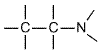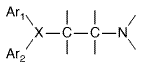| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Histamine, Bradykinin, and Their Antagonists
Overview
|
This chapter describes the physiological role and pathophysical consequences of histamine release and provides a summary of the therapeutic use of histamine H1-receptor antagonists. H2-receptor antagonists are discussed in detail in Chapter 37: Agents Used for Control of Gastric Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease in the context of prevention and treatment of peptic ulcers, their principal therapeutic application. The identity and role of H2-receptor subtypes are described briefly, as are the newly developed H3 agonists and antagonists, although none has been approved by the U.S. Food and Drug Administration (FDA) for clinical use to date. The second part of the chapter describes the physiology and pathophysiology of the kinins and kallidins, a subset of autacoids that contribute to the inflammatory response. The identification of at least two distinct receptors for kinins, designated B1 and B2, allows for the development of selective receptor antagonists, which also are discussed. Serotonin (5-hydroxytryptamine; 5-HT), another autacoid often considered in the same context as histamine and the kinin and kallidin agents, is discussed in detail in Chapter 11: 5-Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists. |
Histamine
|
History The history of When Dale and Laidlaw (1910, 1911) subjected histamine to intensive pharmacological study, they discovered that it stimulated a host of smooth muscles and had an intense vasodepressor action. Remarkably, they pointed out that the immediate signs displayed by a sensitized animal when injected with a normally inert protein closely resemble those of poisoning by histamine. These comments anticipated by many years the discovery of the presence of histamine in the body and its release during immediate hypersensitivity reactions and upon cellular injury. It was not until 1927 that Best et al. isolated histamine from very fresh samples of liver and lung, thereby establishing that this amine is a natural constituent of the body. Demonstrations of its presence in a variety of other tissues soon followedhence the name histamine after the Greek word for tissue, histos. Meanwhile, Lewis and his colleagues had amassed evidence that a substance with the properties of histamine ('H-substance') was liberated from the cells of the skin by injurious stimuli, including the reaction of antigen with antibody (Lewis, 1927). Given the chemical evidence of histamine's presence in the body, there remained little impediment to supposing that Lewis' 'H-substance' was histamine itself. It is now evident that endogenous histamine plays a role in the immediate allergic response and is an important regulator of gastric acid secretion. More recently, a role for histamine as a modulator of neurotransmitter release in the central and peripheral nervous systems also has emerged. Early suspicions that histamine acts through more than one receptor have been borne out, and it is clear that there are at least three distinct classes of receptors for histamine, designated H1 (Ash and Schild, 1966), H2 (Black et al., 1972), and H3 (Arrang et al., 1983). H1 receptors are blocked selectively by the classical 'antihistamines' (such as pyrilamine) developed around 1940. H2-receptor antagonists were introduced in the early 1970s. The discovery of H2 antagonists has contributed greatly to the resurgence of interest in histamine in biology and clinical medicine (see Chapter 37: Agents Used for Control of Gastric Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease). H3 receptors were originally discovered as a presynaptic autoreceptor on histamine-containing neurons that mediate feedback inhibition of the release and synthesis of histamine. The recent development of selective H3-receptor agonists and antagonists has led to an increased understanding of the importance of H3 receptors in histaminergic neurons in vivo. None of these H3-receptor agonists or antagonists, however, has yet emerged as a therapeutic agent. Renewed interest in clinical use of H1-receptor antagonists has occurred over the past 15 years due to the development of second-generation antagonists, collectively referred to as nonsedating antihistamines. Chemistry Histamine is a hydrophilic molecule comprising an imidazole ring and
an amino group connected by two methylene groups. The pharmacologically
active form at all histamine receptors is the monocationic N
Distribution and Biosynthesis of Histamine Distribution Histamine is widely, if unevenly, distributed throughout the animal
kingdom and is present in many venoms, bacteria, and plants. Almost all
mammalian tissues contain histamine in amounts ranging from less than 1 Synthesis, Storage, and Metabolism Histamine, in the amounts normally ingested or formed by bacteria in
the gastrointestinal tract, is rapidly metabolized and eliminated in the
urine. Every mammalian tissue that contains histamine is capable of
synthesizing it from histidine by virtue of its content of L-histidine decarboxylase. The
chief site of histamine storage in most tissues is the mast cell; in the
blood, it is the basophil. These cells synthesize histamine and store it in
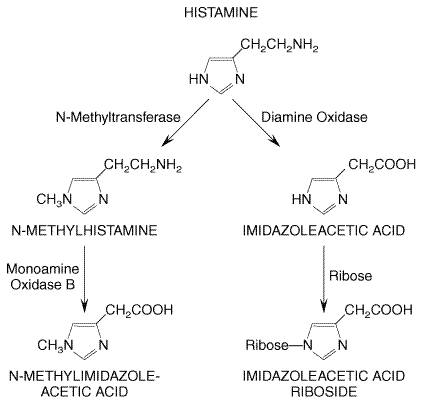
secretory granules. At the secretory granule pH of There are two major paths of histamine metabolism in human beings (Figure 252). The more important of these involves ring methylation to form N-methylhistamine. This is catalyzed by histamine-N-methyltransferase, which is widely distributed. Most of the N-methylhistamine formed is then converted by monoamine oxidase (MAO) to N-methylimidazoleacetic acid. This reaction can be blocked by MAO inhibitors (see Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders). Alternatively, histamine undergoes oxidative deamination catalyzed mainly by the nonspecific enzyme diamine oxidase (DAO), yielding imidazoleacetic acid, which is then converted to imidazoleacetic acid riboside. These metabolites have little or no activity and are excreted in the urine. One important aspect regarding these metabolites, however, is that it has been shown that measurement of N-methylhistamine in urine affords a more reliable index of endogenous histamine production than does measurement of histamine, because it circumvents the problem of artifactually elevated levels of histamine in urine that can arise from the ability of some genitourinary tract bacteria to decarboxylate histidine (Roberts and Oates, 1991). In addition, the metabolism of histamine appears to be altered in patients with mastocytosis such that measurement of histamine metabolites has been shown to be a more sensitive diagnostic indicator of the disease than is measurement of histamine (Keyzer et al., 1983).
Functions of Endogenous Histamine Histamine has important physiological roles. Because histamine is one of the preformed mediators stored in the mast cell, its release as a result of the interaction of antigen with IgE antibodies on the mast cell surface plays a central role in immediate hypersensitivity and allergic responses. The actions of histamine on bronchial smooth muscle and blood vessels account in part for the symptoms of the allergic response. In addition, certain clinically useful drugs can act directly on mast cells to release histamine, thereby explaining some of their untoward effects. Histamine has a major role in the regulation of gastric acid secretion, and its function as a modulator of neurotransmitter release has recently become appreciated. Role in Allergic Responses The principal target cells of immediate hypersensitivity reactions are
mast cells and basophils (Galli, 1993; Schwartz, 1994). As part of the allergic
response to an antigen, reaginic antibodies (IgE) are generated and bind to
the surface of mast cells and basophils via high-affinity Fc
receptors that are specific for IgE. This receptor, Release of Other Autacoids The release of histamine provides only a partial explanation for all of the biological effects that ensue from immediate hypersensitivity reactions. This is because a broad spectrum of other inflammatory mediators is released upon mast cell activation. In addition to activation of phospholipase C and the hydrolysis of inositol phospholipids, stimulation of IgE receptors also activates phospholipase A2, leading to the production of a host of mediators, including platelet-activating factor (PAF) and metabolites of arachidonic acid. Leukotriene D4, which is generated in this way, is a potent contractor of the smooth muscle of the bronchial tree (see Chapters 26: Lipid-Derived Autacoids: Eicosanoids and Platelet-Activating Factor and 28: Drugs Used in the Treatment of Asthma). Kinins also are generated during some allergic responses (see below). Thus, the mast cell secretes a variety of inflammatory compounds in addition to histamine, and each contributes to varying extents to the major symptoms of the allergic response: constriction of the bronchi, decrease in blood pressure, increased capillary permeability, and edema formation (see below). Regulation of Mediator Release The wide variety of mediators released during the allergic response explains the ineffectiveness of drug therapy focused on a single mediator. Considerable emphasis has been placed on the regulation of mediator release from mast cells and basophils, and these cells do contain receptors linked to signaling systems that can enhance or block the IgE-induced release of mediators. Agents that act at muscarinic or Histamine Release by Drugs, Peptides, Venoms, and Other Agents Many compounds, including a large number of therapeutic agents, stimulate the release of histamine from mast cells directly and without prior sensitization. Responses of this sort are most likely to occur following intravenous injections of certain categories of substances, particularly those that are organic bases. Among these bases are amides, amidines, quaternary ammonium compounds, pyridinium compounds, piperidines, alkaloids, and antibiotic bases. Tubocurarine, succinylcholine, morphine, radiocontrast media, and certain carbohydrate plasma expanders also may elicit the response. The phenomenon is one of clinical concern, for it may account for unexpected anaphylactoid reactions. Vancomycin-induced 'red-man syndrome' involving upper body and facial flushing and hypotension may be mediated, at least in part if not entirely, through histamine release (Levy et al., 1987). In addition to therapeutic agents, certain experimental compounds stimulate the release of histamine as their dominant pharmacological characteristic. The archetype is the polybasic substance known as compound 48/80. This is a mixture of low-molecular-weight polymers of p-methoxy-N-methylphenethylamine, of which the hexamer is most active (see Lagunoff et al., 1983). Basic polypeptides often are effective histamine releasers, and their potency generally increases with the number of basic groups over a limited range. Polymyxin B is very active; others include bradykinin and substance P. Since basic polypeptides are released upon tissue injury or are present in venoms, they constitute pathophysiological stimuli to secretion for mast cells and basophils. Anaphylotoxins (C3a and C5a), which are low-molecular-weight peptides that are cleaved from the complement system, may act similarly. Within seconds of the intravenous injection of a histamine liberator, human subjects experience a burning, itching sensation. This effect, most marked in the palms of the hand and in the face, scalp, and ears, is soon followed by a feeling of intense warmth. The skin reddens, and the color rapidly spreads over the trunk. Blood pressure falls, the heart rate accelerates, and the subject usually complains of headache. After a few minutes, blood pressure recovers, and crops of hives usually appear on the skin. Colic, nausea, hypersecretion of acid, and moderate bronchospasm also occur frequently. The effect becomes less intense with successive injections as the mast-cell stores of histamine are depleted. Histamine liberators do not deplete tissues of non-mast-cell histamine. Mechanism All of the above-mentioned histamine-releasing substances can activate the secretory response of mast cells or basophils by causing a rise in intracellular Ca2+. Some are ionophores and transport Ca2+ into the cell; others, such as the anaphylotoxins, appear to act like specific antigens to increase membrane permeability to Ca2+. Still others, such as mastoparan (a peptide from wasp venom), may bypass cell-surface receptors and directly stimulate guanine nucleotidebinding regulatory proteins (G proteins), which then activate phospholipase C (Higashijima et al., 1988). Basic histamine releasers, such as compound 48/80 and polymyxin B, act principally by mobilizing Ca2+ from cellular stores (see Lagunoff et al., 1983). Histamine Release by Other Means Clinical conditions in which release of histamine occurs in response to other stimuli include cold urticaria, cholinergic urticaria, and solar urticaria. Some of these involve specific secretory responses of the mast cells and, indeed, cell-fixed IgE. However, histamine release also occurs whenever there is nonspecific cell damage from any cause. The redness and urticaria that follow scratching of the skin is a familiar example. Gastric Carcinoid Tumors and Increased Proliferation of Mast Cells and Basophils In urticaria pigmentosa (cutaneous mastocytosis), mast cells aggregate in the upper corium and give rise to pigmented cutaneous lesions that urticate when stroked. In systemic mastocytosis, overproliferation of mast cells also is found in other organs. Patients with these syndromes suffer a constellation of signs and symptoms attributable to excessive histamine release, including urticaria, dermographism, pruritus, headache, weakness, hypotension, flushing of the face, and a variety of gastrointestinal effects such as peptic ulceration. Episodes of mast cell activation with attendant systemic histamine release are precipitated by a variety of stimuli, including exertion, emotional upset, and exposure to heat, and from exposure to drugs that release histamine directly or to which patients are allergic. In myelogenous leukemia, excessive numbers of basophils are present in the blood raising its histamine content to high levels, which may contribute to chronic pruritus. Gastric carcinoid tumors secrete histamine, which is responsible for episodes of vasodilation and contributes to the patchy 'geographical' flush (Roberts et al., 1979). Gastric Acid Secretion Histamine is a powerful gastric secretagogue and evokes a copious secretion of acid from parietal cells by acting on H2 receptors. The output of pepsin and intrinsic factor also is increased. However, the secretion of acid also is evoked by stimulation of the vagus nerve and by the enteric hormone gastrin. In addition, there appear to be cells in the gastric mucosa that contain somatostatin, which can inhibit secretion of acid by parietal cells; the release of somatostatin is inhibited by acetylcholine. The interplay among these endogenous regulators has not been precisely defined. However, it is clear that histamine is the dominant physiological mediator of acid secretion because blockade of H2 receptors can not only eradicate acid secretion in response to histamine, but also cause nearly complete inhibition of responses to gastrin or vagal stimulation. This is discussed in more detail in Chapter 37: Agents Used for Control of Gastric Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease. Central Nervous System There is substantial evidence that histamine functions as a neurotransmitter in the CNS. Histamine, histidine decarboxylase, and enzymes that catalyze the degradation of histamine are distributed nonuniformly in the CNS and are concentrated in synaptosomal fractions of brain homogenates. H1 receptors are found throughout the CNS and are densely concentrated in the hypothalamus. Histamine increases wakefulness via H1 receptors (Monti, 1993), explaining the potential for sedation by classical antihistamines. Histamine acting through H1 receptors inhibits appetite (Ookuma et al., 1993). Histamine-containing neurons may participate in the regulation of drinking, body temperature, and the secretion of antidiuretic hormone, as well as in the control of blood pressure and the perception of pain. Both H1 and H2 receptors seem to be involved in these responses (see Hough, 1988). Pharmacological Effects: H1 and H2 Receptors Once released, histamine can exert local or widespread effects on smooth muscles and glands. The autacoid contracts many smooth muscles, such as those of the bronchi and gut, but powerfully relaxes others, including those of small blood vessels. It also is a potent stimulus to gastric acid secretion. Effects attributable to these actions dominate the overall response to histamine; however, there are other effects, such as formation of edema and stimulation of sensory nerve endings. Many of these effects, such as bronchoconstriction and contraction of the gut, are mediated by H1 receptors (Ash and Schild, 1966). Other effects, most notably gastric secretion, are the results of activation of H2 receptors and, accordingly, can be inhibited by H2-receptor antagonists (Black et al., 1972). Some responses, such as the hypotension that results from vascular dilation, are mediated by both H1 and H2 receptors. Histamine Toxicity from Ingestion Histamine has been identified as the toxin in food poisoning from spoiled scombroid fish, such as tuna (Morrow et al., 1991). Bacteria in spoiled scombroid fish, which have a high histidine content, decarboxylate histidine to form large quantities of histamine. Ingestion of the fish causes severe nausea, vomiting, headache, flushing, and sweating. Histamine toxicity, manifested by headache and other symptoms, also can be seen following red wine consumption in persons who possibly have a diminished ability to degrade histamine (Wantke et al., 1994). The symptoms of histamine poisoning can be suppressed by H1 receptor antagonists. Cardiovascular System Histamine characteristically causes dilation of small blood vessels, resulting in flushing, lowered total peripheral resistance, and a fall in systemic blood pressure. In addition, histamine tends to increase capillary permeability. Vasodilation This is the characteristic action of histamine on the vasculature, and it is by far the most important vascular effect of histamine in human beings. Vasodilation involves both H1 and H2 receptors distributed throughout the resistance vessels in most vascular beds; however, quantitative differences are apparent in the degree of dilation that occurs in various beds. Activation of either the H1 or H2 type of histamine receptor can elicit maximal vasodilation, but the responses differ in their sensitivity to histamine, in the duration of the effect, and in the mechanism of their production. H1 receptors have the higher affinity for histamine and mediate a dilator response that is relatively rapid in onset and short lived. By contrast, activation of H2 receptors causes dilation that develops more slowly and is more sustained. As a result, H1 antagonists effectively counter small dilator responses to low concentrations of histamine but only blunt the initial phase of larger responses to higher concentrations of the amine. H2 receptors are located on vascular smooth muscle cells, and the vasodilator effects produced by their stimulation are mediated by cyclic AMP; H1 receptors reside on endothelial cells, and their stimulation leads to the formation of local vasodilator substances (see below). Increased 'Capillary' Permeability This classical effect of histamine on small vessels results in outward passage of plasma protein and fluid into the extracellular spaces, an increase in the flow of lymph and its protein content, and formation of edema. H1 receptors clearly are important for this response; whether or not H2 receptors also participate is uncertain. Increased permeability results mainly from actions of histamine on postcapillary venules, where histamine causes the endothelial cells to contract and separate at their boundaries and thus to expose the basement membrane, which is freely permeable to plasma protein and fluid. The gaps between endothelial cells also may permit passage of circulating cells that are recruited to the tissues during the mast-cell response. Recruitment of circulating leukocytes is promoted by H1-receptormediated upregulation of leukocyte adhesion. This process involves histamine-induced expression of the adhesion molecule P-selectin on the endothelial cells (Gaboury et al., 1995). Triple Response If histamine is injected intradermally, it elicits a characteristic phenomenon known as the 'triple response' (Lewis, 1927). This consists of (1) a localized red spot, extending for a few millimeters around the site of injection, that appears within a few seconds and reaches a maximum in about a minute; (2) a brighter red flush, or 'flare,' extending about 1 cm or so beyond the original red spot and developing more slowly; and (3) a wheal that is discernible in 1 to 2 minutes and occupies the same area as the original small red spot at the injection site. The red spot results from the direct vasodilatory effect of histamine, the flare is due to histamine-induced stimulation of axon reflexes that cause vasodilation indirectly, and the wheal reflects histamine's capacity to increase capillary permeability. Constriction of Larger Vessels Histamine tends to constrict larger blood vessels, in some species more than in others. In rodents, the effect extends to the level of the arterioles and may overshadow dilation of the finer blood vessels. A net increase in total peripheral resistance and an elevation of blood pressure can be observed. Heart Histamine has direct actions on the heart that affect both contractility and electrical events. It increases the force of contraction of both atrial and ventricular muscle by promoting the influx of Ca2+, and it speeds heart rate by hastening diastolic depolarization in the SA node. It also acts directly to slow AV conduction, to increase automaticity, and, in high doses especially, to elicit arrhythmias. With the exception of slowed AV conduction, which involves mainly H1 receptors, all these effects are largely attributable to H2 receptors. If histamine is given intravenously, direct cardiac effects of histamine are not prominent and are overshadowed by baroreceptor reflexes elicited by the reduced blood pressure. Histamine Shock Histamine given in large doses or released during systemic anaphylaxis causes a profound and progressive fall in blood pressure. As the small blood vessels dilate, they trap large amounts of blood, and as their permeability increases, plasma escapes from the circulation. Resembling surgical or traumatic shock, these effects diminish effective blood volume, reduce venous return, and greatly lower cardiac output. Extravascular Smooth Muscle Histamine stimulates, or more rarely relaxes, various smooth muscles. Contraction is due to activation of H1 receptors and relaxation (for the most part) to activation of H2 receptors. Responses vary widely, even in individuals (see Parsons, in Ganellin and Parsons, 1982). Bronchial muscle of guinea pigs is exquisitely sensitive. Minute doses of histamine also will evoke intense bronchoconstriction in patients with bronchial asthma and certain other pulmonary diseases; in normal human beings the effect is much less pronounced. Although the spasmogenic influence of H1 receptors is dominant in human bronchial muscle, H2 receptors with dilator function also are present. Thus, histamine-induced bronchospasm in vitro is potentiated slightly by H2 blockade. In asthmatic subjects in particular, histamine-induced bronchospasm may involve an additional, reflex component that arises from irritation of afferent vagal nerve endings (see Eyre and Chand, in Ganellin and Parsons, 1982; Nadel and Barnes, 1984). The uterus of some species contracts to histamine; in the human uterus, gravid or not, the response is negligible. Responses of intestinal muscle also vary with species and region, but the classical effect is contraction. Bladder, ureter, gallbladder, iris, and many other smooth muscle preparations are affected little or inconsistently by histamine. Exocrine Glands As mentioned above, histamine is an important physiological regulator of gastric acid secretion. This effect is mediated by H2 receptors (see Chapter 37: Agents Used for Control of Gastric Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease). Nerve Endings: Pain, Itch, and Indirect Effects Histamine stimulates various nerve endings. Thus, when released in the epidermis, it causes itch; in the dermis, it evokes pain, sometimes accompanied by itching. Stimulant actions on one or another type of nerve ending, including autonomic afferents and efferents, have been mentioned above as factors that contribute to the 'flare' component of the triple response and to indirect effects of histamine on the bronchi and other organs. In the periphery, neuronal receptors for histamine are generally of the H1 type (see Rocha e Silva, 1978; Ganellin and Parsons, 1982). Mechanism of Action The H1 and H2 receptors have been cloned and shown to belong to the superfamily of G proteincoupled receptors. H1 receptors are coupled to phospholipase C, and their activation leads to formation of inositol-1,4,5-trisphosphate (IP3) and diacylglycerols from phospholipids in the cell membrane; IP3 causes a rapid release of Ca2+ from the endoplasmic reticulum. Diacylglycerols (and Ca2+) activate protein kinase C, while Ca2+ activates Ca2+/calmodulin-dependent protein kinases and phospholipase A2 in the target cell to generate the characteristic response. H2 receptors are linked to the stimulation of adenylyl cyclase and thus to the activation of cyclic AMPdependent protein kinase in the target cell. In a species-dependent manner, adenosine receptors may interact with H1 receptors. In the CNS of human beings, activation of adenosine A1 receptors inhibits second messenger generation via H1 receptors. A possible mechanism for this is interaction (termed cross-talk) between the G proteins to which the A1 and H1 receptors are coupled functionally (Dickenson and Hill, 1993). In the smooth muscle of large blood vessels, bronchi, and intestine, the stimulation of H1 receptors and the resultant IP3-mediated release of intracellular Ca2+ leads to activation of the Ca2+/calmodulin-dependent myosin light chain kinase. This enzyme phosphorylates the 20,000 dalton myosin light chain, with resultant enhancement of cross-bridge cycling and contraction. The effects of histamine on sensory nerves also are mediated by H1 receptors. As mentioned above, the vasodilator effects of histamine are mediated by both H1 and H2 receptors that are located on different cell types in the vascular bed: H1 receptors on the vascular endothelial cells and H2 receptors on smooth muscle cells. Activation of H1 receptors leads to increased intracellular Ca2+, activation of phospholipase A2, and the local production of endothelium-derived relaxing factor, which is nitric oxide (Palmer et al., 1987). Nitric oxide diffuses to the smooth muscle cell, where it activates a soluble guanylyl cyclase and causes the accumulation of cyclic GMP. Stimulation of a cyclic GMPdependent protein kinase and a decrease in intracellular Ca2+ are thought to be involved in the relaxation caused by this cyclic nucleotide. The activation of phospholipase A2 in endothelial cells also leads to the formation of prostaglandins, predominantly prostacyclin (PGI2); this vasodilator makes an important contribution to endothelium-mediated vasodilation in some vascular beds. The mechanism of cyclic AMPmediated relaxation of smooth muscle is
not entirely clear, but it is presumed to involve a decrease in intracellular
Ca2+ (see Taylor et al., 1989). Cyclic AMPmediated actions
in the heart, mast cells, basophils, and other tissues also are understood
incompletely, but the effects of histamine that are mediated by H2
receptors obviously would be produced in the same fashion as those resulting
from stimulation of Clinical Uses The practical applications of histamine are limited to uses as a diagnostic agent. Histamine (histamine phosphate) is used to assess nonspecific bronchial hyperreactivity in asthmatics and as a positive control injection during allergy skin testing. |
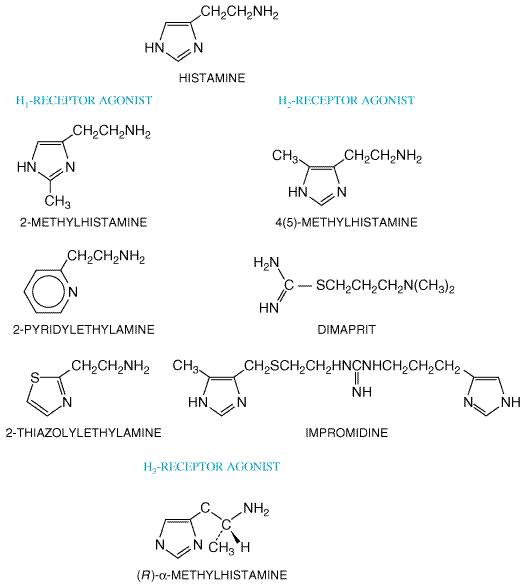
H1-Receptor Antagonists
|
Although antagonists that act selectively at the three types of histamine receptors have been developed, this discussion is confined to the properties and clinical uses of H1 antagonists. Specific H2 antagonists (e.g., cimetidine, ranitidine) are used extensively in the treatment of peptic ulcers; these are discussed in Chapter 37: Agents Used for Control of Gastric Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease. The properties of agonists and antagonists at H3 receptors are discussed later in this chapter. Such agents are not yet available for clinical use. History Histamine-blocking activity was first detected in 1937 by Bovet and Staub in one of a series of amines with a phenolic ether function. The substance, 2-isopropyl-5-methylphenoxy-ethyldiethyl-amine, protected guinea pigs against several lethal doses of histamine, antagonized histamine-induced spasm of various smooth muscles, and lessened the symptoms of anaphylactic shock. This drug was too toxic for clinical use, but by 1944, Bovet and his colleagues had described pyrilamine maleate, which is still one of the most specific and effective histamine antagonists of this category. The discovery of the highly effective histamine antagonists diphenhydramine and tripelennamine soon followed (see Bovet, 1950; Ganellin, in Ganellin and Parsons, 1982). In the 1980s, nonsedating H1-histaminereceptor antagonists were developed for treatment of allergic diseases. By the early 1950s, many compounds with histamine-blocking activity were available to physicians, but they uniformly failed to inhibit certain responses to histamine, most conspicuously gastric acid secretion. The discovery by Black and colleagues of a new class of drugs that blocked histamine-induced gastric acid secretion provided new pharmacological tools with which to explore the functions of endogenous histamine. This discovery ushered in a major new class of therapeutic agents, the H2 receptor antagonists, including cimetidine (TAGAMET), famotidine (PEPCID), nizatidine (AXID), and ranitidine (ZANTAC) (see Chapter 37: Agents Used for Control of Gastric Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease). StructureActivity Relationship All of the available H1 receptor antagonists are
reversible, competitive inhibitors of the interaction of histamine with H1
receptors. Like histamine, many H1 antagonists contain a substituted
ethylamine moiety,
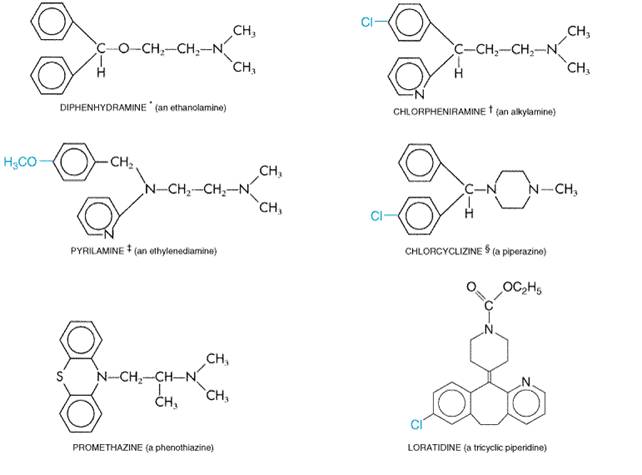
where Ar is aryl and X is a nitrogen or carbon atom or a CO ether linkage to the beta-aminoethyl side chain. Sometimes the two aromatic rings are bridged, as in the tricyclic derivatives, or the ethylamine may be part of a ring structure (Figure 253). (see Ganellin, in Ganellin and Parsons, 1982.)
Pharmacological Properties Most H1 antagonists have similar pharmacological actions and therapeutic applications and can be discussed together conveniently. Their effects are largely predictable from knowledge of the responses to histamine that involve interaction with H1 receptors. Smooth Muscle H1 antagonists inhibit most responses of smooth muscle to histamine. Antagonism of the constrictor action of histamine on respiratory smooth muscle is easily shown in vivo or in vitro. In guinea pigs, for example, death by asphyxia follows quite small doses of histamine, yet the animal may survive a hundred lethal doses of histamine if given an H1 antagonist. In the same species, striking protection also is afforded against anaphylactic bronchospasm. This is not so in human beings, where allergic bronchoconstriction appears to be caused by a variety of mediators such as leukotrienes and platelet activating factor (see Chapter 26: Lipid-Derived Autacoids: Eicosanoids and Platelet-Activating Factor). Within the vascular tree, the H1 antagonists inhibit both the vasoconstrictor effects of histamine and, to a degree, the more rapid vasodilator effects that are mediated by H1 receptors on endothelial cells. Residual vasodilation reflects the involvement of H2 receptors on smooth muscle and can be suppressed only by the concurrent administration of an H2 antagonist. Effects of the histamine antagonists on histamine-induced changes in systemic blood pressure parallel these vascular effects. Capillary Permeability H1 antagonists strongly block the action of histamine that results in increased capillary permeability and formation of edema and wheal. Flare and Itch The flare component of the triple response and the itching caused by intradermal injection of histamine are two different manifestations of the action of histamine on nerve endings. H1 antagonists suppress both. Exocrine Glands Gastric secretion is not inhibited at all by H1 antagonists, and they suppress histamine-evoked salivary, lacrimal, and other exocrine secretions with variable responses. The atropine-like properties of many of these agents, however, may contribute to lessened secretion in cholinergically innervated glands and reduce ongoing secretion in, for example, the respiratory tree. Immediate Hypersensitivity Reactions: Anaphylaxis and Allergy During hypersensitivity reactions, histamine is one of many potent autacoids released (see above), and its relative contribution to the ensuing symptoms varies widely with species and tissue. The protection afforded by histamine antagonists thus also varies accordingly. In human beings, some phenomena, such as edema formation and itch, are effectively suppressed. Others, such as hypotension, are less so. This may be explained by the existence of other mast-cell mediators, specifically prostaglandin D2, also contributing to the vasodilation (Roberts et al., 1980). Bronchoconstriction is reduced little, if at all (see Dahln et al., 1983). Central Nervous System The first-generation H1 antagonists can both stimulate and depress the CNS. Stimulation occasionally is encountered in patients given conventional doses, who become restless, nervous, and unable to sleep. Central excitation also is a striking feature of poisoning, which commonly results in convulsions, particularly in infants. Central depression, on the other hand, is the usual accompaniment of therapeutic doses of the older H1 antagonists. Diminished alertness, slowed reaction times, and somnolence are common manifestations. Some of the H1 antagonists are more likely to depress the CNS than others, and patients vary in their susceptibility and responses to individual drugs. The ethanolamines (e.g., diphenhydramine; see Figure 253) are particularly prone to cause sedation. The second-generation ('nonsedating') H1 antagonists (e.g., loratadine, cetirizine, fexofenadine) are largely excluded from the brain when given in therapeutic doses, because they do not cross the bloodbrain barrier appreciably. Their effects on objective measures of sedation such as sleep latency, EEG, and standardized performance tests are similar to those of placebo (Simons and Simons, 1994). Because of the sedation that occurs with first-generation antihistamines, these drugs cannot be tolerated or used safely by many patients. Thus, the availability of nonsedating antihistamines has been an important advance that allows the general use of these agents. An interesting and useful property of certain H1 antagonists is the capacity to counter motion sickness. This effect was first observed with dimenhydrinate and subsequently with diphenhydramine (the active moiety of dimenhydrinate), various piperazine derivatives, and promethazine. The latter drug has perhaps the strongest muscarinic blocking activity among these agents and is among the most effective of the H1 antagonists in combating motion sickness (see below). Since scopolamine is the most potent drug for the prevention of motion sickness (see Chapter 7: Muscarinic Receptor Agonists and Antagonists), it is possible that the anticholinergic properties of certain H1 antagonists are largely responsible for this effect. Anticholinergic Effects Many of the first-generation H1 antagonists tend to inhibit responses to acetylcholine that are mediated by muscarinic receptors. These atropine-like actions are sufficiently prominent in some of the drugs to be manifest during clinical usage (see below). The second-generation H1 antagonists have no effect on muscarinic receptors. Local Anesthetic Effect Some H1 antagonists have local anesthetic activity, and a few are more potent than procaine. Promethazine (PHENERGAN) is especially active. However, the concentrations required for this effect are several orders higher than those that antagonize histamine. Absorption, Fate, and Excretion The H1 antagonists are well absorbed from the gastrointestinal tract. Following oral administration, peak plasma concentrations are achieved in 2 to 3 hours and effects usually last 4 to 6 hours; however, some of the drugs are much longer acting (Table 251). Extensive studies of the metabolic fate of the older H1 antagonists are limited. Diphenhydramine, given orally, reaches a maximal concentration in the blood in about 2 hours, remains at about this level for another 2 hours, and then falls exponentially with a plasma elimination half-time of about 4 to 8 hours. The drug is widely distributed throughout the body, including the CNS. Little, if any, is excreted unchanged in the urine; most appears there as metabolites. Other first-generation H1 antagonists appear to be eliminated in much the same way (see Paton and Webster, 1985). Information on the concentrations of these drugs achieved in the skin and mucous membranes is lacking. However, significant inhibition of 'wheal-and-flare' responses to the intradermal injection of histamine or allergen may persist for 36 hours or more after treatment with some longer-acting H1 antagonists, even when concentrations of the drugs in plasma are very low. Such results emphasize the need for flexibility in the interpretation of the recommended dosage schedules (see Table 251); less frequent dosage may suffice. Doxepin, a tricyclic antidepressant (see Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders), is one of the most potent antihistamines available; it is about 800 times more potent than diphenhydramine (Sullivan 1982; Richelson, 1979). This may account for the observation that doxepin can be effective in the treatment of chronic urticaria when other antihistamines have failed; it also is available as a topical preparation. Like many other drugs that are metabolized extensively, H1 antagonists are eliminated more rapidly by children than by adults and more slowly in those with severe liver disease. H1-receptor antagonists are among the many drugs that induce hepatic microsomal enzymes, and they may facilitate their own metabolism (see Paton and Webster, 1985; Simons and Simons, 1988). The second-generation H1 antagonist loratadine is rapidly absorbed from the gastrointestinal tract and metabolized in the liver to an active metabolite by the hepatic microsomal P450 system (Simons and Simons, 1994). Consequently, metabolism of this drug can be affected by competition for the P450 enzymes by other drugs. Two other second-generation H1 antagonists that had been marketed previously, astemizole and terfenadine, also underwent P450 metabolism to active metabolites. Both of these drugs were found in rare cases to induce a potentially fatal arrhythmia, torsades de pointes, when their metabolism was impaired, such as by liver disease or drugs that inhibit the 3A family of P450 enzymes. This led to the withdrawal of terfenadine and astemizole from the market in 1998 and 1999. Loratadine, cetirizine (the active metabolite of hydroxyzine), fexofenadine (the active metabolite of terfenadine), and azelastine lack the propensity to prolong repolarization and induce torsades de pointes (DuBuske, 1999). Cetirizine, loratadine, and fexofenadine are all well absorbed and are excreted mainly in the unmetabolized form. Cetirizine and loratadine are primarily excreted into the urine, whereas fexofenadine is primarily excreted in the feces (Brogden and McTavish, 1991; Spencer et al., 1993; Barnes et al., 1993; Russell et al., 1998). Side Effects Sedation and Other Common Adverse Effects The side effect with the highest incidence in the first-generation H1 antagonists, which is not a feature of the second-generation agents, is sedation. Although sedation may be a desirable adjunct in the treatment of some patients, it may interfere with the patient's daytime activities. Concurrent ingestion of alcohol or other CNS depressants produces an additive effect that impairs motor skills (Roehrs et al., 1993). Other untoward reactions referable to central actions include dizziness, tinnitus, lassitude, incoordination, fatigue, blurred vision, diplopia, euphoria, nervousness, insomnia, and tremors. The next most frequent side effects involve the digestive tract and include loss of appetite, nausea, vomiting, epigastric distress, and constipation or diarrhea. Their incidence may be reduced by giving the drug with meals. H1 antagonists appear to increase appetite and cause weight gain in rare patients. Other side effects that apparently are caused by the antimuscarinic actions of some of the first-generation H1-receptor antagonists include dryness of the mouth and respiratory passages, sometimes inducing cough, urinary retention or frequency, and dysuria. These effects are not observed with second-generation H1 antagonists. Mutagenicity Results of one short-term study (Brandes et al., 1994) with an unconventional mouse model indicated that melanoma and fibrosarcoma tumor lines had an increased rate of growth when injected into mice receiving certain H1 antagonists. However, conventional studies with animals and clinical experience do not suggest carcinogenicity for H1-receptor antagonists (Food and Drug Administration, 1994). Other Adverse Effects Drug allergy may develop when H1 antagonists are given orally, but more commonly it results from topical application. Allergic dermatitis is not uncommon; other hypersensitivity reactions include drug fever and photosensitization. Hematological complications such as leukopenia, agranulocytosis, and hemolytic anemia are very rare. Teratogenic effects have been noted in response to piperazine compounds, but extensive clinical studies have not demonstrated any association between the use of such H1 antagonists and fetal anomalies in human beings. Since H1 antagonists interfere with skin tests for allergy, they must be withdrawn well before such tests are performed. In acute poisoning with H1 antagonists, their central excitatory effects constitute the greatest danger. The syndrome includes hallucinations, excitement, ataxia, incoordination, athetosis, and convulsions. Fixed, dilated pupils with a flushed face, together with sinus tachycardia, urinary retention, dry mouth, and fever, lend the syndrome a remarkable similarity to that of atropine poisoning. Terminally, there is deepening coma with cardiorespiratory collapse and death, usually within 2 to 18 hours. Treatment is along general symptomatic and supportive lines. Available H1 Antagonists Below are summarized the therapeutic and side effects of a number of H1 antagonists, based on their chemical structure. Representative preparations are listed in Table 251. Dibenzoxepin Tricyclics (Doxepin) Doxepin is the only drug in this class. Doxepin is marketed as a tricyclic antidepressant (see Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders). However, it also is a remarkably potent H1 antagonist. It can cause drowsiness and is associated with anticholinergic effects. Doxepin is much better tolerated by patients who have depression than by those who do not. In nondepressed patients, sometimes even very small doses, e.g., 20 mg, may be poorly tolerated because of disorientation and confusion. Ethanolamines (Prototype: Diphenhydramine) The drugs in this group possess significant antimuscarinic activity and have a pronounced tendency to induce sedation. About half of those who are treated with conventional doses of these drugs experience somnolence. The incidence of gastrointestinal side effects, however, is low with this group. Ethylenediamines (Prototype: Pyrilamine) These include some of the most specific H1 antagonists. Although their central effects are relatively feeble, somnolence occurs in a fair proportion of patients. Gastrointestinal side effects are quite common. Alkylamines (Prototype: Chlorpheniramine) These are among the most potent H1 antagonists. The drugs are not so prone as some H1 antagonists to produce drowsiness and are among the more suitable agents for daytime use; but again, a significant proportion of patients do experience sedation. Side effects involving CNS stimulation are more common in this than in other groups. First-Generation Piperazines The oldest member of this group, chlorcyclizine, has a more prolonged action and produces a comparatively low incidence of drowsiness. Hydroxyzine is a long-acting compound that is widely used for skin allergies; its considerable CNS-depressant activity may contribute to its prominent antipruritic action. Cyclizine and meclizine have been used primarily to counter motion sickness, although promethazine and diphenhydramine (dimenhydrinate) are more effective (as is scopolamine; see below). Second-Generation Piperazines (Cetirizine) Cetirizine is the only drug in this class. It has minimal anticholinergic effects. It also has negligible penetration into the brain but is associated with a somewhat higher incidence of drowsiness than the other second-generation H1 antagonists. Phenothiazines (Prototype: Promethazine) Most drugs of this class are H1 antagonists and also possess considerable anticholinergic activity. Promethazine, which has prominent sedative effects, and its many congeners are now used primarily for their antiemetic effects (see Chapter 38: Prokinetic Agents, Antiemetics, and Agents Used in Irritable Bowel Syndrome). First-Generation Piperidines (Cyproheptadine, Phenindamine) Cyproheptadine is unique in that it has both antihistamine and antiserotonin activity. Cyproheptadine and phenindamine cause drowsiness and also have significant anticholinergic effects. Second-Generation Piperidines (Prototype: Terfenadine) As mentioned, terfenadine and astemizole were early marketed H1 antagonists in this class but have since been withdrawn because they induced the potentially fatal arrhythmia, torsades de pointes. The drugs currently marketed in this class, which are devoid of this side effect, are loratadine and fexofenadine. These agents are highly selective for H1 receptors and are devoid of significant anticholinergic actions. These agents also penetrate poorly into the CNS. Taken together, these properties appear to account for the low incidence of side effects of piperidine agents. Therapeutic Uses H1 antagonists have an established and valued place in the symptomatic treatment of various immediate hypersensitivity reactions. In addition, the central properties of some of the series are of therapeutic value for suppressing motion sickness or for sedation. Diseases of Allergy H1 antagonists are most useful in acute types of allergy that present with symptoms of rhinitis, urticaria, and conjunctivitis. Their effect, however, is confined to the suppression of symptoms attributable to the histamine released by the antigenantibody reaction. In bronchial asthma, histamine antagonists have limited beneficial effects and are not useful as sole therapy (see Chapter 28: Drugs Used in the Treatment of Asthma). In the treatment of systemic anaphylaxis, in which autacoids other than histamine play major roles, the mainstay of therapy is epinephrine, with histamine antagonists having only a subordinate and adjuvant role. The same is true for severe angioedema, in which laryngeal swelling constitutes a threat to life. Other allergies of the respiratory tract are more amenable to therapy
with H1 antagonists. The best results are obtained in seasonal
rhinitis and conjunctivitis (hay fever, pollinosis), in which these drugs
relieve the sneezing, rhinorrhea, and itching of eyes, nose, and throat. A
gratifying response is obtained in most patients, especially at the beginning
of the season when pollen counts are low; however, the drugs are less effective
when the allergens are in abundance, when exposure to them is prolonged, and
when nasal congestion has become prominent. Topical preparations of
antihistamines such as levocabastine (LIVOSTIN) have been shown to be effective
in allergic conjunctivitis and rhinitis (Janssens and Vanden Bussche, 1991).
A topical ophthalmic preparation of this agent is available in the Certain of the allergic dermatoses respond favorably to H1 antagonists. Benefit is most striking in acute urticaria, although the itching in this condition is perhaps better controlled than are the edema and the erythema. Chronic urticaria is less responsive, but some benefit may occur in a fair proportion of patients. Furthermore, the combined use of H1 and H2 antagonists is effective for some individuals if therapy with an H1 antagonist has failed. As mentioned above, doxepin is sometimes effective in the treatment of chronic urticaria that is refractory to other antihistamines. Angioedema also is responsive to treatment with H1 antagonists, but the paramount importance of epinephrine in the severe attack must be reemphasized, especially in the life-threatening involvement of the larynx (see Chapter 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists). Here, however, it may be appropriate to administer additionally an H1 antagonist by the intravenous route. H1 antagonists also have a place in the treatment of pruritus. Some relief may be obtained in many patients suffering atopic dermatitis and contact dermatitis (although topical corticosteroids are more effective) and in such diverse conditions as insect bites and ivy poisoning. Various other pruritides without an allergic basis sometimes respond to antihistamine therapy, usually when the drugs are applied topically but sometimes when they are given orally. However, the possibility of producing allergic dermatitis with local application of H1 antagonists must be recognized. Again, doxepin may be more effective in suppressing histamine-mediated symptoms in the skin, in this case pruritus, than are other antihistamines. Since these drugs inhibit allergic dermatoses, they should be withdrawn well before skin testing for allergies. The urticarial and edematous lesions of serum sickness respond to H1 antagonists, but fever and arthralgia often do not. Many drug reactions attributable to allergic phenomena respond to therapy with H1 antagonists, particularly those characterized by itch, urticaria, and angioedema; reactions of the serum-sickness type also respond to intensive treatment. However, explosive release of histamine generally calls for treatment with epinephrine, with H1 antagonists being accorded a subsidiary role. Nevertheless, prophylactic treatment with an H1 antagonist may suffice to reduce symptoms to a tolerable level when a drug known to be a histamine liberator is to be given. Common Cold Despite persistent popular belief, H1 antagonists are without value in combating the common cold. The weak anticholinergic effects of the older agents may tend to lessen rhinorrhea, but this drying effect may do more harm than good, as may their tendency to induce somnolence. Motion Sickness, Vertigo, and Sedation Although scopolamine, given orally, parenterally, or transdermally, is the most effective of all drugs for the prophylaxis and treatment of motion sickness, some H1 antagonists are useful in a broad range of milder conditions and offer the advantage of fewer adverse effects. These drugs include dimenhydrinate and the piperazines (e.g., cyclizine, meclizine). Promethazine, a phenothiazine, is more potent and more effective and its additional antiemetic properties may be of value in reducing vomiting, but its pronounced sedative action usually is disadvantageous. Whenever possible, the various drugs should be administered an hour or so before the anticipated motion. Dosing after the onset of nausea and vomiting rarely is beneficial. Some H1 antagonists, notably dimenhydrinate and meclizine, are often of benefit in vestibular disturbances, such as Meniere's disease, and in other types of true vertigo. Only promethazine has usefulness in treating the nausea and vomiting subsequent to chemotherapy or radiation therapy for malignancies; however, other effective antiemetic drugs are available (see Chapter 38: Prokinetic Agents, Antiemetics, and Agents Used in Irritable Bowel Syndrome). Diphenhydramine can be used to reverse the extrapyramidal side effects caused by phenothiazines. The anticholinergic actions of this agent also can be utilized in the early stages of treatment of patients with Parkinson's disease (see Chapter 22: Treatment of Central Nervous System Degenerative Disorders), but it is less effective than other agents such as trihexyphenidyl (ARTANE The tendency of certain of the H1-receptor antagonists to produce somnolence has led to their use as hypnotics. H1 antagonists, principally diphenhydramine, often are present in various proprietary remedies for insomnia that are sold over the counter. While these remedies generally are ineffective in the recommended doses, some sensitive individuals may derive benefit. The sedative and mild antianxiety activities of hydroxyzine and diphenhydramine have contributed to their use as weak anxiolytics. |
H3-ReceptorMediated Actions: Agonists and Antagonists
|
Originally the H3 receptor was
described as a presynaptic receptor present on histaminergic nerve terminals
in the CNS that exerted feedback regulation of histamine synthesis and
release (Arrang et al., 1983). Since then, H3 receptors have been
found to function in a wide variety of tissues as feedback inhibitors not
only of histamine but also of other neurotransmitters, including
acetylcholine, dopamine, norepinephrine, and serotonin (Leurs et al., 1998).
Like H1 and H2 receptors, H3 receptors are G
proteincoupled receptors; their occupation results in a decrease of Ca2+
influx into the cell. (R)- Many early H3 antagonists such as impromidine and burimamide had mixed effects, since they also were agonists for the H2 receptor. Thioperamide was the first specific H3 antagonist available experimentally (Timmerman, 1990). This compound is still the most widely used H3 antagonist and has potent pharmacological properties (see below). Other H3 antagonists being developed include the competitive inhibitor clobenpropit and the irreversible inhibitor N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline (EEDQ). H3 receptors are known to function as feedback inhibitors in a wide variety of organ systems. In the CNS, H3-receptor agonists cause sedation by opposing H1-induced wakefulness (Monti, 1993). In the gastrointestinal tract, H3 receptors antagonize H1-induced ileal contraction as well as downregulate histamine (and thus gastrin) levels through autoregulatory actions in the gastric mucosa (Hollande et al., 1993). The H1-bronchoconstrictor response is opposed by an H3-bronchodilatory response in the pulmonary tree. Ishikawa and Sperelakis (1987) first documented the existence of H3 receptors in the cardiovascular system. These authors documented that H3-receptor agonists depressed perivascular sympathetic neurotransmission and caused vasodilation in the guinea pig mesenteric arteries. Subsequently, H3 receptors were discovered on sympathetic nerve terminals in the human saphenous vein, where H3-receptor agonists inhibited sympathetic outflow and norepinephrine release (Molderings et al., 1992). In addition to interference with sympathetic vasoconstriction, H3 receptors also have been shown to have negative chronotrophic effects in the atria. H3 receptors probably have minimal effects in baseline normal states but may inhibit norepinephrine release during stresses such as ischemia (Imamura et al., 1994). Currently, much attention is focused on the therapeutic potential of ligands of the H3 receptor in a variety of pathological situations. Agonists have potential use as gastroprotective, antiinflammatory, and anticonvulsant agents and in the treatment of septic shock, heart failure, and myocardial infarction. Antagonists have potential use in treating obesity, cognitive dysfunction, and attention-deficithyperactivity disorder in children (Leurs et al., 2000). A number of potent, selective agonists and antagonists of H3 receptors have been developed, but none has yet been approved for clinical use. |
Bradykinin and Kallidin and Their Antagonists
|
A variety of factors including tissue damage, allergic reactions, viral infections, and other inflammatory events activate a series of proteolytic reactions that generate bradykinin and kallidin in the tissues (see Wachtfogel et al., 1993). These peptides are autacoids that act locally to produce pain, vasodilation, increased vascular permeability, and the synthesis of prostaglandins. Thus, they constitute a subset of the large number of mediators that contribute to the inflammatory response. During the past several years, a number of interesting discoveries have been made concerning kinins and their receptors. Kinin metabolites that were formerly considered inactive degradation products now are considered potent mediators of inflammation and pain. These peptides interact with specific receptors whose presence is induced by tissue injury. Based on this information, novel avenues for therapeutic intervention in chronic inflammatory conditions may be possible. History In the 1920s and 1930s, Frey and his associates Kraut and Werle characterized a hypotensive substance in urine and showed that similar material could be obtained from saliva, plasma, and a variety of tissues. Since the pancreas was a rich source, they named this material kallikrein after an old Greek synonym for that organ, kallikras. By 1937, Werle, Gtze, and Keppler had established that kallikreins generate a pharmacologically active substance from some inactive precursor present in plasma. In 1948, Werle and Berek named the active substance kallidin and showed it to be a polypeptide cleaved from a plasma globulin that they termed kallidinogen (see Werle, 1970). Interest in the field intensified when Rocha e Silva and associates (1949) reported that trypsin and certain snake venoms acted on plasma globulin to produce a substance that lowered blood pressure and caused a slowly developing contraction of the gut. Because of this slow response, they named this substance bradykinin, a term derived from the Greek words bradys, meaning 'slow,' and kinein, meaning 'to move.' In 1960, the nonapeptide bradykinin was isolated by Elliott and coworkers and synthesized by Boissonnas and associates. Shortly thereafter, kallidin was found to be a decapeptidebradykinin with an additional lysine residue at the amino terminus. These substances are members of a group of polypeptides with related chemical structures and pharmacological properties that are widely distributed in nature. For the whole group, the generic term kinins has been adopted, and kallidin and bradykinin are referred to as plasma kinins. In 1970, Ferreira et al. reported the isolation of a bradykinin-potentiating factor from the venom of the Brazilian snake, Bothrops, and Ondetti et al. (1971) subsequently reported the isolation of angiotensin converting-enzyme (ACE) inhibitors from the same venom. Later, it was shown that ACE and kininase II are the same enzyme (Erdos, 1977). ACE inhibitors (see Chapter 31: Renin and Angiotensin) now are widely used in the treatment of hypertension, diabetic nephropathy, congestive heart failure, and postmyocardial infarction. In 1980, Regoli and Barab divided the kinin receptors into B1 and B2 classes based on the rank order of potency of kinin analogs. The B1 and B2 receptors have now been cloned. The development of first-generation kinin-receptor antagonists occurred in the mid-1980s (Vavrek and Stewart, 1985). Second-generation, receptor-specific kinin antagonists were developed in the early 1990s. These antagonists have led to increasing understanding of the actions of kinins. The development of a B2-receptor 'knockout' mouse (Borkowski et al., 1995) has furthered our understanding of the role of bradykinin in the regulation of cardiovascular homeostasis. The Endogenous KallikreinKininogenKinin System Synthesis and Metabolism of Kinins Bradykinin is a nonapeptide (see Table 252). Kallidin has an
additional lysine residue at the amino-terminal position and is sometimes
referred to as lysyl-bradykinin. The two peptides are cleaved from
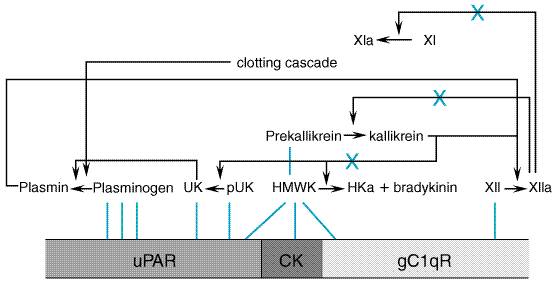
Kallikreins Bradykinin and kallidin are cleaved from high- and
low-molecular-weight kininogens by plasma or tissue kallikrein, respectively.
Plasma kallikrein and tissue kallikrein are distinct enzymes, and they are
activated by different mechanisms (Bhoola et al., 1992). Plasma prekallikrein
is an inactive protein of about 88,000 daltons that is bound in a 1:1 complex
with its substrate, HMW kininogen. The cascade is restrained by the protease
inhibitors present in plasma. Among the most important are the inhibitor of
the activated first component of complement (C1-INH) and The human tissue kallikrein family includes three members: true tissue kallikrein (hKLK1), prostate-specific antigen (PSA, hKLK3), and a PSA-like proteinase (hKLK2). Only true tissue kallikrein exhibits kininogenase activity. Compared to plasma kallikrein, tissue kallikrein is a smaller protein (molecular mass of 29,000 daltons). It is synthesized as a preproprotein in the epithelial cells or secretory cells of a number of tissues including salivary glands, pancreas, prostate, and distal nephron. Tissue kallikrein is also expressed in human neutrophils. It acts locally near its site of origin (Fukushima et al., 1985; Evans et al., 1988). The synthesis of tissue prokallikrein is regulated by a number of factors, including aldosterone in the kidney and salivary gland and androgens in certain other glands. The secretion of the tissue prokallikrein also may be regulated; for example, its secretion from the pancreas is enhanced by stimulation of the vagus nerve (see Proud and Kaplan, 1988; Margolius, 1989). The activation of tissue prokallikrein to kallikrein requires proteolytic cleavage. In human beings, the sequence of these activation events is not well delineated (Bhoola et al., 1992). Kininogens The two substrates for the kallikreins, HMW and LMW kininogen, are products of a single gene that arise by alternative processing of mRNA. HMW and LMW kininogen have been divided into functional domains. The HMW kininogen contains 626 amino acid residues; the internal bradykinin sequence of 9 amino acid residues, domain 4, connects an amino-terminal 'heavy chain' sequence (362 amino acids) containing domains 1 through 3 and a carboxyl-terminal 'light chain' sequence (255 amino acids) containing domains D5H and D6. LMW kininogen is identical to the larger form of the protein from the amino terminus through the bradykinin sequence; its short light chain differs (Takagaki et al., 1985). HMW kininogen is cleaved by plasma and tissue kallikrein to yield bradykinin and kallidin, respectively. LMW kininogen is a substrate only for the tissue kallikrein and the product is kallidin (see Nakanishi, 1987). In addition to serving as precursors of bradykinin and kallidin, the kininogens inhibit cysteine proteinase, inhibit thrombin binding, and exhibit antiadhesive and profibrinolytic properties. Metabolism The decapeptide kallidin is about as active as the nonapeptide bradykinin and need not be converted to the latter to exert its characteristic effects. Some conversion of kallidin to bradykinin occurs as the amino-terminal lysine residue is removed by a plasma aminopeptidase. However, this reaction is slow relative to the rate of inactivation by hydrolysis at the carboxyl terminus. The minimal effective structure required to elicit the classical responses is that of the nonapeptide (Figure 255).
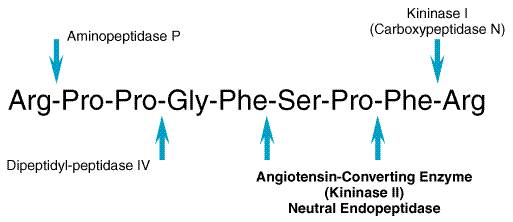
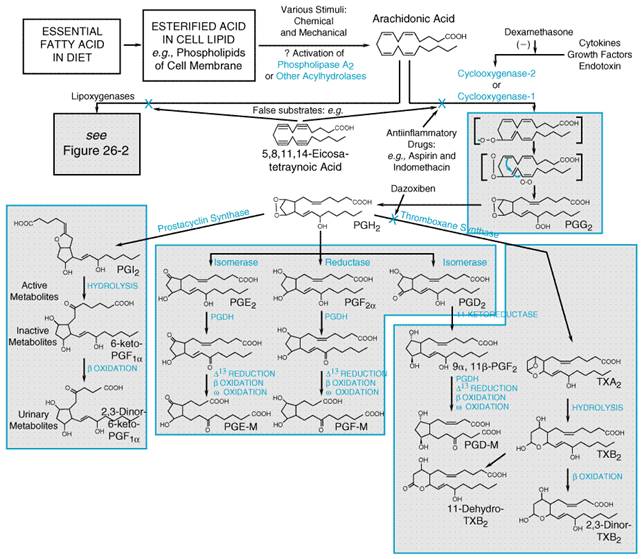
The kinins have an evanescent existencetheir half-life in plasma is only about 15 seconds. Moreover, in a single passage through the pulmonary vascular bed some 80% to 90% of the kinins may be destroyed (see Ryan, 1982). Plasma concentrations of bradykinin have been difficult to define because of its short half-life. Inadequate inhibition of kininogenases or kininases in the blood can lead to artifactual formation or degradation of bradykinin during blood collection. For this reason, physiological concentrations of bradykinin have been reported to range from picomolar to femtomolar (Pellacani et al., 1992). The principal catabolizing enzyme in the lung and in other vascular beds is the dipeptidyl carboxypeptidase kininase II, known in another context as angiotensin converting enzyme (see Chapter 31: Renin and Angiotensin). Removal of the carboxyl-terminal dipeptide abolishes kinin-like activity. Neutral endopeptidase also inactivates kinins by removing the carboxyl-terminal dipeptide. A slower-acting enzyme, arginine carboxypeptidase (carboxypeptidase-N; kininase I), removes the carboxyl-terminal arginine residue producing des-Arg9-bradykinin and des-Arg10-kallidin (Table 252), which are themselves potent B1-kinin receptor agonists (Burch and Kyle, 1992; Trifilieff et al., 1993). A familial carboxypeptidase-N deficiency has been described in which affected individuals with low levels of this enzyme display angioedema or urticaria (see below) (Mathews et al., 1980). Finally, aminopeptidase-P inactivates bradykinin by cleaving the aminoterminus arginine, rendering bradykinin susceptible to further cleavage by dipeptidyl peptidase IV. Bradykinin Receptors There are at least two distinct receptors for kinins, which have been designated B1 and B2 (Regoli and Barab, 1980). The classical bradykinin receptor, now designated the B2 receptor, selectively binds bradykinin and kallidin (see Table 252) and is constitutively present in most normal tissues. B2 receptors mediate the majority of the effects of bradykinin and kallidin in the absence of inflammation. The B1 receptor selectively binds to the carboxy-terminal des-Arg metabolites of bradykinin and kallidin (see Table 252) and is less prevalent than the B2 receptor in most tissues. B1 receptors are present in normal vascular smooth muscle. B1 receptors are upregulated by inflammation and by cytokines, endotoxins, and growth factors (Regoli and Barab, 1980; Dray and Perkins, 1993). During physiological insults such as trauma, tissue damage, or inflammation, B1 receptor effects may predominate. The signaling mechanisms of B1 receptors are less well characterized than are those of B2 receptors. The B2 receptor is a G proteincoupled
7-transmembrane-domain receptor that activates phospholipase A2
and phospholipase C, apparently via interaction with distinct G
proteins. Kinin-induced phospholipase C activation through a G Based on the inability of B1 and B2 antagonists to compete for specific bradykinin binding in guinea pig trachea, the existence of a B3 receptor has been suggested (Farmer et al., 1989; Farmer and DeSiato, 1994). In addition, the presence of B4 and B5 receptors on opossum esophageal smooth muscle cells has been suggested. However, studies with more potent kinin antagonists have not supported the existence of the B3, B4, or B5 receptors. These studies indicate that the guinea pig bronchoconstriction proposed as a B3-receptor effect actually may represent previously unappreciated functions of the B2 receptor (Regoli et al., 1993). Functions and Pharmacology of Kallikreins and Kinins The availability of newer and more specific bradykinin antagonists and the generation of bradykinin-receptor 'knockout' mice have led to significant advances in our understanding of the roles of the kinins. Of current interest is the role of these compounds in diverse areas such as pain, inflammation and chronic inflammatory diseases, the cardiovascular system, and reproduction. Pain The kinins are powerful algesic agents that cause an intense, burning pain when applied to the exposed base of a blister. Bradykinin excites primary sensory neurons and provokes the release of neuropeptides such as substance P, neurokinin A, and calcitonin generelated peptide (Geppetti, 1993). In acute pain, B2 receptors mediate bradykinin algesia. This pain is significantly reduced by B2 antagonists but not by B1 antagonists. The pain of chronic inflammation appears to involve increased numbers of B1 receptors. Inflammation Injected kinins mimic inflammation. Measurement of the components of the kinin cascade and the effects of bradykinin antagonists indicates that kinins participate in a variety of inflammatory diseases. Plasma kinins increase permeability in the microcirculation. The effect, like that of histamine and serotonin in some species, is exerted on the small venules and involves separation of the junctions between endothelial cells. This, together with an increased hydrostatic pressure gradient, causes edema. Such edema, coupled with stimulation of nerve endings (see below), results in a 'wheal-and-flare' response to intradermal injections in human beings. Bradykinin is formed, and there is depletion of the components of the
kinin cascade during episodes of swelling, laryngeal edema, and abdominal
pain in hereditary angioedema (Proud and Kaplan, 1988). B1
receptors on inflammatory cells such as macrophages can elicit production of
the inflammatory mediators IL-1 and tumor necrosis factor Respiratory Disease The kinins have been implicated in the pathophysiology of allergic airway disorders such as asthma and rhinitis. Inhalation or intravenous injection of kinins causes bronchospasm in asthmatic patients but not in normal individuals. Similarly, nasal challenge with bradykinin induces sneezing and serious glandular secretions in patients with allergic rhinitis. Bradykinin-induced bronchoconstriction is blocked by anticholinergic agents but not by antihistamines or cyclooxygenase inhibitors. A bradykinin B2-receptor antagonist also has been shown to improve pulmonary function in patients with severe asthma. Repeated inhalation of bradykinin results in an attenuated response, decreasing the bronchoconstriction in response to bradykinin as well as that in response to adenosine 5' monophosphate (Polosa et al., 1992). Cardiovascular System The kallikrein-kinin system was first implicated in the regulation of blood pressure in the 1920s and 1930s when Frey and Werke identified kallikrein as a hypotensive substance in urine. Since then, numerous investigators have reported that urinary kallikrein concentrations are decreased in individuals with high blood pressure. In experimental animals and human beings, infusion of bradykinin causes vasodilation and lowers blood pressure. Bradykinin causes vasodilation through B2-receptor-dependent effects on endothelial nitric oxide, prostacyclin, and the poorly characterized endothelium-derived hyperpolarizing factor (Vanhoutte, 1989). The availability of specific bradykinin antagonists and genetically altered animals has greatly enhanced our understanding of the role of endogenous bradykinin in the regulation of blood pressure (Madeddu, 1993; Madeddu et al., 1997). Basal blood pressure is normal in B2-receptor antagonisttreated or B2-receptor knockout animals. However, these animals exhibit an exaggerated blood- pressure response to salt loading or activation of the reninangiotensin system. These data suggest that the endogenous kallikreinkinin system plays a minor role in the regulation of blood pressure under normal circumstances, but it may play an important role in hypertensive states. In addition to causing vasodilation, the kallikreinkinin system appears to exert a number of cardioprotective effects. Bradykinin contributes to the protective effect of preconditioning the heart against ischemia and reperfusion injury (Linz and Schlkens, 1992). In the presence of endothelial cells, bradykinin prevents vascular smooth muscle cell growth and proliferation. Bradykinin stimulates tPA release from the vascular endothelium and may inhibit thrombin (Brown et al., 1999; Hasan et al., 1996). Through these mechanisms, bradykinin may contribute to the endogenous defense against cardiovascular events such as myocardial infarction and stroke. Kinins also may increase sympathetic outflow via central and peripheral nervous mechanisms (Dominiak et al., 1992; Schwieler and Hjemdahl, 1992; Madeddu, 1993). These findings suggest that kinins may mediate hypertension in some circumstances via the sympathetic nervous system, though this remains speculative. Kidney Renal kinins act as paracrine hormones to regulate urine volume and composition (Saitoh et al., 1995). Kallikrein is synthesized and secreted by the connecting cells of the distal nephron. Tissue kininogen and kinin receptors are present in the cells of the collecting duct. Like other vasodilators, kinins increase renal blood flow. Bradykinin also causes natriuresis by inhibiting sodium reabsorption at the cortical collecting duct. Renal kallikreins are increased by treatment with mineralocorticoids, ACE inhibitors, and neutral endopeptidase inhibitors. Other Effects The rat uterus is especially sensitive to contraction by kinins through the B2 receptor. Kinins also function in the male reproductive system in areas such as spermatogenesis and in promoting sperm motility, possibly through a B2 receptor on the sperm membrane (Schill and Miska, 1992). Kinins promote dilation of the fetal pulmonary artery, closure of the ductus arteriosus, and constriction of the umbilical vessels, all of which occur in the adjustment from fetal to neonatal circulation. The kallikreinkinin system also functions in a wide variety of other areas in the body, serving to mediate edema formation and smooth muscle contraction. The bradykinin-induced, slowly developing contraction of the isolated guinea pig ileum first prompted the name bradykinin. The kinins also have neurochemical effects in the CNS, in addition to their ability to disrupt the bloodbrain barrier and allow increased CNS penetration (see Inamura et al., 1994). Potential Therapeutic Uses Bradykinin contributes to many of the effects of the widely used cardiovascular drugs, the ACE inhibitors. Aprotinin, a nonspecific kalli- krein antagonist, is administered to patients undergoing coronary bypass in order to minimize bleeding and blood transfusion requirements. Kinin agonists have potential value in increasing the delivery of chemotherapeutic agents beyond the bloodbrain barrier. Based on the physiology outlined above, kinin antagonists are being tested in a number of inflammatory conditions. Kallikrein Inhibitors Aprotinin TRASYLOL) is a natural proteinase inhibitor obtained from bovine lung. Aprotinin inhibits many of the mediators of the inflammatory response, fibrinolysis, and thrombin generation following cardiopulmonary bypass surgery, including kallikrein and plasmin. In several placebo-controlled, double-blind studies, administration of aprotinin during bypass reduced requirements for blood products in patients undergoing coronary artery bypass grafting (Levy et al., 1995). Aprotinin is given as a loading dose followed by a continuous infusion during surgery. Hypersensitivity reactions, including anaphylactic or anaphylactoid reactions, may occur with aprotinin. The rate of such reactions is 2.7% in patients who have been previously exposed to aprotinin and higher in patients who have been exposed to aprotinin within the last six months. A test dose of aprotinin is recommended prior to full dosing. Aprotinin can interfere with an activated clotting time, used to determine the effectiveness of heparin anticoagulation. For this reason, alternate methods must be used to determine the degree of anticoagulation in patients treated with aprotinin during cardiopulmonary bypass. In one multicenter study of aprotinin, there was an increased closure rate of saphenous vein grafts in patients treated with aprotinin compared to those treated with placebo; there were no differences in rates of myocardial infarction or death. Angiotensin Converting Enzyme Inhibitors The ACE inhibitors are widely used in the treatment of hypertension and have been shown to reduce mortality in patients with diabetic nephropathy, left ventricular dysfunction, previous myocardial infarction, and coronary artery disease. ACE inhibitors block the conversion of angiotensin I to angiotensin II, a potent vasoconstrictor and growth promoter (see Chapter 31: Renin and Angiotensin). Data from studies using the specific bradykinin B2 antagonist HOE 140 demonstrate that bradykinin also contributes to many of the protective effects of ACE inhibitors. For example, in animal models, administration of HOE 140 attenuates the favorable effects of ACE inhibitors on blood pressure, on myocardial infarct size, and on ischemic preconditioning (Linz and Schlkens, 1992). Bradykinin receptor antagonism also attenuates the blood pressurelowering effects of acute ACE inhibition in human beings (Gainer et al., 1998). The contribution of bradykinin to the effects of ACE inhibitors may result not only from decreased degradation of bradykinin but also from enhanced receptor sensitivity (Marcic et al., 1999). Occasional patients receiving ACE inhibitors have experienced angioedema. This can occur at any time, but often occurs shortly after initiating therapy. This is an effect of ACE inhibitors as a group and is thought to be due to inhibition of kinin metabolism by ACE (Slater et al., 1988). ACE-inhibitor-associated angioedema is more common in blacks than in Caucasians. Severe anaphylactoid reactions can occur in patients taking ACE inhibitors who are undergoing dialysis with polyacrylonitrile AN69 membranes (Schulman et al., 1993; Verresen et al., 1994). In these patients, kinins are produced by activation of factor XII by the negatively charged surface of the polyacrylonitrile AN69 membrane while ACE inhibition diminishes the clearance of these kinins. A more common side effect of ACE inhibitors (especially in women) is a chronic nonproductive cough that dissipates upon cessation of the ACE inhibitor. The finding that angiotensin AT1-receptor-subtype antagonists do not cause cough has been taken as presumptive evidence for the role of bradykinin in ACE inhibitorinduced cough. Preliminary data suggest that bradykinin also may contribute to the effects of the AT1-receptor antagonists. During AT1-receptor blockade, angiotensin II concentrations increase. Renal bradykinin concentrations also increase through effects of angiotensin II on the unopposed AT2 subtype receptor (Carey et al., 2000). Whether or not bradykinin contributes to the clinical effects of the AT1-receptor antagonists remains to be determined. In addition, a new class of antihypertensive agents, the combined ACE/neutral endopeptidase inhibitors, is undergoing testing. To the extent that these drugs inhibit two kinin-degrading enzymes, bradykinin may be expected to contribute significantly to their clinical effects. Bradykinin Antagonists The introduction of a D-aromatic amino acid in place of the proline residue at position seven conferred antagonist activity to bradykinin and blocked the action of angiotensin converting enzyme. The addition of an N-terminal D-arginine residue also increased the half-life of these antagonists by blocking the action of aminopeptidase P. Nevertheless, the early kinin antagonists were partial agonists and had short half-lives due to enzymatic degradation by carboxypeptidase N in vivo. In the early 1990s, a longer-acting, more selective kinin antagonist, HOE 140, was developed by substituting synthetic amino acids at position seven [D-tetrahydroisoquinoline-3-carboxylic acid (Tic)] and position eight [octahydroindole-2-carboxylic acid (Oic)]. The substitution of the Oic residue at position eight blocked degradation by carboxypeptidase P. The availability of HOE 140 has contributed dramatically to our understanding of the role of bradykinin in human health and disease. CP-0127, a 6-Cys substituted, cross-linked analog of bradykinin, has been tested in the treatment of sepsis in humans in a randomized prospective trial (Fein et al., 1997). In a study of 504 patients with systemic inflammatory response syndrome (SIRS) and presumed sepsis, there was no effect of the bradykinin analog on 28-day survival. However, there was an improvement in risk-adjusted survival in a predefined subset of patients with gram-negative sepsis. A small pilot study in patients with edema following head trauma suggests that bradykinin-receptor antagonism may reduce intracranial pressure. The development of orally active, nonpeptide antagonists promises to make bradykinin antagonism therapeutically feasible in the treatment of disease. The first of these, WIN64338, suffered from having muscarinic cholinergic activity. More recently, the nonpeptide antagonist FR173657 has been shown to decrease bradykinin-induced edema and hypotension in animal models. Bradykinin Agonists RMP-7 [H-Arg-Pro-Hyp-Gly-Thi-Ser- Pro-4Me-Tyr( |
Prospectus
|
The refinement of the structurefunction relationships among histamine receptor subtypes has allowed the continued development of H2-selective antagonists for the treatment of peptic ulcers (see Chapter 37: Agents Used for Control of Gastric Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease). The further understanding of the physiological and pathophysiological roles of H3-receptor subtypes in the CNS and elsewhere similarly may permit the development of new and more selective therapeutic tools. The availability of new peptide and nonpeptide bradykinin antagonists provides the tools for further elucidation of the role of the kallikreinkinin system in health and disease. Ongoing clinical trials will determine the efficacy of bradykinin agonists in enhancing the delivery of chemotherapeutic agents across the bloodbrain barrier. Clinical trials will better define the contribution of bradykinin to the cardioprotective effects of ACE inhibitors, AT1-receptor antagonists, and combined ACE/neutral endopeptidase inhibitors. Acknowledgment The authors wish to acknowledge Drs. Kenneth S. Babe, Jr., and William E. Serafin, authors of this chapter in the ninth edition of Goodman and Gilman's The Pharmacological Basis of Therapeutics, some of whose text we have retained in this edition. |
Chapter 26. Lipid-Derived Autacoids: Eicosanoids and Platelet-Activating Factor
Overview
|
Few biological substances have been the focus of such intense research efforts over the past half-century as have lipid-derived autacoids. Two distinct families of autacoids derived from membrane phospholipids have been identified: the eicosanoids, which are formed from certain polyunsaturated fatty acids (principally arachidonic acid), include the prostaglandins, prostacyclin, thromboxane A2, and the leukotrienes; and modified phospholipids, represented by platelet-activating factor (PAF). The eicosanoids are extremely prevalent and have been detected in almost every tissue and body fluid. Their production increases in response to diverse stimuli, and they produce a broad spectrum of biological effects. Although its precursors are widely distributed, PAF is formed by a smaller number of cell types, principally circulating leukocytes and platelets and endothelial cells. However, because of the wide distribution of these cells, the actions of PAF can be manifest in virtually every organ and tissue of the body. These lipids contribute to a number of physiological and pathological processes including inflammation, smooth muscle tone, hemostasis, thrombosis, parturition, and gastrointestinal secretion. Several classes of drugs, most notably the nonsteroidal antiinflammatory agents, owe their therapeutic effects to blockade of the formation of eicosanoids. This chapter reviews the synthesis, metabolism, and mechanism of action of eicosanoids and PAF and also introduces the therapeutic value of selective inhibitors of eicosanoid synthesis and action. The therapeutic role of these inhibitors is expanded upon in Chapter 27: Analgesic-Antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout, concerning antipyretic and antiinflammatory agents, and in Chapter 54: Hematopoietic Agents: Growth Factors, Minerals, and Vitamins, concerning antiplatelet drugs. |
Eicosanoids
|
History In 1930 Kurzrok and Lieb, two American gynecologists, observed that
strips of human uterus relax or contract when exposed to human semen. A few
years later, Goldblatt in Realization that the 'classically known' prostaglandins constitute only a fraction of the physiologically active products of arachidonate metabolism resulted from the discovery of thromboxane A2 (TXA2) (Hamberg et al., 1975), prostacyclin (PGI2) (Moncada et al., 1976), and the leukotrienes (Samuelsson, 1983). The discovery by Vane, Smith, and Willis in 1971 that aspirin and related drugs inhibit prostaglandin biosynthesis provided insight into the mechanism of action of these drugs as well as an important tool for investigation of the role of these autacoids (see Vane, 1971). The families of prostaglandins, leukotrienes, and related compounds
are called eicosanoids because they are derived from 20-carbon
essential fatty acids that contain three, four, or five double bonds:
8,11,14-eicosatrienoic acid (dihomo-
Biosynthesis Hormones, autacoids, and other substances augment the biosynthesis of eicosanoids by interacting with (presumably) plasma membranebound receptors that are coupled to GTP-binding regulatory proteins (G proteins; see Chapter 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect). This results either in the direct activation of phospholipases or in elevated cytosolic concentrations of Ca2+, which also can activate these enzymes. Physical stimuli are believed to cause an influx of Ca2+ by perturbing the cell membrane, thereby activating phospholipase A2. Phospholipase A2 hydrolyzes the sn-2 ester bond of membrane phospholipids (particularly phosphatidylcholine and phosphatidylethanolamine) with the release of arachidonate. Several different phospholipase A2s have been characterized. Cytosolic phospholipase A2 is likely the major phospholipase involved in agonist-stimulated arachidonate release and prostaglandin production (Lin et al., 1992). Other phospholipases, however, may contribute to the release of arachidonic acid in various cell types (Okazaki et al., 1981; Reddy et al., 1997). Once released, a portion of the arachidonate is metabolized rapidly to oxygenated products by several distinct enzyme systems, including cyclooxygenases or one of several lipoxygenases or cytochrome P450s. Products of Cyclooxygenases The prostaglandins and thromboxanes can be considered analogs of the unnatural compounds with the trivial names prostanoic acid and thrombanoic acid, respectively, the structures of which are as follows:
They fall into several main classes, designated by letters and distinguished by substitutions on the cyclopentane ring. Prostaglandins of the E and D series are hydroxy ketones, while the F Synthesis of prostaglandins is accomplished in a stepwise manner by a ubiquitous complex of microsomal enzymes. The first enzyme in this synthetic pathway is prostaglandin endoperoxide synthase, also called fatty acid cyclooxygenase. There are two isoforms of this enzyme, cyclooxygenase-1 and -2, dubbed COX-1, COX-2 (see Smith et al., 1996; DuBois et al., 1998). The former is constitutively expressed in most cells. In contrast, COX-2 is normally not present but may be induced by certain serum factors, cytokines, and growth factors, an effect that is inhibited by treatment with glucocorticoids such as dexamethasone. The cyclooxygenases have two distinct activities: an endoperoxide
synthase activity that oxygenates and cyclizes the unesterified precursor
fatty acid to form the cyclic endoperoxide PGG, and a peroxidase activity
that converts PGG to PGH (see Hamberg et al., 1974). PGG and
PGH are chemically unstable, but they can be transformed enzymatically into a
variety of products, including PGI, TXA, PGE, PGF, or PGD (see Figure
261; Samuelsson et al., 1975; Needleman et al., 1986; Sigal,
1991). Isomerases for the synthesis of PGE2 and PGD2
have been identified. A reductase that catalyzes the conversion of PGH2
to PGF2 The endoperoxide PGH2 also is metabolized into two unstable
and highly active compounds (Figure 261). Thromboxane A2 (TXA2)
is formed by thromboxane synthase; TXA2 breaks down
nonenzymatically (t1/2= 30 seconds) into the stable but
inactive thromboxane B2 (TXB2). PGI2 is
formed from PGH2 by prostacyclin synthase; it is hydrolyzed
nonenzymatically (t1/2= 3 minutes) to the inactive
6-keto-PGF1 Although most tissues are able to synthesize the PGG and PGH intermediates from free arachidonate, their fate varies in each tissue and depends on the complement of enzymes present and on their relative abundance. For example, lung and spleen are able to synthesize the whole range of products. In contrast, platelets contain thromboxane synthase as the principal enzyme that metabolizes PGH, while endothelial cells contain primarily prostacyclin synthase. Products of Lipoxygenases Lipoxygenases are a family of cytosolic enzymes that catalyze the oxygenation of polyenic fatty acids to corresponding lipid hydroperoxides (see Samuelsson, 1983; Needleman et al., 1986; Brash, 1999). The enzymes require a fatty acid substrate with two cis double bonds separated by a methylene group. Arachidonate, which contains several double bonds in this configuration, is metabolized to a number of products with the hydroperoxy group in different positions. For arachidonate, these metabolites are called hydroperoxy-eicosatetraenoic acids (HPETEs). Lipoxygenases differ in their specificity for placing the hydroperoxy group, and tissues differ in the lipoxygenase(s) that they contain (see Brash, 1999). For example, platelets have only 12-lipoxygenase and synthesize 12-HPETE, whereas leukocytes contain both 5-lipoxygenase and 12-lipoxygenase and produce both 5-HPETE and 12-HPETE (see Figure 262). Other lipoxygenases that also catalyze the formation of 15-HPETE and 8-HPETE have been reported in human beings and laboratory animals. The HPETEs are unstable intermediates, analogous to PGG or PGH, and are further metabolized by a variety of enzymes. All HPETEs may be converted to their corresponding hydroxy fatty acid (HETE) either by a peroxidase or nonenzymatically. 12-HPETE also can undergo a catalyzed molecular rearrangement to epoxy-hydroxyeicosatrienoic acids called hepoxilins. Similarly, leukocytes convert 15-HPETE to trihydroxylated metabolites called lipoxins. The 5-lipoxygenase is one of the most important of the lipoxygenases, since it leads to the synthesis of the leukotrienes (LTs) (Figure 262; see Samuelsson, 1983; Samuelsson et al., 1987; Sigal, 1991). As with the prostaglandins, a subscript is used to indicate the number of double bonds in the fatty acid. Arachidonic acid is the precursor of the four series of leukotrienes and 5,8,11,14,17-eicosapentaenoic acid of the five series. When cells are activated, 5-lipoxygenase translocates to the nuclear membrane and associates with 5-lipoxygenase activating protein (FLAP), an integral membrane protein essential for leukotriene biosynthesis. FLAP appears to act as an arachidonic acid transfer protein that presents the substrate to the 5-lipoxygenase (see Brash, 1999). An experimental drug, MK-886, binds to FLAP and blocks leukotriene production. The 5-lipoxygenase catalyzes a two-step reaction: oxygenation of arachidonate at the fifth carbon to form 5-HPETE followed by dehydration of 5-HPETE to an unstable 5,6-epoxide, known as leukotriene A4 (LTA4) (Peters-Golden, 1998; Borgeat and Samuelsson, 1979). LTA4 may be transformed by LTA hydrolase to a 5,12-dihydroxyeicosatetraenoic acid known as leukotriene B4 (LTB4); alternatively, it may be conjugated with reduced glutathione by LTC4 synthase to form LTC4 (Murphy et al., 1979). Leukotriene D4 (LTD4) is produced by the removal of glutamic acid from LTC4, and LTE4 results from the subsequent cleavage of glycine (see Samuelsson, 1983; Piper, 1984; Samuelsson et al., 1987). LTC4, LTD4, and LTE4 often are referred to as cysteinyl leukotrienes. It is now generally accepted that a mixture of LTC4, LTD4, and LTE4 makes up the material originally known as the 'slow-reacting substance of anaphylaxis' (SRS-A), first described by Feldberg and Kellaway (1938). Products of Cytochrome P450 Arachidonate is metabolized by enzymes that contain cytochrome P450 to a variety of metabolites including 19- or 20-hydroxy arachidonate and epoxyeicosatrienoic acids (see Fitzpatrick and Murphy, 1988; Capdevila et al., 2000). While these metabolites have potent vascular, endocrine, renal, and ocular effects, the physiological importance of this pathway remains to be clarified. Other Pathways A nonenzymatic pathway of arachidonate conversion also has been discovered, giving rise to a novel series of agents termed isoprostanes (Morrow et al., 1990). These compounds, while having structures similar to cyclooxygenase-derived PGs, arise in vivo from the free radicalcatalyzed peroxidation of arachidonate independent of the cyclooxygenase. Unlike the cyclooxygenase-derived eicosanoids, the isoprostanes identified to date are formed completely in situ on phospholipids and subsequently released preformed. Consequently, their production is not blocked in vivo by agents that suppress metabolism of free arachidonate, such as aspirin or nonsteroidal antiinflammatory agents, or by agents that suppress expression of the inducible COX-2 enzyme, such as steroidal antiinflammatory drugs. It is postulated that these agents might contribute to the pathophysiology of inflammatory responses insensitive to currently available steroidal or nonsteroidal antiinflammatory agents. Of importance is that this pathway of eicosanoid formation links free radicalmediated tissue injury with bioactive lipid-derived autacoid generation (Morrow et al., 1999). In the brain, arachidonate is coupled to ethanolamine to give
arachidonylethanolamide, also called anandamide (Devane et al.,
1992). A similar reaction occurs with other unsaturated fatty acids. Anandamide
binds to cannabinoid receptors and displays biochemical and behavioral
effects very similar to those of Inhibitors of Eicosanoid Biosynthesis A number of the biosynthetic steps described above can be inhibited by drugs. Inhibition of phospholipase A2 decreases the release of the precursor fatty acid and thus the synthesis of all metabolites derived therefrom. Since phospholipase A2 is activated by Ca2+ and calmodulin, it may be inhibited by drugs that reduce the availability of Ca2+. Glucocorticoids also inhibit phospholipase A2, but they appear to do so indirectly by inducing the synthesis of a group of proteins termed annexins (formerly, lipocortins), which modulate phospholipase A2 activity (Flower, 1990). Glucocorticoids, however, also regulate expression of COX-2, but not of COX-1 (Masferrer et al., 1994; Smith et al., 1996). It is therefore conceivable that therapeutically effective doses of glucocorticoids as antiinflammatory agents correlate more closely with their potency in suppressing cytokine-induced COX-2 expression than with their potency in inhibiting phospholipase. Aspirin and related nonsteroidal antiinflammatory drugs originally were found to prevent the synthesis of prostaglandins from arachidonate in tissue homogenates (Vane, 1971). It is now known that these drugs inhibit cyclooxygenase and, as a result, inhibit the synthesis of PGG2, PGH2, and all that flows therefrom. However, these drugs do not inhibit the metabolism of arachidonate by lipoxygenases. In fact, inhibition of cyclooxygenase theoretically could lead to increased formation of leukotrienes by increasing the amount of arachidonate that is available to the lipoxygenases (see Piper, 1984). Inhibition of cyclooxygenase provides an important basis for understanding many of the therapeutic and other effects of these agents (see Chapter 27: Analgesic-Antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout). COX-1 and -2 differ in their sensitivity to inhibition by certain antiinflammatory drugs (see Vane et al., 1998; Marnett et al., 1999). This observation has led to the recent development of clinically useful agents that selectively inhibit COX-2 (see Chapter 27: Analgesic-Antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout). These drugs show distinct therapeutic advantages over nonselective nonsteroidal antiinflammatory agents, since COX-2 is the predominant cyclooxygenase at sites of inflammation but not at sites such as the gastrointestinal tract. Thus, as has been largely borne out by clinical trials, COX-2 inhibitors are antiinflammatory but do not possess many of the adverse side effects of nonselective cyclooxygenase inhibitors. Since different metabolites of PGH2 sometimes produce opposite biological effects (see below), there are theoretical advantages in compounds that preferentially inhibit one or another of the enzymes that metabolize PGH2 (see Moncada and Vane, 1978). For example, there had been significant interest in the development of agents that inhibit thromboxane synthase and thus block platelet aggregation and induce vasodilation. Although these drugs block thromboxane production in vitro and in vivo, they have largely failed to produce clinical benefits in a wide variety of disorders. The lack of clinical efficacy of these agents may reflect other vasoactive mediators contributing to the pathophysiology of disorders that were studied or that, following thromboxane synthesis inhibition, there is an accumulation of PGH2, which shares some of the biological effects of TXA2. Similar disappointing results have been obtained in clinical studies with combined thromboxane synthase/thromboxane receptor antagonists. Analogs of the natural fatty acid precursors can serve as competitive inhibitors of the formation of both prostaglandins and the products of lipoxygenases. One such inhibitor is the acetylenic analog of arachidonic acid, 5,8,11,14-eicosatetraynoic acid (see Figure 261). Since leukotrienes function as inflammatory mediators, recent efforts have focused on development of leukotriene receptor antagonists and selective inhibitors of lipoxygenase synthesis. Zileuton, an inhibitor of 5-lipoxygenase, has proven to be useful in the treatment of asthma and possibly other inflammatory diseases. In addition, cysteinyl leukotriene receptor antagonists, including zafirlukast, pranlukast, and montelukast, are useful in the therapy of asthma (see Chapter 28: Drugs Used in the Treatment of Asthma). Eicosanoid Catabolism Efficient mechanisms exist for the catabolism and inactivation of most
eicosanoids. About 95% of infused PGE2 is inactivated during one
passage through the pulmonary circulation. Because of the unique position of
the lungs between the venous and arterial circulation, the pulmonary vascular
bed constitutes an important filter for many substances (including some
prostaglandins) that act locally prior to their release into the venous
circulation. Broadly speaking, the enzymatic catabolic reactions are of two
types: an initial (relatively rapid) step, catalyzed by widely distributed
prostaglandin-specific enzymes, wherein prostaglandins lose most of their
biological activity, and a second (relatively slow) step in which these
metabolites are oxidized by enzymes probably identical with those responsible
for the Unlike PGE2, PGD2 is initially reduced in
vivo to the F-ring prostaglandin 9 The metabolism of TXA2 in human beings is inferred from investigation of the fate of TXB2. Although up to twenty metabolites have been identified in urine, by far the most abundant are 2,3-dinor-TXB2 and 11-dehydro-TXB2 (Uedelhoven et al., 1989; see Figure 261). The degradation of PGI2 apparently begins with its
spontaneous hydrolysis in blood to 6-keto-PGF1 The degradation of LTC4 occurs in the lungs, kidney, and
liver (Denzlinger et al., 1986). The initial steps involve its
conversion to LTE4, and this results in a loss in biological
activity. Leukotriene C4 also may be inactivated by oxidation of
its cysteinyl sulfur to a sulfoxide. The principal route of inactivation of
LTB4 is by Pharmacological Properties of Eicosanoids No other autacoids show more numerous and diverse effects than do prostaglandins and other metabolites of arachidonate. It would be overly confusing to present all of the pharmacological effects that have been ascribed to these substances and even more so to delve into the activities of their synthetic analogs. This discussion is limited to activities that are thought to be the most important. Cardiovascular System Prostaglandins In the vascular beds of human beings and most animals, the PGEs are potent vasodilators. The dilation appears to involve arterioles, precapillary sphincters, and postcapillary venules; large veins are not affected by PGEs. However, PGEs are not universally vasodilatory; constrictor effects have been noted at selected sites (see Bergstrm et al., 1968). PGD2 similarly causes both vasodilation and
vasoconstriction; however, in most vascular beds, including the mesenteric,
coronary, and renal, vasodilation occurs at lower concentrations than does
vasoconstriction. An exception is the pulmonary circulation in which PGD2
causes only vasoconstriction. Responses to PGF2 Systemic blood pressure generally falls in response to PGEs, and blood
flow to most organs, including the heart, mesentery, and kidney, is
increased. These effects are particularly striking in some hypertensive
patients. Blood pressure is increased by PGF2 Cardiac output generally is increased by prostaglandins of the E and F series. Weak, direct inotropic effects have been noted in various isolated preparations. In the intact animal, however, increased force of contraction as well as increased heart rate is in large measure a reflex consequence of a fall in total peripheral resistance. Prostaglandin endoperoxides have variable effects in vascular beds. Their major effects are a result of intrinsic vasoconstrictor activity coupled with vasodilation due to rapid conversion to PGI2, which is a vasodilator. PGH2 is rapidly converted to PGI2 during passage through the lungs. The intravenous administration of PGI2 causes prominent hypotension; it is about five times more potent than PGE2 in producing this effect. The reduction in blood pressure is accompanied by a reflex increase in heart rate. The compound relaxes vascular smooth muscle, and it is thought to be a physiological modulator of vascular tone that functions to oppose the actions of vasoconstrictors. Thromboxane A2 Thromboxane A2 is a potent vasoconstrictor. It contracts vascular smooth muscle in vitro (Bhagwat et al., 1985) and is a vasoconstrictor in the whole animal and in isolated vascular beds. Leukotrienes In human beings, LTC4 and LTD4 cause hypotension (see Feuerstein, 1984; Piper, 1984). This may result in part from a decrease in intravascular volume and in cardiac contractility that is secondary to a marked, leukotriene-induced reduction in coronary blood flow. Although LTC4 and LTD4 have little effect on most large arteries or veins, coronary arteries and distal segments of the pulmonary artery are contracted by nanomolar concentrations of these agents (Berkowitz et al., 1984). The renal vasculature is resistant to this constrictor action, but the mesenteric vasculature is not. The leukotrienes have prominent effects on the microvasculature. LTC4 and LTD4 appear to act on the endothelial lining of postcapillary venules to cause exudation of plasma; they are more than a thousandfold more potent than histamine in this regard (see Feuerstein, 1984; Piper, 1984). In higher concentrations, LTC4 and LTD4 constrict arterioles and reduce exudation of plasma. Blood Eicosanoids modify the function of the formed elements of the blood; in some instances, these actions reflect their physiological roles. The prostaglandins and related products modulate platelet function. PGI2 inhibits the aggregation of human platelets in vitro at concentrations between 1 and 10 nM. This fact and the observation that PGI2 is synthesized by the vascular endothelium have led to the suggestion that PGI2 controls the aggregation of platelets in vivo and contributes to the antithrombogenic properties of the intact vascular wall (see Moncada and Vane, 1978). TXA2 is a major product of arachidonate metabolism in platelets (Hamberg et al., 1975) and, as a powerful inducer of platelet aggregation and the platelet release reaction, is a physiological mediator of platelet aggregation. Pathways of platelet aggregation that are dependent on the generation of TXA2 are sensitive to the inhibitory action of aspirin (see Chapters 27: Analgesic-Antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout and 55: Anticoagulant, Thrombolytic, and Antiplatelet Drugs; Moncada and Vane, 1978). LTB4 is a potent chemotactic agent for polymorphonuclear leukocytes, eosinophils, and monocytes; other leukotrienes do not share this action (see Piper, 1984). Its potency is comparable with that of various chemotactic peptides and PAF. In higher concentrations, LTB4 stimulates the aggregation of polymorphonuclear leukocytes and promotes degranulation and the generation of superoxide. LTB4 promotes adhesion of neutrophils to vascular endothelial cells and their transendothelial migration; application of LTB4 to the skin promotes the local accumulation of neutrophils. Prostaglandins inhibit lymphocyte function and proliferation and suppress the immunological response. PGE2 inhibits the differentiation of B lymphocytes into antibody-secreting plasma cells to depress the humoral antibody response. It also inhibits mitogen-stimulated proliferation of T lymphocytes and the release of lymphokines by sensitized T lymphocytes. Smooth Muscle Prostaglandins contract or relax many smooth muscles beside those of the vasculature. The leukotrienes (e.g., LTD4) contract most smooth muscles. Bronchial and Tracheal Muscle In general, PGFs and PGD2 contract and PGEs relax bronchial and tracheal muscle. Although both PGE1 and PGE2 can produce bronchodilation when given to such patients by aerosol, bronchoconstriction sometimes is observed. Prostaglandin endoperoxides and TXA2 constrict human bronchial smooth muscle. PGI2 causes bronchodilation in most species; human bronchial tissue is particularly sensitive, and PGI2 antagonizes bronchoconstriction that is induced by other agents. LTC4 and LTD4 are bronchoconstrictors in many species, including human beings (see Piper, 1984; Drazen and Austen, 1987). They act principally on smooth muscle in peripheral airways and are a thousand times more potent than histamine both in vitro and in vivo. They also stimulate bronchial mucus secretion and cause mucosal edema. Uterus Strips of nonpregnant human uterus are contracted by PGFs and TXA2
but are relaxed by PGEs. The contractile response is most prominent before
menstruation, whereas relaxation is greatest at midcycle (see Bergstrm
et al., 1968). Uterine strips from pregnant women are uniformly
contracted by PGFs and by low concentrations of PGE2; PGI2
and high concentrations of PGE2 produce relaxation. The
intravenous infusion of PGE2 or PGF2 Gastrointestinal Muscle The main longitudinal muscle from stomach to colon is contracted by both PGEs and PGFs, while circular muscle generally relaxes in response to PGEs and contracts in response to PGFs. Prostaglandin endoperoxides, TXA2, and PGI2 produce contraction but are less active than the PGEs or PGFs on gastrointestinal smooth muscle. The leukotrienes have potent contractile effects. Prostaglandins reduce transit times in the small intestine and colon. Diarrhea, cramps, and reflux of bile have been noted in response to oral PGE; these are common side effects (along with nausea and vomiting) in patients given prostaglandins for abortion. Gastric and Intestinal Secretions PGEs and PGI2 inhibit gastric acid secretion stimulated by feeding, histamine, or gastrin. Volume of secretion, acidity, and content of pepsin all are reduced, probably by an action exerted directly on the secretory cells. In addition, these prostaglandins are vasodilators in the gastric mucosa, and PGI2 may be involved in the local regulation of blood flow. Mucus secretion in the stomach and small intestine is increased by PGEs. These effects help to maintain the integrity of the gastric mucosa and are referred to as the cytoprotectant properties of PGEs. Furthermore, PGEs and their analogs inhibit gastric damage caused by a variety of ulcerogenic agents and promote healing of duodenal and gastric ulcers (see Chapter 37: Agents Used for Control of Gastric Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease). PGEs and PGFs stimulate the movement of water and electrolytes into the intestinal lumen. Such effects may underlie the watery diarrhea that follows the oral or parenteral administration of prostaglandins. By contrast, PGI2 does not induce diarrhea; indeed, it prevents that provoked by other prostaglandins. Kidney and Urine Formation Prostaglandins influence renal salt and water excretion by alterations in renal blood flow and by direct effects on renal tubules. PGE2 and PGI2 infused directly into the renal arteries of dogs increase renal blood flow and provoke diuresis, natriuresis, and kaliuresis; there is little change in the rate of glomerular filtration (see Dunn and Hood, 1977). TXA2 decreases renal blood flow, decreases the rate of glomerular filtration, and participates in tubuloglomerular feedback. PGEs inhibit water reabsorption induced by antidiuretic hormone (ADH). PGE2 also inhibits chloride reabsorption in the thick ascending limb of the loop of Henle in the rabbit. In addition, PGI2, PGE2, and PGD2 cause the secretion of renin from the renal cortex, apparently through a direct effect on the granular juxtaglomerular cells. Central Nervous System Although a large number of observations have been made on the effects of prostaglandins in the central nervous system (CNS), evidence for a clear-cut physiological role has yet to emerge. Both stimulant and depressant effects of prostaglandins on the CNS have been reported following their injection into the cerebral ventricles, and the firing rates of individual brain cells may be increased or decreased after iontophoretic application of these agents. The release of PGE2 in the brain likely explains the genesis of pyrogen-induced fever (Coceani and Akarsu, 1998). PGD2 has been proposed as a mediator responsible for sleep (Urade et al., 1999). Afferent Nerves and Pain PGEs cause pain when injected intradermally; these effects are generally not as immediate or intense as those caused by bradykinin or histamine, but they outlast those caused by the other autacoids. PGEs and PGI2 sensitize the afferent nerve endings to the effects of chemical or mechanical stimuli by lowering the threshold of the nociceptors. Hyperalgesia also is produced by LTB4. The release of these prostaglandins and of LTB4 during the inflammatory process thus serves as an amplification system for the pain mechanism (see Moncada et al., 1978). The role of PGE2 and PGI2 in inflammation is discussed in Chapter 27: Analgesic-Antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout. Endocrine System A variety of endocrine tissues respond to prostaglandins. In a number
of species, the systemic administration of PGE2 increases
circulating concentrations of ACTH, growth hormone, prolactin, and the
gonadotropins. Other effects include stimulation of steroid production by the
adrenals, stimulation of insulin release, thyrotropin-like effects on the
thyroid, and LH-like effects on isolated ovarian tissue, causing increased progesterone
secretion from the corpus luteum. This last effect, observed in vitro,
contrasts with the luteolytic effects of prostaglandins in vivo in
many species but not in pregnant women. This property is possessed especially
but not uniquely by PGF2 Lipoxygenase metabolites of arachidonate also have endocrine effects. 12-HETE stimulates the release of aldosterone from the adrenal cortex and mediates a portion of the aldosterone release stimulated by angiotensin II but not that by ACTH. Metabolic Effects PGEs inhibit the basal rate of lipolysis from adipose tissue in vitro and also lipolysis stimulated by exposure to catecholamines or other lipolytic hormones. Such effects also have been noted in vivo in various species, including human beings, but are more capricious. PGEs have some insulin-like effects on carbohydrate metabolism and exert parathyroid hormone-like effects that result in mobilization of Ca2+ from bone in tissue culture. Mechanism of Action of Eicosanoids Prostaglandin Receptor Diversity The diversity of the effects of prostanoids is explained by the existence of a number of distinct receptors that mediate their actions. One scheme for classifying these receptors in platelets and smooth muscle is based primarily on the pattern of effects and the relative potencies of natural and synthetic agonists. This scheme has been largely substantiated by ligand-binding studies, cloning of the receptors, the discovery of relatively selective antagonists, and the development of mice possessing disruptions of genes encoding the eicosanoid receptors (see Coleman et al., 1994; Austin and Funk, 1999; Narumiya et al., 1999). This scheme is summarized in Table 261. The receptors have been named for the natural prostaglandin for which they have the greatest apparent affinity and have been divided into five main types, designated DP (PGD2), FP (PGF2), IP (PGI2), TP (TXA2), and EP (PGE2). The EP receptors have been further subdivided into EP1, EP2, EP3, and EP4, based on physiological and molecular cloning information (Coleman et al., 1994; Narumiya et al., 1999). There are several splice variants of EP3 that appear to have different signal transduction mechanisms (Narumiya et al., 1999). Previous pharmacological studies suggested the existence of a heterogeneous population of TP receptors, and two splice variants have been characterized (Raychowdhury et al., 1994). However, only one TP gene has been found to date, and evidence from targeted disruption of this gene implies that most of the known functions of TXA2 are mediated through products of a single TP locus. Table 261 also indicates the effects of the natural prostaglandins on smooth muscle tone and platelet aggregation when the various prostaglandin receptors are stimulated. Cell Signaling Pathways All prostanoid receptors identified to date are coupled to effector mechanisms through G proteins (see Coleman et al., 1994; Narumiya et al., 1999). Two second-messenger systems have been associated with the action of prostanoids in platelets and smooth musclenamely, stimulation of adenylyl cyclase (enhanced accumulation of cyclic AMP), inhibition of adenylyl cyclase (reduced accumulation of cyclic AMP), and stimulation of phospholipase C (enhanced formation of diacylglycerols and inositol-1,4,5-trisphosphate leading to an increase in cytosolic Ca2+) (see Table 261). The actions of prostanoids have been extensively studied in platelets. The prostaglandin endoperoxides and TXA2 stimulate the TP receptors and thereby activate platelet aggregation, a response associated with activation of phospholipase C. Subsequent release of intracellular Ca2+ promotes aggregation and production of additional TXA2. PGI2 binds to IP receptors and activates adenylyl cyclase in the platelet, resulting in inhibition of aggregation. PGD2 interacts with a distinct receptor (DP) that also stimulates adenylyl cyclase. PGE1 appears to act through IP receptors. Leukotriene Receptors Receptors have been identified for both LTB4 and the cysteinyl leukotrienes LTC4 and LTD4 in various cells and tissues. At least two classes of receptors exist for the cysteinyl leukotrienes and are termed cysLT1 and cysLT2 (Nicosia et al., 1999). The leukotriene receptors are coupled to G proteins, and their activation increases intracellular Ca2+ concentrations. Other Agents Other metabolites of the lipoxygenase and cytochrome P450 pathways (e.g.,
HETEs, epoxyeicosatrienoic acids, lipoxins, hepoxilins) have potent
biological activities, and there is evidence for the existence of receptors
for some of these substances. It is possible that some of these metabolites
also function as intracellular second messengers. Recently, it has been
proposed that certain eicosanoids, including PGI2, the J-series
prostaglandin, 15-deoxy- Receptor Antagonists There are as yet no potent, selective antagonists of prostanoid receptors that are in routine clinical use. Previously, there was significant interest in the development of TP receptor antagonists for use in human disorders associated with excessive TXA2-mediated platelet aggregation or vasoconstriction. Agents developed included sultroban, vapiprost, and others. Although occasionally effective in animal models of human disease such as atherosclerosis or in small clinical trials, these drugs have been supplanted by other, more effective compounds that act to prevent or ameliorate disease via other biochemical pathways. TP antagonists have proven useful in vitro, however, in elucidating the role of TXA2 in cellular processes. Subtype-selective antagonists for EP receptors have been developed. Compounds include SC 19220, AH 6809, and SC 51089 for EP1 receptors and AH 23848B for EP4 receptors. Further clinical development likely will await clarification of which EP receptor subtype prevails in which physiological or pathophysiological setting. Orally active antagonists of leukotriene C4 and D4
have been approved for the treatment of asthma (see Chapter 28: Drugs
Used in the Treatment of Asthma). These agents act by binding to the cysLT1
receptor and include montelukast and zafirlukast. In patients
with mild to moderately severe asthma, they cause bronchodilation, reduce the
bronchoconstriction caused by exercise and exposure to antigen, and decrease
the patient's requirement for the use of Endogenous Prostaglandins, Thromboxanes, and Leukotrienes: Possible Functions in Physiological and Pathological Processes Because eicosanoids can be formed by virtually every cell, it is not
unreasonable to suspect that each pharmacological effect may reflect a
physiological or pathophysiological function. Such suspicions have been
nurtured and presented in countless hypotheses bearing on just about every
bodily function. Further insights into the roles of eicosanoids in biological
processes have been gleaned by the development of mice with targeted
disruptions of genes regulating eicosanoid biosynthesis or action (see
Platelets An area in which there has been considerable interest is the elucidation of the role played by prostaglandin endoperoxides and TXA2 in platelet aggregation and thrombosis and by PGI2 in the prevention of these events. It is generally accepted that stimulation of platelet aggregation leads to activation of membrane phospholipases, with the consequent release of arachidonate and its transformation into prostaglandin endoperoxides and TXA2. These substances induce platelet aggregation. However, this pathway is not the only mechanism for the induction of platelet aggregation, since, for example, thrombin aggregates platelets without the release of arachidonate. However, the importance of the thromboxane pathway in platelets is implied by the fact that aspirin and antagonists of TP receptors inhibit the second phase of platelet aggregation and induce a mild hemostatic defect in human beings (Hamberg et al., 1974; Patrono, 1994). Additionally, the platelet thromboxane pathway is activated markedly in acute coronary artery syndromes, and aspirin is beneficial in the secondary prevention, and in some cases primary prevention, of coronary and cerebrovascular diseases (see Antiplatelet Trialists' Collaboration, 1994; Patrono, 1994). PGI2 that is generated in the vessel wall may be the physiological antagonist of this system; it inhibits platelet aggregation and contributes to the nonthrombogenic properties of the endothelium. According to this concept, PGI2 and TXA2 represent biologically opposite poles of a mechanism for regulating plateletvessel wall interaction and the formation of hemostatic plugs and intraarterial thrombi (see Moncada and Vane, 1978). Reproduction and Parturition Much interest is attached to the possible involvement of prostaglandins in reproductive physiology. Their very high concentrations in human semen, coupled with the substantial absorption of prostaglandins by the vagina, have encouraged speculation that prostaglandins deposited during coitus may facilitate conception by actions on the cervix, uterine body, fallopian tubes, and transport of semen. Although there is some correlation between lowered concentrations of prostaglandins in semen and certain cases of male infertility, the role of the eicosanoids in semen remains obscure. With menstruation, there is disruption of uterine membranes, the release of arachidonate, and stimulation of prostaglandin synthesis. The concentrations of prostaglandins are elevated in menstrual fluid. These prostaglandins are thought to contract uterine and gastrointestinal smooth muscle and sensitize afferent pain fibers and thereby contribute to the symptoms of primary dysmenorrhea. Inhibitors of cyclooxygenase are more effective than narcotic analgesics in relieving the symptoms of this condition. During pregnancy in the human female, the capacity of the fetal membranes to elaborate prostaglandins rises progressively. Concentrations of prostaglandins in blood and amniotic fluid are elevated during labor, but it is not certain whether this is a major determinant of the onset of labor or only serves to sustain uterine contractions that have been initiated by oxytocin. In any event, inhibitors of cyclooxygenase increase the length of gestation, prolong the duration of spontaneous labor, and interrupt premature labor. Additional evidence for a role of prostaglandins in reproduction and parturition is the fact that targeted disruption of the genes encoding both COX enzymes, the EP2 receptor, and the FP receptor in mice results in various defects in reproduction and parturition (see Austin and Funk, 1999). PGF2 Prostaglandins and prostaglandin analogs also are useful in ripening the cervix for delivery and as abortifacients (see below). Vascular and Pulmonary Smooth Muscle Locally generated PGE2 and PGI2 modulate vascular tone. Produced by the vascular endothelium, PGI2 is released by shear stress and by both vasoconstrictor and vasodilator autacoids. PGI2 appears to counteract the effects of circulating vasoconstrictor autacoids, to maintain blood flow to vital organs, and to mediate a portion of the dilation by other autacoids. The importance of these vascular actions is emphasized by the participation of PGI2 and PGE2 in the hypotension associated with septic shock. These prostaglandins also have been implicated in the maintenance of patency of the ductus arteriosus. This hypothesis has been strengthened by the fact that nonsteroidal antiinflammatory drugs induce closure of a patent ductus in neonates (see Chapter 27: Analgesic-Antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout; Coceani et al., 1980). Prostaglandins also may play a role in the maintenance of placental blood flow. A complex mixture of autacoids is released when sensitized lung tissue
is challenged by the appropriate antigen. Various prostaglandins and
leukotrienes are prominent components of this mixture. While both
bronchodilator (PGE2) and bronchoconstrictor (e.g., PGF2 Kidney Prostaglandins modulate renal blood flow and may serve to regulate urine formation by both renovascular and tubular effects. Additional roles in the regulation of the secretion of renin also are likely. The elaboration of PGE2 and PGI2 is increased by factors that reduce renal blood flow (e.g., stimulation of sympathetic nerves and angiotensin). Under these circumstances, inhibitors of cyclooxygenase augment the renovasoconstriction that is produced by such stimuli. In addition, the effects of ADH on the reabsorption of water may be restrained by the concomitant production and action of PGE2. Increased biosynthesis of prostaglandins has been associated with Bartter's syndrome. This is a rare disease characterized by low-to-normal blood pressure, decreased sensitivity to angiotensin, hyperreninemia, hyperaldosteronism, and excessive loss of K+. There also is an increased excretion of prostaglandins in the urine. After long-term administration of cyclooxygenase inhibitors, sensitivity to angiotensin, plasma renin values, and the concentration of aldosterone in plasma return to normal. Although plasma K+ rises, it remains low, and urinary wasting of K+ persists. Whether an increase in prostaglandin biosynthesis is the cause of Bartter's syndrome or a reflection of a more basic physiological defect is not yet known (see Clive, 1995). Inflammatory and Immune Responses Prostaglandins and leukotrienes are released by a host of mechanical, thermal, chemical, bacterial, and other insults, and they contribute importantly to the genesis of the signs and symptoms of inflammation (see Moncada et al., 1978; Samuelsson, 1983). The peptidoleukotrienes have powerful effects on vascular permeability, while LTB4 is a potent chemoattractant for polymorphonuclear leukocytes and can promote exudation of plasma by mobilizing this source of additional inflammatory mediators. Although prostaglandins do not appear to have direct effects on vascular permeability, both PGE2 and PGI2 markedly enhance edema formation and leukocyte infiltration by promoting blood flow in the inflamed region. Moreover, they potentiate the pain-producing activity of bradykinin and other autacoids. However, PGEs inhibit the participation of lymphocytes in delayed hypersensitivity reactions. Moreover, they inhibit the release of hydrolases and lysosomal enzymes from human neutrophils as well as from mouse peritoneal macrophages. The use of cyclooxygenase inhibitors as antiinflammatory agents is a primary focus of Chapter 27: Analgesic-Antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout. Some experimental tumors in animals and certain spontaneous human tumors (medullary carcinoma of the thyroid, renal-cell adenocarcinoma, carcinoma of the breast) are accompanied by increased concentrations of local or circulating prostaglandins, bone metastasis, and hypercalcemia. Since the PGEs have potent osteolytic activity, it has been suggested that they are implicated in some cases of hypercalcemia. Some studies have implicated the effects of prostaglandins and HETEs on either the hematogenous metastasis of tumors or tumor angiogenesis. Pretreatment of animals with COX inhibitors or inhibitors of thromboxane synthase reduces the formation of tumor colonies or new blood vessel formation. There has been significant interest in the role of prostaglandins and the inducible cyclooxygenase, COX-2, in the development of malignancies, particularly colon cancer. Various prostaglandins induce proliferation of colon cancer cells, and COX inhibitors reduce colon tumor formation in experimental animals. In large epidemiological studies, regular use of aspirin is associated with a decreased incidence of colon cancer in human beings. Furthermore, in patients with familial colon polyposis syndromes, cyclooxygenase inhibitors significantly decrease polyp formation (see Williams et al., 1999). Therapeutic Uses The use of eicosanoids or eicosanoid derivatives as therapeutic agents is limited, even though these compounds have been the focus of intense research efforts. Part of the reason for this is that systemic administration of prostanoids is frequently associated with significant adverse side effects. This is not surprising given that eicosanoids have an array of biological activities in diverse cell types and tissues. Another factor limiting the use of these compounds as therapeutic agents is their short half-lives in the circulation. Thus, in some cases, continuous systemic administration of eicosanoids is required to achieve therapeutic efficacy. Despite these limitations, however, several prostanoids are of clinical utility in situations discussed below. Therapeutic Abortion As described above, there has been intense interest in the effects of
the prostaglandins on the female reproductive system. Their action as abortifacients
when given early in pregnancy is established. However, initial hopes that
they might provide a simple, convenient means of postimplantation
'contraception,' perhaps given as a vaginal suppository, have not
been fulfilled. Moreover, the abortifacient action of prostaglandins may be
inconstant and often incomplete and may be accompanied by side effects.
Prostaglandins appear, however, to be of value in missed abortion and molar
gestation, and they have been widely used for the induction of midtrimester
abortion. While PGE2 or PGF2 In the Several studies also have shown that systemic or intra-vaginal administration of the PGE1 analog misoprostol in combination with mifepristone (RU486; Peyron et al., 1993) or methotrexate (Hausknecht, 1995; Christin-Maitre et al., 2000) is highly effective in the termination of early pregnancy. Gastric Cytoprotection The capacity of several prostaglandin analogs to suppress gastric ulceration is a property of therapeutic importance. Of these, misoprostol (CYTOTEC), a PGE1 analog, is available for general use; its structure is as follows:
When given in doses that suppress gastric acid secretion, misoprostol
appears to heal gastric ulcers about as effectively as the H2
antagonists (see Chapter 37: Agents Used for Control of Gastric
Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease);
however, relief of ulcerogenic pain and healing of duodenal ulcers has not
been achieved consistently with misoprostol. In what may be considered
replacement therapy, the drug currently is used primarily for the prevention
of ulcers that often occur during long-term treatment with nonsteroidal
antiinflammatory drugs. In this setting, misoprostol appears to be as
effective as the proton pump inhibitor omeprazole. The major adverse
effect of misoprostol is diarrhea. Although frequently observed, it is mild
and usually does not force discontinuation of therapy. Misoprostol is rapidly
absorbed, with peak blood concentrations occurring at 30 minutes. It is
converted to the active misoprostol acid with a half-time of 30 to 60 minutes
(see Monk and Clissold, 1987; Walt, 1992). Misoprostol is available
for oral administration for the prevention of gastric ulcers in patients who
are at risk for development of such ulcers during long-term therapy with
nonsteroidal anti-inflammatory drugs. The recommended dosage is 200 Impotence PGE1 (alprostadil) may be used in the treatment of impotence. Intracavernous injection of PGE1 causes complete or partial erection in impotent patients who do not have disorders of the vascular system or cavernous body damage. The erection lasts for one to three hours and is sufficient for sexual intercourse. PGE1 is more effective than papaverine. The agent is available as a sterile powder that is reconstituted with water for injections (CARVERJECT Maintenance of Patent Ductus Arteriosus The ductus arteriosus in neonates is highly sensitive to vasodilation
by PGE1. Patency of the ductus may be necessary to maintain in
some neonates with congenital heart disease. For palliative, but not
definitive, therapy to temporarily maintain patency until surgery can be
performed, PGE1 (alprostadil, PROSTIN VR PEDIATRIC) is highly effective. Alprostadil
usually is infused intravenously at an initial rate of 0.05 to 0.1 Primary Pulmonary Hypertension Primary pulmonary hypertension is a rare, idiopathic disease mainly observed in young adults that leads to right heart failure and is frequently fatal. Lung or lung-heart transplantation has been the treatment previously. Long-term therapy with PGI2 (epoprostenol;FLOLAN) recently has been found to be highly effective and has either delayed or avoided the need for transplantation in a number of patients. In addition, many affected individuals have had a marked improvement in symptoms after receiving treatment with PGI2. The agent is administered by continuous intravenous infusion through a central venous catheter using a portable infusion pump. Adverse effects can include nausea, vomiting, headache, and flushing (McLaughlin et al., 1998). |
Platelet-Activating Factor
|
History In 1971, Henson demonstrated that a soluble factor was released from leukocytes and caused platelets to aggregate. Benveniste and his coworkers confirmed these observations and named the substance platelet-activating factor (PAF); their research indicated that the compound was a polar lipid. During this period, Muirhead described an antihypertensive polar renal lipid (APRL) produced by interstitial cells of the renal medulla. Sufficient evidence had accumulated by 1979 to conclude that PAF and APRL were identical. Hanahan and coworkers then synthesized acetylglyceryletherphosphorylcholine (AGEPC) and determined that this phospholipid had chemical and biological properties identical with those of PAF (Demopoulos et al., 1979). Subsequently, the structures of PAF and APRL were determined independently and were found to be identical with that of AGEPC (Hanahan et al., 1980; Polonsky et al., 1980). Of the names for the compound, platelet-activating factor has gained the greatest acceptance, despite the fact that the lipid has many biological actions in addition to those on platelets (Snyder, 1989; Koltai et al., 1991). Chemistry and Biosynthesis. PAF is 1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine. Its structure is as follows:
In contrast to the two long-chain acyl groups that are present in phosphatidylcholine, PAF contains a long-chain alkyl group joined to the glycerol backbone in an ether linkage at position 1 and an acetyl group at position 2. PAF actually represents a family of phospholipids, because the alkyl group at position 1 can vary in length from 12 to 18 carbon atoms. In human neutrophils, PAF consists predominantly of a mixture of the 16- and 18-carbon ethers, but its composition may change when cells are stimulated. Like the eicosanoids, PAF is not stored in cells but is synthesized in response to stimulation. The major pathway by which PAF is generated involves the precursor 1-O-alkyl-2-acyl-glycerophosphocholine, a lipid found in high concentrations in the membranes of many types of cells. The 2-acyl substituents include an abundance of arachidonate. PAF is synthesized from this substrate in two steps (see Figure 263). The first involves the action of phospholipase A2, with the formation of 1-O-alkyl-2-lyso-glycerophosphocholine (lyso-PAF) and a free fatty acid (usually arachidonate) (Chilton et al., 1984). In some cells, this reaction may represent a major source of the arachidonate that is metabolized to prostaglandins and leukotrienes. In the second step, lyso-PAF is acetylated by acetyl coenzyme A in a reaction catalyzed by lyso-PAF acetyltransferase. This represents the rate-limiting step. The synthesis of PAF may be stimulated during antigen-antibody reactions or by a variety of agents, including chemotactic peptides, thrombin, collagen, and other autacoids; PAF also can stimulate its own formation. Both the phospholipase and acetyltransferase are Ca2+-dependent enzymes, and PAF synthesis is regulated by the availability of Ca2+.
The inactivation of PAF also occurs in two steps (see Figure 263; Chilton et al., 1983; Stafforini et al., 1997). Initially, the acetyl group of PAF is removed by PAF acetylhydrolase to form lyso-PAF; this enzyme is present in both cells and plasma. Lyso-PAF is then converted to a 1-O-alkyl-2-acyl-glycerophosphocholine by an acyltransferase. PAF is synthesized by platelets, neutrophils, monocytes, mast cells, eosinophils, renal mesangial cells, renal medullary cells, and vascular endothelial cells. In most instances, stimulation of PAF synthesis results in the release of PAF and lyso-PAF from the cell. However, in some cells (e.g., endothelial cells) PAF is not released and appears to exert its effects intracellularly (Prescott et al., 1990). In addition to the generation of PAF enzymatically, PAF-like molecules can be formed from the oxidative fragmentation of membrane phospholipids (Patel et al., 1992). They are structurally different from PAF in that they contain a fatty acid at the sn-1 position of glycerol joined through an ester bond and various short-chain acyl groups at the sn-2 position. They mimic the structure of PAF closely enough to bind to its receptor and thus elicit the same responses. They are also substrates for PAF acetylhydrolase. An important distinction between PAF and these PAF-like oxidized lipids is that the synthesis of PAF is highly controlled, whereas the oxidized PAF-like phospholipids are produced in an apparently unregulated manner. The role that these latter compounds play in settings of oxidant stress is under investigation. Pharmacological Properties Cardiovascular System PAF is a potent vasodilator, and it lowers peripheral vascular resistance and systemic blood pressure when injected intravenously. PAF-induced vasodilation is independent of effects on the sympathetic innervation or arachidonate metabolism (Sybertz et al., 1985). However, the effects of PAF on the coronary circulation are a mixture of direct and indirect actions. The intracoronary administration of small amounts of PAF increases coronary blood flow by a mechanism that involves the release of a platelet-derived vasodilator. At higher doses, coronary blood flow is decreased by the formation of intravascular aggregates of platelets and/or the formation of TXA2 (Sybertz et al., 1985). The pulmonary vasculature also is constricted by PAF, and a similar mechanism is thought to be involved. Intradermal injection of PAF causes an initial vasoconstriction followed by a typical wheal and flare. PAF increases vascular permeability and promotes the movement of fluid out of the vasculature (McManus et al., 1981). As with substances such as histamine and bradykinin, the increase in permeability is due to contraction of venular endothelial cells, but PAF is thousandfold more potent than histamine or bradykinin. Platelets PAF is a potent stimulator of in vitro platelet aggregation that is accompanied by the release of TXA2 and the granular contents of the platelet; however, PAF does not require the presence of TXA2 or other aggregating agents to produce this effect. The intravenous injection of PAF causes formation of intravascular platelet aggregates and thrombocytopenia. Leukocytes PAF stimulates polymorphonuclear leukocytes to aggregate, to release leukotrienes and lysosomal enzymes, and to generate superoxide. Since LTB4 is more potent in inducing leukocyte aggregation, it may mediate the effects of PAF. Similarly, PAF promotes aggregation of monocytes and degranulation of eosinophils. PAF is a chemotactic factor for eosinophils, neutrophils, and monocytes. It also promotes the adherence of neutrophils to endothelial cells and their diapedesis. When given systemically, PAF causes leukocytopenia, with neutrophils showing the greatest decline. Intradermal injection causes the accumulation of neutrophils and mononuclear cells at the site of injection, and inhaled PAF increases the infiltration of eosinophils into the airways. Smooth Muscle PAF generally contracts gastrointestinal, uterine, and pulmonary smooth muscle. PAF enhances the amplitude of spontaneous uterine contractions; quiescent muscle contracts rapidly in a phasic fashion. These contractions are inhibited by inhibitors of prostaglandin synthesis. PAF does not affect tracheal smooth muscle but contracts the smooth muscle of peripheral airways. Although controversial, most evidence suggests that another autacoid (e.g., LTC4 or TXA2) mediates this effect of PAF. When given by aerosol, PAF increases airway resistance as well as the responsiveness to other bronchoconstrictors (Cuss et al., 1986). This bronchial hyperresponsiveness occurs after a delay of up to three days in human beings and may persist for 1 to 4 weeks. PAF also increases mucus secretion and the permeability of pulmonary microvessels; this results in fluid accumulation in the mucosal and submucosal regions of the trachea and bronchi. Stomach In addition to contracting the fundus of the stomach, PAF is the most potent known ulcerogen. When given intravenously, it causes hemorrhagic erosions of the gastric mucosa that extend into the submucosa. Kidney When infused intrarenally in animals, PAF decreases renal blood flow, glomerular filtration rate, urine volume, and excretion of Na+ (Schlondorff and Neuwirth, 1986). These effects are not due to the formation of platelet aggregates but are the result of a direct action on the renal circulation. PAF also stimulates the release of vasodilator prostaglandins, which tends to counteract the renal vasoconstriction. Mechanism of Action of PAF Extracellular PAF exerts its actions by stimulating a G proteinlinked cell surface receptor that has been detected in the plasma membranes of a number of cell types (Chao and Olson, 1993). Lyso-PAF is inactive, and biological activity is markedly reduced by relatively small changes in structure. The human PAF receptor has been cloned. When stimulated, it activates multiple signaling pathways, including phospholipase A2, phospholipase C, and phospholipase D, with resultant formation of inositol phosphates and diacylglycerol and release of arachidonate (Peplow, 1999). The arachidonate released by PAF is converted to prostaglandins, TXA2, or leukotrienes, which may function as extracellular mediators of the effects of PAF. PAF also may exert actions without exiting the cell. The clearest example is provided by the endothelial cell. Synthesis of PAF is stimulated by a variety of factors, but it is not released extracellularly (McIntyre et al., 1986). Accumulation of PAF intracellularly is associated with the adhesion of neutrophils to the surface of the endothelial cells, apparently because PAF promotes the expression or exposure of surface proteins that recognize and bind neutrophils. Receptor Antagonists Many compounds have been described that selectively inhibit the actions of PAF in vivo and in vitro (Koltai et al., 1991; Negro Alvarez et al., 1997). These drugs inhibit the binding of PAF to its receptor and block its actions selectively. Driving the development of these agents was the expectation that they would be potent antiinflammatory agents that might be useful in the therapy of disorders such as asthma, sepsis, and other diseases in which PAF is postulated to play a role. Two general classes of agents have been studied: (1) those that are natural compounds and include terpenes, lignans, and gliotoxins and (2) synthetic compounds with structures either related or unrelated to PAF. In animal models of sepsis and other diseases, various PAF antagonists gave encouraging results. In human trials, however, the results have been disappointing, with at least three recent studies failing to confirm any reduction of mortality in sepsis with either TCV-309 or BN 52501, two synthetic PAF receptor antagonists (Heller et al., 1998). Inconsistent results have been obtained in trials involving patients with asthma and psoriasis. Thus, after encouraging animal studies, it appears as though currently available PAF antagonists are of little benefit in human disease. Physiological and Pathological Functions of PAF Unlike the eicosanoids, PAF is synthesized by a select assortment of cells; this is presumed to limit its participation in various physiological and pathological processes. Platelets Since PAF is synthesized by platelets and promotes aggregation, it was proposed to be the mediator of cyclooxygenase inhibitorresistant, thrombin-induced aggregation. However, PAF antagonists fail to block thrombin-induced aggregation, even though they prolong bleeding time and prevent thrombus formation in some experimental models. Thus, PAF does not function as an independent mediator of aggregation but contributes to thrombus formation in a manner analogous to TXA2 and ADP. Reproduction and Parturition PAF may be involved in ovulation, implantation, and parturition. Rupture of the follicle is inhibited in experimental animals by the PAF antagonist ginkgolide B (Abisogun et al., 1989); the administration of PAF restores ovulation. Following ovulation and subsequent fertilization, the embryo begins to produce PAF, which promotes platelet aggregation and the release of platelet factors that appear to stimulate activation and implantation of the blastocyst. PAF is found in the amniotic fluid only after labor commences; PAF is thought to contribute to parturition by several mechanisms. It may cause contraction of the myometrium directly, or it may promote the release of PGE2 (and additional PAF) from amnion cells and promote uterine contractions indirectly. In any event, the importance of PAF is indicated by the delay in parturition induced by PAF antagonists in experimental animals. Inflammatory and Allergic Responses PAF is elaborated by leukocytes and mast cells and exerts proinflammatory effects. For example, intradermal injection of PAF duplicates many of the signs and symptoms of inflammation, including increased vascular permeability, hyperalgesia, edema, and infiltration of neutrophils. PAF also produces effects that suggest its importance in asthma. When inhaled, it is a potent bronchoconstrictor, promotes the accumulation of eosinophils in the lung, causes tracheal and bronchial edema, and stimulates the secretion of mucus. Moreover, PAF produces long-lasting bronchial hyperresponsiveness. The plasma concentration of PAF is increased in experimental anaphylactic shock, and the administration of PAF reproduces many of its signs and symptoms, suggesting a role for the autacoid in this condition. In addition, mice overexpressing the PAF receptor exhibit bronchial hyperreactivity and increased lethality when treated with endotoxin (Ishii et al., 1997). Despite the broad implications of these observations, the effects of PAF antagonists in the treatment of inflammatory and allergic disorders have been disappointing (see above). Although they reverse the bronchoconstriction of anaphylactic shock and improve survival, the impact of PAF antagonists on animal models of asthma and inflammation is marginal. Similarly, in patients with asthma, PAF antagonists partially inhibit the bronchoconstriction induced by antigen challenge but not challenges to methacholine, exercise, or inhalation of cold air. These results may reflect the complexity of these pathological conditions and the fact that other mediators likely contribute to the inflammation associated with these disorders. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 5038
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved