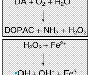| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Treatment of Central Nervous System Degenerative Disorders
Overview
|
The neurodegenerative diseases include common and debilitating disorders such as Parkinson's disease, Alzheimer's disease, Huntington's disease, and amyotrophic lateral sclerosis (ALS). Although the clinical and neuropathological aspects of these disorders are distinct, their unifying feature is that each disorder has a characteristic pattern of neuronal degeneration in anatomically or functionally related regions. Presently available pharmacological treatments for the neurodegenerative disorders are symptomatic and do not alter the course or progression of the underlying disease. The most effective symptomatic therapies are those for Parkinson's disease; a large number of agents from several different pharmacological classes can be used, and, when skillfully applied, these can have a dramatic impact on life span and functional ability. The treatments available for Alzheimer's disease, Huntington's disease, and ALS are less satisfactory but still can make an important contribution to patient welfare. This chapter reviews current therapeutic agents for treatment of the symptoms of neurodegenerative diseases and introduces the reader to research aimed at developing therapeutic agents that alter the course of neurodegenerative diseases by preventing neuronal death or stimulating neuronal recovery. Related material concerning the serotonergic effects of some of the therapeutic agents employed for Parkinson's disease can be found in Chapter 11: 5-Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists, and additional information concerning cholinergic agents that are used in treatment of Alzheimer's disease can be found in Chapters 7: Muscarinic Receptor Agonists and Antagonists and 8: Anticholinesterase Agents. |
Treatment of Central Nervous System Degenerative Disorders: Introduction
|
Neurodegenerative disorders are characterized by progressive and irreversible loss of neurons from specific regions of the brain. Prototypical neurodegenerative disorders include Parkinson's disease (PD) and Huntington's disease (HD), where loss of neurons from structures of the basal ganglia results in abnormalities in the control of movement; Alzheimer's disease (AD), where the loss of hippocampal and cortical neurons leads to impairment of memory and cognitive ability; and amyotrophic lateral sclerosis (ALS), where muscular weakness results from the degeneration of spinal, bulbar, and cortical motor neurons. As a group, these disorders are relatively common and represent a substantial medical and societal problem. They are primarily disorders of later life, developing in individuals who are neurologically normal, although childhood-onset forms of each of the disorders are recognized. PD is observed in more than 1% of individuals over the age of 65 (Tanner, 1992), whereas AD affects as many as 10% of the same population (Evans et al., 1989). HD, which is a genetically determined autosomal dominant disorder, is less frequent in the population as a whole but affects 50% of each generation in families carrying the gene. ALS also is relatively rare but often leads rapidly to disability and death (Kurtzke, 1982). At present, the pharmacological therapy of neurodegenerative disorders is limited to symptomatic treatments that do not alter the course of the underlying disease. Symptomatic treatment for PD, where the neurochemical deficit produced by the disease is well defined, is in general relatively successful, and a number of effective agents are available (Lang and Lozano, 1998; Standaert and Stern, 1993). The available treatments for AD, HD, and ALS are much more limited in effectiveness, and the need for new strategies is particularly acute. |
Selective Vulnerability and Neuroprotective Strategies
|
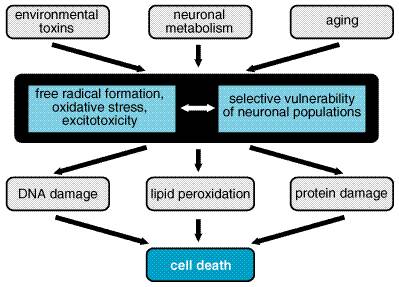
Selective Vulnerability The most striking feature of this group of disorders is the exquisite specificity of the disease processes for particular types of neurons. For example, in PD there is extensive destruction of the dopaminergic neurons of the substantia nigra, while neurons in the cortex and many other areas of the brain are unaffected (Gibb, 1992; Fearnley and Lees, 1994). In contrast, neural injury in AD is most severe in the hippocampus and neocortex, and even within the cortex, the loss of neurons is not uniform but varies dramatically in different functional regions (Arnold et al., 1991). Even more striking is the observation that, in HD, the mutant gene responsible for the disorder is expressed throughout the brain and in many other organs, yet the pathological changes are largely restricted to the neostriatum (Vonsattel et al., 1985; Landwehrmeyer et al., 1995). In ALS, there is loss of spinal motor neurons and the cortical neurons that provide their descending input (Tandan and Bradley, 1985). The diversity of these patterns of neural degeneration has led to the proposal that the process of neural injury must be viewed as the interaction of genetic and environmental influences with the intrinsic physiological characteristics of the affected populations of neurons. These intrinsic factors may include susceptibility to excitotoxic injury, regional variation in capacity for oxidative metabolism, and the production of toxic free radicals as products of cellular metabolism (Figure 221). The factors that convey selective vulnerability may prove to be important targets for neuroprotective agents to slow the progression of neurodegenerative disorders.
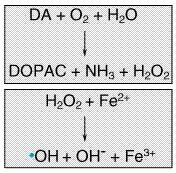
Genetics It has been long suspected that genetics plays an important role in the etiology of neurodegenerative disorders, and recent discoveries have begun to shed light on some mechanisms responsible. HD is transmitted by autosomal dominant inheritance, and the molecular nature of the genetic defect has been identified (discussed below). Most cases of PD, AD, or ALS are sporadic, but families with a high incidence of each of these diseases have been identified, and these studies have begun to yield important clues to the pathogenesis of the disorders. In the case of PD, mutations in three different proteins can lead to autosomal dominant forms of the disease: alpha-synuclein, an abundant synaptic protein; parkin, a ubiquitin hydrolase; and UCHL1, which also participates in ubiquitin-mediated degradation of proteins in the brain (Duvoisin, 1998; Golbe, 1999; Kitada et al., 1998). In AD, mutations in the genes coding for the amyloid precursor protein (APP) and proteins known as the presenilins (which may be involved in APP processing) lead to inherited forms of the disease (Selkoe, 1998). Mutations in the gene coding for copper-zinc superoxide dismutase (SOD1) account for about 2% of the cases of adult-onset ALS (Cudkowicz and Brown, 1996). Although these mutations are rare, their importance extends beyond the families that carry them, because they point to pathways and mechanisms that also may underlie the more common, sporadic cases of these diseases. Genetically determined cases of PD, AD, and ALS are infrequent, but it is likely that an individual's genetic background has an important role in determining the probability of acquiring these diseases. Recent studies of AD have revealed the first of what are likely to be many genetic risk factors for neurodegenerative disorders, in the form of apolipoprotein E (apo E). This protein, well known to be involved in transport of cholesterol and lipids in blood, is found in four distinct isoforms. Although all of the isoforms carry out their primary role in lipid metabolism equally well, individuals who are homozygous for the apo E 4 allele ('4/4') have a much higher lifetime risk of AD than do those homozygous for the apo E 2 allele ('2/2'). The mechanism by which the apo E 4 protein increases the risk of AD is not known, but a secondary function of the protein in metabolism of APP has been suggested (Roses, 1997). Environmental Triggers Infectious agents, environmental toxins, and acquired brain injury have been proposed to have a role in the etiology of neurodegenerative disorders. The role of infection is best documented in the numerous cases of PD that developed following the epidemic of encephalitis lethargica (Von Economo's encephalitis) in the early part of the twentieth century. Most contemporary cases of PD are not preceded by encephalitis, and there is no convincing evidence for an infectious contribution to HD, AD, or ALS. Traumatic brain injury has been suggested as a trigger for neurodegenerative disorders, and in the case of AD there is some evidence to support this view (Cummings et al., 1998). At least one toxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP; discussed in Energy, Metabolism, and Aging), can induce a condition closely resembling PD, but evidence for the widespread occurrence of this or a similar toxin in the environment is lacking (Tanner and Langston, 1990). Excitotoxicity The term excitotoxicity was coined by Olney (1969) to describe the neural injury that results from the presence of excess glutamate in the brain. Glutamate is used as a neurotransmitter by many different neural systems and is believed to mediate most excitatory synaptic transmission in the mammalian brain (see Chapter 12: Neurotransmission and the Central Nervous System). Although glutamate is required for normal brain function, the presence of excessive amounts of glutamate can lead to excitotoxic cell death (Lipton and Rosenberg, 1994). The destructive effects of glutamate are mediated by glutamate receptors, particularly those of the N-methyl-D-aspartate (NMDA) type. Unlike other glutamate-gated ion channels, which primarily regulate the flow of Na+, activated NMDA receptor-channels allow an influx of Ca2+, which in excess can activate a variety of potentially destructive processes. The activity of NMDA receptor-channels is regulated not only by the concentration of glutamate in the synaptic space but also by a voltage-dependent blockade of the channel by Mg2+; thus, entry of Ca2+ into neurons through NMDA receptor-channels requires binding of glutamate to NMDA receptors as well as depolarization of the neuron (e.g., by the activity of glutamate at non-NMDA receptors), which relieves the blockade of NMDA channels by extracellular Mg2+. Excitotoxic injury is thought to make an important contribution to the neural death that occurs in acute processes such as stroke and head trauma (Choi and Rothman, 1990). In the chronic neurodegenerative disorders, the role of excitotoxicity is less certain, but it is thought that regional and cellular differences in susceptibility to excitotoxic injuryconveyed, for example, by differences in types of glutamate receptorsmay contribute to selective vulnerability (Young, 1993). Energy, Metabolism, and Aging The excitotoxic hypothesis provides a link between selective patterns of neuronal injury, the effects of aging, and observations on the metabolic capacities of neurons (Beal et al., 1993). Since the ability of Mg2+ to block the NMDA receptor-channel is dependent on the membrane potential, disturbances that impair the metabolic capacity of neurons will tend to relieve Mg2+ blockade and predispose to excitotoxic injury. The capacity of neurons for oxidative metabolism declines progressively with age, perhaps in part because of a progressive accumulation of mutations in the mitochondrial genome (Wallace, 1992). Patients with PD exhibit several defects in energy metabolism that are even greater than expected for their age, most notably a reduction in the function of complex I of the mitochondrial electron transport chain (Schapira et al., 1990). Additional evidence for the role of metabolic defects in the etiology of neural degeneration comes from the study of patients who inadvertently self-administered MPTP, a 'designer drug' that resulted in symptoms of severe and irreversible parkinsonism (Ballard et al., 1985). Subsequent studies have shown that a metabolite of MPTP induces degeneration of neurons similar to that observed in idiopathic PD and that its mechanism of action appears to be related to an ability to impair mitochondrial energy metabolism in dopaminergic neurons (Przedborski and Jackson-Lewis, 1998). In rodents, neural degeneration similar to that observed in HD can be produced either by direct administration of large doses of NMDA receptor agonists or by more chronic administration of inhibitors of mitochondrial oxidative metabolism, suggesting that disturbances of energy metabolism may underlie the selective pathology of HD as well (Beal et al., 1986, 1993). Oxidative Stress Although neurons depend on oxidative metabolism for survival, a consequence of this process is the production of reactive compounds such as hydrogen peroxide and oxyradicals (Cohen and Werner, 1994). Unchecked, these reactive species can lead to DNA damage, peroxidation of membrane lipids, and neuronal death. Several mechanisms serve to limit this oxidative stress, including the presence of reducing compounds such as ascorbate and glutathione and enzymatic mechanisms such as superoxide dismutase, which catalyzes the reduction of superoxide radicals. Oxidative stress also may be relieved by aminosteroid agents that serve as free radical scavengers. In PD, attention has been focused on the possibility that oxidative stress induced by the metabolism of dopamine may underlie the selective vulnerability of dopaminergic neurons (Jenner, 1998). The primary catabolic pathway of dopamine to 3,4-dihydroxyphenylacetic acid (DOPAC) is catalyzed by monoamine oxidase (MAO) and generates hydrogen peroxide. Hydrogen peroxide, in the presence of ferrous ion, which is relatively abundant in the basal ganglia, may generate hydroxyl free radicals (the Fenton reaction, Figure 222; Olanow, 1990). If the protective mechanisms are inadequate because of inherited or acquired deficiency, the oxyradicals could cause degeneration of dopaminergic neurons. This hypothesis has led to several proposals for therapeutic agents to retard neuronal loss in PD. Two candidates, the free radical scavenger tocopherol (vitamin E) and the MAO inhibitor selegiline (discussed in Selegiline), have been tested in a large-scale clinical trial, but neither was shown to have a substantial neuroprotective effect (The Parkinson Study Group, 1993).
|
Parkinson's Disease
|
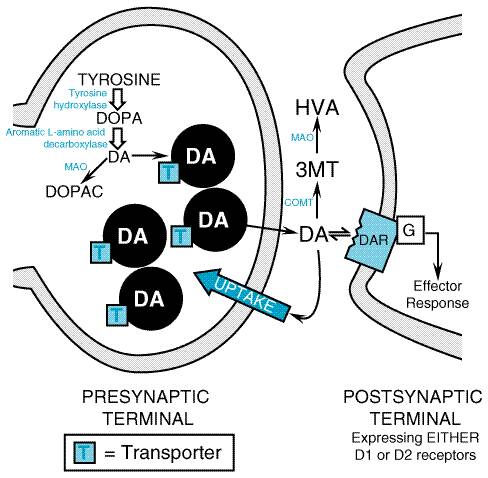
Clinical Overview Parkinsonism is a clinical syndrome comprising four cardinal features: bradykinesia (slowness and poverty of movement), muscular rigidity, resting tremor (which usually abates during voluntary movement), and an impairment of postural balance leading to disturbances of gait and falling. The most common cause of parkinsonism is idiopathic PD, first described by James Parkinson in 1817 as paralysis agitans, or the 'shaking palsy.' The pathological hallmark of PD is a loss of the pigmented, dopaminergic neurons of the substantia nigra pars compacta, with the appearance of intracellular inclusions known as Lewy bodies (Gibb, 1992; Fearnley and Lees, 1994). Progressive loss of dopamine neurons is a feature of normal aging; however, most people do not lose the 70% to 80% of dopaminergic neurons required to cause symptomatic PD. Without treatment, PD progresses over 5 to 10 years to a rigid, akinetic state in which patients are incapable of caring for themselves. Death frequently results from complications of immobility, including aspiration pneumonia or pulmonary embolism. The availability of effective pharmacological treatment has altered radically the prognosis of PD; in most cases, good functional mobility can be maintained for many years, and the life expectancy of adequately treated patients is substantially increased (Diamond et al., 1987). It is important to recognize that several disorders other than PD also may produce parkinsonism, including some relatively rare neurodegenerative disorders, stroke, and intoxication with dopamine receptorblocking drugs. Drugs in common clinical use that may cause parkinsonism include antipsychotics such as haloperidol and thorazine (see Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania) and antiemetics such as prochlorperazine and metoclopramide (see Chapter 38: Prokinetic Agents, Antiemetics, and Agents Used in Irritable Bowel Syndrome). Although a complete discussion of the clinical diagnostic approach to parkinsonism exceeds the scope of this chapter, the distinction between PD and other causes of parkinsonism is important, because parkinsonism arising from other causes usually is refractory to all forms of treatment. Parkinson's Disease: Pathophysiology The primary deficit in PD is a loss of the neurons in the substantia nigra pars compacta that provide dopaminergic innervation to the striatum (caudate and putamen). The current understanding of the pathophysiology of PD can be traced to the classical neurochemical investigations in the 1950s and 1960s, in which a more than 80% reduction in the striatal dopamine content was demonstrated. This parallelled the loss of neurons from the substantia nigra, suggesting that replacement of dopamine could restore function (Cotzias et al., 1969; Hornykiewicz, 1973). These fundamental observations led to an extensive investigative effort to understand the metabolism and actions of dopamine and to learn how a deficit in dopamine gives rise to the clinical features of PD. This effort led to a current model of the function of the basal ganglia that admittedly is incomplete but is still useful. Biosynthesis of Dopamine Dopamine, a catecholamine, is synthesized in the terminals of dopaminergic neurons from tyrosine, which is transported across the bloodbrain barrier by an active process (Figure 223 and Figure 224). The rate-limiting step in the synthesis of dopamine is the conversion of L-tyrosine to L-dihydroxyphenylalanine (L-DOPA), catalyzed by the enzyme tyrosine hydroxylase which is present within catecholaminergic neurons. L-DOPA is converted rapidly to dopamine by aromatic L-amino acid decarboxylase. In dopaminergic nerve terminals, dopamine is taken up into vesicles by a transporter protein; this process is blocked by reserpine, which leads to depletion of dopamine. Release of dopamine from nerve terminals occurs through exocytosis of presynaptic vesicles, a process that is triggered by depolarization leading to entry of Ca2+. Once dopamine is in the synaptic cleft, its actions may be terminated by reuptake through a membrane carrier protein, a process antagonized by drugs such as cocaine. Alternatively, dopamine can be degraded by the sequential actions of MAO and catechol-O-methyltransferase (COMT) to yield two metabolic products, 3,4-dihydroxyphenylacetic acid (DOPAC) and 3-methoxy-4-hydroxyphenylacetic acid (HVA). In human beings, HVA is the primary product of the metabolism of dopamine (Cooper et al., 1996).
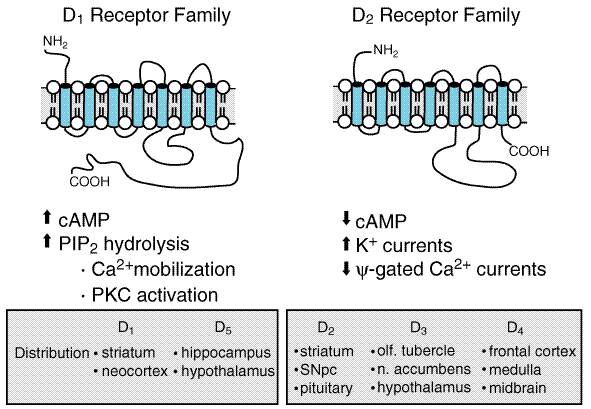
Dopamine Receptors The actions of dopamine in the brain are mediated by a family of
dopamine receptor proteins (Figure 225). Two types of dopamine receptors
were identified in the mammalian brain using pharmacological techniques: D1
receptors, which stimulate the synthesis of the intracellular second
messenger cyclic AMP, and D2 receptors, which inhibit cyclic AMP
synthesis as well as suppress Ca2+ currents and activate
receptor-operated K+ currents. Application of molecular genetics
to the study of dopamine receptors has revealed a more complex receptor
situation than originally envisioned. At present, five distinct dopamine
receptors are known to exist (see Jarvie and Caron, 1993, and Chapter
12: Neurotransmission and the Central Nervous System). The dopamine receptors
share several structural features, including the presence of seven
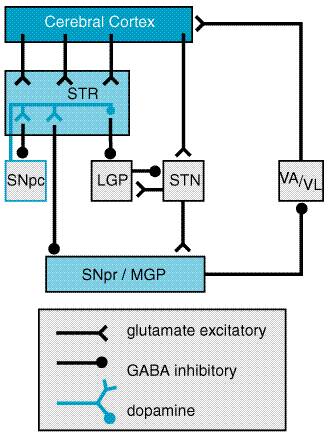
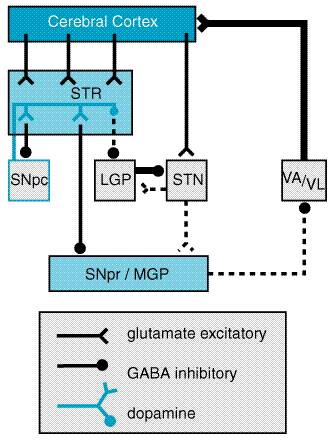
The five dopamine receptors can be divided into two groups on the basis of their pharmacological and structural properties (Figure 225). The D1 and D5 proteins have a long intracellular carboxy-terminal tail and are members of the pharmacologically defined D1 class; they stimulate the formation of cyclic AMP and phosphatidyl inositol hydrolysis. The D2, D3, and D4 receptors share a large third intracellular loop and are of the D2 class. They decrease cyclic AMP formation and modulate K+ and Ca2+ currents. Each of the five dopamine receptor proteins has a distinct anatomical pattern of expression in the brain. The D1 and D2 proteins are abundant in the striatum and are the most important receptor sites with regard to the causes and treatment of PD. The D4 and D5 proteins are largely extrastriatal, while D3 expression is low in the caudate and putamen but more abundant in the nucleus accumbens and olfactory tubercle. Neural Mechanism of Parkinsonism Considerable effort has been devoted to understanding how the loss of dopaminergic input to the neurons of the neostriatum gives rise to the clinical features of PD (for review see Albin et al., 1989; Mink and Thach, 1993; and Wichmann and DeLong, 1993). The basal ganglia can be viewed as a modulatory side loop that regulates the flow of information from the cerebral cortex to the motor neurons of the spinal cord (Figure 226). The neostriatum is the principal input structure of the basal ganglia and receives excitatory glutamatergic input from many areas of the cortex. The majority of neurons within the striatum are projection neurons that innervate other basal ganglia structures. A small but important subgroup of striatal neurons are interneurons that interconnect neurons within the striatum but do not project beyond its borders. Acetylcholine as well as neuropeptides are used as transmitters by the striatal interneurons. The outflow of the striatum proceeds along two distinct routes, identified as the direct and indirect pathways. The direct pathway is formed by neurons in the striatum that project directly to the output stages of the basal ganglia, the substantia nigra pars reticulata (SNpr) and the medial globus pallidus (MGP); these in turn relay to the ventroanterior and ventrolateral thalamus, which provides excitatory input to the cortex. The neurotransmitter of both links of the direct pathway is gamma-aminobutyric acid (GABA), which is inhibitory, so that the net effect of stimulation of the direct pathway at the level of the striatum is to increase the excitatory outflow from the thalamus to the cortex. The indirect pathway is composed of striatal neurons that project to the lateral globus pallidus (LGP). This structure in turn innervates the subthalamic nucleus (STN), which provides outflow to the SNpr and MGP output stage. As in the direct pathway, the first two linksthe projections from striatum to LGP and LGP to STNuse the inhibitory transmitter GABA; however, the final linkthe projection from STN to SNpr and MGPis an excitatory glutamatergic pathway. Thus the net effect of stimulating the indirect pathway at the level of the striatum is to reduce the excitatory outflow from the thalamus to the cerebral cortex.
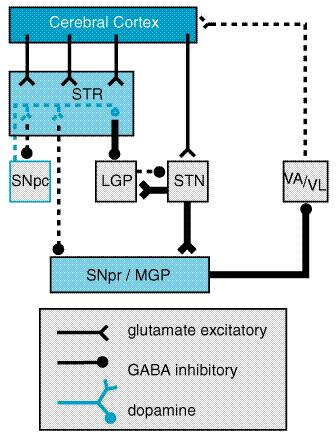
The key feature of this model of basal ganglia function, which accounts for the symptoms observed in PD as a result of loss of dopaminergic neurons, is the differential effect of dopamine on the direct and indirect pathways (Figure 227). The dopaminergic neurons of the substantia nigra pars compacta (SNpc) innervate all parts of the striatum; however, the target striatal neurons express distinct types of dopamine receptors. The striatal neurons giving rise to the direct pathway express primarily the excitatory D1 dopamine receptor protein, while the striatal neurons forming the indirect pathway express primarily the inhibitory D2 type. Thus, dopamine released in the striatum tends to increase the activity of the direct pathway and reduce the activity of the indirect pathway, whereas the depletion that occurs in PD has the opposite effect. The net effect of the reduced dopaminergic input in PD is to increase markedly the inhibitory outflow from the SNpr and MGP to the thalamus and reduce excitation of the motor cortex.
This model of basal ganglia function has important implications for the rational design and use of pharmacological agents in PD. First, it suggests that, to restore the balance of the system through stimulation of dopamine receptors, the complementary effect of actions at both D1 and D2 receptors, as well as the possibility of adverse effects that may be mediated by D3, D4, or D5 receptors, must be considered. Second, it explains why replacement of dopamine is not the only approach to the treatment of PD. Drugs that inhibit cholinergic receptors long have been used for treatment of parkinsonism. Although their mechanisms of action are not completely understood, it seems likely that their effect is mediated at the level of the striatal projection neurons which normally receive cholinergic input from striatal cholinergic interneurons. No clinically useful drugs for parkinsonism are presently available based on actions through GABA and glutamate receptors, even though both have crucial roles in the circuitry of the basal ganglia. However, they represent a promising avenue for drug development (Greenamyre and O'Brien, 1991). Treatment of Parkinson's Disease Commonly used medications for the treatment of PD are summarized in Table 221. Levodopa Levodopa L-DOPA, LARODOPA, DOPAR, L-3,4- dihydroxyphenylalanine), the metabolic precursor of dopamine, is the single most effective agent in the treatment of PD. Levodopa is itself largely inert; its therapeutic as well as adverse effects result from the decarboxylation of levodopa to dopamine. When administered orally, levodopa is rapidly absorbed from the small bowel by an active transport system for aromatic amino acids. Concentrations of the drug in plasma usually peak between 0.5 and 2 hours after an oral dose. The half-life in plasma is short (1 to 3 hours). The rate and extent of absorption of levodopa is dependent upon the rate of gastric emptying, the pH of gastric juice, and the length of time the drug is exposed to the degradative enzymes of the gastric and intestinal mucosa. Competition for absorption sites in the small bowel from dietary amino acids also may have a marked effect on the absorption of levodopa; administration of levodopa with meals delays absorption and reduces peak plasma concentrations. Entry of the drug into the central nervous system (CNS) across the bloodbrain barrier also is an active process mediated by a carrier of aromatic amino acids, and competition between dietary protein and levodopa may occur at this level. In the brain, levodopa is converted to dopamine by decarboxylation, primarily within the presynaptic terminals of dopaminergic neurons in the striatum. The dopamine produced is responsible for the therapeutic effectiveness of the drug in PD; after release, it is either transported back into dopaminergic terminals by the presynaptic uptake mechanism or metabolized by the actions of MAO and COMT (Mouradian and Chase, 1994). In modern practice, levodopa is almost always administered in combination with a peripherally acting inhibitor of aromatic L-amino acid decarboxylase, such as carbidopa or benserazide. If levodopa is administered alone, the drug is largely decarboxylated by enzymes in the intestinal mucosa and other peripheral sites, so that relatively little unchanged drug reaches the cerebral circulation and probably less than 1% penetrates the CNS. In addition, dopamine released into the circulation by peripheral conversion of levodopa produces undesirable effects, particularly nausea. Inhibition of peripheral decarboxylase markedly increases the fraction of administered levodopa that remains unmetabolized and available to cross the bloodbrain barrier and reduces the incidence of gastrointestinal side effects. In most individuals, a daily dose of 75 mg of carbidopa is sufficient to prevent the development of nausea. For this reason, the most commonly prescribed form of carbidopa/levodopa (SINEMET, ATAMET) is the 25/100 form, containing 25 mg of carbidopa and 100 mg of levodopa. With this formulation, dosage schedules of three or more tablets daily provide acceptable inhibition of decarboxylase in most individuals. Occasionally, individuals will require larger doses of carbidopa to minimize gastrointestinal side effects, and administration of supplemental carbidopa (LODOSYN) alone may be beneficial. Levodopa therapy can have a dramatic effect on all the signs and symptoms of PD. Early in the course of the disease, the degree of improvement in tremor, rigidity, and bradykinesia may be nearly complete. In early PD, the duration of the beneficial effects of levodopa may exceed the plasma lifetime of the drug, suggesting that the nigrostriatal dopamine system retains some capacity to store and release dopamine. A principal limitation of the long-term use of levodopa therapy is that, with time, this apparent 'buffering' capacity is lost, and the patient's motor state may fluctuate dramatically with each dose of levodopa. A common problem is the development of the 'wearing off ' phenomenon; each dose of levodopa effectively improves mobility for a period of time, perhaps 1 to 2 hours, but rigidity and akinesia rapidly return at the end of the dosing interval. Increasing the dose and frequency of administration can improve this situation, but this often is limited by development of dyskinesias, excessive and abnormal involuntary movements. Dyskinesias are observed most often when the plasma levodopa concentration is high, although, in some individuals, dyskinesias or dystonia may be triggered when the level is rising or falling. These movements can be as uncomfortable and disabling as the rigidity and akinesia of PD. In the later stages of PD, patients may fluctuate rapidly between being 'off,' having no beneficial effects from their medications, and being 'on' but with disabling dyskinesias, a situation called the 'on/off phenomenon.' Recent evidence has indicated that the induction of on/off phenomena and dyskinesias may be the result of an active process of adaptation to variations in brain and plasma levodopa levels. This process of adaptation is apparently complex, involving not only alterations in the expression of dopamine receptor proteins but also downstream changes in the postsynaptic striatal neurons, including modification of NMDA glutamate receptors (Mouradian and Chase, 1994; Chase, 1998). When levodopa levels are maintained at a constant level by intravenous infusion, dyskinesias and fluctuations are greatly reduced, and the clinical improvement is maintained for up to several days after returning to oral levodopa dosing (Mouradian et al., 1990; Chase et al., 1994). A sustained-release formulation consisting of carbidopa/levodopa in an erodable wax matrix (SINEMET CR) has been marketed in an attempt to produce more stable plasma levodopa levels than can be obtained with oral administration of standard carbidopa/levodopa formulations. This formulation is helpful in some cases, but the absorption of the sustained-release formulation is not entirely predictable. Another technique used to overcome the on/off phenomenon is to sum the total daily dose of carbidopa/levodopa and give equal amounts every 2 hours rather than every 4 or 6 hours. An important unanswered question regarding the use of levodopa in PD is whether this medication alters the course of the underlying disease or merely modifies the symptoms (Agid et al., 1998). Two aspects of levodopa treatment and the outcome of PD are of concern. First, it has been suggested that, if the production of free radicals as a result of dopamine metabolism contributes to the death of nigrostriatal neurons, the addition of levodopa might actually accelerate the process (Olanow, 1990), although no convincing evidence for such an effect has yet been obtained. Second, it is well established that the undesirable on/off fluctuations and wearing off phenomena are observed almost exclusively in patients treated with levodopa, but it is not known if delaying treatment with levodopa will delay the appearance of these effects (Fahn, 1999). In view of these uncertainties, most practitioners have adopted a pragmatic approach, using levodopa only when the symptoms of PD cause functional impairment. In addition to motor fluctuations and nausea, several other adverse effects may be observed with levodopa treatment. A common and troubling adverse effect is the induction of hallucinations and confusion; these effects are particularly common in the elderly and in those with preexisting cognitive dysfunction and often limit the ability to treat parkinsonian symptoms adequately. Conventional antipsychotic agents, such as the phenothiazines, are effective against levodopa-induced psychosis but may cause marked worsening of parkinsonism, probably through actions at the D2 dopamine receptor. A recent approach has been to use the 'atypical' antipsychotic agents, which are effective in the treatment of psychosis but do not cause or worsen parkinsonism (see Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania). The most effective of these are clozapine and quetiapine (Friedman and Factor, 2000). Peripheral decarboxylation of levodopa and release of dopamine into
the circulation may activate vascular dopamine receptors and produce
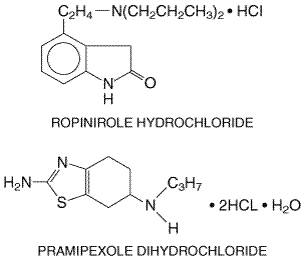
orthostatic hypotension. The actions of dopamine at Dopamine-Receptor Agonists An alternative to levodopa is the use of drugs that are direct agonists of striatal dopamine receptors, an approach that offers several potential advantages. Since enzymatic conversion of these drugs is not required for activity, they do not depend on the functional capacities of the nigrostriatal neurons and thus might be more effective than levodopa in late PD. In addition, dopamine-receptor agonists potentially are more selective in their actions; unlike levodopa, which leads to activation of all dopamine receptor types throughout the brain, agonists may exhibit relative selectivity for different subtypes of dopamine receptors. Most of the dopamine-receptor agonists in current clinical use have durations of action substantially longer than that of levodopa and often are useful in the management of dose-related fluctuations in motor state. Finally, if the hypothesis that free radical formation as a result of dopamine metabolism contributes to neuronal death is correct, then dopamine-receptor agonists have the potential to modify the course of the disease by reducing endogenous release of dopamine as well as the need for exogenous levodopa (Goetz, 1990). Four dopamine-receptor agonists are available for treatment of PD: two older agents bromocriptine (PARLODEL) and pergolide (PERMAX); and two newer, more selective compounds, ropinirole (REQUIP) and pramipexole (MIRAPEX). Bromocriptine and pergolide are both ergot derivatives and share a similar spectrum of therapeutic actions and adverse effects. Bromocriptine is a strong agonist of the D2 class of dopamine receptors and a partial antagonist of the D1 receptors, while pergolide is an agonist of both classes. Ropinirole and pramipexole (Figure 228) have selective activity at D2 class sites (specifically, at the D2 and D3 receptor proteins) and little or no activity at D1 class sites. All four of the drugs are well absorbed orally, and have similar therapeutic actions. Like levodopa, they can relieve the clinical symptoms of PD. The duration of action of the dopamine agonists often is longer than that of levodopa, and they are particularly effective in the treatment of patients who have developed on/off phenomena. All four also may produce hallucinosis or confusion, similar to that observed with levodopa, and may worsen orthostatic hypotension.
The
principal distinction between the newer, more selective agents and the older
ergot derivatives is in their tolerability and speed of titration. Initial
treatment with bromocriptine or pergolide may cause profound hypertension, so
they should be initiated at low dosage. The ergot derivatives also often
induce nausea and fatigue with initial treatment. Symptoms usually are
transient, but they require slow upward adjustment of the dose, over a period
of weeks to months. Ropinirole and pramipexole can be initiated more quickly,
achieving therapeutically useful doses in a week or less. They generally
cause less gastrointestinal disturbance than do the ergot derivatives, but
they can produce nausea and fatigue. Although these properties already have
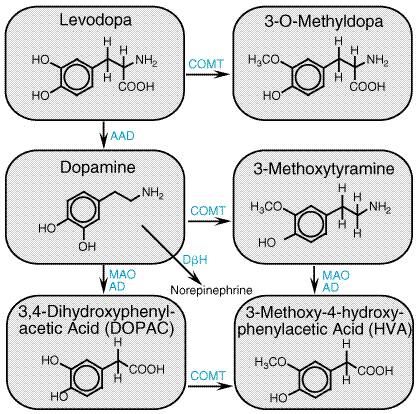
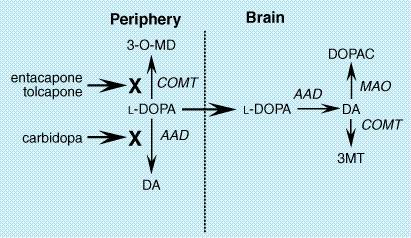
led to widespread adoption of the newer drugs in the The introduction of pramipexole and ropinirole has led to a substantial change in the clinical use of dopamine agonists in PD. Because these selective agonists are well tolerated, they are increasingly used as initial treatment for PD rather than as adjuncts to levodopa. This change has been driven by two factors: (1) the belief that because of their longer duration of action, dopamine agonists may be less likely than levodopa to induce on/off effects and dyskinesias, and (2) the concern that levodopa may contribute to oxidative stress, thereby accelerating loss of dopaminergic neurons. It is important to recognize that, while these concerns are well founded in laboratory experiments, there is at present only limited direct evidence for either of these effects on patients. Two large, controlled clinical trials comparing levodopa to pramipexole or ropinirole as initial treatment of PD recently have revealed a reduced rate of motor fluctuation in patients treated with these agonists, and if confirmed by additional studies, these findings are likely to greatly influence the clinical use of these drugs (Parkinson Study Group, 2000; Rascol et al., 2000). COMT Inhibitors A recently developed class of drugs for the treatment of PD are inhibitors of the enzyme catechol-O-methyltransferase (COMT). COMT and MAO are responsible for the catabolism of levodopa as well as dopamine. COMT transfers a methyl group from the donor S-adenosyl-L-methionine, producing the pharmacologically inactive compounds 3-O-methyl DOPA (from levodopa) and 3-methoxytyramine (from dopamine) (Figure 229). When levodopa is administered orally, nearly 99% of the drug is catabolized and does not reach the brain. The majority is converted by aromatic L-amino acid decarboxylase (AAD) to dopamine, which causes nausea and hypotension. Addition of an AAD inhibitor, such as carbidopa, reduces the formation of dopamine, but increases the fraction of levodopa that is methylated by COMT. The principal therapeutic action of the COMT inhibitors is to block this peripheral conversion of levodopa to 3-O-methyl DOPA, increasing both the plasma half-life of levodopa as well as the fraction of each dose that reaches the central nervous system (Goetz, 1998).
Two
COMT inhibitors presently are available for use in the Selegiline Two isoenzymes of MAO oxidize monoamines. While both isoenzymes (MAO-A and MAO-B) are present in the periphery and inactivate monoamines of intestinal origin, the isoenzyme MAO-B is the predominant form in the striatum and is responsible for the majority of oxidative metabolism of dopamine in the striatum. At low-to-moderate doses (10 mg/day or less), selegiline (ELDEPRYL) is a selective inhibitor of MAO-B, leading to irreversible inhibition of the enzyme (Olanow, 1993). Unlike nonspecific inhibitors of MAO (such as phenelzine, tranylcypromine, and isocarboxazid), selegiline does not inhibit peripheral metabolism of catecholamines; thus, it can be taken safely with levodopa. Selegiline also does not cause the lethal potentiation of catecholamine action observed when patients taking nonspecific MAO inhibitors ingest indirectly acting sympathomimetic amines such as the tyramine found in certain cheeses and wine. Doses of selegiline higher than 10 mg daily can produce inhibition of MAO-A and should be avoided. Selegiline has been used for several years as a symptomatic treatment for PD, although its benefit is fairly modest. The basis of the efficacy of selegiline is presumed to be its ability to retard the breakdown of dopamine in the striatum. With the recent emergence of interest in the potential role of free radicals and oxidative stress in the pathogenesis of PD, it has been proposed that the ability of selegiline to retard the metabolism of dopamine might confer neuroprotective properties. In support of this idea, it was observed that selegiline could protect animals from MPTP-induced parkinsonism by blocking the conversion of MPTP to its toxic metabolite (1-methyl-4-phenylpyridium ion), a transformation mediated by MAO-B. The potential protective role of selegiline in idiopathic PD was evaluated in multicenter randomized trials; these studies showed a symptomatic effect of selegiline in PD, but longer follow-up failed to provide any definite evidence of ability to retard the loss of dopaminergic neurons (Parkinson Study Group, 1993). Selegiline is generally well tolerated in patients with early or mild PD. In patients with more advanced PD or underlying cognitive impairment, selegiline may accentuate the adverse motor and cognitive effects of levodopa therapy. Metabolites of selegiline include amphetamine and methamphetamine, which may cause anxiety, insomnia, and other adverse symptoms. Interestingly, it has been observed that selegiline, like the nonspecific MAO inhibitors, can lead to the development of stupor, rigidity, agitation, and hyperthermia after administration of the analgesic meperidine; the basis of this interaction is uncertain. There also have been reports of adverse effects resulting from interactions between selegiline and tricyclic antidepressants and between selegiline and serotonin-reuptake inhibitors. Muscarinic Receptor Antagonists Antagonists of muscarinic acetylcholine receptors were widely used for the treatment of PD before the discovery of levodopa. The biological basis for the therapeutic actions of anticholinergics is not completely understood. It seems likely that they act within the neostriatum, through the receptors that normally mediate the response to the intrinsic cholinergic innervation of this structure, which arises primarily from cholinergic striatal interneurons. Several muscarinic cholinergic receptors have been cloned (see Chapters 7: Muscarinic Receptor Agonists and Antagonists and 12: Neurotransmission and the Central Nervous System); like the dopamine receptors, these are proteins with seven transmembrane domains that are linked to second-messenger systems by G proteins. Five subtypes of muscarinic receptors have been identified; at least four and probably all five subtypes are present in the striatum, although each has a distinct distribution (Hersch et al., 1994). Several drugs with anticholinergic properties are currently used in the treatment of PD, including trihexyphenidyl (ARTANE, 2 to 4 mg, three times per day), benztropine mesylate (COGENTIN, 1 to 4 mg, two times per day), and diphenhydramine hydrochloride (BENADRYL, 25 to 50 mg, 3 to 4 times per day). All have a modest antiparkinsonian action, which is useful in the treatment of early PD or as an adjunct to dopamimetic therapy. The adverse effects of these drugs are a result of their anticholinergic properties. Most troublesome is sedation and mental confusion, frequently seen in the elderly. They also may produce constipation, urinary retention, and blurred vision through cycloplegia; they must be used with caution in patients with narrow-angle glaucoma. Amantadine Amantadine SYMMETREL), an antiviral agent used for the prophylaxis and treatment of influenza A (see Chapter 50: Antimicrobial Agents: Antiviral Agents (Nonretroviral)), has antiparkinsonian actions. The mechanism of action of amantadine is not clear. It has been suggested that it might alter dopamine release or reuptake; anticholinergic properties also may contribute to its therapeutic actions. Amantadine and the closely related compound memantadine have activity at NMDA glutamate receptors, which may contribute to their antiparkinsonian actions (Stoof et al., 1992). In any case, the effects of amantadine in PD are modest. It is used as initial therapy of mild PD. It also may be helpful as an adjunct in patients on levodopa with dose-related fluctuations. Amantadine usually is administered in a dose of 100 mg twice a day and is well tolerated. Dizziness, lethargy, anticholinergic effects, and sleep disturbance, as well as nausea and vomiting, have been observed occasionally, but even when present these effects are mild and reversible. |
Alzheimer's Disease
|
Clinical Overview AD produces an impairment of cognitive abilities that is gradual in onset but relentless in progression. Impairment of short-term memory usually is the first clinical feature, while retrieval of distant memories is preserved relatively well into the course of the disease. As the condition progresses, additional cognitive abilities are impaired, among them the ability to calculate, exercise visuospatial skills, and use common objects and tools (ideomotor apraxia). The level of arousal or alertness of the patient is not affected until the condition is very advanced, nor is there motor weakness, although muscular contractures are an almost universal feature of advanced stages of the disease. Death, most often from a complication of immobility such as pneumonia or pulmonary embolism, usually ensues within 6 to 12 years after onset. The diagnosis of AD is based on careful clinical assessment of the patient and appropriate laboratory tests to exclude other disorders that may mimic AD; at present, no direct antemortem confirmatory test exists. Pathophysiology AD is characterized by marked atrophy of the cerebral cortex and loss
of cortical and subcortical neurons. The pathological hallmarks of AD are
senile plaques, which are spherical accumulations of the protein Neurochemistry The neurochemical disturbances that arise in AD have been studied
intensively ( Role of The presence of aggregates of Analysis of APP gene structure in pedigrees exhibiting autosomal
dominant inheritance of AD has shown that, in some families, mutations of the
Treatment of Alzheimer's Disease A major approach to the treatment of AD has involved attempts to
augment the cholinergic function of the brain ( Four inhibitors of AChE currently are approved by the United States Food and Drug Administration for treatment of Alzheimer's disease: tacrine (1,2,3,4-tetrahydro-9-aminoacridine; COGNEX), donepezil (ARICEPT) (Mayeux and Sano, 1999), rivastigmine (EXCELON), and galantamine (REMINYL). Tacrine is a potent, centrally acting inhibitor of AChE (Freeman and Dawson, 1991). Studies of oral tacrine in combination with lecithin have confirmed that there is indeed an effect of tacrine on some measures of memory performance, but the magnitude of improvement observed with the combination of lecithin and tacrine is modest at best (Chatellier and Lacomblez, 1990). The side effects of tacrine often are significant and dose-limiting; abdominal cramping, anorexia, nausea, vomiting, and diarrhea are observed in up to one-third of patients receiving therapeutic doses, and elevations of serum transaminases are observed in up to 50% of those treated. Because of the significant side-effect profile, tacrine is not widely used in clinical practice. Donepezil is a selective inhibitor of AChE in the CNS with little effect on AChE in peripheral tissues. It produces modest improvements in cognitive scores in Alzheimer's disease patients (Rogers and Friedhoff, 1988) and has a long half-life (see Appendix II), allowing once-daily dosing. Rivastigmine and galantamine are dosed twice daily and produce a similar degree of cognitive improvement. Adverse effects associated with donepezil, rivastigmine, and galantamine are similar in character but generally less frequent and less severe than those observed with tacrine; they include nausea, diarrhea, vomiting, and insomnia. Donepezil, rivastigmine, and galantamine are not associated with the hepatotoxicity that limits the use of tacrine. Drugs currently under development for treatment of Alzheimer's disease
include additional anticholinesterase agents as well as agents representing
other pharmacological approaches. Memantine, an NMDA-receptor
antagonist, has shown promise in clinical trials of slowing the progression
of AD in patients with moderately severe disease. Antioxidants,
antiinflammatory agents, and estrogens have been studied, but none of these
has established efficacy. The identification of APP and the enzymes involved
in the processing of this protein has opened the door to the development of
antiaggregants, a |
Huntington's Disease
|
Clinical Features HD is a dominantly inherited disorder characterized by the gradual onset of motor incoordination and cognitive decline in midlife. Symptoms develop insidiously, either as a movement disorder manifest by brief jerk-like movements of the extremities, trunk, face, and neck (chorea) or by personality changes, or both. Fine motor incoordination and impairment of rapid eye movements are early features. Occasionally, especially when the onset of symptoms occurs before the age of 20, choreic movements are less prominent; instead, bradykinesia and dystonia predominate. As the disorder progresses, the involuntary movements become more severe, dysarthria and dysphagia develop, and balance is impaired. The cognitive disorder manifests itself first by slowness of mental processing and difficulty in organizing complex tasks. Memory is affected, but affected persons rarely lose their memory of family, friends, and the immediate situation. Such persons often become irritable, anxious, and depressed. Less frequently, paranoia and delusional states are manifest. The outcome of HD is invariably fatal; over a course of 15 to 30 years, the affected person becomes totally disabled and unable to communicate, requiring full-time care; death ensues from the complications of immobility (Hayden, 1981; Harper, 1991, 1992). Pathology and Pathophysiology HD is characterized by prominent neuronal loss in the caudate/putamen of the brain (Vonsattel et al., 1985). Atrophy of these structures proceeds in an orderly fashion, first affecting the tail of the caudate nucleus and then proceeding anteriorly, from medialdorsal to lateralventral. Other areas of the brain also are affected, although much less severely; morphometric analyses indicate that there are fewer neurons in cerebral cortex, hypothalamus, and thalamus. Even within the striatum, the neuronal degeneration of HD is selective. Interneurons and afferent terminals are largely spared, while the striatal projection neurons (the medium spiny neurons) are severely affected. This leads to large decreases in striatal GABA concentrations, whereas somatostatin and dopamine concentrations are relatively preserved (Ferrante et al., 1987; Reiner et al., 1988). Selective vulnerability also appears to underlie the most conspicuous clinical feature of HD, the development of chorea. In most adult-onset cases, the medium spiny neurons that project to LGP and SNpr (the indirect pathway) appear to be affected earlier than those projecting to the MGP (the direct pathway; Albin et al., 1990, 1992). The disproportionate impairment of the indirect pathway increases excitatory drive to the neocortex, producing involuntary choreiform movements (Figure 2210). In some individuals, rigidity rather than chorea is the predominant clinical feature; this is especially common in juvenile-onset cases. In these cases the striatal neurons giving rise to both the direct and indirect pathways are impaired to a comparable degree.
Genetics HD is an autosomal dominant disorder with nearly complete penetrance. The average age of onset is between 35 and 45 years, but the range varies from as early as age two to as late as the mid-eighties. Although the disease is inherited equally from mother and father, more than 80% of those developing symptoms before the age of 20 inherit the defect from the father. This is an example of anticipation, or the tendency for the age of onset of a disease to decline with each succeeding generation, which also is observed in other neurodegenerative diseases with similar genetic mechanisms. Known homozygotes for HD show clinical characteristics identical to the typical HD heterozygote, indicating that the unaffected chromosome does not attenuate the disease symptomatology. Until the discovery of the genetic defect responsible for HD, de novo mutations causing HD were thought to be unusual; but it is clear now that the disease can arise from unaffected parents, especially when one carries an 'intermediate allele,' as described below. The discovery of the genetic mutation responsible for Huntington's disease was the product of an arduous, ten-year, multi-investigator, collaborative effort. In 1993 a region near the telomere of chromosome 4 was found to contain a polymorphic (CAG)n trinucleotide repeat that was significantly expanded in all individuals with HD (Huntington's Disease Collaborative Research Group, 1993). The expansion of this trinucleotide repeat is the genetic alteration responsible for HD. The range of CAG repeat length in normal individuals is between 9 and 34 triplets, with a median repeat length on normal chromosomes of 19. The repeat length in HD varies from 40 to over 100. Repeat lengths of 35 to 39 represent intermediate alleles; some of these individuals develop HD late in life, while others are not affected. Repeat length is correlated inversely with age of onset. The younger the age of onset, the higher the probability of a large repeat number. This correlation is most powerful in individuals with onset before the age of 30; with onset above the age of 30, the correlation is weaker. Thus, repeat length cannot serve as an adequate predictor of age of onset in most individuals. Subsequent work has shown that several other neurodegenerative diseases also arise through expansion of a CAG repeat, including hereditary spinocerebellar ataxias and Kennedy's disease, a rare inherited disorder of motor neurons (Paulson and Fischbeck, 1996). Selective Vulnerability The mechanism by which the expanded trinucleotide repeat leads to the clinical and pathological features of HD is unknown. The HD mutation lies within a gene designated IT15. The IT15 gene itself is very large (10 kilobases) and encodes a protein of approximately 348,000 daltons or 3144 amino acids. The trinucleotide repeat, which encodes the amino acid glutamine, occurs at the 5'-end of IT15 and is followed directly by a second, shorter repeat of (CCG)n, which encodes the amino acid proline. The protein, named huntingtin, does not resemble any other known protein, and the normal function of the protein has not been identified. Mice with a genetic 'knockout' of huntingtin die early in embryonic life, so it must have an essential cellular function. It is thought that the mutation results in a gain of function; that is, the mutant protein acquires a new function or property not found in the normal protein. The HD gene is expressed widely throughout the body. High levels of expression are present in brain, pancreas, intestine, muscle, liver, adrenals, and testes. In brain, expression of IT15 does not appear to be correlated with neuron vulnerability; although the striatum is most severely affected, neurons in all regions of the brain express similar levels of IT15 mRNA (Landwehrmeyer et al., 1995). The ability of the HD mutation to produce selective neural degeneration despite nearly universal expression of the gene among neurons may be related to metabolic or excitotoxic mechanisms. For many years, it has been noted that HD patients are thin, suggesting the presence of a systemic disturbance of energy metabolism. In animal models, agonists for the NMDA subtype of excitatory amino acid receptor can cause pathology similar to that seen in HD when they are injected into the striatum (Beal et al., 1986). More interesting, however, is the fact that inhibitors of complex II of the mitochondrial respiratory chain also can produce HD-like striatal lesionseven when given systemically (Beal et al., 1993). Furthermore, this pathology can be diminished by NMDA-receptor antagonists, suggesting that this is an example of a metabolic impairment giving rise to excitotoxic neuronal injury. Studies employing magnetic resonance imaging (MRI) spectroscopy have provided direct evidence of an alteration in energy metabolism in HD in vivo (Jenkins et al., 1993). Thus, the link between the widespread expression of the gene for the abnormal IT15 protein in HD and the selective vulnerability of neurons in the disease may arise from the interaction of a widespread defect in energy metabolism with the intrinsic properties of striatal neurons, including their capacity and need for oxidative metabolism as well as the types of glutamate receptors present. This hypothesis has a number of potentially important therapeutic implications. It is unlikely that it will be possible in the near future to correct the genetic defect in the brains of individuals with HD, but it may be possible to develop agents that alter metabolic function or protect against excitotoxic injury and thereby arrest or modify the course of the disease. Symptomatic Treatment of Huntington's Disease Practical treatment for symptomatic HD emphasizes the selective use of medications (Shoulson, 1992). No current medication slows the progression of the disease, and many medications can impair function because of side effects. Treatment is needed for patients who are depressed, irritable, paranoid, excessively anxious, or psychotic. Depression can be treated effectively with standard antidepressant drugs with the caveat that those drugs with substantial anticholinergic profiles can exacerbate chorea. Fluoxetine (Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders) is effective treatment for both the depression and the irritability manifest in symptomatic HD. Carbamazepine (Chapter 21: Drugs Effective in the Therapy of the Epilepsies) also has been found to be effective for depression. Paranoia, delusional states, and psychosis usually require treatment with antipsychotic drugs, but the doses required often are lower than those usually used in primary psychiatric disorders. These agents also reduce cognitive function and impair mobility and thus should be used in the lowest doses possible and be discontinued when the psychiatric symptoms are resolved. In individuals with predominantly rigid HD, clozapine (Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania) or carbamazepine may be more effective for treatment of paranoia and psychosis. The movement disorder of HD per se only rarely justifies pharmacological therapy. For those with large-amplitude chorea causing frequent falls and injury, dopamine-depleting agents such as tetrabenazine or reserpine (Chapter 33: Antihypertensive Agents and the Drug Therapy of Hypertension) can be tried, although patients must be monitored for hypotension and depression. Antipsychotic agents also can be used, but these often do not improve overall function because they decrease fine motor coordination and increase rigidity. Many HD patients exhibit worsening of involuntary movements as a result of anxiety or stress. In these situations, judicious use of sedative or anxiolytic benzodiazepines can be very helpful. In juvenile-onset cases where rigidity rather than chorea predominates, dopamine agonists have had variable success in the improvement of rigidity. These individuals also occasionally develop myoclonus and seizures that can be responsive to clonazepam, valproic acid, or other anticonvulsants. |
Amyotrophic Lateral Sclerosis
|
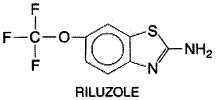
Clinical Features and Pathology ALS is a disorder of the motor neurons of the ventral horn of the spinal cord and the cortical neurons that provide their afferent input. The ratio of males to females affected is approximately 1.5:1 (Kurtzke, 1982). The disorder is characterized by rapidly progressive weakness, muscle atrophy and fasciculations, spasticity, dysarthria, dysphagia, and respiratory compromise. Sensory function generally is spared, as is cognitive, autonomic, and oculomotor activity. ALS usually is progressive and fatal, with most affected patients dying of respiratory compromise and pneumonia after 2 to 3 years, although occasional individuals have a more indolent course and survive for many years. The pathology of ALS corresponds closely to the clinical features: There is prominent loss of the spinal and brainstem motor neurons that project to striated muscles (although the oculomotor neurons are spared) as well as loss of the large pyramidal motor neurons in layer V of motor cortex, which are the origin of the descending corticospinal tracts. In familial cases, Clarke's column and the dorsal horns sometimes are affected (Caroscio et al., 1987; Rowland, 1994). Etiology About 10% of cases of ALS are familial (FALS), usually with an autosomal dominant pattern of inheritance (Jackson and Bryan, 1998). Most of the mutations responsible have not been identified, but an important subset of FALS patients are families with a mutation in the gene for the enzyme superoxide dismutase (SOD1) (Rosen et al., 1993). Mutations in this protein account for about 20% of cases of FALS. Most of the mutations are alterations of single amino acids, but more than 30 different alleles have been found in different kindreds. Transgenic mice expressing mutant human SOD1 develop a progressive degeneration of motor neurons that closely mimics the human disease, providing an important animal model for research and pharmaceutical trials. Interestingly, many of the mutations of SOD1 that can cause disease do not reduce the capacity of the enzyme to perform its primary function, the catabolism of potentially toxic superoxide radicals. Thus, as may be the case in HD, mutations in SOD1 may confer a toxic 'gain of function,' the precise nature of which is unclear. More than 90% of ALS cases are sporadic and are not associated with abnormalities of SOD1 or any other known gene. The cause of the motor neuron loss in sporadic ALS is unknown, but theories include autoimmunity, excitotoxicity, free radical toxicity, and viral infection (Rowland, 1994; Cleveland, 1999). Most of these ideas are not well supported by available data, but there is evidence that glutamate reuptake may be abnormal in the disease, leading to accumulation of glutamate and excitotoxic injury (Rothstein et al., 1992). The only currently approved therapy for ALS, riluzole, is based on these observations. Spasticity and the Spinal Reflex Spasticity is an important component of the clinical features of ALS, in that the presence of spasticity often leads to considerable pain and discomfort and reduces mobility, which already is compromised by weakness. Furthermore, spasticity is the feature of ALS that is most amenable to present forms of treatment. Spasticity is defined as an increase in muscle tone characterized by an initial resistance to passive displacement of a limb at a joint, followed by a sudden relaxation (the so-called clasped-knife phenomenon). Spasticity is the result of the loss of descending inputs to the spinal motor neurons, and the character of the spasticity depends on which nervous system pathways are affected (Davidoff, 1990). Whole repertoires of movement can be generated directly at the spinal cord level; it is beyond the scope of this chapter to describe these in detail. The monosynaptic tendon-stretch reflex is the simplest of the spinal mechanisms contributing to spasticity. Primary Ia afferents from muscle spindles, activated when the muscle is rapidly stretched, synapse directly on motor neurons going to the stretched muscle, causing it to contract and resist the movement. A collateral of the primary Ia afferent synapses on an 'Ia-coupled interneuron' that inhibits the motor neurons innervating the antagonist of the stretched muscle, allowing the contraction of the muscle to be unopposed. Upper motor neurons from the cerebral cortex (the pyramidal neurons) suppress spinal reflexes and the lower motor neurons indirectly by activating the spinal cord inhibitory interneuron pools. The pyramidal neurons use glutamate as a neurotransmitter. When the pyramidal influences are removed, the reflexes are released from inhibition and become more active, leading to hyperreflexia. Other descending pathways from brainstemincluding the rubro-, reticulo-, and vestibulospinal pathways and the descending catecholamine pathwaysalso influence spinal reflex activity. When just the pyramidal pathway is affected, extensor tone in the legs and flexor tone in the arms are increased. When the vestibulospinal and catecholamine pathways are impaired, increased flexion of all extremities is observed and light cutaneous stimulation can lead to disabling whole-body spasms. In ALS, pyramidal pathways are impaired with relative preservation of the other descending pathways, resulting in hyperactive deep-tendon reflexes, impaired fine motor coordination, increased extensor tone in the legs, and increased flexor tone in the arms. The gag reflex often is overactive as well. Treatment of ALS with Riluzole Riluzole (2-amino-6-[trifluoromethoxy]benzothiazole; RILUTEK) is an agent with complex actions in the nervous system (Bryson et al., 1996; Wagner and Landis, 1997). Its structure is as follows:
Riluzole is orally absorbed and highly protein bound. It undergoes extensive metabolism in the liver by both cytochrome P450-mediated hydroxylation and by glucuronidation. Its half-life is about 12 hours. In vitro studies have shown that riluzole has both presynaptic and postsynaptic effects. It inhibits glutamate release, but it also blocks postsynaptic NMDA- and kainate-type glutamate receptors and inhibits voltage-dependent sodium channels. Some of the effects of riluzole in vitro are blocked by pertussis toxin, implicating the drug's interaction with an as-yet unidentified G proteincoupled receptor. In clinical trials, riluzole has modest but genuine effects on the survival of patients with ALS. In the largest trial conducted to date, with nearly 1000 patients, the median duration of survival was extended by about 60 days (Lacomblez et al., 1996). The recommended dose is 50 mg every 12 hours, taken 1 hour before or 2 hours after a meal. Riluzole usually is well tolerated, although nausea or diarrhea may occur. Rarely, riluzole may produce hepatic injury with elevations of serum transaminases, and periodic monitoring of these is recommended. Although the magnitude of the effect of riluzole on ALS is small, it represents a significant therapeutic milestone in the treatment of a disease refractory to all previous treatments. Symptomatic Therapy of Spasticity The most useful agent for the symptomatic treatment of spasticity in
ALS is baclofen (LIORESAL), a GABAB agonist. Initial doses of 5 to 10 mg a day are
recommended, but the dose can be increased to as much as 200 mg a day if
necessary. If weakness occurs, the dose should be lowered. In addition to
oral administration, baclofen also can be delivered directly into the space
around the spinal cord by use of a surgically implanted pump and an
intrathecal catheter. This approach minimizes the adverse effects of the
drug, especially sedation, but it carries the risk of potentially
life-threatening CNS depression and should be used only by physicians trained
in delivering chronic intrathecal therapy. Tizanidine (ZANFLEX) is an agonist of |
Prospectus
|
Although advances in the symptomatic therapy
of the neurodegenerative disorders, particularly PD, have improved the lives
of many patients, the goal of current research is to develop treatments that
can prevent, retard, or reverse neuronal cell death. Promising areas for drug
development are the mechanisms implicated in several of the disorders:
excitotoxicity, defects in energy metabolism, and oxidative stress. Glutamate
antagonists have great potential, but their use is limited by the relatively
nonselective activity of the available agents. Increased knowledge of the
structure and function of glutamate receptor subtypes should make more
selective and useful agents available. Pharmacological reduction of oxidative
stress also is feasible, despite the disappointing results of initial
clinical trials with tocopherol and selegiline. Neural growth factors are
another important area for drug development. Several factors that promote the
differentiation of neurons and the establishment of neural connections during
development have been identified, and these may eventually prove useful in
retarding or reversing neuronal death. A more direct and currently accessible
approach to reversing neuronal loss is surgical transplantation of neurons;
this has been accomplished in PD with a moderate degree of success and has
been proposed as a treatment for other conditions such as AD. In addition to
these general approaches to neurodegeneration, more specific treatments for
the various diseases should become feasible with advances in knowledge of
their etiology. For example, discovery of the role of For further information regarding neurodegenerative diseases for which the drugs discussed in this chapter are useful, the reader is referred to the following chapters in Harrison's Principles of Internal Medicine, 16th ed., McGraw-Hill, New York, 2005: Parkinson's disease, Chapter 351; Alzheimer's disease and Huntington's disease, Chapter 350; and amyotrophic lateral sclerosis, Chapter 353. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 2480
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved