| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
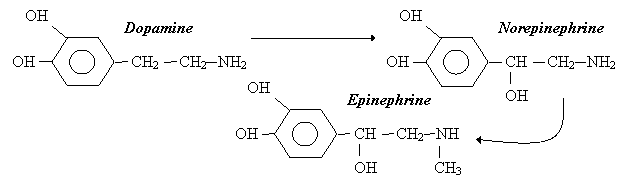
The synthesis of adrenergic transmitters is more complex than cholinergic. Adrenergic neurons transport Tyr into the nerve ending by aromatic aa transporter. Tyr is converted to DOPA by Tyr hydroxylase. This is the rate-limiting step, which is blocked by a Tyr analogue → metyrosine. DOPA is converted to DA, which enters the chromaffin granules through a carrier (blocked by reserpine, resulting in catecholamine depletion). Inside the granule, DA is converted to NE (and in the adrenal medulla NE is further converted to E).
Upon AP, theres Ca2+ influx, resulting in release of NE, ADP & some peptide co-transmitters into the cleft. NE then acts on adrenergic receptors.
Elimination is either by (1) simple diffusion out of the terminal with eventual metabolism in the liver & plasma; (2) by reuptake into the nerve terminal (uptake-1), which takes any phenylethylamine, e.g. amphetamines & DA; or (3) by reaching into neighboring glial & smooth mm cells (uptake-2).
Uptake-1 transporter is blocked by cocaine & tricyclic antidepressants → ↑NE activity in the synapse. NE & E are metabolized by many enzymes (MAO, COMT), forming different metabolites (metanephrines) that are excreted through the kidney. Catecholamine turnover thus can be estimated by analysis of total metabolites in the urine collected along 24hrs.
The monoamines:
Dopamine (DA) centrally acting neurotransmitter, found in:
a. Nigrostriatal system part of the extrapyramidal motor system. ↓DA causes parkinsonism.
b. Mesolimbic-mesocortical system involved in schizophrenia. ↑DA will cause hallucinations.
c. Hypothalamus in the hunger center. ↑DA → inhibits hunger sensation.
d. Tubero-infundibular system along the hypothalamic-pituitary pathway. DA inhibits prolactin secretion.
e. Area postrema DA antagonists can inhibit nausea & vomiting.
Norepinephrine
(NE) involved in
the
Epinephrine (E) hormone secreted from the adrenal medulla, but its central actions are not completely understood.
Serotonin
(5-HT) a central neurotransmitter,
involved in sleep, mood, and food intake.
5-HT reuptake inhibitors are used for depression.
Histamine mainly peripheral action but with some CNS effects, related to sleep.
Adrenergic receptors:
NE acts mainly on α, while E acts both α & β equally.
o
α1: activation will ↑IP3 & DAG. It always leads to smooth
mm. contraction. Sensitivity: NE > E.
(1) vasoconstriction. (2)
glycogenolysis & gluconeogenesis in the liver.
(3) hyperpolarization of intestinal smooth muscles ( peristalsis).
(4) prostate & urogenital smooth muscle contraction, mainly the bladders
trigone & sphincter.
(5) mydriasis (pupil dilation through action on dilatator pupillae m).
o
α2: inhibits adenylyl cyclase ↓cAMP. Found throughout the body & brain. Sensitivity: NE > E.
(1) presynaptic nerve terminals, where it
↓NE release → always inhibitory receptor.
(2) vasoconstriction. (3) platelet aggregation. (4) β-cells of pancreas ↓insulin level.
o
β1: activation will ↑cAMP level. Sensitivity: E > NE. Effects:
(1) HR, conduction &
contractility by positive chronotropic, dromotropic & inotropic effects.
(2) renin secretion by
acting on the juxtaglomerular cells.
o
β2: causes ↑cAMP → smooth mm. relaxation. Sensitivity: only E, NE has no
action.
(1) skeletal mm vessels vasodilation. (2) bronchodilation. (3)
glycogenolysis in skeletal mm.
(4) glycogenolysis & gluconeogenesis in the liver. (5) uterine mm
relaxation.
o β3: lipolysis in adipose tissue.
Indirect sympathomimetics pharmacologically, we cant stimulate NE synthesis at all, but we can stimulate release. This is an AP-independent release, done by drugs of similar structures that will release NE and inhibit MAO enzymes. The increased concentration will eventually result in NE leaving the terminal, the tone:
a. Tyramine found in cheese and red wine and may cause hypertensive crisis when given with MAO-A inhibitors → cheese reaction.
b. Ephedrine & pseudoephedrine they have a direct β2-agonistic effect and are used as nose decongestants & for increasing BP. Ephedrine is the basis for synthesis of the following:
c.
Amphetamine acts on the brain,
increasing DA & NE release → increases activity →
↓tiredness and faster working brain. This allows much easier studying,
but since it only increases short term memory, after the exam all knowledge
will be forgotten.
With repeated administration theres less and less NE release, necessitating
increasing dosage tolerance
develops, inducing sleep for a few days and may also cause depression.
The DA released is involved in the reward mechanism.
d. Ecstasy (MDMA, methylenedioxymethamphetamine) different from amphetamine since it also releases 5-HT in addition to DA & NE. Causes hallucinations & tiredness, probably due to the 5-HT action. It is toxic to the brain, selectively killing serotoninergic neurons.
Reuptake inhibitors
a. Psychostimulant drugs such as cocaine, blocking uptake-1.
b.
Tricyclic antidepressants inhibit
reuptake-1 of 5-HT & NE but not DA.
E.g. imipramine, clomipramine, amytriptyline
& protryptyline, which inhibit both 5-HT & NE reuptake,
and desipramine & nortriptyline that inhibit
only NE uptake.
c.
SSRI (selective serotonin reuptake
inhibitors) increasing synaptic 5-HT,
with some ↑NE.
E.g. fluoxetine (Prozac), paroxetine, fluvoxamine,
sertraline & citalopram.
MAO inhibitors MAO-A metabolizes 5-HT & NE, while MAO-B metabolizes DA.
a. Non selective, irreversible inhibitors not used anymore → iproniazid & pargylin.
b. Selective, irreversible MAO-B inhibitors used for Parkinsonism, DA transmission.
c. Selective, irreversible MAO-A inhibitors clorgylin, not used anymore.
d. Reversible inhibitors of MAO-A (RIMA) moclobemid, which is much safer than irreversible drugs. Moclobemid peripheral action can be replaced by cheese or red wine, since they cause ↑Tyramine. The drug is indicated though for central action. Its not a very good antidepressant, only for mild affective disorders.
Epinephrine quite rarely used as
a drug. It is given parenterally because its metabolized in the GI. The dose
is 0.3-0.5mg subcutaneously or 0.05-0.1mg IV. The drug is light sensitive.
E is short acting, since it is metabolized fast by COMT.
Indicated for anaphylactic shock & angioneurotic edema. It can be given
together with local anesthetics, e.g. lidocaine, since it causes local
vasoconstriction, preventing the spread of the anesthetic into the blood,
increasing its local action.
Side effects: by acting on β1-receptors it can cause
positive inotropic effect, ↑contractility → tachycardia. This
↓ blood supply to the heart → contraindicated for people with heart
problems.
By acting on α1-receptors it causes vasoconstriction →
↓blood supply to visceral organs.
By acting on β2 → vasodilation in vessels supplying
skeletal mm, redistributing the blood flow.
Excessive dosage will cause total vasoconstriction → CNS problems, such
as tremor & anxiety.
NE a potent α1-agonist in the periphery. It has no vasodilator action at all, but causes extreme vasoconstriction. It ↑both the diastolic & systolic BP, which is not good for the heart.
DA has only peripheral actions, since it doesnt cross the BBB. It is given by infusion:
Low (kidney) dose stimulates D1-receptors in the renal artery, leading to vasodilation and can prevent renal failure during shock.
Intermediate dose selective β1-agonist → positive inotropic & chronotropic effects without vasoconstriction. Its the first drug of choice for cardiogenic shock. The only problem with DA is tachycardia that will lead to increase cardiac O2 demand.
High dose the drug loses its β1 selectivity.
Dobutamine a β1 selective agonist, used for treating cardiogenic shock. It is given together with low dose DA to prevent renal failure. Its positive inotropic action is stronger than the chronotropic, thus it causes less severe tachycardia.
Isoproterenol a β1&2 agonist, with no effect on α-receptors at all. It is used for cardiogenic shock but causes tachycardia as a side effect. Its also used in asthma to induce bronchodilation.
Metaproterenol the first β2-selective agonist used for asthma. Might cause tachycardia when taken systemically, but its prevented by local inhalation.
Albuterol & terbutaline these are the most used sympathomimetics for asthma with a long action. When inhaled they cause bronchodilation. They are also used for uterine relaxation, and can prevent painful contractions. It can be life saving since it can prevent uterine rupture caused by improper position of the baby.
Formoterol & salmeterol new β2-agonists, with longer action (12hrs) due to ↑lipid solubility allowing them to dissolve into the smooth m. membranes and continuously supply the adjacent β-receptors. Indicated for asthma.
Some of the drugs are not catecholamines, though similar to epinephrine → they lack the OH group on the aromatic ring. The structural change will lead to increased enzymatic resistance (COMT), resulting in slower metabolism → longer action. They are also less potent than epinephrine.
Phenylephrine & pholedrine selective α1-agonists + indirect release of AP-independent storage by pholedrine. Only indicated for hypotension. They are not effective for orthostatic hypotension.
Drugs
containing imidazoline structures, applied locally, since otherwise theyll
reach postsynaptic a receptors → strong vasoconstriction.
Locally used as nasal decongestants: naphazoline, xylometazoline
(both a1&2), and oxymetazoline → selective α2
agonist.
Side effects if used for long, the rhinitis might become chronic.
Dont use above 3 days.
Tyr hydroxylase inhibitor the best inhibition will be of Tyr kinase, but since we dont have such drugs we can inhibit Tyr hydroxylase instead. This will inhibit the rate-limiting step (Tyr DOPA). The inhibitor is metyrosine (α-methylparatyrosine). It is not used as antihypertensive drug since although it slows transmission, it also ↓DA-ergic transmission.
Monoamine depletors such as reserpine.
It blocks the uptake of biogenic monoamines (NE, DA, 5-HT, etc.) into vesicles,
leading to ineffective transmission. MAO enzymes will eventually metabolize
them in the axoplasm, causing depletion of NE, DA and serotonin both in central
& peripheral neurons.
It is an antihypertensive drug, with long action since drug residues are
attached to the vesicular membrane for many days → risk of orthostatic hypotension. The depletion is dose
dependent.
It is a central drug, thus it causes sedation, depression (due to NE depletion)
and drug induced Parkinson-like syndrome (DA depletion in the striatum).
Adrenergic neuron blockers such as guanethidine,
debrisoquine & bretylium.
They block the release of NE by fixing the neurotransmitter vesicles inside the
axon. Guanethidine uptake into the nerve is also by uptake-1. It causes
transient BP elevation followed by long orthostatic hypotension. Debrisoquine
also causes orthostatic hypotension, but without the transient elevation.
Guanethidine was indicated for glaucoma in the form of eye drops. Due to its
side effects (hypotension, diarrhea & impaired ejaculation) it is rarely
used today. It can induce hypertensive crisis in patients with pheochromocytoma
due to ↑↑catecholamine
release. It has a long t½ (5 days) and the dose varies among
patients → start at low dose and
gradually increase.
False transmitters e.g. the centrally
acting pro-drug α-methylDOPA. The same as DOPA, its a
substrate for DOPA-decarboxylase, forming α-methyl DA α-methylNE. It is a
selective α2-agonist, inhibiting further transmission. Its
indicated as an antihypertensive drug.
All α2-agonists can cause sedation, drowsiness & mental
confusion. The problem is that they are usually given to old people, who
already have decreased mental function and this worsens the situation less in use nowadays. The 1st
metabolite is α-CH3-dopamin, which may block DA effect on the pituitary,
increasing prolactin secretion → hyperprolactinemia.
Centrally acting sympatholytics such as:
a.
Clonidine a selective α2-agonist
that also causes sedation. Since its t½
is 8-12hrs it is given orally in the morning & evening, causing the
patient to sleep all day long.
Clonidine is an antihypertensive drug that only rarely causes orthostatic
hypotension.
It improves glucose tolerance (good for diabetics with HT) and it ↑HDL
& ↓LDL levels.
Withdrawal, mainly after high dosage, will cause a rebound hypertensive crisis
due to ↑↑Sym activity.
Cessation should be done gradually while taking other antiHT drugs.
b.
Guanfacine
resembles clonidine in its profile, but has
different elimination rate (longer t½) → given only once a
day at the evening (since its also a sedative).
It is good for HT patients with sleeping disorders.
Adrenergic neuron killer 6-hydroxydopamine, selectively kills adrenergic neurons in the brain. It is used only in animal experiments and not on men.
Others action on other presynaptic receptors can also cause inhibition, e.g. ACh receptors, opioid receptors, adenosine receptors & D2-receptors.
Haloalkylamines not used since they
block the receptors irreversibly.
E.g. phenoxybenzamine, non-selective, with preference for α1.
Overdose will kill the patient due to excessive vasodilation. It was used in
pheochromocytoma, to ↓the hypertensive
crisis.
Imidazolines the same structure as clonidine (an agonist). The antagonists are:
a. Tolazoline not used since it also has a M & histamine agonistic effects → diarrhea & severe vasodilation. It was used in peripheral vascular diseases, such as Raynauds syndrome & Burgers disease.
b.
Phentolamine a non-selective
α-blocker that is used in pheochromocytoma, since we need to block α1
& α2, both causing vasoconstriction. It might cause
orthostatic hypotension.
By blocking α2 at the presynaptic membrane it blocks the
negative feedback that NE has on the presynaptic terminal → ↑Sym → ↑tachycardia.
Treat this tachycardia with β-blockers.
Indole alkaloids:
a. Yohinbin in low doses it blocks α2 but higher doses will also block α1. Was used for sexual enhancement causing penal vasodilation. When mixed with a very low dose of strychnine it can perception for even further sexual enhancement.
b. Ergot alkaloids can be natural & semisynthetic:
Natural including ergotamine,
ergometrine & ergocryptine. These have 3 main
actions (A) non selective
α blockade; (B) direct smooth mm constriction, by acting on Ca2+
channels long
vasoconstriction, which makes the α blockage irrelevant;
(C) action on 5-HT receptors → can be agonist, partial agonist or
antagonist, depending on the type of 5-HT receptor.
It may cause hallucinations, and overdose will cause ergotism → necrosis
of the hands and legs.
They are indicated for uterine bleeding and during 3rd stage
pregnancy to accelerate birth and constrict the local vessels.
Semisynthetic
A) Dihydroergotamine by saturating a double bond of ergotamine → eliminates the vasoconstriction effect → only α blockade & 5-HTB1/D1-agonism. These 5-HT receptors are present only on meningeal vessels → this is the only place it causes vasoconstriction, acting as anti-migraine drug (for relief).
B) Bromocriptine DA-agonist used for Parkinsons disease & hyperprolactinemia.
C) Methysergide 5HT2-antagonist that is used for migraine prevention. It is not a popular drug due to many side effects.
D) LSD (lysergic acid diethylamide, lysergide) potent serotoninergic drug in low doses (μg). It can be full agonist, partial agonist or antagonist. It mainly causes hallucinations.
Selective α1-blockers
such as prazosin, doxazosin, terazosin
& urapidil. These are vasodilators (antiHT) that also have
anti-atherogenic effect (↑HDL & ↓LDL). Parenteral urapidil can
treat HT crises. They dont cause reflex tachycardia since they act centrally
to counteract the reflex. They may cause orthostatic hypotension, usually seen
only at the beginning of treatment. Later a tolerance to hypotension develops, but
its vasodilating effect is maintained. The hypotension is prevented by starting
with lower dose and gradually increasing.
They can treat HT related to other conditions, such as peripheral vascular
disease, AS (↓LDL), bronchial asthma, COPD (though not acting on the
lungs) & pregnancy.
They are also indicated for prostatic hyperplasia (mainly terazosin), causing mm
relaxation.
The antihypertensive effect is better when combined with β-blockers or
diuretics.
They generally cause negative chronotropic, dromotropic & inotropic effects on the heart. They are different in relation to their selectivity, water solubility, CNS effects and action duration.
A) Non-selective including:
Propranolol the prototype pure antagonist and lipid soluble, which passes the BBB.
Oxprenolol & pindolol are partial β-agonists in certain cases they dont cause much bradycardia; e.g. propranolol will decrease HR from 80 to 30, but a partial agonist will only reach 60 (less severe bradycardia), though they have the same antihypertensive action.
Timolol lipid soluble, only used for glaucoma. b-blockers inhibit aqueous humor production, reducing intraocular pressure. It is indicated for open-angle glaucoma when the Schlemms canal is not blocked.
Nadolol the same as propranolol but water soluble no central effects.
B) β1-selective (cardiac) including:
Metoprolol a lipid soluble pure antagonist that can also be used parenterally.
Atenolol & esmolol water soluble pure antagonists with no CNS effect. Esmolol is very short acting, only given for SVPT.
Betaxolol a pure antagonist that can be used systemically and locally for glaucoma (drops).
Isoprolol long acting β-blocker.
Indications:
HT both by ↓CO and by blocking the RAA effect → ↓AT-II → vasodilation, and aldosterone volume. Centrally acting β-blockers may Sym activity (not important).
Angina pectoris ↓HR &
contractility → ↓O2 demand. Since they decrease HR
→ longer diastole → coronary
perfusion.
They are used for prevention of effort angina, not for Prinzmetal angina.
Supraventricular tachyarrhythmias (AF, Af, ES, SVPT etc.) β-blockers can stop reentry-based arrhythmias. They slow the conduction, allowing normal beats to pass through and take over. Such tachycardia can occur in hyperthyroidism → best to give propranolol.
CHF paradoxical indication, only if the patient is properly treated with anti-CHF drugs, such as ACE-inhibitors, diuretics & cardiac glycosides. Then β-blockers can be added to prolong life due to unknown reason.
HOCM by ↓contractility they reduce the obstruction. It can be treated also by Ca2+ blockers.
Glaucoma by timolol and betaxolol.
Migraine only for prophylaxis by the centrally acting drugs.
Acute stress e.g. stage or exam fright. Only by lipid-soluble drugs. The advantage over other anxiolytics is that they do not cause sedation. It is preferable to use oxprenolol or pindolol, since they dont cause bradycardia.
Essential tremor a fine tremor, prevented by propranolol.
Panic disorders not recommended today since they are very symptomatic. The heart can get used to the drugs and without it, the patient will get tired very easily.
Side effects:
Bradycardia.
AV blocks.
Cardiac decompensation, if the patient is not treated properly for CHF.
Bronchial asthma, especially the non-selective drugs, acting on bronchial β2-receptors.
Peripheral vasoconstriction → causing cold hands & feet.
Hypoglycemia in DM patients → due to blockade of hepatic glycogenolysis & gluconeogenesis.
↑LDL → atherogenic effect.
CNS effects only by lipid soluble drugs → nightmares & depression.
Contraindications bradycardia, AV blocks, asthma & DM.
Labetalol & carvedilol
water soluble drugs with dual action selective α1 & β blockade.
Their advantage over β-blockers is that they dont cause hyperlipidemia
and cold hands & feet.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1078
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved