| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Antimicrobial Agents: Sulfonamides, Trimethoprim-Sulfamethoxazole, Quinolones, and Agents for Urinary Tract Infections
Overview
|
Sulfonamides are used primarily in the treatment of urinary tract infections; in combination with trimethoprim, they also are frequently used for the treatment of otitis, bronchitis, sinusitis, and Pneumocystis carinii pneumonia. Emergence of resistance has limited their usefulness in other settings. The quinolone antibiotics, by virtue of their broad spectrum of antimicrobial activity against aerobic gram-negative bacilli, staphylococci, and gram-negative cocci and their oral bioavailability, are a very important class of antibiotics. Therapeutic uses include treatment of urinary tract infections, prostatitis, several sexually transmitted diseases, osteomyelitis, and bacterial diarrhea. New agents with excellent activity against the organisms of atypical pneumonia, anaerobes, and pneumococci are available for single-agent treatment of pneumonia. Quinolone antibiotics generally are not recommended for use in children or during pregnancy because of their potential to produce arthropathy. The urinary tract antiseptic methenamine is of value for chronic suppressive treatment of urinary tract infections. |
Sulfonamides
|
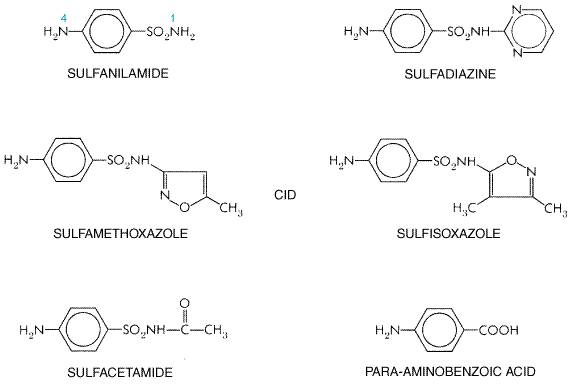
The sulfonamide drugs were the first effective chemotherapeutic agents to be employed systemically for the prevention and cure of bacterial infections in human beings. The considerable medical and public health importance of their discovery and their subsequent widespread use were quickly reflected in the sharp decline in morbidity and mortality figures for treatable infectious diseases. The advent of penicillin and subsequently of other antibiotics has diminished the usefulness of the sulfonamides, and they presently occupy a relatively small place in the therapeutic armamentarium of the physician. However, the introduction in the mid-1970s of the combination of trimethoprim and sulfamethoxazole has resulted in increased use of sulfonamides for the prophylaxis and/or treatment of specific microbial infections. History Investigations at the I. G. Farbenindustrie resulted, in 1932, in a German patent to Klarer and Mietzsch, covering PRONTOSIL and several other azo dyes containing a sulfonamide group. Domagk, a research director of the I. G. working with Klarer and Mietzsch, was aware of the fact that synthetic azo dyes had been studied for their action against streptococci, which prompted him to test the new compounds. He quickly observed that mice with streptococcal and other infections could be protected by PRONTOSIL (Domagk, 1935). The credit for the discovery of the chemotherapeutic value of PRONTOSIL belongs to Domagk, who was awarded the Nobel Prize in Medicine for 1938. In 1933, the first clinical case study was reported by Foerster, who gave PRONTOSIL to a 10-month-old infant with staphylococcal septicemia and obtained a dramatic cure. However, no great attention was paid elsewhere to these epoch-making advances in chemotherapy until the interest of English investigators was aroused. Colebrook and Kenny (1936) as well as Buttle and coworkers (1936) reported their favorable clinical results with PRONTOSIL and its active metabolite, sulfanilamide, in puerperal sepsis and meningococcal infections. These two reports awakened the medical profession to the new field of antibacterial chemotherapy, and experimental and clinical articles soon appeared in great profusion. The development of the carbonic anhydrase inhibitortype diuretics and the sulfonylurea hypoglycemic agents followed from observations made with the sulfonamide antibiotics. Chemistry The term sulfonamide is employed herein as a generic name for derivatives of para-aminobenzenesulfonamide (sulfanilamide); the structural formulas of selected members of this class are shown in nobreak Figure 441. Most of them are relatively insoluble in water, but their sodium salts are readily soluble. The minimal structural prerequisites for antibacterial action are all embodied in sulfanilamide itself. The SO2NH2 group is not essential as such, but the important feature is that the sulfur is directly linked to the benzene ring. The para-NH2 group (the N of which has been designated as N4) is essential and can be replaced only by such radicals as can be converted in vivo to a free amino group. Substitutions made in the amide NH2 group (the N of which has been designated as N1) have variable effects on antibacterial activity of the molecule. However, substitution of heterocyclic aromatic nuclei at N1 yields highly potent compounds.
Effects on Microbial Agents Sulfonamides have a wide range of antimicrobial activity against both gram-positive and gram-negative bacteria. However, resistant strains have become common in recent years, and the usefulness of these agents has diminished correspondingly. In general, the sulfonamides exert only a bacteriostatic effect, and cellular and humoral defense mechanisms of the host are essential for the final eradication of the infection. Antibacterial Spectrum Resistance to sulfonamides is increasingly a problem. Microorganisms
that may be susceptible in vitro to sulfonamides include Streptococcus
pyogenes, Streptococcus pneumoniae, Haemophilus influenzae, Haemophilus
ducreyi, Nocardia, Actinomyces, Calymmatobacterium granulomatis, and Chlamydia
trachomatis. Minimal inhibitory concentrations (MIC) range from 0.1 Although sulfonamides were used successfully for the management of
meningococcal infections for many years, the majority of isolates of Neisseria
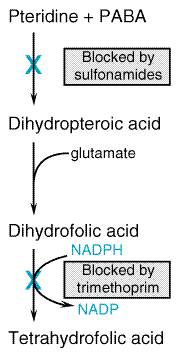
meningitidis of serogroups B and C in the Mechanism of Action Sulfonamides are structural analogs and competitive antagonists of para-aminobenzoic acid (PABA) and thus prevent normal bacterial utilization of PABA for the synthesis of folic acid (pteroylglutamic acid; see Fildes, 1940; Woods, 1940). More specifically, sulfonamides are competitive inhibitors of dihydropteroate synthase, the bacterial enzyme responsible for the incorporation of PABA into dihydropteroic acid, the immediate precursor of folic acid (Figure 442). Sensitive microorganisms are those that must synthesize their own folic acid; bacteria that can utilize preformed folate are not affected. Bacteriostasis induced by sulfonamides is counteracted by PABA competitively. Sulfonamides do not affect mammalian cells by this mechanism, since they require preformed folic acid and cannot synthesize it. They are, therefore, comparable to sulfonamide-insensitive bacteria that utilize preformed folate.
Synergists of Sulfonamides One of the most active agents that exerts a synergistic effect when used with a sulfonamide is trimethoprim (see Bushby and Hitchings, 1968). This compound is a potent and selective competitive inhibitor of microbial dihydrofolate reductase, the enzyme that reduces dihydrofolate to tetrahydrofolate. It is this reduced form of folic acid that is required for one-carbon transfer reactions. The simultaneous administration of a sulfonamide and trimethoprim thus introduces sequential blocks in the pathway by which microorganisms synthesize tetrahydrofolate from precursor molecules. The expectation that such a combination would yield synergistic antimicrobial effects has been realized both in vitro and in vivo (see below). Acquired Bacterial Resistance to Sulfonamides Bacteria resistant to sulfonamides are presumed to originate by random mutation and selection or by transfer of resistance by plasmids (Chapter 43: Antimicrobial Agents: General Considerations). Such resistance, once it is maximally developed, usually is persistent and irreversible, particularly when produced in vivo. Acquired resistance to sulfonamide usually does not involve cross-resistance to chemotherapeutic agents of other classes. The in vivo acquisition of resistance has little or no effect on either virulence or antigenic characteristics of microorganisms. Resistance to sulfonamide is probably the consequence of an altered enzymatic constitution of the bacterial cell; the alteration may be characterized by (1) a lower affinity for sulfonamides by the enzyme that utilizes PABA, dihydropteroate synthase; (2) decreased bacterial permeability or active efflux of the drug; (3) an alternative metabolic pathway for synthesis of an essential metabolite; or (4) an increased production of an essential metabolite or drug antagonist. For example, some resistant staphylococci may synthesize 70 times as much PABA as do the susceptible parent strains. Nevertheless, an increased production of PABA is not a constant finding in sulfonamide-resistant bacteria, and resistant mutants may possess enzymes for folate biosynthesis that are less readily inhibited by sulfonamides. Plasmid-mediated resistance is due to plasmid-encoded, drug-resistant dihydropteroate synthetase. Absorption, Fate, and Excretion Except for sulfonamides especially designed for their local effects in the bowel, this class of drugs is rapidly absorbed from the gastrointestinal tract. Approximately 70% to 100% of an oral dose is absorbed, and sulfonamide can be found in the urine within 30 minutes of ingestion. Peak plasma levels are achieved in 2 to 6 hours, depending on the drug. The small intestine is the major site of absorption, but some of the drug is absorbed from the stomach. Absorption from other sites, such as the vagina, respiratory tract, or abraded skin, is variable and unreliable, but a sufficient amount may enter the body to cause toxic reactions in susceptible persons or to produce sensitization. All sulfonamides are bound in varying degree to plasma proteins, particularly to albumin. The extent to which this occurs is determined by the hydrophobicity of a particular drug and its pKa; at physiological pH, drugs with a high pKa exhibit a low degree of protein binding, and vice versa. Sulfonamides are distributed throughout all tissues of the body. The diffusible fraction of sulfadiazine is uniformly distributed throughout the total body water, while sulfisoxazole is largely confined to the extracellular space. The sulfonamides readily enter pleural, peritoneal, synovial, ocular, and similar body fluids and may reach concentrations therein that are 50% to 80% of the simultaneously determined concentration in blood. Since the protein content of such fluids usually is low, the drug is present in the unbound active form. After systemic administration of adequate doses, sulfadiazine and sulfisoxazole attain concentrations in cerebrospinal fluid that may be effective in meningeal infections. At steady state, the concentration ranges between 10% and 80% of that in the blood. However, because of the emergence of sulfonamide-resistant microorganisms, these drugs are now used only rarely for the treatment of meningitis. Sulfonamides readily pass through the placenta and reach the fetal circulation. The concentrations attained in the fetal tissues are sufficient to cause both antibacterial and toxic effects. The sulfonamides undergo metabolic alterations in vivo, especially in the liver. The major metabolic derivative is the N4-acetylated sulfonamide. Acetylation, which occurs to a different extent with each agent, is disadvantageous, because the resulting products have no antibacterial activity and yet retain the toxic potentialities of the parent substance. Sulfonamides are eliminated from the body partly as the unchanged drug and partly as metabolic products. The largest fraction is excreted in the urine, and the half-life of sulfonamides in the body is thus dependent on renal function. In acid urine, the older sulfonamides are insoluble and may precipitate, causing crystalline deposits that can cause urinary obstruction (see below). Small amounts are eliminated in the feces and in bile, milk, and other secretions. Pharmacological Properties of Individual Sulfonamides The sulfonamides may be classified into three groups on the basis of the rapidity with which they are absorbed and excreted: (1) agents absorbed rapidly and excreted rapidly, such as sulfisoxazole and sulfadiazine; (2) agents absorbed very poorly when administered orally and hence active in the bowel lumen, such as sulfasalazine; (3) sulfonamides employed mainly for topical use, such as sulfacetamide, mafenide, and silver sulfadiazine; and (4) long-acting sulfonamides, sulfadoxine, which are absorbed rapidly but excreted slowly (Table 441). Rapidly Absorbed and Rapidly Eliminated Sulfonamides Sulfisoxazole Early studies of sulfisoxazole (GANTRISIN, others) established that it was a rapidly absorbed and rapidly excreted sulfonamide with excellent antibacterial activity. Since its high solubility eliminates much of the renal toxicity inherent in the use of older sulfonamides, it has essentially replaced the less-soluble agents. Sulfisoxazole is bound extensively to plasma proteins. Following an
oral dose of 2 to 4 g, peak concentrations in plasma of 110 to 250 Sulfisoxazole diolamine is available for topical use in the eye. Sulfisoxazole acetyl is tasteless and hence preferred for oral use in children. Sulfisoxazole also is marketed in a fixed-dose combination with phenazopyridine (sulfisoxazole, 500 mg; phenazopyridine, 50 mg) as a urinary tract antiseptic and analgesic. The urine becomes orange-red soon after ingestion of this mixture because of the presence of phenazopyridine, an orange-red dye. Sulfisoxazole acetyl also is marketed in combination with erythromycin ethylsuccinate (PEDIAZOLE, others) for use in children with otitis media. Fewer than 0.1% of patients receiving sulfisoxazole suffer serious toxic reactions. The untoward effects produced by this agent are similar to those that follow the administration of other sulfonamides, as discussed below. Because of its relatively high solubility in the urine as compared with sulfadiazine, sulfisoxazole only infrequently produces hematuria or crystalluria (0.2% to 0.3%). Despite this, patients taking this drug should ingest an adequate quantity of water. Sulfisoxazole and all sulfonamides that are absorbed must be used with caution in patients with impaired renal function. Like all other sulfonamides, sulfisoxazole may produce hypersensitivity reactions, some of which are potentially lethal. Sulfisoxazole currently is preferred over other sulfonamides by most clinicians when a rapidly absorbed and rapidly excreted sulfonamide is indicated. Sulfamethoxazole Sulfamethoxazole is a close congener of sulfisoxazole, but its rates of enteric absorption and urinary excretion are slower. It is administered orally and employed for both systemic and urinary tract infections. Precautions must be observed to avoid sulfamethoxazole crystalluria because of the high percentage of the acetylated, relatively insoluble form of the drug in the urine. The clinical uses of sulfamethoxazole are the same as those for sulfisoxazole. It also is marketed in fixed-dose combinations with phenazopyridine as a urinary antiseptic and analgesic, and with trimethoprim (see below). Sulfadiazine Sulfadiazine
given orally is rapidly absorbed from the gastrointestinal tract, and peak
blood concentrations are reached within 3 to 6 hours after a single dose.
Following an oral dose of 3 g, peak concentrations in plasma are 50 Sulfadiazine is excreted quite readily by the kidney in both the free and the acetylated form, rapidly at first and then more slowly over a period of 2 to 3 days. It can be detected in the urine within 30 minutes after oral ingestion. About 15% to 40% of the excreted sulfadiazine is in the acetylated form. This form of the drug is excreted more readily than the free fraction, and the administration of alkali accelerates the renal clearance of both forms by further diminishing their tubular reabsorption. In adults and children who are being treated with sulfadiazine, every precaution must be taken to ensure fluid intake adequate to produce a urine output of at least 1200 ml in adults and a corresponding quantity in children. If this cannot be accomplished, sodium bicarbonate may be given to reduce the risk of crystalluria. Poorly Absorbed Sulfonamides Sulfasalazine AZALINE AZULFIDINE) is very poorly absorbed from the gastrointestinal tract. It is used in the therapy of ulcerative colitis and regional enteritis, but relapses tend to occur in about one-third of patients who experience a satisfactory initial response. Corticosteroids are more effective in treating acute attacks, but sulfasalazine is preferred to corticosteroids for treatment of patients mildly or moderately ill with ulcerative colitis. The drug also is being employed as the first approach to treatment of relatively mild cases of regional enteritis and granulomatous colitis. Sulfasalazine is broken down by intestinal bacteria to sulfapyridine, an active sulfonamide that is absorbed and eventually excreted in the urine, and 5-aminosalicylate, which reaches high levels in the feces. There is evidence that this latter compound is the effective agent in inflammatory bowel disease, whereas the sulfapyridine is responsible for most of the toxicity. Toxic reactions include Heinz-body anemia, acute hemolysis in patients with glucose-6-phosphate dehydrogenase deficiency, and agranulocytosis. Nausea, fever, arthralgias, and rashes occur in up to 20% of patients treated with the drug; desensitization has been effective. Sulfasalazine can cause a reversible infertility in males due to changes in sperm number and morphology. There is no evidence that the compound alters the intestinal microflora of persons with ulcerative colitis. Sulfonamides for Topical Use Sulfacetamide Sulfacetamide is the N1-acetyl-substituted derivative of sulfanilamide. Its aqueous solubility (1:140) is approximately 90 times that of sulfadiazine. Solutions of the sodium salt of the drug (ISOPTO CETAMIDE SODIUMSULAMYD, others) are employed extensively in the management of ophthalmic infections. Although topical sulfonamide for most purposes is discouraged because of lack of efficacy and a high risk of sensitization, sulfacetamide has certain advantages. Very high aqueous concentrations are nonirritating to the eye and are effective against susceptible microorganisms. A 30% solution of the sodium salt has a pH of 7.4, whereas the solutions of sodium salts of other sulfonamides are highly alkaline. The drug penetrates into ocular fluids and tissues in high concentration. Sensitivity reactions to sulfacetamide are rare, but the drug should not be used in patients with known hypersensitivity to sulfonamides. Silver Sulfadiazine SILVADENE, others). This drug inhibits the growth in vitro of nearly all pathogenic bacteria and fungi, including some species resistant to sulfonamides. The compound is used topically to reduce microbial colonization and the incidence of infections of wounds from burns. It should not be used to treat an established deep infection. Silver is released slowly from the preparation in concentrations that are selectively toxic to the microorganisms. However, bacteria may develop resistance to silver sulfadiazine. While little silver is absorbed, the plasma concentration of sulfadiazine may approach therapeutic levels if a large surface area is involved. Adverse reactions are infrequent and include burning, rash, and itching. Silver sulfadiazine is considered by most authorities to be one of the agents of choice for the prevention of infection of burns. Mafenide This sulfonamide ( Long-Acting Sulfonamides Sulfadoxine (N1-[5,6-dimethoxy-4-pyrimidiny] sulfanilamide) is a sulfonamide with a particularly long half-life (7 to 9 days). It is utilized in combination with pyrimethamine (500 mg of sulfadoxine plus 25 mg of pyrimethamine as FANSIDAR) for the prophylaxis and treatment of malaria caused by mefloquine-resistant strains of Plasmodium falciparum (see Chapter 40: Drugs Used in the Chemotherapy of Protozoal Infections: Malaria). Because of severe and sometimes fatal reactions, including the StevensJohnson syndrome, the drug should be used for prophylaxis only where the risk of resistant malaria is high. Untoward Reactions to Sulfonamides The untoward effects that follow the administration of sulfonamides are numerous and varied; the overall incidence of reactions is about 5%. Certain forms of toxicity may be related to individual differences in sulfonamide metabolism (Shear et al., 1986). Disturbances of the Urinary Tract Although the risk of crystalluria was relatively high with the older, less soluble sulfonamides, the incidence of this problem is very low with more soluble agents such as sulfisoxazole. Crystalluria has occurred in dehydrated patients with AIDS who were receiving sulfadiazine for Toxoplasma encephalitis. Fluid intake should be such as to ensure a daily urine volume of at least 1200 ml (in adults). Alkalinization of the urine may be desirable if urine volume or pH is unusually low, since the solubility of sulfisoxazole increases greatly with slight elevations of pH. Disorders of the Hematopoietic System Acute Hemolytic Anemia The mechanism of the acute hemolytic anemia produced by sulfonamides is not always readily apparent. In some cases, it has been thought to be a sensitization phenomenon. In other instances, the hemolysis is related to an erythrocytic deficiency of glucose-6-phosphate dehydrogenase activity. Hemolytic anemia is rare after sulfadiazine (0.05%); its exact incidence following therapy with sulfisoxazole is unknown. Agranulocytosis Agranulocytosis occurs in about 0.1% of patients who receive sulfadiazine; it also can follow the use of other sulfonamides. Although return of granulocytes to normal levels may be delayed for weeks or months after sulfonamide is withdrawn, most patients recover spontaneously with supportive care. Aplastic Anemia Complete suppression of bone-marrow activity with profound anemia, granulocytopenia, and thrombocytopenia is an extremely rare occurrence with sulfonamide therapy. It probably results from a direct myelotoxic effect and may be fatal. However, reversible suppression of the bone marrow is quite common in patients with limited bone-marrow reserve (e.g., patients with AIDS or those receiving myelosuppressive chemotherapy). Hypersensitivity Reactions The incidence of other hypersensitivity reactions to sulfonamides is quite variable. Among the skin and mucous membrane manifestations attributed to sensitization to sulfonamide are morbilliform, scarlatinal, urticarial, erysipeloid, pemphigoid, purpuric, and petechial rashes; and erythema nodosum, erythema multiforme of the Stevens-Johnson type, Behet's syndrome, exfoliative dermatitis, and photosensitivity. Drug eruptions occur most often after the first week of therapy but may appear earlier in previously sensitized individuals. Fever, malaise, and pruritus are frequently present simultaneously. The incidence of untoward dermal effects is about 2% with sulfisoxazole, although patients with AIDS manifest a higher frequency of rashes with sulfonamide treatment than do other individuals. A syndrome similar to serum sickness may appear after several days of sulfonamide therapy. Drug fever is a common untoward manifestation of sulfonamide treatment; the incidence approximates 3% with sulfisoxazole. Focal or diffuse necrosis of the liver due to direct drug toxicity or sensitization occurs in fewer than 0.1% of patients. Headache, nausea, vomiting, fever, hepatomegaly, jaundice, and laboratory evidence of hepatocellular dysfunction usually appear 3 to 5 days after sulfonamide administration is started, and the syndrome may progress to acute yellow atrophy and death. Miscellaneous Reactions Anorexia, nausea, and vomiting occur in 1% to 2% of persons receiving sulfonamides, and these manifestations are probably central in origin. The administration of sulfonamides to newborn infants, especially if premature, may lead to the displacement of bilirubin from plasma albumin. In newborn infants, free bilirubin can become deposited in the basal ganglia and subthalamic nuclei of the brain, causing an encephalopathy called kernicterus. Sulfonamides should not be given to pregnant women near term because these drugs pass through the placenta and are secreted in milk. Drug Interactions The most important interactions of the sulfonamides involve those with the oral anticoagulants, the sulfonylurea hypoglycemic agents, and the hydantoin anticonvulsants. In each case, sulfonamides can potentiate the effects of the other drug by mechanisms that appear to involve primarily inhibition of metabolism and, possibly, displacement from albumin. Dosage adjustment may be necessary when a sulfonamide is given concurrently. Sulfonamide Therapy The number of conditions for which the sulfonamides are therapeutically useful and constitute drugs of first choice has been reduced sharply by the development of more effective antimicrobial agents and by the gradual increase in the resistance of a number of bacterial species to this class of drugs. However, the use of sulfonamides has undergone a revival as a result of the introduction of the combination of trimethoprim and sulfamethoxazole. Urinary Tract Infections Since a significant percentage of urinary tract infections in many parts of the world are caused by sulfonamide-resistant microorganisms, these drugs are no longer a therapy of first choice. Trimethoprimsulfamethoxazole, a quinolone, or ampicillin are the preferred agents. However, sulfisoxazole may be used effectively in areas where the prevalence of resistance is not high or when the organism is known to be sensitive. The usual dosage is 2 to 4 g initially followed by 1 to 2 g, orally, four times a day for 5 to 10 days. Patients with acute pyelonephritis with high fever and other severe constitutional manifestations are at risk of bacteremia and shock and should not be treated with a sulfonamide. Nocardiosis Sulfonamides are of value in the treatment of infections due to Nocardia
species. A number of instances of complete recovery from the disease after
adequate treatment with a sulfonamide have been recorded. Sulfisoxazole or sulfadiazine
may be given in dosages of 6 to 8 g daily. Concentrations of sulfonamide in
plasma should be 80 to 160 Toxoplasmosis The combination of pyrimethamine and sulfadiazine is the treatment of choice for toxoplasmosis (Montoya and Remington, 2000). Pyrimethamine is given as a loading dose of 75 mg followed by 25 mg orally per day, with sulfadiazine, 1 g orally every 6 hours, plus folinic acid, 10 mg orally each day, for at least 3 to 6 weeks. Patients should receive at least 2 liters of fluid intake daily to prevent crystalluria during therapy. Use of Sulfonamides for Prophylaxis The sulfonamides exhibit a degree of effectiveness equal to that of oral penicillin in preventing streptococcal infections and recurrences of rheumatic fever among susceptible subjects. Despite the efficacy of sulfonamides for long-term prophylaxis of rheumatic fever, their toxicity and the possibility of infection by drug-resistant streptococci make them less desirable than penicillin for this purpose. They should be used, however, without hesitation in patients who are hypersensitive to penicillin. If untoward responses occur, they usually do so during the first 8 weeks of therapy; serious reactions after this time are rare. White cell counts should be carried out once weekly during the first 8 weeks. |
TrimethoprimSulfamethoxazole
|
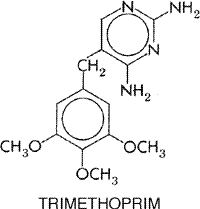
The introduction of trimethoprim in combination with sulfamethoxazole constitutes an important advance in the development of clinically effective antimicrobial agents and represents the practical application of a theoretical consideration; that is, if two drugs act on sequential steps in the pathway of an obligate enzymatic reaction in bacteria (see Figure 442), the result of their combination will be synergistic (see Hitchings, 1961). In much of the world the combination is known as co-trimoxazole. In addition to its combination with sulfamethoxazole (BACTRIM SEPTRA, others), trimethoprim also is available as a single entity preparation (TRIMPEX PROLOPRIM, others). Chemistry Sulfamethoxazole has been discussed earlier in this chapter, and its structural formula is shown in Figure 441. The history of trimethoprim, a diaminopyrimidine, is discussed in Chapter 40: Drugs Used in the Chemotherapy of Protozoal Infections: Malaria. Its structural formula is as follows:
Antibacterial Spectrum The antibacterial spectrum of trimethoprim is similar to that of sulfamethoxazole, although the former drug is usually 20 to 100 times more potent than the latter. Most gram-negative and gram-positive microorganisms are sensitive to trimethoprim, but resistance can develop when the drug is used alone. Pseudomonas aeruginosa, Bacteroides fragilis, and enterococci usually are resistant. There is significant variation in the susceptibility of Enterobacteriaceae to trimethoprim in different geographical locations because of the spread of resistance mediated by plasmids and transposons. The data presented below refer to the antimicrobial activity of the combination of trimethoprim and sulfamethoxazole. Chlamydia diphtheriae and N. meningitidis are susceptible to trimethoprimsulfamethoxazole. Although most S. pneumoniae are susceptible, there has been a disturbing increase in resistance (see below). From 50% to 95% of strains of Staphylococcus aureus, Staphylococcus epidermidis, S. pyogenes, the viridans group of streptococci, E. coli, Proteus mirabilis, Proteus morganii, Proteus rettgeri, Enterobacter species, Salmonella, Shigella, Pseudomonas pseudomallei, Serratia, and Alcaligenes species are inhibited. Also sensitive are Klebsiella species, Brucella abortus, Pasteurella haemolytica, Yersinia pseudotuberculosis, Yersinia enterocolitica, and Nocardia asteroides. Methicillin-resistant strains of S. aureus, although also resistant to trimethoprim or sulfamethoxazole alone, may be susceptible to the combination. A synergistic interaction between the components of the preparation is apparent even when microorganisms are resistant to sulfonamide or are resistant to sulfonamide and moderately resistant to trimethoprim. However, a maximal degree of synergism occurs when microorganisms are sensitive to both components. The activity of trimethoprimsulfamethoxazole in vitro depends on the medium in which it is determined; for example, low concentrations of thymidine almost completely abolish the antibacterial activity (see Symposium, 1973). Mechanism of Action The antimicrobial activity of the combination of trimethoprim and sulfamethoxazole results from its actions on two steps of the enzymatic pathway for the synthesis of tetrahydrofolic acid. As shown earlier in Figure 442, sulfonamide inhibits the incorporation of para-aminobenzoic acid (PABA) into folic acid, and trimethoprim prevents the reduction of dihydrofolate to tetrahydrofolate. The latter is the form of folate essential for one-carbon transfer reactions, for example, the synthesis of thymidylate from deoxyuridylate. Selective toxicity for microorganisms is achieved in two ways. Mammalian cells utilize preformed folates from the diet and do not synthesize the compound. Furthermore, trimethoprim is a highly selective inhibitor of dihydrofolate reductase of lower organisms, with 100,000-fold more drug required to inhibit human reductase than the bacterial enzyme. This is vitally important, since this enzymatic function is a crucial one in all species. The synergistic interaction between sulfonamide and trimethoprim is predictable from their respective mechanisms. There is an optimal ratio of the concentrations of the two agents for synergism, and this is equal to the ratio of the minimal inhibitory concentrations of the drugs acting independently. While this ratio varies for different bacteria, the most effective ratio for the greatest number of microorganisms is 20 parts of sulfamethoxazole to 1 part of trimethoprim. The combination is thus formulated to achieve a sulfamethoxazole concentration in vivo 20 times greater than that of trimethoprim. (See articles by Hitchings, Burchall, and Bushby in Symposium, 1973.) The pharmacokinetic properties of the sulfonamide chosen to be in combination with trimethoprim are thus important, since relative constancy of the concentrations of the two compounds in the body is desired. Bacterial Resistances Bacterial resistance to trimethoprimsulfamethoxazole is a rapidly
increasing problem, although resistance is lower than it is to either of the
agents alone. Resistance often is due to the acquisition of a plasmid that
codes for an altered dihydrofolate reductase. The development of resistance
is a problem for treatment of many different bacterial infections. In a
survey of children with otitis media in Absorption, Distribution, and Excretion The pharmacokinetic profiles of sulfamethoxazole and trimethoprim are closely but not perfectly matched to achieve a constant ratio of 20:1 in their concentrations in blood and tissues. The ratio in blood is often greater than 20:1 and that in tissues is frequently less. After a single oral dose of the combined preparation, trimethoprim is absorbed more rapidly than sulfamethoxazole. The concurrent administration of the drugs appears to slow the absorption of sulfamethoxazole. Peak blood concentrations of trimethoprim usually occur by 2 hours in most patients, while peak concentrations of sulfamethoxazole occur by 4 hours after a single oral dose. The half-lives of trimethoprim and sulfamethoxazole are approximately 11 and 10 hours, respectively. When 800 mg of sulfamethoxazole is given with 160 mg of trimethoprim
(the conventional 5:1 ratio) twice daily, the peak concentrations of the
drugs in plasma are approximately 40 and 2 Trimethoprim is rapidly distributed and concentrated in tissues, and about 40% is bound to plasma protein in the presence of sulfamethoxazole. The volume of distribution of trimethoprim is almost nine times that of sulfamethoxazole. The drug readily enters cerebrospinal fluid and sputum. High concentrations of each component of the mixture also are found in bile. About 65% of sulfamethoxazole is bound to plasma protein. About 60% of administered trimethoprim and from 25% to 50% of administered sulfamethoxazole are excreted in the urine in 24 hours. Two-thirds of the sulfonamide is unconjugated. Metabolites of trimethoprim also are excreted. The rates of excretion and the concentrations of both compounds in the urine are significantly reduced in patients with uremia. Untoward Effects There is no evidence that trimeth-oprimsulfamethoxazole, when given in the recommended doses, induces folate deficiency in normal persons. However, the margin between toxicity for bacteria and that for human beings may be relatively narrow when the cells of the patient are deficient in folate. In such cases, trimethoprimsulfamethoxazole may cause or precipitate megaloblastosis, leukopenia, or thrombocytopenia. In routine use, the combination appears to exert little toxicity. About 75% of the untoward effects involve the skin. These are typical of those known to be produced by sulfonamides, as already described. However, trimethoprimsulfamethoxazole has been reported to cause up to three times as many dermatological reactions as does sulfisoxazole when given alone (5.9%versus 1.7%; Arndt and Jick, 1976). Exfoliative dermatitis, StevensJohnson syndrome, and toxic epidermal necrolysis (Lyell's syndrome) are rare, occurring primarily in older individuals. Nausea and vomiting constitute the bulk of gastrointestinal reactions; diarrhea is rare. Glossitis and stomatitis are relatively common. Mild and transient jaundice has been noted and appears to have the histological features of allergic cholestatic hepatitis. Central nervous system reactions consist of headache, depression, and hallucinations, manifestations known to be produced by sulfonamides. Hematological reactions, in addition to those mentioned above, are various types of anemia (including aplastic, hemolytic, and macrocytic), coagulation disorders, granulocytopenia, agranulocytosis, purpura, HenochSchnlein purpura, and sulfhemoglobinemia. Permanent impairment of renal function may follow the use of trimethoprimsulfamethoxazole in patients with renal disease, and a reversible decrease in creatinine clearance has been noted in patients with normal renal function (Symposium, 1973). Patients with AIDS frequently react adversely when trimethoprimsulfamethoxazole is administered to treat infection due to P. carinii (Gordin et al., 1984). These adverse reactions include rash, neutropenia, Stevens-Johnson syndrome, Sweet's syndrome, and pulmonary infiltrates. It may be possible to continue therapy via rapid oral desensitization (Gluckstein and Ruskin, 1995). Therapeutic Uses Urinary Tract Infections Treatment of uncomplicated lower urinary tract infections with trimethoprimsulfamethoxazole often is highly effective for sensitive bacteria. The preparation has been shown to produce a better therapeutic effect than does either of its components given separately when the infecting microorganisms are of the family Enterobacteriaceae. Single-dose therapy (320 mg of trimethoprim plus 1600 mg of sulfamethoxazole in adults) has been effective in some cases for the treatment of acute uncomplicated urinary tract infections, but a minimum of 3 days of therapy is more likely to be effective (Zinner and Mayer, 2000; Stamm and Hooton, 1993). The combination appears to have special efficacy in chronic and recurrent infections of the urinary tract. Small doses (200 mg of sulfamethoxazole plus 40 mg of trimethoprim per day, or two to four times these amounts once or twice per week) appear to be effective in reducing the number of recurrent urinary tract infections in adult females. This effect may be related to the presence of therapeutic concentrations of trimethoprim in vaginal secretions. Enterobacteriaceae surrounding the urethral orifice may be eliminated or markedly reduced in number, thus diminishing the chance of an ascending reinfection. Trimethoprim also is found in therapeutic concentrations in prostatic secretions, and trimethoprimsulfamethoxazole is often effective for the treatment of bacterial prostatitis. Bacterial Respiratory Tract Infections Trimethoprimsulfamethoxazole is effective for acute exacerbations of chronic bronchitis. Administration of 800 to 1200 mg of sulfamethoxazole plus 160 to 240 mg of trimethoprim twice a day appears to be effective in decreasing fever, purulence and volume of sputum, and sputum bacterial count. Trimethoprimsulfamethoxazole should not be used to treat streptococcal pharyngitis, since it does not eradicate the microorganism. It is effective for acute otitis media in children and acute maxillary sinusitis in adults caused by susceptible strains of H. influenzae and S. pneumoniae. Gastrointestinal Infections The combination is an alternative for fluoroquinolone for treatment of shigellosis, since many strains of the causative agent are now resistant to ampicillin; however, resistance to trimethoprimsulfamethoxazole is increasingly common. It is also a second-line drug (ceftriaxone or a fluoroquinolone is the preferred treatment) for typhoid fever, but resistance is an increasing problem. In adults, trimethoprim-sulfamethoxazole appears to be effective when the dose is 800 mg of sulfamethoxazole plus 160 mg of trimethoprim every 12 hours for 15 days. Trimethoprimsulfamethoxazole appears to be effective in the management of carriers of sensitive strains of Salmonella typhi and other species of Salmonella. One proposed schedule is the administration of 800 mg of sulfamethoxazole plus 160 mg of trimethoprim twice a day for 3 months; however, failures have occurred. The presence of chronic disease of the gallbladder may be associated with a high incidence of failure to clear the carrier state. Acute diarrhea due to sensitive strains of enteropathogenic E. coli can be treated or prevented with either trimethoprim or trimethoprim plus sulfamethoxazole (Hill and Pearson, 1988). However, antibiotic treatment (either trimethoprimsulfamethoxazole or cephalosporin) of diarrheal illness due to enterohemorrhagic E. coli O157:H7 may increase the risk of hemolytic-uremic syndrome, perhaps by increasing the release of shiga toxin by the bacteria (Wong et al., 2000). Infection by Pneumocystis carinii High-dose therapy (trimethoprim, 20 mg/kg per day, plus sulfamethoxazole, 100 mg/kg per day, in three or four divided doses) is effective for this severe infection in patients with AIDS. This combination compares favorably to pentamidine for treatment of this disease. Adjunctive corticosteroids should be given at the onset of antipneumocystis therapy in patients with a PO2 of <70 mm Hg or an alveolar-arterial gradient of >35 mm Hg (Lane et al., 1994). However, the incidence of side effects is high for both regimens (Sattler and Remington, 1981; Lane et al., 1994). Lower-dose oral therapy with 800 mg of sulfamethoxazole plus 160 mg of trimethoprim (given twice daily) has been used successfully in AIDS patients with less severe pneumonia (PO2 >60 mm Hg: Medina et al., 1990). Prophylaxis with 800 mg of sulfamethoxazole and 160 mg of trimethoprim once daily or three times a week is effective in preventing pneumonia caused by this organism in patients with AIDS (Schneider et al., 1992; Gallant et al., 1994). Adverse reactions are less frequent with the lower prophylactic doses of trimethoprimsulfamethoxazole. The most common problems are rash, fever, leukopenia, and hepatitis. Prophylaxis in Neutropenic Patients Several studies have demonstrated the effectiveness of low-dose therapy (150 mg/m2 of body-surface area of trimethoprim and 750 mg/m2 of body-surface area of sulfamethoxazole) for the prophylaxis of infection by P. carinii (see Hughes et al., 1977). In addition, significant protection against sepsis caused by gram-negative bacteria was noted when 800 mg of sulfamethoxazole plus 160 mg of trimethoprim were given twice daily to severely neutropenic patients. The emergence of resistant bacteria may limit the usefulness of trimethoprimsulfamethoxazole for prophylaxis (Gualtieri et al., 1983). Miscellaneous Infections Nocardia infections have been treated successfully with the combination, but failures also have been reported. Although a combination of doxycycline and streptomycin or gentamicin now is considered to be the treatment of choice for brucellosis, trimethoprimsulfamethoxazole may be an effective substitute for the doxycycline combination. Trimethoprimsulfamethoxazole also has been used successfully in the treatment of Whipple's disease, infection by Stenotrophomonas maltophilia, and the intestinal parasites Cyclospora and Isospora. Wegener's granulomatosis may respond, depending on the stage of the disease. |
The Quinolones
|
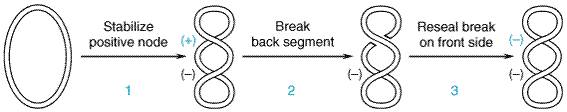
Older members of this class of synthetic antimicrobial agents, particularly nalidixic acid, have been available for the treatment of urinary tract infections for many years. These drugs are of relatively minor significance because of their limited therapeutic utility and the rapid development of bacterial resistance. Against this backdrop, the more recent introduction of fluorinated 4-quinolones, such as ciprofloxacin (CIPRO) and ofloxacin (FLOXIN), represents a particularly important therapeutic advance, since these agents have broad antimicrobial activity and are effective after oral administration for the treatment of a wide variety of infectious diseases. Relatively few side effects appear to accompany the use of these fluoroquinolones, and microbial resistance to their action does not develop rapidly (see Andriole, 1993; Hooper, 2000a). Chemistry The compounds that are currently available for clinical use in the Mechanism of Action The quinolone antibiotics target bacterial DNA gyrase and topoisomerase IV (Drlica and Zhao, 1997). For many gram-positive bacteria (such as S. aureus), topoisomerase IV is the primary activity inhibited by the quinolones (Ng et al., 1996). In contrast, for many gram-negative bacteria (such as E. coli) DNA gyrase is the primary quinolone target (Hooper, 2000a; Alovero et al., 2000). The two strands of double-helical DNA must be separated to permit DNA replication or transcription. However, anything that separates the strands results in 'overwinding' or excessive positive supercoiling of the DNA in front of the point of separation. To combat this mechanical obstacle, the bacterial enzyme DNA gyrase is responsible for the continuous introduction of negative supercoils into DNA. This is an ATP-dependent reaction requiring that both strands of the DNA be cut to permit passage of a segment of DNA through the break; the break is then resealed. The DNA gyrase of E. coli is composed of two
105,000-dalton A subunits and two 95,000-dalton B subunits encoded by the gyrA
and gyrB genes, respectively. The A subunits, which carry out the
strand-cutting function of the gyrase, are the site of action of the
quinolones (Figure 443). The drugs inhibit gyrase-mediated DNA supercoiling
at concentrations that correlate well with those required to inhibit
bacterial growth (0.1 to 10
Topoisomerase
IV also is composed of four subunits encoded by the parC and parE genes in E. coli (Drlica and Zhao, 1997;
Hooper, 2000a). Topoisomerase IV separates interlinked (catenated) daughter
DNA molecules that are the product of DNA replication. Eukaryotic cells do
not contain DNA gyrase. However, they do contain a conceptually and
mechanistically similar type II DNA topoisomerase that removes positive
supercoils from eukaryotic DNA to prevent its tangling during replication.
Quinolones inhibit eukaryotic type II topoisomerase only at much higher
concentrations (100 to 1000 Antibacterial Spectrum The fluoroquinolones are potent bactericidal agents against E. coli
and various species of Salmonella, Shigella, Enterobacter, Campylobacter,
and Neisseria (see Eliopoulos and Eliopoulos, 1993). Minimal
inhibitory concentrations of the fluoroquinolones for 90% of these strains
(MIC90) are usually less than 0.2 Activity against streptococci is limited to a subset of the
quinolones, including grepafloxacin (RAXAR, now withdrawn from the market), levofloxacin
(LEVAQUIN), gatifloxacin (TEQUIN), clinafloxacin, and moxifloxacin
(ALELOX) (Hooper, 2000a; Eliopoulos and
Eliopoulos, 1993). Several intracellular bacteria are inhibited by
fluoroquinolones at concentrations that can be achieved in plasma; these
include species of Chlamydia, Mycoplasma, Legionella, Brucella, and Mycobacterium
(including Mycobacterium tuberculosis; Leysen et al., 1989; Alangaden
and Lerner, 1997). Ciprofloxacin, ofloxacin (FLOXIN), pefloxacin, and sparfloxacin
(ZAGAM) have MIC90 values
from 0.5 to 3 Several of the new fluoroquinolones have activity against anaerobic bacteria, including trovafloxacin (TROVAN), gatifloxacin, moxifloxacin, clinafloxacin, and sitafloxacin (Medical Letter, 2000). Although nalidixic acid (NEGGRAM) and cinoxacin (CINOBAC) are bactericidal to most of the
common gram-negative bacteria that cause urinary tract infections, their
intrinsic activity is limited. Concentrations of nalidixic acid that approach
20 Resistance to quinolones may develop during therapy via mutations in the bacterial chromosomal genes encoding DNA gyrase or topoisomerase IV, or by active transport of the drug out of the bacteria (Oethinger et al., 2000). No quinolone-modifying or inactivating activities have been identified in bacteria (Gold and Moellering, 1996; Ng et al., 1996; Okusu et al., 1996). Resistance has increased after the introduction of fluoroquinolones, especially in Pseudomonas and staphylococci (Pegues et al., 1998; Peterson et al., 1998). Increasing fluoroquinolone resistance also is being observed in Clostridium jejuni, Salmonella, Neisseria gonorrhoeae, and S. pneumoniae (Smith et al., 1999; Centers for Disease Control, 1994b; Thornsberry et al., 1997; Mlbak et al., 1999). Absorption, Fate, and Excretion The quinolones are well absorbed after oral administration and are
widely distributed in body tissues. Peak serum levels of the fluoroquinolones
are obtained within 1 to 3 hours of an oral dose of 400 mg, with peak levels
ranging from 1.1 Routes of elimination differ among the quinolones. Renal clearance predominates for ofloxacin, lomefloxacin, and cinoxacin; pefloxacin, nalidixic acid, sparfloxacin, grepafloxacin, and trovafloxacin are predominantly eliminated nonrenally. Dose adjustments in patients with renal insufficiency are required for cinoxacin, norfloxacin, ciprofloxacin, ofloxacin, enoxacin, and lomefloxacin but not for nalidixic acid, grepafloxacin, trovafloxacin, and pefloxacin. None of the agents is efficiently removed by peritoneal or hemodialysis. A fluoroquinolone other than trovafloxacin, grepafloxacin, or pefloxacin should be used in patients with hepatic failure. Adverse Effects Quinolones and fluoroquinolones are generally well tolerated (Lipsky and Baker, 1999). The most common adverse reactions involve the gastrointestinal tract, with 3% to 17% of patients reporting mostly mild nausea, vomiting, and/or abdominal discomfort. Diarrhea and antibiotic-associated colitis have been unusual. Central nervous system side effects, predominately mild headache and dizziness, have been seen in 0.9% to 11% of patients. Rarely, hallucinations, delirium, and seizures have occurred, predominantly in patients who also were receiving theophylline or a nonsteroidal antiinflammatory drug (see below). Nonsteroidal antiinflammatory drugs may augment displacement of gamma-aminobutyric acid (GABA) from its receptors by the quinolones (Halliwell et al., 1993). Rashes, including photosensitivity reactions, also can occur. All of these agents can produce arthropathy in several species of immature animals. Traditionally, the use of quinolones in children has been contraindicated for that reason. However, children with cystic fibrosis given ciprofloxacin, norfloxacin, and nalidixic acid have had few, and reversible, joint symptoms (Burkhardt et al., 1997). Therefore, in some cases the benefits may outweigh the risks of quinolone therapy in children. Arthralgias and joint swelling have developed in children receiving fluoroquinolones; therefore, these drugs are not generally recommended for use in prepubertal children or pregnant women. Ciprofloxacin, grepafloxacin, pefloxacin, and enoxacin inhibit the metabolism of theophylline, and toxicity from elevated concentrations of the methylxanthine may occur (Schwartz et al., 1988). Concurrent administration of a nonsteroidal antiinflammatory drug may potentiate the central nervous system stimulant effects of the quinolones, with seizures reported in patients receiving enoxacin and fenbufen. Leukopenia, eosinophilia, and mild elevations in serum transaminases rarely occur. Rare but serious hepatic damage, including liver failure resulting in death, was observed with patients receiving trovafloxacin. For this reason, the use of trovafloxacin has been restricted to serious or life-threatening infections where the benefits of therapy outweigh the risk (Public Health Advisory, 1999). Temafloxacin was removed from the market by the manufacturer after approximately 1 out of 5000 prescriptions resulted in hemolysis, renal failure, thrombocytopenia, and/or disseminated intravascular coagulation. These problems have rarely been seen with other quinolones. QTc interval (QT interval, corrected for heart rate) prolongation has been observed for sparfloxacin and for grepafloxacin, which is no longer marketed. Therapeutic Uses Urinary Tract Infections Nalidixic acid and cinoxacin are useful only for urinary tract
infections caused by susceptible microorganisms. The fluoroquinolones are
significantly more potent and have a much broader spectrum of antimicrobial
activity. Norfloxacin is approved for use in the Prostatitis Norfloxacin, ciprofloxacin, and ofloxacin all have been effective in uncontrolled trials for the treatment of prostatitis caused by sensitive bacteria. Fluoroquinolones administered for 4 to 6 weeks appear to be effective in patients not responding to trimethoprimsulfamethoxazole (Hooper and Wolfson, 1991). Sexually Transmitted Diseases Fluoroquinolones lack activity for Treponema pallidum but have activity in vitro against N. gonorrhoeae, C. trachomatis, and Haemophilus ducreyi. It is important to remember that the quinolones are contraindicated in pregnancy. For chlamydial urethritis/cervicitis, a 7-day course of ofloxacin or sparfloxacin is an alternative to a 7-day treatment with doxycycline or a single dose of azithromycin; other available quinolones have not been reliably effective. A single oral dose of a fluoroquinolone such as ofloxacin or ciprofloxacin is effective treatment for gonorrhea and is an alternative to intramuscular ceftriaxone or oral cefixime for this infection, although fluoroquinolone resistance is emerging (Centers for Disease Control, 1994b). Pelvic inflammatory disease has been treated effectively with a 14-day course of ofloxacin combined with an antibiotic with activity against anaerobes (clindamycin or metronidazole) (Centers for Disease Control and Prevention, 1998). Chancroid (infection by H. ducreyi) can be treated with 3 days of ciprofloxacin or enoxacin. Gastrointestinal and Abdominal Infections For traveler's diarrhea (frequently caused by enterotoxigenic E. coli), the quinolones are equal to trimethoprim-sulfamethoxazole in effectiveness, reducing the duration of loose stools by 1 to3 days (DuPont and Ericsson, 1993). Norfloxacin, ciprofloxacin, and ofloxacin given for 5 days all have been effective in the treatment of patients with shigellosis, with even shorter courses effective in many cases (Bennish et al., 1992). Norfloxacin is superior to trimethoprim-sulfamethoxazole in decreasing the duration of diarrhea in cholera (Bhattacharya et al., 1990). Ciprofloxacin and ofloxacin treatment cures most patients with enteric fever caused by S. typhi as well as bacteremic nontyphoidal infections in AIDS patients, and it clears chronic fecal carriage. Shigellosis is effectively treated with either ciprofloxacin or azithromycin (Khan et al., 1997). The in vitro ability of the quinolones to induce the Shiga toxin stx2 gene in E. coli suggests that the quinolones should not be used for Shiga toxin-producing E. coli (Miedouge et al., 2000). Ciprofloxacin and ofloxacin have been less effective in treating episodes of peritonitis occurring in patients on chronic ambulatory peritoneal dialysis, likely due to the higher MICs for these drugs for the coagulase-negative staphylococci that are a common cause of peritonitis in this setting. Respiratory Tract Infections The major limitation of the use of quinolones for the treatment of
community-acquired pneumonia and bronchitis had been the poor in vitro
activity of ciprofloxacin, ofloxacin, and norfloxacin against S.
pneumoniae and anaerobic bacteria. However many of the new
fluoroquinolonesincluding levofloxacin, trovafloxacin, gatifloxacin,
clinafloxacin, and moxifloxacinhave excellent activity against S.
pneumoniae. Initial clinical experience with some of these newer
quinolones shows comparable efficacy to Bone, Joint, and Soft Tissue Infections The treatment of chronic osteomyelitis requires prolonged (weeks to months) antimicrobial therapy with agents active against S. aureus and gram-negative rods. The fluoroquinolones, by virtue of their oral administration and appropriate antibacterial spectrum for these infections, may be appropriately used in some cases (Gentry and Rodriguez-Gomez, 1991). Higher doses of ciprofloxacin are used than for treatment of urinary tract infections; recommended doses are 500 mg every 12 hours or, if severe, 750 mg twice daily. Therapy usually is continued for 7 to 14 days; bone and joint infections may require treatment for 4 to 6 weeks or more. Dosage should be reduced for patients with severely impaired renal function. Ciprofloxacin should not be given to children or pregnant women. Clinical cures have been as high as 75% in chronic osteomyelitis in which gram-negative rods predominated (Hooper, 2000a). Failures have been associated with the development of resistance in S. aureus, P. aeruginosa, and Serratia marcescens. In diabetic foot infections, which are commonly caused by a mixture of bacteria including gram-negative rods, anaerobes, streptococci, and staphylococci, the fluoroquinolones in combination with an agent with antianaerobic activity are a reasonable choice. Ciprofloxacin as sole therapy was effective in 50% of diabetic foot infections (Peterson et al., 1989). Other Infections The quinolones may be used as part of multiple-drug regimens for the
treatment of multidrug-resistant tuberculosis and for the treatment of
atypical mycobacterial infections as well as Mycobacterium avium
complex infections in AIDS (see Chapter 48: Antimicrobial Agents:
Drugs Used in the Chemotherapy of Tuberculosis, Mycobacterium avium
Complex Disease, and Leprosy). In neutropenic cancer patients with fever, the
combination of a quinolone with an aminoglycoside is comparable to |
Antiseptic and Analgesic Agents for Urinary Tract Infections
|
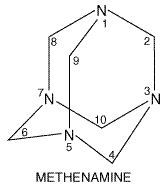
The urinary tract antiseptics inhibit the growth of many species of bacteria. They cannot be used to treat systemic infections because effective concentrations are not achieved in plasma with safe doses. However, because they are concentrated in the renal tubules, they can be used by oral administration to treat infections of the urinary tract. Furthermore, effective antibacterial concentrations reach the renal pelves and the bladder. Treatment with such drugs can be thought of as local therapy in that only in the kidney and bladder, with the rare exceptions mentioned below, are adequate therapeutic levels achieved (see Hooper, 2000b). Methenamine Methenamine is a urinary tract antiseptic that owes its activity to formaldehyde. Chemistry Methenamine is hexamethylenetetramine (hexa-methyleneamine). It has the following structure:
The compound decomposes in water to generate formaldehyde, according to the following reaction: NH4(CH2)6+
6H2O + 4H+ At pH 7.4, almost no decomposition occurs; however, 6% of the theoretical amount of formaldehyde is yielded at pH 6 and 20% at pH 5. Thus, acidification of the urine promotes the formaldehyde-dependent antibacterial action. The reaction is fairly slow, and 3 hours are required to reach 90% of completion (Strom and Jun, 1993). Antimicrobial Activity Nearly all bacteria are sensitive to free formaldehyde at
concentrations of about 20 Pharmacology and Toxicology Methenamine is absorbed orally, but 10% to 30% decomposes in the
gastric juice unless the drug is protected by an enteric coating. Because of
the ammonia produced, methenamine is contraindicated in hepatic
insufficiency. Excretion in the urine is nearly quantitative. When the urine
pH is 6 and the daily urine volume is 1000 to 1500 ml, a daily dose of 2 g
will yield a concentration of 18 to 60 Gastrointestinal distress frequently is caused by doses greater than 500 mg four times a day, even with enteric-coated tablets. Painful and frequent micturition, albuminuria, hematuria, and rashes may result from doses of 4 to 8 g a day given for longer than 3 to 4 weeks. Once the urine is sterile, a high dose should be reduced. Because systemic methenamine has low toxicity at the typically used doses, renal insufficiency does not constitute a contraindication to the use of methenamine alone, but the acids given concurrently may be detrimental. Methenamine mandelate is contraindicated in renal insufficiency. Crystalluria from the mandelate moiety can occur. Methenamine combines with sulfamethizole and perhaps other sulfonamides in the urine, which results in mutual antagonism. Therapeutic Uses and Status Methenamine is not a primary drug for the treatment of acute urinary tract infections, but it is of value for chronic suppressive treatment (Stamm and Hooton, 1993). The agent is most useful when the causative organism is E. coli, but it can usually suppress the common gram-negative offenders and often S. aureus and S. epidermidis as well. Enterobacter aerogenes and Proteus vulgaris are usually resistant. Urea-splitting bacteria (mostly Proteus) make it difficult to control the urine pH. The physician should strive to keep the pH below 5.5. Nitrofurantoin Nitrofurantoin FURADANTIN MACROBID, others) is a synthetic nitrofuran that is used for the prevention and treatment of infections of the urinary tract. Its structural formula is as follows:
Antimicrobial Activity Enzymes capable of reducing nitrofurantoin appear to be crucial for
its activation. Highly reactive intermediates are formed, and these seem to
be responsible for the observed capacity of the drug to damage DNA. Bacteria
reduce nitrofurantoin more rapidly than do mammalian cells, and this is
thought to account for the selective antimicrobial activity of the compound.
Bacteria that are susceptible to the drug rarely become resistant during therapy.
Nitrofurantoin is active against many strains of E. coli and
enterococci. However, most species of Proteus and Pseudomonas
and many of Enterobacter and Klebsiella are resistant.
Nitrofurantoin is bacteriostatic for most susceptible microorganisms at
concentrations of 32 Pharmacology and Toxicity Nitrofurantoin is rapidly and completely absorbed from the
gastrointestinal tract. The macrocrystalline form of the drug is absorbed and
excreted more slowly. Antibacterial concentrations are not achieved in plasma
following ingestion of recommended doses, because the drug is rapidly
eliminated. The plasma half-life is 0.3 to 1 hour; about 40% is excreted
unchanged into the urine. The average dose of nitrofurantoin yields a
concentration in urine of approximately 200 The most common untoward effects are nausea, vomiting, and diarrhea; the macrocrystalline preparation is better tolerated. Various hypersensitivity reactions occasionally occur. These include chills, fever, leukopenia, granulocytopenia, hemolytic anemia [associated with glucose-6-phosphate dehydrogenase deficiency (Gait, 1990)], cholestatic jaundice, and hepatocellular damage. Chronic active hepatitis is an uncommon but serious side effect (Black et al., 1980; Tolman, 1980). Acute pneumonitis with fever, chills, cough, dyspnea, chest pain, pulmonary infiltration, and eosinophilia may occur within hours to days of the initiation of therapy; it usually resolves within hours after discontinuation of the drug. More insidious subacute reactions also may be noted, and interstitial pulmonary fibrosis can occur in patients on chronic medication. This appears to be due to generation of oxygen radicals as a result of redox cycling of the drug in the lung. Elderly patients are especially susceptible to the pulmonary toxicity of nitrofurantoin (see Holmberg et al., 1980). Megaloblastic anemia is rare. Various neurological disorders occasionally are observed. Headache, vertigo, drowsiness, muscular aches, and nystagmus are readily reversible, but severe polyneuropathies with demyelination and degeneration of both sensory and motor nerves have been reported; signs of denervation and muscle atrophy result. Neuropathies are most likely to occur in patients with impaired renal function and in persons on long-continued treatment. Nitrofurantoin-induced polyneuropathy has been reviewed by Toole and Parrish (1973). Certain adverse reactions may be caused by toxic reactive metabolites (Spielberg and Gordon, 1981). The oral dosage of nitrofurantoin for adults is 50 to 100 mg four times a day, with meals and at bedtime. Alternatively, the daily dosage is better expressed as 5 to 7 mg/kg in four divided doses (not to exceed 400 mg). A single 50- to 100-mg dose at bedtime may be sufficient to prevent recurrences. The daily dose for children is 5 to 7 mg/kg, but it may be as low as 1 mg/kg for long-term therapy (Lohr et al., 1977). A course of therapy should not exceed 14 days, and repeated courses should be separated by rest periods. Pregnant women, individuals with impaired renal function (creatinine clearance less than 40 ml per minute), and children less than 1 month of age should not receive nitrofurantoin. Nitrofurantoin is approved only for the treatment of urinary tract infections caused by microorganisms known to be susceptible to the drug. Currently, bacterial resistance to nitrofurantoin is more frequent than resistance to fluoroquinolones or trimethoprimsulfamethoxazole, making nitrofurantoin a second-line agent for treatment of urinary tract infections (Stamm and Hooton, 1993). Nitrofurantoin also is not recommended for treatment of pyelonephritis or prostatitis. However, nitrofurantoin is effective for prophylaxis of recurrent urinary tract infections (Brumfitt and Hamilton-Miller, 1995). Phenazopyridine Phenazopyridine hydrochloride PYRIDIUM, others) is not a urinary antiseptic. However, it does have an analgesic action on the urinary tract and alleviates symptoms of dysuria, frequency, burning, and urgency. The usual dose is 200 mg three times daily. The compound is an azo dye, and the urine is colored orange or red; the patient should be so informed. Gastrointestinal upset is seen in up to 10% of patients and can be reduced by administering the drug with food; overdosage may result in methemoglobinemia. Phenazopyridine also is marketed in combination with sulfisoxazole and sulfamethoxazole. |
Prospectus
|
The most dramatic advance in the past decade for the group of antimicrobial agents reviewed in this chapter has been the introduction of the fluoroquinolones, such as ciprofloxacin. The attractiveness of these agents includes their bioavailability after oral administration, excellent gram-negative spectrum of activity, and relatively few side effects. Promising new fluoroquinolone agents are now being introduced and demonstrate a broader spectrum of antimicrobial activity, including, in some cases, effectiveness against gram-positive and anaerobic infections. The improved activity of newer quinolones against penicillin- and cephalosporin-resistant strains of pneumococcus and the atypical pneumonia organisms is resulting in an increasing role for these agents in the empirical treatment of pneumonia. The development and use of quinolones for the treatment of mycobacterial infections is ongoing (see Chapter 48: Antimicrobial Agents: Drugs Used in the Chemotherapy of Tuberculosis, Mycobacterium avium Complex Disease, and Leprosy). For further information regarding particular infections for which the
antimicrobial agents discussed in this chapter are useful, the reader is
referred to the following chapters of Harrison's Principles of Internal
Medicine, 16th ed., |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3821
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved