| CATEGORII DOCUMENTE |
| Alimentatie nutritie | Asistenta sociala | Cosmetica frumusete | Logopedie | Retete culinare | Sport |
OVULE CU CLOTRIMAZOL 100mg
1. Formula de fabricatie
Administrarea substantelor medicamentoase se face sub forma unor preparate farmaceutice convenabile administrarii pe anumite cai de administrare. Obiectivul formularii medicamentelor il constituie conferirea unor calitati, reproductibile in productia pe scara larga industriala, care sa le permita sa produca un efect terapeutic previzibil. In acest scop trebuie sa se tina seama de proprietatile fizico-chimice si biofarmaceutice ale substantei medicamentoase, excipientilor, adjuvantilor si recipientilor de ambalare a medicamentului. Inaintea formularii substantei medicamentoase intr-un produs farmaceutic se iau in considerare:
proprietatile fizico-chimice ale substantei active si excipientilor, cum ar fi solubilitatea, stabilitatea, coeficientul de repartitie.
proprietatile biofarmaceutice ale substantei medicamentoase incluzind factori care pot influenta biodisponibilitatea.
procedeele tehnologice si aparatura industriala care trebuiesc folosite in vederea transformarii substantei medicamentoase si excipientilor in forme farmaceutice.
considerente terapeutice .
Prepararea unor forme farmaceutice corespunzatoare si eficiente se va face numai cand toti acesti factori au fost luati in considerare si legati unul cu altul. Acesta este principiul fundamental care trebuie sa actioneze in formularea medicamentelor.
La obtinerea ovulelor cu clotrimazol 100mg sunt necesare urmǎtoarele materii prime:
- 4.2 Kg clotrimazol
- 121.8 Kg unt de cacao
Total : 126 Kg
1 ovul = 3 g.0.1g clotrimazol2.9g unt de cacao
1 cutie = 6 ovule .0,6 g clotrimazol..17.4 g unt de cacao
Din cele 126 de kg amestec se va obtine un lot de 7000 de cut
Untul de cacao a fost obtinut in 1692 de W. Homberg si introdus ca masa de supozitoare de Baume in 1762. Este un produs oficializat in toate farmacopeile. Untul de cacao este obtinut prin presarea la cald a semintelor decorticate si torefiate de Theobroma cacao (fam. Sterculiaceae).
Untul de cacao este un amestec de trigliceride ale acizilor grasi saturati (palmitic, stearic, arahidic) si nesaturati (acidul oleic).
Descriere:
- masa grasa solida, galbuie, cu miros de cacao si gust aromat
Solubilitate:
- usor solubil in benzen, cloroform, eter, eter de petrol, putin solubil in alcool, practice insolubil in apa.
Indice de aciditate: cel mult 2.25
Indice de iod:
Indice de saponificare:
Punct de topire: 30-35 ºC
Aciditate-alcalinitate:
- 5,o g unt de cacao se incalzesc pe baia de apa timp de 5 min cu 25 ml apa proaspat fiarta si racita; se raceste si se flitreaza prin hartie de filtru cu porii fini. La 10 ml solutie flitrata se adauga 0.1 ml fenolftaleina-solutie; solutia trebuie sa ramana incolora. La adaugarea de cel mult 0.2 ml hidroxid de sodiu 0.01 mol/l solutia trebuie sa, se coloreze in roz.
Absortia luminii:
- 2,o g unt de cacao se dizolva in 5 ml ciclohexan. Solutia se agit, timp de 3 min cu 3 ml hidroxid de sodium 4 mol/l. Faza organica se separa, se spala de 7 ori cu cate 3 ml apa, se filtreaza prin hartie de flitru cu porii fini pe sulfat de sodiu anhidru si se avapora pe baia de apa. 0,1 g reziduu se dizolva in 10 ml ciclohexan. Absorbanta solutiei determinata la 270 nm trebuie sa fie de cel mult 0.18.
2. Descrierea procesului tehnologic si a echpamentelor folosite
Fazele procesului tehnologic sunt urmatoarele:
- livrarea materiilor prime
- cantarirea materiilor prime, eventual pulverizarea substantelor solide insolubile in excipient;
- topirea excipientilor;
- dispersarea substantelor active;
- turnarea in forme si racirea supozitoarelor;
- divizarea plachetelor, marcarea, gruparea, ambalarea;
- depozitare; expeditie;
- controlul in cursul fabricarii si al produsului finit.
1. Livarea materiilor prime
Subatantele active si excipientii sau alte substante auxiliare sunt livrate din depozitul fabricii in cantitatile inscrise in fisa de fabricare, pentru sarja respectiva care se va prepara.
2. Cantarirea materiilor prime se efectueaza cu balante de precizie.
Se cantaresc cele 0.1 kg de clotrimazol, si 9.9 kg de unt de cacao cu un exces de 20-30% inscrisa in fisa de fabricatie pentru a compensa eventualele pierderi.
3. Topirea excipientilor
Cele 9.9 kg de unt de cacao se topesc in containere din otel inoxidabil, cu peretii dublii, prin care circula vapori de apa supraincalziti. Se topste pana la lichefiere si clarificare dupa care se lasa, sa se raceasca la o temperatura de cateva grade deasupra punctului de solidificare.
4. Dispersarea substantelor active
Dupa o pulverizare fina a celor 0.1 kg de clotrimazol se amesteca cu o cantitate mica de excipient fluid; amestecul se trece de 2-3 ori prin moara coloidala, pentru a se obtine gradul de dispersie.
5. Turnarea in forme si racirea ovulelor
Turnarea in forme se efectueaza mentinand masa la temperatura constanta, controlata.
Se flosesc trei procedee:
a).turnare urmata de racire si conditionare a supozitoarelor
Aceasta metoda se utilizeaza pentru prepararea supozitoarelor vaginale in statiile pilot sau pentru fabricarea unor loturi mici de ovule. Se flosesc aparate care:
-fie actioneaza ca masini de trunare;
-fie actioneaza ca masini de trunare, prevazute si cu tipare in acelasi timp.
Aceste masini pot fi simple si semiautomate, fara sistem de racire. Masina prezinta o cuva de incalzire termostata, cu dispozitiv de agitare si de turnare in forme metalice. Excipientii pentru supozitoare se pot topii in acest rezervor si se amesteca cu substantele active, apoi amestecul fluid este turnat in tipare metalice sau folii preformate din plastomeri.
b). turnarea si racirea in mod continuu, in masina in linie, urmata de conditionare
In aceste aparate, amestecul de substante active cu excipientul se efectueaza intr-o parte a masinii, intr-un tanc cu agitator; el este apoi turnat in tipare metalice, cu ajutorul pompelor dozatoare. Tiparele sunt racite, golite.
c) turnare si conditionare simultana, in mod continuu pe masina in linie
Aceasta se efectueaza cu masini in intregime automatizate si computerizate.
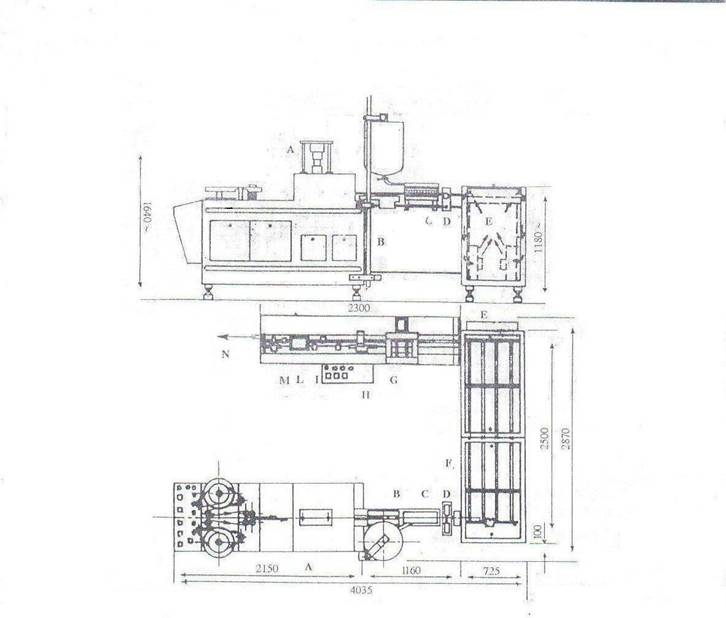
Materialele de ambalare-rolele din aluminiu sau din plastomer: 2 benzi paralele sunt desfasurate pe orizontala si plasate pe doua matrite (A) din metal incalzite la 160 ºC, pentru a lua forma unei jumatati de ovul, dupa care cele doua parti sunt sudate impreuna (B), pentru a forma matrita, lasand deasupra, in varf, un orificiu prin care se injecteaza masa fluida (C).
Masa de ovule se afla intr-un recipient incalzit, sub agitare continua, din care este injectata cu ajutorul a 8-16 pompe dozatoare, deodata in 8-16 alveole, in sir, una dupa alta.
Unele aparate sunt prevazute cu dispozitive de taiere a foliilor in 28-30 unitati lungime (D), pentru a fi cantarite dupa racire (F), in scopul verificarii greutat
Benzile umplute sunt introduse, cu o anumita viteza, in tunelul de racire (E) la 4ºC si, la iesire, ovulele sunt solodificate. Urmeaza inscriptionarea datei (I), codificarea (L), divizarea in 6-10 unitati (M) si ambalarea automata, impreuna cu prospectul in cutii de carton inscriptionate (N).
Randamentul unei astfel de masini ajunge la 30000 supozitoare/ora.
6. Conditionarea ovulelor
Toata productia de supozitoare vaginale a fost modificata prin interventia umana minima, prin utilizarea liniilor automatizate si computerizate: operatorul deschide computerul folosind cheia si masina incepe sa lucreze singura; daca se produce o defectiune, operatorul este imediat anuntat printr-un semnal sonor, aprinderea becului rosu, si masina se opreste.
In industrie se utilizeaza 2 procedee de conditionare primara:
a). procedeul mai vechi discontinuu: dupa racirea si solidificare supozitoarele vaginale sunt scoase din formele metalice si apoi introduce individual in benzi formate din filme de celofan sau aluminiu, acoperite cu un lac termocolant sau din polietilena.
b). procedeul in flux continuu: amestecul fluid ce contine masa de supozitoare vaginale este divizat direct in alveolele preformate constituite din plastomeri termoformabili ca: polietilena, policlorura de vinil, acetat de celuloza.
Acest procedeu este astazi utilizat curent in fabricarea cu masini automate si computerizate a ovulelor; linia de fabricare contine si masina integrata cu dispozitivul de termoformare a blisterelor.
Masinile utilizeaza folii de aluminiu/propilen laminate, lacuite, carora le imprima prin sudare forma alveolelor, iar varful modelelor ramane deschis, pentru a lasa sa intre dozele de umplere. Dupa ce s-a injectat masa de supozitoare, in mod obisnuit, cu pompe cu piston de diferite doze, varfurile sunt sigilate la cald.
Benzile cu supozitoare vaginale sunt apoi trecute incet intr-o statie de racire.
7. Divizarea plachetelor termoformate, marcare, grupare, ambalare
Blisterele sau benzile cu supozitoare sunt imprimate cu data de fabricare si codul sarjei. Se efectueaza crestaturi pe traseul carora va avea loc ruperea ambalajului in vederea scoaterii ovulului, cat si perforatii, care sa permita separarea supozitoarelor vaginale intre ele.
Acestea sunt apoi divizate in numar de 3, 6 sau 10 plahete si introduce impreuna cu prospectul produslui in cutii de carton inscriptionate specific si, eventual, cu accesorii de aplicare.
Cutiile sunt grupate cate 25-50 de bucati si ambalate in pachete, pe care se aplica eticheta produsului.
8. Depozitare, Expeditie
Produsele finite sunt stocate in depozitul sectiei de supozitoare vaginale, in vederea efectuarii buletinului de analiza.
Supozitoarele vaginale trebuie sa fie depozitate in conditii adecvate, ferit de lumina si umiditate, la o temperatura de cel mult 25ºC.
Transportul supozitoarelor vaginale se efectueaza numai cu mijloace acoperite.
3. Metode de analizǎ a ovulelor.
Descriere :
Supozitoarele vaginale (ovulele) au formǎ sfericǎ sau ovoidalǎ, masa de 2 - 4g (ovulele preparate cu unt de cacao sau grǎsimi semisintetice neutre) si masa de 5 - 12g (ovulele preparate cu masǎ gelatinoasǎ).
Aspect. Supozitoarele vaginale trebuie sǎ aibǎ un aspect omogen si sǎ-si pǎstreze forma si consistenta la temperatura camerei.
In sectiune longitudinalǎ, examinate cu lupa (4.5X), nu trebuie sǎ prezinte aglomerǎri de particule, cristale sau bule de aer.
Comportamentul la topire sau dizolvare. Intr-un flacon conic de 100 ml, care contine 50ml apǎ mentinutǎ la temperatura de 37 2C se introduce un supozitor. Flaconul se agitǎ prin usoarǎ rotire o datǎ la 5 min.
Supozitoarele preparate cu baze liposolubile trebuie sǎ se topeascǎ in cel mult 30 min, iar cele preparate cu baze hidrosolubile trebuie sǎ se dizolve in cel mult 1 orǎ. Por rǎmane in flacon particule de substante active si substante auxiliare nedizolvate.
Uniformitatea masei. Se cantǎresc 20 de supozitoare si se calculeazǎ masa medie. Aceleasi supozitoare se cantǎresc individual. Fatǎ de masa medie calculatǎ, masa individualǎ poate sǎ prezinte abaterile procentuale prevǎzute in tabelul I.
Tabelul I
|
Masa medie a supozitorului |
Abatere admisǎ |
|
panǎ la 2g |
10% |
|
2g panǎ la 5g |
7.5% |
|
5g si mai mult de 5g |
5% |
Dozare. Se efectueazǎ comform prevederilor de la monografia respectivǎ. Continutul in substantǎ activǎ pe ovul poate sǎ prezinte fatǎ de valoarea declaratǎ, dacǎ nu se prevede altfel, abaterile procentuale prevǎzute in tabelul
Tabelul II
|
Continutul declarat in substantǎ activǎ pe ovul |
Abaterea admisǎ |
|
panǎ la 10mg |
10% |
|
10mg panǎ la 100mg |
7.5% |
|
100mg si mai mult de 100mg |
5% |
Conservare. In recipiente bine inchise, la cel mult 25C.
Capitolul 3
Regulile de buna practica de fabricatie (RBPF)
pentru produsele medicamentoase
Fabricarea unui medicament trebuie sa se faca in conformitate cu exigentele dosarului de autorizare de punere pe piata, pentru a asigura eficienta terapeutica si siguranta clinica la utilizarea sa de catre pacienti. Compania producatoare de medicamente trebuie sa dispuna de un sistem de asigurare a calitatii (AC) bine pus la punct, corect pus in practica si controlat efectiv, care include conceptul Regulillor de Buna Practica de Fabricatie (RBPF) si Controlul Calitatii (CC). Acest sistem trebuie sa permita o gestionare a calitatii, sau managementul calitatii, adica sa confere si sa pastreze calitatea medicamentului pe toata durata de valabilitate. Asigurarea calitatii este un concept larg, care se refera la tot ceea ce poate sa influenteze calitatea unui produs medicamentos. El reprezinta un ansamblu de masuri luate de catre producatorul de medicamente, pentru a se asigura ca acestea au calitatea ceruta scopului pentru care s-au realizat.
Principiile si
liniile directoare ale acestui concept au fost elaborate in Europa prin
directiva 91/356 din
RBPF constituie unul din elementele sistemului de Asigurare a Calitat Ele garanteaza ca produsele fabricate si controlate dupa standardele de calitate corespunzatoare utilizarii lor si cerute prin autorizatia de punere pe piata sau specificatia produsului. RBPF nu sunt prevederi obligatorii in farmacopee, ci recomandari pentru companiile de medicamente sa indeplineasca obiectivele lor prin mijloace adaptate specificului unitatii si produsele lor.
Cerintele de baza ale BPF sunt:
definirea clara a procedeului de fabricatie si revizuirea lui sistematica pe baza experientei dobandite, pentru a asigura reproductibilitatea tuturor caracteristicilor produsului;
validarea etapelor critice ale procedeului de fabricatie si a schimbarilor semnificative ale acestuia;
asigurarea tuturor mijoacelor necesare pentru aplicarea RBPF si anume: personal calificat si instruit corespunzator; local si spatiu adecvate; echipamente, instalatii si servicii adecvate; materii prime, recipiente de ambalare si etichete corespunzatoare; proceduri si instructiuni aprobate; depozitare si transport corespunzatoare;
redactarea clara, fara ambiguitati a procedurilor si instructiunilor
instruirea personalului pentru efectuarea corecta a procedurilor
inregistrarea manuala sau cu instrumente de inregistrare a tuturor rezultatelor din toate etapele procesului de obtinere a medicamentului, incat produsul sa corespunda specificatiilor, abaterile semnificative se inregistreaza detaliat si se analizeaza
documentele de fabricatie si distributie trebuie sa oglindeasca fidel istoricul complet al unei serii, sa fie exprimate clar si sa se pastreze
distributia produselor medicamentoase nu trebuie sa prejudicieze calitatea lor
reclamatiile eventuale asupra produselor medicamentoase comercializate trebuie examinate, se vor investiga cauzele defectelor semnalate si se vor lua masuri atat privitoare la produse cat si pentru prevenirea repetarii deficientei.produsul farmaceutic trebuie conceput si realizat ca sigur si eficient;
calitatea nu poate fi asigurata doar prin inspectarea sau testarea produsului farmaceutic finitS
deficientele in formularea si prepararea medicamentului cat si in controlul calitatii sale, nu pot fi corectate prin inspectare sau testare.
calitatea produsului depinde de numerosi factori, dar se pot evidentia patru categorii de baza care pot fi surse de eroare sau variatii in procedeul de preparare;
materiile prime si recipientele de ambalare ( diferiti furnizori, diferite loturi de la un furnizor, diferentele de lot );
aparatura ( echipamentele ) si facilitati ( diferitele masini pentru un singur procedeu, diferente intre masini, uzura acestora, intretinere preventiva necorespunzatoare, conditii de lucru necorespunzatoare );
procedee (de preparare si control) neclare sau nespecifice, necorespunzatoare, neglijenta intamplatoare, diferente intre fabrici;
personal ( insuficienta pregatire si intelegere sau motivatie; lipsa de interes; neatentie, oboseala; slaba comunicare si cooperare)
fiecare etapa a procesului tehnologic trebuie controlata in timpul derularii fluxului tehnologic (control interfazic) si sa existe asigurarea ca produsul finit va intruni toate conditiile de calitate prevazute in autorizatia de punere pe piata. Acest obiectiv justifica necesitatea validarii procedeelor. Obiectivul este de a monitoriza la fiecare serie (lot) procedeul de fabricatie pentru a fi siguri ca urmeaza o cale corecta. Rezultatul va fi uniformitatea si reproductibilitatea la toate seriile, la fiecare unitate dozata care la randul lor asigura eficienta si siguranta clinica a produsului
rezultatele testarilor unui lot trebuie exprimate cu date precise, specifice, numeric si nu prin admis sau respins
atat procedurile de preparare cat si cele de control trebuie validate. Validarea dovedeste prin documentare stiintifica ca procedeul este sub control, are productibilitate si robustete, nefiind perturbat de mici schimbari care pot interveni pe parcursul derularii sale si ofera incredere ca prin el se sting obiectivele scontate.
ca medicamentele sunt proiectate si produse tinand cont de exigentele Regulilor de Buna Practica de Fabricatie si a Regulilor de Buna Practica de Laborator;
ca operatiile care alcatuiesc fluxul de fabricatie, RBPF si controlul calitatii in timpul fluxului de productie si al produsului finit sunt descrise clar;
ca responsabilitatile manageriale sunt definite clar;
ca reglementarile privind fabricarea, aprovizionarea si utilizarea materiilor prime si a materialelor de ambalare sunt corecte;
efectuarea controalelor necesare pentru produsele intermediare, a controalelor interfazice si a validarilor;
fabricarea si controlarea produselor finite conform procedurilor stabilite;
interdictia distribuirii produselor medicamentoase fara certificatul, dat de persoana autorizata, ca fiecare serie de productie a fost fabricata si controlata conform cerintelor din autorizatia de punere pe piata si a altor reglementari privind productia, controlul si eliberarea produselor medicamentoase;
ca au fost luate masurile corespunzatoare de depozitare, expediere si manipulare ulterioara a produselor medicamentoase in conditii care asigura mentinerea calitatii acestora in timpul perioadei de valabilitate;
existenta unei proceduri de autoinspectie si/sau audit de calitate care evalueaza in mod regulat aplicarea si eficacitatea sistemului unitatii si productiei lor.
Contolul calitatii face parte din RBPF. El se refera la prelevarea probelor, specificatii (norme de calitate), controlul de laborator si de procedurile de organizare, documentare si eliberare a rezultatelor analizelor. Prin acestea este garantata efectuarea analizelor necesare, iar materiile prime, materialele de ambalare si produsele finite nu sunt eliberate pentru a fi utilizate, vandute sau furnizate, pana cand calitatea lor nu a fost declarata ca fiind corespunzatoare..Cerintele fundamentale pentru controlul calitatii:
existenta instalatiilor adecvate, a personalului calificat si a procedurilor aprobate pentru: prelevarea probelor, controlul si analiza materiilor prime, a materialelor de ambalare, a produselor intermediare, vrac si finite, iar unde este cazul si supravegherea conditiilor de mediu conform scopurilor RBPF;
prelevarea de catre personalul departamentului de control a calitatii, conform metodelor aprobate, a probelor de materii prime, articole de ambalare produse intermediare, vrac si finite;
validarea metodelor de control;
inregistrarea manuala sau cu instrumente de inregistrare a tuturor procedurilor cerute, pentru a putea dovedi efectuarea lor reala, precum si obligativitatea inregistrarii si investigarii amanuntite a modificarilor produse;
respectarea compozitiei calitative si cantitative a produselor finite, conform celor inscrise in autorizatia de punere pe piata; materiile prime si produsele finite trebuie sa aiba puritatea ceruta si sa fie corect ambalate si etichetate;
inregistrarile rezultatelor controlului cantitatii materiilor prime, a materialelor de ambalare, a produselor trebuie facuta critic in raport cu documentele de fabricatie si sa se efectueze o estimare a abaterilor fata de procedurile stabilite;
interdictia distribuirii seriilor produsului medicamentos care nu au certificatul de calitate al persoanei autorizate, in conformitate cu autorizatia de punere pe piata;
obligativitatea pastrarii de contraprobe in cantitate suficienta din materialele prime si din produsele medicamentoase finite, care sa permita un control ulterior daca este necesar; contraprobele din produsul finit se pastreaza in ambalajul lor final, cu exceptia ambalajelor deosebit de mari.
RBPF descriu mijloace de aplicare specifice: personalul, localurile si echipamentele, documentatia, productia, controlul calitatii, contractul de fabricatie si control, reclamatii si retragerea produsului, autoinspectia.
In afara acestor prevederi sectorizate si generale, exista anexe referitoare la fabricatia unor produse particulare: fabricatia produselor medicamentoase sub forma de aerosoli sub presiune pentru inhalat prezentate in recipiente cu valva dozatoare; fabricatia lichidelor, cremelor, unguentelor, sisteme computerizate; fabricatia produselor radiofarmaceutice; fabricatia produselor biologice de uz uman; fabricatia produselor medicamentoase de origine vegetala; fabricatia gazelor medicinale; utilizarea radiatiilor ionizante in fabricatia produselor medicamentoase; fabricatia produselor pentru investigatie clinica; fabricatia produselor medicamentoase derivate din sange sau plasma umana; produse homeopate.
BIBLIOGRAFIE
1.Iuliana Popovici, Dumitru Lupuleasa,Tehnologie farmaceutica Vol.I I
Ed.Polirom 2008
2.Sorin Leucuta, Tehnologie farmaceutica industiala , Ed.Dacia 2001
3.E.Hateganu , Chimie farmaceutica Ed. Medicala 2006.
4.E.Hateganu, Chimie terapeutica Ed.Medicala 2008.
5.Farmacopeea Romana Editia X.
6.Farmacopeea Romana Editia IX.
7.Florentina Roncea Tehnologie farmaceutica -note de curs.
8. D. Lupuleasa , Obtinerea industriala a medicamentelor - note de curs.
Identificarea clotrimazolului:
- identificare rapid : B
- identificare secundar : A,C,D
A: punct de topire: 141 - 145C
B: spectrul in infrarosu, comparat cu spectrul obtinut cu clotrimazol CRS
C: examinarea inaintea pulveriz rii in lumina ultraviolet la 254nm
Teste:
Aspectul solutiei
Se dizolv 1.25 g in alcool si se dilueaz la 25 ml cu acelasi solvent. Solutia trebuie s fie limpede si s nu fie mai intens colorat decat solutia de referint BY6.
(2-clorfenil)-difenilmetanol
Examinare in strat cromatografic utilizand silicagel GF254 ca substant de contrast.
Solutia test (a) : se dizolv 0.50g substant ce urmeaz a fi examinat in alcool si se dilueaz la 5 ml cu acelasi solvent.
Solutia test (b) : se dilueaz 1 ml din solutia test (a) la 10 ml cu alcool.
Solutia de referint (a) : se dizolv 50 mg clotrimazol CRS in alcool si se dilueaz la 5 ml cu acelasi solvent.
Solutia de referint (b) : se dizolv 10 mg (2-clorfenil)-difenilmetanol in alcool si se dilueaz la 5 ml cu acelasi solvent. Se dilueaz un ml solutie cu 10 ml alcool.
Saturarea atmosferei din camera de developare se efectueaz folosind un amestec de 0.5 volume solutie concentrat de amoniac, 10 volume de propranolol si 90 volumede toluen.
Pe linia de start a placii cromatografice se aplica cate 10μl din fiecare din solutiile de mai sus. Placa cromatografica se introduce in vasul cromatografic cu developant, se lasa pina cind acesta a migrat pe o distanta de aproximativ 15 cm de la linia de start, se scoate, se usuca la aer. Se pulverizeaz cu 10 %V/V sol H2SO4 in alcool si se usuc la 100-105C pentru 30 minute. Orice pat de (2-clorfenil)-difenilmetanol obtinut cu solutie test a nu este mai intens colorat decat pata obtinut cu solutie de referint b.
Imidazolul
Examinare in strat cromatografic utilizand silicagel GR ca substant de contrast.
Solutia test (a) : se dizolv 0.50g substant ce urmeaz a fi examinat in alcool si se dilueaz la 10 ml cu acelasi solvent.
Solutia de referint : se dizolv 10 mg imidazol in alcool si se dilueaz la 10 ml cu acelasi solvent. Se dilueaz 1 ml solutie cu 10 ml alcool.
Saturarea atmosferei din camera de developare se efectueaz folosind un amestec de 0.5 volume solutie concentrat de amoniac, 10 volume de propranolol si 90 volumede toluen. Pe linia de start a placii cromatografice se aplica cate 10μl din fiecare din solutiile de mai sus. Placa cromatografica se introduce in vasul cromatografic cu developant, se lasa pina cind acesta a migrat pe o distanta de aproximativ 15 cm de la linia de start, se scoate, se usuca la aer. La fundul tancului cromatografic se aseaz un disc de evaporare ce contine un amestec de 1 volumac hidrocloric, 1 volum apa si 2 volume de solutie 15 g/l permanganat de potasiu. Se inchide tancul si se las 15 minute. Se aseaz placa uscat in tanc si se inchide. Se las placa in contact cu vaporii de clor pentru 5 minute. Se scoate placa se aseaz in curent de aer cald, inainte ca excesul de clor s fie indep rtat si ca stratul din apropierea punctului de aplicare sǎ nu dea o coloratie albastrǎ cu o picǎturǎ de KI si solutie de amidon. Orice patǎ coresunzǎtoare imidazolului obtinutǎ cu solutie test nu este mai intensǎ decat pata obtinutǎ cu solutia de referintǎ.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3790
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved