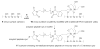| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Nanomedicine and Protein Misfolding Diseases
Abstract
Misfolding and self assembly of proteins in nano-aggregates of different sizes and morphologies (nano-ensembles, primarily nanofilaments and nano-rings) is a complex phenomenon that can be facilitated, impeded, or prevented, by interactions with various intracellular metabolites, intracellular nanomachines controlling protein folding and interactions with other proteins. A fundamental understanding of molecular processes leading to misfolding and self-aggregation of proteins involved in various neurodegenerative diseases will provide critical information to help identify appropriate therapeutic routes to control these processes. An elevated propensity of misfolded protein conformation in solution to aggregate with the formation of various morphologies impedes the use of traditional physical chemical approaches for studies of misfolded conformations of proteins. In our recent alternative approach, the protein molecules were tethered to surfaces to prevent aggregation and AFM force spectroscopy was used to probe the interaction between protein molecules depending on their conformations. It was shown that formation of filamentous aggregates is facilitated at pH values corresponding to the maximum of rupture forces. In this paper, a novel surface chemistry was developed for anchoring of amyloid β (Aβ) peptides at their N-terminal moieties. The use of the site specific immobilization procedure allowed to measure the rupture of Aβ - Aβ contacts at single molecule level. The rupture of these contacts is accompanied by the extension of the peptide chain detected by a characteristic elasto-mechanical component of the force-distance curves. Potential applications of the nanomechanical studies to understanding the mechanisms of development of protein misfolding diseases are discussed.
Introduction
Misfolding and self assembly of proteins in nano-aggregates of different sizes and morphologies (nano-ensembles, primarily nanofilaments and nano-rings) is a common thread linking a number of important human health problems. Particularly, recent studies highlighted increasing recognition of the public health importance of protein deposition diseases, including neurodegenerative disorders such as Parkinsons disease, Downs syndrome, Alzheimers and Huntingtons diseases, systemic and localized amyloidoses, transmissible encephalopathies and several other neurodegenerative disorders (1). The first and perhaps most important elements in most neurodegenerative processes are misfolded and aggregated proteins. These are inducers of cellular stress and activators of immunity in neurodegenerative diseases and affect neuronal dysfunction and loss. Altogether, the accumulation of abnormal protein nano-ensembles exerts toxicity by disrupting intracellular transport, overwhelming protein degradation pathways, and/or disturbing vital cell functions. In addition, the formation of inclusion bodies is known to represent a major problem in the production of recombinant therapeutic proteins ( ). Formulation of these therapeutic proteins into delivery systems and their in vivo delivery are often complicated by protein association ( ). Finally, since protein refolding is frequently accompanied by the transient association of partially folded intermediates, the propensity to self-assemble in nano-ensembles is considered a general characteristic of the majority of partially folded proteins ( - ). Thus, protein folding abnormalities and subsequent events underlie a multitude of pathologies and difficulties with protein therapeutic applications. Current demographic trends indicate that need for macromolecule therapeutics for age-related and other degenerative disorders and will be at the forefront of future medical developments. The field of medicine therefore can be dramatically advanced by establishing a fundamental understanding of key factors leading to misfolding and self-assembly of the various protein folding pathologies.
Misfolded conformations of proteins differ from folded and other aberrant protein conformations by their increased propensity to interact with each other leading to the formation of nano-aggregates. The structure of individual protein molecules within well ordered aggregates can be partially elucidated by traditional structural techniques, including X-ray crystallography, NMR, circular dichroism, fluorescence, and IR spectroscopies (reviewed in (8, )). However, none of these techniques is capable of sensing the misfolded conformation of the protein prior to aggregation. Apparently, the conformation of misfolded protein preceding the aggregation is different from what it is in aggregates, but to what extent it is different causing the disease is not clear. The vast majority of current experimental approaches to analyze protein misfolding and aggregation are based on traditional, ensemble techniques that do not allow investigators to distinguish between conformational changes in individual protein molecules and the changes induced by protein-protein interaction. This leaves open the question of the effect of these factors on folding/misfolding of an individual protein molecule. Current tools do not allow for the measurement of protein interaction forces or the kinetics of interconversion among different protein conformations. The ability to measure these parameters is critical to achieve a quantitative understanding of protein misfolding and aggregation at the nanoscale level. Thus, new experimental tools and approaches are crucial for understanding the protein misfolding phenomenon.
It is well established that AFM force spectroscopy is an important method for probing the stability of protein conformations (10) and can provide high resolution structural information ( , ). In this context, we have recently demonstrated that force spectroscopy is capable of probing misfolded protein conformations ( ). To measure single molecule interactions proteins were anchored on the mica surface and the AFM probe, and the interaction between the tethered molecules was measured after bringing them together by approaching the tip to the surface. The major advantage of probing single molecules is to enable intimate detection and quantitative characterization of conformational states without interference from neighboring molecules. Using this approach, we were able to monitor directly the strength of the interprotein interaction depending on the protein conformation. The data obtained show that the interaction between all the proteins increases sharply with a decrease in pH. AFM imaging showed that at pH values corresponding to maximum interprotein interaction, the rate of protein aggregation increases dramatically.
We extended these studies by introducing a novel surface chemistry that allowed us to attach amyloid β peptides selectively at N-termini and in so doing to detect the interactions between individual peptide molecules. These studies illustrate the power of nanomechanical tools for unraveling molecular mechanisms of protein misfolding and fibrils formation.
Materials and Methods
Procedure for the Synthesis of Maleimide-Pentaethylene Glycol-Silatrane (Maleimide-PEG-Silatrane)
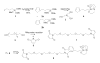
Maleimide-PEG-silatrane 8 was prepared as shown in Scheme 1. 3-Cyclopentadienylpropyltrimethoxysilane 2 was prepared as an inseparable mixture of two double bond isomers, 2a and 2b by reaction of one equivalent 3-iodopropyltrimethoxysilane 1 (Gelest) with one equivalent of sodium cyclopentadienide in tetrahydrofuran (Aldrich) at room temperature ( ). For convenience, only isomer 2a and its derivatives are shown in subsequent reactions. Heating a solution of 2 and one equivalent of triethanolamine 3 in sodium methoxide-methanol followed by evaporation of the solvent led to cyclopentadiene silatrane 4 ( ). Separately, bis-maleimide 7 was prepared from chemically pure pentaethylene glycol 5 by way of a bis Mitsunobu reaction with maleimide ( ) ( ). Silatrane 4 undergoes a rapid Diels-Alder reaction when treated with a 2-fold excess of bis-maleimide 7 giving the desired maleimide-PEG-silatrane 8. Unlike the lower trialkoxysilanes, silatrane 8 is reasonably stable in water solution. 1H NMR spectra of maleimide-PEG-silatrane 8 in D2O at ambient temperature with pD values ranging from 2 to 7 indicate that 90-95% of the starting 8 remains after 48 h.
Amyloid β immobilization.
Aβ peptide modified with additional cysteine at N-terminus was purchased from AnaSpec (SanJose, CA) and was used without further purification. The peptide powder was dissolved in 1 ml of dd water and 20 μl aliquots were stored at −80C until use. Buffered solution (Na2CO3, pH 9.8) of the protein was treated with 10 mM solution of TCEP-HCl (Tris(2-carboxyethyl)phosphine- hydrochloride) at 25C for 10 min to reduce thiols of the protein. In order to immobilize protein the functionalization of mica surface and AFM tips was performed prior to the treatment with peptide. The immobilization chemistry is shown in Scheme 2. A freshly cleaved mica surface (cartoon 9) or UV cleaned AFM tips were immersed in an aqueous solution of silatrane 8 (167 μM) for 3 hours and then rinsed with dd water, leading to a surface containing immobilized maleimide units as depicted in cartoon 10. Surface 10 was then treated with buffered solution of the amyloid peptide-cys (11.3 μM) for 1 hour after reduction of thiols. The terminal cysteine residue reacts with the maleimide units on the surface leading to the covalently immobilized peptide as shown in cartoon 11. Unreacted maleimide moieties were quenched with buffered (Na2CO3, pH 9.8) 10 mM β -mercaptoethanol solution by 10 min treatment. Mica and tips with attached Aβ peptide were left overnight in Na2CO3 buffer, pH 9.8 to prevent peptide aggregation and dissociate aggregates which possibly formed at early stages of surface modification ( ).
Measurements of Aβ - Aβ interactions.
Force curves were collected on Nanoscope Multimode AFM with IIIa controller equipped with the Picoforce controller (Veeco Metrology Inc., Santa Barbara, CA) and on Molecular Force Probe 3D instrument (MFP 3D, Asylum Research Inc., Santa Barbara, CA). The ramp size used was 300 nm with a 0.25 Hz frequency, an application force of 50 pN for both instruments. The noise level for force curves was obtained by using the averaging procedure in which the mean force value was calculated over 10 consecutive data points of the force-distance curve that was subtracted from the value obtained for the next set calculated after shifting the 10 data point interval to another data point. Silicon nitride cantilevers with nominal values of spring constants in the range of 0.01-0.04 N/m were used. Spring constants for each cantilever were obtained using thermal method (17) with the MFP-3D instrument (Asylum Research, CA). Extension and retraction curves were acquired initially in Na2CO3 buffer with 180mM NaCl pH 9.8. The buffers were exchanged to lower pH each time starting with the buffer at pH 9.8. The other buffers, appearing in the order used, were pH 5.6: 20mM phosphate buffer saline (PBS), pH 3.7: 10mM sodium acetate, and pH 2.0: 50mM KCl and 13mM HCl. NaCl was added to each buffer to keep constant the ionic strength, 180mM.
RESULTS AND DISCUSSION
To achieve this goal and to measure the interprotein interactions, we used the AFM force spectroscopy approach, in which proteins to be anchored to the substrate surface and the AFM tip (Fig. 1). We have recently tested this idea using the immobilization approach in which protein was covalently linked to amino-functionalized mica and Si3N4 AFM probes via glutaraldehyde crosslinking ( ). Although this procedure allowed us to study various proteins using the same immobilization strategy, glutaraldehyde reaction takes place at all amine-containing moieties (e.g., lysine and arginine) in addition to the protein N-termini. To avoid this ambiguity in the protein tethering that can complicate the single molecule analysis of rupture events, in this paper we used an alternative approach, in which Aβ peptide was bound to the surface at N-terminal only. We took advantage of the fact that Aβ peptide does not contain cysteins and attached this aminoacid to the N-terminus of the peptide allowing for the use of thiol-specific chemistry for the protein immobilization. We selected N-terminus because N-terminal part of the peptide is not critically involved in various conformational transitions of this peptide including the formation of β-sheet conformations within amyloid fibrils (e.g., ( )). We have recently proposed the surface chemistry based on the use of silatrane for functionalization of mica for imaging various biological samples ( , ). An important feature of the silatrane chemistry is that immobilization of such a large molecule as an oligonucleotide can be performed in one step if silatrane with attached oligonucleotide is used. We synthesized maleimide-PEG-silatrane (MAS; see Methods section, Scheme 1) to functionalize mica (MAS mica) and the AFM tip surfaces with maleimide capable of binding SH-terminated peptide (Methods section, Scheme 2).
|
|
Figure 1 Schemes illustrating the rupture of the protein in folded (low affinity, green arrows) and misfolded conformations (high affinity with multiple contact points-red wave shape). |
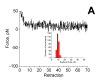
Aβ peptide was immobilized on both MAS mica and the tip surfaces and the interaction between the peptide molecules was measured by a series of approach-retraction cycles. The results for measuring interprotein interaction at pH 9.8 and 5.6 did not reveal clear rupture events. This finding is consistent with our earlier observations (13). One of the typical force-distance curves for the experiments performed at pH5.6 is shown in Fig. 2A. There are some variations (steps) along the curves that were measured as described in Methods. The maximal steps for each force-distance curve were plotted as histogram shown as an insert in Fig. 2A. The force rupture values are centered around the value 102 pN that is close to the noise level for the force-distance curves for the cantilevers with the spring constants used in these experiments. Similar analysis for the data obtained at pH 9.8 provided the value 92 pN.
|
Figure 2A A typical force curve obtained for probing the Aβ-Aβ interaction at pH 5.6. The histogram summarizing the rupture force measurements is inserted into the figure. |
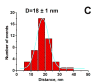
The data for pH3.7 are dramatically different. One of the force-distance curves is shown in Fig. 2B. An intensive peak at the beginning of the force curve (a green arrow) corresponding to the short-range adhesion forces between the tip and mica surfaces typically observed at acidic pH values ( , ) is accompanied by a rupture event (indicated with a red arrowhead) with the step size ~70 pN that is substantially larger the noise level. The histogram for the data obtained from the analysis of series of such events is shown as insert (i) in Fig. 2B. The mean value for this distribution is 452 pN. Note that control experiments with Aβ-peptide at the mica surface or the tip only did not show these types of interactions. Given a finite size of the MAS-PEG linkers and similar size of the peptide (ca. 15 nm total), it is reasonable to attribute the second peak to the rupture of a single Aβ-Aβ pair. Additional evidence supporting this interpretation comes from the analysis of this particular part of the force-distance curve. The profile of this curve is typical for stretching of polymer of various kinds, and this section can be fitted by the worm like chain (WLC). The WLC fit of this force distance curve is shown as black smooth curve to the left of the rupture event above the arrowhead. Such an analysis allowed us to measure the entire stretching range. The value 15.8 nm was obtained for the experiment shown in Fig. 2B. The histogram for the measurements of this value performed over ~50 various rupture events is shown in Fig. 2C. The mean value is 181 nm. Note that this number is close to the expected value (15 nm) for a full extension of the N-terminus of Aβ peptide that is not involved in the peptide folding and 5 PEG moieties of the MAS linker. Additional proofs for these estimates were obtained from the experiments with stretching of the surface immobilized Aβ peptide via MAS chemistry with the tip terminated with glutaraldehyde. During the tip-surface contact covalent bonds between the residues with free amino groups of the peptide (the closest to the N-terminal is Lys-16) and the tip immobilized glutaraldehyde can be formed allowing the stretching the Aβ peptide molecule between the anchors. The stretching experiments showed the elastic-type profile of the force curve with the stretch value 7.8-8.5 nm; this value is very close to the half of the stretching effect observed for stretching Aβ-Aβ pairs.
|
Figure 2B A force curve for the Aβ-Aβ interaction at pH 3.7. The arrowhead (red) points to the Aβ-Aβ rupture event. Insert (i) shows the histogram for the Aβ-Aβ rupture force values. Black line above the arrowhead (more ) |
|
|
Figure 2C The histogram of the extension distance for the Aβ-Aβ dimer rupture measured from the force curves obtained at pH 3.7 |
Similar force spectroscopy data were obtained for pulling experiments for pH2.0 and the mean value for the rupture forces was 411 pN (Fig. 2C) that is very close to the value obtained at pH 3.7. Overall, the data on the pH-dependent interaction obtained are consistent with our earlier data ( ) suggesting the Aβ-peptide misfolding at acidic pH, but here we were able to measure the interactions between individual Aβ-Aβ pairs.
According to the solid state NMR data ( , ), 1-40 Aβ peptides within the fibril adopt parallel, in-register β structures in which residues 1224 and 3040 adopt β-strand backbone conformations. Parallel orientation of these monomer units provides the stability of the amyloid fibrils. If misfolded conformation of Aβ peptides is close to the peptide conformation in fibrils, the force spectroscopy data provide the quantitative measure for the intermolecular interaction stabilizing the fibrils. Recently Kellermayer et al ( ) applied AFM force spectroscopy to analysis forces between amyloid β molecules within amyloid fibrils. In this approach, the fibril is firmly attached to the surface and the AFM tip pulls the filament from the fibril after making a strong contact to the fibril. These experiments provided a minimal force ~30 pN for unzipping one 1-40 Aβ- peptide from the fibril. This value is very close to 45 pN rupture force obtained in our single molecule force spectroscopy experiments suggesting that misfolded Aβ-peptide conformations is close to one within the amyloid fibril. It is instructive to compare the rupture forces measured in this paper values with the stability of other systems in order to obtain a better understanding about the nature of forces stabilizing Aβ-Aβ pairs. The value ~40pN was obtained in the paper ( ) for the 15 bp DNA duplex rupture in the pulling experiments with a very similar approach-retraction rate. This force is rather close to ~45 pN value for the rupture of Aβ-Aβ interaction, opening a speculation on the similarities of forces for stabilization of DNA helix and the Aβ-Aβ duplex.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 983
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved