| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists
Overview
|
This chapter deals with the diverse physiological roles of 5-hydroxytryptamine (5-HT, serotonin) as a neurotransmitter in the central nervous system, as a regulator of smooth muscle function in the cardiovascular and gastrointestinal systems, and as a regulator of platelet function. Molecular cloning has revealed an unexpected diversity of receptor subtypes, which fall into four structural and functional families. Three subtype families (5-HT1, 5-HT2, and 5-HT4) are coupled via G proteins to a variety of enzymatic and electrical effector systems; the 5-HT3 receptor, in marked contrast, serves as a 5-HTgated ion channel. This chapter covers 5-HT-receptor agonists and antagonists including new ones emerging as a result of the use of recombinant receptors as tools to screen for subtype-selective agents. Experimental models used to test for drugs that alter complex behaviors, such as compulsion, aggression, anxiety, depression, and sleep-wake cycles, also are described. New subtype-selective 5-HT-receptor agonists already have demonstrated therapeutic effectiveness in the acute treatment of migraine headaches and in anxiety (see Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders); subtype-selective antagonists have proven to be effective for treatment of various gastrointestinal disorders (see Chapter 38: Prokinetic Agents, Antiemetics, and Agents Used in Irritable Bowel Syndrome). Serotonin actions in vivo also can be regulated by pharmacological agents that control its availability as a neurotransmitter, such as selective inhibitors of neuronal reuptake of serotonin. These agents have proven to be efficacious in depression and anxiety disorders and are discussed in Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders. |
5-Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists: Introduction
|
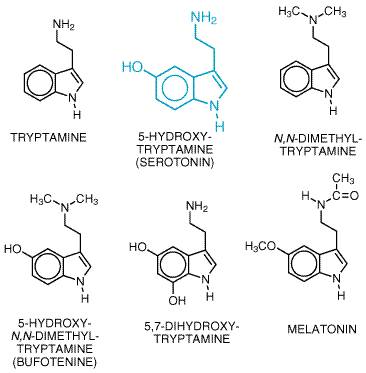
5-Hydroxytryptamine (5-HT, serotonin) has been recognized for more than 50 years as an effector on various types of smooth muscle and, subsequently, as an agent that enhances platelet aggregation and as a neurotransmitter in the central nervous system (CNS). 5-HT is found in high concentrations in enterochromaffin cells throughout the gastrointestinal tract, in platelets, and in specific regions of the CNS. Although implicated in the regulation of a number of physiological processes and their malfunction, the exact sites and modes of action of 5-HT remain ill-defined and elusive. These ambiguities are probably, in large part, the consequence of the large number of 5-HT-receptor subtypes defined initially by pharmacological analyses and confirmed by cDNA cloning. The availability of cloned receptors is allowing the development of subtype-selective drugs and elucidation of the various actions of 5-HT at a molecular level. Furthermore, an increasing number of therapeutic goals can be approached by drugs that target one or more of the subtypes of serotonin receptors. History In the 1930s, Erspamer began to study the distribution of enterochromaffin cells, which stained with a reagent for indoles. The highest concentrations were found in gastrointestinal mucosa, followed by platelets and the CNS (Erspamer, 1966). Page and his colleagues at the Cleveland Clinic were the first to isolate and chemically characterize a vasoconstrictor substance released from platelets in clotting blood (Rapport et al., 1948). This substance was called serotonin (Page, 1976) and was shown to be identical to the indole isolated by Erspamer. The discovery of biosynthetic and degradative pathways (Udenfriend, 1959) and clinical interest in the pressor effects of 5-HT (Sjoerdsma, 1959) led to the hypothesis that the symptoms of patients with tumors of intestinal enterochromaffin cells (carcinoid syndrome) are the result of abnormally high production of 5-HT. Several hundred milligrams of 5-HT and its metabolites may be excreted within a 24-hour period in patients with carcinoid tumors. The gross effects of 5-HT, produced in excess in malignant carcinoid, give some indication of the actions of 5-HT. For example, these patients may display psychotic behaviors similar to those produced by lysergic acid diethylamide (LSD). Several naturally occurring hallucinogenic tryptamine-like substances were identified from animal and plant sources, suggesting that these substances may be formed in vivo and could explain the abnormal behavior of carcinoid patients. An early explanation of the action of 5-HT was obtained with the parasitic liver fluke, Fasciola hepatica (Mansour, 1979). Exposure of the fluke to 5-HT elicited a marked increase in motility and a concomitant increase in cyclic AMP formation. Both effects were blocked by LSD. The increased motility was mediated by regulation of glycolysis at its rate-limiting step, phosphofructokinase, via a cyclic AMPdependent phosphorylation of the enzyme. The 5-HT receptor involved appears to be unrelated to mammalian receptors linked to adenylyl cyclase. Such detailed explanations of the neurohumoral effects of 5-HT have not yet been achieved in mammals. In the mid-1950s, it was suggested that 5-HT may function as a neurotransmitter in the mammalian brain (see Brodie and Shore, 1957). 5-HT appeared early in the evolution of the plant and animal kingdoms. This may explain why there are so many 5-HT-receptor subtypes (Peroutka and Howell, 1994). Recent cloning of 5-HT-receptor subtypes has revealed that some drugs previously considered to be subtype-selective have high affinities for multiple molecularly defined receptors (see Table 111; see also Sjoerdsma and Palfreyman, 1990, for additional details about the discovery and effects of 5-HT). Source and Chemistry 5-HT, 3-(
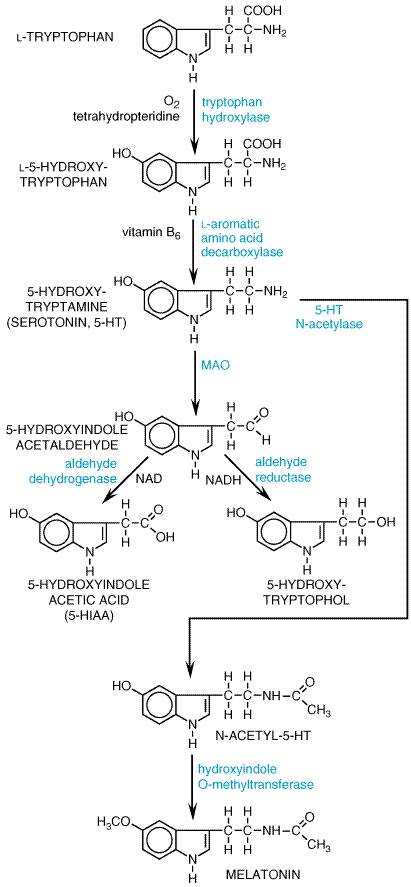
Synthesis and Metabolism 5-HT is synthesized by a two-step pathway from the essential amino acid tryptophan (see Figure 112). Tryptophan hydroxylase, a mixed-function oxidase that requires molecular oxygen and a reduced pteridine cofactor for activity, is the rate-limiting enzyme in the pathway. The active uptake of tryptophan is the first step in the synthesis of 5-HT in the brain. Unlike tyrosine hydroxylase, tryptophan hydroxylase is not regulated by end-product inhibition, although regulation by phosphorylation is common to both enzymes. Brain tryptophan hydroxylase is not saturated with substrate; consequently, the amount of tryptophan in the brain influences the synthesis of 5-HT. Tryptophan is actively transported into the brain by a carrier that also transports other large neutral and branch-chain amino acids. The levels of tryptophan in the brain are influenced not only by its plasma concentration but also by the plasma concentrations of other amino acids that compete for the brain uptake carrier. The enzyme that converts L-5-hydroxytryptophan to 5-HT, aromatic L-amino acid decarboxylase, is widely distributed and has a broad substrate specificity. A longstanding debate about whether or not L-5-hydroxytryptophan decarboxylase and L-dopa decarboxylase are identical enzymes was clarified when cDNA cloning confirmed that a single gene product decarboxylates both amino acids. 5-Hydroxytryptophan is not detected in the brain, because the amino acid is rapidly decarboxylated. Therefore, it is not possible to alter levels of 5-HT in the brain by manipulating the levels of 5-hydroxytryptophan. The principal route of metabolism of 5-HT involves monoamine oxidase (MAO) forming 5-hydroxyindole acetic acid (5-HIAA) by a two-step process (see Figure 112). The aldehyde formed by the action of MAO is converted to 5-HIAA by an ubiquitous enzyme, aldehyde dehydrogenase. An alternative route, reduction of the acetaldehyde to an alcohol, 5-hydroxytryptophol, is normally insignificant. 5-HIAA is actively transported out of the brain by a process that is sensitive to the nonspecific transport inhibitor probenecid. Since 5-HIAA accounts for nearly 100% of the metabolism of 5-HT in brain, the turnover rate of brain 5-HT is estimated by measuring the rate of rise of 5-HIAA after administration of probenecid. 5-HIAA from brain and peripheral sites of 5-HT storage and metabolism is excreted in the urine along with small amounts of 5-hydroxytryptophol sulfate or glucuronide conjugates. The usual urinary excretion of 5-HIAA by a normal adult is 2 to 10 mg daily. Larger amounts are excreted by patients with malignant carcinoid, providing a reliable diagnostic test for the disease. The massive amounts of pyridine nucleotides and tryptophan that are required for 5-HT synthesis in patients with malignant carcinoid may produce symptoms of niacin and tryptophan deficiencies. Ingestion of ethyl alcohol results in elevated amounts of NADH, which diverts 5-hydroxyindoleacetaldehyde from the oxidative route to the reductive pathway (see Figure 112). This tends to increase the excretion of 5-hydroxytryptophol and correspondingly reduces the excretion of 5-HIAA. Two isoforms of monoamine oxidase (MAO-A and -B) were distinguished
initially on the basis of substrate and inhibitor specificities. Both
isoforms have been cloned, and the properties of the cloned enzymes are
consistent with the pharmacological profiles established previously (Shih,
1991; see Chapter 10: Catecholamines, Sympathomimetic Drugs, and
Adrenergic Receptor Antagonists). MAO-A preferentially metabolizes 5-HT and norepinephrine;
clorgyline is a specific inhibitor of this enzyme. MAO-B prefers Other minor pathways of metabolism of 5-HT, such as sulfation and O-or N-methylation, have been suggested. The latter reaction could lead to formation of an endogenous psychotropic substance, 5-hydroxy-N,N-dimethyltryptamine (bufotenine; see Figure 111). However, other methylated indoleamines such as N,N-dimethyltryptamine and 5-methoxy-N,N-dimethyltryptamine are far more active hallucinogenic agents and are more likely candidates as endogenous psychotomimetics. In addition to metabolism by MAO, a Na+-dependent, carrier-mediated uptake process exists and is involved in terminating the action of 5-HT. The 5-HT transporter is localized in the outer membrane of serotonergic axon terminals (where it terminates the action of 5-HT in the synapse) and in the outer membrane of platelets (where it takes up 5-HT from the blood). This uptake system is the only way that platelets acquire 5-HT, as they do not have the enzymes required for synthesis of 5-HT. The 5-HT transporter, as well as other monoamine transporters, has been cloned (see Chapter 12: Neurotransmission and the Central Nervous System). The deduced amino acid sequence and predicted membrane topology place the amine transporters in a family clearly distinct from the transport proteins that concentrate amines in intracellular storage vesicles. Furthermore, the vesicular transporter is a nonspecific amine carrier, while the 5-HT transporter and the other amine transporters are highly specific. Neither pharmacological studies nor cDNA cloning has provided evidence to support the existence of multiple 5-HT transporters. Recent studies have found that the 5-HT transporter is regulated by phosphorylation with subsequent internalization (Ramamoorthy and Blakely, 1999), providing an unexpected mechanism for dynamic regulation of serotonergic transmission. |
Physiological Functions of Serotonin
|
Multiple 5-HT Receptors Early studies of peripheral tissues advanced the hypothesis that the multiple actions of 5-HT are explained by an interaction with more than one 5-HT-receptor subtype. Extensive pharmacological characterizations, as well as recent cloning of receptor cDNAs, have provided ample support for this hypothesis. The multiple 5-HT-receptor subtypes cloned to date are the largest of all known neurotransmitter receptor families. The 5-HT-receptor subtypes are expressed in distinct but often overlapping patterns (Palacios et al., 1990) and are coupled to different transmembrane-signaling mechanisms (see Table 111). Four 5-HT-receptor families with defined functions, 5-HT1 through 5-HT4, currently are recognized. The 5-HT1, 5-HT2, and 5-HT47 receptor families are members of the superfamily of G proteincoupled receptors with a predicted membrane topology composed of an extracellular N-terminal segment linked to an intracellular C terminus by seven transmembrane-spanning segments (see Chapters 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship between Drug Concentration and Effect and 12: Neurotransmission and the Central Nervous System). The 5-HT3 receptor, on the other hand, is a ligand-gated ion channel that gates Na+ and K+ and has a predicted membrane topology akin to that of the nicotinic cholinergic receptor (see Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia). History of 5-HT Receptor Subtypes Gaddum and Picarelli (1957) in a pioneering paper proposed the
existence of two 5-HT-receptor subtypes, which they termed M and Dreceptors.
M receptors were believed to be located on parasympathetic nerve endings,
controlling the release of acetylcholine, whereas D receptors were thought to
be located on smooth muscle. Although results of subsequent studies in both
the periphery and brain were consistent with the notion of multiple subtypes
of 5-HT receptor, the radioligand-binding studies of Peroutka and Snyder
(1979) provided the first definitive evidence for two distinct recognition
sites for 5-HT. 5-HT1 receptors had a high affinity for [3H]-5-HT,
while 5-HT2 receptors had a low affinity for [3H]-5-HT
and a high affinity for [3H]-spiperone. Subsequently, high
affinity for 5-HT was used as a primary criterion for classifying a receptor
subtype as a member of the 5-HT1 receptor family. This
classification strategy proved to be invalid; for example, a receptor expressed
in the choroid plexus was named the 5-HT1C receptor because it was
the third receptor shown to have a high affinity for 5-HT. However, based on
its pharmacological properties, second-messenger function, and deduced amino
acid sequence, the 5-HT1C receptor clearly belonged to the 5-HT2
receptor family and recently has been renamed the 5-HT2C receptor.
The current, widely accepted classification scheme (Hoyer et al., 1994)
proposes seven subfamilies of 5-HT receptors (see Table 111). It is
likely that further modifications of this scheme will be required. For
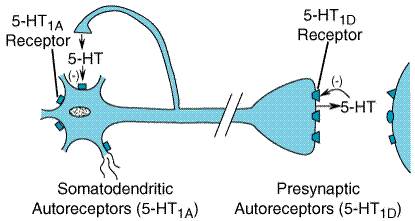
example, convincing evidence suggests that the 5-HT1D 5-HT1 Receptors All five members of the 5-HT1-receptor subfamily are negatively coupled to adenylyl cyclase. At least one 5-HT1-receptor subtype, the 5-HT1A receptor, also activates a receptor-operated K+ channel and inhibits a voltage-gated Ca2+ channel, a common property of receptors coupled to the pertussis toxinsensitive Gi/Go family of G proteins (Limbird, 1988). The 5-HT1A receptor is found in the raphe nuclei of the brainstem, where it functions as an inhibitory, somatodendritic autoreceptor on cell bodies of serotonergic neurons (Figure 113). Another subtype, the 5-HT1D receptor (and its rat homolog, 5-HT1B), functions as an autoreceptor on axon terminals, inhibiting 5-HT release. 5-HT1D receptors, abundantly expressed in the substantia nigra and basal ganglia, may regulate the firing rate of dopamine-containing cells and the release of dopamine at axonal terminals.
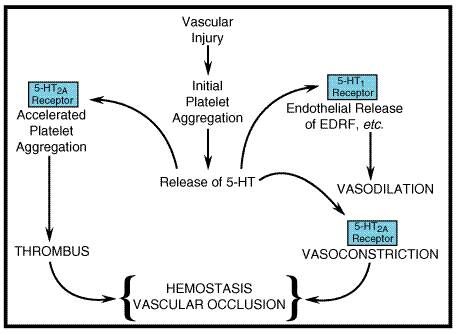
5-HT2 Receptors The three subtypes of 5-HT2 receptors are linked to phospholipase C with the generation of two second messengers, diacylglycerol (which activates protein kinase C) and inositol trisphosphate (which releases intracellular stores of Ca2+). The 5-HT2-receptor subtypes couple to pertussis toxininsensitive G proteins, such as Gq and G11. For the 5-HT2A-receptor subtype, however, coupling to pertussis toxinsensitive proteins (Gi/Go) versus insensitive proteins (Gq, and perhaps others) varies in different cellular preparations. 5-HT2A receptors are broadly distributed in the CNS, primarily in serotonergic terminal areas. High densities of 5-HT2A receptors are found in prefrontal cortex, claustrum, and platelets. 5-HT2A receptors in the gastrointestinal tract are thought to correspond to the D subtype of 5-HT receptor described by Gaddum and Picarelli (1957). 5-HT2B receptors originally were described in stomach fundus. The expression of 5-HT2B receptor mRNA is highly restricted in the CNS. 5-HT2C receptors have a very high density in the choroid plexus, an epithelial tissue that is the primary site of cerebrospinal fluid production. Despite the high density of 5-HT2C receptors in choroid plexus, the role of these receptors is unknown. The 5-HT2C receptor has been shown to be regulated by RNA editing, a posttranscriptional event that alters expression of the genetic code at the level of RNA (Burns et al., 1997). Multiple receptor isoforms with alterations of as many as three amino acids within the second intracellular loop are predicted, and these edited isoforms have modified G proteincoupling efficiency. 5-HT3 Receptors The 5-HT3 receptor is unique, being the only monoamine neurotransmitter receptor that is known to function as a ligand-operated ion channel. The 5-HT3 receptor corresponds to Gaddum and Picarelli's M receptor (Richardson et al., 1985). Activation of 5-HT3 receptors elicits a rapidly desensitizing depolarization mediated by the gating of cations. These receptors are located on parasympathetic terminals in the gastrointestinal tract, including vagal and splanchnic afferents. In the CNS, a high density of 5-HT3 receptors is found in the nucleus tractus solitarii and in the area postrema. 5-HT3 receptors in both the gastrointestinal tract and the CNS participate in the emetic response, providing an anatomical basis for the antiemetic property of 5-HT3-receptor antagonists. Most ligand-operated ion channels are composed of multiple subunits; however, the original, cloned 5-HT3-receptor subunit is able to form functional channels that gate cations when expressed in Xenopus oocytes or in cultured cells (Maricq et al., 1991). Nevertheless, extensive pharmacological and physiological evidence obtained in tissues and in intact animals clearly suggests the existence of multiple components of 5-HT3 receptors. Recently, splice variants of the 5-HT3 receptor have been identified, perhaps explaining the observed functional diversity. 5-HT4 Receptors 5-HT4 receptors are widely distributed throughout the body. In the CNS, the receptors are found on neurons of the superior and inferior colliculi and in the hippocampus. In the gastrointestinal tract, 5-HT4 receptors are located on neurons (e.g., myenteric plexus) as well as on smooth muscle and secretory cells. The 5-HT4 receptor is thought to evoke secretion in the alimentary tract and to facilitate the peristaltic reflex. 5-HT4 receptors activate adenylyl cyclase, leading to a rise in intracellular levels of cyclic AMP (Hegde and Eglen, 1996). The latter effect may explain the utility of prokinetic benzamides in gastrointestinal disorders (see Chapter 38: Prokinetic Agents, Antiemetics, and Agents Used in Irritable Bowel Syndrome). Additional Cloned 5-HT Receptors Two other cloned receptors, 5-HT6 and 5-HT7 receptors, are linked to activation of adenylyl cyclase. Multiple splice variants of the 5-HT7 receptor have been found, although functional distinctions are not clear. The absence of selective agonists and antagonists has foiled definitive studies of the role of the 5-HT6 and 5-HT7 receptors. Circumstantial evidence suggests that 5-HT7 receptors play a role in smooth muscle relaxation both in the gut and the vasculature. The atypical antipsychotic drug clozapine has a high affinity for 5-HT6 and 5-HT7 receptors. It remains to be determined whether or not this property is related to the broader effectiveness of clozapine compared to conventional antipsychotic drugs. Clozapine appears to be effective in many patients who do not respond to conventional antipsychotic drugs (see Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania). Two subtypes of the 5-HT5 receptor have been cloned; although the 5-HT5A receptor recently has been shown to inhibit adenylyl cyclase, functional coupling of the cloned 5-HT2B receptor has not yet been described. Sites of 5-HT Action 5-HT has a major role in the regulation of gastrointestinal motility; it is stored and secreted by enterochromaffin cells and by platelets. Although peripheral stores account for most of the 5-HT in the body, this monoamine also acts as a neurotransmitter in the CNS. Enterochromaffin Cells Enterochromaffin cells, identified histologically, are located in the gastrointestinal mucosa, with the highest density found in the duodenum. These cells synthesize 5-HT from tryptophan and store 5-HT and other autacoids, such as the vasodilator peptide substance P and other kinins. Basal release of enteric 5-HT is augmented by mechanical stretching, such as that caused by food or the administration of hypertonic saline, and also by efferent vagal stimulation. 5-HT probably has an additional role in stimulating motility via the myenteric network of neurons, located between the layers of smooth muscle (Gershon, 1991; see also Chapter 38: Prokinetic Agents, Antiemetics, and Agents Used in Irritable Bowel Syndrome). The greatly enhanced secretion of 5-HT and other autacoids in malignant carcinoid leads to a multitude of cardiovascular, gastrointestinal, and CNS abnormalities. In addition, the synthesis of large amounts of 5-HT by carcinoid tumors may result in tryptophan and niacin deficiencies (pellagra). Platelets Platelets differ from other formed elements of blood in expressing mechanisms for uptake, storage, and endocytotic release of 5-HT. 5-HT is not synthesized in platelets but is taken up from the circulation and stored in secretory granules by active transport, similar to the uptake and storage of norepinephrine by sympathetic nerve terminals (see Chapters 6 and 12). Thus, Na+-dependent transport across the surface membrane of platelets is followed by uptake into dense core granules via an electrochemical gradient generated by a H+-translocating ATPase. A gradient of 5-HT as high as 1000:1 with an internal concentration of 0.6 M in the dense core storage vesicles can be maintained by platelets. Measuring the rate of Na+-dependent 5-HT-uptake by platelets provides a sensitive assay for 5-HT-uptake inhibitors. The main function of platelets is to plug holes in injured endothelial cells. Conversely, the functional integrity of the endothelium is critical for the action of platelets (Furchgott and Vanhoutte, 1989). The endothelial surface is exposed constantly to platelets, because the shear forces of circulating blood favor centrifugal stratification of platelets (Gibbons and Dzau, 1994). Release of endothelium-derived relaxing factor (EDRF; nitric oxide and perhaps other components) antagonizes the vasoconstrictor action of thromboxane and 5-HT (Furchgott and Vanhoutte, 1989; Figure 114). The net effect of platelet aggregation is critically determined by the functional status of the endothelium (Hawiger, 1992; Ware and Heistad, 1993). When platelets make contact with injured endothelium, they release substances that promote platelet adhesion and release of 5-HT, including ADP, thrombin, and thromboxane A2 (see Chapters 26 and 55). 5-HT binding to platelets 5-HT2A receptors elicits a weak aggregation response that is markedly enhanced in the presence of collagen. If the damaged blood vessel is injured to a depth where vascular smooth muscle is exposed, 5-HT exerts a direct constrictor effect, thereby promoting hemostasis. Locally released autacoids (thromboxane A2, kinins, and vasoactive peptides) enhance this action. In atherosclerosis, thrombus formation is potentiated by the destruction of endothelium and, therefore, a deficiency of EDRF. An amplification cycle involving 5-HT in thrombus formation is induced. In addition to atherosclerosis, a cycle of this kind may be involved in other vascular diseases including Raynaud's phenomenon and coronary vasospasm.
Cardiovascular System The classical response of blood vessels to 5-HT is contraction, particularly in the splanchnic, renal, pulmonary, and cerebral vasculatures. This response also occurs in bronchial smooth muscle. 5-HT also induces a variety of responses by the heart that are the result of activation of 5-HT-receptor subtypes, stimulation or inhibition of autonomic activity, or dominance of reflex responses to 5-HT (Saxena and Villaln, 1990). Thus, 5-HT has positive inotropic and chronotropic actions on the heart that may be blunted by simultaneous stimulation of afferent nerves from baroreceptors and chemoreceptors. An effect on vagus nerve endings elicits the Bezold-Jarisch reflex, causing extreme bradycardia and hypotension. The local response of arterial blood vessels to 5-HT also may be inhibitory, the result of the release of EDRF and prostaglandins and blockade of norepinephrine release from sympathetic nerves. On the other hand, 5-HT amplifies the local constrictor action of norepinephrine, angiotensin II, and histamine, which reinforce the hemostatic response to 5-HT (see Gershon, 1991). Gastrointestinal Tract Enterochromaffin cells in the mucosa appear to be the location of the synthesis and most of the storage of 5-HT in the body and are the source of circulating 5-HT. 5-HT released from these cells enters the portal vein and is subsequently metabolized by MAO-A in the liver (Gillis, 1985). 5-HT that survives oxidation in the liver is rapidly removed by the endothelium of lung capillaries and then inactivated by MAO. 5-HT released by mechanical or vagal stimulation also acts locally to regulate gastrointestinal function. Motility of gastric and intestinal smooth muscle may be either enhanced or inhibited (Dhasmana et al., 1993) via at least six subtypes of 5-HT receptors (Table 112). The stimulatory response occurs at nerve endings on longitudinal and circular enteric muscle (5-HT4), at postsynaptic cells of the enteric ganglia (5-HT3 and 5-HT1P), and by direct effects of 5-HT on the smooth muscle cells (5-HT2A in intestine and 5-HT2B in stomach fundus). In esophagus, 5-HT acting at 5-HT4 receptors causes either relaxation or contraction, depending on the species. Abundant 5-HT3 receptors on vagal and other afferent neurons and on enterochromaffin cells play a pivotal role in emesis (Grunberg and Hesketh, 1993). Serotonergic terminals have been described in the myenteric plexus. Release of enteric 5-HT occurs in response to acetylcholine, noradrenergic nerve stimulation, increases in intraluminal pressure, and lowered pH (Gershon, 1991), triggering peristaltic contraction. Central Nervous System A multitude of brain functions are influenced by 5-HT, including sleep, cognition, sensory perception, motor activity, temperature regulation, nociception, appetite, sexual behavior, and hormone secretion. All of the cloned 5-HT receptors are expressed in the brain, often in overlapping areas. Although patterns of 5-HT receptor expression in individual neurons have not been defined, it is likely that multiple 5-HT receptor subtypes with similar or opposing actions are expressed in individual neurons, leading to a tremendous diversity of actions. The principal cell bodies of 5-HT neurons are located in raphe nuclei of the brainstem and project throughout the brain and spinal cord (see Chapter 12: Neurotransmission and the Central Nervous System). In addition to being released at discrete synapses, evidence suggests that release of serotonin also occurs at sites of axonal swelling, termed varicosities, which do not form distinct synaptic contacts (Descarries et al., 1990). 5-HT released at nonsynaptic varicosities is thought to diffuse to outlying targets, rather than acting on discrete synaptic targets. Such nonsynaptic release with an ensuing widespread influence of 5-HT is consistent with a longstanding belief that 5-HT acts as a neuromodulator as well as a neurotransmitter (see Chapter 12: Neurotransmission and the Central Nervous System). Serotonergic nerve terminals contain all of the proteins needed to synthesize 5-HT from L-tryptophan (see Figure 112). Newly formed 5-HT is rapidly accumulated in synaptic vesicles, where it is protected from MAO. 5-HT released by nerve-impulse flow is reaccumulated into the presynaptic terminal by a Na+-dependent carrier, the 5-HT transporter. Presynaptic reuptake is a highly efficient mechanism for terminating the action of 5-HT released by nerve-impulse flow. MAO localized in postsynaptic elements and surrounding cells rapidly inactivates 5-HT that escapes reuptake. Electrophysiology The physiological consequences of 5-HT release vary with the brain area and the neuronal element involved, as well as with the population of 5-HT receptor subtype(s) expressed (see Aghajanian, 1995). 5-HT has direct excitatory and inhibitory actions (Table 113), which may occur in the same preparation but with distinct temporal patterns. For example, in hippocampal neurons, 5-HT elicits hyperpolarization mediated by 5-HT1A receptors followed by a slow depolarization mediated by 5-HT4 receptors. 5-HT1A receptorinduced membrane hyperpolarization and reduction in input resistance is the result of an increase in K+ conductance. These ionic effects, which are blocked by pertussis toxin, are independent of cyclic AMP, suggesting that 5-HT1A receptors couple directly, via Gi-like G proteins, to receptor-operated K+ channels (Andrade et al., 1986). Somatodendritic 5-HT1A receptors on raphe cells also elicit a K+-dependent hyperpolarization. The G protein involved is pertussis toxinsensitive, but the K+ current apparently is different from the current elicited at postsynaptic 5-HT1A receptors in the hippocampus. The precise signaling mechanism involved in inhibition of 5-HT release by the 5-HT1D autoreceptor is not known, although inhibition of voltage-gated calcium channels likely contributes to the mechanism. Slow depolarization induced by 5-HT2A-receptor activation in areas such as the prefrontal cortex, nucleus accumbens, and facial motor nucleus involves a decrease in K+ conductance (Aghajanian et al., 1987). A second, distinct mechanism involving Ca2+-activated membrane currents enhances neuronal excitability and potentiates the response to excitatory signals such as glutamate. The role of the phosphoinositide hydrolysis signaling cascade in these physiological actions of 5-HT2A receptors has not been clearly defined. It appears that in areas where 5-HT1 and 5-HT2A receptors coexist, the effect of 5-HT reflects a combination of the two opposing responses, with a prominent 5-HT1 receptormediated hyperpolarization and an opposing 5-HT2A receptormediated depolarization. When 5-HT2A receptors are blocked, hyperpolarization is enhanced. In many cortical areas, 5-HT2A receptors are localized on GABAergic interneurons and on pyramidal cells. Activation of interneurons enhances GABA (gamma-aminobutyric acid) release, which secondarily slows the firing rate of pyramidal cells. Thus, there is the potential for the 5-HT2A receptor to regulate differentially cortical pyramidal cells, depending on the specific target cells (interneurons versus pyramidal cells). 5-HT2C receptors have been shown to depress a K+ current in Xenopus oocytes expressing the cloned receptor mRNA; a similar action has not been definitively identified in the brain. The 5-HT4 receptor, which is coupled to activation of adenylyl cyclase, also elicits a slow neuronal depolarization mediated by a decrease in K+ conductance. It is not clear why two distinct 5-HT receptor families linked to different signaling pathways are capable of eliciting a common neurophysiological action. Yet another receptor, the 5-HT1P receptor, elicits a slow depolarization. This receptor, which couples to activation of adenylyl cyclase, is restricted to the enteric nervous system and has a unique pharmacological profile (Gershon, 1991). The fast depolarization elicited by 5-HT3 receptors reflects direct gating of an ion channel intrinsic to the receptor structure itself. The 5-HT3 receptorinduced inward current has the characteristics of a cation-selective ligand-operated channel. Membrane depolarization is mediated by simultaneous increases in Na+ and K+ conductance (Higashi and Nishi, 1982). Patchclamp analyses confirmed that the 5-HT3 receptor functions as a receptorion channel complex, comparable to the nicotinic cholinergic receptor. 5-HT3 receptors have been characterized in the CNS and in sympathetic ganglia, primary afferent parasympathetic and sympathetic nerves, enteric neurons, and neuronally derived clonal cell lines, such as NG108-15 cells. The pharmacological properties of 5-HT3 receptors, which are different from those of other 5-HT receptors, suggest that multiple 5-HT3 receptor subtypes may exist and may correspond to different combinations of subunits (see Chapter 12: Neurotransmission and the Central Nervous System). Behavior The behavioral alterations elicited by drugs that interact with 5-HT receptors are extremely diverse. Many animal behavioral models for initial assessment of agonist and antagonist properties of drugs depend on aberrant motor or reflex responses, such as startle reflexes, hind-limb abduction, head twitches, and other stereotypical behaviors. Operant behavioral paradigms, such as drug discrimination, provide models of specific 5-HT receptor activation and are useful for exploring the action of CNS-active drugs, including agents that interact with 5-HT. For example, investigations of the mechanism of action of hallucinogenic drugs have relied heavily on drug discrimination (as discussed below). The following discussion focuses on animal models that may relate to pathological conditions in human beings and will not attempt to cover the voluminous literature dealing with 5-HT and behavior. see Glennon and Lucki, 1988; Zifa and Fillion, 1992; and Koek et al., 1992, for excellent reviews on this topic. Sleep-Wake Cycle Control of the sleep-wake cycle is one of the first behaviors in which a role for 5-HT was identified. Following the pioneering work in cats by Mouret et al. (1967), many studies showed that depletion of 5-HT with p-chlorophenylalanine elicited insomnia, which was reversed by administration of the 5-HT precursor 5-hydroxytryptophan. Conversely, treatment with L-tryptophan or with nonselective 5-HT agonists accelerated sleep onset and prolonged total sleep time. 5-HT antagonists were reported to both increase and decrease slow-wave sleep, probably reflecting interacting or opposing roles for subtypes of 5-HT receptors (for review, see Wauquier and Dugovic, 1990). One relatively consistent finding reported in human beings as well as in laboratory animals is an increase in slow-wave sleep following administration of a selective 5-HT2A/2C receptor antagonist such as ritanserin. Aggression and Impulsivity Results of studies in laboratory animals and in human beings suggest that 5-HT serves a critical role in aggression and impulsivity. Many human studies reveal a correlation between low cerebrospinal fluid 5-HIAA and violent impulsivity and aggression (Brown and Linnoila, 1990). As an example, low 5-HIAA is associated with violent suicide acts but not with suicidal ideation per se (Virkkunen et al., 1995). As with so many of the effects of 5-HT, pharmacological studies of aggressive behavior in laboratory animals have not been definitive, although a role for 5-HT is suggested. Two genetic studies have reinforced and amplified this notion. The 5-HT1B receptor was the first 5-HT receptor to be investigated using gene targeting via homologous recombination to eliminate the gene encoding the 5-HT1B receptor protein in mice (Saudou et al., 1994). These so-called 5-HT1B'knockout' mice develop extreme aggression, suggesting either a role for 5-HT1B receptors in the development of neuronal pathways important in aggression or a direct role in the mediation of aggressive behavior. A genetic study in human beings identified a point mutation in the gene coding for MAO-A that was associated with extreme aggressiveness and mental retardation (Brunner et al., 1993), and this has been confirmed in mutant mice lacking MAO-A (Cases et al., 1995). These genetic studies add credence to the proposition that abnormalities in 5-HT are correlated with aggressive behaviors. Anxiety and Depression The effects of 5-HT-active drugs, like the selective serotonin-reuptake inhibitors (SSRIs), in anxiety and depressive disorders strongly suggest an effect of 5-HT in the neurochemical mediation of these disorders. However, 5-HT-related drugs with clinical effects in anxiety and depression have varied effects in classical animal models of these disorders, depending on factors such as the experimental paradigm as well as animal species and strain. For example, the effective anxiolytic buspirone (BUSPAR, see Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders), a 5-HT1A-receptor partial agonist, does not reduce anxiety in classical approach-avoidance paradigms that were instrumental in development of anxiolytic benzodiazepines. However, buspirone and other 5-HT1A-receptor agonists are effective in other animal behavioral tests used to predict anxiolytic effects (Barrett and Vanover, 1993). Further, recent studies in 5-HT1A-receptor 'knockout' mice suggest a role for this receptor in anxiety and, possibly, depression (Parks et al., 1998; Ramboz et al., 1998). Agonists of certain 5-HT receptors, including 5-HT2A, 5-HT2C, and 5-HT3 receptors [e.g., m-chlorophenylpiperazine (mCPP)], have been shown to have anxiogenic properties in laboratory-animal and human studies. Similarly, these receptors have been implicated in the animal models of depression, such as learned helplessness. An impressive finding in human beings with depression is the abrupt reversal of the antidepressant effects of drugs, such as SSRIs, by manipulations that rapidly reduce the amount of 5-HT in brain. These approaches include administration of p-chlorophenylalanine or a tryptophan-free drink containing large quantities of neutral amino acids (Delgado et al., 1990). Curiously, this kind of 5-HT depletion has not been shown to worsen depression or induce depression in nondepressed subjects, suggesting that the continued presence of 5-HT is required to maintain the effects of these drugs. This clinical finding adds credence to somewhat less convincing neurochemical findings that suggest a role for 5-HT in the pathogenesis of depression. Pharmacological Manipulation of the Amount of 5-HT in Tissues Experimental strategies for evaluating the role of 5-HT depend on techniques that manipulate tissue levels of 5-HT or block 5-HT receptors. Until recently, manipulation of the levels of endogenous 5-HT was the most commonly used strategy, because the actions of 5-HT antagonists were poorly understood. Tryptophan hydroxylase, the rate-limiting enzyme in 5-HT synthesis, is a vulnerable site. A diet low in tryptophan reduces the concentration of brain 5-HT; conversely, ingestion of a tryptophan load increases levels of 5-HT in the brain. In addition, administration of a tryptophan hydroxylase inhibitor causes a profound depletion of 5-HT. The most widely used selective tryptophan hydroxylase inhibitor is p-chlorophenylalanine, which acts irreversibly. p-Chlorophenylalanine produces profound, long-lasting depletion of 5-HT levels with no change in levels of catecholamines. p-Chloroamphetamine
and other halogenated amphetamines promote 5-HT release from platelets and
neurons. A rapid release of 5-HT is followed by a prolonged and selective
depletion of 5-HT in brain. The halogenated amphetamines are valuable
experimental tools and two of them, fenfluramine and dexfenfluramine,
were used clinically to reduce appetite. These drugs were withdrawn from the Another highly specific mechanism for altering synaptic availability of 5-HT is inhibition of presynaptic reaccumulation of neuronally released 5-HT. SSRIs, such as fluoxetine (PROZAC), potentiate the action of 5-HT released by neuronal activity. When coadministered with L-5-hydroxytryptophan, SSRIs elicit a profound activation of serotonergic responses. SSRIs are one of the newest and most widely used treatments for endogenous depression (see Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders). Sibutramine (MERIDIA), an inhibitor of the reuptake of 5-HT, norepinephrine, and dopamine, is used as an appetite suppressant in the management of obesity. The drug is converted to two active metabolites that probably account for its therapeutic effects. Which neurotransmitter is primarily responsible for sibutramine's effects in obese patients is unclear. Nonselective treatments that alter 5-HT levels include MAO inhibitors and reserpine. MAO inhibitors block the principal route of degradation, thereby increasing levels of 5-HT, whereas reserpine treatment releases intraneuronal stores with subsequent depletion of 5-HT. These treatments profoundly alter levels of 5-HT throughout the body. However, because comparable changes occur in the levels of catecholamines, reserpine and MAO inhibitors are of limited utility as research tools. Both, at one time or another, have been useful in the treatment of mental diseases: reserpine as an antipsychotic drug (see Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania) and MAO inhibitors as antidepressants (see Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders). |
5-HT-Receptor Agonists and Antagonists
|
5-HT-Receptor Agonists Direct-acting 5-HT-receptor agonists have widely different chemical
structures, as well as diverse pharmacological properties (see Table
114). This diversity is not surprising in light of the number of
5-HT-receptor subtypes. 5-HT1A receptorselective agonists have
helped elucidate the functions of this receptor in the brain and have
resulted in a new class of antianxiety drugs including buspirone, gepirone,
and ipsaperone (see Chapter 19: Drugs and the Treatment of
Psychiatric Disorders: Depression and Anxiety Disorders). 5-HT1D
receptorselective agonists, such as sumatriptan, have unique
properties that result in constriction of intracranial blood vessels. Sumatriptan
was first in a series of new serotonin-receptor agonists available for
treatment of acute migraine attacks (see below). Other such agents now
FDA-approved in the 5-HT-Receptor Agonists and Migraine Migraine headache afflicts 10% to 20% of the population, producing a
morbidity estimated to be approximately sixty-four million workdays per year
in the The therapy of headaches classified as migraine is complicated by the variability of the responses among and within individual patients and by the lack of a firm experimental foundation of the pathophysiology of the syndrome. The efficacy of antimigraine drugs varies with the absence or presence of aura, duration of the headache, its severity and intensity, and as yet undefined environmental and genetic factors (Deleu et al., 1998). A rather vague and inconsistent pathophysiological characteristic of migraine is the spreading depression of neural impulses from a focal point of vasoconstriction followed by vasodilation (Olesen et al., 1981). However, it is unlikely that vasoconstriction followed by vasodilation (spreading depression) or vasodilation alone accounts for the local edema and focal tenderness often observed in migraine patients. Consistent with the hypothesis that 5-HT is a key mediator in the pathogenesis of migraine, 5-HT-receptor agonists have become the mainstay for acute treatment of migraine headaches. This hypothesis is based on evidence obtained in laboratory experiments and on the following evidence obtained in human beings: (1) Plasma and platelet concentrations of 5-HT vary with the different phases of the migraine attack. (2) Urinary concentrations of 5-HT and its metabolites are elevated during most migraine attacks. (3) Migraine may be precipitated by agents such as reserpine and fenfluramine that release biogenic amines, including serotonin, from intracellular storage sites. 5-HT1-Receptor Agonists: the Triptans The introduction of sumatriptan (IMITREX), zolmitriptan (ZOMIG), naratriptan (AMERGE), and rizatriptan (MAXALT and MAXALT-MLT) in the therapy of migraine has led to significant progress in preclinical and clinical research on migraine. At the scientific level, the selective pharmacological effects of these agents, dubbed the triptans, at 5-HT1 receptors have led to new insights into the pathophysiology of migraine. At the clinical level, the drugs are effective, acute antimigraine agents. Their ability to decrease, rather than exacerbate, the nausea and vomiting of migraine is an important advance in the treatment of the condition. History The development of sumatriptan was the first experimentally based
approach to identify and develop a novel therapy for migraine. In 1972, Humphrey
and colleagues initiated a long-term project aimed at identifying novel
therapeutic agents in the treatment of migraine (see Humphrey et
al., 1990). The goal of this project was to develop selective
vasoconstrictors of the extracranial circulation based on the theories of the
etiology of migraine prevalent in the early 1970s. Humphrey and his
colleagues focused on the identification of 5-HT receptors in the carotid
vasculature based on the evidence that the efficacy of traditional
antimigraine drugs such as ergotamine derived from their ability to induce
vasoconstriction of the carotid arteriovenous anastomoses, presumably via
their effects on 5-HT receptors (Saxena, 1978). The synthesis of many novel
tryptamine analogs was followed by determination of their actions on in
vitro vascular preparations and in intact animals. Sumatriptan, first
synthesized in 1984, potently contracted the dog isolated saphenous vein (Humphrey
et al., 1988), a vessel believed to contain the novel 5-HT receptor
located in the carotid circulation. Sumatriptan became available for clinical
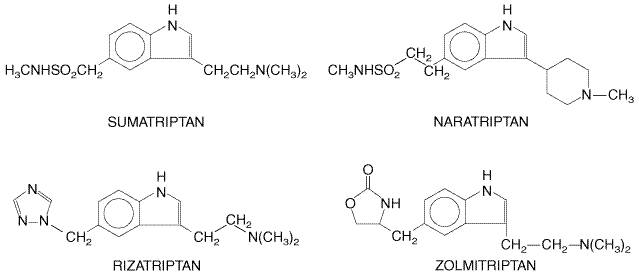
use in the Chemistry The triptans are derivatives of indole, with substituents on the 3 and 5 positions. Their structures are given in Figure 115.
Pharmacological Properties In contrast to ergot alkaloids (see below), the pharmacological
effects of the triptans appear to be limited to the 5-HT1 family
of receptors, providing evidence that this receptor subclass plays an
important role in the acute relief of a migraine attack. The triptans are
much more selective agents than are ergot alkaloids in that they interact
potently with 5-HT1D and 5-HT1B receptors and have a
low or no affinity for other subtypes of 5-HT receptors. The triptans are
essentially inactive at Mechanism of Action Two hypotheses have been proposed to explain the efficacy of 5-HT1B/1D receptor agonists in migraine. One hypothesis implicates the ability of these receptors to cause constriction of intracranial blood vessels including arteriovenous anastomoses. According to a prominent pathophysiological model of migraine, as yet unknown events lead to the abnormal dilation of carotid arteriovenous anastomoses in the head. As much as 80% of carotid arterial blood flow has been reported to be 'shunted'via these anastomoses, located mainly in the cranial skin and ears, diverting blood from the capillary beds, thus producing cerebral ischemia and hypoxia. Based on this model, an effective antimigraine agent would close the shunts and restore blood flow to the brain. Indeed, ergotamine, dihydroergotamine, and sumatriptan share the ability to produce this vascular effect with a pharmacological specificity that mirrors the effects of these agents on 5-HT1B and 5-HT1D receptor subtypes (den Boer et al., 1991). An alternative hypothesis concerning the significance of one or more 5-HT1 receptors in migraine pathophysiology relates to the observation that both 5-HT1B and 5-HT1D receptors serve as presynaptic autoreceptors, modulating neurotransmitter release from neuronal terminals (see Figure 113). 5-HT1 agonists may block the release of proinflammatory neuropeptides at the level of the nerve terminal in the perivascular space. Indeed, ergotamine, dihydroergotamine, and sumatriptan are able to block the development of neurogenic plasma extravasation in dura mater that follows depolarization of perivascular axons following capsaicin injection or unilateral electrical stimulation of the trigeminal nerve (Moskowitz, 1992). The ability of potent 5-HT1-receptor agonists to inhibit endogenous neurotransmitter release in the perivascular space could account for their efficacy in the acute treatment of migraine. Absorption, Fate, and Excretion When given subcutaneously, sumatriptan reaches its peak plasma concentration in approximately 12 minutes. Following oral administration, peak plasma concentrations occur within 1 to 2 hours. Bioavailability following the subcutaneous route of administration is approximately 97%; after oral administration or nasal spray, bioavailability is only 14% to 17%. The elimination half-life is approximately 1 to 2 hours. Sumatriptan is metabolized predominantly by MAO-A, and its metabolites are excreted in the urine. Zolmitriptan reaches its peak plasma concentration 1.5 to 2 hours after oral administration. Its bioavailability is about 40% following oral ingestion. Zolmitriptan is converted to an active N-desmethyl metabolite, which has severalfold higher affinity for 5-HT1B and 5-HT1D receptors than does the parent drug. Both the metabolite and the parent drug have half-lives of 2 to 3 hours. Naratriptan, administered orally, reaches its peak plasma concentration in 2 to 3 hours and has an absolute bioavailability of about 70%. It is the longest acting of the triptans, having a half-life of about 6 hours. Fifty percent of an administered dose of naratriptan is excreted unchanged in the urine, and about 30% is excreted as products of cytochrome P450 oxidation. Rizatriptan has an oral absolute bioavailability of about 45% and reaches peak plasma levels within 1 to 1.5 hours after oral ingestion of tablets of the drug. An orally disintegrating dosage form has a somewhat slower rate of absorption, yielding peak plasma levels of the drug 1.6 to 2.5 hours after administration. The principal route of metabolism of rizatriptan is via oxidative deamination by MAO-A. Plasma protein-binding of the triptans ranges from about 14% (sumatriptan, rizatriptan) to 30% (naratriptan). Adverse Effects and Contraindications Rare but serious cardiac events have been associated with the administration of 5-HT1 agonists, including coronary artery vasospasm, transient myocardial ischemia, atrial and ventricular arrhythmias, and myocardial infarction. Most such events have occurred in patients with risk factors for coronary artery disease. In general, however, only minor side effects are seen with the triptans in the acute treatment of migraine. As many as 83% of patients experience at least one side effect after subcutaneous injection of sumatriptan (Simmons and Blakeborough, 1994). After subcutaneous injection, a majority of patients report transient mild pain, stinging, or burning sensations at the site of injection. The most common side effect of sumatriptan nasal spray is a bitter taste. Orally administered triptans can cause paresthesia; asthenia and fatigue; flushing; feelings of pressure, tightness, or pain in the chest, neck, and jaw; drowsiness; dizziness; nausea; and sweating. The triptans are contraindicated in patients who have a history of ischemic or vasospastic coronary artery disease, cerebrovascular or peripheral vascular disease, or other significant cardiovascular diseases. These drugs also are contraindicated in patients with uncontrolled hypertension. Naratriptan is contraindicated in patients with severe renal or hepatic impairment. Rizatriptan should be used with caution in patients with renal or hepatic disease, but it is not contraindicated in such patients. Sumatriptan, rizatriptan, and zolmitriptan are contraindicated in patients who are taking monoamine oxidase inhibitors. Use in Treatment of Migraine The triptans are effective in the acute treatment of migraine (with or without aura), but are not intended for use in prophylaxis of migraine. Treatment with these agents should begin as soon as possible after onset of a migraine attack. Oral dosage forms of the triptans are the most convenient to use, but they may not be practical in patients experiencing nausea and vomiting with migraine attack. Approximately 70% of individuals report significant headache relief from a subcutaneous dose of sumatriptan. This dose may be repeated once within a 24-hour period if the first dose does not relieve the headache. An oral formulation and a nasal spray of sumatriptan also are available. The onset of action is as early as 15 minutes with the nasal spray. The recommended oral dose of sumatriptan is 25 to 100 mg, which may be repeated after 2 hours up to a total dose of 200 mg over a 24-hour period. When administered by nasal spray, from 5 to 20 mg of sumatriptan is recommended. The dose can be repeated after 2 hours up to a maximum dose of 40 mg over a 24-hour period. Zolmitriptan is given orally in a 1.25- to 2.5-mg dose, which can be repeated after 2 hours, up to a maximum dose of 10 mg over 24 hours, if the migraine attack persists. Naratriptan is given orally in a 1 to 2.5-mg dose, which should not be repeated until 4 hours after the previous dose. The maximum dose over a 24-hour period should not exceed 5 mg. The recommended oral dose of rizatriptan is 5 to 10 mg. The dose can be repeated after 2 hours up to a maximum dose of 30 mg over a 24-hour period. The safety of treating more than 3 or 4 headaches over a 30-day period with triptans has not been established. Because triptans may cause an acute, although usually small, increase in blood pressure, they should not be given to individuals with uncontrolled hypertension. Triptans should not be used concurrently with (or within 24 hours of) an ergot derivative (see below) nor should more than one triptan be used concurrently or within 24 hours of each other. Ergot and the Ergot Alkaloids The dramatic effect of ergot ingested during pregnancy has been recognized for more than 2000 years. Early in the 20th century, the isolation and chemical identification of the active principles of ergot were accomplished, and detailed study of their biological activity was begun. The elucidation of the constituents of ergot and their complex actions was an important chapter in the evolution of modern pharmacology. The ergot alkaloids are therefore discussed here, even though the very complexity of their actions limits their therapeutic uses (Table 115). The pharmacological effects of the ergot alkaloids are varied and complex; however, in general, the effects result from their actions as partial agonists or antagonists at adrenergic, dopaminergic, and serotonergic receptors (see also Chapter 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists). The spectrum of effects depends on the agent, dosage, species, tissue, physiological and endocrinological state, and experimental conditions. History Ergot is the product of a fungus (Claviceps purpurea) that grows on rye and other grains. The contamination of an edible grain by a poisonous, parasitic fungus spread death for centuries. As early as 600 B C., an Assyrian tablet alluded to a 'noxious pustule in the ear of grain.' Written descriptions of ergot poisoning first appeared in the Middle Ages. Strange epidemics were described in which the characteristic symptom was gangrene of the feet, legs, hands, and arms. In severe cases, the tissue became dry and black and mummified limbs separated off without loss of blood. Limbs were said to be consumed by the holy fire, blackened like charcoal with agonizing burning sensations. The disease was called holy fire or St. Anthony's fire in honor of the saint at whose shrine relief was said to be obtained. The relief that followed migration to the shrine of St. Anthony was probably real, for the sufferers received a diet free of contaminated grain during their sojourn at the shrine. The symptoms of ergot poisoning were not restricted to limbs. A frequent complication of ergot poisoning was abortion. Indeed, ergot was known as an obstetrical herb before it was identified as the cause of St. Anthony's fire. Chemistry The ergot alkaloids can all be considered to be derivatives of the
tetracyclic compound 6-methylergoline (Table 116). The naturally occurring
alkaloids contain a substituent in the beta configurations at position 8 and
a double bond in ring D. The natural alkaloids of therapeutic interest are
amide derivatives of d-lysergic acid. The first pure ergot alkaloid, ergotamine,
was obtained in 1920, followed by the isolation of ergonovine in 1932.
Numerous semisynthetic derivatives of the ergot alkaloids have been prepared
by catalytic hydrogenation of the natural alkaloids, e.g., dihydroergotamine.
Another synthetic derivative, bromocriptine (2-bromo- Absorption, Fate, and Excretion The pharmacokinetic properties of the ergot alkaloids have been reviewed by Perrin (1985). The oral administration of ergotamine by itself results in undetectable systemic drug concentrations, because of extensive first-pass metabolism. Bioavailability after sublingual administration also is poor and often is inadequate for therapeutic purposes. Although the concurrent administration of caffeine is said to improve both the rate and extent of absorption, the bioavailability of ergotamine still is probably less than 1%. The bioavailability after administration of rectal suppositories is greater. Ergotamine is metabolized in the liver by largely undefined pathways, and 90% of the metabolites are excreted in the bile. Only traces of unmetabolized drug can be found in urine and feces. Ergotamine produces vasoconstriction that endures for 24 hours or longer, despite a plasma half-life of approximately 2 hours. Dihydroergotamine is much less completely absorbed and is eliminated more rapidly than ergotamine, presumably due to its rapid hepatic clearance. Ergonovine and methylergonovine are rapidly absorbed after oral administration and reach peak concentrations in plasma within 60 to 90 minutes that are more than tenfold those achieved with an equivalent dose of ergotamine. An uterotonic effect can be observed within 10 minutes after oral administration of 0.2 mg of ergonovine to women postpartum. Judging from the relative duration of action, ergonovine is metabolized and/or eliminated more rapidly than is ergotamine. The half-life of methylergonovine in plasma ranges between 0.5 and 2 hours. Use in the Treatment of Migraine Ergot derivatives were first found to be effective antimigraine agents
in the 1920s, and they continue to be a class of therapeutic agents used for
the acute relief of migraine; however, ergot alkaloids are nonselective
pharmacological agents in that they interact with numerous neurotransmitter
receptors, including 5-HT1 and 5-HT2 receptors as well
as adrenergic and dopaminergic receptors. For example, the ergot alkaloid dihydroergotamine
can compete potently for radioligand binding to a variety of receptor
subpopulations, including all known 5-HT1 receptors as well as a
number of other biogenic amine receptors, such as 5-HT2A, 5-HT2B,
D2 dopamine, and The use of ergot alkaloids for migraine should be restricted to
patients having frequent, moderate migraine or infrequent, severe migraine
attacks. As with other medications used to abort an attack, the patient
should be advised to take ergot preparations as soon as possible after the
onset of a headache. Gastrointestinal absorption of ergot alkaloids is
erratic, perhaps explaining the large variation in patient response to these
drugs. Accordingly, currently available preparations in the Adverse Effects and Contraindications Nausea and vomiting, due to a direct effect on CNS emetic centers, occur in approximately 10% of patients after oral administration of ergotamine and in about twice that number after parental administration. This side effect is problematic, since nausea and sometimes vomiting are part of the symptomatology of a migraine headache. Leg weakness is common, and muscle pains, which occasionally are severe, may occur in the extremities. Numbness and tingling of fingers and toes are other reminders of the ergotism that this alkaloid may cause. Precordial distress and pain suggestive of angina pectoris, as well as transient tachycardia or bradycardia, also have been noted, presumably as a result of coronary vasospasm induced by ergotamine. Localized edema and itching may occur in an occasional hypersensitive patient, but usually do not necessitate interruption of ergotamine therapy. In the event of acute or chronic poisoning (ergotism), treatment consists of complete withdrawal of the offending drug and symptomatic measures. The latter include attempts to maintain adequate circulation by agents such as anticoagulants, low-molecular-weight dextran, and potent vasodilator drugs, such as intravenous sodium nitroprusside. Dihydroergotamine has lower potency than does ergotamine as an emetic and as a vasoconstrictor and oxytocic. Ergot alkaloids are contraindicated in women who are or may become pregnant, because the drugs may cause fetal distress and miscarriage. Ergot alkaloids also are contraindicated in patients with peripheral vascular disease, coronary heart disease, hypertension, impaired hepatic or renal function, and sepsis. Ergot alkaloids should not be taken within 24 hours of the use of the triptans and should not be used concurrently with other drugs that can cause vasocontriction. Use of Ergot Alkaloids in Postpartum Hemorrhage All of the natural ergot alkaloids markedly increase the motor activity of the uterus. After small doses, contractions are increased in force or frequency, or both, but are followed by a normal degree of relaxation. As the dose is increased, contractions become more forceful and prolonged, resting tonus is dramatically increased, and sustained contracture can result. Although this characteristic precludes their use for induction or facilitation of labor, it is quite compatible with their use postpartum or after abortion to control bleeding and maintain uterine contraction. The gravid uterus is very sensitive, and small doses of ergot alkaloids can be given immediately postpartum to obtain a marked uterine response, usually without significant side effects. In current obstetric practice, ergot alkaloids are used primarily to prevent postpartum hemorrhage. Although all natural ergot alkaloids have qualitatively the same effect on the uterus, ergonovine is the most active and also less toxic than ergotamine. For these reasons ergonovine and its semisynthetic derivative methylergonovine have replaced other ergot preparations as uterine-stimulating agents in obstetrics. D-Lysergic Acid Diethylamide (LSD) Of the many drugs that are nonselective 5-HT agonists, LSD is the most
remarkable. This ergot derivative profoundly alters human behavior, eliciting
perception disturbances, such as sensory distortion (especially visual and
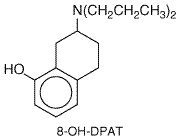
auditory), and hallucinations at doses as low as 1 LSD was synthesized in 1943 by Albert Hoffman, who discovered its unique properties when he accidentally ingested the drug. The chemical precursor, lysergic acid, occurs naturally in a fungus that grows on wheat and rye, but it is devoid of hallucinogenic actions. LSD contains an indolealkylamine moiety embedded within its structure, and early investigators postulated that it would interact with 5-HT receptors. Extensive studies have shown that LSD interacts with brain 5-HT receptors as an agonist/partial agonist. LSD mimics 5-HT at 5-HT1A autoreceptors on raphe cell bodies, producing a marked slowing of the firing rate of serotonergic neurons. In the raphe, LSD and 5-HT are equieffective; however, in areas of serotonergic axonal projections (such as visual relay centers), LSD is far less effective than is 5-HT (Aghajanian et al., 1987). This nonuniform action in cell body and target areas may explain the abnormal visual responses that LSD produces. In drug discrimination, an animal behavioral model thought to reflect the subjective effects of abused drugs, the discriminative stimulus effects of LSD and other hallucinogenic drugs appear to be mediated by activation of 5-HT2A receptors (Glennon, 1990). Consistent with these behavioral results, analyses of receptor-linked phosphoinositide hydrolysis show that LSD and other hallucinogenic drugs act as partial or full agonists at 5-HT2A and 5-HT2C receptors. An important unanswered question is whether or not the agonist property of hallucinogenic drugs at 5-HT2C receptors contributes to the behavioral alterations. LSD also interacts potently with many other 5-HT receptors, including recently cloned receptors whose functions have not yet been determined. The hallucinogenic phenethylamine derivatives such as 1-(4-bromo-2,5-dimethoxyphenyl)-2-aminopropane, on the other hand, are selective 5-HT2A/2C-receptor agonists. Promising signs of progress in understanding the actions of hallucinogens are the results of clinical investigations of hallucinogens. It is now possible to test in human beings the hypotheses developed in animal models. For example, PET imaging studies (Vollenweider et al., 1997) revealed that administration of the hallucinogen psilocybin mimics the pattern of brain activation found in schizophrenic patients experiencing hallucinations. Consistent with results of animal studies, this action of psilocybin is blocked by pretreatment with 5-HT2A/2C antagonists (Vollenweider et al., 1998). 8-Hydroxy-(2-N,N-Dipropylamino)-Tetraline (8-OH-DPAT) This prototypic selective 5-HT1A receptor agonist is a valuable experimental tool. The structure of 8-OH-DPAT is given below.
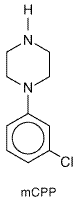
8-OH-DPAT does not interact with other members of the 5-HT1 receptor subfamily or with 5-HT2, 5-HT3, or 5-HT4 receptors. 8-OH-DPAT reduces the firing rate of raphe cells by activating 5-HT1A autoreceptors and inhibits neuronal firing in terminal fields (e.g., hippocampus) by direct interaction with postsynaptic 5-HT1A receptors. A series of long-chain arylpiperazines, such as buspirone, gepirone, and ipsapirone, are selective partial agonists at 5-HT1A receptors. Other closely related arylpiperazines act as 5-HT1A-receptor antagonists. Buspirone, the first clinically available drug in this series, has been effective in the treatment of anxiety (see Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders). It has been postulated that the sedative properties of the benzodiazepines, which buspirone does not have, may explain why patients usually prefer the benzodiazepines to relieve anxiety. Other arylpiperazines (gepirone and ipsapirone) are being developed for treatment of depression as well as anxiety. m-Chlorophenylpiperazine (mCPP) The in vivo actions of mCPP primarily reflect activation of 5-HT1B and/or 5-HT2A/2C receptors, although this agent is not subtype-selective in radioligand-binding studies in vitro. mCPP (structure below) is an active metabolite of the antidepressant drug trazodone (DESYREL
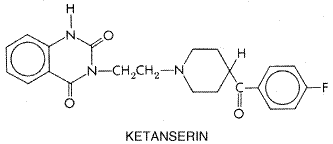
mCPP has been extensively employed to probe brain 5-HT function in human beings. The drug alters a number of neuroendocrine parameters and elicits profound behavioral effects, with anxiety as a prominent symptom (Murphy, 1990). mCPP elevates cortisol and prolactin secretion, probably via a combination of 5-HT1-and 5-HT2A/2C-receptor activation. It also increases growth-hormone secretion, apparently by a 5-HT-independent mechanism. 5-HT2A/2C receptors appear to mediate at least part of the anxiogenic effects of mCPP, as 5-HT2A/2C-receptor antagonists attenuate mCPP-induced anxiety. Animal studies suggest a greater involvement of the 5-HT2C receptor in anxiogenic actions of mCPP. 5-HT-Receptor Antagonists The properties of 5-HT-receptor antagonists also vary widely. Ergot alkaloids and related compounds tend to be nonspecific 5-HT-receptor antagonists; however, a few ergot derivatives such as metergoline bind preferentially to members of the 5-HT2-receptor family. A number of selective antagonists for 5-HT2A/2C and 5-HT3 receptors are currently available. Members of these drug classes have widely different chemical structures, with no common structural motif predictably conveying high affinity. Ketanserin is the prototypic 5-HT2A-receptor antagonist (see below). A large series of 5-HT3-receptor antagonists are being explored for treatment of various gastrointestinal disturbances (see Chapter 38: Prokinetic Agents, Antiemetics, and Agents Used in Irritable Bowel Syndrome). Ondansetron (ZOFRAN), dolasetron (ANZEMET), and granisetron (KYTRIL), all 5-HT3-receptor antagonists, have proven to be highly efficacious in the treatment of chemotherapy-induced nausea (Grunberg and Hesketh, 1993; see also Chapter 38: Prokinetic Agents, Antiemetics, and Agents Used in Irritable Bowel Syndrome). Clinical effects of 5-HT-related drugs often exhibit a significant delay in onset. This is particularly the case with drugs used to treat affective disorders such as anxiety and depression (see Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders). This delayed onset has generated considerable interest in potential adaptive changes in 5-HT-receptor density and sensitivity after chronic drug treatments. Laboratory studies have documented agonist-promoted receptor subsensitivity and down-regulation of the 5-HT-receptor subtypes, a compensatory response common to many neurotransmitter systems. However, an unusual adaptive process, antagonist-induced down-regulation of 5-HT2C receptors, takes place in rats and mice after chronic treatment with receptor antagonists (Sanders-Bush, 1990). The mechanism of this paradoxical regulation of 5-HT2A/2C receptors has generated considerable interest, since many clinically effective drugs, including clozapine, ketanserin, and amitriptyline, exhibit this unusual property. These drugs, as well as several other 5-HT2A/2C-receptor antagonists, possess negative intrinsic activity, reducing constitutive (spontaneous) receptor activity in a cell line expressing the 5-HT2C-receptor cDNA (Barker et al., 1994). This property of negative intrinsic activity is contrary to classical concepts, where receptor antagonists are thought to block the action of an agonist but have no effect alone. Another group of 5-HT2A/2C-receptor antagonists was found to act in the classical manner. It is not known if these differences in the properties of 5-HT2A/2C-receptor antagonists are clinically significant. Ketanserin Ketanserin SUFREXAL) (structure below) opened a new
era in 5-HT-receptor pharmacology. Ketanserin potently blocks 5-HT2A
receptors, less potently blocks 5-HT2C receptors, and has no
significant effect on 5-HT3 or 5-HT4 receptors or any
members of the 5-HT1-receptor family. It is important to note,
however, that ketanserin also has high affinity for
Ketanserin lowers blood pressure in patients with hypertension,
causing a reduction comparable to that seen with Chemical relatives of ketanserin such as ritanserin are more
selective 5-HT2A-receptor antagonists with low affinity for Atypical Antipsychotic Drugs Clozapine CLOZARIL), a 5-HT2A/2C-receptor antagonist, represents a new class of atypical antipsychotic drugs with reduced incidence of extrapyramidal side effects, compared to the classical neuroleptics, and a greater efficacy for reducing negative symptoms of schizophrenia (see Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania). Clozapine also has a high affinity for subtypes of dopamine receptors. One of the newest strategies for the design of additional atypical antipsychotic drugs is to combine 5-HT2A/2C and dopamine D2 receptorblocking actions in the same molecule (Leysen et al., 1993). Risperidone (RISPERDAL), for example, is a potent 5-HT2A-and D2-receptor antagonist. Low doses of risperidone have been reported to attenuate negative symptoms of schizophrenia with a low incidence of extrapyramidal side effects. Extrapyramidal effects are commonly seen, however, with doses of risperidone in excess of 6 mg/day. Other atypical antipsychotic agentsquetiapine (SEROQUEL) and olanzapine (ZYPREXA)act on multiple receptors, but their antipsychotic effects are thought to be due to antagonism of dopamine and serotonin. Methysergide Methysergide SANSERT; 1-methyl-d-lysergic acid butanolamide) is a congener of methylergonovine (see Table 116). Methysergide blocks 5-HT2A and 5-HT2C receptors but appears to have partial agonist activity in some preparations. Methysergide inhibits the vasoconstrictor and pressor effects of 5-HT as well as the actions of 5-HT on various types of extravascular smooth muscle. It has been found to both block and mimic the central effects of 5-HT. Methysergide is not selective (it also interacts with 5-HT1 receptors), but its therapeutic effects appear primarily to reflect blockade of 5-HT2 receptors. Although methysergide is an ergot derivative, it has only weak vasoconstrictor and oxytocic activity. Methysergide has been used for the prophylactic treatment of migraine and other vascular headaches, including Horton's syndrome. It is without benefit when given during an acute migraine attack. The protective effect takes 1 to 2 days to develop and disappears slowly when treatment is terminated. This might be due to the accumulation of an active metabolite of methysergide, methylergometrine, which is more potent than the parent drug. Methysergide also has been used to combat diarrhea and malabsorption in patients with carcinoid tumors and may be beneficial in the postgastrectomy dumping syndrome. Both of these conditions have a 5-HTmediated component. However, methysergide is not effective against other substances (e.g., kinins) that also are released by carcinoid tumors. For this reason, the preferred agent to treat malabsorption in carcinoid patients is a somatostatin analog, octreotide acetate (SANDOSTATIN), which inhibits the secretion of all the mediators released by the carcinoid tumors. Side effects of methysergide are usually mild and transient, although
drug withdrawal is infrequently required to reverse more severe reactions.
Common side effects consist of gastrointestinal disturbances, including
heartburn, diarrhea, cramps, nausea, and vomiting, and symptoms related to vasospasm-induced
ischemia (numbness and tingling of extremities, pain in the extremities, low
back and abdominal pain). Effects attributable to central actions include
unsteadiness, drowsiness, weakness, lightheadedness, nervousness, insomnia,
confusion, excitement, hallucinations, and even frank psychotic episodes.
Reactions suggestive of vascular insufficiency have been observed in a few
patients, as well as exacerbation of angina pectoris. A potentially serious
complication of prolonged treatment is inflammatory fibrosis, giving rise to
various syndromes, including retroperitoneal fibrosis, pleuropulmonary
fibrosis, and coronary and endocardial fibrosis. Usually the fibrosis
regresses after drug withdrawal, although persistent cardiac valvular damage
has been reported. Because of this danger, other drugs are preferred for the
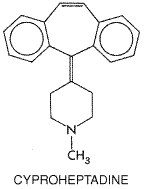
prophylactic treatment of migraine. Cyproheptadine The structure of cyproheptadine (PERIACTIN; see below) resembles that of the phenothiazine histamine H1-receptor antagonists, and, indeed, it is an effective H1-receptor antagonist. Cyproheptadine also has prominent 5-HT-blocking activity on smooth muscle by virtue of its binding to 5-HT2A receptors. In addition, it has weak anticholinergic activity and possesses mild central depressant properties.
Cyproheptadine shares the properties and uses of other H1-receptor antagonists (see Chapter 25: Histamine, Bradykinin, and Their Antagonists). It is effective in controlling skin allergies, particularly the accompanying pruritus, and appears to be useful in cold urticaria. In allergic conditions, the action of cyproheptadine as a 5-HT-receptor antagonist is irrelevant, since 5-HT2A receptors are not involved in human allergic responses. Some physicians recommend cyproheptadine to counteract the sexual side effects of selective 5-HT-reuptake inhibitors such as fluoxetine and sertraline (see Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders). The 5-HT-blocking actions of cyproheptadine explain its value in the postgastrectomy dumping syndrome, intestinal hypermotility of carcinoid, and migraine prophylaxis. Cyproheptadine is not, however, a preferred treatment for these conditions. Side effects of cyproheptadine include those common to other H1-receptor antagonists, such as drowsiness. Weight gain and increased growth in children have been observed and attributed to an interference with regulation of the secretion of growth hormone. |
Prospectus
|
The availability of molecular reagents, such as cDNA clones coding for 5-HT-receptor subtypes and 5-HT-selective neurotransmitter transporters (Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders), as well as genetically altered mice, will enhance the development of more selective therapeutic agents. It is now known that 5-HT-receptor subtypes have varying degrees of constitutive/spontaneous activity. Moreover, 5-HT receptor antagonists exist that either simply block receptor occupancy by agonists (antagonists) or stabilize unproductive receptor conformations as well as block agonist occupancy (inverse agonists). Even though there is only scant evidence for constitutive activity in vivo, drug development has been further refined by focusing on reduction of preexisting constitutive neuronal activity as opposed to blockade of excess neurotransmitter action. Improved experimental models for behavioral dysfunctions of complex origin, such as anxiety, depression, aggression, compulsivity, and others, already have revealed therapeutic outcomes that can be achieved by simultaneously blocking multiple receptor populations. Development of animal models that reflect physiological mechanisms influencing, for example, sleep, sex, appetite, emotions, sensory and pain perception, motor control, and digestion in human beings should permit further elucidation of receptor subpopulations that can be targeted to relieve dysfunctions in these complex processes. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3244
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved