| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Gene Therapy
Overview
|
Advances in molecular and cellular biology have described the proteins that mediate many disease processes, while DNA technology provides ready access to the genes that control these events. The size, complexity, and cellular inaccessibility of these proteins make their delivery or modification by conventional pharmacological means impossible. Conceptually, gene therapy can overcome these barriers by the selective introduction of recombinant DNA into tissues so that the biologically active proteins can be synthesized within the cells whose function is to be altered. As such, delivery of recombinant DNA has become a central issue in all gene therapy strategies. Beyond delivery technologies, there exist many therapeutic paradigms that utilize DNA and other nucleic acids as drugs. Although originally envisioned as a treatment for inherited disorders, gene therapy has found applications in acquired illnesses such as cancer and infectious diseases. This chapter provides an introduction to the therapeutic issues and current strategies being explored to apply gene therapy to this wide range of diseases. |
Gene Therapy: Introduction
|
The modern era of molecular medicine has been highlighted by revolutionary accomplishments in genetics, genomics, and human molecular biology. There is extraordinary optimism that medicine will soon benefit from the development of new therapeutic technologies to directly target human genes, referred to as gene therapy. Growth of this discipline, which has emerged in the past decade, is evidenced by the exponential expansion of the medical and scientific literature devoted to this topic. There are five new biomedical journals that focus exclusively on the subject of gene therapy or nucleic acid drug development, and there are countless books and monographs on the subject. More than 300 clinical trials involving gene transfer in patients have been approved (Rosenberg et al., 2000), and the first nucleic acid drug, an antisense oligonucleotide (fomivirsen), has been approved by the United States Food and Drug Administration (FDA). Despite the tremendous advances over the past decade, gene therapy remains largely investigational. Many substantial obstacles still must be overcome in developing safe and effective nucleic acid delivery strategies that will promote long-lasting, tissue-specific expression of genetic material. This chapter divides the subject of gene therapy into three general themes: technologies for gene delivery, therapeutic paradigms, and disease targets. |
Gene Transfer Technologies
|
The ideal DNA delivery system would be one that could accommodate a broad size range of inserted DNA, could be produced easily in a concentrated form, and could be targeted to specific types of cells. Furthermore, such a system would provide long-term gene expression and would be nontoxic and nonimmunogenic. Such a DNA delivery system does not yet exist, and none of the available technologies for in vivo gene transfer is without significant limitations. A variety of viral and nonviral technologies are in development for use in human gene therapy. Table 51 compares the general features, advantages, and disadvantages of the more commonly employed gene transfer methodologies. Obstacles to Gene Therapy The therapeutic applications of gene transfer technology increase with each discovery of a new cellular process. At present, the ability to develop clinically efficacious therapies from scientifically sound principles is limited by several problems that, to some extent, plague all gene therapy strategies. For the foreseeable future, gene therapy is limited to somatic cells (nongerm-line cells). How these cells in a given tissue are targeted by the DNA delivery method has been an area of intense interest. Once the gene has been successfully transferred, the duration of transgene expression becomes important. Finally, the DNA vector itself must be analyzed for its potential to cause unwanted side effects (Jolly, 1994). DNA Delivery and Pharmacokinetics The delivery of exogenous DNA, and its subsequent processing by target cells, require the introduction of new pharmacokinetic paradigms beyond those that describe the conventional medicines in use today (see Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination). With in vivo gene transfer, one must account for the fate of the DNA vector itself (volume of distribution, rate of clearance into tissues, etc.), as well as for the consequences of altered gene expression and protein function. A multicompartmental model to describe these events in a quantitative fashion has been developed (Ledley and Ledley, 1994). Several processes that must be considered include: (1) the distribution of the DNA vector following in vivo administration; (2) the fraction of vector taken up by the target cell population; (3) the trafficking of the genetic material within cellular organelles; (4) the rate of degradation of the DNA; (5) the level of mRNA produced; (6) the stability of the mRNA produced; (7) the amount and stability of the protein produced; and (8) the compartmentalization of the transcribed protein within the cell, or its secretory fate. It is conceivable, although yet to be realized, that each of these events may be incorporated into the design of the gene transfer system in a rational way so as to tailor the gene transfer to the specific requirements of the disease being treated. Duration of Expression of Transferred Gene The length of time over which the transferred gene will function is of tremendous importance. In the treatment of inherited diseases, it is desirable to have stable gene expression over many years. In contrast, in the treatment of malignancy, long-term production of a therapeutic protein may be unnecessary and could have deleterious consequences. Vectors that integrate the transferred DNA into the chromosomes of the recipient cell have the greatest potential for long-term expression. Retroviral vectors and adeno-associated viral vectors have integrating functions. The persistence of the transgene DNA in the genome of the recipient cell does not, however, guarantee long-term gene expression in that cell. The production of the intended mRNA and protein may decline because of inactivation of the transgene promoter even though the DNA persists (Bestor, 2000). In some circumstances, loss of transgene expression may occur because of loss of the transduced cell by host immune processes (see Jolly, 1994). Adverse Consequences of Heterologous Gene Expression Along with factors that limit gene transfer and expression, there are potential adverse consequences that may arise as a result of successful gene transfer. As with any new drug, it will be impossible to predict these events in advance of more clinical experience. Nonetheless, some specific events can be anticipated independent of the transgene employed. Because, in most circumstances, gene transfer will result in the synthesis of a new protein, the possibility of an immune response must be considered. A severe immune response could inactivate a secreted product (as is seen in hemophilia patients receiving factor VIII replacement therapy) or lead to an 'autoimmune' response to transduced tissues. In some circumstances, the DNA vector itself may be immunogenic, as has been demonstrated for adenovirus vectors. An immune response to the vector may limit the duration of its effectiveness or preclude its readministration. Pathological events may arise from viral vector replication. Significant efforts have been directed toward the design of viral vectors that are unable to replicate (replication-incompetent) in the target cell. This has been achieved by the deletion of specific genes from the viral genome that are necessary for viral replication (see Figure 51). In order to produce the virus, it must then be grown in vitro in a cell specifically designed to provide those functions removed from the virus. By these means, replication-incompetent retroviruses, adenoviruses, adeno-associated viruses, and herpes viruses have been produced. This approach does not completely eliminate replicative potential in all circumstances. The virus may overcome the deletion of replication machinery by the use of unidentified host-cell factors or, theoretically, by recombination in the patient with wild-type viruses. Fortunately, these latter events have not been reported so far.
Ethical and Regulatory Issues Substantial attention has been directed toward ethical matters associated with gene therapy (Juengst and Walters, 1999). The major issues include concerns regarding the balance of risks and benefits to subjects enrolled in experimental gene transfer research, the selection and protection of research subjects, and the ethics of human germ-line gene transfer. As discussed below, assurance of patient safety has become a central issue in the regulatory process governing gene therapy research. Gene transfer into the human germ line, although potentially feasible, carries great moral implications. The potential to alter the genetic constitution of future generations raises enormous public concern that eugenic practices could evolve by which society selects against individuals with specific genotypes. Concern also has been raised that gene transfer techniques would be used for 'frivolous' purposes, such as cosmetic alterations or other enhancements that are unrelated to disease treatment. Continued public debate as well as discussions among scientists and ethicists are critical to the success and widespread acceptance of gene therapy as a standard treatment option. Arising from public and governmental concern for the safety and
ethical implications of gene therapy, strict regulatory processes have
evolved (Wivel and Anderson, 1999). In the early 1980s, federal oversight in
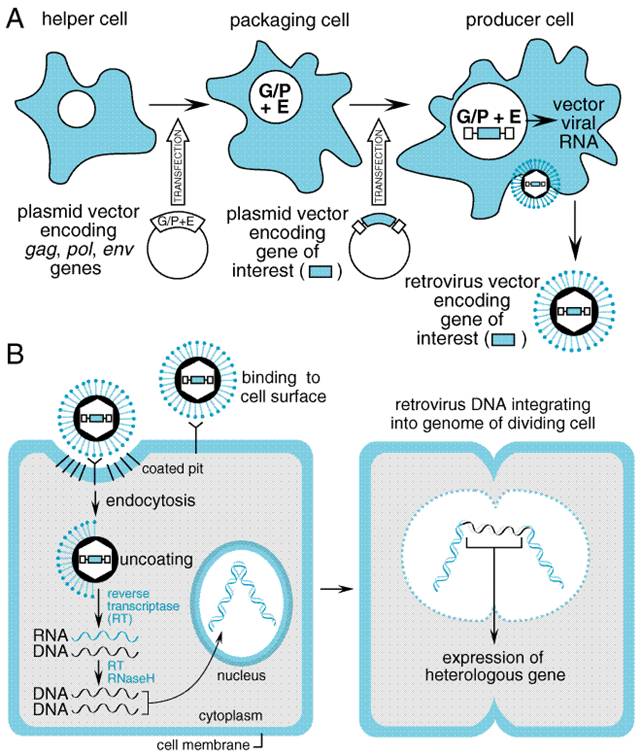
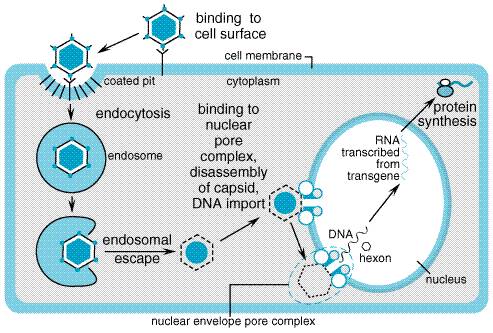
the Viral Vectors The natural life cycle of mammalian viruses has made them a logical starting point for the design of therapeutic gene-transfer vehicles. Viruses transfer and express exogenous genetic material during infection of host cells. In the simplest analysis, a virus consists of a nucleic acid genome encapsulated in a particle that can be taken up by the target cell, leading to the expression of virally encoded genes. For viral vectors to be useful, several viral functions must be altered. In most applications, the virus is rendered replication-incompetent to prevent uncontrolled spread of the transgene and must have some element of its own genome removed to allow for insertion of the transgene. Beyond this, additional modifications are dependent on the specific virus. Viral vectors have been used extensively in preclinical research and are the basis for the majority of gene therapy clinical trials now under way. Several important aspects of the viral life cycle and other biological features must be considered before selecting a vector for a specific application (Robbins and Ghivizzani, 1998). A critical determinant of the success of viral-based gene transfer is the ability of the virus to infect target cells (tropism) and to express a heterologous gene. Tropism is determined in part by the expression of specific cell-surface receptors on the host cell that provide attachment for the infecting virus and facilitate its uptake. Expression of a heterologous gene requires entry of the viral genome into the host cell nucleus followed by proper transcription and translation of its sequences. Several additional factors govern whether expression in the infected cell will be transient or long lasting. Finally, several aspects of genetic engineering and production of the viral vector impinge on its utility as a gene transfer vehicle. The principal viral vectors employed in current clinical gene transfer trials, or that appear promising for future trials, are derived from retroviruses, adenovirus, adeno-associated virus, herpes simplex virus, and lentiviruses. The following sections describe the basic biological features of each viral vector relevant to their use in gene therapy applications. Specific uses of individual vectors are described in more detail in later sections of this chapter. Retroviruses Retroviruses are small RNA viruses that can infect and replicate exclusively within dividing cells and are capable of integrating their genome into the host cell DNA. Therefore, retroviral vectors offer the potential for long-term expression in a limited range of target cell types. Most retroviral vectors have been derived from the Moloney murine leukemia virus (MMLV) and have been engineered to avoid expression of native viral genes, thereby preventing host immune responses against infected cells. Because of their requirement for cell division, retroviral vectors have been mostly used for ex vivo gene transfer (see below) or for the investigational treatment of cancer. Life Cycle Retroviruses are composed of an RNA genome that is packaged in an envelope derived from host cell membrane along with viral proteins. Three viral genes (gag, pol, env) are required for replication and packaging. For a retrovirus to effect gene expression, it must first reverse transcribe its positive-strand RNA genome into double-stranded DNA, which is then integrated into the host cell DNA. This process is mediated by the reverse transcriptase and integrase proteins contained in the retrovirus particle. Breakdown of the host cell nuclear envelope during mitosis is necessary for entry of the virus into the nucleus. The integrated provirus is able to use host cell machinery to carry out transcription of viral mRNAs and their subsequent processing and translation into viral proteins. The virus completes its life cycle by synthesizing new positive-strand RNA genomes from the integrated provirus. An encapsidation signal (psi) within the RNA mediates the organization of the viral genomic RNA and proteins into particles that bud from the cell surface. Vector Design and Production Retroviral vectors are constructed from the proviral form of the virus. The gag, pol, and env genes are removed to make room for the gene(s) of therapeutic interest and to eliminate the replicative functions of the virus (see Figure 51 for a strategic overview). Up to 8 kb of heterologous DNA can be incorporated into the retroviral vector. Because all virally encoded mRNAs are eliminated from the recombinant retrovirus, no viral proteins are produced by retroviral vectors. This removes any potential viral-encoded antigens that might lead to an immune response to the transduced cells. Along with the gene of therapeutic interest, sequences containing promoter and enhancer functions also may be included with the transgene to facilitate its efficient expression and, in some circumstances, to provide for tissue-specific expression in vivo. Alternatively, the native promoter and enhancer functions contained in the long terminal repeat (LTR) of the virus may be used for this purpose. After deletion of the genes encoding viral structural proteins and proteins that mediate viral replication, these viruses can be produced only in specially engineered viral packaging cell lines that are capable of providing these proteins (see Figure 51). The packaging cell line is optimally constructed by stably inserting the deleted viral genes (gag, pol, and env) into the cell in such a manner that these genes will reside on different chromosomes within the packaging cell. This strategy decreases the likelihood of a recombination event occurring that produces an intact viral genome that could be packaged into a replication-competent virus. The packaging cell line is used to construct a retroviral producer cell line that will generate replication-incompetent retrovirus containing the gene(s) of interest. This is done by introducing the recombinant proviral DNA into the packaging cell line. The recombinant proviral DNA is in the form of plasmid DNA containing the LTR sequences flanking a small portion of the gag gene that contains the encapsidation sequence and the genes of interest; this is transfected into the packaging cell line using standard techniques for DNA transfer. Several versions of this basic design have been employed to decrease the likelihood of recombinant events that could lead to the production of replication-competent virus (Jolly, 1994). The ability of the virus to infect a specific cell type is determined to a large extent by interactions between the viral envelope protein (encoded by env) and a corresponding cell-surface receptor. The MMLV envelope is ecotropic, which means that infection is restricted to the cells of a particular species, in this case mouse. An envelope affording a broader infection range is available by using the env gene from the 4070A strain of murine leukemia virus. This envelope gene has amphotropic specificity and can promote the infection of human, murine, and other mammalian cells. Modifications of the envelope protein can be achieved through a phenomenon known as pseudotyping, whereby the retrovirus incorporates alternative envelope proteins during viral packaging. For example, the glycoprotein (dubbed G protein, but not to be confused with G proteins involved in signal transduction; see Chapter 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect) of vesicular stomatitis virus (VSV-G) has been shown to incorporate efficiently into MMLV retrovirus particles (Chen et al., 1996). Incorporation of VSV-G expands the host range of the vector and improves the efficiency of infection. In addition, pseudotyping with VSV-G improves retroviral vector stability, which allows the pseudotyped virus to be concentrated by ultracentrifugation to higher titers. One drawback of using VSV-G is its toxicity to the mammalian cells that are used for viral packaging. To some extent, this toxicity can be avoided by using packaging cell lines that have inducible VSV-G expression (Iida et al., 1996). Retrovirus vectors pseudotyped with other envelope proteins, such as those derived from the Gibbon ape leukemia virus (Gallardo et al., 1997) and the lymphocytic choriomeningitis virus (Miletic et al., 1999), may be less toxic to mammalian host cells with preservation of the advantages of VSV-G pseudotyping. General Clinical Applications The clinical administration of retroviruses has been accomplished most frequently by the ex vivo transduction of patients' cells, and by the direct injection of virus into tissue. The ex vivo approach requires the isolation and maintenance in tissue culture of the cells, infection with the viral vector, and subsequent reimplantation into the patient. This approach was used to modify lymphocytes and hematopoietic cells in the treatment of adenosine deaminase deficiency (Parkman et al., 2000) and hyperlipidemia (Grossman et al., 1994); the same strategy also was used to express immune modulatory agents in tumor cells (Lode and Reisfeld, 2000). Direct in vivo delivery of retroviral vectors has been utilized largely in the treatment of solid tumors (Gomez-Navarro et al., 1999). Safety The use of retroviral vectors has raised several important safety issues. One concern is that because the virus integrates into the target cell DNA (an attractive feature for long-term expression) and because integration occurs in a nearly random fashion, integration could be mutagenic (insertional mutagenesis). For example, undesired mutations might occur if insertion of the retroviral DNA altered the function of a gene that regulates cell growth. Although replication-competent retroviruses have tumorigenic potential, this has not been observed with the replication-incompetent vectors that are in use as gene transfer agents. Lentiviruses The lentiviruses are a subset of retroviruses that can infect dividing and nondividing cells (Buchschacher, Jr. and Wong-Staal, 2000). The best-studied lentivirus is the human immunodeficiency virus (HIV)-1, and gene transfer vectors derived from this viral genome have potential advantages over previous retroviral vectors. In particular, they show promise in their ability to efficiently transduce hematopoietic stem cells (Miyoshi et al., 1999). These vectors also are capable of establishing long-term expression. However, because of their lineage, substantial biosafety concerns need to be addressed before lentiviral vectors advance toward use in clinical trials (Amado and Chen, 1999). Life Cycle The biology of lentiviruses is similar to that of retroviruses (Tang et al., 1999). The major difference enabling lentiviruses to infect nondividing cells is the ability of its viral preintegration complex to interact with and be transported by the nuclear membrane. This preintegration complex consists of transcribed viral DNA, integrase, and the matrix protein encoded by the gag gene. The matrix protein contains a localization sequence that enables the complex to dock with a nucleopore. Transport into the nucleus of a nondividing cell then provides the viral genome the opportunity to integrate into the host cell DNA. Vector Design and Production Lentiviral vectors derived from HIV-1 are rendered replication-incompetent by deletion of various accessory genes and by the use of packaging cell lines in which components necessary for assembling virus particles are provided by separate genetic elements (Srinivasakumar and Schuening, 1999). This greatly minimizes the possibility of recombination events occurring during vector production that, theoretically, could generate a self-replicating virus. In addition, deletion of the tat gene and deletions in the viral LTR region also reduce the likelihood that a replication-competent lentivirus will emerge during production or in vivo. Both actively dividing and nondividing cells can be infected by lentiviral vectors. This includes hematopoietic stem cells and terminally differentiated cells such as muscle, neurons, hepatocytes, and retinal photoreceptors. However, cells may need to be stimulated to enter the G1 phase of the cell cycle before they can be transduced with lentivirus (Park et al., 2000). The lentivirus env gene can be replaced by pseudotyping with VSV-G or another appropriate viral envelope protein to facilitate a broader host range (Li et al., 1998). Long-term expression of lentiviral-encoded transgenes has been demonstrated within the central nervous systems of experimental animals. Stable and efficient gene delivery also has been demonstrated in the retina. Expression of lentivirus-encoded transgenes is associated with little or no inflammation or signs of tissue pathology. Safety Given the lineage of lentiviral vectors derived from HIV-1, there has been appropriate concern about the possibility of recombination events leading to the development of a replication-competent virus (Amado and Chen, 1999). Conceivably, a self-replicating lentiviral vector could be harmful by undergoing insertional mutagenesis or by acquiring characteristics of the parent HIV-1. Concerns also have been raised regarding the consequences of HIV infection of an individual treated previously with a lentiviral vector. Theoretically, the wild-type HIV virus could enable mobilization of the gene-transfer vector by acting as a helper virus. This theoretical phenomenon in fact could be an advantage for using lentiviral vectors to treat HIV infection with antiHIV gene therapy. Through improved vector design and production, these and other concerns can be addressed. Adenoviruses Adenoviruses are double-stranded, linear DNA viruses that replicate independently of host cell division. Adenoviral vectors possess several attractive features that have encouraged their development for clinical use. They are capable of transducing a broad spectrum of human tissues, including respiratory epithelium, vascular endothelium, cardiac and skeletal muscle, peripheral and central nervous tissue, hepatocytes, the exocrine pancreas, and many tumor types. Over 40 serotypes of human adenoviruses are known, and the clinical spectrum of human adenoviral infections is well described (Horwitz, 1990). Most if not all adults have had prior exposure to adenovirus and are seropositive for antiadenovirus antibodies when tested by sensitive methods. Efficient gene transfer and transgene expression can be obtained in dividing and nondividing cells. Several routes of administration can be used, including intravenous, intrabiliary, intraperitoneal, intravesicular, intracranial, and intrathecal injection, and direct injection into the target organ parenchyma. The multiple routes of administration provide flexibility in targeting based on anatomical boundaries. There are two significant disadvantages to adenovirus-based vectors. First, because the virus remains episomal after host cell infection, long-term expression generally does not occur. Second, adenoviral infection induces both cellular and humoral immune responses that eliminate virally transduced cells and reduce the efficacy of repeat administration. This immune response also may account for adverse effects of adenoviral gene transfer. Life Cycle Infection by adenovirus begins with binding of the fiber protein,
which extends from the icosahedral capsid, to the coxsackievirus and
adenovirus receptor (CAR) on the host cell surface (Figure 52). Following
attachment, an interaction between a tripeptide motif (Arg-Gly-Asp) in the
penton base occurs with cell-surface integrins (
Since the E1 genes are involved intimately in adenovirus replication, their removal renders the virus replication-incompetent or, at the very least, severely crippled with respect to replication. Due to the complexity of the virus, it has been more difficult to remove all adenoviral genes, as is done with retroviral vectors. The expression of adenoviral proteins with the currently employed adenoviral vectors leads to both a cellular and a humoral immune response to recombinant adenoviral vectors. In some instances, this may limit the utility of this vector both in terms of host immune response to adenovirally transduced cells and with respect to readministration of the vector. Vector Design and Production Although several adenoviral serotypes are known, serotypes 2 and 5 have been used most extensively for vector construction. First-generation adenoviral vectors were engineered by deletion of the E1 and E3 regions in the viral genome. These deletions render the virus replication-incompetent and allow the insertion of foreign DNA of up to 7.5 kb in length. Second-generation adenoviral vectors feature additional deletions of the E2 and E4 regions, modifications that help reduce antigenicity but limit viral gene expression in infected cells. More extensive removal of viral genes results in helper-dependent adenoviral vectors that should be much less likely to induce an immune response and have greatly increased carrying capacity (Kochanek, 1999). However, helper-dependent adenoviral vector systems appear to be less stable in vivo, and there are limitations for producing high-titer virus. Large amounts of adenoviral vector can be produced by growing the recombinant virus in a packaging cell line (typically, human embryonic kidney 293 cells) engineered to express E1 proteins that complement the E1-deficient viral genome. The virus is isolated by lysing the infected packaging cells and purifying the crude lysate by cesium chloride density-gradient centrifugation, a procedure that not only separates the virus from other tissue culturederived substances but also concentrates the virus to very high titers (over 1013 particles per ml). The purified virus is remarkably stable in a variety of aqueous buffers and can be frozen for a prolonged period of time without loss of activity. Host Cell Range Adenoviruses can infect a broad range of dividing and nondividing cells. Their broad host range can be attributed to the near ubiquitous expression of cell-surface receptors that can mediate adenovirus recognition and uptake. However, some cells express low levels of the CAR or present the receptor at an inaccessible cellular location. Modifying the tropism of adenoviral vectors also is feasible (Wickham, 2000). Immunological strategies have been used in which a dual-specific antibody directed at both the viral fiber and a target cell surface protein simultaneously neutralizes the intrinsic targeting of the virus and redirects its attachment to a specific cell type. The fiber protein and its terminal knob domain also can be genetically engineered to redirect or enhance attachment of the virus (Douglas et al., 1999). Finally, an adapter protein strategy can be used in which recombinant CAR is fused to a ligand such as epidermal growth factor (EGF), and the fusion protein facilitates specific attachment of the virus to cells expressing the EGF receptor (Dmitriev et al., 2000). General Clinical Applications There are numerous clinical trials in progress utilizing adenoviral vectors for gene transfer in both inherited and acquired conditions. The lack of long-term expression and resulting immune reaction to infected cells pose substantial obstacles to the treatment of lifelong inherited conditions. The episomal nature of the adenovirus genome ultimately limits the duration of gene expression in tissues with active cell division, such as bone marrow and epithelia, because each round of target cell division after gene transfer is not accompanied by replication of the transgene. The use of replication-incompetent and replication-competent adenoviral vectors also may have utility in the treatment of cancer (see below). Safety The main adverse effects of adenoviral vectors relate to the immune
reaction elicited by infected cells. Safety concerns over use of adenoviral
vectors were heightened following a highly publicized death that occurred
during a clinical trial ( Adeno-Associated Viruses Adeno-associated viruses (AAV) are small, nonenveloped, single-stranded DNA viruses that have many attributes desirable in a gene-transfer vector. They are nonpathogenic, can stably transduce nondividing cells with high efficiency, and can be engineered to carry heterologous genes without requiring the potential immunogenic expression of viral proteins. The major limitations of AAV as a gene delivery vector are its limited DNA-carrying capacity and shortcomings in producing high-titer virus. Clinical investigations using AAV vectors are commencing, and there are indications that this gene-transfer vehicle may be well suited to a variety of gene therapy applications (Monahan and Samulski, 2000). Life Cycle Adeno-associated virus has a helper-dependent life cycle, meaning that viral replication requires genetic elements from another viral genome. The virus has two distinct phases to its life cycle. In the absence of helper virus (adenovirus), the wild-type virus will infect a host cell, integrate into the host cell genome, and remain latent for an extended period of time. In the presence of adenovirus, the lytic phase of the virus is induced and leads to active virus replication. Structurally, the AAV genome is composed of two open reading frames (called rep and cap) flanked by inverted terminal repeat (ITR) sequences. The rep region encodes four proteins that mediate AAV replication, viral DNA transcription, and endonuclease functions used in host genome integration. The rep genes are the only AAV sequences required for viral replication. The cap sequence encodes structural proteins that form the viral capsid. The ITRs contain the viral origins of replication, provide encapsidation signals, and participate in viral DNA integration. The function of many of these proteins and the overall biology of the virus have been studied largely in wild-type viruses (Kotin, 1994). Infection begins by attachment of the virus to its principal
cell-surface receptor, heparan sulfate proteoglycan (Summerford and Samulski,
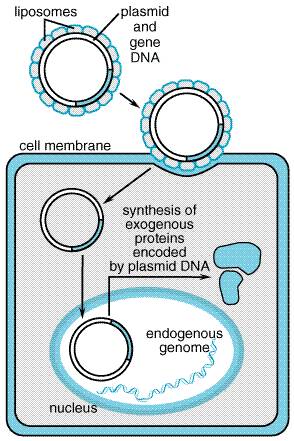
1998). Additional cofactors, fibroblast growth factor receptor type 1, and Vector Design and Production Current AAV vectors can be produced using a system of three recombinant plasmids (Xiao et al., 1998). The main vector plasmid contains a transgene located between the two ITRs. A second plasmid provides rep and cap, and a third plasmid carries essential elements from the adenoviral genome necessary for viral packaging. This triple plasmid strategy obviates the need for coinfection of producer cells by wild-type adenovirus. Because of the small size of the AAV genome, the DNA-carrying capacity is limited to 5.2 kb. This clearly limits the size of the DNA cargo and restricts the capacity of the vector to carry important promoter/enhancer elements to direct gene expression in the target cell. Doubling the vector capacity may be feasible by use of a dual-vector system, in which two halves of a transgene are assembled in vivo from two separate AAV vectors that combine into a circular concatamer during transduction (Sun et al., 2000; Yan et al., 2000). Using this approach, it is conceivable to assemble larger genes or to include important regulatory elements that are too large to fit within a single vector molecule (Duan et al., 2000). Currently, the major limitations for production of recombinant AAV relate to difficulties in obtaining high-titer virus and in quantifying the viral titer. Recombinant AAV can infect a wide range of host cells. Preclinical studies have demonstrated efficient gene transfer into skeletal muscle, the central nervous system, lung, liver, gastrointestinal tract, and eye. General Clinical Applications The clinical experience with AAV-based gene delivery is growing, and clinical trials exploiting delivery to lung and muscle are in progress. This vector appears to be well suited for establishing long-term expression in muscle, heart, the central nervous system, and other tissues. Early results from clinical trials using AAV vectors to express factor IX ectopically in skeletal muscle for the treatment of hemophilia appear to be successful (see below). The ability of this vector system to achieve long-term gene expression without concomitant host immune reactions or cytotoxicity suggests that it may be an appropriate delivery vehicle for the treatment of certain inherited conditions. Safety AAV is a nonpathogenic virus, and early experiences with this viral vector have demonstrated the absence of significant host immune responses. Formerly, there was concern about contamination of AAV vector by the helper adenovirus, but this has been alleviated by newer production schemes (Xiao et al., 1998). Finally, recombinant AAV vectors may integrate randomly into the genome, and there is concern regarding the possibility of insertional mutagenesis. The site-specific integration of wild-type AAV on chromosome 19 may require that specific viral sequences be retained in the vector (Rivadeneira et al., 1998). Herpes Simplex Virus-1 The herpes simplex virus (HSV) is a large (152-kb), double-stranded DNA virus that replicates in the nucleus of infected cells and exhibits a broad host cell range. The virus can infect dividing and nondividing cells and persist in a nonintegrated state. Large (20- to 30-kb) sequences of foreign DNA can be inserted into the viral genome by homologous recombination, or through insertion/deletion mutagenesis. Herpes simplex virus-1 has a natural tropism for neuronal tissues, and its potential utility as a gene transfer vehicle for treating neurological conditions including Parkinson's disease and brain cancer has been recognized (Fink and Glorioso, 1997; Simonato et al., 2000). The major drawbacks of using HSV-1 are its cytotoxicity and the occurrence of transgene silencing. Life Cycle The natural biology of HSV-1 infection involves both lytic and latent phases. During primary infection, virus attaches to and penetrates epithelial cells (skin or mucosa) and replicates. Assembled viral capsids exit the infected cell and simultaneously acquire an envelope by budding through the plasma membrane. Virus particles then fuse with peripheral sensory nerves in the vicinity of the primary infection and are transported retrograde in the nerve axon to its cell body. Virus attachment is mediated through heparan sulfate moieties on the target cell surface (Laquerre et al., 1998). Once in the neuronal cell body, the virus can continue in the lytic phase or enter a latent phase. The latent phase is characterized by silencing of lytic genes and expression of a discrete set of latency-associated transcripts (LATs) driven by two latency-associated promoters (LAP1, LAP2). Wild-type virus can reenter the lytic phase and spread viral progeny to the original site of primary infection or into the central nervous system. Vector Design and Production Replication-incompetent HSV-I has been engineered by deleting several genes essential for the lytic phase, in particular, the immediate-early genes ICP4, ICP22, and ICP27 (Krisky et al., 1998). Deletion of these genes also produces vector that exhibits less cytotoxicity and more prolonged transgene expression in cultured cells. Helper virus-free packaging systems have been developed that permit production of vector capable of transducing neuronal cells in vivo without cytopathic effects (Fraefel et al., 1996), although viral titer may be somewhat limited. There also are descriptions of more efficient approaches for engineering recombinant HSV-1 vector that enable insertion of two independent transgene expression cassettes into a single virus (Krisky et al., 1997). A significant problem with HSV-1 vectors is the difficulty in achieving long-term expression due to transgene silencing. Use of the latency-active promoters to drive gene expression, or linking transgenes to an internal ribosome entry site (IRES) inserted downstream of the latency-associated transcript regulatory sequences, appears to have good prospects for improving the longevity of HSV-1mediated expression (Goins et al., 1999; Lachmann and Efstathiou, 1997; Marshall et al., 2000). HSV-1 is capable of infecting a broad range of human cells, but its predilection for neurons is most notable. Modifying the tropism of HSV-1 may permit more specific targeting of the vector. Deletion of viral glycoprotein genes that are responsible for cell attachment produces an entry-incompetent HSV-1, which can be complemented by alternative attachment proteins (Anderson et al., 2000). General Applications Because of their DNA-carrying capacity, HSV-1 vectors may provide a vehicle for delivering particularly large gene cargoes. For example, a complete dystrophin cDNA (14 kb) has been introduced successfully into cultured muscle cells from mice with experimental muscular dystrophy (Akkaraju et al., 1999). Replication-competent HSV-1 vectors are being developed for treatment of brain and other neoplasms (Martuza, 2000). Safety The major safety concern for HSV-1 vectors is their cytotoxicity. Newer production schemes that eliminate helper virus and additional genetic engineering to remove cytopathic genes may reduce this liability. Nonviral DNA Delivery Strategies A variety of nonviral approaches to mediate cellular uptake of exogenous DNA have been developed and tested. These include naked plasmid DNA, DNA-liposome complexes, DNA-protein complexes, and DNA-coated gold particles. Production is generally easier and less expensive than with viral vectors. However, in general, low efficiency of transduction and transient expression are substantial limitations to their usefulness in gene therapy. Longer-lasting expression may be possible by engineering the gene of interest with transposons, naturally occurring, mobile genetic elements that can integrate into chromosomal DNA (Yant et al., 2000). Uncomplexed Plasmid DNA Surprisingly, purified DNA (or mRNA) can be injected directly into tissues, resulting in transient gene expression. This has been best illustrated in muscle tissue, where direct injection of uncomplexed (naked) DNA is most effective (Wolff et al., 1992). Skin also is capable of expressing plasmid DNA delivered either by direct injection (Hengge et al., 1996) or by other physical methods including ballistic transfection using DNA-coated gold beads (Lin et al., 2000). Expression of an antigenic protein in either skin or muscle using this approach may have utility for immunization (Davis et al., 1995), and clinical trials are addressing the effectiveness and safety of this strategy for vaccination against infectious diseases (Le et al., 2000; Tacket et al., 1999). Plasmid DNA injection of muscle also may prove useful for the ectopic synthesis of therapeutic proteins such as erythropoietin (Tripathy et al., 1996). DNA-Coated Gold Particles Plasmid DNA can be affixed to gold particles (approximately 1 micron in diameter) and then 'shot' into accessible cells. The DNA is coprecipitated onto the gold particle and then propelled, using an electric spark or pressurized gas as the motive force. This 'gene gun' can be used to accelerate the DNA-coated particles into superficial cells of the skin (epidermis) or into skin tumors (melanomas). Gene expression lasts only a few days, which may be more a function of the cells targeted (e.g., skin cells that are sloughed) than the method of delivery. Gene-gun delivery is ideally suited to gene-mediated immunization (Haynes et al., 1996), where only brief expression of antigen is necessary to achieve an immune response. Liposomes Liposomes are either unilamellar or multilamellar spheres that are manufactured using a variety of lipids. They are capable of delivering DNA to the interior of cells. The premise is that by surrounding hydrophilic molecules with hydrophobic molecules, agents otherwise impermeable to cell membranes might be escorted into the cell. A diagram illustrating the presumed mechanism for liposome-plasmid transfection is given in Figure 53. Proteins and other nonlipid molecules can be incorporated into the lipid membranes. Because the substance to be delivered must be encapsulated within the liposomes, the manufacturing process is complex. Also, most DNA constructs used for gene therapy are large compared to the liposome, so encapsulation efficiency is very low. For convenience, liposomes are classified as either anionic or cationic, based on their net negative or positive charge, respectively.
Anionic Liposomes The first in vivo delivery of a gene using liposomes involved transfer of insulin complexed with anionic liposomes into rats (Soriano et al., 1983). The transfected rats had increased circulating levels of insulin and decreased blood glucose concentrations. In spite of this early success, there are significant drawbacks to the use of anionic liposomes for delivering DNA. These structures, when given intravenously, primarily target the reticuloendothelial cells of the liver, making them of little use for other cell targets. Various proteins can be inserted into the external layer of liposomes to alter their in vivo behavior, including cell-selective delivery. This approach can enable liposomes given intravenously to evade the reticuloendothelial system. Protein ligands or antibodies to cell surface molecules incorporated into the liposome surface also can target liposomes to specific cell-surface receptors on desired cell populations (Wu and Wu, 1987). Cationic Liposomes In vivo, cationic liposomes have properties quite different from those of anionic liposomes. Intravenous injection of cationic complexes has been shown to effect transgene expression in most organs if the liposome-DNA complex is injected into the afferent blood supply to the organ. In addition, the liposome-DNA complexes can be administered by intraairway injection or aerosol to target lung epithelium. In experimental animals, neither intravenous injection nor aerosol delivery of cationic liposome-plasmid complexes appears to be toxic (Canonico et al., 1994). DNA-Protein Conjugates Cell-specific DNA-delivery systems have been developed that utilize unique cell-surface receptors on the target cell (Michael and Curiel, 1994). By attaching the ligand recognized by such a receptor to the transgene DNA, the DNA-ligand complex becomes selectively bound to and internalized into the target cell (Wu and Wu, 1987). These molecular conjugate vectors are attractive, because they potentially offer cell-specific gene transfer without the attendant problems of viral vectors. Initial model systems focused on developing effective means of attaching the DNA to the ligand using polycations, antibody-antigen complexes, and biotin-streptavidin linkers. Poly-L-lysine (PLL), a polycation, has been widely used, as it can be easily coupled to a variety of protein ligands by chemical cross-linking methods. When the PLL-ligand adduct is mixed with plasmid DNA, macromolecular complexes form in which the DNA is electrostatically bound to the PLL-ligand molecules. These toroidal structures (50 to 100 nm in diameter) present ligands to the cell-surface receptor that are efficiently endocytosed. The transferrin receptor (Zenke et al., 1990), the asialoorosomucoid receptor (Wu and Wu, 1987), and cell-surface carbohydrates (Batra et al., 1994) also have been used to demonstrate the potential of ligand-mediated gene delivery. The asialoorosomucoid receptor is of particular interest, because it is found almost exclusively on hepatocytes and therefore might be useful in mediating gene transfer into the liver. |
Therapeutic Paradigms
|
The transfer of nucleic acid molecules into living cells has many diverse clinical applications. Although originally conceived as a treatment of inherited disorders, gene therapy and the use of nucleic acid drugs have been deployed for the treatment of a wide variety of acquired diseases, including cancer and infections. This section contains discussions of many of these different strategies and illustrates their potential applications. Gene Therapy for Inherited Disorders Gene therapy can be applied to the treatment of inherited disorders, especially those transmitted by recessive inheritance. In this situation, a genetic defect typically ablates expression of the normal, functional gene product (loss of function). Strategies to restore gene function require high-efficiency delivery of the normal gene sequence to tissues that are affected by the inherited condition. This approach has been referred to as 'gene replacement,' although generally no attempt is made to remove the native, mutant gene. A more appropriate term describing this approach might be 'gene augmentation.' Furthermore, inserting additional copies of a normal gene into a tissue expressing a condition transmitted by a dominant mode of inheritance will not necessarily overcome the cellular defect. This is especially true when a dominantnegative disease mechanism occurs. Use of this paradigm is illustrated for several disease conditions later in this chapter. Correcting a genetic deficiency requires that the inserted gene
product be expressed in sufficient amounts to achieve a therapeutic effect;
the threshold for this effect varies widely among genetic diseases. Often,
this can be estimated from clinical observations comparing the severity of
the disease with the extent of the gene deficiency. This is illustrated by
the hemophilias, where the extent of bleeding complications is roughly
proportional to the extent of the deficiency in circulating coagulation
factors. Such estimates are not possible in other disorders such as cystic
fibrosis, where the amount of cystic fibrosis transport regulator (CFTR) gene
expression in the airway and in other epithelial cells that is necessary to
achieve therapeutic benefit is not known. These issues become more complex in
diseases where gene expression must be carried out in a highly regulated fashion.
This can be illustrated by the thalassemias, which arise from defects in the
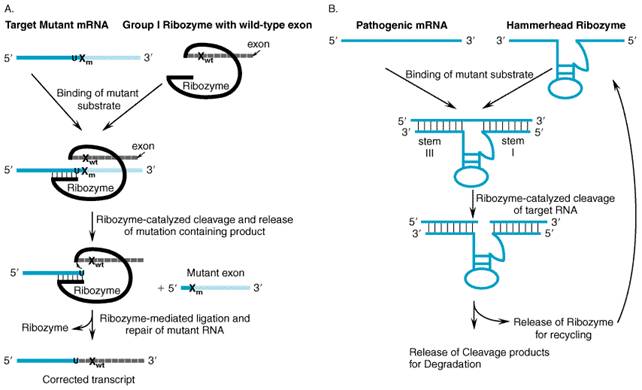
synthesis of hemoglobin Genetic Repair Strategies Several approaches have been developed to repair genetic errors directly rather than complementing the defect with a functional allele. Genetic repair strategies offer several potential advantages, such as a concomitant decrease in the production of deleterious gene products and the increased likelihood that expression of the targeted gene remains under appropriate physiologic control. This paradigm conceptually is well suited for the treatment of inherited conditions arising from dominant-negative mutations. One strategy is RNA repair, which enlists catalytic RNA molecules (ribozymes) capable of mediating trans-splicing reactions. With this approach, a defective portion of an mRNA molecule is replaced with the corresponding wild-type sequence. A second strategy focuses on repairing the mutation at the genomic level by inducing mechanisms using specialized oligonucleotides composed of both RNA and DNA residues. In both cases, the repaired gene or transcript remains under the transcriptional control of the native gene. RNA Repair by Trans-Splicing Ribozymes RNA enzymes or ribozymes have been the focus of much study since their discovery in the early 1980s. Numerous biochemical experiments have been performed to elucidate the mechanisms of how certain RNA molecules can form active sites and perform catalysis. More recently, the study of ribozymes has attracted increased attention because of the potential usefulness of these RNA enzymes for a variety of gene therapy applications. Ribozymes The first discovered ribozyme was the self-splicing group I intron from Tetrahymena thermophila (T. thermophila). The reaction mediated by this RNA enzyme has been extensively characterized, and the mechanism by which it excises itself from precursor ribosomal RNAs (pre-rRNA) without the aid of proteins is well understood (Cech, 1993). The second ribozyme to be recognized was the RNA subunit of RNase P. RNase P catalyzes the removal of upstream sequences on precursor-tRNAs to produce mature 5' ends on tRNA molecules in a wide variety of cell types (Symons, 1992). A second class of catalytic introns (group II) has been discovered in the organelle genomes of several lower organisms (Frank and Pace, 1998). In addition to the splicing reaction, the group II intron also has the ability to insert itself into double-stranded DNA with assistance from a multifunctional, intron-encoded protein. Several other catalytic RNA motifs have been discovered that naturally are associated with plant and human pathogens. The hammerhead and hairpin ribozymes are derived from satellite RNAs from plant viroid and virusoids, and the hepatitis delta virus (HDV) ribozyme is derived from a short, single-stranded RNA virus found in some patients with hepatitis B virus. Each of these small RNA enzymes catalyzes a self-cleavage reaction that is believed to play a major role in the replication of these single-stranded RNA pathogens (Symons, 1992). Ribozymes differ in size and catalytic mechanism. Each type of ribozyme adopts a characteristic secondary and tertiary structure that is required for it to assemble a catalytic center and perform catalysis (Cech, 1992). Ribozymes derived from group I introns and RNase P are typically greater than 200 nucleotides in length; both cleave target RNAs to generate products with 3'-hydroxyl and 5'-phosphate termini. By contrast, hammerhead, hairpin, and the HDV ribozymes are only 30 to 80 nucleotides in length and form cleavage products with 2', 3' cyclic phosphate and 5'-hydroxyl termini. All of these self-cleaving ribozymes have been reengineered so that they can cleave other target RNA molecules in trans in a sequence-specific manner. This ability to cleave specifically targeted RNAs has led to much speculation about the potential utility of trans-cleaving ribozymes as inhibitors of gene expression (Rossi, 1999). Such trans-splicing ribozymes may prove to be effective at repairing defective cellular transcripts by cleaving off mutant nucleotides and ligating on functional RNA sequences (see Figure 54) (Sullenger, 1999).
Ribozyme-mediated RNA repair may be useful for treating a variety of diseases. As mentioned above, the Tetrahymena group I ribozyme can catalyze a trans-splicing reaction. In the first example of this application, the group I ribozyme from T. thermophila was reengineered to repair truncated lacZ transcripts via targeted trans-splicing in Escherichia coli (Sullenger and Cech, 1994) and in mammalian cells (Jones et al., 1996). In both settings, the ribozyme spliced restorative sequences onto mutant lacZ target RNAs with high fidelity and maintained the open reading frame for translation of the repaired transcripts. In a subsequent study, the efficiency of RNA repair was monitored; the ribozyme was able to revise up to 50% of the truncated lacZ transcripts when ribozyme and lacZ substrate encoding plasmids were cotransfected into mammalian fibroblasts (Jones and Sullenger, 1997). More recently, three studies have demonstrated that group I ribozymes
can successfully amend faulty transcripts that are associated with common
genetic diseases and cancer. In one study, a trans-splicing ribozyme
was demonstrated capable of repairing transcripts associated with myotonic
dystrophy (Phylactou et al., 1998). Other investigations have employed
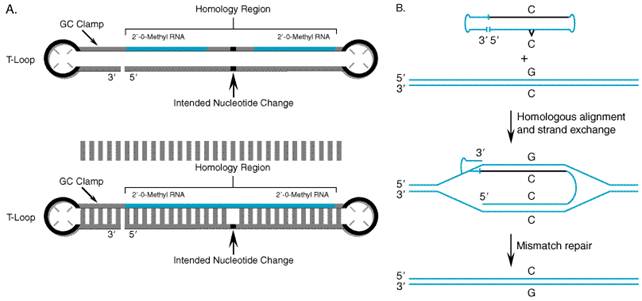
RNA repair to correct sickle RNA/DNA Oligonucleotides (Chimeraplasty) Disease-producing mutations may be repaired by harnessing the DNA mismatch repair machinery in cells. Chimeric RNA/DNA oligonucleotides have been successfully employed to repair mutant sequences at the genomic level in several animal models of disease. Insights gleaned from studies of homologous recombination led Kmiec and colleagues to pioneer this novel form of gene repair, termed chimeraplasty (Yoon et al., 1996). Chimeric oligonucleotides consist of a self-complementary sequence composed of both DNA and modified RNA residues (2'-O-methyl ribonucleotides) that form a duplex structure flanked by two polythymidine hairpin loops (Figure 55). The presence of 2'-O-methyl RNA residues provides an increased binding affinity with the target sequence and nuclease resistance.
Design of Chimeric Oligonucleotides The conventional chimeric oligonucleotide targeting sequence is made up of two stretches of ten modified ribonucleotide residues interrupted by five deoxynucleotide residues, with the central DNA base designed to mismatch with the targeted mutation (Figure 55). At the point of this mismatch, the oligonucleotide sequence serves as a template to guide the DNA repair machinery in correcting the mutation. Chimeric oligonucleotides also can direct the insertion and deletion of single nucleotides. While the typical length for the homologous targeting sequence is 25 bases, it has been shown that the frequency of nucleotide correction increases with the length of this region (Gamper et al., 2000). Mechanism of Action The exact mechanism of action of chimeric RNA/DNA oligonucleotides is not fully understood at this time, but much has been learned using a human extract assay system (Cole-Strauss et al., 1999; Gamper et al., 2000). The targeted gene-repair reaction appears to require both the cellular homologous recombination and mismatch repair mechanisms. Studies suggest that the key intermediate is a complement-stabilized D-loop, a four-stranded joint molecule in which the two strands of the chimeric oligonucleotide are base-paired with the complementary target sequence (Figure 55). Previous chimeraplasty studies were carried out using a molecule that contained the desired mismatch on both strands of the oligonucleotide. It is now thought that the chimeric RNA/DNA strand stabilizes the interaction with the target sequence, and only the DNA strand serves as a template for mismatch repair; thus, the latter is the only strand requiring the mismatch. It also is believed that the activity of the molecule is increased if the chimeric strand consists of only RNA bases rather than an RNA/DNA mixture. Chimeric RNA/DNA oligonucleotides were investigated initially by
targeting episomally expressed plasmid DNA, and subsequently were shown to
repair genomic DNA mutations in cultured cells. In vitro model systems
have included the sickle cell hemoglobin In vivo
gene repair was first demonstrated in the Gunn rat model of Crigler-Najjar
syndrome type I, which is caused by a frameshift mutation in the gene
encoding UDP-glucuronosyltransferase. The mutation causes loss of enzyme
activity and hyperbilirubinemia (Kren et al., 1999). A chimeric
oligonucleotide was designed for the site-directed insertion of the missing
base in the genomic DNA encoding this protein, and it was delivered
intravenously to the liver of affected rats as a complex with lactosylated
polyethyleneimine ( Other investigations have demonstrated the feasibility of in vivo chimeraplasty to repair mutations in the tyrosinase gene of albino BALB/c mice, and in the dystrophin genes of both mouse and golden retriever models of Duchenne muscular dystrophy (Alexeev et al., 2000; Bartlett et al., 2000b; Rando et al., 2000). Current challenges to deploying chimeric RNA/DNA oligonucleotides for
human gene therapy include the need for optimal delivery strategies and
cost-effective, synthesis and purification schemes (Ye et al., 1998).
Most of the delivery effort has been focused on using charged liposomes and
synthetic polymers including Gene Inactivation Strategies Approaches for inhibiting the expression of specific genes has become an important therapeutic paradigm for gene-based therapy of many acquired diseases. Antisense oligonucleotides and certain classes of ribozymes are in use or in various stages of development to treat a wide range of disease processes including viral infections and cancer. In fact, a specific antisense oligonucleotide designed for the treatment of ocular cytomegalovirus (CMV) infection was the first nucleic acid drug to receive FDA approval for clinical use. Antisense Oligonucleotides Short, synthetic, single-stranded stretches of DNA complementary to an
mRNA can block the expression of specific target genes at the level of
translation (Galderisi et al., 1999; Ma et al., 2000).
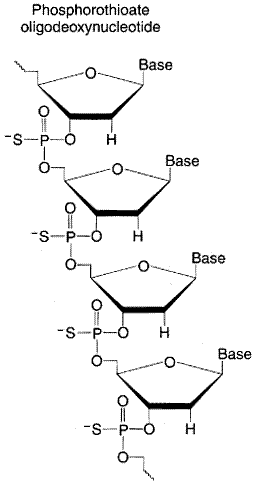
First-generation antisense oligonucleotides contained phosphorothioate
linkages in which one of the nonbridging oxygen atoms on the phosphate is
replaced with sulfur to render the molecule nuclease resistant (see
structure below). Second-generation antisense oligonucleotides incorporate
alternative intramolecular linkages and also feature mixtures of
deoxyribonucleotides and chemically substituted ribonucleotides. Newer
antisense oligonucleotides may offer improved pharmacokinetic and safety
profiles because of reduced nonspecific interactions (Agrawal and Kandimalla,
2000). Mechanism of Action Antisense oligonucleotides exert their effects by sequence-specific
and sequence-nonspecific mechanisms (Galderisi et al., 1999; Ma et
al., 2000). The principal sequence-specific mechanism involves
hybridizing to a target mRNA through Watson-Crick complementary base pairing.
This interaction impedes ribosome-mediated translation of mRNA into protein
and invites the activity of RNase H, an enzyme that cleaves RNA in the
context of an RNA/DNA duplex. The typical length of antisense
oligonucleotides (15 to 20 bases) is sufficiently long to achieve highly
sequence-specific gene targeting. There also are nonspecific effects of
antisense oligonucleotides that are independent of the targeted mRNA
sequence. Because of their polyanionic nature, antisense oligonucleotides may
bind to a variety of proteins and can activate, complement, or interfere with
the coagulation cascade. The latter effects may cause prolongation of plasma
clotting times in human subjects (Sheehan and Lan, 1998). Antisense
oligonucleotides that contain certain sequence motifs such as
cytosine-guanosine dinucleotides (CpG) and guanosine triplets (GGG) can
induce significant immune responses, including modulation of T-cell function
and the induction of several cytokines, including interleukins, interferon- Pharmacokinetics Dose-dependent increases in steady-state plasma levels of antisense oligonucleotides generally have been observed in early-phase clinical trials when the drugs were administered intravenously or subcutaneously (Levin, 1999). Pharmacokinetics are generally independent of the oligonucleotide sequence and length. After intravenous administration, antisense oligonucleotides are excreted mainly in the urine within 24 hours. In experimental animals, detectable levels may be found in most tissues except the brain for up to 48 hours. The elimination half-life of oligonucleotides administered intraocularly is more prolonged (de Smet et al., 1999). Fomivirsen Antisense oligonucleotides are primarily in development as antiviral and antineoplastic agents. The first gene-based therapeutic agent to receive FDA approval is fomivirsen (VITRAVENE), a 21-base-pair phosphorothioate oligonucleotide targeted to mRNA transcribed from the immediate-early transcriptional unit of human cytomegalovirus (CMV) (Nichols and Boeckh, 2000; Perry and Balfour, 1999). Fomivirsen is indicated for the local (intravitreal) treatment of CMV retinitis for patients with acquired immunodeficiency syndrome. For patients with advanced drug-refractory and sight-threatening disease, weekly fomivirsen treatment can significantly delay disease progression. The most common adverse effects of treatment include increased intraocular pressure and mild to moderate intraocular inflammation, both of which tend to be transient and easily reversible with topical corticosteroids. Other Antisense Oligonucleotides Several other antisense oligonucleotides are in early clinical trials for the treatment of various hematologic and solid organ neoplasms (Cotter, 1999; Yuen and Sikic, 2000). Antisense oligonucleotides in development for the treatment of chronic myelogenous leukemia are being targeted to a hybrid mRNA sequence that arises because of a chromosomal translocation resulting in over-expression of mRNA encoding a chimeric oncogene (bcr/abl). Other agents in development are designed to block expression of bcl-2, a protein that inhibits apoptosis and may play a significant role in the development of various lymphoid malignancies. Additional antineoplastic targets for antisense oligonucleotides include mRNAs encoding protein kinases (c-raf, protein kinase C), hematopoietic cell transcription factors (c-myb), and other proteins known to be involved in tumorigenesis or tumor suppression (h-ras, p53). Results from early clinical trials with these compounds suggest that they are generally well tolerated, and their clinical efficacy is promising. Trans-Cleaving Ribozymes Several investigations have demonstrated the ability of various engineered ribozymes to cleave a specific target RNA sequence in vitro (James and Gibson, 1998; Kijima et al., 1995). Most work has been done using hammerhead or hairpin ribozymes, which are considerably smaller than group I intron-derived ribozymes. These molecules can be designed to bind to specific RNA sequences and execute a cleavage reaction that can inactivate the target transcript (Figure 54). This strategy for gene inactivation has been applied for the experimental treatment of various viral infections, including HIV-1 and hepatitis B virus (Menke and Hobom, 1997; Wands et al., 1997) and malignant conditions caused by the expression of dominant oncogenes (Irie et al., 1997). Trans-cleaving ribozymes have two theoretical advantages compared with other RNA-based inhibition strategies: (1) transcript cleavage results in the direct, irreversible inactivation of the target RNA, and (2) because a single ribozyme can catalyze multiple cleavage reactions and thus destroy multiple transcripts, relatively few ribozyme molecules may be required to inhibit a given target gene effectively. Inhibition of HIV by Ribozymes Ribozymes may be able to cleave viral target RNAs at a number of stages in the viral life cycle. Potential RNA targets include incoming genomic RNAs, early viral mRNAs, late viral mRNAs and full-length genomic RNAs that are being encapsidated. Although cleavage of incoming RNAs would prevent viral integration and therefore be highly effective in protecting cells, the fact that HIV-genomic RNAs are encapsidated within a viral core may make these transcripts difficult for ribozymes to access. Therefore, cleavage of early viral transcripts may prove to be the most attractive strategy for conferring resistance to HIV. Hammerhead and hairpin ribozymes are particularly well suited for this purpose because of their small size, simple secondary structure, and the ease with which they can be manipulated to target specific HIV substrate RNAs for cleavage. Several studies have suggested that ribozymes can inhibit HIV replication in cell culture experiments when cells are challenged with a very low HIV inoculum. In the first application of this approach to inhibit HIV replication, an anti-gag hammerhead ribozyme was generated that specifically cleaved gag-encoding RNAs in vitro and inhibited HIV replication in a human T-cell line (Sarver et al., 1990). Subsequently, such trans-cleaving ribozymes have been designed to target a variety of highly conserved sequences throughout the HIV genome and have been shown to inhibit viral replication to varying degrees in a number of tissue culture studies. Moreover, certain trans-cleaving ribozymes have been demonstrated to inhibit the replication of diverse viral strains as well as clinical isolates in primary T-cell cultures (Poeschla and Wong-Staal, 1994). Comparisons between catalytically active and inactive forms of these anti-HIV ribozymes have demonstrated that maximal inhibition of virus replication is usually associated with catalytic activity and is not due simply to the antisense property of these anti-HIV ribozymes. To assess the activity of ribozymes in more clinically relevant settings, human peripheral blood lymphocytes have been stably transduced with a hairpin ribozyme targeting the U5 region of the HIV genome. These cells were shown to resist challenge by both HIV molecular clones and clinical isolates (Leavitt et al., 1994). More recently, macrophage-like cells that differentiated from hematopoietic stem/progenitor cells from fetal cord blood and were stably transduced with a hairpin ribozyme targeted at the 5' leader sequence resisted infection by a macrophage-tropic virus (Yu et al., 1995). Transduction of pluripotent hematopoietic stem cells with HIV-resistance genes may represent an avenue to continually generate cells that are resistant to HIV infection. Such stem cells differentiate into monocytes and macrophages, the major targets of HIV infection. Clinical trials to assess the safety and efficacy of this strategy in HIV-infected patients have begun (Wong-Staal et al., 1998). Cleavage by Ribozymes of Dominant Oncogenes Neoplastic transformation often is associated with the expression of mutant oncogenes. Because ribozymes can be designed to inhibit the expression of specific gene products, their potential as antineoplastic agents has been exploited. For example, hammerhead ribozymes have been reported to suppress the tumorigenic properties of various neoplastic cells harboring activated human ras genes (Scharovsky et al., 2000), and the bcr/abl fusion transcript that arises in chronic myelogenous leukemia (James and Gibson, 1998). In vitro experiments have shown that the 8500 nucleotide bcr/abl transcript can be cleaved efficiently by a hammerhead ribozyme targeted to the fusion point (James et al., 1996). In CML blast crisis cell lines, expression of ribozymes targeted at bcr/abl mRNA has been demonstrated to decrease the production of p210bcr/abl and bcr/abl transcripts and to reduce cell proliferation (Shore et al., 1993). Similar results have been reported using an anti-bcr/abl ribozyme based on the structure of RNase P (Cobaleda and Sanchez-Garcia, 2000). Insertional Gene Inactivation by Group II Introns A recent study suggests that group II introns also are useful for targeted gene inactivation (Guo et al., 2000). Two targets important for HIV infection, the human chemokine receptor CCR5 gene and regions of the HIV-1 proviral DNA, were targeted by a modified group II intron. The coding sequences of both genes could be disrupted in human cells by the insertion of the intron sequence at a defined point. This work, while still preliminary, provides another novel approach for gene inactivation. Ectopic Synthesis of Therapeutic Proteins Deficiencies of a variety of growth factors and peptide hormones are potentially amenable to treatment using the paradigm of ectopic gene expression. This approach involves delivery of a gene to evoke expression of a circulating protein from a tissue that normally does not synthesize the product. In experimental animals and early clinical trials, this strategy has been used successfully to induce expression of coagulation factors (factor VIII, IX) growth factors (IGF-1, erythropoietin), and peptide hormones (growth hormone, growth hormonereleasing hormone). In some cases, it is desirable to induce continuous secretion of a therapeutic protein (e.g., factor IX), while in other situations gene expression needs to be under strict regulation (e.g., erythropoietin). Skeletal muscle has become the most frequently used tissue for ectopic production of therapeutic proteins (MacColl et al., 1999). Skeletal muscle is a large and stable cell mass that can be conveniently accessed by intramuscular injection. In several preclinical studies, both viral and nonviral gene-transfer techniques have been demonstrated to efficiently transduce skeletal muscle with few adverse effects. There are numerous potential advantages to this strategy of delivering therapeutic proteins using ectopic gene expression. In general, this approach is less expensive and more convenient than delivery of recombinant proteins or plasma-derived concentrates. There also is a greatly reduced risk of transmission of blood-borne diseases, such as hepatitis and HIV infection, that are associated with treatment of hemophilia, for example. Successful use of this paradigm has been demonstrated for the experimental treatment of hemophilia, delivery of human growth hormone for the treatment of congenital dwarfism, and ectopic production of erythropoietin for treatment of chronic anemia, as outlined below. Factor IX Hemophilia A and B arise because of congenital deficiency of the coagulation factors VIII and IX, respectively. Preclinical studies using mice and hemophilic dogs have demonstrated the efficacy of an AAV vector encoding factor IX to promote sustained skeletal muscle expression of this coagulation factor sufficient to improve the clinical phenotype (Herzog et al., 1999). Early clinical experience using an AAV vector expressing human factor IX driven by the CMV immediate early promoter in patients with severe hemophilia B has been described (Kay et al., 2000). This study demonstrated modest changes in circulating factor IX and reduced requirement for factor IX protein infusions in a small group of treated patients. Improvements in the attainable expression level may be possible using alternative promoters, including muscle-specific enhancer/promoter sequences (Hagstrom et al., 2000). Erythropoietin Erythropoietin (EPO) insufficiency occurs most commonly in chronic renal failure. Frequent injection of recombinant EPO is necessary to maintain adequate hematocrit levels in chronic dialysis patients. The major disadvantage of this therapy is cost. Preclinical studies have demonstrated the utility of inducing erythropoietin production ectopically in skeletal muscle (Tripathy et al., 1996) or skin (Klinman et al., 1999). This has been accomplished by intramuscular or intradermal injection of plasmid DNA or viral vector encoding human EPO under the control of a constitutively active promoter such as the CMV immediate-early promoter. Experimental animals have been shown to maintain elevated plasma EPO levels and elevated hematocrits for several months after treatment. Under physiological conditions, EPO secretion by the kidney is tightly
regulated, and therefore an inducible expression system is desirable for
therapeutic applications. Such a system has been developed using an
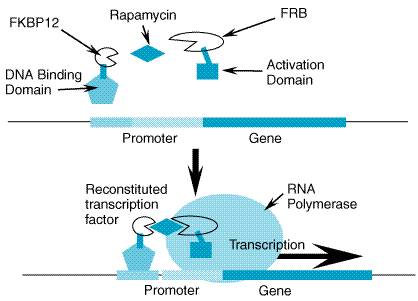
artificial, sirolimus (rapamycin)-regulated transcription factor (Figure 56)
(Ye et al., 1999). The immunosuppressant sirolimus is an orally
administered drug (see Chapter 53: Immunomodulators: Immunosuppressive
Agents, Tolerogens, and Immunostimulants) capable of interacting with two
proteins, FK506 binding protein-12 (FKBP12) and FKBP12-rapamycin-associated
protein (FRAP). The inducible expression system for regulated expression of
EPO consists of three molecular components: (1) the transcriptional
activation domain from the p65 subunit of nuclear protein kappa B (NF
Other Growth Factors and Hormones Long-term, regulated expression of human growth hormone has been
achieved in mice after intramuscular injection of AAV vectors encoding this
gene (Rivera et al., 1999). These studies utilized the sirolimus-inducible
system described above for EPO. In these experiments, sirolimus induced
significant elevations of serum human growth hormone in a dose-dependent
manner with a lag time between drug administration and first measurable serum
growth-hormone level of approximately three hours. Growth-hormone levels in
blood peaked approximately one day after sirolimus administration in this
study. Growth hormonereleasing hormone (GHRH) also can be expressed
ectopically in porcine muscle following direct injection of plasmid DNA
encoding this peptide under transcriptional control of a skeletal muscle Insulin-like growth factor I (IGF-1) also has been expressed ectopically in skeletal muscle. IGF-1 is critical for the growth of skeletal muscle and other tissues. Intramuscular injection of an AAV virus encoding IGF-l into mice resulted in sustained increases in muscle mass and muscle strength for up to 27 months (Barton-Davis et al., 1998). These effects prevented age-related changes in muscular function, suggesting that this strategy might be of benefit in diseases where skeletal muscle damage secondary to aging is accelerated. Cancer Gene Therapy It is generally accepted that cancer arises because of an accumulation of multiple molecular genetic defects that culminate in a cellular phenotype characterized by unregulated growth. Based on this knowledge, a variety of gene therapy strategies have been developed as potential new therapies for cancer (Gomez-Navarro et al., 1999). Indeed, more than half of all approved gene therapy trials relate to cancer treatment. For some neoplastic conditions, gene therapy may provide an effective treatment alternative to conventional therapies, while in other circumstances, it may serve as an adjuvant treatment. Cancer is an extraordinarily complex disease process, and a variety of gene-based treatment strategies have been conceived (see Chapter 52: Antineoplastic Agents). Current knowledge of the role of protooncogenes and tumor suppressor genes in the genesis of malignancy has stimulated the development of gene therapy tactics directed at ablating or restoring such genes, respectively. In other strategies, cancer cells are endowed with the ability to convert a systemically delivered prodrug to a toxic metabolite (cell-targeted suicide), or are targeted for destruction by replicating viral vectors (viral-mediated oncolysis). Conversely, transfer of drug-resistance genes into normal cells may provide chemoprotection during high-dose antineoplastic drug treatment. Finally, immune system modulation can activate anticancer defense mechanisms. Each of these strategies has advantages and disadvantages as well as unique obstacles to its successful deployment. The successful development of effective gene-based treatments for cancer faces several challenges. To eradicate a malignancy, any treatment strategy needs to affect every neoplastic cell. Because most cancers exert their morbidity and mortality through metastatic spread, gene delivery methods must be capable of reaching widespread anatomical locations. Target cell specificity is another important obstacle for gene-based treatment of cancer. An ideal vector would target only malignant cells and have no effect on normal cells. A variety of approaches are under development to exploit unique molecular markers of tumor cells or to use transcriptional targeting strategies through the use of tumor-specific gene promoters (Curiel, 1999; Nettelbeck et al., 2000). Finally, the genetic complexity of cancer implies that multiple approaches may be needed to achieve ultimate success. Oncogene Inactivation Several oncogenic proteins have been identified and associated with various malignancies (Park, 1998). A variety of strategies are under development to block expression of these oncogenic proteins in malignant cells by interfering with either transcription or translation. The most commonly applied approach in clinical trials to date has been the use of antisense strategies (see above) (Gewirtz et al., 1998). Suboptimal delivery of antisense molecules to malignant cells and the variable efficacy of specific antisense molecules for any given target gene are substantial obstacles to the success of this approach. Transcription of oncogenes also can be inhibited using the adenoviral gene E1A, which interferes with the transcription of erbB-2, a strategy useful in treating cancers that overexpress this oncogenic protein, such as breast and ovarian cancers (Gomez-Navarro et al., 1999). Augmentation of Tumor Suppressor Genes More than twenty-four tumor-suppressor genes have been identified, and mutations in these genes have been associated with a variety of neoplastic conditions (Fearon, 1998). This knowledge has stimulated efforts to develop techniques to replace or repair defective tumor-suppressor genes in malignant cells. Several clinical trials are under way to deliver p53 using adenoviral vectors to a variety of cancers (Gomez-Navarro et al., 1999). Similarly, viral vectors have been utilized to introduce the retinoblastoma gene and the breast cancer gene BRCA1 into bladder and ovarian cancers, respectively. Preclinical studies have demonstrated success with this approach, but not in all cases. In some situations, this approach will fail, because the mutant gene exhibits a dominant-negative effect on the normal gene. To circumvent this problem for p53 gene therapy, a genetic repair strategy (see previous section) rather than a gene-augmentation approach could be more effective (Watanabe and Sullenger, 2000). Cell-Targeted Suicide Conversion of a prodrug to a toxic metabolite by genetically engineering tumor cells is an attractive way to create an artificial difference between normal and neoplastic tissue (Springer and Niculescu-Duvaz, 2000). This can be achieved by the expression of a gene that confers a dominant, negatively selectable phenotype to the cancer cell, such as cell death imparted by expression of a prodrug-metabolizing enzyme. A variety of enzymes are capable of performing such a function, and they typically kill cells by activation of a relatively nontoxic prodrug to a cytotoxic form (Table 52). Greater selectivity in killing malignant cells will be obtained by transferring a gene that is not normally found in human beings (e.g., HSV-thymidine kinase), rather than by overexpressing an endogenous gene (e.g., deoxycytidine kinase). The prototype for this approach utilizes the HSV-1 thymidine kinase gene (HSV-TK) given in combination with the prodrug ganciclovir (Morris et al., 1999). The HSV-TK phosphorylates ganciclovir in a manner distinct from mammalian thymidine kinase. Phosphorylated ganciclovir is ultimately incorporated into DNA and inhibits DNA synthesis and transcription. The efficacy and safety of this approach is being tested in several clinical trials involving multiple malignancies. The major obstacle is selective delivery of HSV-TK to neoplastic cells. Normal cells, especially hepatic cells, also can be rendered ganciclovir-sensitive if transduced by HSV-TK. Another important limitation of this approach is the need to transduce all tumor cells with the prodrug-metabolizing enzyme. This obstacle is circumvented in part due to the 'bystander effect,' a phenomenon in which generation of the toxic drug by transduced tumor cells facilitates killing of neighboring nontransduced tumor cells by local dissemination of the compound. With the HSV-TK/ganciclovir system, the bystander effect requires functional gap junctions, possibly to enable cell-to-cell spread of the toxic metabolite, which is not diffusable across cell membranes. Combinations of two or more prodrug-metabolizing enzymes may offer enhanced efficacy by virtue of synergy among different toxic metabolites acting through different mechanisms. The major limitation of this approach has been the requirement to deliver the prodrug-metabolizing enzyme locally at a dose sufficient to transduce most or all tumor cells without systemic dissemination. The emergence of drug-resistant, malignant cell populations also is a potential barrier to this approach. Chemoprotection Gene transfer approaches can be utilized to confer greater drug tolerance to normal bone marrow cells in patients undergoing high-dose chemotherapy. The mechanisms by which cancer cells are able to survive the cytotoxic effects of chemotherapy are well described for a number of chemotherapeutic agents (see Chapter 52: Antineoplastic Agents). Although these genes limit the effectiveness of many chemotherapy regimens, it is possible that they might be exploited to protect normal tissues from the toxic effects of chemotherapy. The MDR-1 gene encoding the multidrug transporter protein (also known as P-glycoprotein) has received much attention in this regard. This transmembrane protein transports a wide variety of chemotherapeutic agents (e.g., doxorubicin, vinca alkaloids, epipodophyllotoxins, and paclitaxel) and other drugs out of cells, thus protecting them from the agents' toxic effects (Gottesman et al., 1994). Many cancers display a dose-dependent sensitivity to chemotherapy, whereby larger doses of chemotherapy lead to greater tumor regression and improved survival (see Chapter 52: Antineoplastic Agents). This is best illustrated by testicular cancers, which are highly curable when treated aggressively. Unfortunately, toxicity to normal tissues, especially the bone marrow, limits the use of larger doses of chemotherapy in many cancers. To overcome this, autologous bone marrow transplantation has been employed to rescue the bone marrow from the toxic effects of high-dose chemotherapy. Capitalizing on this concept, an investigational gene therapy strategy has been developed whereby the MDR-1 gene would be used to render the bone marrow resistant to the toxic effects of the chemotherapy (Aran et al., 1999). Clinical trials have demonstrated the safety and feasibility of MDR-1 gene transfer to bone marrow stem cells and peripheral blood hematopoietic progenitor cells in patients undergoing high-dose chemotherapy for advanced cancer (Cowan et al., 1999; Devereux et al., 1998; Hanania et al., 1996; Hesdorffer et al., 1998; Moscow et al., 1999). All studies employed replication-incompetent retroviral vectors to transduce cells ex vivo under varying cell culture conditions. In general, the transduction efficiency observed using this approach and the level of engraftment success was low. However, the use of cytokine preconditioning and inclusion of fibronectin cell-adhesion domains in stem-cell cultures prior to viral transfection results in significantly greater transduction efficiency and longer expression in the engrafted marrow cells (Abonour et al., 2000). Virus-Mediated Oncolysis Certain viruses, including adenovirus and HSV-1, can infect and lyse tumor cells (Alemany et al., 2000; Heise and Kirn, 2000). In most gene therapy applications, the ability of the virus to replicate in the host cell is disabled. By contrast, oncolysis can be accomplished by enabling the virus to replicate selectively within tumor cells. The use of oncolytic viruses in combination with other gene-based antineoplastic strategies has emerged as a promising addition to the multidimensional treatment of cancer (Hermiston, 2000). Selective replication of a virus in tumor cells leads to cell lysis and to local dissemination of infective viral progeny to neighboring cancer cells. This phenomenon provides amplification of the initial viral dose. Because cell lysis is the ultimate goal of this treatment, it is not necessary for these viral vectors to establish long-term transgene expression in the targeted host cell. Most investigational uses of this strategy have utilized replication-competent adenovirus and HSV-1. Two general strategies have been employed to engineer viral vectors capable of replicating in tumor cells and not in normal cells. First, a viral gene required for replication, such as the adenovirus E1A gene, can be driven by a tumor-specific promoter. The other approach involves deleting viral genes whose functions are required for replication in normal cells, but that are not required for replication in neoplastic cells. For example, the adenovirus E1B-55kD gene is required for efficient replication in normal cells expressing an active p53 protein, but virus lacking E1B-55kD (Onxy-015) will replicate within and lyse malignant cells lacking functional p53 (Dix et al., 2000). In a phase I clinical trial using intratumoral injection of Onxy-015 in patients with recurrent head and neck cancer, this viral vector was tolerated well and produced evidence of tumor necrosis at the site of viral injection (Ganly et al., 2000). In a similar strategy, deletion of the gene encoding ribonucleotide reductase in the HSV genome produces a virus that can be replicated selectively in mammalian tumor cells that overexpress this enzyme. Although HSV has a natural tropism for neuronal tissues, studies have demonstrated effectiveness of HSV-based oncolytic vectors for nonneuronal malignancies (Yoon et al., 2000). Perhaps the greatest utility of replication-selective viruses that target malignant cells will come by coupling this approach with other gene-based cancer treatments. In particular, replication-competent adenoviruses and HSV vectors have been developed that carry one or two prodrug-metabolizing enzymes capable of sensitizing tumor cells to chemotherapy. Aghi et al. (1999) have demonstrated that an engineered, replication-competent HSV vector that expresses rat CYP2B1 and HSV-TK confers sensitivity to cyclophosphamide and ganciclovir, respectively, to cultured glioma cells. Similarly, Rogulski et al. (2000) utilized double-suicide gene therapy delivered by a replication-competent adenovirus to cervical carcinoma xenografts in mice. In this case, the double-suicide gene combination included HSV-TK/ganciclovir and cytosine deaminase/5-fluorocytosine. This approach dramatically potentiated the efficacy of radiation therapy. Virus-mediated oncolysis also may enhance antitumor immune responses. These immune responses may include reactions not only to the viral component but also to specific tumor antigens that are released following oncolysis (Agha-Mohammadi and Lotze, 2000). Several observations suggest that these additional immune enhancements may enable eradication of metastases following local tumor treatment. Additional effects on the immune response also can be achieved by engineering the viral vector to direct expression of various cytokines. Immunomodulation Most cancer cells exhibit poor immunogenicity, and the neoplastic state itself may be associated with specific impairments in mounting effective antitumor immune responses. Various cytokines can enhance immunity against cancer cells, and this observation has stimulated the development of gene-based approaches to modulate the immune reaction in malignancy (Agha-Mohammadi and Lotze, 2000). Ectopic Cytokine Expressions A variety of cytokines have been shown to decrease tumor growth when
ectopically expressed in tumor cells or in their microenvironment (Tepper and
Mule, 1994). Tumor cells engineered to secrete certain cytokines have been
observed to be less able to form tumors when implanted in syngeneic hosts,
whereas their in vitro growth is unaffected, suggesting that host
factors are induced in response to the cytokines that decrease
tumorigenicity. Some immunostimulatory agents do not alter the growth rate of
the tumor initially, but lead to immunity against tumor growth if the animal
is later challenged with wild-type tumor cells. It is apparent that
genetically engineered tumor cells elicit a variety of host immune responses
depending on the immunomodulatory agent employed. For example, secretion of
interleukin-4 (IL-4) by a tumor cell elicits a strong local inflammatory
response without any effect on distant tumor cells or on tumor cells
administered an exogenous vector at later times. In contrast,
granulocyte/macrophage colony-stimulating factor (GM-CSF) has little effect
on the tumorigenicity, but evokes a pronounced antitumor immunity (Dranoff et
al., 1993). Greater therapeutic efficacy may be achievable by transducing
tumor cells with multiple cytokine genes. Various combinations of the
interleukins, interferon- Immune Enhancement Other approaches aimed at increasing the immune response to cancer cells have been developed. One such approach is to express on the surface of cancer cells highly immunogenic molecules, such as allotypic MHC antigens. Alternatively, rather than express an exogenous 'rejection' antigen, tumor cells may be modified so that the endogenous, weakly immunogenic, tumor-associated antigens are better recognized. It has been long known that additional 'costimulatory' pathways distinct from the T-cell receptor are needed to achieve T-cell activation (see Chapter 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants). The molecules B7-1 (CD 80) and B7-2 (CD 86) stimulate one such pathway. The B7s, whose expression normally is limited to antigen-presenting cells and other specialized immune effector cells, engage specific receptors (CD-28 and CTLA-4) on the T-cell surface in concert with antigen binding to the T-cell receptor. Subsequently, T-cell activation, cell proliferation, and cytokine production ensue and can lead to the elaboration of antitumor immunity. The absence of a costimulatory signal at the time of T-cell receptor engagement is not a neutral event; rather, it results in the development of tumor-specific anergy, not mere failure to activate the T cell. Thus, the simple presence of antigens in tumor cells would be expected to produce an immune-tolerant state rather than an immune-responsive state if costimulatory events do not take place. In effect, this is what is seen in most clinical situations where human tumors grow apparently unimpeded by host immune mechanisms. When some tumor cells are provided with costimulatory molecules, effective T-cell activation takes place. This has been demonstrated by ectopic expression of B7 on tumor cells; these genetically engineered tumor cells then are used to stimulate an immune response to the parental tumor cell line. DNA Vaccines Vaccination against both infectious and noninfectious diseases is possible using DNA-encoded antigens (Gurunathan et al., 2000; Kowalczyk and Ertl, 1999). Skin or muscle can be inoculated easily with a bacterial plasmid vector carrying a gene coding for an antigenic protein. Following transcription and translation of the gene in host cells, the antigen induces a multifaceted immune reaction featuring both humoral and cell-mediated responses. The ability to stimulate T-helper cell and cytotoxic T-lymphocyte responses provides a potential advantage over conventional protein-based vaccines that do not have these effects. Two additional advantages of DNA vaccines are the relatively low production costs and the lack of risks associated with attenuated pathogens. Furthermore, unmethylated plasmid sequences containing purine-purine-C-G-pyrimidine-pyrimidine sequence motifs stimulate lymphocyte proliferation and cytokine release and thus act as potent adjuvants (Roman et al., 1997; Sato et al., 1996). The major limitations of DNA vaccines include the relatively weak humoral immune response, the theoretical risks of insertional mutagenesis, and provocation of an autoimmune response. DNA vaccines are being evaluated in both preclinical and clinical studies for the treatment of a wide range of infections (viral, bacterial, parasitic) as well as certain acquired diseases such as malignancy and chronic allergic conditions. The safety and feasibility of using a DNA vaccination strategy to immunize against HIV-1 infection recently has been demonstrated (Boyer et al., 2000). |
Disease Targets for Gene Therapy
|
This section highlights the use of gene therapy for treatment of various inherited conditions affecting the immune system, hematopoietic system, liver, lung, and skeletal muscle. Many other organ systems and dozens of other genetic disorders that are not discussed also are potential targets for gene therapy. The few topics presented here should illustrate some of the major issues and obstacles facing treatment of other disease targets. Immunodeficiency Disorders Gene therapy for the treatment of congenital immunodeficiency
disorders illustrates the use of ex vivo gene transfer into
hematopoietic stem cells. Three distinct immunodeficiency syndromes, adenosine
deaminase deficiency ( Adenosine Deaminase Deficiency The first genetic disorder to be clinically treated with gene therapy
was Because of concerns that use of mature T lymphocytes for treatment of X-Linked Severe Combined Immunodeficiency In the most common form of SCID, mutations in the gene encoding the
cytokine receptor Chronic Granulomatous Disease This disorder is caused by genetic defects in one of four genes encoding subunits of the respiratory-burst oxidase, a superoxide-generating enzyme complex present within phagocytic leukocytes. A defect in this system results in an inability to fight bacterial and fungal infections and can be life threatening. Genetic correction of a small fraction of circulating phagocytes would suffice to provide clinical benefit. This is based on the observation that unaffected female carriers of this X-linked trait have as few as 5% oxidase-positive neutrophils. Allogenic bone marrow transplantation is the standard treatment, although there are many conceivable advantages of a gene-based treatment strategy. Two of the genes that cause CGD (gp91phox and p47phox) are targeted in gene therapy trials (Kume and Dinauer, 2000). In a phase I clinical trial involving five adult patients with p47phox deficiency, intravenous infusions of peripheral blood stem cells transduced ex vivo with a retroviral vector carrying normal p47phox were successful in generating functionally corrected granulocytes (Malech et al., 1997). Persistence of the corrected cell phenotype was demonstrated for up to six months after the infusion, although the quantity of functional cells was probably insufficient for clinical benefit. Further work is needed to enhance the efficiency of gene transfer into stem cells and to demonstrate more long-lasting effects. Liver Disease The liver can be afflicted with a variety of metabolic, infectious, and neoplastic diseases for which specific molecular interventions can be envisioned. Potential applications are made more feasible by the existence of multiple methods for effecting gene transfer to the liver. Molecular conjugates, adenoviral vectors, liposomes, and retroviral vectors all have been used for hepatocyte gene transfer (Shetty et al., 2000). For in vivo gene transfer, the liver is accessible by a number of routes, including direct injection, intravenous, and intrabiliary administration of vectors. Ex vivo strategies can be implemented by partial surgical resection of the liver, isolation of hepatocytes, and in vitro hepatocyte transduction. The genetically modified cells can then be reimplanted into the liver. The liver can be targeted selectively for gene transfer by exploiting unique hepatocyte surface receptors that are capable of mediating receptor-mediated endocytosis (Smith and Wu, 1999). Specific ligands that are recognized by the asialoglycoprotein receptor can be coupled to DNA typically in combination with a polymer such as polylysine, or liposomes. Envelope proteins on retroviral vectors also have been genetically modified to incorporate peptide sequences from hepatocyte-targeted proteins such as human hepatocyte growth factor (Nguyen et al., 1998). Some viral vectors may have a natural proclivity to target the liver. Rapid hepatic uptake of adenoviral vectors administered intravenously accounts for approximately 90% of the delivered dose. The adenovirus knob protein (terminal domain of the fiber protein, Figure 52) appears responsible for this phenomenon (Zinn et al., 1998). Familial Hypercholesterolemia Patients with familial hypercholesterolemia have an inherited deficiency or dysfunction of the low-density lipoprotein (LDL) receptor and, as a consequence, develop extremely high plasma levels of cholesterol and arteriosclerosis at a very early age (see Chapter 36: Drug Therapy for Hypercholesterolemia and Dyslipidemia). The genetic defect manifests itself as a diminished ability of the liver to clear LDL particles from the blood, and serum lipid levels provide a convenient marker of the disease. Although pharmacological interventions have had limited success, correction of the hepatic dysfunction by orthotopic liver transplantation leads to normalization of blood lipid levels and slowing of arterial disease progression. This clinical observation suggested that if the liver could be genetically modified to express the LDL receptor, the same benefits might be achieved. The Watanabe heritable hyperlipidemic rabbit has served as an ideal animal model, demonstrating that this approach does lead to persistent reductions in serum LDL (Chowdhury et al., 1991). Several patients now have been treated in a clinical trial using an ex vivo DNA-delivery approach and retrovirus to introduce the LDL receptor gene into hepatocytes isolated from the patients following partial hepatectomy (Grossman et al., 1994). This study demonstrated the feasibility, safety, and potential efficacy of ex vivo hepatic gene therapy. Hemophilia A Inherited deficiency of coagulation factor VIII leads to a lifelong risk of spontaneous hemorrhage that can be debilitating and potentially life-threatening. Standard treatment includes frequent infusions of plasma-derived factor VIII that carries the risk of blood-borne infection as well as significant inconvenience. Hemophilia A is well suited for gene therapy, because factor VIII levels in blood are therapeutic over an extended range, and even modest (5% of normal) levels of the protein can ameliorate the major morbidity associated with this disease (Kay and High, 1999). Unlike hemophilia B and factor IX deficiency, there has been little success in applying the paradigm of ectopic synthesis (see above). This is explained by the fact that the full-length factor VIII-coding region is more than 7 kb, and this has restricted vector selection to retroviruses and adenovirus (Balague et al., 2000; VandenDriessche et al., 1999). However, it also is feasible to engineer a recombinant AAV vector carrying a truncated (B domain-deleted) form of factor VIII (Chao et al., 2000). The B domain of factor VIII can be deleted without impairing the pro-coagulant activity of the protein, and the truncated gene (4.4 kb) is well within the carrying capacity of recombinant AAV vectors. Long-term hepatic expression in experimental animals has been achieved using a variety of viral vectors carrying factor VIII (Kaufman, 1999). Delivery can be achieved intravenously, although gene transfer to organs other than liver, including spleen and lungs, can occur. In addition, ex vivo approaches also have been used. Human bone marrow stromal cells have been transduced with factor VIII retroviral vectors and then transplanted into the spleens of immunodeficient mice (Chuah et al., 2000). Although circulating factor VIII levels in the mice rose significantly after engraftment, transgene silencing prevented long-term expression. A major issue facing gene therapy of hemophilia is the possibility of developing inhibitory antibodies against the transgene. The development of inhibitory antibodies also is a common sequela of protein-based therapy of both hemophilia A and B. Choice of gene transfer vector, dose, and target tissue are likely factors that will influence the propensity for developing antibodies (Kaufman, 1999). Hemoglobinopathies Sickle cell disease and the thalassemias are common single-gene disorders associated with substantial morbidity and mortality. In theory, these disorders should be amenable to ex vivo gene transfer into hematopoietic stem cells which then would be used to reconstitute a patient's bone marrow with cells expressing a specific transferred gene. However, major challenges remain in developing vectors capable of achieving long-term and therapeutic expression levels of transferred globin genes (Emery and Stamatoyannopoulos, 1999; Persons and Nienhuis, 2000). In addition, gene-repair strategies utilizing either trans-splicing ribozymes or chimeric RNA/DNA oligonucleotides have been utilized in vitro (see above). The most successful in vivo approach to date has been the use
of retroviral vectors that carry the The transient and poor expression of Preselection of successfully transduced cells reduces the incidence of gene silencing and of age-dependent reduction in expression levels and may partially circumvent these problems. This approach is presumably successful because hematopoietic stem cells in which the transferred gene is not initially silenced are selected. Two approaches for preselecting transduced stem cells have been conceived. Kalberer et al. (2000) have described the successful utilization of ex vivo preselection of transduced stem cells on the basis of expression of a marker protein. An alternative approach for preselecting hematopoietic cells capable of longer-term expression of the transgene involves use of a coexpressed drug-resistance gene enabling negative-selection strategies (Emery and Stamatoyannopoulos, 1999). Lung Diseases From the perspective of organ specificity, the lung provides an opportunity for highly specific delivery of gene transfer vectors to the respiratory epithelium through the bronchial airways. Clinical trials of gene therapy for lung disease most often have used aerosol systems to achieve topical delivery of gene delivery vectors (Ennist, 1999). However, despite its accessibility, respiratory epithelium strongly resists invasion by foreign particles, including viral and nonviral delivery systems. There are multiple barriers to transducing respiratory epithelial cells by the aerosol route (Boucher, 1999). These barriers include a mucous clearance mechanism that may remove vectors from the airways, a glycocalyx that may block binding to cell surface receptors, and finally an apical cell membrane that expresses a low density of receptors for viral vectors and exhibits a low rate of endocytosis. Several gene delivery vectors have been applied to treating inherited lung disease (Albelda et al., 2000). Adenoviral vectors are uniquely suited for gene therapy of lung disease because of their natural tropism for respiratory epithelium. However, several studies have demonstrated that adenoviral vectors are inefficient delivery vehicles because of their transient expression and propensity to provoke immune responses (Welsh, 1999). Adeno-associated viral vectors may offer the advantages of more stable expression of the transduced gene and less inflammation. Cystic Fibrosis The gene responsible for cystic fibrosis (CFTR) was discovered more than ten years ago, and there have been multiple efforts to develop safe and effective vectors for introducing this gene into the respiratory epithelium of patients suffering from this disease (Boucher, 1999). The results from several phase I clinical trials have been published, and most of these involved use of adenoviral vectors to transduce either nasal or pulmonary epithelium. In general, adenoviral vectors are capable of achieving gene transfer, but this is inefficient as judged by the small proportion of transduced cells, and the transient nature of gene expression typically lasting only several days (Grubb et al., 1994; Zuckerman et al., 1999). In addition, immune responses attenuate the effectiveness of subsequent vector administrations (Harvey et al., 1999). Adeno-associated viral vectors for delivering CFTR are now in clinical trials. Preclinical studies demonstrated long-term transgene expression with little immune response in experimental systems. The results of one phase I clinical trial targeting maxillary sinus epithelium in ten cystic-fibrosis patients has been reported (Drapkin et al., 2000). The results from this study are encouraging, but much work remains to evaluate the long-term safety and efficacy of this vector. Liposomal vectors also are under evaluation in cystic fibrosis. The results of three clinical trials have been published and some evidence of effective gene transfer is evident. However, like adenoviral gene transfer in respiratory epithelium, liposome-mediated gene delivery results in transient expression (Albelda et al., 2000). Several novel strategies for increasing the efficiency of viral gene transfer to respiratory tissues are being tested. Because most receptors for viral vectors are located on the basolateral membrane of these cells, experimental approaches that disrupt epithelial tight junctions have been tested and demonstrate an increased efficiency of gene transfer applied from the apical cell surface (Boucher, 1999). However, it is unlikely that these strategies can be adopted safely for clinical use. In another approach, modifications of the vector have been engineered to facilitate specific interactions with apical cell-membrane receptors. Modified vectors that target apical purine-nucleotide receptors using monoclonal antibodies (Boucher, 1999), or that target the urokinase/plasminogen-activator receptor by employing small peptide ligands, have been tested and appear to improve gene transfer efficiency in vitro (Drapkin et al., 2000).
Deficiency of Currently, recombinant Skeletal Muscle A variety of inherited disorders of muscle including Duchenne muscular dystrophy and the limb girdle muscular dystrophies are prime targets for the development of gene-based therapies. Mature skeletal muscle presents several unique opportunities and obstacles for gene delivery (Hartigan-O'Connor and Chamberlain, 2000). This tissue can be transduced locally by the direct injection of naked plasmid DNA or DNA carried by viral and nonviral vectors. In addition to these in vivo gene transfer methods, myoblast-mediated ex vivo gene transfer has been demonstrated to be an alternative delivery strategy (Floyd et al., 1998). Systemic delivery of genes to muscle is complicated by the large mass
of this tissue and a permeability barrier, consisting of the vascular
endothelium and extracellular matrix, that blocks access of gene delivery
vectors to the myocyte membrane from the vasculature space. A variety of
strategies have been developed to breach this permeability barrier. One
particularly successful approach has been the employment of the inflammatory
mediator histamine. In one study, the hindlimb vasculature of cardiomyopathic
hamsters was sequentially perfused with a vasodilator (papaverine),
histamine, and an AAV vector carrying either a marker gene (e.g.,
encoding Skeletal muscle presents other obstacles to infection by certain viral vectors. Adenovirus has a low infectivity of mature skeletal muscle because of the low expression levels of the CAR and integrin molecules that are necessary for attachment and internalization of the virus (Nalbantoglu et al., 1999). This low level of infectivity is maturation-dependent, with transduction being most efficient in immature muscle fibers (Acsadi et al., 1994). By contrast, gene transfer using HSV vectors is not maturation-dependent (van Deutekom et al., 1998). Experimental use of adenovirus to transduce skeletal muscle has been successful, but expression of transgenes has been transient in most cases because of the immune reaction induced by the viral infection. Adeno-associated virus also has been demonstrated to be capable of efficient and stable transduction of adult skeletal muscle (Fisher et al., 1997). In some animal studies, transgene expression was demonstrated to persist for as long as two years. Duchenne Muscular Dystrophy Duchenne muscular dystrophy is an X-linked, recessive muscle disease caused by the absence of the cytoskeletal protein, dystrophin. The dystrophin-coding sequence is large (14 kb), precluding the use of viral vectors with limited carrying capacity, such as AAV and first-generation adenoviral vectors. Newer adenoviral vectors capable of carrying the complete dystrophin gene sequence have been tested in experimental models of muscular dystrophy, such as the mdx mouse, and have been shown to be effective in transducing muscle cells when delivered locally (Clemens et al., 1996). In addition to the full-length dystrophin gene, viral vectors carrying the related gene for utrophin have been demonstrated to correct the dystrophic phenotype in mdx mice (Rafael et al., 1998). Limb Girdle Muscular Dystrophy The limb girdle muscular dystrophies are a group of related inherited
disorders that include four genetically distinct forms caused by mutations in
the |
Prospectus
|
Human gene therapy remains experimental, but there are several indications that, in the second decade of its evolution, this strategy will emerge as a safe and effective alternative to existing conventional therapies for many inherited and some acquired diseases. Already, some cancer gene therapy protocols are being tested in phase III clinical trials to compare their efficacy and safety with those of standard therapy. Additional antisense oligonucleotide drugs for the treatment of viral infections, including HIV, and possibly for malignancy likely will be approved for clinical use in the near future. The prospects for routine use of gene transfer to achieve ectopic expression of clotting factors and erythropoietin for the treatment of hemophilia and chronic anemia, respectively, in the near future seems promising. The development of safer, more efficient gene transfer vehicles is critical to the success of most gene therapy paradigms, and advances in this field are tightly coupled to improvements in vector design, manufacture, and efficacy. These advances likely will bring feasibility to other, more heroic, measures such as in utero gene therapy. Finally, for progress to be made, it is critical that safety be assured to patients participating in the investigational phases of gene therapy development, and that the ethical concerns of the public be carefully considered and respected. The demand for gene therapy to treat inherited diseases will certainly grow with the continuing discovery of new genetic conditions and their molecular defects. Gene therapy also could emerge as an important modality for treating more common disorders as we increase our appreciation for the role of allelic variation in conferring disease susceptibility. The need for improved gene transfer technologies and new therapeutic paradigms will only increase as physicians enter the era of genetic medicine. Acknowledgment The authors wish to acknowledge Drs. Stephen L. Eck and James M. Wilson, authors of this chapter in the ninth edition of Goodman and Gilman's The Pharmacological Basis of Therapeutics, some of whose text has been retained. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1363
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved