| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Antihypertensive Agents and the Drug Therapy of Hypertension
Overview
|
Arterial pressure is the product of cardiac output and peripheral vascular resistance. Drugs lower pressure by actions on either the peripheral resistance or the cardiac output or both. The cardiac output may be reduced by drugs that either inhibit myocardial contractility or decrease ventricular filling pressure. Many of the antihypertensive drugs that affect adrenergic receptors, the renin-angiotensin system, Ca2+channels, and Na+ and water balance are also discussed in Chapters 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia, 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists, 29: Diuretics, 31: Renin and Angiotensin, 32: Drugs Used for the Treatment of Myocardial Ischemia, and 34: Pharmacological Treatment of Heart Failure. The pharmacology of antihypertensive agents that are not discussed elsewhere is presented here; in addition, the properties of all of the major drugs that are particularly relevant to their use in hypertension are reviewed, and an overview of the therapy of hypertension is provided. |
Antihypertensive Agents and the Drug Therapy of Hypertension: Introduction
|
Hypertension is the most common
cardiovascular disease. As many as 43 million adults in the Elevated arterial pressure causes pathological changes in the vasculature and hypertrophy of the left ventricle. As a consequence, hypertension is the principal cause of stroke, leads to disease of the coronary arteries with myocardial infarction and sudden cardiac death, and is a major contributor to cardiac failure, renal insufficiency, and dissecting aneurysm of the aorta. Hypertension is defined conventionally as blood pressure At very severe levels of hypertension (systolic The presence of certain target organ changes confers on a patient a worse prognosis than that for a patient with the same level of blood pressure lacking these findings. Thus, retinal hemorrhages, exudates, and discedema indicate a far worse short-term prognosis for a given level of blood pressure. Left ventricular hypertrophy defined by electrocardiogram, or more accurately by echocardiography, is associated with a substantially worse long-term outcome that includes a higher risk of sudden cardiac death. The risk of cardiovascular disease, disability, and death in hypertensive patients also is increased markedly by concomitant cigarette smoking and by elevated low-density lipoprotein; the coexistence of hypertension with these risk factors increases cardiovascular morbidity and mortality to an extent that is supraadditive. Robust evidence from multiple controlled trials indicates that pharmacological treatment of patients with diastolic pressures of 95 mm Hg or greater will reduce morbidity, disability, and mortality from cardiovascular disease. Effective antihypertensive therapy will almost completely prevent the hemorrhagic strokes, cardiac failure, and renal insufficiency due to hypertension. There is a marked reduction in total strokes. Moreover, several recent clinical trials suggest that reduction of diastolic blood pressure to 85 mm Hg confers a greater therapeutic benefit than reduction to 90 mm Hg, particularly in patients with diabetes (Hansson et al., 1998). The usual approach to patients with diastolic blood pressure in the range of 85 to 94 mm Hg is to use nonpharmacological therapy as an initial strategy. Because blood pressures in this range predict a clear increase in cardiovascular risk, the recommendations for nonpharmacological therapy should be accompanied by careful observation; in addition to the value of regular follow-up visits to maintain surveillance of blood pressure, they also afford an opportunity to assist and support patients in their efforts to achieve the changes in lifestyle required for effective nonpharmacological reduction in blood pressure. Antihypertensive drugs can be classified according to their sites or mechanisms of action (see Table 331). As arterial pressure is the product of cardiac output and peripheral vascular resistance, it can be lowered by actions of drugs on either the peripheral resistance or the cardiac output, or both. Drugs may reduce the cardiac output by either inhibiting myocardial contractility or decreasing ventricular filling pressure. Reduction in ventricular filling pressure may be achieved by actions on the venous tone or on blood volume via renal effects. Drugs can reduce peripheral resistance by acting on smooth muscle to cause relaxation of resistance vessels or by interfering with the activity of systems that produce constriction of resistance vessels (e.g., the sympathetic nervous system). The hemodynamic consequences of long-term treatment with antihypertensive agents are presented in Table 332, which also provides a framework for potential complementary effects of concurrent therapy with two or more drugs. The simultaneous use of drugs with similar mechanisms of action and hemodynamic effects often produces little additional benefit. However, concurrent use of drugs from different classes is a strategy for achieving effective control of blood pressure while minimizing dose-related adverse effects. |
Diuretics
|
One of the earliest strategies for the management of hypertension was to alter Na+ balance by restriction of salt in the diet. Pharmacological alteration of Na+ balance became practical in the 1950s with the development of the orally active thiazide diuretics (see Chapter 29: Diuretics). These and related diuretic agents have antihypertensive effects when used alone, and they enhance the efficacy of virtually all other antihypertensive drugs. The exact mechanism for reduction of arterial blood pressure by diuretics is not certain. Initially, the drugs decrease extracellular volume and cardiac output. However, the hypotensive effect is maintained during long-term therapy because of reduced vascular resistance; cardiac output returns to pretreatment values and extracellular volume remains somewhat reduced. Because of the persistent reduction in vascular resistance, some investigators have postulated that the diuretics have a direct effect on vascular smooth muscle that is independent of their saluretic effect. However, substantial data indicate that this is not the case. Thus, anephric patients and nephrectomized animals do not show a reduction in blood pressure when given diuretics (Bennett et al., 1977); a high salt intake or an infusion of saline (but not dextran) to counteract the net negative Na+ balance produced by diuretics reverses the antihypertensive effect; during effective therapy, plasma volume remains about 5% below pretreatment values and the plasma renin activity remains elevated, confirming a persistent reduction in body Na+ (Shah et al., 1978); diuretics do not relax vascular smooth muscle in vitro; and the hemodynamic effects of the diuretics to reduce vascular resistance are reproduced by restriction of salt (Freis, 1983). Potential mechanisms for reduction of vascular resistance by a persistent, albeit small, reduction in body Na+ include a decrease in interstitial fluid volume; a fall in smooth muscle Na+ concentration that may secondarily reduce the intracellular Ca2+ concentration, such that the cells are more resistant to contractile stimuli; and a change in the affinity and response of cell surface receptors to vasoconstrictor hormones (Insel and Motulsky, 1984). Benzothiadiazines and Related Compounds Benzothiadiazines ('thiazides') and related diuretics make
up the most frequently used class of antihypertensive agents in the Regimen for Administration of the Thiazide-Class Diuretics in Hypertension When a thiazide-class diuretic is utilized as the sole antihypertensive drug (monotherapy), it should be administered in a low dose. Further, there is mounting evidence that the administration of these diuretics in the long-term treatment of hypertension should be in conjunction with a K+-sparing agent. Antihypertensive effects can be achieved in many patients with as little as 12.5 mg of chlorthalidone (HYGROTON) or hydrochlorothiazide (HYDRODIURIL) daily. This should be the initial dose in most elderly patients who do not require urgent reduction of pressure. Furthermore, when used as monotherapy, the maximal daily dose of thiazide-class diuretics usually should not exceed 25 mg of hydrochlorothiazide or chlorthalidone (or equivalent). Even though more diuresis can be achieved with higher doses of these diuretics, abundant evidence indicates that doses higher than this are not required for monotherapy of hypertension and probably are not as safe. A large study comparing 25 and 50 mg hydrochlorothiazide daily in an elderly population did not show a greater decrease in blood pressure with the larger dose (MRC Working Party, 1987). In the randomized, controlled trials of antihypertensive therapy in the elderly (SHEP Cooperative Research Group, 1991; Dahlf et al., 1991; MRC Working Party, 1992) that demonstrate the best outcomes in cardiovascular morbidity and mortality, 25 mg of hydrochlorothiazide or chlorthalidone was the maximum dose given; if greater reduction of blood pressure than achieved with this dose was required, treatment with a second drug was initiated. With respect to safety, a case-control study (Siscovick et al., 1994) found a dose-dependent increase in the occurrence of sudden death at doses of hydrochlorothiazide greater than 25 mg daily. This finding supports the hypothesis engendered by a retrospective analysis of the Multiple Risk Factor Intervention Trial (Multiple Risk Factor Intervention Trial Research Group, 1982), suggesting that increased cardiovascular mortality is associated with higher diuretic doses. Further, the drug-specific metabolic effects of the thiazide-class diuretics, as well as the side effects perceived by patients, are to some degree dose-dependent, providing additional reasons not to administer more than the 25-mg dose of hydrochlorothiazide/chlorthalidone required to achieve nearly maximum blood pressure reduction. Taken together, clinical studies to date indicate that, if adequate blood pressure reduction is not achieved with the 25-mg daily dose of hydrochlorothiazide or chlorthalidone, a second drug should be added rather than increasing the dose of diuretic. When used in the treatment of hypertension, thiazide-class diuretics usually should be administered in conjunction with a K+-sparing agent. Attenuation of the kaliuretic effect of the thiazide-class diuretics can be achieved by drugs that block the Na+ channels in the late distal tubule and collecting duct (amiloride and triamterene), or by inhibition of aldosterone action (spironolactone). Oral K+ supplementation in the usual doses is not as effective as these K+-sparing agents. At this time, the data from clinical trials most strongly support the use of amiloride in combination with a thiazide-class diuretic. In the two large clinical trials that demonstrate the best results in terms of cardiovascular morbidity and mortality (Dahlf et al., 1991; MRC Working Party, 1992), amiloride was the drug employed together with hydrochlorothiazide, used in a ratio of 1 mg amiloride/10 mg hydrochlorothiazide. Angiotensin converting enzyme inhibitors and angiotensin receptor antagonists will attenuate diuretic-induced loss of potassium to some degree, and this is a consideration if a second drug is required to achieve further blood pressure reduction beyond that attained with the diuretic alone. Because the diuretic and hypotensive effects of these drugs are greatly enhanced when they are given in combination, care should be taken to initiate combination therapy with low doses of each of these drugs. Administration of angiotensin converting enzyme inhibitors or angiotensin receptor antagonists together with other K+-sparing agents or with K+ supplements requires caution; combination of K+-sparing agents with each other or with K+ supplementation can cause serious hyperkalemia in occasional patients. In contrast with the limitation on dose of thiazide-class diuretics used as monotherapy, the treatment of severe hypertension that is unresponsive to three or more drugs may require larger doses of the thiazide-class diuretics. Indeed, hypertensive patients may become refractory to drugs that block the sympathetic nervous system or to vasodilator drugs, because these drugs engender a state in which the blood pressure is very volume-dependent. Therefore, it is appropriate to consider the use of thiazide-class diuretics in doses of 50 mg of daily hydrochlorothiazide equivalent when treatment with appropriate combinations and doses of three or more drugs fails to yield adequate control of the blood pressure. Dietary Na+ restriction is a valuable adjunct to the management of such refractory patients and will minimize the dose of diuretic that is required. This can be achieved by a modest restriction of Na+ intake to 2 g daily. A more strict Na+ restriction is not feasible for most patients. Since the degree of K+ loss relates to the amount of Na+ delivered to the distal tubule, such restriction of Na+ can minimize the development of hypokalemia and alkalosis. The effectiveness of the thiazide class of drugs as diuretic or antihypertensive agents is progressively diminished when the glomerular filtration rate falls below 30 ml/min. One exception is metolazone, which retains efficacy in patients with this degree of renal insufficiency. Most patients will respond to the thiazide class of diuretics with a reduction in blood pressure within 2 to 4 weeks, although a minority will not achieve maximum reduction in arterial pressure for up to 12 weeks on a given dose. Therefore, doses should not be increased more often than every 2 to 4 weeks. Although the blood pressure of patients who have suppressed plasma renin activity is almost uniformly sensitive to diuretics in the thiazide class, most other patients also respond. There is no way to predict the antihypertensive response from the duration or severity of the hypertension in a given patient, although diuretics are unlikely to be effective as a sole therapy in patients with severe hypertension. Since the effect of the thiazide class of diuretics is additive with that of other antihypertensive drugs, combination regimens that include these diuretics are common and rational. Diuretics also have the advantage of minimizing the retention of salt and water that is commonly caused by vasodilators and some sympatholytic drugs. Adverse Effects and Precautions The adverse effects of diuretics are discussed in Chapter 29: Diuretics. Some of these are effects that determine whether patients can tolerate and comply with diuretic treatment. Sexual impotence is the most common troublesome side effect of the thiazide-class diuretics, and physicians should inquire specifically regarding its occurrence in conjunction with treatment with these drugs. Gout may be a consequence of the hyperuricemia induced by these diuretics. The occurrence of either of these adverse effects is a reason for considering alternative approaches to therapy. Muscle cramps also are related to diuretic therapy in a dose-dependent manner. Other effects of the thiazide-class diuretics are laboratory observations that are of concern primarily because they are putative surrogate markers for adverse drug effects on morbidity and mortality. Surrogate markers are factors that, in epidemiological studies, have been found to correlate with disease outcomes and therefore are used in investigations of drug therapy as a surrogate of the actual effect of a drug on disease outcome. Thus, reduction in systolic blood pressure is an example of a surrogate marker for the reduction of stroke by antihypertensive therapy that has been extensively validated (SHEP Cooperative Research Group, 1991). In contrast, the epidemiological evidence linking frequent ventricular ectopic depolarizations to sudden cardiac death had suggested that these ventricular arrhythmias might be surrogate markers for a beneficial effect of antiarrhythmic drugs on sudden cardiac death. However, reduction of ventricular ectopic depolarizations and nonsustained ventricular tachycardia by the antiarrhythmic drugs encainide and flecainide was associated with an increase in sudden cardiac death, demonstrating the fallibility of using surrogate markers to predict the outcome of a specific pharmacological intervention (CAST Investigators, 1989). Accordingly, the effect of drugs on surrogate markers cannot be considered as convincing evidence; rather, surrogate markers form the basis for hypotheses that require testing with controlled clinical trials. Until such trials are carried out, however, the effect of drugs on surrogate markers that predict adverse outcomes do cause concern and influence decisions regarding dose. The effects of diuretic drugs on several surrogate markers for adverse outcomes merit consideration. The K+ depletion produced by thiazide-class diuretics is dose-dependent over a wide range of doses and is variable among individuals, such that a subset of patients may become substantially K+-depleted on diuretic drugs. Even small doses given chronically, however, lead to some K+ depletion. There are two types of ventricular arrhythmia that are thought to be enhanced by K+ depletion. One of these is polymorphic ventricular tachycardia (torsades de pointes), which is induced by a number of drugs, including quinidine. Such drug-induced polymorphic ventricular tachycardia is an arrhythmia initiated by abnormal ventricular repolarization, and it is markedly enhanced by drugs that produce K+ depletion, which elicits abnormal repolarization (see Chapter 35: Antiarrhythmic Drugs). Accordingly, thiazide diuretics should not be given together with drugs that can cause polymorphic ventricular tachycardia. The most important concern regarding K+ depletion is its
influence on ischemic ventricular fibrillation, the leading cause of sudden
cardiac death and a major contributor to cardiovascular mortality in treated
hypertensive patients. Studies in experimental animals have demonstrated that
K+ depletion lowers the threshold for electrically induced
ventricular fibrillation in the ischemic myocardium and also increases
spontaneous ischemic ventricular fibrillation (Curtis and Hearse, 1989; Yano et
al., 1989). A case-control study of hypertensive patients has found a
positive correlation between diuretic dose and sudden cardiac death and an
inverse correlation between the use of adjunctive K+-sparing
agents and sudden cardiac death (Siscovick et al., 1994). One
controlled clinical trial has demonstrated a significantly greater occurrence
of sudden cardiac death in patients treated with 50 mg of hydrochlorothiazide
daily in comparison with the The thiazide class of diuretics elevates the levels of low-density lipoprotein (LDL) and increases the ratio of LDL/high-density lipoprotein (HDL). A linkage between increased LDL and increased coronary heart disease has been demonstrated in epidemiological studies and in investigations of lipid-lowering drugs, justifying, the consideration that LDL is a surrogate marker for coronary heart disease morbidity and mortality. Whether the increase in the LDL/HDL ratio caused by diuretics is in fact predictive of increased coronary heart disease in patients on diuretics is not presently known, and until adequate morbidity and mortality trials comparing antihypertensive drugs are completed, the effect of the thiazide-class diuretics on plasma lipids can be a basis for concern but not for definitive recommendations regarding the choice of these drugs relative to other antihypertensive agents. In epidemiological studies, left ventricular hypertrophy is a powerful predictor of an increase in cardiac death in hypertensive patients. The thiazide-class diuretics are less effective in reducing left ventricular hypertrophy than are other antihypertensive drugs such as the angiotensin converting enzyme inhibitors (Dahlf et al., 1992). The thiazide-class diuretics increase glycosylated hemoglobin in patients with diabetes mellitus (Gall et al., 1992). This finding, taken together with evidence that angiotensin converting enzyme inhibitors delay the deterioration of renal function in diabetic patients, suggests that thiazide-class diuretics are not the first drugs of choice in the monotherapy of hypertensive patients with diabetes mellitus. All of the thiazide-like drugs cross the placenta, but they have not been found to have direct adverse effects on the fetus. However, if administration of a thiazide is begun during pregnancy, there is a risk of transient volume depletion that may result in placental hypoperfusion. Since the thiazides appear in breast milk, they should be avoided by nursing mothers. The Choice of a Thiazide-Type Diuretic as the Initial Drug in the Treatment of Hypertension Few issues in hypertension are more controversial than whether or not patients should be placed on a diuretic as the initial or only drug for the treatment of hypertension (Joint National Committee, 1997; Tobian et al., 1994). A definitive answer to this question awaits the completion of a large clinical trial (the ALLHAT trial) conducted by the National Institutes of Health comparing a thiazide-class diuretic with other antihypertensive drugs as the initial or only therapeutic agents. In the interim, decisions will be made based on interpretations of the available data. There are several general types of data, from which interpretations have been inferred, but none provides convincing evidence. One set of data is the group of metabolic effects of the thiazide-class of diuretics discussed above: the increase in LDL, depletion of K+, impairment of diabetes control, and a reduction of left ventricular hypertrophy that is less than achieved with other drugs. The case-control study demonstrating a dose-dependent linkage of these diuretics to sudden cardiac death strengthens the inference that K+ depletion is not benign (Siscovick et al., 1994). Another type of data is that obtained from the clinical trials that have established the benefit of antihypertensive therapy. In controlled clinical trials conducted before 1991 in predominantly middle-aged patients, the beneficial effect of antihypertensive therapy on coronary heart disease and cardiac death was found to be less than the effect on stroke. Long-term prospective observational studies predict that a decrease of 5 to 6 mm Hg in diastolic pressure should lead to a 35% to 40% reduction in stroke; indeed, an overview of 14 randomized trials of pharmacological therapy (Collins et al., 1990) reveals that the prevalence of fatal plus nonfatal stroke is lowered by 42% (p<0.0002) as a consequence of that change in diastolic pressure. Whereas the observational studies predict a 20% to 25% reduction in fatal plus nonfatal coronary heart disease with a difference of 5 to 6 mm Hg in diastolic pressure, reduction of pressure to this degree with antihypertensive treatment lowered total coronary heart disease by only 14% (p<0.01). Fatal stroke was lowered by 45% (p<0.0001), whereas fatal coronary heart disease was reduced by only 11% (not significant) during antihypertensive treatment. The failure of antihypertensive therapy to yield the expected reduction in coronary heart disease, and particularly coronary heart disease mortality, has led to considerable speculation, including the possibility that the diuretic drugs that were the primary agents used in these trials may impose adverse effects on coronary heart disease, including sudden cardiac death. These disappointing results in terms of coronary heart disease were obtained in trials on mostly middle-aged patients that predominantly employed hydrochlorothiazide or chlorthalidone in doses up to 50 mg without a K+-sparing agent. Subsequent to this meta-analysis of the 14 controlled trials, 2 trials
in elderly patients that used a low dose of hydrochlorothiazide (25 mg daily)
together with the K+-sparing agent amiloride demonstrated more
favorable trends in reducing coronary mortality, lowering it by 50% (Dahlf et
al., 1991) and 40% (MRC Working Party, 1992). In the latter study, the
reduction in stroke and coronary events was significant on diuretic therapy,
whereas no significant reductions in these endpoints were seen in a
comparison group randomized to the The following approach to selection of a diuretic as the initial drug is derived from the data above. In elderly patients (age 65 or greater), selection of a thiazide-class diuretic as the initial drug is rational if it is given in doses in the range of 12.5 to 25 mg of hydrochlorothiazide (or equivalent) daily and together with a K+-sparing agent. This is based on the high level of efficacy in the clinical trials with such regimens, the relatively low profile of side effects, and the fact that elderly patients are likely to have a good response to diuretic therapy. In patients under age 65, a greater individualization of choice of the
initial antihypertensive drug seems warranted. The earlier clinical trials
(conducted in this age group with high doses of the thiazide-class drug given
without K+-sparing agents) provide neither reassurance regarding
the thiazide-class diuretics nor rejection of them. Individualization of
initial drug choice can consider several factors. In diabetic patients,
angiotensin converting enzyme inhibitors attenuate the decline in renal
function, making these drugs preferable to diuretics, which may impair glucose
tolerance in diabetes. Other drugs, particularly the angiotensin converting
enzyme inhibitors, are more effective than diuretics in reducing left
ventricular mass in patients with left ventricular hypertrophy. Caucasian
race, age less than 65, and a high or normal renin status at baseline predict
an antihypertensive response to an angiotensin converting enzyme inhibitor or
a Other Diuretic Antihypertensive Agents The thiazide-type diuretics are more effective antihypertensive agents
than are the loop diuretics, such as furosemide and bumetanide, in patients
who have normal renal function (Ram et al., 1981). This differential
effect is most likely related to the short duration of action of loop
diuretics, such that a single daily dose does not cause a significant net
loss of Na+ for an entire 24-hour period. The spectacular efficacy
of the loop diuretics in producing a rapid and profound natriuresis is a
potential detriment for the treatment of hypertension. When a loop diuretic
is given twice daily, the acute diuresis can be excessive and lead to more
side effects than occur with a slower-acting, milder thiazide diuretic. The
loop diuretics produce hypercalciuria, rather than the hypocalciuria
associated with the thiazides. However, other metabolic consequences of the
thiazides are shared with the loop diuretics, including hypokalemia,
hyperuricemia, glucose intolerance, and potentially adverse effects on plasma
concentrations of lipids. Although spironolactone in doses up to 100 mg per day is equivalent to hydrochlorothiazide in its hypotensive effect (Jeunemaitre et al., 1988), higher doses produce an unacceptable incidence of side effects (Schrijver and Weinberger, 1979). Spironolactone may be particularly useful for individuals with clinically significant hyperuricemia, hypokalemia, or glucose intolerance, and it is the agent of choice for management of primary aldosteronism. In contrast to thiazide diuretics, spironolactone does not affect plasma concentrations of Ca2+ or glucose. The effects of spironolactone on plasma lipids have not been studied extensively, but data indicate that the changes in triglycerides, LDL cholesterol, and total cholesterol are less than those seen with the thiazides. However, spironolactone may decrease the concentration of HDL cholesterol (Falch and Schreiner, 1983). The other K+-sparing diuretics, triamterene and amiloride, are used primarily to reduce the kaliuresis and potentiate the hypotensive effect of a thiazide (DeCarvalho et al., 1980; Multicenter Diuretic Cooperative Study Group, 1981). These agents should be used cautiously with frequent measurements of K+ concentrations in plasma in patients predisposed to hyperkalemia. Patients taking spironolactone, amiloride, or triamterene should be cautioned regarding the possibility that concurrent use of K+-containing 'salt substitutes' could produce hyperkalemia. Renal insufficiency is a relative contraindication to the use of K+-sparing diuretics. Diuretic-Associated Drug Interactions Since the antihypertensive effects of diuretics are frequently
additive with those of other antihypertensive agents, a diuretic commonly is
used in combination with other drugs. As discussed above, the concurrent
administration of diuretics with quinidine and other drugs that cause
polymorphic ventricular tachycardia greatly increases the risk of this
drug-induced arrhythmia. The K+- and Mg2+-depleting
effects of the thiazide-like and loop diuretics also can potentiate
arrhythmias that arise from digitalis toxicity. Corticosteroids can amplify
the hypokalemia produced by the diuretics. All diuretics can decrease the
clearance of Li+, resulting in increased plasma concentrations of
Li+ and potential toxicity (Amdisen, 1982). Nonsteroidal
antiinflammatory drugs (see Chapter 27: Analgesic-Antipyretic and
Antiinflammatory Agents and Drugs Employed in the Treatment of Gout) that
inhibit the synthesis of prostaglandins reduce the antihypertensive effects
of diuretics. It is not known if this interaction is due to Na+
retention resulting from blockade of the natriuretic effect of the diuretic
by the antiinflammatory agent or is related to inhibition of vascular
synthesis of prostaglandins (Webster, 1985). Based on recent studies, it
would appear that the effects of selective COX-2 inhibitors on renal
prostaglandin synthesis and function are similar to those of the nonselective
nonsteroidal antiinflammatory drugs. Nonsteroidal antiinflammatory drugs, |
Sympatholytic Agents
|
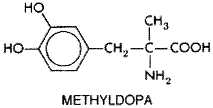
Since the demonstration in 1940 that bilateral excision of the thoracic sympathetic chain could lower blood pressure, the search for effective chemical sympatholytic agents has been intensive. Many compounds were tolerated poorly because they produced symptomatic orthostatic hypotension, sexual dysfunction, diarrhea, and fluid retention, with subsequent reduction of the antihypertensive effect. However, newer agents and rational combinations of these drugs with diuretics and vasodilators have overcome many of these difficulties. The subgroups of sympatholytic agents are shown in Table 331. Methyldopa Methyldopa ALDOMET) is a centrally acting antihypertensive agent. It is a prodrug that exerts its antihypertensive action via an active metabolite. Methyldopa (
Because A body of evidence supports the conclusion that methyldopa acts in the
brain via an active metabolite to lower blood pressure (Bobik et
al., 1988; Granata et al., 1986; Reid, 1986). In experimental
animals, the hypotensive effect of methyldopa is blocked by DOPA
decarboxylase inhibitors that have access to the brain, but not by inhibitors
that are excluded from the central nervous system (CNS). The hypotensive
effect also is abolished by inhibitors of dopamine Pharmacological Effects Methyldopa reduces vascular resistance without causing much change in cardiac output or heart rate in younger patients with uncomplicated essential hypertension. In older patients, however, cardiac output may be decreased as a result of a reduction in heart rate and stroke volume; this is secondary to relaxation of veins and a reduction in preload. The fall in arterial pressure is maximal 6 to 8 hours after an oral or intravenous dose. Although the decrease in supine blood pressure is less than that in the upright position, symptomatic orthostatic hypotension is less common with methyldopa than with drugs that act exclusively on peripheral adrenergic neurons or autonomic ganglia; this is because methyldopa attenuates but does not completely block baroreceptor-mediated vasoconstriction. For this reason, it is well tolerated during surgical anesthesia. Any severe hypotension is reversible with volume expansion. Renal blood flow is maintained and renal function is unchanged during treatment with methyldopa. Plasma concentrations of norepinephrine fall in association with the reduction in arterial pressure, and this reflects the decrease in sympathetic tone. Renin secretion also is reduced by methyldopa, but this is not a major effect of the drug and is not necessary for its hypotensive effects. Salt and water often are gradually retained with prolonged use of methyldopa, and this tends to blunt the antihypertensive effect. This has been termed 'pseudotolerance,' and it can be overcome with concurrent use of a diuretic. Of interest, treatment with methyldopa may reverse left ventricular hypertrophy within 12 weeks without any apparent relationship to the degree of change of arterial pressure (Fouad et al., 1982). Absorption, Metabolism, and Excretion Since methyldopa is a prodrug that is metabolized in the brain to the active form, its concentration in plasma has less relevance for its effects than is true for many other drugs. When administered orally, methyldopa is absorbed by an active amino acid transporter. Peak concentrations in plasma occur after 2 to 3 hours. The drug is distributed in a relatively small apparent volume (0.4 liter/kg) and is eliminated with a half-life of about 2 hours. The transport of methyldopa into the CNS is apparently also an active process (Bobik et al., 1986). Methyldopa is excreted in the urine primarily as the sulfate conjugate (50% to 70%) and as the parent drug (25%). The remaining fraction is excreted as other metabolites, including methyldopamine, methylnorepinephrine, and O-methylated products of these catecholamines (Campbell et al., 1985). The half-life of methyldopa is prolonged to 4 to 6 hours in patients with renal failure. In spite of its rapid absorption and short half-life, the peak effect of methyldopa is delayed for 6 to 8 hours, even after intravenous administration, and the duration of action of a single dose is usually about 24 hours; this permits once- or twice-daily dosing (Wright et al., 1982). The discrepancy between the effects of methyldopa and the measured concentrations of the drug in plasma is most likely related to the time required for transport into the CNS, conversion to the active metabolites, and accumulation of these metabolites in central adrenergic neurons. Patients with renal failure are more sensitive to the antihypertensive effect of methyldopa, but it is not known if this is due to alteration in excretion of the drug or to an increase in transport into the CNS. Adverse Effects and Precautions In addition to lowering blood pressure, the active metabolites of methyldopa
act on Methyldopa also produces some adverse effects that are not related to its pharmacological action. Hepatotoxicity, sometimes associated with fever, is an uncommon but potentially serious toxic effect of methyldopa. Prompt diagnosis of hepatotoxicity requires a low threshold for considering the drug as a cause for hepatitis-like symptoms (e.g., nausea, anorexia) and screening for hepatotoxicity (e.g., with determination of gamma-glutamyl transpeptidase or alanine aminotransferase) at about 3 weeks and again at about 3 months following initiation of treatment with this drug. The incidence of methyldopa-induced hepatitis is unknown, but about 5% of patients will have transient increases in alanine aminotransferase activity in plasma. Hepatic dysfunction usually is reversible with prompt discontinuation of the drug, but it will recur if methyldopa is given again, and a few cases of fatal hepatic necrosis have been reported. Hepatitis may occur only after long-term therapy with methyldopa, but it usually appears within 3 months of starting the drug. It is advisable to avoid the use of methyldopa in patients with hepatic disease. Methyldopa can cause hemolytic anemia. At least 20% of patients who receive methyldopa for a year develop a positive Coombs test (antiglobulin test) that is due to autoantibodies directed against the Rh locus on the patients' erythrocytes. The development of a positive Coombs test per se, however, is not an indication to stop treatment with methyldopa; 1% to 5% of these patients will develop a hemolytic anemia which requires prompt discontinuation of the drug. The Coombs test may remain positive for as long as a year after discontinuation of methyldopa, but the hemolytic anemia usually resolves within a matter of weeks. Severe hemolysis may be attenuated by treatment with glucocorticoids. Adverse effects that are even more rare include leukopenia, thrombocytopenia, red cell aplasia, lupus erythematosuslike syndrome, lichenoid and granulomatous skin eruptions, myocarditis, retroperitoneal fibrosis, pancreatitis, diarrhea, and malabsorption. Therapeutic Uses Methyldopa is an effective antihypertensive agent when given in conjunction with a diuretic. It is generally well tolerated by patients with ischemic heart disease and by those with diastolic dysfunction, in whom it reduces left ventricular mass. However, frequent side effects and the potential for immunological abnormalities and organ toxicity are such that it is not used as the initial drug in monotherapy but is reserved for patients in whom it may have special value. Methyldopa is the preferred drug for treatment of hypertension during pregnancy based on its effectiveness and safety for both mother and fetus. The usual initial dose of methyldopa is 250 mg twice daily, and there is little additional effect with doses above 2 g per day. Administration of a single daily dose of methyldopa at bedtime minimizes sedative effects, but administration twice daily may be required for some patients. A parenteral preparation of the ethyl ester of methyldopa, methyldopate hydrochloride (ALDOMET), also is available. It is usually given by intermittent intravenous infusion of 250 to 500 mg every 6 hours. The rate of deesterification of the methyldopate is variable among patients, and the doses given intravenously may deliver less methyldopa to the circulation than the same dose given orally. Clonidine, Guanabenz, and Guanfacine The detailed pharmacology of the Pharmacological Effects The When guanabenz was first introduced, there was considerable interest
in observations that the drug could be natriuretic in experimental animals.
However, studies in human subjects have given variable results. With
long-term therapy, there is usually a small loss of weight with no clinically
significant changes in salt and water balance, suggesting that the 'pseudotolerance'
(Na+ retention) seen with methyldopa and guanadrel may not occur
with guanabenz. Nonetheless, the antihypertensive effects of diuretics and
guanabenz are additive. If individuals are given guanabenz after a salt load,
the drug has a natriuretic effect, and a new steady-state of Na+
balance is attained by 1 week. This short-term effect is thought to be
related to a reduction in renal sympathetic stimulation, with a consequent
reduction in Na+ reabsorption in the proximal nephron (Gehr et
al., 1986). Guanabenz also has been shown to cause a water diuresis in
some situations, which may be due to inhibition of the release and the renal
actions of vasopressin (Strandhoy, 1985). Stimulation of renal Adverse Effects and Precautions Although the Sudden discontinuation of clonidine and related Treatment of the withdrawal syndrome depends on the urgency of
reducing the arterial blood pressure. In the absence of hypertensive
encephalopathy, patients can be treated with their usual dose of
antihypertensive drug, which should reduce the pressure within 2 hours. If a
more rapid effect is required, sodium nitroprusside or a combination of an Because perioperative hypertension has been described in patients when
clonidine was withdrawn the night before surgery, surgical patients who are
being treated with an Adverse drug interactions with Overdosage with an Therapeutic Uses The Clonidine also has been used in hypertensive patients for the
diagnosis of pheochromocytoma. The lack of suppression of the plasma
concentration of norepinephrine to less than 500 pg/ml 3 hours after an oral
dose of 0.3 mg of clonidine suggests the presence of such a tumor. A
modification of this test, wherein overnight urinary excretion of
norepinephrine and epinephrine is measured after administration of a 0.3-mg
dose of clonidine at bedtime, may be useful when results based on plasma
norepinephrine concentrations are equivocal (MacDougall et al., 1988).
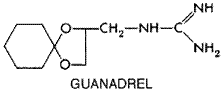
Other uses for Guanadrel Guanadrel HYLOREL) specifically inhibits the function of peripheral postganglionic adrenergic neurons. The structure of guanadrel, which contains the strongly basic guanidine group, is as follows:
Locus and Mechanism of Action Guanadrel is targeted uniquely to the peripheral adrenergic neuron, where it inhibits sympathetic function. The drug reaches its site of action by active transport into the neuron by the same transporter that is responsible for the reuptake of norepinephrine (see Chapter 6: Neurotransmission: The Autonomic and Somatic Motor Nervous Systems). In the neuron, guanadrel is concentrated within the neurosecretory vesicles, where it replaces norepinephrine. During chronic administration, guanadrel acts as a 'substitute neurotransmitter,' in that it is present in storage vesicles, it depletes the normal transmitter, and it can be released by stimuli that normally release norepinephrine. This replacement of norepinephrine with an inactive transmitter is probably the principal mechanism of its neuron-blocking action. When given intravenously, guanadrel initially can release norepinephrine in an amount sufficient to increase arterial blood pressure. This does not occur with oral administration, since norepinephrine is released only slowly from the vesicles under this circumstance and is degraded within the neuron by monoamine oxidase. Nonetheless, because of the potential for norepinephrine release, guanadrel is contraindicated in patients with pheochromocytoma. During adrenergic neuron blockade with guanadrel, effector cells become supersensitive to norepinephrine. The supersensitivity is similar to that produced by postganglionic sympathetic denervation. Pharmacological Effects Essentially all of the therapeutic and adverse effects of guanadrel result from sympathetic blockade. The antihypertensive effect is achieved by a reduction in peripheral vascular resistance that results from inhibition of sympathetically mediated vasoconstriction. Thus, the arterial pressure is reduced modestly in the supine position when sympathetic activity is normally low, but the pressure can fall to a greater extent during situations where reflex sympathetic activation is a mechanism for maintaining arterial pressure, such as assumption of the upright posture, exercise, and depletion of plasma volume. Renal blood flow and glomerular filtration rate are modestly decreased during therapy with guanadrel, but this is without clinical consequence; renin secretion is not reduced. Plasma volume often becomes expanded, which may diminish the antihypertensive efficacy of guanadrel and require administration of diuretic to restore the antihypertensive effect. Absorption, Distribution, Metabolism, and Excretion Guanadrel is rapidly absorbed, leading to maximal levels in plasma at
1 to 2 hours. Because guanadrel must be transported into and accumulate in
adrenergic neurons, the maximum effect on blood pressure is not seen until 4
to 5 hours. Although the Guanadrel is cleared from the body by both renal and nonrenal disposition. Its elimination is impaired in patients with renal insufficiency; total-body clearance was reduced by 4- to 5-fold in a group of patients with a clearance of creatinine averaging 13 ml per minute. Adverse Effects Guanadrel produces undesirable effects that are related entirely to sympathetic blockade. Symptomatic hypotension during standing, exercise, ingestion of alcohol, or hot weather is the result of the lack of sympathetic compensation for these stresses. A general feeling of weakness and lassitude is partially, but not entirely, related to postural hypotension. Rarely, guanadrel can precipitate congestive heart failure in patients with limited cardiac reserve as a result of drug-induced fluid retention. Sexual dysfunction usually presents as delayed or retrograde ejaculation. Diarrhea also may occur. Because guanadrel is actively transported to its site of action, drugs that block neuronal uptake of norepinephrine will inhibit the effect of guanadrel. Such drugs include the tricyclic antidepressants, cocaine, chlorpromazine, ephedrine, phenylpropanolamine, and amphetamine. Therapeutic Uses Because of the availability of a number of drugs that lower blood pressure without producing orthostatic hypotension, guanadrel is not employed in the monotherapy of hypertension, and is used chiefly as an additional agent in patients who have not achieved a satisfactory antihypertensive effect on two or more other agents. The usual starting dose is 10 mg daily, and side effects can be minimized by not exceeding 20 mg daily. Reserpine Reserpine
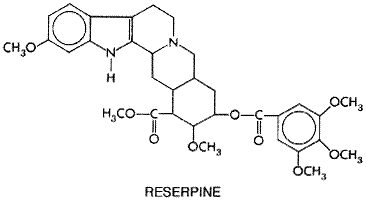
is an alkaloid extracted from the root of Rauwolfia serpentina
(Benth.), a climbing shrub indigenous to
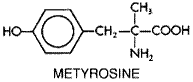
Locus and Mechanism of Action Reserpine binds tightly to storage vesicles in central and peripheral adrenergic neurons, and the drug remains at such sites for prolonged periods of time (Giachetti and Shore, 1978). The storage vesicles are rendered dysfunctional as a result of their interaction with reserpine, and nerve endings lose their ability to concentrate and store norepinephrine and dopamine. Catecholamines leak into the cytoplasm, where they are destroyed by intraneuronal monoamine oxidase, and little or no active transmitter is discharged from nerve endings when they are depolarized. A similar process occurs at storage sites for 5-hydroxytryptamine. Reserpine-induced depletion of biogenic amines correlates with evidence of sympathetic dysfunction and antihypertensive effects. Recovery of sympathetic function requires synthesis of new storage vesicles, which takes days to weeks after discontinuation of the drug. Since reserpine depletes amines in the CNS as well as in the peripheral adrenergic neuron, it is probable that its antihypertensive effects are related to both a central and a peripheral action; it is certain that many of the side effects of reserpine are related to its effects in the CNS. Pharmacological Effects Both cardiac output and peripheral vascular resistance are reduced during long-term therapy with reserpine. Orthostatic hypotension may occur but does not usually cause symptoms. Heart rate and renin secretion fall. Salt and water are retained, which commonly results in 'pseudotolerance.' Absorption, Metabolism, and Excretion Few data are available on the pharmacokinetic properties of reserpine because of the lack of an assay capable of detecting low concentrations of the drug or its metabolites. Reserpine that is bound to isolated storage vesicles cannot be removed by dialysis, indicating that the binding is not in equilibrium with the surrounding medium. Because of the irreversible nature of reserpine binding, the amount of drug in plasma is unlikely to bear any consistent relationship to drug concentration at the site of action. Reserpine is entirely metabolized, and none of the parent drug is excreted unchanged. Toxicity and Precautions Most of the adverse effects of reserpine are due to its effect on the CNS. Sedation and inability to concentrate or perform complex tasks are the most common adverse effects. More serious is the occasional psychotic depression that can lead to suicide. Depression usually appears insidiously over many weeks or months and may not be attributed to the drug because of the delayed and gradual onset of symptoms. Reserpine must be discontinued at the first sign of depression, and the drug should never be given to patients with a history of depression. Reserpine-induced depression may last several months after the drug is discontinued. Depression appears to be uncommon, but not unknown, with doses of 0.25 mg per day or less. Other side effects include nasal stuffiness and exacerbation of peptic ulcer disease, which is uncommon with small oral doses. Therapeutic Uses Reserpine was the sympatholytic drug used in the landmark Veterans Administration Cooperative Study that demonstrated the beneficial effects of treatment of hypertension (Veterans Administration Cooperative Study Group on Antihypertensive Agents, 1967, 1970), but with the availability of newer drugs that are both effective and well tolerated, the use of reserpine has diminished because of its CNS side effects. However, in comparative studies, low doses of reserpine given concurrently with a diuretic were as well tolerated as combinations of a diuretic with propranolol or methyldopa. The major advantage of reserpine is that it is much less expensive than other antihypertensive drugs. Reserpine is used once daily with a diuretic, and several weeks are necessary to achieve a maximum effect. The daily dose should be limited to 0.25 mg or less, and as little as 0.05 mg per day may be efficacious when a diuretic is also used. Metyrosine Metyrosine DEMSER) is (-)-
Metyrosine is used as an adjuvant to phenoxybenzamine and other
Locus and Mechanism of Action Antagonism of Pharmacological Effects The Renal blood flow is reduced in the short term by most Adverse Effects and Precautions The adverse effects of
Sudden discontinuation of some Nonsteroidal antiinflammatory drugs such as indo-methacin can blunt
the antihypertensive effect of propranolol and probably other Epinephrine can produce severe hypertension and bradycardia when a
nonselective Therapeutic Uses The
The development of drugs that selectively block Pharmacological Effects Initially, the Adverse Effects The use of doxazosin as monotherapy for hypertension increases the
risk for developing congestive heart failure. There is every reason to assume
that this is a 'class effect' and represents an adverse effect of
all of the A major precaution regarding the use of the Therapeutic Uses Because Combined Labetalol NORMODYNE,
TRANDATE) (see
Chapter 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor
Antagonists) is an equimolar mixture of four stereoisomers. One isomer is an Carvedilol COREG) (see Chapter 10:
Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists)
is a |
Vasodilators
|
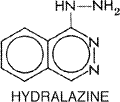
Hydralazine Hydralazine APRESOLINE) was one of the first orally
active antihypertensive drugs to be marketed in the
Locus and Mechanism of Action Hydralazine causes direct relaxation of arteriolar smooth muscle. The molecular mechanism of this effect is not known. It is not a dilator of capacitance vessels (e.g., the epicardial coronary arteries) and does not relax venous smooth muscle. Hydralazine-induced vasodilation is associated with powerful stimulation of the sympathetic nervous system, which results in increased heart rate and contractility, increased plasma renin activity, and fluid retention; all of these effects counteract the antihypertensive effect of hydralazine. Although most of the sympathetic activity is due to a baroreceptor-mediated reflex, hydralazine may stimulate the release of norepinephrine from sympathetic nerve terminals and augment myocardial contractility directly (Azuma et al., 1987). Pharmacological Effects Most of the effects of hydralazine are confined to the cardiovascular
system. The decrease in blood pressure after administration of hydralazine is
associated with a selective decrease in vascular resistance in the coronary,
cerebral, and renal circulations, with a smaller effect in skin and muscle.
Because of preferential dilation of arterioles over veins, postural
hypotension is not a common problem; hydralazine lowers blood pressure
equally in the supine and upright positions. Although hydralazine lowers
pulmonary vascular resistance, the greater increase in cardiac output can
cause mild pulmonary hypertension. It is difficult to predict which patients
will respond in this manner, but the increase in cardiac output can be
attenuated by the use of Absorption, Metabolism, and Excretion Hydralazine is N-acetylated in the bowel and/or the liver. The
rate of acetylation is genetically determined; about half of the people in
the The peak concentration of hydralazine in plasma and the peak hypotensive effect of the drug occur within 30 to 120 minutes of ingestion. Although its half-life in plasma is about an hour, the duration of the hypotensive effect of hydralazine can last as long as 12 hours. There is no clear explanation for this discrepancy. Toxicity and Precautions Two types of side effects occur after the use of hydralazine. The
first, which are extensions of the pharmacological effects of the drug,
include headache, nausea, flushing, hypotension, palpitation, tachycardia,
dizziness, and angina pectoris. Myocardial ischemia occurs because of the
increased oxygen demand imposed by the baroreflex-induced stimulation of the
sympathetic nervous system and also because hydralazine does not dilate the
epicardial coronary arteries; thus, the arteriolar dilation it produces may
cause a 'steal' of blood flow away from the ischemic region.
Following parenteral administration to patients with coronary artery disease,
the myocardial ischemia may be sufficiently severe and protracted to cause
frank myocardial infarction. For this reason, parenteral administration of
hydralazine is contraindicated in hypertensive patients with coronary artery
disease and inadvisable for most hypertensive patients over 40 years old. In
addition, if the drug is used alone, there may be salt retention with
development of high-output congestive heart failure. These symptoms were
common during the early clinical use of hydralazine; because tachyphylaxis
developed, the daily dose of the drug was frequently increased to 400 to 1000
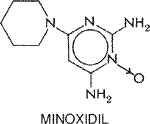
mg. When combined with a The second type of side effect is caused by immunological reactions, of which the drug-induced lupus syndrome is the most common. Administration of hydralazine also can result in an illness that resembles serum sickness, hemolytic anemia, vasculitis, and rapidly progressive glomerulonephritis. The mechanism of these autoimmune reactions is unknown, but hydralazine has been shown to inhibit methylation of DNA and induce self-reactivity in T cells (Cornacchia et al., 1988). The drug-induced lupus syndrome usually occurs after at least 6 months of continuous treatment with hydralazine, and its incidence is related to dose, sex, acetylator phenotype, and race (Perry, 1973). In one study, after three years of treatment with hydralazine, drug-induced lupus occurred in 10.4% of patients who received 200 mg daily, 5.4% who received 100 mg daily, and none who received 50 mg daily (Cameron and Ramsay, 1984). The incidence is four times higher in women than in men, and the syndrome is seen more commonly in Caucasians than in African Americans. The rate of conversion to a positive antinuclear antibody test is faster in slow acetylators than in rapid acetylators, suggesting that the native drug or a nonacetylated metabolite is responsible. However, since the majority of patients with positive antinuclear antibody tests do not develop the drug-induced lupus syndrome, hydralazine need not be discontinued unless clinical features of the syndrome appear. These features are similar to those of other drug-induced lupus syndromes and consist mainly of arthralgia, arthritis, and fever. Pleuritis and pericarditis may be present, and pericardial effusion can occasionally cause cardiac tamponade. Discontinuation of the drug is all that is necessary for most patients with the hydralazine-induced lupus syndrome, but symptoms may persist in a few patients and administration of corticosteroids may be necessary. Hydralazine also can produce a pyridoxine-responsive polyneuropathy. The mechanism appears to be related to the ability of hydralazine to combine with pyridoxine to form a hydrazone. This side effect is very unusual with doses up to 200 mg per day. Therapeutic Uses Hydralazine generally is not used as the sole drug for the long-term treatment of hypertension because of the development of tachyphylaxis secondary to an increase in cardiac output and fluid retention. In addition, the drug should be used with the greatest of caution in elderly patients and in hypertensive patients with coronary artery disease because of the possibility of precipitation of myocardial ischemia. The usual oral dosage of hydralazine is 25 to 100 mg twice daily. Twice-daily administration is as effective as administration four times a day for control of blood pressure, regardless of acetylator phenotype. The maximum recommended dose of hydralazine is 200 mg per day to minimize drug-induced lupus syndrome. Slow acetylators show a better response to this dosage than do fast acetylators because of the greater bioavailability of the drug. Hydralazine has been used widely to treat hypertension that occurs during pregnancy. However, the drug should be used cautiously during early pregnancy, since hydralazine can combine with DNA and cause a positive Ames test (Williams et al., 1980). Parenteral administration of hydralazine has been used for the treatment of hypertensive emergencies in pregnancy, but is not recommended for the treatment of hypertensive emergencies in patients in the age range for coronary artery disease. The drug is contraindicated for the short-term production of hypotension in patients with dissecting aortic aneurysm or in those with symptomatic ischemic heart disease. Minoxidil The discovery in 1965 of the hypotensive action of minoxidil (LONITEN) was a significant advance in the treatment of hypertension, since the drug has proven to be efficacious in patients with the most severe and drug-resistant forms of hypertension. The chemical structure of minoxidil is as follows:
Locus and Mechanism of Action Minoxidil is not active in vitro but must be metabolized by hepatic sulfotransferase to the active molecule, minoxidil N-O sulfate (McCall et al., 1983); the formation of this active metabolite is a minor pathway in the metabolic disposition of minoxidil. Minoxidil sulfate relaxes vascular smooth muscle in isolated systems where the parent drug is inactive. Minoxidil sulfate activates the ATP-modulated potassium channel. By opening potassium channels in smooth muscle and thereby permitting potassium efflux, it causes hyperpolarization and relaxation of smooth muscle (Leblanc et al., 1989). Pharmacological Effects Minoxidil produces arteriolar vasodilation with essentially no effect on the capacitance vessels; the drug resembles hydralazine and diazoxide in this regard. Minoxidil increases blood flow to skin, skeletal muscle, the gastrointestinal tract, and the heart more than to the CNS. The disproportionate increase in blood flow to the heart may have a metabolic basis, in that administration of minoxidil is associated with a reflex increase in myocardial contractility and in cardiac output. The cardiac output can increase markedly, as much as three- to fourfold. The principal determinant of the elevation in cardiac output is the action of minoxidil on peripheral vascular resistance to enhance venous return to the heart; by inference from studies with other drugs, the increased venous return probably results from enhancement of flow in the regional vascular beds with a fast time constant for venous return to the heart (Ogilvie, 1985). The adrenergically mediated increase in myocardial contractility contributes to the increased cardiac output, but is not the predominant causal factor. The effects of minoxidil on the kidney are complex. Minoxidil is a renal vasodilator, but systemic hypotension produced by the drug occasionally can decrease renal blood flow. However, in the majority of patients who take minoxidil for the treatment of hypertension, renal function improves, especially if renal dysfunction is secondary to hypertension (Mitchell et al., 1980). Minoxidil is a very potent stimulator of renin secretion; this effect is mediated by a combination of renal sympathetic stimulation and activation of the intrinsic renal mechanisms for regulation of renin release. Absorption, Metabolism, and Excretion Minoxidil is well absorbed from the gastrointestinal tract. Although peak concentrations of minoxidil in blood occur 1 hour after oral administration, the maximal hypotensive effect of the drug occurs later, possibly because formation of the active metabolite is delayed. Only about 20% of the absorbed drug is excreted unchanged in the urine, and the main route of elimination is by hepatic metabolism. The major metabolite of minoxidil is the glucuronide conjugate at the N-oxide position in the pyrimidine ring. This metabolite is less active than minoxidil, but it persists longer in the body. The extent of biotransformation of minoxidil to its active metabolite, minoxidil N-O sulfate, has not been evaluated in human beings. Minoxidil has a half-life in plasma of 3 to 4 hours, but its duration of action is 24 hours or occasionally even longer. It has been proposed that persistence of minoxidil in vascular smooth muscle is responsible for this discrepancy. However, without knowledge of the pharmacokinetic properties of the active metabolite, an explanation for the prolonged duration of action cannot be given. Adverse Effects and Precautions The adverse effects of minoxidil are predictable and can be divided into three major categories: fluid and salt retention, cardiovascular effects, and hypertrichosis. Retention of salt and water results from increased proximal renal
tubular reabsorption, which is in turn secondary to reduced renal perfusion
pressure and to reflex stimulation of renal tubular The cardiac consequences of the baroreceptor-mediated activation of
the sympathetic nervous system during minoxidil therapy are similar to those
seen with hydralazine; there is an increase in heart rate, myocardial
contractility, and myocardial oxygen consumption. Thus, myocardial ischemia
can be induced by minoxidil in patients with coronary artery disease. The
cardiac sympathetic responses are attenuated by concurrent administration of
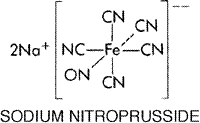
a The increased cardiac output evoked by minoxidil has particularly adverse consequences in those hypertensive patients who have left ventricular hypertrophy and diastolic dysfunction. Such poorly compliant ventricles respond suboptimally to increased volume loads, with a resulting increase in left ventricular filling pressure. This probably is a major contributor to the increased pulmonary artery pressure seen with minoxidil (and hydralazine) therapy in hypertensive patients, and is compounded by the retention of salt and water caused by minoxidil. Cardiac failure can result from minoxidil therapy in such patients; the potential for this complication can be reduced but not prevented by effective diuretic therapy. Pericardial effusion is an uncommon but serious complication of minoxidil. Although more commonly described in patients with cardiac failure and renal failure, pericardial effusion can occur in patients with normal cardiovascular and renal function. Mild and asymptomatic pericardial effusion is not an indication for discontinuing minoxidil, but the situation should be monitored closely to avoid progression to tamponade. Effusion usually clears when the drug is discontinued, but it will recur if treatment with minoxidil is resumed (Reichgott, 1981). Flattened and inverted T waves frequently are observed in the electrocardiogram following the initiation of minoxidil treatment. These are not ischemic in origin and are seen with other drugs that activate potassium channels. These drugs accelerate myocardial repolarization, shorten the refractory period, and one of them, pinacidil, lowers the ventricular fibrillation threshold and increases spontaneous ventricular fibrillation in the setting of myocardial ischemia (Chi et al., 1990). The effect of minoxidil on the refractory period and ischemic ventricular fibrillation has not been investigated; whether or not such findings enhance the risk of ventricular fibrillation in human myocardial ischemia is unknown. Hypertrichosis occurs in all patients who receive minoxidil for an extended period and is probably a consequence of potassium channel activation. Growth of hair occurs on the face, back, arms, and legs and is particularly offensive to women. Frequent shaving or depilatory agents can be used to manage this problem. Topical minoxidil (ROGAINE) is now marketed for the treatment of male-pattern baldness. The topical use of minoxidil can cause measurable cardiovascular effects in some individuals (Leenen et al., 1988). Other side effects of the drug are rare and include rashes, StevensJohnson syndrome, glucose intolerance, serosanguinous bullae, formation of antinuclear antibodies, and thrombocytopenia. Therapeutic Uses Minoxidil is best reserved for the treatment of severe hypertension that responds poorly to other antihypertensive medications (Campese, 1981). It has been used successfully in the treatment of hypertension in both adults and children. Minoxidil should never be used alone; it must be given concurrently with a diuretic to avoid fluid retention and with a sympatholytic drug (usually a receptor antagonist) to control reflex cardiovascular effects. The drug usually is administered either once or twice a day, but some patients may require more frequent dosage for adequate control of blood pressure. The initial daily dose of minoxidil may be as little as 1.25 mg, which can be increased gradually to 40 mg in one or two daily doses. Sodium Nitroprusside Although sodium nitroprusside has been known since 1850 and its hypotensive effect in human beings was described in 1929, its safety and usefulness for the short-term control of severe hypertension were not demonstrated until the mid-1950s. Several investigators subsequently demonstrated that sodium nitroprusside also was effective in improving cardiac function in patients with left ventricular failure (see Chapter 34: Pharmacological Treatment of Heart Failure). The structural formula of sodium nitroprusside is as follows:
Locus and Mechanism of Action Nitroprusside is a nitrovasodilator. It is metabolized by blood vessels to its active metabolite, nitric oxide. Nitric oxide activates guanylyl cyclase, leading to the formation of cyclic GMP and vasodilation (Murad, 1986). The metabolic activation of nitroprusside is catalyzed by a different nitric oxidegenerating system than that for nitroglycerin, probably accounting for the difference in the potency of these drugs at different vascular sites and the fact that tolerance develops to nitroglycerin but not to nitroprusside (Kowaluk et al., 1992). Pharmacological Effects Nitroprusside dilates both arterioles and venules, and the hemodynamic response to its administration results from a combination of venous pooling and reduced arterial impedance. Because of its effect on venules, the hypotensive effect of sodium nitroprusside is greater when the patient is upright. In subjects with normal left ventricular function, venous pooling affects cardiac output more than does the reduction of afterload; cardiac output thus tends to fall. In contrast, in patients with severely impaired left ventricular function and diastolic ventricular distention, the reduction of arterial impedance is the predominant effect, leading to a rise in cardiac output (see Chapter 34: Pharmacological Treatment of Heart Failure). Sodium nitroprusside is a nonselective vasodilator, and regional distribution of blood flow is little affected by the drug. In general, renal blood flow and glomerular filtration are maintained, and plasma renin activity increases. Unlike minoxidil, hydralazine, diazoxide, and other arteriolar vasodilators, sodium nitroprusside usually causes only a modest increase in heart rate and an overall reduction in myocardial demand for oxygen. Absorption, Metabolism, and Excretion Sodium nitroprusside is an unstable molecule that decomposes under strongly alkaline conditions and when exposed to light. The drug must be given by continuous intravenous infusion to be effective. Its onset of action is within 30 seconds; the peak hypotensive effect occurs within 2 minutes, and when the infusion of the drug is stopped, the effect disappears within 3 minutes. The metabolism of nitroprusside by smooth muscle is initiated by its reduction, which is followed by the release of cyanide and then nitric oxide (Bates et al., 1991; Ivankovich et al., 1978). Cyanide is further metabolized by liver rhodanase to thiocyanate, which is eliminated almost entirely in the urine. The mean elimination half-time for thiocyanate is 3 days in patients with normal renal function, and it can be much longer in patients with renal insufficiency. Toxicity and Precautions The short-term side effects of nitroprusside are due to excessive
vasodilation, with hypotension and the consequences thereof. Close monitoring
of blood pressure and the use of a continuous variable-rate infusion pump
will prevent an excessive hemodynamic response to the drug in the majority of
cases. Less commonly, toxicity may result from conversion of nitroprusside to
cyanide and thiocyanate. Toxic accumulation of cyanide leading to severe
lactic acidosis can occur usually if sodium nitroprusside is infused at a
rate greater than 5 Nitroprusside can worsen arterial hypoxemia in patients with chronic obstructive pulmonary disease because the drug interferes with hypoxic pulmonary vasoconstriction and therefore promotes mismatching of ventilation with perfusion. Rebound hypertension may occur after abrupt cessation of short-term nitroprusside infusions (Packer et al., 1979); this may be caused by persistently elevated concentrations of renin in the plasma. Therapeutic Uses Sodium nitroprusside is used primarily to treat hypertensive
emergencies, but the drug can be used in many situations when short-term
reduction of cardiac preload and/or afterload is desired. Thus, nitroprusside
has been used to lower blood pressure during acute aortic dissection, to
increase cardiac output in congestive heart failure (see Chapter 34:
Pharmacological Treatment of Heart Failure), and to decrease myocardial
oxygen demand after acute myocardial infarction. In addition, nitroprusside
is the drug most often used to induce controlled hypotension during
anesthesia in order to reduce bleeding in surgical procedures. In the
treatment of acute aortic dissection, it is important to administer a Sodium nitroprusside is available in vials that contain 50 mg. The
contents of the vial should be dissolved in 2 to 3 ml of 5% dextrose in
water. Addition of this solution to 250 to 1000 ml of 5% dextrose in water
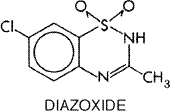
produces a concentration of 50 to 200 Diazoxide Diazoxide HYPERSTAT IV) is used in the treatment of hypertensive emergencies. Sodium nitroprusside is the drug of choice for this indication, but diazoxide maintains a place in the treatment of hypertensive emergencies in situations in which accurate infusion pumps are not available and/or close monitoring of blood pressure is not feasible. The drug is a benzothiadiazine derivative, like the thiazide diuretics, but it does not cause diuresis, apparently because it lacks a sulfonamido group. Its structural formula is as follows:
Mechanism of Action and Pharmacological Effects Diazoxide hyperpolarizes arterial smooth muscle cells by activating ATP-sensitive K+ channels; this causes relaxation of the vascular smooth muscle (Standen et al., 1989). The effect of the drug in vivo is exclusively arteriolar, with negligible effect on capacitance vessels. This produces substantial reflex activation of the sympathetic nervous system. Cardiac output may double from stimulation of heart rate and myocardial contractility. The avid retention of salt and water is probably a result of stimulation of renal sympathetic nerves and changes in intrarenal hemodynamics, as with other arteriolar vasodilators. Diazoxide increases coronary blood flow, and cerebral and renal blood flows are maintained by autoregulation. Renin secretion is enhanced, and the combination of an increased cardiac output, salt and water retention, and elevated concentrations of angiotensin II counteract the antihypertensive effects of diazoxide. Absorption, Metabolism, and Excretion Although well absorbed orally, diazoxide is administered only intravenously for the treatment of severe hypertension. Approximately 20% to 50% of the drug is eliminated as such by the kidney, and the rest is metabolized in the liver to the 3-hydroxymethyl and 3-carboxy derivatives (Pruitt et al., 1974). Although the plasma half-life of diazoxide is 20 to 60 hours, the duration of the hypotensive response to the drug is variable and can be as short as 4 hours or as long as 20 hours; the development of a brisk rise in renin secretion may antagonize the early hypotensive effect of diazoxide. The main indication for the use of diazoxide is for the treatment of
hypertensive emergencies. Injection of an intravenous bolus lowers blood
pressure within 30 seconds, and a maximum effect is achieved within 3 to 5
minutes. Although initial recommendations were to administer a 300-mg bolus
of diazoxide, excessive hypotension with resultant cerebral and
cardiovascular damage has resulted from this practice. Hypotension can be
minimized by the administration of a 'minibolus' of 50 to 150 mg at
intervals of 5 to 15 minutes until the desired blood pressure is achieved (Wilson
and Vidt, 1978). Diazoxide also can be given by slow intravenous infusion at
a rate of 15 to 30 mg per minute (Garrett and Kaplan, 1982). Prior
administration of a Adverse Effects and Precautions The most common side effects caused by diazoxide are myocardial
ischemia, salt and water retention, and hyperglycemia. Myocardial ischemia
may be precipitated or aggravated by diazoxide, and it results from the
reflex adrenergic stimulation of the heart and from increased flow to
nonischemic regions that 'steal' blood flow from the regions
supplied by stenotic vessels. Retention of fluid can be avoided by
restriction of salt and water. The routine use of diuretic agents with
diazoxide is not recommended because patients with malignant hypertension are
frequently volume-depleted. Hyperglycemia results from diazoxide's capacity
to inhibit the secretion of insulin from pancreatic |
Ca2+-Channel Antagonists
|
Ca2+-channel blocking agents are
an important group of drugs for the treatment of hypertension. The general
pharmacology of these drugs is presented in Chapter 32: Drugs Used for the
Treatment of Myocardial Ischemia; their use in heart failure is discussed in Chapter
34: Pharmacological Treatment of Heart Failure; and their use in cardiac
arrhythmia is covered in Chapter 35: Antiarrhythmic Drugs. The logic behind
their use in hypertension comes from the understanding that fixed
hypertension is the result of increased peripheral vascular resistance. Since
contraction of vascular smooth muscle is dependent on the free intracellular
concentration of Ca2+, inhibition of transmembrane movement of Ca2+
should decrease the total amount of Ca2+ that reaches intracellular
sites. Indeed, all of the Ca2+-channel blockers lower blood
pressure by relaxing arteriolar smooth muscle and decreasing peripheral
vascular resistance (Lehmann et al., 1983). As a consequence of a
decrease in peripheral vascular resistance, the Ca2+-channel
blockers evoke a baroreceptor-mediated sympathetic discharge. In the case of
the dihydropyridines, mild to moderate tachycardia ensues from the adrenergic
stimulation of the sinoatrial node, whereas tachycardia is minimal to absent
with verapamil and diltiazem because of the direct negative chronotropic
effect of these two drugs. The increased adrenergic stimulation of the heart
serves to counter the negative inotropic effect of Ca2+-channel
blockers such as verapamil, diltiazem, and nifedipine; the
importance of this compensatory support of myocardial contractility should be
considered in decisions regarding possible concurrent use of In considering the cardiovascular effects of the Ca2+-channel blockers, it is essential to evaluate both the hemodynamic effects in the normal heart and the interaction of these drugs with cardiac disease, given that both cardiac failure and coronary artery diseases are important consequences of hypertension and that left ventricular hypertrophy is a harbinger for sudden cardiac death in hypertensive patients. As a consequence of the peripheral vasodilation, Ca2+-channel blockers may increase venous return, which will result in an increased cardiac output except in the case of those that exert substantial negative inotropic effects (e.g., verapamil and diltiazem). The increased venous return is not as great as with minoxidil or hydralazine, but is a consideration in the management of patients with diastolic dysfunction due to hypertensive cardiomyopathy who are at risk of left ventricular failure. The Ca2+-channel blockers do not improve the diastolic function of the ventricle. Although earlier noninvasive studies had demonstrated that peak filling rates of the left ventricle of hypertensive patients were shortened by Ca2+-channel blockers, direct hemodynamic evaluation of ventricular function has demonstrated that verapamil causes an increase in left ventricular end-diastolic pressure, an undesirable hemodynamic consequence that occurs in conjunction with, and probably as a contributor to, the acceleration of peak filling rate (Nishimura et al., 1993). In addition to these findings that Ca2+-channel blockers do not improve and have the potential for worsening the hemodynamics in diastolic dysfunction, the long-term effects of the Ca2+-channel blockers on left ventricular hypertrophy, a major contributor to diastolic dysfunction, should be considered. An overview of all trials evaluating the effects of antihypertensive agents on left ventricular mass concludes that, although Ca2+-channel blockers do reduce left ventricular mass and do so more effectively than diuretics, they are less effective than angiotensin converting enzyme inhibitors and methyldopa (Dalhf et al., 1993). Based on the sum of this evidence, Ca2+-channel blockers probably are not the first choice as the initial drug in the treatment of patients whose hypertension is accompanied by left ventricular hypertrophy nor as the predominant drug in a combination for their treatment. All Ca2+-channel blockers are equally effective when used
alone for the treatment of mild to moderate hypertension; and in comparative
trials, Ca2+-channel blockers are as effective in lowering blood
pressure as The presence of ischemic heart disease in conjunction with
hypertension raises a special set of concerns regarding some of the Ca2+-channel
blockers. Dihydropyridine Ca2+-channel blockers do not improve
survival in patients following myocardial infarction. Neither verapamil nor diltiazem
improves mortality in the entire group of postinfarction patients. Data
suggesting that diltiazem may exert a favorable effect on mortality in the
subset of patients who exhibit no abnormality in systolic ventricular
function does not form a strong basis for its use in such patients because of
the post hoc selection of this group for analysis (Multicenter
Diltiazem Postinfarction Trial Research Group, 1988). This is in contrast to
the clear benefit to the survival of patients after myocardial infarction
that is conferred by treatment with In diabetic patients with hypertension, the available evidence favors an angiotensin converting enzyme inhibitor as the initial antihypertensive drug (Estacio et al., 1998), following which it is appropriate to consider adding a Ca2+-channel blocker if a second drug is required. The profile of adverse reactions to the Ca2+-channel
blockers varies among the drugs in this class, but only a small fraction of
patients discontinue these drugs because of perceived adverse reactions. The dihydropyridines
cause the highest incidence of vascular side effects. Approximately 10% of
patients receiving the standard formulation of nifedipine (immediate-release
capsules) develop headache, flushing, dizziness, and peripheral edema.
Dizziness and flushing are much less of a problem with the sustained-release
formulations and with the dihydropyridines having a long half-life and
relatively constant concentrations of drug in plasma. The edema usually is not
the result of fluid retention; it most likely results from increased
hydrostatic pressure in the lower extremities owing to precapillary dilation
and reflex postcapillary constriction. Contraction of the lower esophageal
sphincter is inhibited by the Ca2+-channel blockers. Accordingly,
all Ca2+-channel blockers can cause gastroesophageal reflux.
Constipation is a common side effect of verapamil, but it occurs less
frequently with other Ca2+-channel blockers. Inhibition of
sinoatrial node function by diltiazem and verapamil can lead to bradycardia
and even sinoatrial node arrest, particularly in patients with sinoatrial
node dysfunction; this effect is exaggerated by concurrent use of Oral administration of nifedipine as an approach to urgent reduction of blood pressure has been abandoned. Sublingual administration does not achieve the maximum plasma concentration any more quickly than does oral administration. Moreover, in the absence of deleterious consequences of high arterial pressure, data do not support the rapid lowering of blood pressure. Short-acting, parenterally administered agents should be used in the setting of hypertensive emergency. Nifedipine is a suboptimal choice for treatment of hypertensive pulmonary edema; nitroprusside produces a greater reduction in left ventricular end-diastolic pressure than do equihypotensive doses of nifedipine (Aroney et al., 1991), and the magnitude of nitroprusside's pharmacological effect can be regulated more effectively. There is no place in the treatment of hypertension for the use of nifedipine or other dihydropyridine Ca2+-channel blockers with short half-lives when administered in a standard (immediate release) formulation, because of the oscillation in blood pressure and concurrent surges in sympathetic reflex activity within each dosage interval. Ca2+-channel blockers are versatile drugs with proven
efficacy in all types of patients (Kiowski et al., 1985). They seem to
be especially efficacious in low-renin hypertension. Compared with other
classes of antihypertensive agents, there is a greater frequency of achieving
blood pressure control with Ca2+-channel blockers as monotherapy
in elderly subjects and in African Americans, population groups in which the
low renin status is more prevalent. Long-acting dihydropyridine Ca2+-channel
blockers have been found to reduce cardiovascular mortality in older patients
(Staessen et al., 1997). The efficacy of Ca2+-channel
blockers is enhanced by the concomitant use of an angiotensin converting enzyme
inhibitor, methyldopa, or Significant drugdrug interactions may be encountered when Ca2+-channel blockers are used to treat hypertension. Verapamil blocks the drug transporter, P-glycoprotein. Both the renal and hepatic disposition of digoxin occur via this transporter. Accordingly, verapamil inhibits the elimination of digoxin and other drugs that are cleared from the body by the P-glycoprotein (see Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination) (Pedersen et al., 1981). When used with quinidine, Ca2+-channel blockers may cause excessive hypotension, particularly in patients with idiopathic hypertrophic subaortic stenosis. Ca2+-channel blockers should not be used in patients with
SA or AV nodal abnormalities or in patients with overt congestive heart
failure. These drugs usually are safe, however, in hypertensive patients with
asthma, hyperlipidemia, diabetes mellitus, and renal dysfunction. Unlike |
Angiotensin Converting Enzyme Inhibitors
|
Angiotensin II is an important regulator of cardiovascular function (see Chapter 31: Renin and Angiotensin). The ability to reduce levels of angiotensin II with orally effective inhibitors of angiotensin converting enzyme (ACE) represents an important advance in the treatment of hypertension. Captopril (CAPOTEN) was the first such agent to be developed for the treatment of hypertension. Since then, enalapril (VASOTEC), lisinopril (PRINIVIL), quinapril (ACCUPRIL), ramipril (ALTACE), benazepril (LOTENSIN), moexipril (UNIVASC), fosinopril (MONOPRIL), trandolapril (MAVIK), and perindopril (ACEON) also have become available. These drugs have proven to be very useful for the treatment of hypertension because of their efficacy and their very favorable profile of side effects, which enhances compliance. The angiotensin converting enzyme inhibitors appear to confer a special advantage in the treatment of patients with diabetes, slowing the development of diabetic glomerulopathy. They also have been shown to be effective in slowing the progression of other forms of chronic renal disease, such as glomerulosclerosis, and many of these patients also have hypertension. An angiotensin converting enzyme inhibitor is probably the preferred initial agent in the treatment of hypertensive patients with left ventricular hypertrophy. Patients with hypertension and ischemic heart disease are candidates for treatment with angiotensin converting enzyme inhibitors; this includes treatment in the immediate postmyocardial infarction period which has been shown to lead to improved ventricular function and reduced morbidity and mortality (see also Chapter 34: Pharmacological Treatment of Heart Failure). The endocrine consequences of inhibiting the biosynthesis of angiotensin II are of importance in a number of facets of hypertension treatment. Because angiotensin converting enzyme inhibitors blunt the normal aldosterone response to Na+ loss, the normal role of aldosterone to oppose diuretic-induced natriuresis is diminished. Thus, angiotensin converting enzyme inhibitors enhance the efficacy of diuretic drugs. This means that even very small doses of diuretics may substantially improve the antihypertensive efficacy of angiotensin converting enzyme inhibitors; and on the other end of the spectrum, the use of high doses of diuretics together with angiotensin converting enzyme inhibitors may lead to excessive reduction in blood pressure and to Na+ loss in some patients. The attenuation of aldosterone production by the angiotensin
converting enzyme inhibitors also influences K+ homeostasis. There
is only a very small and clinically unimportant rise in serum K+
when angiotensin converting inhibitors are used alone in patients with normal
renal function. However, substantial retention of K+ can occur in
some patients with renal insufficiency. Furthermore, the potential for
developing hyperkalemia should be considered when angiotensin converting
enzyme inhibitors are used with other drugs that can cause K+
retention; these include the K+-sparing diuretics (amiloride, triamterene,
spironolactone), nonsteroidal antiinflammatory drugs, K+
supplements, and There are several cautions in the use of angiotensin converting enzyme inhibitors in patients with hypertension. Angioedema is an infrequent but serious and potentially fatal adverse effect of all of the angiotensin converting enzyme inhibitors. Thus, patients starting treatment with these drugs should be explicitly warned to discontinue their use with the advent of any signs of angioedema. The angiotensin converting enzyme inhibitors should not be used during pregnancy, a fact that should be communicated to patients of childbearing age. In most patients there is no appreciable change in glomerular filtration rate following the administration of a converting enzyme inhibitor. However, in renovascular hypertension, the glomerular filtration rate is maintained as the result of increased resistance in the postglomerular arteriole caused by angiotensin II. Accordingly, in patients with bilateral renal artery stenosis or stenosis in a sole kidney, the administration of a converting enzyme inhibitor will reduce the filtration fraction and cause a substantial reduction in glomerular filtration rate. Converting enzyme inhibitors lower the blood pressure to some extent in most patients with hypertension. Following the initial dose of a converting enzyme inhibitor, there may be a considerable fall in blood pressure in some patients; this response to the initial dose is a function of pretreatment plasma renin activity. The potential for a large initial drop in blood pressure is the reason for using a low dose for initiating therapy. On continuing treatment, there usually is a progressive fall in blood pressure that in most patients does not reach a maximum for about one week. The level of blood pressure seen during chronic treatment is not strongly correlated with the level of pretreatment plasma renin activity. Young and middle-aged Caucasian patients have a higher probability of responding to the angiotensin converting enzyme inhibitors. Elderly African-American patients as a group are more resistant to the hypotensive effect of these drugs, but concurrent use of a diuretic in low doses overcomes this relative resistance. These drugs are discussed in detail in Chapter 31: Renin and Angiotensin. |
Angiotensin IIReceptor Antagonists
|
The importance of angiotensin II in regulating cardiovascular function has led to the development of nonpeptide antagonists of the angiotensin II receptor for clinical use. Losartan (COZAAR), candesartan (ATACAND), irbesartan (AVAPRO), valsartan (DIOVAN), telmisartan (MICARDIS), and eprosartan (TEVETEN) have been approved for the treatment of hypertension. By preventing effects of angiotensin II, these agents relax smooth muscle and thereby promote vasodilation, increase renal salt and water excretion, reduce plasma volume, and decrease cellular hypertrophy. Angiotensin IIreceptor antagonists also theoretically overcome some of the disadvantages of ACE inhibitors, which not only prevent conversion of angiotensin I to angiotensin II but also prevent ACE-mediated degradation of bradykinin and substance P. Cough, an adverse effect of ACE inhibitors, has not been associated with angiotensin IIreceptor antagonists. Angioedema occurs rarely. Two distinct subtypes of angiotensin II receptors have been cloned, designated as type 1 (AT1) and type 2 (AT2). The AT1 angiotensin II receptor subtype is located predominantly in vascular and myocardial tissue and also in brain, kidney, and adrenal glomerulosa cells, which secrete aldosterone (see Chapter 31: Renin and Angiotensin). The AT2 subtype of angiotensin II receptor is found in the adrenal medulla, kidney, and in the CNS, and may play a role in vascular development (Horiuchi et al., 1999). Because the AT1 receptor mediates feedback inhibition of renin release, renin and angiotensin II concentrations are increased during AT1-receptor antagonism. The clinical consequences of increased angiotensin II effects on an uninhibited AT2 receptor are unknown; however, emerging data suggest that the AT2 receptor may elicit antigrowth and antiproliferative responses. Adverse Effects and Precautions The adverse effects of AT1-receptor antagonists may be considered in the context of those known to be associated with the ACE inhibitors. ACE inhibitors cause problems of two major types, those related to a diminished level of angiotensin II and those due to molecular actions independent of abrogating the function of angiotensin II. Adverse effects of ACE inhibitors that result from inhibiting angiotensin II-related functions (see above) occur also with AT1-receptor antagonists. These include hypotension, hyperkalemia, and reduced renal function, including that associated with bilateral renal artery stenosis and stenosis in the artery of a solitary kidney. Hypotension is most likely to occur in patients in whom the blood pressure is highly dependent on angiotensin II, including those with volume depletion (e.g., with diuretics), renovascular hypertension, cardiac failure, and cirrhosis; in such patients initiation of treatment with low doses and attention to blood volume is essential. Hyperkalemia will occur only in conjunction with other factors that alter K+ homeostasis, such as renal insufficiency, ingestion of excess K+, and the use of drugs that promote K+ retention. In contrast with ACE inhibitors, the AT1-receptor antagonists do not cause cough. Angioedema has been reported, but it is not clear whether or not the rate of angioedema in patients taking the AT1-receptor antagonists is any higher than that in the general population. Hepatic dysfunction has been reported with the AT1-receptor antagonists. AT1-receptor antagonists should not be administered during the second or third trimester of pregnancy and should be discontinued as soon as pregnancy is detected. Although it is not yet known whether or not AT1-receptor antagonists are secreted in human breast milk, significant amounts are detected in the milk of animals; consequently, AT1-receptor antagonists should not be administered to patients who are breast-feeding. Therapeutic Uses When given in adequate doses, the AT1-receptor antagonists appear to be as effective as ACE inhibitors in the treatment of hypertension. As with ACE inhibitors, these drugs may be less effective in African-American and low-renin patients. The full effect of AT1-receptor antagonists on blood pressure typically is not observed until 3 to 6 weeks after the initiation of therapy. If blood pressure is not controlled by an AT1-receptor antagonist alone, a low dose of a hydrochlorothiazide or other diuretic may be added. In several randomized, double-blind studies of patients with mild to severe hypertension, the addition of hydrochlorothiazide to an AT1-receptor antagonist produced significant additional reductions in blood pressure in patients who demonstrated an insufficient response to hydrochlorothiazide alone. A smaller initial dosage is preferred for patients who have already received diuretics and therefore have an intravascular volume depletion, and for other patients whose blood pressure is highly dependent on angiotensin II. Ongoing clinical trials should shed light on the relative efficacy of ACE inhibitors and AT1-receptor antagonists in patients with diabetic nephropathy, coronary artery disease, and left ventricular dysfunction (Pitt et al., 1999a). Given the different mechanisms by which they act, there is no assurance that the effects of ACE inhibitors and antagonists of the AT1 receptor will be equivalent. |
Nonpharmacological Therapy of Hypertension
|
Nonpharmacological approaches to the reduction of blood pressure generally are advisable as the initial approach to treatment of patients with diastolic blood pressures in the range of 90 to 95 mm Hg. Further, these approaches will augment the effectiveness of pharmacological therapy in patients with higher levels of blood pressure. Also, for patients with diastolic blood pressures in the range of 85 to 90 mm Hg, the epidemiological data on cardiovascular risks support the institution of nonpharmacological therapy. To maintain compliance with a therapeutic regimen, the intervention should not lessen the quality of life. All drugs have side effects. If minor alterations of normal activity or diet can reduce blood pressure to a satisfactory level, the complications of drug therapy can be avoided. In addition, nonpharmacological methods to lower blood pressure allow the patient to participate actively in the management of his or her disease. Reduction of weight, restriction of salt, and moderation in the use of alcohol may reduce blood pressure and improve the efficacy of drug treatment. In addition, regular isotonic exercise also lowers blood pressure in hypertensive patients. Smoking per se does not cause hypertension. However, smokers do have a higher incidence of malignant hypertension (Isles et al., 1979), and smoking is a major risk factor for coronary heart disease. Hypertensive patients have an exceptionally great incentive to stop smoking. Consumption of caffeine can raise blood pressure and elevate plasma concentrations of norepinephrine, but long-term consumption of caffeine causes tolerance to these effects and has not been associated with the development of hypertension. An increased intake of Ca2+ has been reported by some investigators to lower blood pressure. The mechanism of this effect is not understood, but suppression of the secretion of parathyroid hormone apparently is involved. However, supplemental Ca2+ does not lower blood pressure when populations of hypertensive subjects are studied. Although it is possible that there are some hypertensive patients who have a hypotensive response to Ca2+, there is no easy way to identify such individuals. Supplemental use of Ca2+ for this purpose cannot be recommended at the present time (Kaplan, 1988). Reduction of Body Weight Obesity and hypertension are closely associated, and the degree of obesity is positively correlated with the incidence of hypertension. Obese hypertensives may lower their blood pressure by losing weight regardless of a change in salt consumption (Maxwell et al., 1984). The mechanism by which obesity causes hypertension is unclear, but increased secretion of insulin in obesity could result in insulin-mediated enhancement of renal tubular reabsorption of Na+ and an expansion of extracellular volume. Obesity also is associated with increased activity of the sympathetic nervous system; this is reversed by weight loss. Maintenance of weight loss is difficult for many. A combination of aerobic physical exercise and dietary counseling may enhance compliance. Sodium Restriction Severe restriction of salt will lower the blood pressure in most hospitalized hypertensive patients; this treatment method was advocated prior to the development of effective antihypertensive drugs (Kempner, 1948). However, severe salt restriction is not practical from a standpoint of compliance. Several studies have shown that moderate restriction of salt intake to approximately 5 g per day (2 g Na+) will, on average, lower blood pressure by 12 mm Hg systolic and 6 mm Hg diastolic. The higher the initial blood pressure, the greater the response. In addition, subjects over 40 years of age are more responsive to the hypotensive effect of moderate restriction of salt (Grobbee and Hofman, 1986). Even though not all hypertensive patients respond to restriction of salt, this intervention is benign and can easily be advised as an initial approach in all patients with mild hypertension. An additional benefit of salt restriction is improved responsiveness to some antihypertensive drugs. Alcohol Restriction Consumption of alcohol can raise blood pressure, but it is unclear how much alcohol must be consumed to observe this effect (MacMahon et al., 1984). Heavy consumption of alcohol increases the risk of cerebrovascular accidents but not coronary heart disease (Kagan et al., 1985). In fact, small amounts of ethanol have been found to protect against the development of coronary artery disease. The mechanism by which alcohol raises blood pressure is unknown, but it may involve increased transport of Ca2+ into vascular smooth muscle cells. Excessive intake of alcohol also may result in poor compliance with antihypertensive regimens. All hypertensive patients should be advised to restrict consumption of ethanol to no more than 30 ml per day. Physical Exercise Increased physical activity lowers rates of cardiovascular disease in men (Paffenbarger et al., 1986). It is not known if this beneficial effect is secondary to an antihypertensive response to exercise. Lack of physical activity is associated with a higher incidence of hypertension (Blair et al., 1984). Although consistent changes in blood pressure are not always observed, meticulously controlled studies have demonstrated that regular isotonic exercise reduces both systolic and diastolic blood pressures by approximately 10 mm Hg (Nelson et al., 1986). The mechanism by which exercise can lower blood pressure is not clear, but several hemodynamic and humoral changes have been documented. Regular isotonic exercise reduces blood volume and plasma catecholamines and elevates plasma concentrations of atrial natriuretic factor. The beneficial effect of exercise can occur in subjects who demonstrate no change in body weight or salt intake during the training period. Relaxation and Biofeedback Therapy The fact that long-term stressful stimuli can cause sustained hypertension in animals has given credence to the possibility that relaxation therapy will lower blood pressure in some hypertensive patients. A few studies have generated positive results but, in general, relaxation therapy has inconsistent and modest effects on blood pressure (Jacob et al., 1986). In addition, the long-term efficacy of such treatment has been difficult to demonstrate, presumably in part because patients must be highly motivated to respond to relaxation and biofeedback therapy. Only those few patients with mild hypertension who wish to use this method should be encouraged to try, and these patients should be closely followed and receive pharmacological treatment if necessary. Potassium Therapy There is a positive correlation between total body Na+ and blood pressure and a negative correlation between total body K+ and blood pressure in hypertensive patients (Lever et al., 1981). In addition, dietary intake, plasma concentrations, and urinary excretion of K+ are reduced in various populations of hypertensive subjects. Increased intake of K+ might reduce blood pressure by increasing excretion of Na+, suppressing renin secretion, causing arteriolar dilation (possibly by stimulating Na+, K+-ATPase activity and decreasing intracellular concentrations of Ca2+), and impairing responsiveness to endogenous vasoconstrictors. In hypertensive rats, supplementation with K+ decreases blood pressure and reduces the incidence of stroke, irrespective of blood pressure (Tobian, 1986). In mildly hypertensive patients, oral K+ supplements of 48 mmol per day reduce both systolic and diastolic blood pressure (Siani et al., 1987). Supplementation with K+ also may protect against ventricular ectopy and stroke (Khaw and Barrett-Connor, 1987). Based on all of these data, it seems prudent to use a high-K+ diet in conjunction with moderate restriction of Na+ in the nonpharmacological treatment of hypertension. However, a high-K+ diet should not be recommended for patients on angiotensin converting enzyme inhibitors. |
Prospectus
|
The most anticipated development in
antihypertensive therapy is the new knowledge expected from clinical trials
comparing the effectiveness of drugs on the important endpoints of morbidity
and mortality. One clinical trial addressing these endpoints has been
launched by the National Heart, Lung, and Blood Institute (Davis et al.,
1996). It is the Antihypertensive and Lipid Lowering treatment to prevent
Heart Attack Trial (ALLHAT) and is comparing the outcomes of treatment with a
thiazide-class diuretic (chlorthalidone), an angiotensin converting enzyme
inhibitor (lisinopril), a Ca2+-channel blocker (amlodipine), and
an A growing recognition of the contribution of the renin-angiotensin-aldosterone system (RAAS) to the development and progression of hypertensive end-organ damage promises to bring a number of studies examining new strategies for interrupting the RAAS. For example, studies comparing the efficacy of ACE inhibition, AT1-receptor antagonism, and the combination of ACE inhibition and AT1-receptor antagonism are ongoing. The finding that the addition of an aldosterone antagonist decreases mortality in patients with congestive heart failure treated with an ACE inhibitor (Pitt et al., 1999b) has sparked a renewed interest in aldosterone antagonists in the treatment of hypertension. These studies, if they are successful, could profoundly influence the approach to treating hypertension. For further discussion of hypertension, see Chapter 230 in Harrison's
Principles of Internal Medicine, 16th ed., |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1914
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved