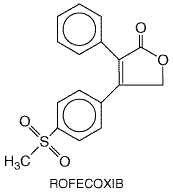
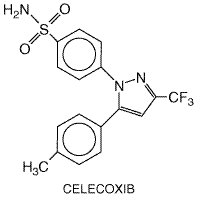
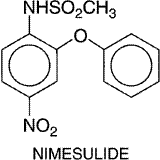
to inhibit the activation of NF The vast majority of NSAIDs listed in Table 271 are organic acids and, in contrast to aspirin, act as reversible, competitive inhibitors of cyclooxygenase activity. Even the nonacidic parent drug, nabumetone, is converted to an active acetic acid derivative in vivo. As organic acids, the compounds generally are well absorbed orally, highly bound to plasma proteins, and excreted either by glomerular filtration or by tubular secretion. In contrast to aspirin, whose duration of action is determined by the rate of synthesis of new cyclooxygenase enzyme, the duration of action of all other NSAIDs, which are reversible inhibitors of cyclooxygenase, is primarily related to the pharmacokinetic clearance of the drugs from the body. NSAIDs can be roughly divided into two groups, those with short (<6 hours) and those with long (>10 hours) half-lives (Brooks and Day, 1991). Because aspirin and other NSAIDs are organic acids, they accumulate at sites of inflammation, which is an attractive pharmacokinetic property of drugs intended as antiinflammatory agents. Most NSAIDs developed before the availability of selective COX-2 inhibitors inhibit both COX-1 and COX-2 with little selectivity or have modest selectivity for the constitutive COX-1 isoform. The hope that it would be possible to retain the antiinflammatory effects of aspirin-like drugs with a lower ulcerogenic potential has propelled efforts to design NSAIDs with greater selectivity for COX-2 versus COX-1 (Meade et al., 1993; Mitchell et al., 1993; Massferrer et al., 1994b; O'Neill et al., 1994). These efforts have led to the recent introduction of highly selective COX-2 inhibitors (rofecoxib and celecoxib) and the recognition that two previously marketed NSAIDs that have very low gastric toxicity (etodolac and nimesulide) also have a high degree of selectivity for inhibition of COX-2. The relative COX-isozyme selectivity of most of the NSAIDs available has been described in detail (Warner et al., 1999). There is good evidence that therapeutic doses of aspirin and other NSAIDs reduce prostaglandin biosynthesis in human beings, and there is a reasonably good rank order correlation between the potency of these drugs as inhibitors of cyclooxygenase and their antiinflammatory activity (Vane and Botting, 1987). There are some exceptions to this, but these exceptions may in part be attributed to the experimental conditions used, which do not always mimic the in vivo situation. For example, potencies of compounds to inhibit purified enzyme compared to enzymes contained in cells sometimes have been found to be different (Mitchell et al., 1993). Moreover, in vitro conditions do not take into account factors such as binding of the drugs to plasma proteins. Furthermore, in addition to inhibiting cyclooxygenase, some drugs have been found to exert other effects that are antiinflammatory (Yamamoto et al., 1999; Yin et al., 1998). Nonetheless, many findings are consistent with inhibition of prostaglandin synthesis as the principal basis for the therapeutic actions of NSAIDs. An example of other lines of evidence linking cyclooxygenase
inhibition to antiinflammatory activity is the high degree of
stereoselectivity among several pairs of enantiomers of Acetaminophen, which is a very weak antiinflammatory agent, is a weak inhibitor of cyclooxygenase. Moreover, acetaminophen appears to inhibit the enzyme only in an environment that is low in peroxide (e.g., the hypothalamus; see Marshall et al., 1987; Hanel and Lands, 1982), which may in part explain the poor antiinflammatory activity of acetaminophen, since sites of inflammation usually contain increased concentrations of peroxides generated by leukocytes. Shared Therapeutic Activities and Side Effects of NSAIDs Therapeutic Effects All NSAIDs, including selective COX-2 inhibitors (Morrison et al., 1999; Malmstrom et al., 1999), are antipyretic, analgesic, and antiinflammatory. One important exception is acetaminophen, which is antipyretic and analgesic but is largely devoid of antiinflammatory activity. This can be explained by the fact that acetaminophen effectively inhibits cyclooxygenases in the brain but not at sites of inflammation in peripheral tissues (see above). When employed as analgesics, these drugs usually are effective only against pain of low-to-moderate intensity, such as dental pain. Although their maximal effects are much lower, they lack the unwanted effects of the opioids on the central nervous system (CNS), including respiratory depression and the development of physical dependence. NSAIDs do not change the perception of sensory modalities other than pain. Chronic postoperative pain or pain arising from inflammation is particularly well controlled by NSAIDs, whereas pain arising from the hollow viscera usually is not relieved. As antipyretics, NSAIDs reduce the body temperature in febrile states. The fact that selective COX-2 inhibitors are effective antipyretic agents indicates that the COX isoform predominantly involved in thermoregulation is COX-2. NSAIDs find their chief clinical application as antiinflammatory agents in the treatment of musculoskeletal disorders, such as rheumatoid arthritis, osteoarthritis, and ankylosing spondylitis. Chronic treatment of patients with rofecoxib and celecoxib has been shown to be effective in suppressing inflammation without the gastric toxicity that is associated with treatment with nonselective NSAIDs (Simon et al., 1998, 1999; Bensen et al., 1999; Emery et al., 1999; Hawkey et al., 2000; Schnitzer et al., 1999; Ehrich et al., 1999; Laine et al., 1999). In general, NSAIDs provide only symptomatic relief from the pain and inflammation associated with the disease and do not arrest the progression of pathological injury to tissue. Some other uses of NSAIDs also depend upon their capacity to block prostaglandin biosynthesis. Prostaglandins have been implicated in the maintenance of patency of the ductus arteriosus, and indomethacin and related agents have been used in neonates to close the ductus when it has remained patent. On the other hand, administration of nonselective NSAIDs to pregnant women can cause premature contraction of the ductus in utero. In fetal lambs, production of vasodilatory prostaglandins by both COX-1 and COX-2 appear to participate in maintaining patency of the ductus arteriosus (Clyman et al., 1999). Although it remains to be established which isoform(s) is involved in maintaining patency of the fetal ductus in utero in human beings, it is prudent to exercise caution in the use of selective COX-2 inhibitors in pregnant women. The release of prostaglandins by the endometrium during menstruation may be a cause of severe cramps and other symptoms of primary dysmenorrhea; treatment of this condition with NSAIDs has met with considerable success (see Shapiro, 1988). A recent study revealed that the selective COX-2 inhibitor rofecoxib is as effective as the nonselective NSAID sodium naproxen in the treatment of dysmenorrhea (Morrison et al., 1999). Therefore, it is anticipated that there will be an increase in the use of selective COX-2 inhibitors for this condition. Prostaglandin D2 released from mast cells in large amounts has been found to be the major mediator of severe episodes of vasodilation and hypotension in patients with systemic mastocytosis. Treatment of these patients with antihistamines alone is usually ineffective, whereas addition of an NSAID usually leads to effective prevention of these episodes (Roberts et al., 1980; Roberts and Oates, 1991; Metcalf, 1991). Prostaglandin E2 also has been implicated in the humoral hypercalcemia associated with some neoplasms, and treatment with NSAIDs can effectively suppress serum calcium levels in some cancer patients (Brenner et al., 1982; Robertson, 1981). Bartter's syndrome is characterized hypokalemia, hyperreninemia, hyperaldosteronism, juxtaglomerular hyperplasia, normotension, and resistance to the pressor effect of angiotensin II. Excessive production of renal prostaglandins has been implicated in the pathogenesis of some of the metabolic abnormalities in this syndrome, and NSAIDs have been found to be useful in the treatment of this disorder (Dunn, 1981). A rare, complex syndrome occurs in infants resembling a Bartter's syndromelike tubulopathy with systemic features including fever, diarrhea, and osteopenia with hypercalciuria. This syndrome is associated with marked overproduction of prostaglandin E2, and it has been termed hyperprostaglandin E syndrome. Most of these abnormalities can be effectively controlled with long-term treatment with indomethacin (Seyberth et al., 1987). An important area where the use of NSAIDs is emerging is in the prevention of colon cancer. Epidemiological studies suggested that frequent use of aspirin is associated with a striking reduction (approximately 50%) in the incidence of colon cancer (Thun et al., 1991; Giovannucci et al., 1995). Interestingly, this reduction occurred with ingestion of as little as four to six 325-mg tablets per week. These observations have stimulated intense investigation into the mechanism(s) involved in the reduction of colon cancer incidence. NSAIDs, in particular sulindac sulfide, have been found to suppress significantly polyp formation in patients with familial polyposis coli and in mice bearing a mutation in the same APC gene. Whether or not these effects of NSAIDs are due to a cyclooxygenase-independent effect of NSAIDs has been questioned (Wu, 2000). Nevertheless, there is a compelling body of evidence suggesting that the effect of NSAIDs on colon cancer is mediated by inhibition of COX-2, which is strikingly upregulated in colon tumors (Gupta and Dubois, 1998). Controlled, randomized, prospective trials currently are under way to evaluate aspirin and selective COX-2 inhibitors as chemopreventive agents for sporadic colon cancer. Large doses of niacin (nicotinic acid) effectively lower serum cholesterol levels, reduce LDL, and raise HDL (see Chapter 36: Drug Therapy for Hypercholesterolemia and Dyslipidemia). However, niacin is poorly tolerated because it induces intense flushing. This flushing has been shown to be mediated by a release of prostaglandin D2 from the skin, which can be inhibited by treatment with low doses of aspirin (Morrow et al., 1989; Morrow et al., 1992). Side Effects of NSAID Therapy In addition to sharing many therapeutic activities, NSAIDs share several unwanted side effects, outlined in Table 272 (see also Borda and Koff, 1992). The most common is a propensity to induce gastric or intestinal ulceration that sometimes can be accompanied by anemia from the resultant blood loss. The notable exception to this is that highly selective COX-2 inhibitors lack the propensity to cause gastric ulceration. Patients who use nonselective NSAIDs on a chronic basis have about three times greater relative risk for serious adverse gastrointestinal events compared to nonusers (Gabriel et al., 1991). Nonselective NSAIDs vary considerably in their tendency to cause such erosions and ulcers (see individual sections). Gastric damage by these agents can be brought about by at least two distinct mechanisms. Although local irritation by orally administered drugs allows back diffusion of acid into the gastric mucosa and induces tissue damage, parenteral administration also can cause damage and bleeding, correlated with inhibition of the biosynthesis of gastric prostaglandins, especially PGI2 and PGE2, that serve as cytoprotective agents in the gastric mucosa (see articles by Ivey and by Isselbacher, in Symposium, 1988a). These eicosanoids inhibit acid secretion by the stomach, enhance mucosal blood flow, and promote the secretion of cytoprotective mucus in the intestine; inhibition of their synthesis may render the stomach more susceptible to damage. All of the NSAIDs discussed in this chapter, with the exception of p-aminophenol derivatives and the highly selective COX-2 inhibitors, have a strong tendency to cause gastrointestinal side effects ranging from mild dyspepsia and heartburn to ulceration of the stomach or duodenum, sometimes with fatal results. Administration of the PGE1 analog misoprostol along with these NSAIDs can be beneficial in the prevention of duodenal and gastric ulceration produced by these drugs (Graham et al., 1993). It also is possible that enhanced generation of lipoxygenase products contributes to ulcerogenicity in patients treated with NSAIDs and that there may be an association with Helicobacter pylori infection (see Borda in Borda and Koff, 1992). Other side effects of these drugs that result from blockade of the synthesis of endogenous prostaglandins and thromboxane A2 include disturbances in platelet function, the prolongation of gestation or spontaneous labor, premature closure of the patent ductus, and changes in renal function. Platelet function is impaired because NSAIDs prevent the formation by the platelets of thromboxane A2 (TXA2), a potent aggregating agent. This accounts for the ability of these drugs to increase the bleeding time. Aspirin is a particularly effective inhibitor of platelet function, because, as discussed above, the irreversible effects of aspirin on cyclooxygenase activity require new platelet production for restoration of enzyme activity. This 'side effect' has been exploited in the prophylactic treatment of thromboembolic disorders (see Chapter 55: Anticoagulant, Thrombolytic, and Antiplatelet Drugs). Acute administration of 400 mg and 800 mg of the selective COX-2 inhibitor celecoxib to human beings has been found to suppress PGI2 production by about 80%, without inhibiting TXA2 production and platelet aggregation (McAdam et al., 1999). Similar results were obtained with rofecoxib. This finding suggests that COX-2 is a major source of PGI2 production in vivo. Since an important action of PGI2 is thought to be suppression of platelet activation, alteration of the TXA2/PGI2 ratio that accompanies selective inhibition of COX-2 is theoretically prothrombotic. It remains to be determined whether or not this possibility is clinically relevant. Nonetheless, it may be prudent to consider these findings when choosing an NSAID for the treatment of patients who are particularly prone to thrombotic events. Prolongation of gestation by NSAIDs has been demonstrated in both experimental animals and women. Prostaglandins of the E and F series are potent uterotropic agents, and their biosynthesis by the uterus increases dramatically in the hours before parturition. This increase in prostanoid production is thought to result from induction of COX-2 expression (Slater et al., 1999). Prostaglandins are thus postulated to have a major role in the initiation and progression of labor and delivery. Accordingly, some NSAIDs have been used as tocolytic agents to inhibit preterm labor, including selective COX-2 inhibitors (Sawdy et al., 1997). However, as mentioned previously, administration of nonselective COX inhibitors can cause premature closure of the ductus arteriosus in utero, and evidence obtained in fetal lambs suggests that selective COX-2 inhibitors also may cause this effect. Clinically relevant, adverse effects on renal function have been well recognized with the use of nonselective NSAIDs; recent evidence suggests that selective COX-2 inhibitors also have the propensity to cause such effects (Brater, 1999). NSAIDs have little effect on renal function in normal human subjects, presumably because the production of vasodilatory prostaglandins has only a minor role in sodium-replete individuals. However, these drugs decrease renal blood flow and the rate of glomerular filtration in patients with congestive heart failure, hepatic cirrhosis with ascites, chronic renal disease, or in those who are hypovolemic (see Clive and Stoff, 1984; Patrono and Dunn, 1987; Oates et al., 1988; Wilson and Carruthers in Borda and Koff, 1992); acute renal failure may be precipitated under these circumstances. In individuals with these clinical conditions, renal perfusion is more dependent than in normal individuals upon prostaglandins that cause vasodilation and thus oppose the increased vasoconstrictive influences of norepinephrine and angiotensin II that result from the activation of pressor reflexes. In addition to their hemodynamic effects in the kidney, NSAIDs promote the retention of salt and water by reducing the prostaglandin-induced inhibition of both the reabsorption of chloride and the action of antidiuretic hormone. This may cause edema in some patients who are treated with NSAIDs; it also may reduce the effectiveness of antihypertensive regimens (see Patrono and Dunn, 1987; Oates et al., 1988). These drugs promote hyperkalemia by several mechanisms, including enhanced reabsorption of K+ as a result of decreased availability of Na+ at distal tubular sites and suppression of the prostaglandin-induced secretion of renin. The latter effect may account in part for the usefulness of NSAIDs in the treatment of Bartter's syndrome, which is characterized by hypokalemia, hyperreninemia, hyperaldosteronism, juxtaglomerular hyperplasia, normotension, and resistance to the pressor effect of angiotensin II. Excessive production of renal prostaglandins may play an important part in the pathogenesis of this syndrome. Although nephropathy is uncommonly associated with the long-term use
of individual NSAIDs, the abuse of analgesic mixtures has been linked to the
development of renal injury, including papillary necrosis and chronic
interstitial nephritis (see Kincaid-Smith, 1986). The injury often is
insidious in onset, is usually manifest initially as reduced tubular function
and concentrating ability, and may progress to irreversible renal
insufficiency if misuse of analgesics continues. Females are involved more
frequently than are males, and often there is a history of recurring urinary
tract infection. Emotional disturbances are common, and other drugs may be
abused concurrently. Despite numerous clinical observations and experimental
studies in laboratory animals and human beings, insights concerning the
mechanisms underlying NSAID-fostered renal injury are lacking. Phenacetin was
suggested to be the nephrotoxic component of older analgesic mixtures
(commonly, aspirinphenacetincaffeine, or 'APC') and, therefore,
was removed from these products. Although the incidence of analgesic
nephropathy in some countries has subsequently declined, this has not been a
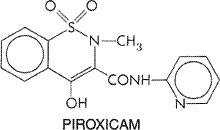
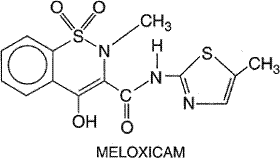
universal result, especially in Certain individuals display intolerance to aspirin and most NSAIDs; this is manifest by symptoms that range from vasomotor rhinitis with profuse watery secretions, angioneurotic edema, generalized urticaria, and bronchial asthma to laryngeal edema and bronchoconstriction, flushing, hypotension, and shock. Although less common in children, this syndrome may occur in 10% to 25% of patients with asthma, nasal polyps, or chronic urticaria and can occur when these patients receive even small amounts (<80 mg) of aspirin. A subset of patients with mastocytosis also exhibits adverse reactions with the use of aspirin. Despite the resemblance to anaphylaxis, this reaction does not appear to be immunological in nature. These reactions are not limited to aspirin. Almost without exception, an individual who exhibits intolerance to aspirin also will react when given any of the other NSAIDs, despite their chemical diversity. Although nonacetylated salicylates and acetaminophen are less likely to produce these reactions in individuals who react to other NSAIDs, they can produce severe reactions in some, especially if high doses are administered. [Such patients also may react if they ingest tartrazine (FD&C Yellow No. 5 dye), which is found in many foods and beverages.] The underlying mechanism for this hypersensitivity reaction to NSAIDs is not known, but a common factor appears to be the ability of the drugs to inhibit cyclooxygenase activity. This has prompted the hypothesis that the reaction reflects the diversion of arachidonic acid metabolism toward the formation of increased amounts of leukotrienes and other products of lipoxygenase pathways. This view is as yet unproven, and it does not explain why only a minority of patients with asthma or other predisposing conditions display the reaction. Even so, results in a small number of patients suggest that blockade of 5-lipoxygenase with the drug zileuton may prevent symptoms and signs of aspirin intolerance (Israel et al., 1993). Hypersensitivity to aspirin is a contraindication to therapy with any of the drugs discussed in this chapter; administration of any one of these could provoke a life-threatening reaction reminiscent of anaphylactic shock (see above). Choice of an NSAID in Varying Clinical Situations The choice of an agent as an antipyretic or analgesic is seldom a problem. It is in the field of rheumatology that the decision becomes complex (see Brooks and Day, 1991). The choice among NSAIDs for the treatment of arthritides is largely empirical. A drug may be chosen and given for a week or more; if the therapeutic effect is adequate, treatment should be continued unless toxicity occurs. Large variations are possible in the response of individuals to different NSAIDs, even when the drugs are structurally similar members of the same chemical family. Thus, a patient may do well on one propionic acid derivative (such as ibuprofen) but not on another. Initially, fairly low doses of the agent chosen should be prescribed to determine the effect of the drug and patient tolerance. When the patient has problems sleeping because of pain or morning stiffness, a larger single dose of the drug may be given at night. A week is generally long enough to determine the effect of a given drug. If the drug is effective, treatment should be continued, reducing the dose if possible and stopping it altogether if it is no longer necessary. Side effects usually appear in the first weeks of therapy, although gastric ulceration usually takes much longer to develop. If the patient does not achieve therapeutic benefit from one NSAID, another compound should be tried, since, as noted above, there is a marked variation in the response of individuals to different but closely related drugs. Discussion of principles of the use of NSAIDs also is provided in earlier reviews (Symposium, 1983a; Lewis and Furst, 1987). For mild arthropathies, the scheme outlined above, together with rest and physical therapy, probably will be effective. However, patients with a more debilitating disease may not respond adequately. In such cases, more aggressive therapy should be initiated with aspirin or another agent. It is best to avoid continuous combination therapy with more than one NSAID; there is little evidence of extra benefit to the patient, and the incidence of side effects generally is additive. The choice of drugs for children is considerably restricted, and only drugs that have been extensively tested in children should be used. This commonly means that only aspirin, naproxen, or tolmetin should be prescribed. However, the association of Reye's syndrome in children with the administration of aspirin for the treatment of febrile viral illnesses precludes its use in this setting. Although some controversy remains regarding whether there is a causative link between aspirin use in children and the development of Reye's syndrome, the epidemiologic evidence for this was so compelling that labeling of aspirin and aspirin-containing medications to indicate Reye's syndrome as a risk in children was mandated in 1986 (see Hurwitz, 1989). The use of aspirin in children has declined dramatically, and Reye's syndrome has almost disappeared (Belay et al., 1999; Monto, 1999). Acetaminophen has not been implicated in Reye's syndrome and can be substituted for aspirin for antipyresis in children. The use of any of the NSAIDs in pregnant women generally is not recommended. If such a drug must be given to a pregnant woman, low doses of aspirin are probably the safest. Although toxic doses of salicylates cause teratogenic effects in animals, there is no evidence to suggest that salicylates in moderate doses have teratogenic effects on the human fetus. In any case, aspirin and other NSAIDs should be discontinued prior to the anticipated time of parturition to avoid complications such as prolongation of labor, increased risk of postpartum hemorrhage, and intrauterine closure of the ductus arteriosus. Many NSAIDs are highly bound to plasma proteins and thus may displace certain other drugs from the binding sites. Such interactions can occur in patients given salicylates or other NSAIDs together with warfarin, sulfonylurea hypoglycemic agents, or methotrexate; the dosage of such agents may require adjustment, or concurrent administration should be avoided. The problem with warfarin is accentuated, because almost all NSAIDs suppress normal platelet function. Numerous other drug interactions are observed with NSAIDs (see Brooks and Day, 1991). For the seriously debilitated patient who cannot tolerate these drugs or in whom they are not adequately effective, other forms of therapy should be considered. Gold, hydroxychloroquine, and penicillamine are discussed in a separate section of this chapter. Other relevant drugs include immunosuppressive agents (Chapter 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants) and glucocorticoids (Chapter 60: Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones). A final important consideration in the selection of an NSAID for a patient is the cost of therapy, particularly since these agents frequently are used on a long-term basis. Generally speaking, aspirin is very inexpensive; the cost of the newer, nonselective NSAIDs and selective COX-2 inhibitors drugs is much higher. |
The Salicylates
|
Despite the introduction of many new drugs, aspirin
(acetylsalicylic acid) is still the most widely prescribed
analgesic-antipyretic and antiinflammatory agent and is the standard for the
comparison and evaluation of the others. Prodigious amounts of the drug are
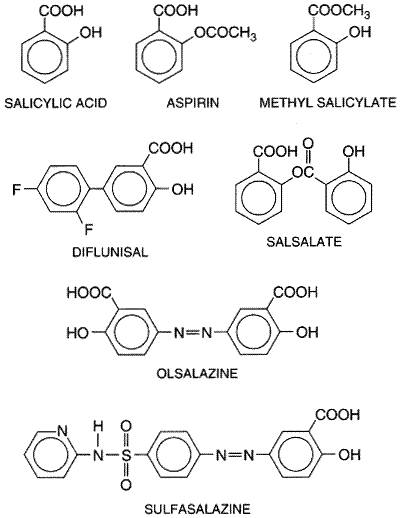
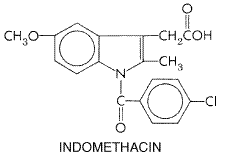
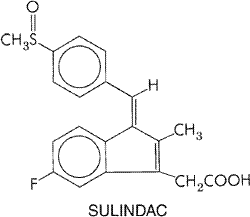
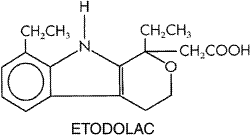
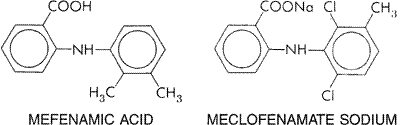
consumed in the Chemistry Salicylic acid (orthohydroxybenzoic acid) is so irritating that it can be used only externally; therefore, various derivatives of this acid have been synthesized for systemic use. These comprise two large classes, namely, esters of salicylic acid obtained by substitution in the carboxyl group and salicylate esters of organic acids, in which the carboxyl group of salicylic acid is retained and substitution is made in the hydroxyl group. For example, aspirin is an ester of acetic acid. In addition, there are salts of salicylic acid. The chemical relationships can be seen from the structural formulas shown in Figure 271.
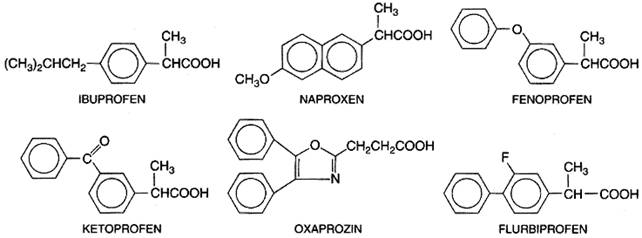
Structure-Activity Relationships Salicylates generally act by virtue of their content of salicylic acid, although some of the unique effects of aspirin are due to its capacity to acetylate proteins, as described earlier. Substitutions on the carboxyl or hydroxyl groups change the potency or toxicity of salicylate agents. The ortho position of the hydroxyl group is an important feature for the action of salicylate. The effects of simple substitutions on the benzene ring have been extensively studied, and new salicylate derivatives still are being synthesized. A difluorophenyl derivative, diflunisal, also is available for clinical use. Pharmacological Properties Analgesia As noted above, the types of pain usually relieved by salicylates are those of low intensity that arise from integumental structures rather than from viscera, especially headache, myalgia, and arthralgia. The salicylates are more widely used for pain relief than are any other classes of drugs. Long-term use does not lead to tolerance or addiction, and toxicity is lower than that of opioid analgesics. The salicylates alleviate pain by virtue of a peripheral action; direct effects on the CNS also may be involved. Antipyresis As discussed above, salicylates usually lower elevated body temperatures rapidly and effectively. However, moderate doses that produce this effect also increase oxygen consumption and metabolic rate. In toxic doses, these compounds have a pyretic effect that results in sweating; this enhances the dehydration that occurs in salicylate intoxication (see below). Miscellaneous Neurological Effects In high doses, salicylates have toxic effects on the CNS, consisting
of stimulation (including convulsions) followed by depression. Confusion,
dizziness, tinnitus, high-tone deafness, delirium, psychosis, stupor, and
coma may occur. The tinnitus and hearing loss caused by salicylate poisoning
are due to increased labyrinthine pressure or an effect on the hair cells of
the cochlea, perhaps secondary to vasoconstriction in the auditory
microvasculature. Tinnitus is typically observed at plasma salicylate
concentrations of 200 to 450 Salicylates induce nausea and vomiting, which result from stimulation
of sites that are accessible from the cerebrospinal fluid (CSF), probably in
the medullary chemoreceptor trigger zone. In human beings, centrally induced
nausea and vomiting generally appear at plasma salicylate concentrations of
about 270 Respiration The effects of salicylate on respiration are important, because they contribute to the serious acidbase balance disturbances that characterize poisoning by this class of compounds. Salicylates stimulate respiration directly and indirectly. Full therapeutic doses of salicylates increase oxygen consumption and CO2 production (especially in skeletal muscle); these effects are a result of salicylate-induced uncoupling of oxidative phosphorylation. The increased production of CO2 stimulates respiration. The increased alveolar ventilation balances the increased CO2 production, and thus plasma CO2 tension (PCO ) does not change. The initial increase in alveolar ventilation is characterized mainly by an increase in depth of respiration and only a slight increase in rate. If the respiratory response to CO2 has been depressed by the administration of a barbiturate or an opioid, salicylates will cause a marked increase in plasma PCO and respiratory acidosis. Salicylate directly stimulates the respiratory center in the medulla.
This results in marked hyperventilation, characterized by an increase in
depth and a pronounced increase in rate. Patients with salicylate poisoning
may have prominent increases in respiratory minute volume, and respiratory
alkalosis ensues. Plasma salicylate concentrations of 350 A depressant effect of salicylate on the medulla appears after high doses or after prolonged exposure. Toxic doses of salicylates cause central respiratory depression as well as circulatory collapse secondary to vasomotor depression. Since enhanced CO2 production continues, respiratory acidosis ensues. AcidBase Balance and Electrolyte Pattern Therapeutic doses of salicylate produce definite changes in the acidbase balance and electrolyte pattern. The initial event, as discussed above, is respiratory alkalosis. Compensation for the respiratory alkalosis is achieved by increased renal excretion of bicarbonate, which is accompanied by increased Na+ and K+ excretion; plasma bicarbonate is thus lowered, and blood pH returns toward normal. This is the stage of compensated respiratory alkalosis. This stage is most often seen in adults given intensive salicylate therapy and seldom proceeds further. Subsequent changes in acidbase status generally occur only when toxic doses of salicylates are ingested by infants and children and occasionally after large doses in adults. In infants and children, the phase of respiratory alkalosis may not be observed, since the child with salicylate intoxication is rarely seen early enough. Instead, the stage of acidbase toxicity usually presented clinically is characterized by a decrease in blood pH, a low plasma bicarbonate concentration, and a normal or nearly normal plasma PCO ; except for the PCO value, these changes resemble those of metabolic acidosis. However, in reality there is a combination of respiratory acidosis and metabolic acidosis produced as follows: the enhanced production of CO2 outstrips its alveolar excretion because of direct salicylate-induced depression of respiration; consequently, plasma PCO increases and blood pH decreases. Since the concentration of bicarbonate in plasma already is low because of increased renal bicarbonate excretion, the acidbase status at this stage is essentially an uncompensated respiratory acidosis. Superimposed, however, is a true metabolic acidosis caused by accumulation of acids as a result of three processes. First, toxic concentrations of salicylates displace about 2 to 3 meq per liter of plasma bicarbonate. Second, vasomotor depression caused by toxic doses of salicylate impairs renal function with consequent accumulation of strong acids of metabolic origin, namely, sulfuric and phosphoric acids. Third, organic acids accumulate secondary to salicylate-induced derangement of carbohydrate metabolism, especially pyruvic, lactic, and acetoacetic acids. The series of events that produces acidbase disturbances in salicylate intoxication also causes alterations of water and electrolyte balance. The low plasma PCO leads to decreased renal tubular reabsorption of bicarbonate and increased renal excretion of Na+, K+, and water. In addition, water is lost by salicylate-induced sweating and hyperventilation; dehydration rapidly occurs. Since more water than electrolyte is lost through the lungs and by sweating, the dehydration is associated with hypernatremia. Prolonged exposure to high doses of salicylate also causes depletion of K+ due to both renal and extrarenal factors. Cardiovascular Effects Ordinary therapeutic doses of salicylates have no important direct cardiovascular actions. The peripheral vessels tend to dilate after large doses because of a direct effect on their smooth muscle. Toxic amounts depress the circulation both directly and by central vasomotor paralysis. In patients given large doses of sodium salicylate or aspirin, such as the doses used in acute rheumatic fever, the circulating plasma volume increases (about 20%), the hematocrit falls, and cardiac output and work are increased. Consequently, in patients with clear evidence of carditis, such alterations can cause congestive failure and pulmonary edema. High doses of salicylates also can produce noncardiogenic pulmonary edema, particularly in older patients who are ingesting salicylates regularly over a prolonged duration. Gastrointestinal Effects The ingestion of salicylate may result in epigastric distress, nausea, and vomiting. The mechanism of the emetic effect is discussed above. Salicylate also may cause gastric ulceration; exacerbation of peptic ulcer symptoms (heartburn, dyspepsia), gastrointestinal hemorrhage, and erosive gastritis all have been reported in patients on high-dose therapy but also may occur even when low doses are administered. These adverse effects occur primarily with acetylated salicylate (aspirin). This is because nonacetylated salicylates are much weaker cyclooxygenase inhibitors than aspirin, because they lack the ability to acetylate the enzyme and thus irreversibly inhibit its activity. Aspirin-induced gastric bleeding sometimes is painless and, if
unrecognized, may lead to an iron-deficiency anemia. The daily ingestion of 4
or 5 g of aspirin, a dose that produces plasma salicylate concentrations in
the usual range for antiinflammatory therapy (120 to 350 Hepatic and Renal Effects Salicylates can cause hepatic injury. This usually occurs in patients
treated with high doses of salicylate that result in plasma salicylate
concentrations above 150 Salicylates can cause retention of salt and water as well as acute reduction of renal function in patients with congestive heart failure, renal disease, or hypovolemia (see above). Although long-term use of salicylates alone rarely is associated with nephrotoxicity, the prolonged and excessive ingestion of analgesic mixtures containing salicylates in combination with other compounds can produce papillary necrosis and interstitial nephritis. Uricosuric Effects The effects of salicylates on uric acid excretion are markedly dependent on dose (see'Uricosuric Agents,' below). Low doses (1 or 2 g per day) may decrease urate excretion and elevate plasma urate concentrations; intermediate doses (2 or 3 g per day) usually do not alter urate excretion; large doses (over 5 g per day) induce uricosuria and lower plasma urate levels. Such large doses are poorly tolerated. Even small doses of salicylate can block the effects of probenecid and other uricosuric agents that decrease tubular reabsorption of uric acid. Effects on the Blood Ingestion of aspirin by healthy individuals causes a prolongation of the bleeding time. For example, a single dose of 0.65 g of aspirin (2 tablets) approximately doubles the mean bleeding time of normal persons for a period of 4 to 7 days. This effect is due to irreversible acetylation of platelet cyclooxygenase and the consequent reduced formation of TXA2 until production of unmodified platelets from megakaryocyte precursors occurs. Patients with severe hepatic damage, hypoprothrombinemia, vitamin K deficiency, or hemophilia should avoid aspirin because the inhibition of platelet hemostasis can result in hemorrhage. If conditions permit, aspirin therapy should be stopped at least 1 week prior to surgery; care also should be exercised in the use of aspirin during long-term treatment with oral anticoagulant agents because of the possible danger of blood loss from the gastric mucosa as well as from hemorrhage at other sites. However, aspirin is used for the prophylaxis of thromboembolic disease, especially in the coronary and cerebral circulation (see Willard et al., 1992; Patrono, 1994; see Chapter 55: Anticoagulant, Thrombolytic, and Antiplatelet Drugs). Salicylates do not ordinarily alter the leukocyte or platelet count, the hematocrit, or the hemoglobin content. However, doses of 3 to 4 g per day markedly decrease plasma iron concentration and shorten erythrocyte survival time. Aspirin is included among the drugs that can cause a mild degree of hemolysis in individuals with a deficiency of glucose-6-phosphate dehydrogenase. Effects on Rheumatic, Inflammatory, and Immunological Processes and on Connective Tissue Metabolism For almost 100 years, the salicylates have retained their preeminent position in the treatment of the rheumatic diseases. Although they suppress the clinical signs and even improve the histological picture in acute rheumatic fever, subsequent tissue damage such as cardiac lesions and other visceral involvement is unaffected. In addition to their action on prostaglandin biosynthesis, the mechanism of action of the salicylates in rheumatic disease also may involve effects on other cellular and immunological processes in mesenchymal and connective tissues. Because of the known relationship between rheumatic fever and immunological processes, attention has been directed to the capacity of salicylates to suppress a variety of antigenantibody reactions. These include the inhibition of antibody production, of antigenantibody aggregation, and of antigen-induced release of histamine. Salicylates also induce a nonspecific stabilization of capillary permeability during immunological insults. The concentrations of salicylates needed to produce these effects are high, and the relationship of these effects to the antirheumatic efficacy of salicylates is unclear. Salicylates also can influence the metabolism of connective tissue, and these effects may be involved in their antiinflammatory action. For example, salicylates can affect the composition, biosynthesis, or metabolism of connective tissue mucopolysaccharides in the ground substance that provides barriers to spread of infection and inflammation. Metabolic Effects The salicylates have multiple effects on metabolic processes, some of which already have been discussed. Only a few pertinent aspects will be presented here. In general, these effects are minimal at usual recommended doses. Oxidative Phosphorylation The uncoupling of oxidative phosphorylation by salicylate is similar to that induced by 2,4-dinitrophenol. The effect may occur with doses of salicylate used in the treatment of rheumatoid arthritis and can result in the inhibition of a number of adenosine triphosphate (ATP)-dependent reactions. Other consequences include the salicylate-induced increase in oxygen uptake and carbon dioxide production described above, the depletion of hepatic glycogen, and the pyretic effect of toxic doses of salicylate. Salicylate in toxic doses may decrease aerobic metabolism as a result of inhibition of various dehydrogenases, by competing with the pyridine nucleotide coenzymes, and inhibition of some oxidases that require nucleotides as coenzymes, such as xanthine oxidase. Carbohydrate Metabolism Large doses of salicylates may cause hyperglycemia and glycosuria and deplete liver and muscle glycogen; these effects partly are explained by the release of epinephrine. Such doses also reduce aerobic metabolism of glucose, increase glucose-6-phosphatase activity, and promote the secretion of glucocorticoids. Nitrogen Metabolism Salicylate in toxic doses causes a significant negative nitrogen balance, characterized by an aminoaciduria. Adrenocortical activation may contribute to the negative nitrogen balance by enhancing protein catabolism. Fat Metabolism Salicylates reduce lipogenesis by partially blocking incorporation of acetate into fatty acids; they also inhibit epinephrine-stimulated lipolysis in fat cells and displace long-chain fatty acids from binding sites on human plasma proteins. The combination of these effects leads to increased entry and enhanced oxidation of fatty acids in muscle, liver, and other tissues, and to decreased plasma concentrations of free fatty acids, phospholipid, and cholesterol; the oxidation of ketone bodies also is increased. Endocrine Effects Very large doses of salicylate stimulate steroid secretion by the adrenal cortex through an effect on the hypothalamus and transiently increase plasma concentrations of free adrenocorticosteroids by displacement from plasma proteins. However, it is clear that the antiinflammatory effects of salicylate are independent of these effects. Long-term administration of salicylate decreases thyroidal uptake and clearance of iodine but increases oxygen consumption and rate of disappearance of thyroxine and triiodothyronine from the circulation. These effects probably are due to the competitive displacement by salicylate of thyroxine and triiodothyronine from transthyretin and the thyroxine-binding globulin in plasma and usually are of minimal clinical significance. Salicylates and Pregnancy There is no evidence that moderate therapeutic doses of salicylates are teratogenic in human beings; however, babies born to women who ingest salicylates for long periods may have significantly reduced weights at birth. There also is an increase in perinatal mortality, anemia, antepartum and postpartum hemorrhage, prolonged gestation, and complicated deliveries. These effects occur when aspirin is administered during the third trimester, and thus its use during this period should be avoided. As mentioned previously, administration of NSAIDs during the third trimester of pregnancy also can cause premature closure of the ductus arteriosus. Local Irritant Effects Salicylic acid is quite irritating to skin and mucosa and destroys epithelial cells. The keratolytic action of the free acid is employed for the local treatment of warts, corns, fungal infections, and certain types of eczematous dermatitis. The tissue cells swell, soften, and desquamate. Methyl salicylate (oil of wintergreen) is irritating to both skin and gastric mucosa and is used only externally. Pharmacokinetics and Metabolism Aspirin and other salicylates have several unique pharmacokinetic features that must be considered in patients receiving these drugs. Absorption Orally ingested salicylates are absorbed rapidly, partly from the stomach but mostly from the upper small intestine. Appreciable concentrations are found in plasma in less than 30 minutes; after a single dose, a peak value is reached in about 1 hour and then gradually declines. Rate of absorption is determined by many factors, particularly the disintegration and dissolution rates if tablets are given, the pH at the mucosal surfaces, and gastric emptying time. Salicylate absorption occurs by passive diffusion primarily of nondissociated salicylic acid or acetylsalicylic acid across gastrointestinal membranes and hence is influenced by gastric pH. Even though salicylate is more ionized as the pH is increased, a rise in pH also increases the solubility of salicylate and thus dissolution of the tablets. The overall effect is to enhance absorption. As a result, there is little meaningful difference between the rates of absorption of sodium salicylate, aspirin, and the numerous buffered preparations of salicylates. The presence of food delays absorption of salicylates. Rectal absorption of salicylate usually is slower than oral absorption and is incomplete and unreliable; rectal administration therefore is not advisable when high plasma concentrations of the drug are required. Salicylic acid is rapidly absorbed from the intact skin, especially when applied in oily liniments or ointments, and systemic poisoning has occurred from its application to large areas of skin. Methyl salicylate is likewise speedily absorbed when applied cutaneously; its gastrointestinal absorption may be delayed many hours, and, therefore, gastric lavage should be performed even in cases of poisoning that are seen late. Distribution After absorption, salicylate is distributed throughout most body tissues and most transcellular fluids, primarily by pH-dependent passive processes. Salicylate is actively transported by a low-capacity, saturable system out of the CSF across the choroid plexus. The drug readily crosses the placental barrier. The volumes of distribution of usual doses of aspirin and sodium
salicylate in normal subjects average about 170 ml/kg of body weight; at high
therapeutic doses, this volume increases to about 500 ml/kg because of
saturation of binding sites on plasma proteins. Ingested aspirin mainly is
absorbed as such, but some enters the systemic circulation as salicylic acid,
because of hydrolysis by esterases in the gastrointestinal mucosa and the
liver. Aspirin can be detected in the plasma only for a short time as a
result of hydrolysis in plasma, liver, and erythrocytes; for example, 30
minutes after a dose of 0.65 g, only 27% of the total plasma salicylate is in
the acetylated form. As a result, plasma concentrations of aspirin are always
low and rarely exceed 20 At concentrations encountered clinically, from 80% to 90% of the
salicylate is bound to plasma proteins, especially albumin; the proportion of
the total that is bound declines as plasma concentrations are increased. In
addition, hypoalbuminemia, as may occur in rheumatoid arthritis, is
associated with a proportionately higher level of free salicylate in the
plasma. Salicylate competes with a variety of compounds for plasma protein
binding sites; these include thyroxine, triiodothyronine, penicillin, phenytoin,
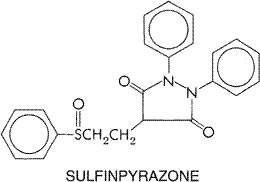
sulfinpyrazone, bilirubin, uric acid, and other NSAIDs, such as naproxen. Aspirin
is bound to a more limited extent; however, it acetylates human plasma
albumin in vivo by reaction with the Biotransformation and Excretion The biotransformation of salicylate takes place in many tissues, but particularly in the hepatic endoplasmic reticulum and mitochondria. The three chief metabolic products are salicyluric acid (the glycine conjugate), the ether or phenolic glucuronide, and the ester or acyl glucuronide. In addition, a small fraction is oxidized to gentisic acid (2,5-dihydroxybenzoic acid) and to 2,3-dihydroxybenzoic and 2,3,5-trihydroxybenzoic acids; gentisuric acid, the glycine conjugate of gentisic acid, also is formed. Salicylates are excreted in the urine as free salicylic acid (10%), salicyluric acid (75%), salicylic phenolic (10%) and acyl glucuronides (5%), and gentisic acid (<1%). However, excretion of free salicylate is extremely variable and depends upon both the dose and the urinary pH. In alkaline urine, more than 30% of the ingested drug may be eliminated as free salicylate, whereas in acidic urine this may be as low as 2%. The plasma half-life for aspirin is approximately 15 minutes; that for salicylate is 2 to 3 hours in low doses and about 12 hours at usual antiinflammatory doses. The half-life of salicylate may be as long as 15 to 30 hours at high therapeutic doses or when there is intoxication. Thus, small increases in dose can result in disproportionate increases in plasma levels of salicylate. Failure to recognize this phenomenon can lead to salicylate toxicity. This dose-dependent elimination is the result of the limited ability of the liver to form salicyluric acid and the phenolic glucuronide, and a larger proportion of unchanged drug is excreted in the urine at higher doses. Aspirin is one of the NSAIDs for which plasma level determinations can provide a means to monitor therapy and toxicity. The plasma concentration of salicylate is increased by conditions that decrease glomerular filtration rate or reduce its secretion by the proximal tubule, such as renal disease or the presence of inhibitors that compete for the transport system (e.g., probenecid). Changes in urinary pH also have significant effects on salicylate excretion; for example, the clearance of salicylate is about four times as great at pH 8.0 as at pH 6.0, and it is well above the glomerular filtration rate at pH 8.0. High rates of urine flow decrease tubular reabsorption, whereas the opposite is true in oliguria. The conjugates of salicylic acid with glycine and glucuronic acid do not readily back diffuse across the renal tubular cells. Their excretion, therefore, is both by glomerular filtration and proximal tubular secretion and is not pH dependent. Diflunisal, a difluorophenyl derivative of salicylic acid (see Figure 271) is almost completely absorbed after oral administration, and peak concentrations occur in plasma within 2 to 3 hours. It is extensively bound to plasma albumin (99%). Diflunisal appears in the milk of lactating women. About 90% of the drug is excreted as glucuronides, and its rate of elimination is dependent upon dosage. At the usual analgesic dose (500 to 750 mg per day) the plasma half-life ranges between 8 and 12 hours. (For reviews, see Davies, 1983; van Winzum et al., in Symposium, 1983a.) Therapeutic Uses There are many systemic and a few local uses of the salicylates. Several are based on tradition and empirical results rather than on a clear understanding of the mechanism of therapeutic benefit. Salicylates are used commonly to treat inflammation in a wide variety of settings, including rheumatoid and other types of arthritis, musculoskeletal injury, and acute rheumatic fever. Therapy often is of a symptomatic nature in terms of alleviating fever, pain, and other signs of inflammation. Systemic Uses The two most commonly used preparations of salicylate for systemic effects are sodium salicylate and aspirin (acetylsalicylic acid). The dose of salicylate depends on the condition being treated. Other salicylates are available for systemic use. These include salsalate (salicylsalicylic acid;DISALCID, others), which is hydrolyzed to salicylic acid during and after absorption. Sodium thiosalicylate (injection), choline salicylate (oral liquid; ARTHROPAN), and magnesium salicylate (tablets; MAGAN, others) also are available. A combination of choline and magnesium salicylates (TRILISATE, others) also is available. Diflunisal is discussed below. Antipyresis Antipyretic therapy is reserved for patients in whom fever in itself may be deleterious and for those who experience considerable relief when a fever is lowered. Little is known about the relationship between fever and the acceleration of inflammatory or immune processes; it may at times be a protective physiological mechanism. The course of the patient's illness may be obscured by the relief of symptoms and the reduction of fever from the use of antipyretic drugs. The antipyretic dose of salicylate for adults is 325 to 650 mg orally every 4 hours; for children, 50 to 75 mg/kg per day is given in four to six divided doses, not to exceed a total daily dose of 3.6 g. The route of administration nearly always is oral; parenteral administration is rarely necessary. The rectal administration of aspirin suppositories may be necessary in infants or when oral medication is not retained. Analgesia Salicylate is valuable for the nonspecific relief of certain types of pain, for example, headache, arthritis, dysmenorrhea, neuralgia, and myalgia. For this purpose, it is prescribed in the same doses and manner as for antipyresis. Rheumatoid Arthritis Although aspirin is regarded as the standard with which other drugs should be compared for the treatment of rheumatoid arthritis, many clinicians favor the use of drugs other than aspirin because of a lower incidence of side effects, in particular gastrointestinal effects. In addition to the analgesia that allows more effective therapeutic exercises, there is improvement in appetite, a feeling of well-being, and a reduction in the inflammation in joint tissues and surrounding structures. Damage to joints is the most difficult aspect of rheumatoid arthritis to manage, and any agent that reduces the inflammation is important in lessening or delaying the development of crippling diseases. Salicylates and other NSAIDs can be shown to produce objectively measurable antiinflammatory changes when given in large doses for long periods to patients with active rheumatoid disease. Large doses of salicylates, such as those used for rheumatic fever (4 to 6 g daily), are advised, but some patients respond well to less. The majority of patients with rheumatoid arthritis can be controlled
with salicylates alone or with other NSAIDs. Some patients with progressive
or resistant disease require therapy with more toxic drugs, sometimes termed second-line
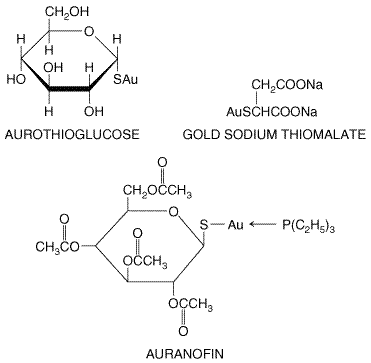
drugs, such as gold salts, hydroxychloroquine, penicillamine,
glucocorticoids, or immunosuppressive agents, in particular methotrexate. In
the Other Uses Because of the potent and long-lasting effect of low doses of aspirin on platelet function, this drug is used in the treatment or prophylaxis of diseases associated with platelet hyperaggregability, such as coronary artery disease and postoperative deep-vein thrombosis (see Patrono, 1994 and Chapter 55: Anticoagulant, Thrombolytic, and Antiplatelet Drugs). The maximal effectiveness of such therapy appears to depend upon selective blockade of TXA2 synthesis by platelets without preventing production of PGI2 by endothelial cells (see Chapters 26: Lipid-Derived Autacoids: Eicosanoids and Platelet-Activating Factor and 55: Anticoagulant, Thrombolytic, and Antiplatelet Drugs). Although the optimal dosage to prevent thrombotic events has not been firmly established, selective antiplatelet action appears to be best achieved when the dose of aspirin is 40 to 80 mg per day, while higher doses also inhibit PGI2 production. A relative excess of TXA2 over PGI2 has been implicated in the genesis of preeclampsia and hypertension induced by pregnancy (see Lubbe, 1987). The administration of 60 to 100 mg of aspirin per day to pregnant women who have a high risk of developing hypertension lowers the incidence of hypertension and also may prevent preeclampsia in patients with higher blood pressure (Imperiale and Petrulis, 1991; Sibai et al., 1993). Relationship of Plasma Salicylate Concentration to Therapeutic Effect and Toxicity For optimal antiinflammatory effect for patients with rheumatic
diseases, plasma salicylate concentrations of 150 to 300 The plasma concentration of salicylate generally is little affected by other drugs, but concurrent administration of aspirin lowers the concentrations of indomethacin, naproxen, ketoprofen, and fenoprofen, at least in part by displacement from plasma proteins. Important adverse interactions of aspirin with warfarin and methotrexate are mentioned above. Other interactions of aspirin include the antagonism of spironolactone-induced natriuresis and the blockade of the active transport of penicillin from CSF to blood. Local Uses Inflammatory Bowel Disease Mesalamine (5-aminosalicylic acid) is a salicylate that is used for its local effects in the treatment of inflammatory bowel disease. The drug is not effective orally because it is poorly absorbed and is inactivated before reaching the lower intestine. It is currently available as a suppository and rectal suspension enema (ROWASA) for treatment of mild-to-moderate proctosigmoiditis; two oral formulations that deliver drug to the lower intestine, olsalazine (sodium azodisalicylate, a dimer of 5-aminosalicylate linked by an azo bond; DIPENTUM) and mesalamine formulated in a pH-sensitive polymer-coated oral preparation (ASACOL), have been efficacious in treatment of inflammatory bowel disease, in particular ulcerative colitis. Sulfasalazine (salicylazosulfapyridine; AZULFIDINE) contains mesalamine linked covalently to sulfapyridine (see Chapter 44: Antimicrobial Agents: Sulfonamides, Trimethoprim-Sulfamethoxazole, Quinolones, and Agents for Urinary Tract Infections; Figure 271); it is poorly absorbed after oral administration, but it is cleaved to its active components by bacteria in the colon. The drug is of benefit in the treatment of inflammatory bowel disease, principally because of the local actions of mesalamine. Sulfasalazine and more recently olsalazine also have been used in the treatment of rheumatoid arthritis and ankylosing spondylitis (see Symposium, 1988b; Felson et al., 1992). Toxic Effects of Salicylates As a result of their wide use and ready availability, salicylates frequently are the cause of intoxication. Poisoning or serious intoxication often occurs in children and is sometimes fatal. The drugs should not be viewed as harmless household remedies. Salicylate Intoxication The fatal dose varies with the preparation of salicylate. From 10 to 30 g of sodium salicylate or aspirin has caused death in adults, but much larger amounts (130 g of aspirin, in one case) have been ingested without fatal outcome. The lethal dose of methyl salicylate (oil of wintergreen, sweet birch oil, gautheria oil, betula oil) is considerably less than that of sodium salicylate. As little as 4 ml (4.7 g) of methyl salicylate may be fatal in children. Symptoms and Signs Mild chronic salicylate intoxication is termed salicylism. When
fully developed, the syndrome includes headache, dizziness, ringing in the
ears, difficulty in hearing, dimness of vision, mental confusion, lassitude,
drowsiness, sweating, thirst, hyperventilation, nausea, vomiting, and
occasionally diarrhea. A more severe degree of salicylate intoxication is
characterized by more pronounced CNS disturbances (including generalized
convulsions and coma), skin eruptions, and marked alterations in acidbase
balance. Fever is usually prominent, especially in children. Dehydration
often occurs as a result of hyperpyrexia, sweating, vomiting, and the loss of
water vapor during hyperventilation. Gastrointestinal symptoms often are
present; about 50% of individuals with plasma salicylate concentrations of
more than 300 A prominent feature of salicylate intoxication is the disturbance in acidbase balance and electrolyte composition of the plasma described above. The most severe metabolic disturbances, in particular acidosis, occur in infants and very young children who become intoxicated. Hemorrhagic phenomena occasionally are seen during salicylate poisoning, the mechanism and significance of which have been discussed. Petechial hemorrhages are a prominent postmortem feature. Thrombocytopenic purpura is a rare complication. While hyperglycemia may occur during salicylate intoxication, hypoglycemia may be a serious consequence of toxicity in young children. It should be seriously considered in any young child with coma, convulsions, or cardiovascular collapse. Severe toxic encephalopathy may be a prominent feature of salicylate poisoning and may be difficult to differentiate from rheumatic encephalopathy. As poisoning progresses, central stimulation is replaced by increasing depression, stupor, and coma. Cardiovascular collapse and respiratory insufficiency ensue, and terminal asphyxial convulsions and pulmonary edema sometimes appear. Death usually results from respiratory failure after a period of unconsciousness. Salicylate toxicity in adults may not be diagnosed readily, because such patients usually become intoxicated from their therapeutic regimen; there is no history of acute overdosage. Prominent features of toxicity in this group are noncardiogenic pulmonary edema, nonfocal neurological abnormalities, and laboratory findings that include acidbase abnormalities, unexplained ketosis, and a prolonged prothrombin time. Symptoms of poisoning by methyl salicylate differ little from those described for aspirin. Central excitation, intense hyperpnea, and hyperpyrexia are prominent features. The odor of the drug can easily be detected on the breath and in the urine and vomitus. Poisoning by salicylic acid differs only in the increased prominence of gastrointestinal symptoms due to the marked local irritation. Treatment Salicylate poisoning represents an acute medical emergency, and death may result despite all recommended procedures. The treatment is directed at cardiovascular and respiratory support and correction of acidbase abnormalities plus use of measures to accelerate excretion of salicylate. Salicylate medication is withdrawn as soon as intoxication is suspected. Blood should be obtained for plasma salicylate determinations and acidbase and electrolyte studies. The salicylate concentration is reasonably well correlated with clinical severity, when corrected for the duration of the intoxication, and is of value in assessing the type of therapy to be instituted. Since absorption of salicylate from the gastrointestinal tract may be delayed for many hours after an overdose, measures to reduce such absorption always should be employed. Use of activated charcoal is the currently preferred method to accomplish this. Hyperthermia and dehydration are the immediate threats to life, and the initial therapy must be directed to their correction and to the maintenance of adequate renal function. Adequate amounts of intravenous fluids must be given promptly. The type and amount of solutions to be employed depend upon the interpretation of the laboratory data on acidbase balance. If the patient presents with an acidosis, correction of the low blood pH is essential, especially since acidosis results in a shift of salicylate from plasma into brain and other tissues. Bicarbonate solution should be infused intravenously in sufficient quantity to maintain alkaline diuresis. Correction of ketosis and hypoglycemia by administration of glucose also is essential for complete control of the metabolic acidosis; however, the ketosis clears only slowly. If K+ deficiency occurs during salicylate intoxication, it should be treated by adding the cation to the intravenous fluids once it has been determined that urine formation is adequate. Plasma transfusion may be beneficial, especially if shock intervenes. Hemorrhagic phenomena may necessitate blood transfusion and vitamin K (phytonadione). Measures to rid the body of salicylate rapidly should be undertaken
immediately. Forced diuresis with alkalinizing solution appears not to be
better than alkali alone. In severe intoxication, hemodialysis is the most
effective measure available for the removal of salicylate and for the
correction of the electrolyte and acidbase disturbances. Hemodialysis should
be considered in patients with salicylate concentrations above 1000 Aspirin Hypersensitivity Aspirin hypersensitivity or intolerance is discussed above. It is important to recognize this syndrome even though it is rather uncommon, since the administration of aspirin and many other NSAIDs may result in severe and possibly fatal reactions. The nonacetylated salicylates appear to be considerably less apt to produce these reactions than are aspirin and other NSAIDs. Treatment of such responses does not differ from that ordinarily employed in acute anaphylactic reactions; epinephrine is the drug of choice. Diflunisal Diflunisal (DOLOBID) is a difluorophenyl derivative of salicylic acid (see Figure 271); it is not converted to salicylic acid in vivo. Diflunisal is more potent than aspirin in antiinflammatory tests in animals and appears to be a competitive inhibitor of cyclooxygenase. However, it is largely devoid of antipyretic effects, perhaps because of poor penetration into the CNS. The drug has been used primarily as an analgesic in the treatment of osteoarthritis and musculoskeletal strains or sprains; in these circumstances it is about three to four times more potent than aspirin. The usual initial dose is 500 to 1000 mg, followed by 250 to 500 mg every 8 to 12 hours. For rheumatoid arthritis or osteoarthritis, 250 to 500 mg is administered twice daily; maintenance dosage should not exceed 1.5 g per day. Diflunisal does not produce auditory side effects and appears to cause fewer and less intense gastrointestinal and antiplatelet effects than does aspirin. |
Para-Aminophenol Derivatives: Acetaminophen
|
Acetaminophen (paracetamol; N-acetyl-p-aminophenol; TYLENOL, others) is the active metabolite of phenacetin, a so-called coal tar analgesic. Acetaminophen is an effective alternative to aspirin as an analgesic-antipyretic agent; however, unlike aspirin, its antiinflammatory activity is weak and thus it is not a useful agent to treat inflammatory conditions. Because acetaminophen is well tolerated, lacks many of the side effects of aspirin, and is available without prescription, it has earned a prominent place as a common household analgesic. However, acute overdosage causes fatal hepatic damage, and the number of self-poisonings and suicides with acetaminophen has grown alarmingly in recent years. In addition, many individuals, physicians included, seem unaware of the poor antiinflammatory activity of acetaminophen. History Acetanilide is the parent member of this group of drugs. It was introduced
into medicine in 1886 under the name of antifebrin by Cahn and Hepp, who had
accidentally discovered its antipyretic action. However, acetanilide proved
to be excessively toxic. In the search for less toxic compounds,
para-aminophenol was tried in the belief that the body oxidized acetanilide
to this compound. Toxicity was not lessened, however, and a number of
chemical derivatives of para-aminophenol were then tested. One of the more
satisfactory of these was phenacetin (acetophenetidin). It was introduced
into therapy in 1887 and was extensively employed in analgesic mixtures until
it was implicated in analgesic-abuse nephropathy (see above).
Phenacetin no longer is available in the Acetaminophen was first used in medicine by von Mering in 1893. However, it has gained popularity only since 1949, after it was recognized as the major active metabolite of both acetanilide and phenacetin. Pharmacological Properties Acetaminophen has analgesic and antipyretic effects that do not differ significantly from those of aspirin. However, as mentioned, it has only weak antiinflammatory effects. Minor metabolites contribute significantly to the toxic effects of acetaminophen. The pharmacological properties of acetaminophen have been reviewed by Clissold (1986). The failure of acetaminophen to exert antiinflammatory activity may be attributed to the fact that acetaminophen is only a weak inhibitor of cyclooxygenase in the presence of the high concentrations of peroxides that are found in inflammatory lesions. In contrast, its antipyretic effect may be explained by its ability to inhibit cyclooxygenase in the brain, where peroxide tone is low (Marshall et al., 1987; Hanel and Lands, 1982). Further, acetaminophen does not inhibit neutrophil activation as do other NSAIDs (Abramson and Weissmann, 1989). Single or repeated therapeutic doses of acetaminophen have no effect on the cardiovascular and respiratory systems. Acidbase changes do not occur, nor does the drug produce the gastric irritation, erosion, or bleeding that may occur after administration of salicylates. Acetaminophen has no effects on platelets, bleeding time, or the excretion of uric acid. Pharmacokinetics and Metabolism Acetaminophen is rapidly and almost completely absorbed from the gastrointestinal tract. The concentration in plasma reaches a peak in 30 to 60 minutes, and the half-life in plasma is about 2 hours after therapeutic doses. Acetaminophen is relatively uniformly distributed throughout most body fluids. Binding of the drug to plasma proteins is variable; only 20% to 50% may be bound at the concentrations encountered during acute intoxication. After therapeutic doses, 90% to 100% of the drug may be recovered in the urine within the first day, primarily after hepatic conjugation with glucuronic acid (about 60%), sulfuric acid (about 35%), or cysteine (about 3%); small amounts of hydroxylated and deacetylated metabolites also have been detected. Children have less capacity for glucuronidation of the drug than do adults. A small proportion of acetaminophen undergoes cytochrome P450mediated N-hydroxylation to form N-acetyl-benzoquinoneimine, a highly reactive intermediate. This metabolite normally reacts with sulfhydryl groups in glutathione. However, after ingestion of large doses of acetaminophen, the metabolite is formed in amounts sufficient to deplete hepatic glutathione (see below). Therapeutic Uses Acetaminophen is a suitable substitute for aspirin for analgesic or antipyretic uses; it is particularly valuable for patients in whom aspirin is contraindicated (e.g., those with peptic ulcer) or when the prolongation of bleeding time caused by aspirin would be a disadvantage. The conventional oral dose of acetaminophen is 325 to 1000 mg (650 mg rectally); the total daily dose should not exceed 4000 mg. For children, the single dose is 40 to 480 mg, depending upon age and weight; no more than five doses should be administered in 24 hours. A dose of 10 mg/kg also may be used. Toxic Effects In recommended therapeutic dosage, acetaminophen usually is well tolerated. Skin rash and other allergic reactions occur occasionally. The rash is usually erythematous or urticarial, but sometimes it is more serious and may be accompanied by drug fever and mucosal lesions. Patients who show hypersensitivity reactions to the salicylates only rarely exhibit sensitivity to acetaminophen. In a few isolated cases, the use of acetaminophen has been associated with neutropenia, thrombocytopenia, and pancytopenia. The most serious adverse effect of acute overdosage of acetaminophen is a dose-dependent, potentially fatal hepatic necrosis (see Thomas, 1993). Renal tubular necrosis and hypoglycemic coma also may occur. The mechanism by which overdosage with acetaminophen leads to hepatocellular injury and death involves its conversion to a toxic reactive metabolite (see also Chapter 4: Principles of Toxicology and Treatment of Poisoning). Minor pathways of acetaminophen elimination are via conjugation with glucuronide and sulfate. The major pathway of metabolism is via cytochrome P450s to the intermediate, N-acetyl-para-benzoquinonimine, which is very electrophilic. Under normal circumstances, this intermediate is eliminated by conjugation with glutathione (GSH) and then further metabolized to a mercapturic acid and excreted into the urine. However, in the setting of acetaminophen overdose, hepatocellular levels of GSH become depleted. Two consequences ensue as result of depletion of GSH. Since GSH is an important factor in antioxidant defense, hepatocytes are rendered highly susceptible to oxidant injury. Depletion of GSH also allows the reactive intermediate to bind covalently to cell macromolecules, leading to dysfunction of enzymatic systems. Hepatotoxicity In adults, hepatotoxicity may occur after ingestion of a single dose of 10 to 15 g (150 to 250 mg/kg) of acetaminophen; doses of 20 to 25 g or more are potentially fatal. Alcoholics can have hepatotoxicity with much lower doses, even with doses in the therapeutic range. The mechanism of this effect is discussed above (see also Chapter 4: Principles of Toxicology and Treatment of Poisoning). Symptoms that occur during the first 2 days of acute poisoning by acetaminophen may not reflect the potential seriousness of the intoxication. Nausea, vomiting, anorexia, diaphoresis, and abdominal pain occur during the initial 24 hours and may persist for a week or more. Clinical indications of hepatic damage become manifest within 2 to 4 days of ingestion of toxic doses. Plasma aminotransferases are elevated (sometimes markedly so), and the concentration of bilirubin in plasma may be increased; in addition, the prothrombin time is prolonged. Perhaps 10% of poisoned patients who do not receive specific treatment develop severe liver damage; of these, 10% to 20% eventually die of hepatic failure. Acute renal failure also occurs in some patients. Biopsy of the liver reveals centrilobular necrosis with sparing of the periportal area. In nonfatal cases, the hepatic lesions are reversible over a period of weeks or months. Severe liver damage (with levels of aspartate aminotransferase
activity in excess of 1000 IU per liter of plasma) occurs in 90% of patients
with plasma concentrations of acetaminophen greater than 300
Early diagnosis is vital in the treatment of overdosage with acetaminophen, and methods are available for the rapid determination of concentrations of the drug in plasma. However, therapy should not be delayed while awaiting laboratory results if the history suggests a significant overdosage. Vigorous supportive therapy is essential when intoxication is severe. Gastric lavage should be performed in all cases, preferably within 4 hours of the ingestion. The principal antidotal treatment is the administration of sulfhydryl
compounds, which probably act, in part, by replenishing hepatic stores of
glutathione. N-acetylcysteine (MUCOMYST MUCOSIL) is effective when given orally
or intravenously. An intravenous form is available in |
Indomethacin, Sulindac, and Etodolac
|
Indomethacin was the product of a laboratory
search for drugs with antiinflammatory properties. It was introduced in 1963
for the treatment of rheumatoid arthritis and related disorders. Although
indomethacin is used widely and is effective, toxicity often limits its use. Sulindac
was developed in an attempt to find a less toxic but effective congener of
indomethacin. The development, chemistry, and pharmacology of both drugs have
been reviewed by Rhymer and Gengos (in Symposium, 1983a) and by Shen (in
Rainsford, 1985a). Etodolac is one of the more recent antiinflammatory
drugs approved for use in the Indomethacin Chemistry The structural formula of indomethacin, a methylated indole derivative, is shown below:
Pharmacological Properties Indomethacin has prominent antiinflammatory and analgesic-antipyretic properties similar to those of the salicylates. The antiinflammatory effects of indomethacin are evident in patients with rheumatoid and other types of arthritis, including acute gout. Although indomethacin is more potent than aspirin, doses that are tolerated by patients with rheumatoid arthritis usually do not produce effects that are superior to those of salicylate. Indomethacin has analgesic properties distinct from its antiinflammatory effects, and there is evidence for both a central and a peripheral action; it also is an antipyretic. Indomethacin is a potent inhibitor of the cyclooxygenases; it also inhibits the motility of polymorphonuclear leukocytes. Like many other NSAIDs, indomethacin uncouples oxidative phosphorylation at supratherapeutic concentrations and depresses the biosynthesis of mucopolysaccharides. Pharmacokinetics and Metabolism Indomethacin is rapidly and almost completely absorbed from the
gastrointestinal tract after oral ingestion. The peak concentration in plasma
is attained within 1 to 2 hours in the fasting subject but may be somewhat
delayed when the drug is taken after meals. The concentrations in plasma
required for an antiinflammatory effect have not been definitely determined
but are probably less than 1 Indomethacin is converted primarily to inactive metabolites, including those formed by O-demethylation (about 50%), conjugation with glucuronic acid (about 10%), and N-deacylation. Some of these metabolites are detectable in plasma, and free and conjugated metabolites are eliminated in the urine, bile, and feces. There is enterohepatic cycling of the conjugates and probably of indomethacin itself. Between 10% and 20% of the drug is excreted unchanged in the urine, in part by tubular secretion. The half-life in plasma is variable, perhaps because of enterohepatic cycling, but averages about 2.5 hours. Drug Interactions The total plasma concentration of indomethacin plus its inactive
metabolites is increased by concurrent administration of probenecid, possibly
because of reduced tubular secretion of the former. However, it has not been
determined whether or not the dosage of indomethacin must be adjusted when
the two drugs are employed together. Indomethacin does not interfere with the
uricosuric effect of probenecid. Indomethacin is said not to modify the
effect of the oral anticoagulant agents. However, concurrent administration
could be hazardous because of the increased risk of gastrointestinal
bleeding. Indomethacin antagonizes the natriuretic and antihypertensive
effects of furosemide; the antihypertensive effects of thiazide diuretics, Therapeutic Uses Because of the high incidence and severity of side effects associated with long-term administration, indomethacin (INDOCIN) is not commonly used for therapy as an analgesic or antipyretic. However, it has proven to be useful as an antipyretic in certain settings (e.g., Hodgkin's disease) when the fever has been refractory to other agents. Clinical trials of indomethacin as an antiinflammatory agent have been reviewed by Rhymer and Gengos (in Symposium, 1983a). The majority of these trials have demonstrated that indomethacin relieves pain, reduces swelling and tenderness of the joints, increases grip strength, and decreases the duration of morning stiffness. In these actions, the drug is superior to placebo, and estimates of its potency relative to salicylates vary between 10 and 40 times higher. Overall, about two-thirds of patients benefit from treatment with indomethacin, typically with treatment initiated at 25 mg two or three times daily. However, if 75 to 100 mg of the drug fails to provide benefit within 2 to 4 weeks, alternative therapy must be considered. The incidence and severity of side effects with indomethacin can limit its therapeutic utility; however, since the side effects of indomethacin appear to be better tolerated when taken at night, a useful way to take advantage of the effectiveness of indomethacin and to minimize untoward side effects is to give a large single dose (up to 100 mg) at bedtime, perhaps in combination with other and better-tolerated NSAIDs for daytime therapy. This enables the patient to obtain a better-quality sleep, reduces the severity and length of morning stiffness, and provides good analgesia until midmorning. Indomethacin often is more effective than aspirin in the treatment of ankylosing spondylitis and osteoarthrosis. It also is very effective in the treatment of acute gout, although it is not uricosuric. Patients with Bartter's syndrome have been treated successfully with indomethacin, as well as with other inhibitors of prostaglandin synthesis. The results are frequently dramatic; however, the condition of the patients may deteriorate rapidly when therapy is discontinued, and the long-term therapy necessary to control the disease requires administration of a drug that is better tolerated. Indomethacin has at least two uses in obstetrics and neonatal medicine. It can be used as a tocolytic agent to suppress uterine contractions in women with preterm labor. In addition, cardiac failure in neonates caused by a patent ductus arteriosus may be controlled by the administration of indomethacin. A typical regimen involves the intravenous administration of 0.1 to 0.2 mg/kg every 12 hours for three doses. Successful closure can be expected in more than 70% of neonates who are treated with the drug. Such therapy is indicated primarily in premature infants who weigh between 500 and 1750 g, who have a hemodynamically significant patent ductus arteriosus, and in whom other supportive maneuvers have been attempted. Unexpectedly, treatment with indomethacin also may decrease the incidence and severity of intraventricular hemorrhage in low birth weight neonates (Ment et al., 1994). The principal limitation of treating neonates is renal toxicity, and therapy is stopped if the output of urine falls below 0.6 ml/kg per hour. Renal failure, enterocolitis, thrombocytopenia, or hyperbilirubinemia contraindicates the use of indomethacin. Toxic Effects A very high percentage (35% to 50%) of patients receiving usual therapeutic doses of indomethacin experience untoward symptoms, and about 20% must discontinue its use. Most adverse effects are dose related. Gastrointestinal complaints and complications consist of anorexia, nausea, and abdominal pain. Single ulcers or multiple ulceration of the entire upper gastrointestinal tract, sometimes with perforations and hemorrhage, have been reported. Occult blood loss may lead to anemia in the absence of ulceration. Acute pancreatitis also has been reported. Diarrhea may occur and is sometimes associated with ulcerative lesions of the bowel. Hepatic involvement is rare, although some fatal cases of hepatitis and jaundice have been reported. The most frequent CNS effect (indeed, the most common side effect) is severe frontal headache, occurring in 25% to 50% of patients who take the drug for long periods. Dizziness, vertigo, light-headedness, and mental confusion also are frequent. Severe depression, psychosis, hallucinations, and suicide have occurred. Hematopoietic reactions include neutropenia, thrombocytopenia, and, rarely, aplastic anemia. As is common with other nonselective inhibitors of the cyclooxygenases, platelet function is impaired, and reactions predictably occur in patients who exhibit hypersensitivity reactions to aspirin. Indomethacin should not be used in pregnant women, nursing mothers, persons operating machinery, or patients with psychiatric disorders, epilepsy, or Parkinsonism. Indomethacin also is contraindicated in individuals with renal disease or ulcerative lesions of the stomach or intestines. Sulindac Chemistry Sulindac is closely related to indomethacin; its structural formula is as follows:
Sulindac is essentially a prodrug. Little if any of the antiinflammatory activity is due to the parent drug, sulindac sulfoxide; most of its pharmacological activity resides in its sulfide metabolite. Pharmacological Properties Sulindac exhibits the classical activities of NSAIDs. In all tests, sulindac is less than half as potent as indomethacin. Because sulindac is a prodrug, it appears to be either inactive or relatively weak in in vitro tests because it is not metabolized to its active sulfide metabolite. The sulfide metabolite is more than 500 times more potent than sulindac as an inhibitor of cyclooxygenase. These observations may help to explain the somewhat lower incidence of gastrointestinal toxicity of sulindac as compared with indomethacin, since the gastric or intestinal mucosa is not exposed to high concentrations of an active drug during oral administration. Nevertheless, gastrointestinal toxicity is more common with sulindac than with many other NSAIDs. Sulindac also may be unusual in that some clinical studies indicate that it does not alter the urinary excretion of prostaglandins or alter renal function, perhaps because of the kidney's ability to regenerate the parent sulfoxide from active sulfide metabolites (see Wilson and Carruthers in Borda and Koff, 1992). However, 'renal-sparing' is apparently only relative and dose dependent (Waslen et al., 1989; Kulling et al., 1995); the drug therefore must be used with caution in patients who are dependent upon the synthesis of prostaglandins in the kidney for maintenance of renal function. Pharmacokinetics and Metabolism The metabolism and pharmacokinetics of sulindac are complex and vary enormously among species. After oral administration in human beings, about 90% of the drug is absorbed. Peak concentrations of sulindac in plasma are attained within 1 to 2 hours, while those of the sulfide metabolite occur about 8 hours after the oral administration of sulindac. Sulindac undergoes two major biotransformations in addition to conjugation reactions. It is oxidized to the sulfone and then reversibly reduced to the sulfide. It is this latter metabolite that is the active moiety, although all three compounds are found in comparable concentrations in human plasma. The half-life of sulindac itself is about 7 hours, but the active sulfide has a half-life of as long as 18 hours. Sulindac and its metabolites undergo extensive enterohepatic circulation. Sulindac and the sulfone and sulfide metabolites are all extensively bound to plasma protein. Little of the sulfide or its conjugates is found in urine. The principal components that are excreted in the urine are the sulfone and its conjugate, which account for nearly 30% of an administered dose; sulindac and its conjugates account for about 20%. Up to 25% of an oral dose may appear as metabolites in the feces. Therapeutic Uses Sulindac CLINORIL) has been used mainly for the treatment of rheumatoid arthritis, osteoarthrosis, and ankylosing spondylitis. The drug also has been used with success in the treatment of acute gout. The analgesic and antiinflammatory effects exerted by sulindac (400 mg per day) are comparable to those achieved with aspirin (4 g per day), ibuprofen (1200 mg per day), and indomethacin (125 mg per day) (see Rhymer, in Symposium, 1983a). Although dosage should be optimized for each individual, the most common dosage for adults is 150 to 200 mg twice a day. The drug usually is given with food to reduce gastric discomfort, although this may delay absorption and reduce concentration in plasma. Sulindac, like indomethacin, has been used for tocolytic therapy. A novel use of sulindac is as treatment to reduce the number and size of adenomas in the large bowel in patients with familial adenomatous polyposis (Giardiello et al., 1993). Its effectiveness in this situation may be attributed in part to the fact that reduction of sulindac to its active sulfide is mediated primarily by microflora in the gut acting on sulindac excreted in the bile (Strong et al., 1985). Toxic Effects Although the incidence of toxicity is lower than with indomethacin, untoward reactions to sulindac are common. Gastrointestinal side effects are seen in nearly 20% of patients, although these are generally mild. Abdominal pain and nausea are the most frequent complaints. CNS side effects are seen in up to 10% of patients, with drowsiness, dizziness, headache, and nervousness being those most frequently reported. Skin rash and pruritus occur in 5% of patients. Transient elevations of hepatic enzymes in plasma are less common. Etodolac Etodolac is an inhibitor of cyclooxygenase and possesses antiinflammatory activity. However, there is an unusually large difference between doses that produce antiinflammatory effects and those that cause gastric irritation in experimental animals. This can be explained by the fact that etodolac has been shown to be a selective inhibitor of COX-2 (Warner et al., 1999). This selectivity also explains the findings that chronic treatment of human beings with etodolac does not reduce gastric mucosal prostaglandin production, and the incidence of gastric toxicity is not different from placebo and is much less than seen with naproxen (Laine et al., 1995). The drug appears to be uricosuric. The structure of etodolac is as follows:
Pharmacokinetics and Metabolism Etodolac is rapidly and well absorbed orally, and it is about 99% bound to plasma proteins. It is actively metabolized by the liver to various metabolites that are largely excreted in the urine. The drug may undergo enterohepatic circulation in human beings; its half-life in plasma is about 7 hours. Therapeutic Uses A single oral dose (200 to 400 mg) of etodolac (LODINE) provides postoperative analgesia that typically lasts for 6 to 8 hours. Etodolac also is effective in the treatment of osteoarthritis and rheumatoid arthritis. A sustained-release preparation (LODINE XL) is available, allowing once-a-day administration. Toxic Effects As mentioned above, gastric toxicity is much lower than with nonselective COX inhibitors. About 5% of patients who have taken the drug for up to 1 year discontinue treatment because of side effects, which include skin rashes and CNS effects. |
The Fenamates
|
The fenamates are a family of NSAIDs that are derivatives of N-phenylanthranilic acid. They include mefenamic, meclofenamic, and flufenamic acids. Although the biological activity of this group of drugs was discovered in the 1950s, the fenamates have not gained widespread clinical acceptance. Therapeutically, they have no clear advantages over several other NSAIDs and frequently cause side effects, such as diarrhea. As an analgesic agent, mefenamic acid (PONSTEL) has been used to relieve pain arising from rheumatic conditions, soft tissue injuries, other painful musculoskeletal conditions, and dysmenorrhea. Toxicity limits its usefulness, and it appears to offer no advantage over other analgesic agents. As antiinflammatory agents, mefenamic acid and meclofenamate sodium have been tested mainly in short-term trials in the treatment of osteoarthritis and rheumatoid arthritis and appear to offer no advantage over other NSAIDs. These drugs are not recommended for use in children or pregnant women. Mefenamic acid and meclofenamate are the only members of the series
available in the Chemistry Mefenamic acid and meclofenamate are both N-substituted phenylanthranilic acids. Their structures are as follows:
Pharmacological Properties The fenamates have antiinflammatory, antipyretic, and analgesic properties. In tests of analgesia, mefenamic acid was the only fenamate to display a central as well as a peripheral action. The fenamates appear to owe these properties primarily to their capacity to inhibit cyclooxygenase. Unlike the other NSAIDs, some of the fenamates (especially meclofenamic acid) also may antagonize certain effects of prostaglandins. Pharmacokinetic Properties Peak concentrations in plasma are reached in 0.5 to 2 hours after a single oral dose of meclofenamate and in 2 to 4 hours for mefenamic acid. The two agents have similar half-lives in plasma (2 to 4 hours). In human beings, approximately 50% of a dose of mefenamic acid is excreted in the urine, primarily as the conjugated 3-hydroxymethyl metabolite and the 3-carboxyl metabolite and its conjugates. Twenty percent of the drug is recovered in the feces, mainly as the unconjugated 3-carboxyl metabolite. Toxic Effects and Precautions The most common side effects (occurring in approximately 25% of all patients) involve the gastrointestinal system. Usually these take the form of dyspepsia or upper gastrointestinal discomfort, although diarrhea, which may be severe and associated with steatorrhea and inflammation of the bowel, also is relatively common. A potentially serious side effect seen in isolated cases is a hemolytic anemia, which may be of an autoimmune type. The fenamates are contraindicated in patients with a history of gastrointestinal disease. If diarrhea or skin rash appears, the drug should be stopped at once. The physician and patient should watch for signs of hemolytic anemia. |
Tolmetin, Ketorolac, and Diclofenac
|
Tolmetin and ketorolac are structurally related heteroaryl acetic acid derivatives with different pharmacological features. Diclofenac is a phenylacetic acid derivative that was developed specifically as an antiinflammatory agent. Tolmetin Tolmetin is an antiinflammatory, analgesic, and antipyretic agent that
was introduced into clinical practice in the
Pharmacological Properties Tolmetin is an effective antiinflammatory agent that also exerts antipyretic and analgesic effects. Like most of the other drugs considered in this chapter, tolmetin causes gastric erosions and prolongs bleeding time. The pharmacology of tolmetin has been reviewed by Ehrlich (in Symposium, 1983a) and by Wong (in Rainsford, 1985b). Pharmacokinetics and Metabolism Tolmetin is rapidly and completely absorbed after oral administration. Peak concentrations are achieved 20 to 60 minutes after oral administration, and the half-life in plasma is about 5 hours. Accumulation of the drug in synovial fluid begins within 2 hours and persists for up to 8 hours after a single oral dose. After absorption, tolmetin is extensively (99%) bound to plasma proteins. Virtually all of the drug can be recovered in the urine after 24 hours; some is unchanged but most is conjugated or otherwise metabolized. The major metabolic transformation involves oxidation of the para-methyl group to a carboxylic acid. Therapeutic Uses Tolmetin (tolmetin sodium;TOLECTIN) is approved in the Toxic Effects Side effects occur in 25% to 40% of patients who take tolmetin, and 5% to 10% discontinue use of the drug. Gastrointestinal side effects are the most common, with epigastric pain (15% incidence), dyspepsia, nausea, and vomiting being the chief manifestations. Gastric and duodenal ulceration also have been observed. CNS side effects, including nervousness, anxiety, insomnia, drowsiness, and visual disturbance, are less common and are said to be neither as frequent nor as severe as those caused by indomethacin. Similarly, the incidence of tinnitus, deafness, and vertigo is less than with aspirin. Ketorolac Ketorolac is a potent analgesic but only a moderately effective antiinflammatory drug. It is one of the few NSAIDs approved for parenteral administration. The structure of ketorolac is given below:
Pharmacological Properties Ketorolac inhibits prostaglandin biosynthesis. It has antipyretic, antiinflammatory, and analgesic activity, but in assays of inflammation it has greater systemic analgesic than antiinflammatory activity. Unlike opioid agonists, ketorolac is not associated with tolerance, withdrawal effects, or respiratory depression. Ketorolac also has antiinflammatory activity when topically administered in the eye. Ketorolac inhibits platelet aggregation and promotes gastric ulceration. The pharmacology of ketorolac has been reviewed (Buckley and Brogden, 1990). Pharmacokinetics and Metabolism Ketorolac is rapidly absorbed whether given orally or intramuscularly, achieving peak plasma concentration in 30 to 50 minutes. Oral bioavailability is about 80%. Almost totally bound to plasma proteins, it is excreted with an elimination half-life of 4 to 6 hours. Urinary excretion accounts for about 90% of eliminated drug, with about 10% excreted unchanged and the remainder as a glucuronidated conjugate. The rate of elimination is reduced in the elderly and in patients with renal failure. Therapeutic Uses Ketorolac (administered as the tromethamine salt TORADOL) is used for postoperative pain, as an alternative to opioid agents, and is administered intramuscularly, intravenously, or orally. Typical intramuscular doses are 30 to 60 mg; intravenous doses are 15 to 30 mg; and oral doses are 5 to 30 mg. Ketorolac probably should not be used for obstetric analgesia. The drug is indicated only for short-term treatment of pain (no longer than 5 days) and should not be used for minor or chronic pain. Topical ketorolac may be useful for inflammatory conditions in the eye and is approved for the treatment of seasonal allergic conjunctivitis and ocular inflammation. Toxic Effects Side effects occur about twice as often with ketorolac as with placebo. These side effects include somnolence, dizziness, headache, gastrointestinal pain, dyspepsia and nausea, and pain at the site of injection. Diclofenac Diclofenac is an antiinflammatory agent approved for several uses in
the
Pharmacological Properties Diclofenac has analgesic, antipyretic, and antiinflammatory activities. It is an inhibitor of cyclooxygenase, and its potency is substantially greater than that of indomethacin, naproxen, or several other agents. In addition, diclofenac appears to reduce intracellular concentrations of free arachidonate in leukocytes, perhaps by altering the release or uptake of the fatty acid. Pharmacokinetics and Metabolism Diclofenac is rapidly and completely absorbed after oral administration; peak concentrations in plasma are reached within 2 to 3 hours. Administration with food slows the rate but does not alter the extent of absorption. There is a substantial first-pass effect, such that only about 50% of diclofenac is available systemically. The drug is extensively bound to plasma proteins (99%), and its half-life in plasma is 1 to 2 hours. Diclofenac accumulates in synovial fluid after oral administration, which may explain the duration of therapeutic effect that is considerably longer than the plasma half-life. Diclofenac is metabolized in the liver by a cytochrome P450 isozyme of the CYP2C subfamily to 4-hydroxydiclofenac, the principal metabolite, and other hydroxylated forms; after glucuronidation and sulfation, the metabolites are excreted in the urine (65%) and bile (35%). Therapeutic Uses Diclofenac sodium is approved in the Toxic Effects Diclofenac produces side effects in about 20% of patients, and approximately 2% of patients discontinue therapy as a result. Gastrointestinal effects are the most common; bleeding and ulceration or perforation of the intestinal wall have been observed. Elevation of hepatic aminotransferase activities in plasma occurs in about 15% of patients. Although usually moderate, these values may increase more than threefold in a small percentage of patientsoften those who are being treated for osteoarthritis. The elevations in aminotransferase usually are reversible. Another member of this phenylacetic acid family of NSAIDs, bromfenac, was withdrawn from the market because of its association with severe, irreversible liver injury in some patients. Therefore, aminotransferase activities should be evaluated during the first 8 weeks of therapy with diclofenac, and the drug should be discontinued if abnormal values persist or if other signs or symptoms develop. Other untoward responses to diclofenac include CNS effects, skin rashes, allergic reactions, fluid retention and edema, and rarely, impairment of renal function. The drug is not recommended for children, nursing mothers, or pregnant women. |
Propionic Acid Derivatives
|
Arylpropionic acid derivatives represent a group of effective, useful NSAIDs. They may offer significant advantages over aspirin and indomethacin for many patients, since they usually are better tolerated. Nevertheless, propionic acid derivatives share all of the detrimental features of the entire class of drugs. Furthermore, their rapid proliferation in number and the heavy promotion of these drugs make it difficult for the physician to choose rationally among members of the group and between propionic acid derivatives and the more established agents. The similarities among drugs in this class (and certain of the others discussed above) are far more striking than are the differences. The approved indications for the use of one or another of the propionic acid derivatives include the symptomatic treatment of rheumatoid arthritis, osteoarthritis, ankylosing spondylitis, and acute gouty arthritis; they also are used as analgesics, for acute tendinitis and bursitis, and for primary dysmenorrhea. Information regarding dosage forms and usual antiinflammatory doses is shown in Table 273. Clinical studies indicate that the propionic acid derivatives are comparable to aspirin for the control of the signs and symptoms of rheumatoid arthritis and osteoarthritis. In patients with rheumatoid arthritis, there is a reduction in joint swelling, pain, and duration of morning stiffness. By objective measurements, strength, mobility, and stamina are improved. In general, the intensity of untoward effects is less than that associated with the ingestion of indomethacin or high doses of aspirin. However, aspirin is less expensive than most of the propionic derivatives for those who can tolerate it. Ibuprofen, naproxen, flurbiprofen, fenoprofen, ketoprofen, and oxaprozin are
described individually below. These drugs currently are available in the Ibuprofen was the first member of the propionic acid class of NSAIDs
to come into general use, so experience with this drug is greater. It is
available for sale without a prescription in the
Pharmacological Properties The pharmacodynamic properties of the propionic acid derivatives do not differ significantly. All are effective cyclooxygenase inhibitors, although there is considerable variation in their potency. For example, naproxen is approximately 20 times more potent than aspirin, while ibuprofen, fenoprofen, and aspirin are roughly equipotent as cyclooxygenase inhibitors. All of these agents alter platelet function and prolong bleeding time, and it should be assumed that any patient who is intolerant of aspirin also will experience a severe reaction after administration of one of these drugs. Some of the propionic acid derivatives have prominent inhibitory effects on leukocyte function; naproxen is particularly potent in this regard. While the compounds do vary in potency, this is not of obvious clinical significance. All are effective antiinflammatory agents in various experimental animal models of inflammation; all have useful antiinflammatory, analgesic, and antipyretic activities in human beings. Although all of these compounds can cause gastric toxicity in patients, these are usually less severe than with aspirin. It is difficult to find data on which to base a rational choice among the members of the propionic acid derivatives, if in fact one can be made. However, in relatively small clinical studies that compared the activity of several members of this group, patients preferred naproxen in terms of analgesia and relief of morning stiffness (see Huskisson, in Symposium, 1983a; Hart and Huskisson, 1984). With regard to side effects, naproxen was the best tolerated, followed by ibuprofen and fenoprofen. There was considerable interpatient variation in the preference for a single drug and also between the designations of the best and the worst drug. Unfortunately, it is probably impossible to predict a priori which drug will be most suitable for any given individual. Nevertheless, more than 50% of patients with rheumatoid arthritis probably will achieve adequate symptomatic relief from the use of one or another of the propionic acid derivatives, and many clinicians favor their use instead of aspirin in such patients. Drug Interactions The potential adverse drug interactions of particular concern with propionic acid derivatives result from their high degree of binding to albumin in plasma. However, the propionic acid derivatives do not alter the effects of the oral hypoglycemic drugs or warfarin. Nevertheless, the physician should be prepared to adjust the dosage of warfarin because these drugs impair platelet function and may cause gastrointestinal lesions. Ibuprofen Ibuprofen is supplied as tablets containing 200 to 800 mg; only the 200-mg tablets (ADVIL, NUPRIN, others) are available without a prescription. For rheumatoid arthritis and osteoarthritis, daily doses of up to 3200 mg in divided portions may be given, although the usual total dose is 1200 to 1800 mg. It also may be possible to reduce the dosage for maintenance purposes. For mild-to-moderate pain, especially that of primary dysmenorrhea, the usual dosage is 400 mg every 4 to 6 hours as needed. The drug may be given with milk or food to minimize gastrointestinal side effects. Ibuprofen has been discussed in detail by Kantor (1979) and by Adams and Buckler (in Symposium, 1983a). Pharmacokinetics and Metabolism Ibuprofen is rapidly absorbed after oral administration, and peak concentrations in plasma are observed after 15 to 30 minutes. The half-life in plasma is about 2 hours. Ibuprofen is extensively (99%) bound to plasma proteins, but the drug occupies only a fraction of the total drug-binding sites at usual concentrations. Ibuprofen passes slowly into the synovial spaces and may remain there in higher concentration as the concentrations in plasma decline. In experimental animals, ibuprofen and its metabolites pass easily across the placenta. The excretion of ibuprofen is rapid and complete. More than 90% of an ingested dose is excreted in the urine as metabolites or their conjugates. The major metabolites are a hydroxylated and a carboxylated compound. Toxic Effects Ibuprofen has been used in patients with a history of gastrointestinal intolerance to other NSAIDs. Nevertheless, therapy usually must be discontinued in 10% to 15% of patients because of intolerance to the drug. Gastrointestinal side effects are experienced by 5% to 15% of patients taking ibuprofen; epigastric pain, nausea, heartburn, and sensations of 'fullness' in the gastrointestinal tract are the usual difficulties. However, the incidence of these side effects is less with ibuprofen than with aspirin or indomethacin. Other side effects of ibuprofen have been reported less frequently. They include thrombocytopenia, skin rashes, headache, dizziness and blurred vision, and, in a few cases, toxic amblyopia, fluid retention, and edema. Patients who develop ocular disturbances should discontinue the use of ibuprofen. Ibuprofen is not recommended for use by pregnant women, or by those who are breast-feeding their infants. Naproxen The pharmacological properties and therapeutic uses of naproxen (ALEVE, NAPROSYN, others) have been reviewed by Segre (in Symposium, 1983a), Allison and colleagues (in Rainsford, 1985b), and Todd and Clissold (1990). Pharmacokinetics and Metabolism Naproxen is fully absorbed when administered orally. The rapidity, but not the extent, of absorption is influenced by the presence of food in the stomach. Peak concentrations in plasma occur within 2 to 4 hours and are somewhat more rapid after the administration of naproxen sodium. Absorption may be accelerated by the concurrent administration of sodium bicarbonate or reduced by magnesium oxide or aluminum hydroxide. Naproxen also is absorbed rectally, but peak concentrations in plasma are achieved more slowly. The half-life of naproxen in plasma is about 14 hours; this value is increased about twofold in elderly subjects and may necessitate adjustment of dosage. Metabolites of naproxen are almost entirely excreted in the urine. About 30% of the drug undergoes 6-demethylation, and most of this metabolite, as well as naproxen itself, is excreted as the glucuronide or other conjugates. Naproxen is almost completely (99%) bound to plasma proteins following normal therapeutic doses. Naproxen crosses the placenta and appears in the milk of lactating women at approximately 1% of the maternal plasma concentration. Toxic Effects Although the incidence of gastrointestinal and CNS side effects is about equal to that caused by indomethacin, naproxen is better tolerated in both regards. Gastrointestinal complications have ranged from relatively mild dyspepsia, gastric discomfort, and heartburn to nausea, vomiting, and gastric bleeding. CNS side effects range from drowsiness, headache, dizziness, and sweating to fatigue, depression, and ototoxicity. Less common reactions include pruritus and a variety of dermatological problems. A few instances of jaundice, impairment of renal function, angioneurotic edema, thrombocytopenia, and agranulocytosis have been reported. Fenoprofen The pharmacological properties and therapeutic uses of fenoprofen (NALFON) have been reviewed by Burt and coworkers (Symposium, 1983a). Pharmacokinetics and Metabolism Oral doses of fenoprofen are readily, but incompletely (85%) absorbed. The presence of food in the stomach retards absorption and lowers peak concentrations in plasma, which are usually achieved within 2 hours. The concomitant administration of antacids does not seem to alter the concentrations that are achieved. After absorption, fenoprofen is almost completely (99%) bound to plasma albumin. The drug is extensively (>90%) metabolized and excreted almost entirely in the urine. Fenoprofen undergoes metabolic transformation to the 4-hydroxy analog. The glucuronic acid conjugate of fenoprofen itself and 4-hydroxy fenoprofen are formed in almost equal amounts and together account for 90% of the excreted drug. The half-life of fenoprofen in plasma is about 3 hours. Toxic Effects The most frequently reported side effects have been gastrointestinal ones; abdominal discomfort and dyspepsia occur in about 15% of patients. These side effects are almost always less intense than with equieffective doses of aspirin and force discontinuation of therapy in a small percentage of patients. Other side effects include skin rash and, less frequently, CNS effects such as tinnitus, dizziness, lassitude, confusion, and anorexia. Ketoprofen Ketoprofen ORUDIS ORUVAIL) shares the pharmacological properties of other propionic acid derivatives; these have been reviewed by Harris and Vvra (in Rainsford, 1985b) and Vvra (in Lewis and Furst, 1987). Although it is a cyclooxygenase inhibitor, ketoprofen is said to stabilize lysosomal membranes and may antagonize the actions of bradykinin. Pharmacokinetics and Metabolism Ketoprofen is rapidly absorbed after oral administration and maximal concentrations in plasma are achieved within 1 to 2 hours; food reduces the rate but not the extent of absorption. The drug is extensively bound to plasma proteins (99%), and it has a half-life in plasma of about 2 hours; slightly longer half-lives are observed in elderly subjects. Ketoprofen is conjugated with glucuronic acid in the liver, and the conjugate is excreted in the urine. Patients with impaired renal function eliminate the drug more slowly. Toxic Effects Dyspepsia and other gastrointestinal side effects have been observed in about 30% of patients, but these side effects are generally mild and are less frequent than those in patients treated with aspirin; untoward effects are reduced when the drug is taken with food, milk, or antacids. Ketoprofen can cause fluid retention and increased plasma concentrations of creatinine. These effects are generally transient and occur in the absence of symptoms, but they are more common in patients who are receiving diuretics or in those over the age of 60. Renal function should be monitored in such patients. Flurbiprofen The pharmacological properties, therapeutic indications, and adverse
effects of flurbiprofen (ANSAID) are similar to those of other
antiinflammatory derivatives of propionic acid (see Smith et al.,
in Rainsford, 1985b). Flurbiprofen also has been used in trials in Oxaprozin Oxaprozin DAYPRO) is unique among propionic acid derivatives because it can be administered once daily. Its other pharmacological properties, adverse effects, and therapeutic uses are similar to those of other propionic acid derivatives (see Todd and Brogden, 1986). Oxaprozin is well absorbed orally, with peak plasma concentrations achieved in 3 to 6 hours. The drug is metabolized in the liver and primarily eliminated by urinary excretion. The half-life is 40 to 60 hours, and this increases with age. |
Enolic Acids (Oxicams)
|
Piroxicam Piroxicam is one of the oxicam derivatives, a class of enolic acids that have antiinflammatory, analgesic, and antipyretic activity. In recommended doses, piroxicam appears to be the equivalent of aspirin, indomethacin, or naproxen for the long-term treatment of rheumatoid arthritis or osteoarthritis. It may be tolerated better than aspirin or indomethacin. The principal advantage of piroxicam is its long half-life, which permits the administration of a single daily dose. The pharmacological properties and therapeutic uses of piroxicam have been reviewed by Wiseman (see Rainsford, 1985b), and by Lombardino and Wiseman (in Lewis and Furst, 1987). The structural formula of piroxicam is as follows:
Pharmacological Properties Piroxicam is an effective antiinflammatory agent; it is about equal in potency to indomethacin as an inhibitor of prostaglandin biosynthesis in vitro. Piroxicam also can inhibit activation of neutrophils independent of its ability to inhibit the cyclooxygenase; hence, additional modes of antiinflammatory action have been proposed, including inhibition of proteoglycanase and collagenase in cartilage (Abramson and Weissman, 1989; Lombardino and Wiseman, in Lewis and Furst, 1987). Piroxicam exerts antipyretic and analgesic effects in experimental animals and human beings. As with other NSAIDs, piroxicam can cause gastric erosions and it prolongs bleeding time. Pharmacokinetics and Metabolism Piroxicam is completely absorbed after oral administration; peak concentrations in plasma occur within 2 to 4 hours. Antacids do not alter the rate or extent of absorption, but food may alter the rate. There is enterohepatic cycling of piroxicam, and estimates of the half-life in plasma have been variable; a mean value appears to be about 50 hours. After absorption, piroxicam is extensively (99%) bound to plasma proteins. At steady state (e.g., after 7 to 12 days), concentrations of piroxicam in plasma and synovial fluid are approximately equal. Less than 5% of the drug is excreted in the urine unchanged. The major metabolic transformation in human beings is cytochrome P450-mediated hydroxylation of the pyridyl ring (predominantly by an isozyme of the CYP2C subfamily), and this inactive metabolite and its glucuronide conjugate account for about 60% of the drug excreted in the urine and feces. Therapeutic Uses Piroxicam FELDENE) is approved in the Toxic Effects The reported incidence of adverse effects in patients who take piroxicam is about 20%; approximately 5% of patients stop using the drug because of side effects. Gastrointestinal reactions are the most common; the incidence of peptic ulcer is less than 1%. Piroxicam and some other NSAIDs can reduce the renal excretion of lithium to a clinically significant extent. Meloxicam Another oxicam, meloxicam (MOBIC), recently was approved by the FDA for use in treating osteoarthritis. Its pharmacokinetics have been described in detail (Turck et al., 1996). The structure of meloxicam is given below:
The recommended dose for meloxicam is 7.5 mg once daily for osteoarthritis; in severe cases, this dose can be increased to 15 mg. The recommended dose for cases of rheumatoid arthritis is 15 mg once daily. Meloxicam has been suggested to be a selective COX-2 inhibitor based on in vitro studies. However, when tested in vivo in human beings, its selectivity to inhibit COX-2 compared to COX-1 was only about 10-fold, and there was some inhibition of platelet COX-1-mediated thromboxane production after oral treatment with both 7.5 mg/day and 15 mg/day (Panara et al., 1999). In clinical trials, less gastrointestinal side effects had been found with meloxicam compared to nonselective COX inhibitors. However, in a study where endoscopy scores of gastric injury were evaluated, there was significantly less gastric injury compared to piroxicam (20 mg/day) in subjects treated with 7.5 mg/day of meloxicam but not with 15 mg/day (Patoia et al., 1996). Collectively, these findings suggest that the extent of inhibition of COX-1 with meloxicam is largely a function of dose and interindividual variability of drug levels. Further clinical trials and post-marketing clinical experience will be needed to assess whether currently reported short-term benefits in suppression of inflammatory responses without gastrointestinal complications are paralleled by long-term efficacy without toxicity. Other Oxicams A number of other oxicam derivatives are under study or in use outside
of the |
Nabumetone
|
Nabumetone is an antiinflammatory agent
approved in 1991 for use in the
Clinical trials with nabumetone (RELAFEN) have indicated substantial efficacy in the treatment of rheumatoid arthritis and osteoarthritis, with a relatively low incidence of side effects. The dose typically is 1000 mg given once daily. The drug also appears to be effective in the short-term treatment of soft tissue injuries. Pharmacological Properties Nabumetone is a weak inhibitor of cyclooxygenase in vitro, but it is an active antiinflammatory drug that possesses antipyretic and analgesic activities. In experimental animals, nabumetone appears to cause less gastric damage than do other antiinflammatory agents. Pharmacokinetics and Metabolism Nabumetone is absorbed rapidly and is converted in the liver to one or more active metabolites, principally 6-methoxy-2-naphthylacetic acid, a potent inhibitor of cyclooxygenase. This metabolite is inactivated by O-demethylation in the liver, is then conjugated before excretion, and is eliminated with a half-life of about 24 hours. Toxic Effects Side effects of treatment with nabumetone include lower bowel complaints, skin rash, headache, dizziness, heartburn, tinnitus, and pruritus. The incidence of gastrointestinal ulceration appears to be lower with nabumetone than with other NSAIDs (Scott and Palmer, 2000). This may result, in part, from the fact that nabumetone is a prodrug, and an active compound is metabolically generated only after absorption of the administered drug. Differential inhibition of cyclooxygenases is unlikely, because the active metabolite of nabumetone, 6-methoxy-2-naphthylacetic acid, is not a selective inhibitor of COX-2 (Patrignani et al., 1994). |
Pyrazolon Derivatives
|
This group of drugs includes phenylbutazone,
oxyphenbutazone, antipyrine, aminopyrine, and dipyrone. These
drugs have been in clinical use for many years. With the exception of
antipyrine, which is used in analgesic otic drop preparations, these agents
are not available in the |
Diaryl Substituted Furanones
|
The only member of this class currently available is rofecoxib (VIOXX), a selective COX-2 inhibitor that was introduced in 1999. Details of its pharmacodynamics, pharmacokinetics, therapeutic efficacy, and toxicity have been reviewed by Scott and Lamb (1999). Chemistry The structure of rofecoxib is as follows:
Pharmacological Properties Rofecoxib exhibits antiinflammatory, antipyretic, and analgesic activities. These properties have been attributed to selective inhibition of COX-2. At therapeutic concentrations in human beings, rofecoxib does not inhibit COX-1 and does not alter platelet function. The incidence of gastric ulceration seen with endoscopy during treatment with rofecoxib (25 mg and 50 mg daily) is significantly less than that in subjects treated with ibuprofen (2400 mg daily). Clinical studies have not ruled out some increase in the occurrence of ulcers with rofecoxib compared to placebo. Fecal blood loss, however, has not been found to be significantly different from placebo. Rofecoxib appears to significantly inhibit endogenous production of prostaglandins in human beings, as do other selective COX-2 inhibitors (McAdam et al., 1999; Cullen et al., 1998). Pharmacokinetics and Metabolism Rofecoxib is readily absorbed following oral administration and is highly bound to plasma proteins. Metabolism of rofecoxib occurs primarily by cytosolic reductases producing dihydro derivatives. Most of the drug is excreted in the urine as metabolites; 14% is excreted in the feces as unchanged drug. There appears to be saturable metabolism at therapeutic doses. The effective half-life is approximately 17 hours. Renal insufficiency does not alter the pharmacokinetics of the drug, but it is not recommended for use in patients with advanced renal disease, because the safety of the drug in this patient population has not been established. Limited information is available regarding the influence of significant hepatic insufficiency on the pharmacokinetics of the drug. Significant interactions have been identified with rifampin, methotrexate, and warfarin but not with ketoconazole, prednisone/prednisolone, oral contraceptives, or digoxin. Toxic Effects Whether or not the greater safety of rofecoxib regarding gastric injury is sustained during long-term treatment remains to be established. Effects attributed to inhibition of prostaglandin production in the kidney, hypertension and edema, occur with nonselective COX inhibitors and also with rofecoxib. Therefore, rofecoxib should be used with caution in patients with hypertension and congestive heart failure. Whether or not patients with aspirin hypersensitivity reactions also exhibit hypersensitivity reactions following administration of selective COX-2 inhibitors has not been investigated. Until the safety of selective COX-2 inhibitors in this patient population has been established, the use of rofecoxib in these patients is contraindicated. Therapeutic Uses Rofecoxib is approved by the United States Food and Drug Administration for the treatment of osteoarthritis, acute pain in adults, and dysmenorrhea. The drug has efficacy similar to that of nonselective COX inhibitors in reducing dental pain, postoperative pain, and pain associated with primary dysmenorrhea. The recommended starting dose for osteoarthritis is 12.5 mg once daily, increasing to a maximum of 25 mg once daily if necessary. For acute pain and dysmenorrhea, 50 mg per day is the recommended dose; treatment at this dose for more than 5 days has not been studied. |
Diaryl Substituted Pyrazoles
|
The only member of this class currently
available is celecoxib (CELEBREX). It is one of the selective COX-2 inhibitors and was approved for
marketing in the Chemistry The structure of celecoxib is as follows:
Pharmacokinetics and Metabolism The rate of absorption after oral administration is moderate, with peak plasma levels occurring after 2 to 4 hours; the extent of absorption is not known. Celecoxib is extensively bound to plasma proteins. Little drug is excreted unchanged; most is excreted as carboxylic acid and glucuronide metabolites in the urine and feces. The elimination half-life is approximately 11 hours. Plasma concentrations are lower in patients with renal insufficiency, in whom there is a 47% increase in apparent clearance. Plasma concentrations are increased by approximately 40% to 180% in patients with mild and moderate hepatic impairment, respectively. Significant interactions occur with fluconazole and lithium but not with ketoconazole or methotrexate. Celecoxib is metabolized by CYP2C9, so clinical vigilance is necessary during coadministration of other substrates or inhibitors of this enzyme. Pharmacologic Properties, Toxic Effects, and Therapeutic Uses The pharmacological properties and toxic effects of celecoxib are
essentially the same as those of rofecoxib (see above). Celecoxib is
approved in the |
Other Nonsteroidal Antiinflammatory Drugs
|
A large number of antiinflammatory agents are under development or clinical study. Although many are members of classes of drugs discussed above, others have novel structures and apparently different mechanisms of action. Two such agents are apazone and nimesulide. Apazone (Azapropazone) Apazone is an NSAID that is antiinflammatory, analgesic, and
antipyretic but is only a weak inhibitor of cyclooxygenase. In addition,
apazone is a potent uricosuric agent and is particularly useful for the
treatment of acute gout. The antiinflammatory effects of apazone may be due
in part to an ability of the drug to inhibit neutrophil migration,
degranulation, and superoxide production (Mackin et al., 1986). The
drug is not currently available in the
Apazone has been used for the treatment of rheumatoid arthritis, osteoarthritis, and gout. Clinical experience to date suggests that apazone is well tolerated. Mild gastrointestinal side effects (nausea, epigastric pain, dyspepsia) and skin rashes occur in about 3% of patients, while CNS effects (headache, vertigo) are reported less frequently. The overall incidence of untoward reactions is probably 6% to 10%. Because apazone is an inhibitor of cyclooxygenase, albeit less active than other NSAIDs, all precautions discussed above for the group are applicable. Nimesulide Nimesulide is a sulfonanilide compound (see Symposium, 1993b)
which is not available in the
Nimesulide is antiinflammatory, analgesic, and antipyretic. It exerts some actions in addition to inhibiting cyclooxygenase that may contribute to its antiinflammatory effects; it inhibits neutrophil activation and exhibits antioxidant properties. Nimesulide has been found to be a selective COX-2 inhibitor in vivo in human beings at clinically recommended doses (Cullen et al., 1998). Consistent with this is the finding that nimesulide is associated with a very low incidence of adverse side effects, especially in the gastrointestinal tract (Rainsford, 1999; Bjarnason and Thjodleifsson, 1999). |
Gold
|
Gold, in elemental form, has been employed for centuries as an antipruritic to relieve the itching palm. In more modern times, the observation by Robert Koch in 1890 that gold inhibited Mycobacterium tuberculosisin vitro led to trials in arthritis and lupus erythematosus, thought by some to be tuberculous manifestations. Later observations of success in treating chronic arthritis stimulated interest in gold therapy (chrysotherapy). At present, gold is employed in the treatment of rheumatoid arthritis; usually it is reserved for patients with progressive disease who do not obtain satisfactory relief from therapy with NSAIDs. However, gold compounds are among the agents that are used in an attempt to arrest the progress of the disease and to induce remissions; these are sometimes called disease-modifying drugs, although this is probably a misnomer (Edmonds et al., 1993). Since degenerative lesions do not regress once formed, there is an increasing tendency to attempt to induce remission early in the course of the disease. Such therapy is often initiated with gold, which although potentially beneficial, causes a high incidence of toxicity (Felson et al., 1992; Cash and Klippel, 1994). Chemistry The significant preparations of gold are all compounds in which the gold is attached to sulfur. The more water-soluble compounds employed in therapy contain hydrophilic groups in addition to the aurothio group. The structural formulas of aurothioglucose, gold sodium thiomalate, and auranofin are as follows:
Monovalent gold has a relatively strong affinity for sulfur, weak affinities for carbon and nitrogen, and almost no affinity for oxygen, except in chelates. The high affinity for sulfur and the inhibitory effect of gold salts on various enzymes have suggested that the therapeutic effects of gold salts might derive from inhibition of sulfhydryl systems. However, other sulfhydryl inhibitors do not appear to have therapeutic actions in common with gold. Pharmacological Properties Gold compounds can suppress or prevent, but not cure, experimental arthritis and synovitis due to a number of infectious and chemical agents. Gold compounds have minimal antiinflammatory effects in other circumstances and cause only a gradual reduction of the signs and symptoms of inflammation associated with rheumatoid arthritis. Although many effects of these drugs have been observed, which, if any, are related to the therapeutic effects of gold in rheumatoid arthritis is unknown. Perhaps the best hypotheses relate to the capacity of gold compounds to inhibit the maturation and function of mononuclear phagocytes and of T cells, thereby suppressing immune responsiveness. Decreased concentrations of rheumatoid factor and immunoglobulins often are observed in patients who are treated with gold. In experimental animals, gold is sequestered in organs that are rich in mononuclear phagocytes, and it selectively accumulates in the lysosomes of type A synovial cells and other macrophages within the inflamed synovium of patients who are treated with gold compounds. Moreover, the administration of gold thiomalate to animals depresses the migration and phagocytic activity of macrophages in inflammatory exudates, and chrysotherapy reduces the augmented phagocytic capacity of blood monocytes from patients with rheumatoid arthritis. Other mechanisms of action of gold compounds have been suggested, but none is generally accepted. These include inhibition of prostaglandin synthesis, interference with complement activation, cross-linking of collagen, and inhibition of the activity of lysosomal and other enzymes, including protein kinase C, in T cells. Absorption, Distribution, and Excretion Aurothioglucose and Gold Sodium Thiomalate These more water-soluble gold compounds are rapidly absorbed after intramuscular injection, and peak concentrations in blood are reached in 2 to 6 hours. These agents are absorbed erratically when administered orally. Tissue distribution depends not only on the type of compound administered but also on the time after administration and probably on the duration of treatment. Early in the course of therapy, several percent of the total body content of gold is in the blood, where it is first bound (about 95%) to albumin. The concentration in synovial fluid eventually reaches about half that in plasma. With continued therapy, the concentration of gold in the synovium of affected joints is about ten times that of skeletal muscle, bone, or fat. Gold deposits also are found in macrophages of many tissues, as well as in proximal tubular epithelium, seminiferous tubules, hepatocytes, and adrenocortical cells. The pharmacokinetic properties of gold in these compounds are complex and vary with the dose and the duration of treatment. The plasma half-life is about 7 days for a 50-mg dose. With successive doses, the half-life lengthens, and values of weeks or months may be observed after prolonged therapy, reflecting the avid binding of gold in tissues. After a cumulative dose of 1 g of gold, about 60% of the amount administered is retained in the body. After termination of treatment, urinary excretion of gold can be detected for as long as a year, even though concentrations in blood fall to the normal trace amounts in about 40 to 80 days. Substantial quantities of gold have been found in the liver and skin of patients many years after the cessation of therapy. The excretion of gold is 60% to 90% renal and 10% to 40% fecal, the latter probably mostly by biliary secretion. Sulfhydryl agents, such as dimercaprol, penicillamine, and N-acetylcysteine, increase the excretion of gold. Auranofin Auranofin is a more hydrophobic gold-containing compound that is absorbed more readily after oral administration (to the extent of about 25%). Steady-state concentrations of gold in plasma are proportional to the doses administered and are reached after 8 to 12 weeks of treatment. Therapeutic doses of auranofin (6 mg per day) lead to concentrations of gold in plasma that typically are lower than those achieved with conventional parenteral therapy, and the accumulation of gold during a 6-month course of treatment with auranofin is only about 20% of that found with injectable gold compounds. Studies in animals suggest that auranofin binds to tissues to a lesser extent than does gold sodium thiomalate. After cessation of treatment, the half-life of gold in the body is about 80 days. Auranofin is predominantly excreted in the feces. Toxic Effects The most common toxic effects associated with the therapeutic use of gold are those that involve the skin and the mucous membranes, usually of the mouth. These occur in about 15% of all patients. While clearly dose-related, these effects do not correlate well with the concentration of gold in plasma. Cutaneous reactions may vary in severity from simple erythema to severe exfoliative dermatitis. Lesions of the mucous membranes include stomatitis, pharyngitis, tracheitis, gastritis, colitis, and vaginitis; glossitis is fairly common. A gray-to-blue pigmentation (chrysiasis) may occur in the skin and mucous membranes, especially in areas exposed to light. In 5% to 10% of patients receiving gold, kidney function also may be affected. Transient and mild proteinuria occurs in more than 50% of patients during therapy. Heavy albuminuria and microscopic hematuria occur in 1% to 3% of cases. The site of damage is usually the proximal tubules. In addition, a gold-induced nephrosis can occur; the predominant lesion is membranous glomerulonephritis that is usually reversible by cessation of treatment. Severe blood dyscrasias also may occur. Thrombocytopenia is observed in about 1% of patients. Most often this appears to be an immunological disturbance that results in an accelerated degradation of platelets. Occasionally the thrombocytopenia is a consequence of effects upon the bone marrow. In either case, withdrawal of the drug usually leads to recovery, but fatalities have occurred. Leukopenia, agranulocytosis, and aplastic anemia also may occur; aplastic anemia is rare but often fatal. Auranofin appears to be better tolerated than are the injectable gold compounds, and the incidence and severity of mucocutaneous and hematological side effects are less. However, auranofin produces a high incidence of gastrointestinal disturbances, which lead to discontinuation of therapy by about 5% of patients receiving the drug. About half of patients have a change in bowel habits (more frequent or loose stools often associated with abdominal cramping). Proteinuria is less common with auranofin than with parenteral preparations, and the incidence of nephrotoxicity also may be less. Gold may cause a variety of other severe toxic reactions, including encephalitis, peripheral neuritis, hepatitis, pulmonary infiltrates, and nitritoid (vasomotor) crisis. Fortunately, these reactions are infrequent and, when encountered, usually result from failure to discontinue therapy when earlier, less serious symptoms occur. Avoidance and Treatment Regular examination of the skin, buccal mucosa, urine, and blood, including cell and platelet counts, should be made. It is the practice in many arthritis clinics to initiate therapy with small doses of gold and to increase the dose gradually. Although untoward effects are not eliminated by this procedure, the severity of the reactions that occur early is somewhat reduced. If an untoward response occurs, therapy should be withheld until the adverse effect subsides completely. If the reaction is a rash or stomatitis, antihistamines and glucocorticoids may be administered, the latter systemically and/or topically. Glucocorticoids also are also indicated in gold-induced nephrosis. If the reaction to gold therapy is not serious, injections of parenteral gold preparations may be cautiously resumed 2 or 3 weeks after the toxic reaction has subsided. Maintenance dosage should be two-thirds to three-fourths that previously planned. However, many experts decline to use the drug again once toxicity has occurred. For auranofin, a decrease in dosage also can be attempted, but therapeutic responses may not be obtained. If a severe reaction to gold occurs or if the above-mentioned steps fail to control the toxic effects, treatment with dimercaprol and glucocorticoid should be instituted. Dimercaprol chelates gold and the chelate is then excreted. Accordingly, the administration of dimercaprol may shorten a therapeutic remission induced by gold. Therapeutic Uses Gold compounds find their chief therapeutic application in rheumatoid arthritis. In part because these compounds can cause serious toxicity, and, in the case of oral gold, perhaps less efficacy, the use of gold compounds as second-line therapy for rheumatoid arthritis has declined in recent years (Cash and Klippel, 1994). At present, gold is used in early, active arthritis, particularly for disease that progresses despite an adequate regimen of NSAIDs, rest, and physical therapy. Both subjective and objective manifestations of rheumatoid arthritis are improved. Gold compounds often arrest, at least temporarily, the progression of the disease in involved joints; prevent involvement of unaffected joints; improve grip strength and morning stiffness; and decrease the erythrocyte sedimentation rate and abnormal plasma glycoprotein and fibrinogen levels. Gold should not be used if the disease is mild, and it usually is of little benefit when the disease is advanced. The optimal intramuscular dosage schedule for the treatment of rheumatoid arthritis is still debated. The usual dose is 10 mg of aurothioglucose (SOLGANAL) or gold sodium thiomalate, in the first week as a test dose, followed by 25 mg in the second and third weeks. Thereafter, either 25 to 50 mg (gold sodium thiomalate) or 50 mg (aurothioglucose) is administered at weekly intervals until the cumulative dose reaches 1 g. A favorable response may not be evident for a few months. If a remission occurs, treatment is continued, but the dose is reduced or the dosage interval is increased. For oral therapy of active rheumatoid arthritis, the daily dosage is 3 to 6 mg of auranofin (RIDAURA), which is given in one or two portions; some patients may require 9 mg daily in three divided doses. This higher dosage should not be instituted until the lower dosage has been given for 6 months, and therapy should be discontinued after 3 additional months if the response is still inadequate. Although patients have been maintained successfully on auranofin for several years, the optimal duration of therapy has not been determined. Therapy with gold is sometimes beneficial in juvenile rheumatoid
arthritis, palindromic rheumatism, psoriatic arthritis, Sjgren's syndrome,
nondisseminated lupus erythematosus, and pemphigus. Except for injectable
preparations in the treatment of juvenile forms of arthritis, the use of gold
in these conditions has not been approved in the Contraindications Gold therapy is contraindicated in patients with renal disease, hepatic dysfunction or a history of infectious hepatitis, or hematological disorders. Gold should not be readministered to patients who have developed severe hematological or renal toxicity during a course of chrysotherapy; auranofin should not be administered after the occurrence of several additional gold-induced disorders, including pulmonary fibrosis, necrotizing enterocolitis, and exfoliative dermatitis. Gold is contraindicated during pregnancy or breast-feeding. Patients who recently have had radiation should not receive gold because of its depressant action on hematopoietic tissue. Concomitant use of antimalarials, immunosuppressants, penicillamine, or dipyrone is contraindicated because of the potential of these drugs to cause blood dyscrasias. Urticaria, eczema, and colitis also are considered to be contraindications to the use of gold. Finally, gold is poorly tolerated by elderly individuals. |
Other Drugs for Rheumatoid Arthritis
|
In addition to nonsteroidal antiinflammatory agents and gold, other drugs also are used for the treatment of rheumatoid arthritis. These include immunosuppressive agents [e.g., cyclosporine, azathioprine (see Chapter 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants), leflunomide, and the folate antagonist, methotrexate (see Chapters 52: Antineoplastic Agents and 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants)], glucocorticoids (see Chapter 60: Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones), penicillamine, and hydroxychloroquine. With the exception of glucocorticoids, sulfasalazine, and, perhaps, methotrexate, these drugs do not possess antiinflammatory or analgesic properties. In general, their therapeutic effects become evident only after several weeks or months of treatment. They are reserved for patients who are refractory to therapeutic regimens that include rest, physiotherapy, and NSAIDs. Although glucocorticoids often can produce dramatic symptomatic improvement, these agents do not arrest the progress of rheumatoid arthritis and are used only as adjuvants to other treatment because of their long-term toxicity (see Chapter 60: Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones). Immunosuppressants sometimes relieve joint inflammation, but each of these drugs has its unique and significant toxicities (see Chapter 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants). Of the cytotoxic immunosuppressants, only azathioprine and low oral doses of methotrexate have been approved for the treatment of rheumatoid arthritis. Methotrexate appears to be a particularly useful drug for second-line therapy in rheumatoid arthritis (Felson et al., 1992). Cyclosporine also has been shown to be effective in many patients, but its use is commonly associated with nephrotoxicity, especially in patients receiving concomitant treatment with NSAIDs (see Chapter 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants and Faulds et al., 1993). Even though their mechanisms of action are not understood, hydroxychloroquine and penicillamine are useful, orally effective alternatives to gold in the treatment of patients with early, mild, and nonerosive disease. Penicillamine is more apt to produce serious toxicity, including various cutaneous lesions, blood dyscrasias, and a number of autoimmune syndromes (see Chapter 67: Heavy Metals and Heavy-Metal Antagonists). Hydroxychloroquine shares the toxicity of other 4-aminoquinoline antimalarials (see Chapter 40: Drugs Used in the Chemotherapy of Protozoal Infections: Malaria). Of greatest concern during the long-term treatment of rheumatoid arthritis is the danger of producing irreversible retinal damage. The risk of corneal deposits and ocular toxicity appears to be less for hydroxychloroquine than for chloroquine at the usual antirheumatic doses (200 to 400 mg daily). Even so, ophthalmological examinations should be performed before treatment is begun and every 3 to 6 months thereafter. |
Drugs Employed in the Treatment of Gout
|
An acute attack of gout occurs as a result of an inflammatory reaction to crystals of sodium urate (the end product of purine metabolism in human beings) that are deposited in the joint tissue. The inflammatory response involves local infiltration of granulocytes, which phagocytize the urate crystals. Lactate production is high in synovial tissues and in the leukocytes associated with the inflammatory process, and this favors a local decrease in pH that fosters further deposition of uric acid. Deposition of urate crystals occurs in patients with hyperuricemia, which is caused by increased production or decreased excretion of uric acid. Several therapeutic strategies can be used to counter attacks of gout. Uricosuric drugs increase the excretion of uric acid, thus reducing concentrations in plasma. Colchicine, although associated with a high frequency of toxicity, is specifically efficacious in gout, probably secondary to an effect on the mobility of granulocytes. Allopurinol is a selective inhibitor of the terminal steps of the biosynthesis of uric acid. Although prostaglandins may be implicated in the pain and inflammation, there is no evidence that they contribute to the pathogenesis of gout; nevertheless, nonsalicylate-containing NSAIDs afford symptomatic relief, and some of them are uricosuric as well. The pharmacology of NSAIDs is described in a previous section. Discussion in this section is limited to colchicine, allopurinol, and the uricosuric agents. Colchicine Colchicine is a unique antiinflammatory agent in that it is largely effective only against gouty arthritis. It provides dramatic relief of acute attacks of gout, and is an effective prophylactic agent against such attacks. History Colchicine is an alkaloid of Colchicum autumnale (autumn
crocus, meadow saffron). Although the poisonous action of colchicum was known
to Dioscorides, preparations of the plant were not recommended for pain of
articular origin until the sixth century A.D. Colchicum was introduced for the therapy of
acute gout by von Strck in 1763, and its specificity for this syndrome soon
resulted in its incorporation in a number of 'gout mixtures'
popularized by charlatans. Benjamin Franklin, himself a sufferer from gout,
is reputed to have introduced colchicum therapy in the Chemistry The structural formula of colchicine is as follows:
The structureactivity relationship of colchicine and related agents has been discussed by Wallace (1961). Pharmacological Properties The antiinflammatory effect of colchicine in acute gouty arthritis is relatively selective for this disorder. Colchicine is only occasionally effective in other types of arthritis; it is not an analgesic and does not provide relief of other types of pain. Colchicine is an antimitotic agent and is widely employed as an experimental tool in the study of cell division and function. Effect in Gout Colchicine does not influence the renal excretion of uric acid or its concentration in blood. By virtue of its ability to bind to tubulin, colchicine interferes with the function of the mitotic spindles and causes depolymerization and disappearance of the fibrillar microtubules in granulocytes and other motile cells. This action is apparently the basis for the beneficial effect of colchicine, namely, the inhibition of the migration of granulocytes into the inflamed area and a decreased metabolic and phagocytic activity of granulocytes. This reduces the release of lactic acid and proinflammatory enzymes that occurs during phagocytosis and breaks the cycle that leads to the inflammatory response. Neutrophils exposed to urate crystals ingest them and produce a glycoprotein, which may be the causative agent of acute gouty arthritis. Injected into joints, this substance produces a profound arthritis that is histologically indistinguishable from that caused by direct injection of urate crystals. Colchicine appears to prevent the elaboration by leukocytes of this glycoprotein. Effect on Cell Division Colchicine can arrest plant and animal cell division in vitro and in vivo. Mitosis is arrested in metaphase, due to failure of spindle formation. Cells with the highest rates of division are affected earliest. High concentrations may completely prevent cells from entering mitosis, and they often die. The action also is characteristic of the vinca alkaloids (vincristine and vinblastine), podophyllotoxin, and griseofulvin. Other Effects Colchicine inhibits the release of histamine-containing granules from mast cells, the secretion of insulin from beta cells of pancreatic islets, and the movement of melanin granules in melanophores. Although it is questionable whether or not these effects occur at clinically achieved concentrations of colchicine, all of these processes may involve the translocation of granules by the microtubular system. Colchicine also exhibits a variety of other pharmacological effects. It lowers body temperature, increases the sensitivity to central depressants, depresses the respiratory center, enhances the response to sympathomimetic agents, constricts blood vessels, and induces hypertension by central vasomotor stimulation. It enhances gastrointestinal activity by neurogenic stimulation but depresses it by a direct effect, and alters neuromuscular function. Pharmacokinetics and Metabolism Colchicine is rapidly absorbed after oral administration, and peak concentrations occur in plasma by 0.5 to 2 hours. Large amounts of the drug and metabolites enter the intestinal tract in the bile and intestinal secretions, and this fact, plus the rapid turnover of intestinal epithelium, probably explains the prominence of intestinal manifestations in colchicine poisoning. The kidney, liver, and spleen also contain high concentrations of colchicine, but it is apparently largely excluded from heart, skeletal muscle, and brain. The drug can be detected in leukocytes and in the urine for at least 9 days after a single intravenous dose. Colchicine is metabolized to a mixture of compounds in vitro. Most of the drug is excreted in the feces; however, in normal individuals, 10% to 20% of the drug is excreted in the urine. In patients with liver disease, hepatic uptake and elimination are reduced and a greater fraction of the drug is excreted in the urine. Toxic Effects The most common side effects reflect the action of colchicine on the rapidly proliferating epithelial cells in the gastrointestinal tract, especially in the jejunum. Nausea, vomiting, diarrhea, and abdominal pain are the most common and earliest untoward effects of colchicine overdosage. To avoid more serious toxicity, administration of the drug should be discontinued as soon as these symptoms occur. There is a latent period of several hours or more between the administration of the drug and the onset of symptoms. This interval is not altered by dosage or route of administration. For this reason, and because of individual variation, adverse effects may be unavoidable during an initial course of medication with colchicine. However, since patients often remain relatively consistent in their response to a given dose of the drug, toxicity can be reduced or avoided during subsequent courses of therapy by reducing the dose. The drug is equally effective when given intravenously; the onset of the therapeutic effect may be faster, and the gastrointestinal side effects may be almost completely avoided. In acute poisoning with colchicine, there is hemorrhagic gastroenteritis, extensive vascular damage, nephrotoxicity, muscular depression, and an ascending paralysis of the CNS. Colchicine produces a temporary leukopenia that is soon replaced by a leukocytosis, sometimes due to a striking increase in the number of basophilic granulocytes. The site of action is apparently directly on the bone marrow. Myopathy and neuropathy also have been noted with colchicine treatment, especially in patients with decreased renal function. Long-term administration of colchicine entails some risk of agranulocytosis, aplastic anemia, myopathy, and alopecia; azoospermia also has been described. Therapeutic Uses Colchicine provides dramatic relief from acute attacks of gout. The effect is sufficiently selective that the drug has been used for diagnostic purposes, but the test is not infallible. Colchicine also has an established role to prevent and to abort acute attacks of gout. However, its toxicity and the availability of alternative agents that are less toxic have substantially lessened its usefulness. Acute Attacks When colchicine is given promptly within the first few hours of an attack, fewer than 5% of patients fail to obtain relief. Pain, swelling, and redness abate within 12 hours and are completely gone within 48 to 72 hours. Although for many years colchicine was administered orally, current practice is to administer the drug intravenously (see Wallace and Singer, 1988). Although a number of regimens have been used, a single dose of 2 mg, diluted in 10 to 20 ml of 0.9% sodium chloride solution, usually is adequate; a total dose of 4 mg should not be exceeded. To avoid cumulative toxicity, treatment with colchicine should not be repeated within 7 days. Great care should be exercised in prescribing colchicine for elderly patients, and for those with cardiac, renal, hepatic, or gastrointestinal disease. In these patients and in those who do not tolerate or respond to colchicine, indomethacin or another NSAID is preferred. Prophylactic Uses For patients with chronic gout, colchicine has established value as a prophylactic agent, especially when there is frequent recurrence of attacks. Prophylactic medication also is indicated upon initiation of long-term medication with allopurinol or the uricosuric agents, since acute attacks often increase in frequency during the early months of such therapy. The prophylactic dose of colchicine depends upon the frequency and severity of prior attacks. As small an oral dose as 0.5 mg two to four times a week may suffice; as much as 1.8 mg per day may be required by some patients. Colchicine should be taken in larger abortive doses immediately upon the first twinge of articular pain or the appearance of any prodrome of an acute attack. Before and after surgery in patients with gout, colchicine should be given for 3 days (0.5 or 0.6 mg, three times a day); this greatly reduces the very high incidence of acute attacks of gouty arthritis precipitated by operative procedures. Daily administration of colchicine is useful for the prevention of attacks of familial Mediterranean fever (familial paroxysmal polyserositis) and for prevention and treatment of amyloidosis in such patients (Zemer et al., 1991). Colchicine appears to benefit patients with primary biliary cirrhosis in terms of improvement of liver function tests and perhaps of survival (Warnes, 1991). Colchicine also has been employed to treat a variety of skin disorders, including psoriasis and Behet's syndrome. Allopurinol Allopurinol is effective for the treatment of both the primary hyperuricemia of gout and that secondary to hematological disorders or antineoplastic therapy. In contrast to the uricosuric agents that increase the renal excretion of urate, allopurinol inhibits the terminal steps in uric acid biosynthesis. Since overproduction of uric acid is a contributing factor in most patients with gout and a characteristic of most types of secondary hyperuricemia, allopurinol represents a rational approach to therapy. History The introduction of allopurinol by Hitchings, Elion, and associates provides an elegant example of the development of a drug on a rational biochemical basis. Originally synthesized as a candidate for an antineoplastic agent, allopurinol was found to lack antimetabolite activity, but it proved to be a substrate for and an inhibitor of xanthine oxidase. Allopurinol delays inactivation of mercaptopurine by xanthine oxidase and reduces the plasma concentration and renal excretion of uric acid. Subsequent clinical study for treatment of gout by Rundles and coworkers was successful and quickly confirmed. Chemistry and Pharmacological Properties Allopurinol, an analog of hypoxanthine, has the following structural formula:
Both allopurinol and its primary metabolite, oxypurinol (alloxanthine), are inhibitors of xanthine oxidase. Inhibition of this enzyme accounts for the major pharmacological effects of allopurinol. In human beings, uric acid is formed primarily by the xanthine oxidasecatalyzed oxidation of hypoxanthine and xanthine. At low concentrations, allopurinol is a substrate for and competitive inhibitor of the enzyme; at high concentrations, it is a noncompetitive inhibitor. Oxypurinol, the metabolite of allopurinol formed by the action of xanthine oxidase, is a noncompetitive inhibitor of the enzyme; the formation of this compound, together with its long persistence in tissues, is responsible for much of the pharmacological activity of allopurinol. Inhibition of uric acid biosynthesis reduces its plasma concentration and urinary excretion and increases the plasma concentrations and renal excretion of the more soluble oxypurine precursors. In the absence of allopurinol, the urinary content of purines is almost solely uric acid. During treatment with allopurinol, the urinary purines are divided among hypoxanthine, xanthine, and uric acid. Since each has its independent solubility, the concentration of uric acid in plasma is reduced without exposing the urinary tract to an excessive load of uric acid and the likelihood of calculus formation. By lowering the uric acid concentration in plasma below its limit of solubility, allopurinol facilitates the dissolution of tophi and prevents the development or progression of chronic gouty arthritis. The formation of uric acid stones virtually disappears with therapy, and this prevents the development of nephropathy. Although it appears that gouty nephropathy can be reversed by allopurinol if administered before renal function is severely compromised, there is little evidence of improvement in advanced renal disease. The incidence of acute attacks of gouty arthritis may increase during the early months of therapy as a consequence of mobilization of tissue stores of uric acid. Coadministration of colchicine helps suppress such acute attacks. Following reduction of excess tissue stores of uric acid, the incidence of acute attacks decreases. Tissue deposition of xanthine and hypoxanthine usually does not occur during allopurinol therapy because the renal clearance of the oxypurines is rapid; their plasma concentrations are only slightly increased and do not exceed their solubility. Although xanthine constitutes about 50% of total oxypurine excreted in the urine and is relatively insoluble, xanthine stone formation during allopurinol therapy has occurred only occasionally in patients with very high uric acid production prior to treatment. The risk can be minimized by alkalinization of the urine and by increasing the daily fluid intake during the administration of allopurinol. In some patients, the allopurinol-induced increase in excretion of oxypurines is less than the reduction in uric acid excretion; this disparity is primarily a result of reutilization of oxypurines and feedback inhibition of de novo purine biosynthesis. Pharmacokinetics and Metabolism Allopurinol is absorbed relatively rapidly after oral ingestion, and peak plasma concentrations are reached within 60 to 90 minutes. About 20% is excreted in the feces in 48 to 72 hours, presumably as unabsorbed drug. Allopurinol is rapidly cleared from plasma with a half-time of 1 to 2 hours, primarily by conversion to oxypurinol. Less than 10% of a single dose or about 30% of the drug ingested during long-term medication is excreted unchanged in the urine. Oxypurinol is slowly excreted in the urine by the net balance of glomerular filtration and probenecid-sensitive tubular reabsorption. The plasma half-life of oxypurinol is 18 to 30 hours in patients with normal renal function and increases in proportion to the reduction of glomerular filtration in patients with renal impairment. Allopurinol and its active metabolite oxypurinol are distributed in total tissue water, with the exception of brain, in which their concentration is about one-third that in other tissues. Neither compound is bound to plasma proteins. The plasma concentrations of the two compounds do not correlate well with therapeutic or toxic effects. Drug Interactions Allopurinol increases the half-life of probenecid and enhances its uricosuric effect, while probenecid increases the clearance of oxypurinol, thereby increasing dose requirements of allopurinol. Allopurinol decreases metabolism and clearance of mercaptopurine (and its derivative azathioprine); thus the dosage of mercaptopurine and azathioprine should be reduced when coadministered with allopurinol. Allopurinol also may interfere with the hepatic inactivation of other drugs, including the oral anticoagulant agents. Although the effect is variable and of clinical significance only in some patients, increased monitoring of prothrombin activity is recommended in patients receiving both medications. Whether the increased incidence of skin rash in patients receiving concurrent allopurinolampicillin medication, compared with that observed when these agents are administered individually, should be ascribed to allopurinol or to hyperuricemia remains to be established. Hypersensitivity reactions have been reported in patients with compromised renal function, especially those who are receiving a combination of allopurinol and a thiazide diuretic. The concomitant administration of allopurinol and theophylline leads to increased accumulation of an active metabolite of theophylline, 1-methylxanthine; the concentration of theophylline in plasma also may be increased (see Chapter 28: Drugs Used in the Treatment of Asthma). Therapeutic Uses Allopurinol ZYLOPRIM, ALOPRIM, others) is available for oral use and provides effective therapy for both the primary hyperuricemia of gout and that secondary to polycythemia vera, myeloid metaplasia, or other blood dyscrasias. Allopurinol is contraindicated in patients who have exhibited serious adverse effects or hypersensitivity skin rash from the medication, nursing mothers, and children, except those with malignancy or certain inborn errors of purine metabolism. In gout, allopurinol generally is used in the severe chronic forms characterized by one or more of the following conditions: gouty nephropathy, tophaceous deposits, renal urate stones, impaired renal function, or hyperuricemia not readily controlled by the uricosuric drugs. The aim of therapy is to reduce the plasma uric acid concentration
below 6 mg/dl (equivalent to 360 Allopurinol also is administered prophylactically to reduce the hyperuricemia and to prevent urate deposition or renal calculi in patients with leukemias, lymphomas, or other malignancies, particularly when antineoplastic or radiation therapy is initiated. A dose of 600 to 800 mg daily for 2 to 3 days is advisable, together with a high fluid intake. In children with secondary hyperuricemias associated with malignancies, the usual daily dose is 150 to 300 mg, depending upon age. Allopurinol inhibits the enzymatic inactivation of mercaptopurine and its derivative azathioprine by xanthine oxidase. Thus, when allopurinol is used concomitantly with oral mercaptopurine or azathioprine, dosage of the antineoplastic agent must be reduced to one-fourth to one-third of the usual dose (see Chapter 52: Antineoplastic Agents). The risk of bone-marrow suppression also is increased when allopurinol is administered with cytotoxic agents that are not metabolized by xanthine oxidase, particularly cyclophosphamide. The iatrogenic hyperuricemia sometimes induced by the thiazides and other drugs can be prevented or reversed by concurrent allopurinol medication, although this is rarely necessary. Allopurinol also is useful in lowering the high plasma concentrations of uric acid in patients with Lesch-Nyhan syndrome and thereby prevents the complications resulting from hyperuricemia; there is no evidence that it alters the progressive neurological and behavioral abnormalities characteristic of the disease. Toxic Effects Allopurinol is well tolerated by most patients. The most common adverse effects are hypersensitivity reactions. They may occur even after months or years of medication. The effects usually subside within a few days after medication is discontinued. Serious reactions preclude further use of the drug. Attacks of acute gout may occur more frequently during the initial months of allopurinol medication and may require concurrent prophylactic therapy with colchicine (see above). The cutaneous reaction caused by allopurinol is predominantly a pruritic, erythematous, or maculopapular eruption, but occasionally the lesion is urticarial or purpuric. In rare patients, toxic epidermal necrolysis or Stevens-Johnson syndrome occurs, which can be fatal. This risk for Stevens-Johnson syndrome is primarily limited to the first 2 months of treatment (Roujeau et al., 1995). Fever, malaise, and muscle aching also may occur. Such effects are noted in about 3% of patients with normal renal function but more frequently in those with renal impairment. Since the onset of skin rash may be followed by severe hypersensitivity reactions, allopurinol should be discontinued by patients who develop such rashes. Transient leukopenia or leukocytosis and eosinophilia are rare reactions but may require cessation of therapy. Hepatomegaly and elevated levels of aminotransferase activities in plasma and progressive renal insufficiency also may occur. |
Uricosuric Agents
|
A uricosuric agent is a drug that increases the rate of excretion of uric acid. There is perhaps no other class of therapeutic agents for which the observations in their entirety appear so inconsistent and at times contradictory. This results from the complexity of the transport mechanisms, as well as the marked species variation of individual mechanisms and their sensitivity to drug action. Birds, reptiles, and some mammals demonstrate net secretion of urate; in some mammalian species both net secretion and net reabsorption can be observed; and in others, including human beings, net reabsorption is found almost invariably. In human beings and in other species that demonstrate net reabsorption, the reabsorptive process is mediated by a specific transporter and it is inhibitable. Finally, in all species that have been studied thoroughly, the major transport mechanism, either secretion or reabsorption, is opposed by a smaller flux operating in the opposite direction; that is, there is bidirectional transport. As a consequence of all these factors, a drug that is uricosuric in one species may produce urate retention in another; within one species a drug may cause either urate retention or uricosuria, depending on the dose; and one uricosuric drug may either add to or inhibit the action of another. In human beings, uric acid is largely reabsorbed; the amount excreted is usually about 10% of that filtered. Studies with proximal tubule brush-border membranes indicate that the first step in reabsorption is the uptake of urate from tubular fluid by a transporter that can act as an anion exchanger. Thus, urate in the tubular fluid can be exchanged for either an organic or an inorganic anion moving in the opposite direction. It has been suggested that the anionic compositions of luminal and intracellular fluids are such that reabsorption of urate is favored. The exit step for urate at the basolateral membrane also is mediated by an anion exchanger. Uricosuric drugs, when present in the lumen or when tested in isolated brush-border members, compete with urate for the brush-border transporter, thereby inhibiting its reabsorption via the urateanion exchanger system. The paradoxical effect of uricosuric agents refers to the fact that, depending on dosage, a drug may either decrease or increase the excretion of uric acid. Decreased excretion usually occurs at a low dosage, while increased excretion is observed at a higher dosage. Not all agents show this phenomenon. With some drugs, such as salicylate, the biphasic effect may be seen within the normal dosage range. Two mechanisms for a drug-induced decrease in excretion of urate have been advanced; they are not mutually exclusive. The first presumes that the small secretory movement of urate is mediated by a mechanism that is thought to be extremely sensitive to low concentrations of compounds such as salicylate. Higher concentrations may inhibit urate reabsorption in the usual manner. The second proposal suggests that the urate-retaining anionic drug gains access to the intracellular fluid by an independent mechanism and promotes reabsorption of urate across the brush border by anion exchange. There are two mechanisms by which one drug may nullify the uricosuric action of another. First, the drug may inhibit the secretion of the uricosuric agent, thereby denying it access to its site of action, the luminal aspect of the brush border. Second, the inhibition of urate secretion by one drug may counterbalance the inhibition of urate reabsorption by the other (Fanelli and Weiner, 1979). There are situations in which two uricosuric agents administered together almost completely nullify each other's actions (see, for example, Yet al., 1963). In such an instance, one of the drugs (A) must have a strong paradoxical action. Drug B inhibits the secretion of A, thereby preventing its uricosuric action but not its urate-retaining action. The latter effect balances the uricosuric action of drug B. There are a great many compounds that have uricosuric activity, but
only a few are prescribed for this purpose. Probenecid and sulfinpyrazone
are the two uricosuric drugs available in the Probenecid History Probenecid was developed as a result of a planned approach to achieve a specific objective. When penicillin was first introduced, it was in critically short supply and the rapid renal excretion of the antibiotic was thus of practical significance. For this reason, Beyer and associates began a study to find an organic acid that would depress the tubular secretion of penicillin in the manner described above. The first compound to be evaluated clinically was carinamide. It proved to be effective, but the drug was secreted by the renal tubules fairly rapidly and it was necessary to give frequent doses. This problem was overcome with the discovery of probenecid (Beyer et al., 1951). Chemistry Probenecid is a highly lipid-soluble benzoic acid derivative (Ka 3.4) with the following structural formula:
Pharmacological Actions Inhibition of Inorganic Acid Transport The actions of probenecid are confined largely to inhibition of the transport of organic acids across epithelial barriers. This is most important for the renal tubule, in which tubular secretion of many drugs and drug metabolites is inhibited. The renal action of probenecid reduces the concentrations of certain compounds in urine and raises them in plasma. This is a desirable therapeutic effect in the case of penicillin and related antibiotics that have a beneficial systemic action, but it may be undesirable with an agent such as nitrofurantoin when it is employed as a urinary antiseptic. When tubular secretion of a substance is inhibited, its final concentration in the urine is determined by the degree of filtration, which in turn is a function of binding to plasma protein, and by the degree of reabsorption. The significance of each of these factors varies widely with different compounds. Uric acid is the only important endogenous compound whose excretion is known to be increased by probenecid. This results from inhibition of its reabsorption (see above). The uricosuric action of probenecid is blunted by the administration of salicylates. Inhibition of Transport of Miscellaneous Substances Probenecid inhibits the tubular secretion of a number of drugs, such as methotrexate and the active metabolite of clofibrate, but there is no clinical indication for the coadministration of probenecid in most instances. Probenecid inhibits renal secretion of the glucuronides of NSAIDs such as naproxen, ketoprofen, and indomethacin and thereby can increase plasma concentrations of such compounds. However, these metabolites are inactive. In the case of a number of endogenous or exogenous organic acids whose rate of excretion is determined for diagnostic purposes, misleading values may be obtained if the patient is receiving probenecid. Inhibition of Monoamine Transport to CSF Probenecid inhibits the transport of 5-hydroxyindoleacetic acid (5-HIAA) and other acidic metabolites of cerebral monoamines from the subarachnoid space to the plasma. The transport of drugs such as penicillin G also may be affected. Inhibition of Biliary Excretion Since probenecid and some of its metabolites may be secreted into the bile, it is not surprising that probenecid depresses the biliary secretion of other compounds, including the diagnostic agents indocyanine green and sulfobromophthalein (BSP). The inhibition of biliary secretion also has implications in the use of rifampin for the treatment of tuberculosis. Higher concentrations of the antibiotic are achieved in plasma if probenecid is administered concurrently. Absorption, Fate, and Excretion Probenecid is completely absorbed after oral administration. Peak concentrations in plasma are reached in 2 to 4 hours. The half-life of the drug in plasma is dose-dependent and varies from less than 5 hours to more than 8 hours over the therapeutic range. Between 85% and 95% of the drug is bound to plasma albumin. The small unbound portion gains access to the glomerular filtrate; a much larger portion is actively secreted by the proximal tubule. The high lipid solubility of the undissociated form results in virtually complete absorption by back diffusion unless the urine is markedly alkaline. A small amount of probenecid glucuronide appears in the urine. It is also hydroxylated to metabolites that retain their carboxyl function and have uricosuric activity. Toxic Effects Probenecid is well tolerated by most patients. Some degree of gastrointestinal irritation is experienced by at least 2% of patients; the incidence is considerably higher after large doses. Caution is advised in administering probenecid to patients with a history of peptic ulcer. Most reports place the incidence of hypersensitivity reactions, usually mild skin rashes, between 2% and 4%. More serious hypersensitivity reactions occur, but they are rare. The appearance of a rash during the concurrent administration of probenecid and penicillin G or a congener presents the physician with an awkward diagnostic dilemma. Huge overdosage of probenecid results in stimulation of the central nervous system, convulsions, and death from respiratory failure. Therapeutic Use Probenecid is marketed for oral administration. In the treatment of chronic gout, 250 mg is given twice daily for 1 week, following which 500 mg is administered twice daily. In some patients it may be necessary to increase the daily dosage gradually to a maximum of 2 g, given in four divided portions. Liberal fluid intake should be maintained throughout therapy because of the tendency of probenecid to produce uric acid stones. For this reason, probenecid should not be used in gouty patients with nephrolithiasis or with overproduction of uric acid. In addition, an acute gouty attack may be precipitated in up to 20% of gouty patients treated with probenecid alone. Thus, concomitant therapy should include colchicine or an NSAID. To block the renal excretion of penicillin effectively, a total daily dose of 2 g is employed in adults. This is administered in four divided doses. For children weighing less than 50 kg, an initial dose of 25 mg/kg is followed by maintenance doses of 10 mg/kg given four times daily. Adjunct in Penicillin Therapy The oral administration of probenecid in conjunction with penicillin G results in higher and more prolonged concentrations of the antibiotic in plasma than when penicillin is given alone. The elevation in the plasma level is at least twofold and sometimes much greater. Although the reduction of a daily dose of penicillin G from 1 million to 500,000 units has very little significance, a reduction by 50% or more may be of importance for convenience in the treatment of resistant infections that may require the administration of penicillin G in very large doses. This combined regimen also may be useful to minimize the amount of K+ that is administered to some patients who receive very large doses of penicillin. Probenecid also is included in certain regimens that can be completed during one visit to the physician for the treatment and prophylaxis of gonococcal infections (see Chapter 46: Antimicrobial Agents: The Aminoglycosides). Sulfinpyrazone History Despite its therapeutic efficacy as an antiinflammatory and uricosuric agent, phenylbutazone (see above) had undesirable side effects severe enough to preclude its continuous use. For this reason, a number of congeners were evaluated for uricosuric and antiinflammatory activity. One of these, in which a phenylthioethyl configuration replaces the butyl side chain of the parent compound, displayed promising activity. When the metabolites of the new compound were studied, it was found that side chain oxidation in vivo led to the formation of the sulfoxide, sulfinpyrazone, which was a potent uricosuric agent. Chemistry The chemical structure of sulfinpyrazone is as follows:
It is a strong organic acid (Ka 2.8) that readily forms soluble salts. Pharmacological Actions Sulfinpyrazone in sufficient dosage is a potent inhibitor of the renal tubular reabsorption of uric acid. As with other uricosuric agents, small doses may reduce the excretion of uric acid. Like probenecid, sulfinpyrazone reduces the renal tubular secretion of many other organic anions. The drug may induce hypoglycemia by inhibiting the metabolism of the sulfonylurea oral hypoglycemic agents; hepatic metabolism of warfarin also is impaired. The uricosuric action of sulfinpyrazone is additive to that of probenecid but is mutually antagonistic to that of salicylates (Yet al., 1963). Sulfinpyrazone lacks the antiinflammatory and analgesic properties of its congener, phenylbutazone. The inhibitory effect of sulfinpyrazone on platelet function is discussed in Chapter 55: Anticoagulant, Thrombolytic, and Antiplatelet Drugs. Absorption, Fate, and Excretion Sulfinpyrazone is well absorbed after oral administration. It is strongly bound to plasma albumin (98% to 99%) and displaces other anionic drugs that have their highest affinity for the same binding site (site I) (Sudlow et al., 1975). The half-life of the drug in plasma after its intravenous injection is about 3 hours. After oral administration, however, its uricosuric effect may persist for as long as 10 hours. Although little sulfinpyrazone is available for filtration at the glomerulus, it is secreted by the proximal tubule and undergoes little passive back diffusion. Approximately half of the orally administered dose appears in the urine within 24 hours. Most of the drug (90%) in the urine is unchanged; the remainder is eliminated as the N1-p-hydroxyphenyl metabolite, which also is a potent uricosuric substance. Toxic Effects Gastrointestinal irritation occurs in 10% to 15% of all patients receiving sulfinpyrazone, and occasionally a patient may require discontinuance of its use. Gastric distress is lessened when the drug is taken in divided doses with meals. Sulfinpyrazone should be given to patients with a history of peptic ulcer only with the greatest caution. Hypersensitivity reactions, usually a rash with fever, do occur, but less frequently than with probenecid. The severe blood dyscrasias and salt and water retention, hazards of phenylbutazone therapy, have not been observed during sulfinpyrazone therapy. However, depression of hematopoiesis has been demonstrated experimentally, and periodic blood-cell counts therefore are advised during prolonged therapy. Therapeutic Use Sulfinpyrazone ANTURANE) is available for oral administration. For the treatment of chronic gout, the initial dosage is 100 to 200 mg given twice daily. After the first week, the dosage may be gradually increased until a satisfactory lowering of plasma uric acid is achieved and maintained. This may require from 200 to 800 mg per day, divided in two to four doses and preferably given with meals or milk; a liberal fluid intake should be maintained. Larger doses are poorly tolerated and unlikely to produce a further uricosuric effect in the resistant patient. Benzbromarone This is a potent uricosuric agent that is used in
The drug is readily absorbed after oral ingestion, and peak concentrations in blood are achieved in about 4 hours. It is metabolized to the monobromine and dehalogenated derivatives, both of which have uricosuric activity, and is excreted primarily in the bile. The uricosuric action is blunted by aspirin or sulfinpyrazone. No paradoxical retention of urate has been observed. At clinically effective doses, there is no effect on the synthesis of urate. Therefore, benzbromarone probably reduces the concentration of urate in plasma solely by inhibiting its tubular reabsorption. Benzbromarone is of interest as a member of a newer chemical class of uricosuric agents. It is a potent and reversible inhibitor of the urateanion exchanger in the proximal tubule (Dan and Koga, 1990). As the micronized powder it is effective in a single daily dose of 40 to 80 mg, which makes it significantly more potent than other uricosuric drugs. It may be useful clinically in patients who are either allergic or refractory to other drugs used for the treatment of gout or in patients with renal insufficiency. Preparations that combine allopurinol and benzbromarone are more effective than either drug alone in lowering serum uric acid levels, in spite of the fact that benzbromarone lowers plasma levels of oxypurinol, the active metabolite of allopurinol. |
Treatment of Gout and Hyperuricemia
|
The use of probenecid and sulfinpyrazone for the mobilization of uric acid in chronic gout is well established. In about two-thirds of patients, these agents cause uric acid to be excreted at a rate sufficient to exceed that of formation and thereby promptly lower the plasma uric acid concentration. Continuous oral administration to patients with tophaceous gout approximately doubles the daily excretion of urates, prevents the formation of new tophi, and causes gradual shrinkage, or even disappearance, of old tophi. In gouty arthritis, there is a reduction in the swelling of chronically enlarged joints, and a dramatic degree of rehabilitation may be achieved in patients who suffer severe pain and limitation of joint movement. In patients who do not respond well to uricosuric agents because of impaired renal function, allopurinol is especially useful, as described above. In patients with gouty nephropathy, allopurinol offers an additional advantage over the uricosuric agents in that the daily excretion of uric acid is reduced rather than increased. Its administration is compatible with the simultaneous use of the uricosuric agents if necessary. Neither the uricosuric agents nor allopurinol alters the course of acute attacks of gout or supplants the use of antiinflammatory agents in their management. Indeed, the acute attacks may increase in frequency or severity during the early months of therapy when urate is being mobilized from affected joints. Therefore, therapy with uricosuric agents should not be initiated during an acute attack but may be continued if already begun. Colchicine in small doses (0.5 to 1.8 mg per day) may be administered at this period to reduce the frequency of attacks. When an acute attack occurs, it is treated with an antiinflammatory drug such as indomethacin or naproxen. The use of salicylates is contraindicated both because they can elevate uric acid levels and they antagonize the action of probenecid and sulfinpyrazone. In the treatment of gout, the uricosuric drugs are given continually in the lowest dose that will maintain satisfactory plasma uric acid concentrations. Since the pKa of uric acid is 5.6 and the solubility of the undissociated form is very low, maintaining the output of a large volume of alkaline urine minimizes its intrarenal deposition. This precaution is essential during the early weeks of therapy when uric acid excretion is large, especially in patients with a history of renal disease associated with the passage of urate stones or gravel. Eventual improvement in renal function in patients with gouty nephropathy has been reported, but it is uncommon. The use of allopurinol permits a more favorable prognosis in such patients. Acute attacks of gout can be treated effectively with colchicine or a
nonsalicylate-containing NSAID, as discussed above. Because of the greater
frequency of toxicity with colchicine, use of NSAIDs is the preferred
treatment of acute gout. After the acute arthritis has responded to therapy,
the patient should be evaluated in order to select a rational regimen for
long-term management. Elevated concentrations of uric acid in plasma and the
observation of crystals of urate in the aspirated fluid from an affected
joint establish the diagnosis of hyperuricemia and symptomatic gout. When
evaluated on a diet that is low in purines, patients with hyperuricemia can
be categorized with regard to quantities of uric acid excreted in the urine.
About 80% to 90% of such individuals excrete less than 600 mg of uric acid
daily; the remainder excrete more than this amount due to excessive synthesis
of urate. The former group can be managed effectively with uricosuric agents;
the latter, however, is logically treated with allopurinol. If deposits of
urate are evident as tophi, renal stones, or renal insufficiency, allopurinol
is the preferred drug. During the first several months of treatment with
allopurinol, colchicine may be given simultaneously to prevent acute attacks
of gout. Patients with mild-to-moderate hyperuricemia (7 to 9 mg/dl;
equivalent of 420 to 530 Drug-induced hyperuricemia most commonly is caused by diuretics (see Chapter 29: Diuretics); acute attacks of gout are only rarely caused by such agents. However, hyperuricemia that accompanies chemotherapy or radiotherapy for various neoplasms may be considerably more severe and usually is treated prophylactically with allopurinol and hydration. |
Prospectus
|
Nonsteroidal antiinflammatory drugs are efficacious in providing symptomatic relief, but all available agents have associated, and sometimes severe, toxicity. These agents have been highly useful for treatment of acute, self-limited inflammatory conditions. However, their ability to modify disease progression in chronic inflammatory settings is not well documented and remains an area of continuing controversy. In contrast is the efficacy of agents, such as allopurinol, in the treatment of patients with gout in whom not only is there a regression of signs and symptoms but also an arrest of disease progression. Advances in understanding the pathobiology of the inflammatory process have suggested several novel approaches for development of drugs to block this process. These include: (1) cytokine inhibitors, (2) inhibitors of cell adhesion molecules, (3) phospholipase A2 inhibitors, (4) inhibitors of lipoxygenase and leukotriene receptors, and (5) isoform specificinhibitors of cyclooxygenase. Agents that modify the production or action of 'proinflammatory' cytokines, such as IL-1, TNF, IL-8, and others, are under study. Multiple approaches are in development or clinical trial, including use of antibodies or antibody fragments, molecules to block cytokine generation, and endogenous (e.g., IL-1ra) and synthetic receptor antagonists. The molecular cloning of receptors for many of the cytokines may provide structure-based therapeutic agents. For example, tenidap sodium, a drug already in clinical studies, appears to be an IL-1synthesis inhibitor and/or IL-1receptor antagonist, although it probably has other activities as well. Antagonists to various peptides that contribute to cytokine-mediated responses (e.g., substance P, bradykinin) also are in development. Inhibition of cell adhesion molecules is a fertile area for development of new types of antiinflammatory agents. Multiple approaches are under investigation. These include soluble fragments of receptors to bind cell adhesion molecules and use of antibodies, peptides, and carbohydrate moieties to block cell adhesion molecules (see Bevilacqua and Nelson, 1993; Rao et al., 1994). Most available NSAIDs have been directed against cyclooxygenase.
Although some agents have been developed that inhibit lipoxygenase or
leukotriene receptors, the possibility of developing agents that block both
proteins by structural modification of known cyclooxygenase inhibitors is
being pursued. In addition, efforts continue to identify agents that will be
directed against lipases involved in generation of free arachidonic acid or
their regulatory proteins. The goal is to develop compounds whose antiinflammatory
activity will resemble the glucocorticoids but whose toxicity will be less
frequent and severe than that of the steroids (see Bomalaski and One of the most important advancements in antiinflammatory drugs has been the identification of selective inhibitors of COX-2, the inflammation-induced form of the enzyme. The notion that blockade of COX-1 is responsible for many of the side effects of currently available NSAIDs while blockade of COX-2 mediates the antiinflammatory activity of the drugs has spurred efforts to develop COX-2-specific agents. In addition to the COX-2-selective drugs discussed in this chapter, several other second-generation agents with more favorable pharmacokinetics and/or higher selectivity are under development. The currently available drugs appear to have a high degree of safety, especially in regards to gastric toxicity. Postmarketing surveillance and further studies should shed light on whether or not the safety of these drugs is sustained during chronic long-term therapy and whether or not the ability of these agents to significantly inhibit prostacyclin production might have clinical relevance in some situations. It is important to remember that inflammation represents a series of homeostatic events that have evolved to aid in our survival in the face of pathogens and tissue injury. Viewed in this context, 'better' antiinflammatory therapy runs the risk of blocking such events and thereby doing more harm than good. Beyond the global issue of survival, blockade of physiologically important mechanisms (such as prostaglandin-, leukotriene-, cell adhesion molecule-, or cytokine-mediated events) likely will be associated with some degree of cellular and organ system toxicity. Thus, it may be difficult, or impossible, to avoid toxicity with antiinflammatory drugs targeted against such mechanisms. For further discussion of rheumatoid arthritis, osteoarthritis, and
gout, see Chapters 301, 312, and 313, respectively, in Harrison's
Principles of Internal Medicine, 16th ed., Acknowledgment The authors wish to acknowledge Dr. Paul A. Insel, the author of this chapter in the ninth edition of Goodman and Gilman's The Pharmacological Basis of Therapeutics, some of whose text we have retained in this edition. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 2868
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved