| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Antimicrobial Agents:
Penicillins, Cephalosporins, and Other ![]() -Lactam Antibiotics
-Lactam Antibiotics
Overview
|
The cephalosporin antibiotics are classified by generation, with the
first-generation agents having gram-positive and modest gram-negative
activity; the second generation having somewhat better activity against gram
negatives and including some agents with antianaerobe activity; the third
generation with activity against gram-positive organisms and much more
activity against the Enterobacteriaceae, with a subset active against P.
aeruginosa; and the fourth generation with a spectrum similar to the
third, but having increased stability to hydrolysis by
Bacterial resistance against the |
The Penicillins
|
The penicillins constitute one of the most important groups of antibiotics. Although numerous other antimicrobial agents have been produced since the first penicillin became available, these still are widely used, major antibiotics, and new derivatives of the basic penicillin nucleus still are being produced. Many of these have unique advantages, such that members of this group of antibiotics are presently the drugs of choice for a large number of infectious diseases. History The history of the brilliant research that led to the discovery and
development of penicillin has been recorded by the chief participants. (SeeFleming,
1946; Florey, 1946, 1949; Abraham, 1949; Chain, 1954.) In 1928, while
studying Staphylococcus variants in the laboratory at St. Mary's
Hospital in A decade later, penicillin was developed as a systemic therapeutic
agent by the concerted research of a group of investigators at A vast research program soon was initiated in the The deep-fermentation procedure for the biosynthesis of penicillin marked a crucial advance in the large-scale production of the antibiotic. From a total production of a few hundred million units a month in the early days, the quantity manufactured rose to over 200 trillion units (nearly 150 tons) by 1950. The first marketable penicillin cost several dollars per 100,000 units; today, the same dose costs only a few cents. Chemistry The basic structure of the penicillins, as shown in Figure 451,
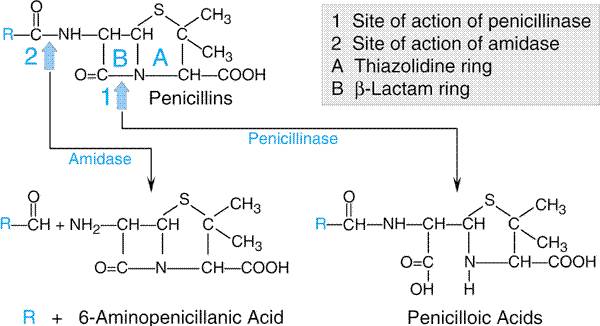
consists of a thiazolidine ring (A) connected to a
Semisynthetic Penicillins The discovery that 6-aminopenicil-break lanic acid could be obtained
from cultures of Penicillium chrysogenum that were depleted of
side-chain precursors led to the development of the semisynthetic
penicillins. Side chains can be added that alter the susceptibility of the
resultant compounds to inactivating enzymes ( Unitage of Penicillin The international unit of penicillin is the specific penicillin
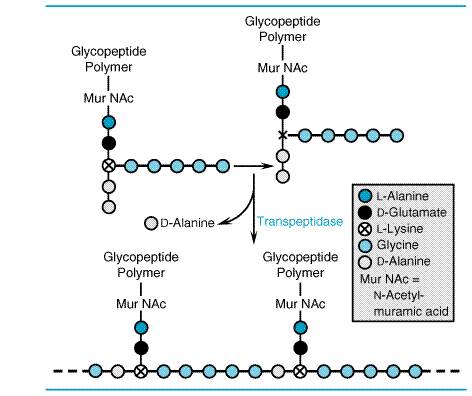
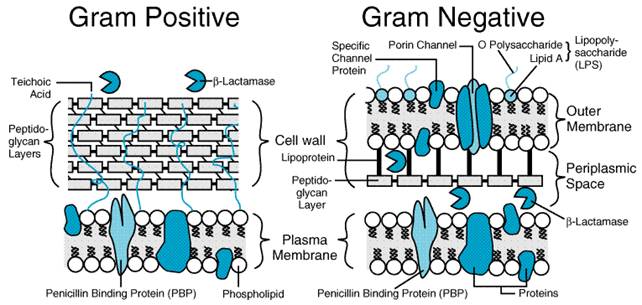
activity contained in 0.6 Mechanism of Action of the Penicillins and Cephalosporins The The cell walls of bacteria are essential for their normal growth and development. Peptidoglycan is a heteropolymeric component of the cell wall that provides rigid mechanical stability by virtue of its highly cross-linked latticework structure (Figure 452). In gram-positive microorganisms, the cell wall is 50 to 100 molecules thick, but it is only 1 or 2 molecules thick in gram-negative bacteria (Figure 453). The peptidoglycan is composed of glycan chains, which are linear strands of two alternating amino sugars (N-acetylglucosamine and N-acetylmuramic acid) that are cross-linked by peptide chains.
The biosynthesis of the peptidoglycan involves about 30 bacterial enzymes and may be considered in three stages. The first stage, precursor formation, takes place in the cytoplasm. The product, uridine diphosphate (UDP)-acetylmuramyl-pentapeptide, called a 'Park nucleotide' after its discoverer (Park and Strominger, 1957), accumulates in cells when subsequent synthetic stages are inhibited. The last reaction in the synthesis of this compound is the addition of a dipeptide, D-alanyl-D-alanine. Synthesis of the dipeptide involves prior racemization of L-alanine and condensation catalyzed by D-alanyl-D-alanine synthetase. D-Cycloserine is a structural analog of D-alanine and acts as a competitive inhibitor of both the racemase and the synthetase (seeChapter 48: Antimicrobial Agents: Drugs Used in the Chemotherapy of Tuberculosis, Mycobacterium avium Complex Disease, and Leprosy). During reactions of the second stage, UDP-acetylmuramyl-pentapeptide and UDP-acetylglucosamine are linked (with the release of the uridine nucleotides) to form a long polymer. The third and final stage involves the completion of the cross-link.
This is accomplished by a transpeptidation reaction that occurs outside the
cell membrane. The transpeptidase itself is membrane-bound. The terminal glycine
residue of the pentaglycine bridge is linked to the fourth residue of the
pentapeptide (D-alanine),
releasing the fifth residue (also D-alanine) (Figure 452). It is this last step in peptidoglycan
synthesis that is inhibited by the Although inhibition of the transpeptidase described above is
demonstrably important, there are additional, related targets for the actions
of penicillins and cephalosporins; these are collectively termed penicillin-binding
proteins (PBPs; Spratt, 1980; Ghuysen, 1991). All bacteria have several
such entities; for example, S. aureus has four PBPs, while Escherichia
coli has at least seven. The PBPs vary in their affinities for different Mechanisms of Bacterial Resistance to Penicillins and Cephalosporins Although most or all bacteria contain PBPs,
Other
instances of bacterial resistance to the
Bacteria
can destroy In general, gram-positive bacteria produce a large amount of Other Factors That Influence the Activity of The density of the bacterial population and the age of an infection
influence the activity of The presence of proteins and other constituents of pus, low pH, or low
oxygen tension does not appreciably decrease the ability of Classification of the Penicillins and Summary of Their Pharmacological Properties It is useful to classify the penicillins according to their spectra of antimicrobial activity (seeTable 451; Chambers, 2000).
Although the pharmacological properties of the individual drugs are discussed in detail below, certain generalizations are useful. Following absorption of orally administered penicillins, these agents are widely distributed throughout the body. Therapeutic concentrations of penicillins are achieved readily in tissues and in secretions such as joint fluid, pleural fluid, pericardial fluid, and bile. However, only low concentrations of these drugs are found in prostatic secretions, brain tissue, and intraocular fluid, and penicillins do not penetrate living phagocytic cells to a significant extent. Concentrations of penicillins in cerebrospinal fluid (CSF) are variable, but are less than 1% of those in plasma when the meninges are normal. When there is inflammation, concentrations in CSF may increase to as much as 5% of the plasma value. Penicillins are eliminated rapidly, particularly by glomerular filtration and renal tubular secretion, such that their half-lives in the body are short; values of 30 to 90 minutes are typical. Concentrations of these drugs in urine thus are high. Penicillin G and Penicillin V Antimicrobial Activity The antimicrobial spectra of penicillin G(benzylpenicillin) and penicillin V (the phenoxymethyl derivative) are very similar for aerobic gram-positive microorganisms. However, penicillin G is five to ten times more active against Neisseria species sensitive to penicillins and against certain anaerobes. Penicillin G has activity against a variety of species of
gram-positive and gram-negative cocci, although many bacteria previously
sensitive to the agent are now resistant. Most streptococci (but not
enterococci) are very susceptible to the drug; concentrations of less than
0.01 Whereas most strains of S. aureus were highly sensitive to similar concentrations of penicillin G when this agent was first employed therapeutically, more than 90% of strains of staphylococci isolated from individuals inside or outside of hospitals are now resistant to penicillin G. Most strains of Staphylococcus epidermidis also are resistant to penicillin. Unfortunately, penicillinase-producing strains of gonococci that are highly resistant to penicillin G have become widespread. With rare exceptions, meningococci are quite sensitive to penicillin G. Although the vast majority of strains of Corynebacterium
diphtheriae are sensitive to penicillin G, some are highly resistant.
This also is true for Bacillus anthracis. Most anaerobic
microorganisms, including Clostridium species, are highly sensitive. Bacteroides
fragilis is an exception; many strains are now resistant because of
elaboration of Absorption Oral Administration of Penicillin G About one-third of an orally administered dose of penicillin G is
absorbed from the intestinal tract under favorable conditions. Gastric juice
at pH 2 rapidly destroys the antibiotic. The decrease in gastric acid
production with aging accounts for better absorption of penicillin G from the
gastrointestinal tract of older individuals. Absorption is rapid, and maximal
concentrations in blood are attained in 30 to 60 minutes. The peak value is
approximately 0.5 U/ml (0.3 Oral Administration of Penicillin V The virtue of penicillin V in comparison with penicillin G is that it
is more stable in an acidic medium, and therefore is better absorbed from the
gastrointestinal tract. On an equivalent oral-dose basis, penicillin V (K+
salt PEN-VEE
K, V-CILLIN K,
others) yields plasma concentrations two to five times greater than those
provided by penicillin G. The peak concentration in the blood of an adult
after an oral dose of 500 mg is nearly 3 Parenteral Administration of Penicillin G After intramuscular injection, peak concentrations in plasma are reached within 15 to 30 minutes. This value declines rapidly, since the half-life of penicillin G is 30 minutes. Many means for prolonging the sojourn of the antibiotic in the body and thereby reducing the frequency of injections have been explored. Probenecid blocks renal tubular secretion of penicillin, but it is rarely used for this purpose (see below). More commonly, repository preparations of penicillin G are employed. The two such compounds currently favored are penicillin G procaine (DURACILLIN, A.S., WYCILLIN, others) and penicillin G benzathine (BICILLIN L-A, PERMAPEN). Such agents release penicillin G slowly from the area in which they are injected and produce relatively low but persistent concentrations of antibiotic in the blood. Penicillin Gprocaine suspension is an aqueous preparation of the crystalline salt that is only 0.4% soluble in water. Procaine combines with penicillin mol for mol; a dose of 300,000 U thus contains approximately 120 mg of procaine. When large doses of penicillin G procaine are given (e.g., 4.8 million units), procaine may reach toxic concentrations in the plasma. If the patient is believed to be hypersensitive to procaine, 0.1 ml of 1% solution of procaine should be injected intradermally as a test. The anesthetic effect of the procaine accounts in part for the fact that injections of penicillin G procaine are virtually painless. The injection of 300,000 U of penicillin Gprocaine produces a peak
concentration in plasma of about 0.9 Penicillin G benzathine suspension is the aqueous suspension of the salt obtained by the combination of 1 mol of an ammonium base and 2 mol of penicillin G to yield N,N'-dibenzylethylenediamine dipenicillin G. The salt itself is only 0.02% soluble in water. The long persistence of penicillin in the blood after a suitable intramuscular dose reduces cost, need for repeated injections, and local trauma. The local anesthetic effect of penicillin G benzathine is comparable to that of penicillin G procaine. Penicillin G benzathine is absorbed very slowly from intramuscular
depots and produces the longest duration of detectable antibiotic of all the
available repository penicillins. For example, in adults, a dose of 1.2
million units given intramuscularly produces a concentration in plasma of
0.09 Distribution Penicillin G is distributed widely throughout the body, but the concentrations in various fluids and tissues differ widely. Its apparent volume of distribution is about 0.35 liter/kg. Approximately 60% of the penicillin G in plasma is reversibly bound to albumin. Significant amounts appear in liver, bile, kidney, semen, joint fluid, lymph, and intestine. While probenecid markedly decreases the tubular secretion of the penicillins, this is not the only factor responsible for the elevated plasma concentrations of the antibiotic that follow its administration. Probenecid also produces a significant decrease in the apparent volume of distribution of the penicillins. Cerebrospinal Fluid Penicillin does not readily enter the CSF when the meninges are normal. However, when the meninges are acutely inflamed, penicillin penetrates into the CSF more easily. Although the concentrations attained vary and are unpredictable, they are usually in the range of 5% of the value in plasma and are therapeutically effective against susceptible microorganisms. Penicillin and other organic acids are secreted rapidly from the CSF into the bloodstream by an active transport process. Probenecid competitively inhibits this transport and thus elevates the concentration of penicillin in CSF. In uremia, other organic acids accumulate in the CSF and compete with penicillin for secretion; the drug occasionally reaches toxic concentrations in the brain and can produce convulsions. Excretion Under normal conditions, penicillin G is rapidly eliminated from the body, mainly by the kidney but in small part in the bile and by other routes. Approximately 60% to 90% of an intramuscular dose of penicillin G in aqueous solution is eliminated in the urine, largely within the first hour after injection. The remainder is metabolized to penicilloic acid. The half-time for elimination of penicillin G is about 30 minutes in normal adults. Approximately 10% of the drug is eliminated by glomerular filtration and 90% by tubular secretion. Renal clearance approximates the total renal plasma flow. The maximal tubular secretory capacity for penicillin in the normal male adult is about 3 million units (1.8 g) per hour. Clearance values are considerably lower in neonates and infants because of incomplete development of renal function; as a result, after doses proportionate to surface area, the persistence of penicillin in the blood is several times as long in premature infants as in children and adults. The half-life of the antibiotic in children less than 1 week old is 3 hours; by 14 days of age it is 1.4 hours. After renal function is fully established in young children, the rate of renal excretion of penicillin G is considerably more rapid than in adults. Anuria increases the half-life of penicillin G from a normal value of 0.5 hour to about 10 hours. When renal function is impaired, 7% to 10% of the antibiotic may be inactivated each hour by the liver. Patients with renal shutdown who require high-dose therapy with penicillin can be treated adequately with 3 million units of aqueous penicillin G followed by 1.5 million units every 8 to 12 hours. The dose of the drug must be readjusted during dialysis and the period of progressive recovery of renal function. If, in addition to renal failure, hepatic insufficiency also is present, the half-life will be prolonged even further. Therapeutic Uses Pneumococcal Infections Penicillin G remains the agent of choice for the management of
infections caused by sensitive strains of S. pneumoniae. However,
strains of pneumococci resistant to usual doses of penicillin G are being
more frequently isolated in several countries, including the Pneumococcal Pneumonia Until it is highly likely or established that the infecting isolate of pneumococcus is penicillin-sensitive, pneumococcal pneumonia should be treated with a third-generation cephalosporin or with vancomycin. For parenteral therapy of sensitive isolates of pneumococci, penicillin G or penicillin G procaine is favored. Although oral treatment with 500 mg of penicillin V given every 6 hours for treatment of pneumonia due to penicillin-sensitive isolates has been used with success in this disease, it cannot be recommended for routine initial use because of the existence of resistance. Therapy should be continued for 7 to 10 days, including 3 to 5 days after the patient's temperature has returned to normal. Pneumococcal Meningitis Until it is established that the infecting pneumococcus is penicillin-sensitive, pneumococcal meningitis should be treated with a combination of vancomycin and a third-generation cephalosporin (John, 1994; Catalan et al., 1994). Some authorities advocate a third-generation cephalosporin plus rifampin. Prior to the appearance of penicillin resistance, penicillin treatment reduced the death rate in this disease from nearly 100% to about 25%. The recommended therapy is 20 million to 24 million units of penicillin G daily by constant intravenous infusion or divided into boluses given every 2 to 3 hours. The usual duration of therapy is 14 days. Streptococcal Infections Streptococcal Pharyngitis (Including Scarlet Fever) This is the most common disease produced by Streptococcus pyogenes
(group A Streptococcal Pneumonia, Arthritis, Meningitis, and Endocarditis While uncommon, these conditions should be treated with penicillin G when they are caused by S. pyogenes; daily doses of 12 million to 20 million units are administered intravenously for 2 to 4 weeks. Such treatment of endocarditis should be continued for a full 4 weeks. Infections Caused by Other Streptococci The viridans streptococci are the most common cause of
infectious endocarditis. These are nongroupable Enterococcal endocarditis is one of the few diseases that is optimally treated with two antibiotics. The recommended therapy for penicillin- and aminoglycoside-sensitive enterococcal endocarditis is 20 million units of penicillin G or 12 grams of ampicillin daily, administered intravenously in combination with an aminoglycoside. Therapy usually should be continued for 6 weeks, but selected patients with a short duration of illness (less than 3 months) have been treated successfully in 4 weeks (Wilson et al., 1984). Infections with Anaerobes Many anaerobic infections are caused by mixtures of microorganisms.
The majority are sensitive to penicillin G. An exception is the B.
fragilis group, in which up to 75% of strains may be resistant to high
concentrations of this antibiotic. Pulmonary and periodontal infections (with
the exception of Staphylococcal Infections The vast majority of staphylococcal infections are caused by microorganisms that produce penicillinase. A patient with a staphylococcal infection who requires treatment with an antibiotic should receive one of the penicillinase-resistant penicillinsfor example, nafcillin, oxacillin, or methicillin. So-called methicillin-resistant staphylococci are resistant to penicillin G, all of the penicillinase-resistant penicillins, and the cephalosporins. Isolates occasionally may appear to be sensitive to various cephalosporins in vitro, but resistant populations arise during therapy and lead to failure (Chambers et al., 1984). Vancomycin is the drug of choice for infections caused by these bacteria, although reduced susceptibility to vancomycin has now been observed (Centers for Disease Control and Prevention, 1997). Ciprofloxacin also may be effective, although prolonged therapy often leads to the emergence of ciprofloxacin-resistant S. aureus. Meningococcal Infections Penicillin G remains the drug of choice for meningococcal disease.
Patients should be treated with high doses of penicillin given intravenously,
as described for pneumococcal meningitis. Penicillin-resistant strains of N.
meningitides have been reported in Gonococcal Infections Gonococci gradually have become more resistant to penicillin G, and penicillins are no longer the therapy of choice, unless it is known that gonococcal strains in a particular geographical area are susceptible. Uncomplicated gonococcal urethritis is the most common infection, and a single intramuscular injection of 250 mg of ceftriaxone is the recommended treatment (Sparling and Handsfield, 2000). Gonococcal arthritis, disseminated gonococcal infections with skin lesions, and gonococcemia should be treated with ceftriaxone, 1 g daily given either intramuscularly or intravenously for 7 to 10 days. Ophthalmia neonatorum also should be treated with ceftriaxone for 7 to 10 days (25 to 50 mg/kg per day intramuscularly or intravenously). Syphilis Therapy of syphilis with penicillin G is highly effective. Primary, secondary, and latent syphilis of less than 1 year's duration may be treated with penicillin G procaine (2.4 million units per day intramuscularly) plus probenecid (1.0 g per day orally) for 10 days or with 1 to 3 weekly intramuscular doses of 2.4 million units of penicillin G benzathine (3 doses in patients with HIV). Patients with late latent syphilis, neurosyphilis, or cardiovascular syphilis may be treated with a variety of regimens. Since these diseases are potentially lethal and their progression can be halted (but not reversed), intensive therapy with 20 million units of penicillin G daily for 10 days is recommended. Infants with congenital syphilis discovered at birth or during the postnatal period should be treated for at least 10 days with 50,000 U/kg daily of aqueous penicillin G in two divided doses or 50,000 U/kg of procaine penicillin G in a single daily dose (seeTramont, 2000). The majority (70% to 90%) of patients with secondary syphilis develop the Jarisch-Herxheimer reaction. This also may be seen in patients with other forms of syphilis. Several hours after the first injection of penicillin, chills, fever, headache, myalgias, and arthralgias may develop. The syphilitic cutaneous lesions may become more prominent, edematous, and brilliant in color. Manifestations usually persist for a few hours, and the rash begins to fade within 48 hours. It does not recur with the second or subsequent injections of penicillin. This reaction is thought to be due to release of spirochetal antigens, with subsequent host reactions to the products. Aspirin gives symptomatic relief, and therapy with penicillin should not be discontinued. Actinomycosis Penicillin G is the agent of choice for the treatment of all forms of actinomycosis. The dose should be 12 million to 20 million units of penicillin G intravenously per day for 6 weeks. Some physicians continue therapy for 2 to 3 months with oral penicillin V (500 mg four times daily). Surgical drainage or excision of the lesion may be necessary before cure is accomplished. Diphtheria There is no evidence that penicillin or any other antibiotic alters the incidence of complications or the outcome of diphtheria; specific antitoxin is the only effective treatment. However, penicillin G eliminates the carrier state. The parenteral administration of 2 to 3 million units per day in divided doses for 10 to 12 days eliminates the diphtheria bacilli from the pharynx and other sites in practically 100% of cases. A single daily injection of penicillin G procaine for the same period produces about the same results. Anthrax Penicillin G is the agent of choice in the treatment of all clinical forms of anthrax. However, strains of Bacillus anthracis resistant to this antibiotic have been recovered from human infections. When penicillin G is used, the dose should be 12 million to 20 million units per day. Clostridial Infections Penicillin G is the agent of choice for gas gangrene; the dose is in the range of 12 million to 20 million units per day, given parenterally. Adequate debridement of the infected areas is essential. Antimicrobial drugs probably have no effect on the ultimate outcome of tetanus. Debridement and administration of human tetanus immune globulin may be indicated. Penicillin is administered, however, to eradicate the vegetative forms of the bacteria that may persist. Fusospirochetal Infections Gingivostomatitis, produced by the synergistic action of Leptotrichia buccalis and spirochetes that are present in the mouth, is readily treatable with penicillin. For simple 'trench mouth,' 500 mg of penicillin V given every 6 hours for several days is usually sufficient to clear the disease. Rat-Bite Fever The two microorganisms responsible for this infection, Spirillum minor
in the Orient and Streptobacillus moniliformis in Listeria Infections Penicillin G or ampicillin with or without gentamicin are regarded as the drugs of choice in the management of infections due to L. monocytogenes. The recommended dose of penicillin G is 15 million to 20 million units parenterally per day for at least 2 weeks. When endocarditis is the problem, the dose is the same, but the duration of treatment should be no less than 4 weeks. Lyme Disease Although a tetracycline is the usual drug of choice for early disease, amoxicillin is effective; the dose is 500 mg three times daily for 21 days. Severe disease is treated with a third-generation cephalosporin or 20 million units of intravenous penicillin G daily for 14 days. Erysipeloid The causative agent of this disease, Erysipelothrix rhusiopathiae, is sensitive to penicillin. The uncomplicated infection responds well to a single injection of 1.2 million units of penicillin G benzathine. When endocarditis is present, penicillin G, 12 million to 20 million units per day, has been found to be effective; therapy should be continued for 4 to 6 weeks. Prophylactic Uses of the Penicillins Demonstration of the effectiveness of penicillin in eradicating microorganisms was quickly, and quite naturally, followed by attempts to prove that it also was effective in preventing infection in susceptible hosts. As a result, the antibiotic has been administered in almost every situation in which a risk of bacterial invasion has been present. As prophylaxis has been investigated under controlled conditions, it has become clear that penicillin is highly effective in some situations, useless and potentially dangerous in others, and of questionable value in still others (seeChapter 43: Antimicrobial Agents: General Considerations). Streptococcal Infections The administration of penicillin to individuals exposed to Streptococcus pyogenes affords protection from infection. The oral ingestion of 200,000 units of penicillin G or penicillin V twice a day or a single injection of 1.2 million units of penicillin G benzathine is effective. Indications for this type of prophylaxis include outbreaks of streptococcal disease in closed populations, such as boarding schools or military bases. Patients with extensive deep burns are at high risk of severe wound infections with S. pyogenes; several days of 'low-dose' prophylaxis appears to be effective in reducing the incidence of this complication. Recurrences of Rheumatic Fever The oral administration of 200,000 units of penicillin G or penicillin V every 12 hours produces a striking decrease in the incidence of recurrences of rheumatic fever in susceptible individuals. Because of the difficulties of compliance, parenteral administration is preferable, especially in children. The intramuscular injection of 1.2 million units of penicillin G benzathine once a month yields excellent results. In cases of hypersensitivity to penicillin, sulfisoxazole or sulfadiazine, 1 g twice a day for adults, also is effective; for children weighing under 27 kg, the dose is halved. Prophylaxis must be continued throughout the year. The duration of such treatment is an unsettled question. It has been suggested that prophylaxis should be continued for life, because instances of acute rheumatic fever have been observed in the fifth and sixth decades. However, the necessity for such prolonged prophylaxis has not been established and may be unnecessary for certain young adults judged to be low risk for recurrence (Berrios et al., 1993). Syphilis Prophylaxis for a contact with syphilis consists of a course of therapy as described for primary syphilis. A serological test for syphilis should be performed at monthly intervals for at least 4 months thereafter. Surgical Procedures in Patients with Valvular Heart Disease About 25% of cases of subacute bacterial endocarditis follow dental extractions. This observation, together with the fact that up to 80% of persons who have teeth removed experience a transient bacteremia, emphasizes the potential importance of chemoprophylaxis for those who have congenital or acquired valvular heart disease of any type and need to undergo dental procedures. Since transient bacterial invasion of the bloodstream occurs occasionally after surgical procedures (e.g., tonsillectomy and genitourinary and gastrointestinal procedures) and during childbirth, these too are indications for prophylaxis in patients with valvular heart disease. Whether the incidence of bacterial endocarditis actually is altered by this type of chemoprophylaxis remains to be determined. Detailed recommendations for both adults and children with valvular heart disease have been formulated (seeDajani et al., 1990; Durack, 2000). The Penicillinase-Resistant Penicillins The penicillins described in this section are resistant to hydrolysis by staphylococcal penicillinase. Their appropriate use should be restricted to the treatment of infections that are known or suspected to be caused by staphylococci that elaborate the enzymethe vast majority of strains of this bacterium that are encountered in the hospital or in the general community. These drugs are much less active than is penicillin G against other penicillin-sensitive microorganisms, including non-penicillinase-producing staphylococci. The role of the penicillinase-resistant penicillins as the agents of
choice for most staphylococcal disease may be changing with the increasing
incidence of isolates of so-called methicillin-resistant microorganisms. As
commonly used, this latter term denotes resistance of these bacteria to all
of the penicillinase-resistant penicillins and cephalosporins. Such strains
are usually resistant as well to the aminoglycosides, tetracyclines, erythromycin,
and clindamycin. Vancomycin is considered to be the drug of choice for such
infections. Some physicians use a combination of vancomycin and rifampin,
especially for life-threatening infections and those involving foreign
bodies. Methicillin-resistant S. aureus contains an additional high
molecular weight PBP with a very low affinity for The Isoxazolyl Penicillins: Oxacillin, Cloxacillin, and Dicloxacillin These three congeneric semisynthetic penicillins are similar pharmacologically and are thus conveniently considered together. Their structural formulas are shown in Table 451. All are relatively stable in an acidic medium and are adequately absorbed after oral administration. All are markedly resistant to cleavage by penicillinase. These drugs are not substitutes for penicillin G in the treatment of diseases amenable to it. Furthermore, because of variability in intestinal absorption, oral administration is not a substitute for the parenteral route in the treatment of serious staphylococcal infections that require a penicillin unaffected by penicillinase. Pharmacological Properties The isoxazolyl penicillins are potent inhibitors of the growth of most
penicillinase-producing staphylococci. This is their valid clinical use. Dicloxacillin
(PATHOCIL, others) is the most active, and
many strains of S. aureus are inhibited by concentrations of 0.05 to
0.8 These agents are rapidly but incompletely (30% to 80%) absorbed from
the gastrointestinal tract. Absorption of the drugs is more efficient when
they are taken on an empty stomach; preferably they are administered one hour
before or two hours after meals to ensure better absorption. Peak
concentrations in plasma are attained by 1 hour and approximate 5 to 10 The isoxazolyl penicillins are rapidly excreted by the kidney. Normally, about one-half of any of these drugs is excreted in the urine in the first 6 hours after a conventional oral dose. There also is significant hepatic elimination of these agents in the bile. The half-lives for all are between 30 and 60 minutes. Intervals between doses of oxacillin, cloxacillin, and dicloxacillin do not have to be altered for patients with renal failure. The above-noted differences in plasma concentrations produced by the isoxazolyl penicillins are related mainly to differences in rate of urinary excretion and degree of resistance to degradation in the liver. Nafcillin This semisynthetic penicillin is highly resistant to penicillinase and has proven effective against infections caused by penicillinase-producing strains of S. aureus. Its structural formula is shown in Table 451. Pharmacological Properties Nafcillin (UNIPEN, NALLPEN,
others) is slightly more active than oxacillin against penicillin Gresistant
S. aureus (most strains are inhibited by 0.06 to 2 Nafcillin is inactivated to a variable degree in the acidic medium of
the gastric contents. Its absorption after oral administration is irregular,
regardless of whether the drug is taken with meals or on an empty stomach.
Consequently, although oral preparations are available, injectable preparations
should be used because of the variable absorption of nafcillin from the
gastrointestinal tract. The peak plasma concentration is about 8 The Aminopenicillins: Ampicillin, Amoxicillin, and Their Congeners These agents have similar antibacterial activity and a spectrum that
is broader than the antibiotics heretofore discussed. They are all destroyed
by Antimicrobial Activity Ampicillin and the related aminopenicillins are bactericidal for both
gram-positive and gram-negative bacteria. The meningococci and L.
monocytogenes are sensitive to the drug. Many pneumococcal isolates have
varying levels of resistance to ampicillin. Penicillin-resistant strains
should be considered ampicillin/amoxicillin-resistant. H. influenzae
and the viridans group of streptococci exhibit varying degrees of
resistance. Enterococci are about twice as sensitive to ampicillin, on a
weight basis, as they are to penicillin G (MIC for ampicillin averages 1.5
mg/ml). Although most strains of N. gonorrhoeae, E. coli, P.
mirabilis, Salmonella, and Shigella were highly susceptible when
ampicillin was first used in the early 1960s, an increasing percentage of
these species is now resistant. From 30% to 50% of E. coli, a
significant number of P. mirabilis, and practically all species of Enterobacter
are presently insensitive. Resistant strains of Salmonella (plasmid
mediated) have been recovered with increasing frequency in various parts of
the world. Most strains of Shigella are now resistant. Most strains of
Pseudomonas, Klebsiella, Serratia, Acinetobacter, and
indole-positive Proteus also are resistant to this group of
penicillins; these antibiotics are less active against B. fragilis
than is penicillin G. However, concurrent administration of a Ampicillin This drug is the prototypical agent of the group. Its structural formula is shown in Table 451. Pharmacological Properties Ampicillin (OMNIPEN, POLYCILLIN, others) is stable in acid and is well
absorbed after oral administration. An oral dose of 0.5 g produces peak
concentrations in plasma of about 3 Amoxicillin This drug, a penicillinase-susceptible semi-synthetic penicillin, is a close chemical and pharmacological relative of ampicillin (seeTable 451). The drug is stable in acid and is designed for oral use. It is more rapidly and completely absorbed from the gastrointestinal tract than is ampicillin, which is the major difference between the two. The antimicrobial spectrum of amoxicillin is essentially identical to that of ampicillin, with the important exception that amoxicillin appears to be less effective than ampicillin for shigellosis. Peak concentrations of amoxicillin (AMOXIL, others) in plasma are two to two
and one-half times greater for amoxicillin than for ampicillin after oral
administration of the same dose; they are reached at 2 hours and average
about 4 Therapeutic Indications for the Aminopenicillins Upper Respiratory Infections Ampicillin and amoxicillin are active against S. pyogenes and
many strains of S. pneumoniae and H. influenzae, which are
major upper respiratory bacterial pathogens. The drugs constitute effective
therapy for sinusitis, otitis media, acute exacerbations of chronic
bronchitis, and epiglottitis caused by sensitive strains of these organisms.
Amoxicillin is the most active of all the oral Urinary Tract Infections Most uncomplicated urinary tract infections are caused by Enterobacteriaceae, and E. coli is the most common species; ampicillin often is an effective agent, although resistance is increasingly common. Enterococcal urinary tract infections are treated effectively with ampicillin alone. Meningitis Acute bacterial meningitis in children is most frequently due to S. pneumoniae or N. meningitidis. Since 20% to 30% of strains of S. pneumoniae now may be resistant to this antibiotic, ampicillin is not indicated for single-agent treatment of meningitis. Ampicillin has excellent activity against L. monocytogenes, a cause of meningitis in immunocompromised persons. Thus, the combination of ampicillin and vancomycin plus a third-generation cephalosporin is a rational regimen for empiric treatment of suspected bacterial meningitis. Salmonella Infections Disease associated with bacteremia, disease with metastatic foci, and the enteric fever syndrome (including typhoid fever) respond favorably to antibiotics. A fluoroquinolone or ceftriaxone is considered by some to be the drug of choice, but the administration of trimethoprimsulfamethoxazole or high doses of ampicillin (12 g per day for adults) also is effective. In some geographical areas, resistance to ampicillin is common. The typhoid carrier state has been eliminated successfully in patients without gallbladder disease with ampicillin, trimethoprimsulfamethoxazole, or ciprofloxacin. Antipseudomonal Penicillins: The Carboxypenicillins and the Ureidopenicillins The carboxypenicillins, carbenicillin and ticarcillin
and their close relatives, are active against some isolates of P.
aeruginosa and certain indole-positive Proteus species that are
resistant to ampicillin and its congeners. They are ineffective against most
strains of S. aureus, Enterococcus faecalis, Klebsiella, and L.
monocytogenes.B. fragilis is susceptible to high concentrations of these
drugs, but penicillin G is actually more active on the basis of weight. The
ureidopenicillins, mezlocillin and piperacillin, have superior
activity against P. aeruginosa compared to carbenicillin and
ticarcillin. In addition, mezlocillin and piperacillin are useful for
treatment of infections with Klebsiella. The carboxypenicillins and
the ureidopenicillins are sensitive to destruction by Carbenicillin and Carbenicillin Indanyl Carbenicillin This drug is a penicillinase-susceptible derivative of 6-aminopenicillanic acid. Its structural formula is shown in Table 451. Carbenicillin was the first penicillin with activity against P. aeruginosa and some Proteus strains that are resistant to ampicillin. It has been superseded by ticarcillin or piperacillin for most uses (see below). Preparations of carbenicillin may cause adverse effects in addition to those that follow use of the other penicillins (see below). Congestive heart failure may result from the administration of excessive Na+. Hypokalemia may occur because of obligatory excretion of cation with the large amount of nonreabsorbable anion (carbenicillin) presented to the distal renal tubule. The drug interferes with platelet function, and bleeding may occur because of abnormal aggregation of platelets. Carbenicillin Indanyl Sodium (GEOCILLIN This congener is the indanyl ester of carbenicillin; it is acid-stable and is suitable for oral administration. After absorption, the ester is rapidly converted to carbenicillin by hydrolysis of the ester linkage. The antimicrobial spectrum of the drug is therefore that of carbenicillin. Although relatively low concentrations of carbenicillin are achieved in plasma, the active moiety is excreted rapidly in the urine. Thus, the only use of this drug is for the management of urinary tract infections caused by Proteus species other than P. mirabilis and by P. aeruginosa. Ticarcillin (TICAR This semisynthetic penicillin (Table 451) is very similar to carbenicillin, but it is two to four times more active against P. aeruginosa. Ticarcillin is inferior to piperacillin and mezlocillin for the treatment of serious infections caused by Pseudomonas. Mezlocillin This ureidopenicillin is more active against Klebsiella than is carbenicillin; its activity against Pseudomonasin vitro is similar to that of ticarcillin. It is more active than ticarcillin against Enterococcus faecalis. Mezlocillin sodium (MEZLIN) is available as a powder to be dissolved for injection and contains about 2 mEq of Na+ per gram. The usual dose for adults is 6 to 18 g per day, divided into four to six portions. Mezlocillin and piperacillin (below) are excreted in bile to a significant degree. In the absence of biliary tract obstruction, high concentrations of mezlocillin in bile are achieved by intravenous administration. Piperacillin Piperacillin (PIPRACIL) extends the spectrum of ampicillin to include most strains of P.
aeruginosa, Enterobacteriaceae (non- Therapeutic Indications These penicillins are important agents for the treatment of patients
with serious infections caused by gram-negative bacteria. Such patients
frequently have impaired immunological defenses, and their infections often
are acquired in the hospital. Many authorities feel that a Untoward Reactions to Penicillins Hypersensitivity Reactions Hypersensitivity reactions are by far the most common adverse effects noted with the penicillins, and these agents probably are the most common cause of drug allergy. Allergic reactions complicate between 0.7% and 4% of all treatment courses. There is no convincing evidence that any single penicillin differs from the group in its potential for causing true allergic reactions. In approximate order of decreasing frequency, manifestations of allergy to penicillins include maculopapular rash, urticarial rash, fever, bronchospasm, vasculitis, serum sickness, exfoliative dermatitis, StevensJohnson syndrome, and anaphylaxis (Anonymous, 1988; Weiss and Adkinson, 2000). The overall incidence of such reactions to the penicillins varies from 0.7% to 10% in different studies. Hypersensitivity reactions may occur with any dosage form of penicillin; allergy to one penicillin exposes the patient to a greater risk of reaction if another is given. On the other hand, the occurrence of an untoward effect does not necessarily imply repetition on subsequent exposures. Hypersensitivity reactions may appear in the absence of a previous known exposure to the drug. This may be caused by unrecognized prior exposure to penicillin in the environment (e.g., in foods of animal origin or from the fungus producing penicillin). Although elimination of the antibiotic usually results in rapid clearing of the allergic manifestations, they may persist for 1 or 2 weeks or longer after therapy has been stopped. In some cases, the reaction is mild and disappears even while the use of penicillin is continued; in others, it necessitates immediate cessation of penicillin treatment. In a few instances, it is necessary to interdict the future use of penicillin because of the risk of death, and the patient should be so warned. It must be stressed that fatal episodes of anaphylaxis have followed the ingestion of very small doses of this antibiotic or skin testing with minute quantities of the drug. Penicillins and breakdown products of penicillins act as haptens after
their covalent reaction with proteins. The most important antigenic
intermediate of penicillin appears to be the penicilloyl moiety, which is
formed when the Antipenicillin antibodies are detectable in virtually all patients who have received the drug and in many who have never knowingly been exposed to it (Klaus and Fellner, 1973). Recent treatment with the antibiotic induces an increase in major-determinant-specific antibodies that are skin sensitizing. The incidence of positive skin reactors is three to four times higher in atopic than in nonatopic individuals. Clinical and immunological studies suggest that immediate allergic reactions are mediated by skin-sensitizing or IgE antibodies, usually of minor-determinant specificities. Accelerated and late urticarial reactions usually are mediated by major-determinantspecific skin-sensitizing antibodies. The recurrent-arthralgia syndrome appears to be related to the presence of skin-sensitizing antibodies of minor-determinant specificities. Some maculopapular and erythematous reactions may be due to toxic antigen-antibody complexes of major determinantspecific IgM antibodies. Accelerated and late urticarial reactions to penicillin may terminate spontaneously because of the development of blocking antibodies. Skin rashes of all types may be caused by allergy to penicillin. Scarlatiniform, morbilliform, urticarial, vesicular, and bullous eruptions may develop. Purpuric lesions are uncommon and are usually the result of a vasculitis; thrombocytopenic purpura may occur very rarely. HenochSchnlein purpura with renal involvement has been a rare complication. Contact dermatitis is observed occasionally in pharmacists, nurses, and physicians who prepare penicillin solutions. Fixed-drug reactions also have occurred.More severe reactions involving the skin are exfoliative dermatitis and exudative erythema multiforme of either the erythematopapular or vesiculobullous type; these lesions may be very severe and atypical in distribution and constitute the characteristic StevensJohnson syndrome. The incidence of skin rashes appears to be highest following the use of ampicillin, being about 9%; rashes follow the administration of ampicillin in nearly all patients with infectious mononucleosis. When allopurinol and ampicillin are administered concurrently, the incidence of rash also increases. Ampicillin-induced skin eruptions in such patients may represent a 'toxic' rather than a truly allergic reaction. Positive skin reactions to the major and minor determinants of penicillin sensitization may be absent. The rash may clear even while administration of the drug is continued. The most serious hypersensitivity reactions produced by the penicillins are angioedema and anaphylaxis. Angioedema with marked swelling of the lips, tongue, face, and periorbital tissues, frequently accompanied by asthmatic breathing and 'giant hives,' has been observed after topical, oral, or systemic administration of penicillins of various types. Acute anaphylactic or anaphylactoid reactions induced by various preparations of penicillin constitute the most important immediate danger connected with their use. Among all drugs, the penicillins are most often responsible for this type of untoward effect. Anaphylactoid reactions may occur at any age. Their incidence is thought to be 0.004% to 0.04% in persons treated with penicillins (Kucers and Bennett, 1987). About 0.001% of patients treated with these agents die from anaphylaxis. It has been estimated that there are at least 300 deaths per year due to this complication of therapy. About 70% have had penicillin previously, and one-third of these reacted to it on a prior occasion. Anaphylaxis has most often followed the injection of penicillin, although it also has been observed after oral ingestion of the drug and even has resulted from the intradermal instillation of a very small quantity for the purpose of testing for the presence of hypersensitivity. The clinical pictures that develop vary in severity. The most dramatic is sudden, severe hypotension and rapid death. In other instances, bronchoconstriction with severe asthma; abdominal pain, nausea, and vomiting; extreme weakness and a fall in blood pressure; or diarrhea and purpuric skin eruptions have characterized the anaphylactic episodes. Serum sickness varies from mild fever, rash, and leukopenia to severe arthralgia or arthritis, purpura, lymphadenopathy, splenomegaly, mental changes, electrocardiographic abnormalities suggestive of myocarditis, generalized edema, albuminuria, and hematuria. It is mediated by IgG antibodies. This reaction usually appears after penicillin treatment has been continued for 1 week or more; it may be delayed, however, until 1 or 2 weeks after the drug has been stopped. Serum sickness caused by penicillin may persist for a week or longer. Vasculitis of the skin or other organs may be related to hypersensitivity to penicillin. The Coombs reaction frequently becomes positive during prolonged therapy with a penicillin or cephalosporin, but hemolytic anemia is rare. Reversible neutropenia may occur. It is not known if this is truly a hypersensitivity reaction; it has been noted with all of the penicillins and has been seen in up to 30% of patients treated with 8 to 12 g of nafcillin for longer than 21 days. The bone marrow shows an arrest of maturation. Fever may be the only evidence of a hypersensitivity reaction to the penicillins. It may reach high levels and be maintained, remittent, or intermittent; chills occasionally occur. The febrile reaction usually disappears within 24 to 36 hours after administration of the drug is stopped but may persist for days. Eosinophilia is an occasional accompaniment of other allergic reactions to penicillin. At times, it may be the sole abnormality, and eosinophils may reach levels of 10% to 20% or more of the total number of circulating white blood cells. Interstitial nephritis rarely may be produced by the penicillins; methicillin has been implicated most frequently. Hematuria, albuminuria, pyuria, renal-cell and other casts in the urine, elevation of serum creatinine, and even oliguria have been noted. Biopsy shows a mononuclear infiltrate with eosinophilia and tubular damage. IgG is present in the interstitium. This reaction usually is reversible. Management of the Patient Potentially Allergic to Penicillin Evaluation of the patient's history is the most practical way to avoid the use of penicillin in patients who are at the greatest risk of adverse reaction. The majority of patients who give a history of allergy to penicillin should be treated with a different type of antibiotic. Unfortunately there is no available means to confirm a history of penicillin allergy. Skin testing for IgE-mediated immediate-type responses is compromised by the lack of a commercially available minor determinant mixture and the inability of skin tests using major and minor penicillin determinants to predict confidently allergic reactions to synthetic penicillins. Radioallergosorbent tests (RAST) for IgE antipenicilloyl determinants suffer from the same limitations as skin tests (Weiss and Adkinson, 2000). Desensitization occasionally is recommended for patients who are allergic to penicillin and who must receive the drug. This procedure consists of administering gradually increasing doses of penicillin in the hope of avoiding a severe reaction and should be performed only in an intensive-care setting. This may result in a subclinical anaphylactic discharge and the binding of all IgE before full doses are administered. Penicillin may be given in doses of 1, 5, 10, 100, and 1000 U intradermally in the lower arm, with 60-minute intervals between doses. If this is well tolerated, then 10,000 U and 50,000 U may be given subcutaneously. Desensitization also may be accomplished by the oral administration of penicillin (Sullivan et al., 1982). When full doses are reached, penicillin should not be discontinued and then restarted, since immediate reactions may recur (seeWeiss and Adkinson, 2000, for details). The patient should be observed constantly during the desensitizing procedure, an intravenous line must be in place, and epinephrine and equipment and expertise for artificial ventilation must be on hand. It must be emphasized that this procedure may be dangerous, and its efficacy is unproven. Patients with life-threatening infections (e.g., endocarditis or meningitis) may be continued on penicillin despite the development of a maculopapular rash, although alternative antimicrobial agents should be used whenever possible. The rash often clears as therapy is continued. This is thought to be due to the development of blocking antibodies of the IgG class. The rash may be treated with antihistamines or adrenocorticosteroids, although there is no evidence that this therapy is efficacious. Rarely, exfoliative dermatitis with or without vasculitis develops in these patients if therapy with penicillin is continued. Other Adverse Reactions The penicillins have minimal direct toxicity. Apparent toxic effects that have been reported include bone marrow depression, granulocytopenia, and hepatitis. The last-named effect is rare but is most commonly seen following the administration of oxacillin and nafcillin (Kirkwood et al., 1983). The administration of penicillin G, carbenicillin, piperacillin, or ticarcillin has been associated with a potentially significant defect of hemostasis that appears to be due to an impairment of platelet aggregation; this may be caused by interference with the binding of aggregating agents to platelet receptors (Fass et al., 1987). Most common among the irritative responses to penicillin are pain and sterile inflammatory reactions at the sites of intramuscular injectionsreactions that are related to concentration. Serum transaminases and lactic dehydrogenase may be elevated as a result of local damage to muscle. In some individuals who receive penicillin intravenously, phlebitis or thrombophlebitis develops. Many persons who take various penicillin preparations by mouth experience nausea, with or without vomiting, and some have mild-to-severe diarrhea. These manifestations often are related to the dose of the drug. When penicillin is injected accidentally into the sciatic nerve,
severe pain occurs and dysfunction in the area of distribution of this nerve
develops and persists for weeks. Intrathecal injection of penicillin G may
produce arachnoiditis or severe and fatal encephalopathy. Because of this,
intrathecal or intraventricular administration of penicillins should be
avoided. The parenteral administration of large doses of penicillin G
(greater than 20 million units per day, or less with renal insufficiency) may
produce lethargy, confusion, twitching, multifocal myoclonus, or localized or
generalized epileptiform seizures. These are most apt to occur in the
presence of renal insufficiency, localized lesions of the central nervous
system (CNS), or hyponatremia. When the concentration of penicillin G in CSF
exceeds 10 Injection of penicillin Gprocaine may result in an immediate reaction, characterized by dizziness, tinnitus, headache, hallucinations, and sometimes seizures. This is due to the rapid liberation of toxic concentrations of procaine. It has been reported to occur in 1 of 200 patients receiving 4.8 million units of penicillin G procaine to treat their venereal disease. Reactions Unrelated to Hypersensitivity or Toxicity Regardless of the route by which the drug is administered, but most strikingly when it is given by mouth, penicillin changes the composition of the microflora by eliminating sensitive microorganisms. This phenomenon is usually of no clinical significance, and the normal microflora is reestablished shortly after therapy is stopped. In some persons, however, superinfection results from the changes in flora. Pseudomembranous colitis, related to overgrowth and production of a toxin by Clostridium difficile, has followed oral and, less commonly, parenteral administration of penicillins. |
The Cephalosporins
|
History and Source Cephalosporium acremonium, the first source of the cephalosporins, was isolated in 1948 by Brotzu from the sea near a sewer outlet off the Sardinian coast. Crude filtrates from cultures of this fungus were found to inhibit the in vitro growth of S. aureus and to cure staphylococcal infections and typhoid fever in human beings. Culture fluids in which the Sardinian fungus was cultivated were found to contain three distinct antibiotics, which were named cephalosporin P, N, and C. With the isolation of the active nucleus of cephalosporin C, 7-aminocephalosporanic acid, and with the addition of side chains, it became possible to produce semisynthetic compounds with antibacterial activity very much greater than that of the parent substance. (For a historical review and discussion of the biochemistry of the cephalosporins, seeAbraham, 1962; Flynn, 1972.) Chemistry Cephalosporin C contains a side chain derived from D Cephalosporin C can be hydrolyzed by acid to 7-aminocephalosporanic
acid. This compound subsequently has been modified by the addition of
different side chains to create a whole family of cephalosporin antibiotics.
It appears that modifications at position 7 of the The cephamycins are similar to the cephalosporins, but have a methoxy
group at position 7 of the Mechanism of Action Cephalosporins and cephamycins inhibit bacterial cell-wall synthesis in a manner similar to that of penicillin. This is discussed in detail above. Classification The explosive growth of the cephalo-sporins during the past decade has
taxed the best of memories and makes a system of classification most
desirable. Although cephalosporins may be classified by their chemical
structure, clinical pharmacology, resistance to Classification by generations is based on general features of
antimicrobial activity (seeKarchmer, 2000). The first-generation
cephalosporins, epitomized by cephalothin and cefazolin, have
good activity against gram-positive bacteria and relatively modest activity
against gram-negative microorganisms. Most gram-positive cocci (with the
exception of enterococci, methicillin-resistant S. aureus, and S.
epidermidis) are susceptible. Most oral cavity anaerobes are sensitive,
but the Bacteroides fragilis group is resistant. Activity against Moraxella
catarrhalis, E. coli, K. pneumoniae, and P. mirabilis
is good. The second-generation cephalosporins have somewhat increased
activity against gram-negative microorganisms, but are much less active than
the third-generation agents. A subset of second-generation agents (cefoxitin,
cefotetan, and cefmetazole) also is active against the B.
fragilis group. Third-generation cephalosporins generally are less
active than first-generation agents against gram-positive cocci, but they are
much more active against the Enterobacteriaceae, including Mechanisms of Bacterial Resistance to the Cephalosporins Resistance to the cephalosporins may be related to inability of the
antibiotic to reach its sites of action; to alterations in the
penicillin-binding proteins (PBPs) that are targets of the cephalosporins,
such that the antibiotics bind with lower affinity; or to bacterial enzymes ( The most prevalent mechanism of resistance to cephalosporins is
destruction of the cephalosporins by hydrolysis of the It is important to remember that none of the cephalosporins has reliable activity against the following bacteria: penicillin-resistant S. pneumoniae, methicillin-resistant S. aureus, methicillin-resistant S. epidermidis and other coagulase-negative staphylococci, Enterococcus, L. monocytogenes, Legionella pneumophila, Legionella micdadei, C. difficile, Xanthomonas maltophilia, Campylobacter jejuni, and Acinetobacter species. General Features of the Cephalosporins Cephalexin, cephradine, cefaclor, cefadroxil, loracarbef, cefprozil, cefixime, cefpodoxime proxetil, ceftibuten, and cefuroxime axetil are absorbed after oral administration and can be given by this route. Cephalothin and cephapirin cause pain when given by intramuscular injection and thus are usually used only intravenously. The other agents can be administered intramuscularly or intravenously. Cephalosporins are excreted primarily by the kidney; dosage thus
should be altered in patients with renal insufficiency. Probenecid slows the
tubular secretion of most cephalosporins. Cefpiramide (not yet available in
the Several cephalosporins penetrate into CSF in sufficient concentration to be useful for the treatment of meningitis. These include cefuroxime, cefotaxime, ceftriaxone, cefepime, and ceftizoxime (see'Therapeutic Uses,' below). Cephalosporins also cross the placenta, and they are found in high concentrations in synovial and pericardial fluid. Penetration into the aqueous humor of the eye is relatively good after systemic administration of third-generation agents, but penetration into the vitreous humor is poor. There is some evidence that concentrations sufficient for therapy of ocular infections due to gram-positive and certain gram-negative microorganisms can be achieved after systemic administration. Concentrations in bile are usually high, with those achieved after administration of cefoperazone and cefpiramide being the highest. Specific Agents First-Generation Cephalosporins Cephalothin
is not well absorbed orally and is available only for parenteral
administration. Because of pain on intramuscular injection, it usually is
given intravenously. Since, among the cephalosporins, cephalothin is the most
impervious to attack by staphylococcal The antibacterial spectrum of cefazolin is similar to that of cephalothin.
Although cefazolin is more active against E. coli and Klebsiella
species, it is somewhat more sensitive to staphylococcal Cephalexin
is available for oral administration, and it has the same antibacterial
spectrum as the other first-generation cephalosporins. However, it is
somewhat less active against penicillinase-producing staphylococci. Oral
therapy with cephalexin results in peak concentrations in plasma of 16 Cephradine is similar in structure to cephalexin, and its activity in vitro is almost identical. Cephradine is not metabolized and, after rapid absorption from the gastrointestinal tract, is excreted unchanged in the urine. Cephradine can be administered orally, intramuscularly, or intravenously. When administered orally, it is difficult to distinguish cephradine from cephalexin; some authorities feel that these two drugs can be used interchangeably. Because cephradine is so well absorbed, the concentrations in plasma are nearly equivalent after oral or intramuscular administration. Cefadroxil is the para-hydroxy analog of cephalexin. Concentrations of cefadroxil in plasma and urine are at somewhat higher levels than are those with cephalexin. The drug may be orally administered once or twice a day for the treatment of urinary tract infections. Its activity in vitro is similar to that of cephalexin. Second-Generation Cephalosporins Cefamandole
is more active than the first-generation cephalosporins against certain
gram-negative microorganisms. It contains the methyl-tetrazole-thiomethyl
(MTT) group at position 3, which is associated with disulfiram-like
reactions, hypoprothrombinemia, and inhibition of vitamin K activation.
Cefamandole and other second-generation cephalosporins have a broader
spectrum than do the first-generation agents and are active against Enterobacter
species, indole-positive Proteus species, and Klebsiella
species. Strains of H. influenzae containing the plasmid Cefoxitin
is a cephamycin produced by Streptomyces lactamdurans. It is resistant
to some Cefaclor
is used orally. The concentrations in plasma after oral administration are
about 50% of those achieved after an equivalent oral dose of cephalexin.
However, cefaclor is more active against H. influenzae and Moraxella
catarrhalis, although some Loracarbef
is an orally administered carbacephin, similar in activity to cefaclor, that
is more stable against some Cefuroxime
is very similar to cefamandole in structure and antibacterial activity in
vitro (Smith and LeFrock, 1983), although it lacks the MTT group and its
attendant toxicities and is somewhat more resistant to Cefuroxime axetil is the 1-acetyloxyethyl ester of cefuroxime. Thirty to fifty percent of an oral dose is absorbed, and the drug is then hydrolyzed to cefuroxime; resultant concentrations in plasma are variable. Cefonicid has antimicrobial activity in vitro similar to that of cefamandole. The half-life of the drug is about 4 hours, and administration once daily has been effective for certain infections caused by susceptible microorganisms (Gremillion et al., 1983). Cefotetan
is a cephamycin, and, like cefoxitin, it has good activity against B.
fragilis. It also is effective against several other species of Bacteroides,
and it is slightly more active than cefoxitin against gram-negative aerobes.
After an intramuscular dose of 1 g, peak plasma concentrations of cefotetan
average 70 Ceforanide is similar in structure and antimicrobial activity to cefamandole; however, it is less active against strains of H. influenzae (Barriere and Mills, 1982). Its half-life is about 2.6 hours and it is administered parenterally every 12 hours. Cefprozil is an orally administered agent more active than first-generation cephalosporins against penicillin-sensitive streptococci, E. coli, P. mirabilis, Klebsiella spp., and Citrobacter spp. It has a serum half-life of 1.2 to 1.4 hours (Barriere, 1992). Third-Generation Cephalosporins Cefotaxime
is highly resistant to many (but not the extended spectrum) of the bacterial Ceftizoxime has a spectrum of activity in vitro very similar to that of cefotaxime, except that it is less active against S. pneumoniae and more active against B. fragilis (Haas et al., 1995). The half-life is somewhat longer, 1.8 hours, and the drug can thus be administered every 8 to 12 hours for serious infections. Ceftizoxime is not metabolized, and 90% is recovered in urine (Neu et al., 1982). Ceftriaxone has activity in vitro very similar to that of ceftizoxime and cefotaxime. A half-life of about 8 hours is the outstanding feature. Administration of the drug once or twice daily has been effective for patients with meningitis (Del Rio et al., 1983; Brogden and Ward, 1988), while dosage once a day has been effective for other infections (Baumgartner and Glauser, 1983). About half the drug can be recovered from the urine; the remainder appears to be eliminated by biliary secretion. A single dose of ceftriaxone (125 to 250 mg) is effective in the treatment of urethral, cervical, rectal, or pharyngeal gonorrhea, including disease caused by penicillinase-producing microorganisms (Rajan et al., 1982; Handsfield and Murphy, 1983). Cefixime
is orally administered and, compared to orally administered second-generation
agents, is less active against gram-positive cocci and more active against
Enterobacteriaceae and Cefpodoxime proxetil is an orally administered third-generation agent very similar in activity to cefixime, except that is it slightly more active against S. aureus. It has a serum half-life of 2.2 hours. Third-Generation Cephalosporins with Good Activity Against Pseudomonas Cefoperazone
is less active than cefotaxime against gram-positive microorganisms and less
active than cefotaxime or moxalactam against many species of gram-negative
bacteria. It is more active than both of these agents against P.
aeruginosa, but less active than ceftazidime. Unfortunately, resistant
strains may emerge on treatment. Activity against B. fragilis is
similar to that of cefotaxime. Cefoperazone is slightly less stable with Ceftazidime is one-quarter to one-half as active by weight against gram-positive microorganisms as is cefotaxime. Its activity against the Enterobacteriaceae is very similar, but its major distinguishing feature is excellent activity against Pseudomonas and other gram-negative bacteria. Ceftazidime has poor activity against B. fragilis (Hamilton-Miller and Brumfitt, 1981). Its half-life in plasma is about 1.5 hours, and the drug is not metabolized. Ceftazidime is more active in vitro against Pseudomonas than is cefoperazone or piperacillin (Edmond et al., 1999; Sahm et al., 1999). Fourth-Generation Cephalosporins Cefepime
and cefpirome are fourth-generation cephalosporins. Cefepime is
available for use in the Adverse Reactions Hypersensitivity reactions to the cephalosporins are the most common
side effects (seePetz, 1978), and there is no evidence that any single
cephalosporin is more or less likely to cause such sensitization. The
reactions appear to be identical to those caused by the penicillins, and this
may be related to the shared Because of the similarity in structure of the penicillins and cephalosporins, patients who are allergic to one class of agents may manifest cross-reactivity when a member of the other class is administered. Immunological studies have demonstrated cross-reactivity in as many as 20% of patients who are allergic to penicillin (seeLevine, 1973), but clinical studies indicate a much lower frequency (about 1%) of such reactions (Saxon et al., 1984). There are no skin tests that can reliably predict whether a patient will manifest an allergic reaction to the cephalosporins. Patients with a history of a mild or a temporally distant reaction to penicillin appear to be at low risk of rash or other allergic reaction following the administration of a cephalosporin. However, patients who have had a recent severe, immediate reaction to a penicillin should be given a cephalosporin with great caution, if at all. A positive Coombs reaction appears frequently in patients who receive large doses of a cephalosporin. Hemolysis is not usually associated with this phenomenon, although it has been reported. Cephalosporins have produced rare instances of bone-marrow depression, characterized by granulocytopenia (Kammer, 1984). The cephalosporins have been implicated as potentially nephrotoxic
agents, although they are not nearly as toxic to the kidney as are the
aminoglycosides or the polymyxins (Barza, 1978). Renal tubular necrosis has
followed the administration of cephaloridine in doses greater than 4 g per
day; this agent is no longer available in the Therapeutic Uses The cephalosporins are widely used and therapeutically important antibiotics. Unfortunately, a wide array of bacteria are resistant to their activity. Clinical studies have shown cephalosporins to be effective as both therapeutic and prophylactic agents (Donowitz and Mandell, 1988). The first-generation cephalosporins are excellent agents for skin and soft tissue infections due to S. aureus and S. pyogenes. A single dose of cefazolin just before surgery is the preferred prophylaxis for procedures in which skin flora are the likely pathogens. For colorectal surgery where prophylaxis for intestinal anaerobes is desired, the second-generation agents cefoxitin or cefotetan are preferred. The second-generation cephalosporins have been displaced by third-generation agents for many infections. The second-generation agents have inferior activity against penicillin-resistant S. pneumoniae compared to either the third-generation agents or ampicillin, and therefore should not be used for empirical treatment of meningitis or pneumonia. The oral second-generation cephalosporins can be used to treat respiratory tract infections, although they are suboptimal for treatment of penicillin-resistant S. pneumoniae. In situations where facultative gram-negative bacteria and anaerobes are involved, such as intraabdominal infections, pelvic inflammatory disease, and diabetic foot infection, cefoxitin and cefotetan have been shown to be effective. The third-generation cephalosporins, either with or without aminoglycosides, have been considered to be the drugs of choice for serious infections caused by Klebsiella, Enterobacter, Proteus, Providencia, Serratia, and Haemophilus species. Ceftriaxone is now the therapy of choice for all forms of gonorrhea and for severe forms of Lyme disease. The third-generation cephalosporins cefotaxime or ceftriaxone (as part of a 3-drug combination with vancomycin and ampicillin) are used for the initial treatment of meningitis in nonimmunocompromised adults and children older than 3 months (pending identification of the causative agent) because of their antimicrobial activity, good penetration into CSF, and record of clinical successes. They are the drugs of choice for the treatment of meningitis caused by H. influenzae, sensitive S. pneumoniae, N. meningitidis, and gram-negative enteric bacteria. Cefotaxime has failed in the treatment of meningitis due to resistant S. pneumoniae (Friedland and McCracken, 1994). Ceftazidime plus an aminoglycoside is the treatment of choice for Pseudomonas meningitis. Third-generation cephalosporins, however, lack activity against L. monocytogenes and penicillin-resistant pneumococci, which may cause meningitis. The antimicrobial spectrum of cefotaxime and ceftriaxone is excellent for the treatment of community-acquired pneumonia, i.e., that caused by pneumococci (achievable serum concentrations exceed minimal inhibitory concentrations for many or most penicillin-resistant isolates), H. influenzae, or S. aureus. The fourth-generation cephalosporins are indicated for the empirical
treatment of nosocomial infections where antibiotic resistance due to
extended-spectrum |
Other ![]() -Lactam
Antibiotics
-Lactam
Antibiotics
|
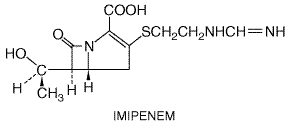
Important therapeutic agents with a Carbapenems Carbapenems are Imipenem Imipenem is marketed in combination with cilastatin, a drug that inhibits the degradation of imipenem by a renal tubular dipeptidase. Source and Chemistry Imipenem is derived from a compound produced by Streptomyces cattleya. The compound thienamycin is unstable, but imipenem, the N-formimidoyl derivative, is stable. The structural formula of imipenem is as follows:
Antimicrobial Activity Imipenem, like other The activity of imipenem is excellent in vitro for a wide
variety of aerobic and anaerobic microorganisms. Streptococci (including
penicillin-resistant S. pneumoniae), enterococci (excluding Enterococcus
faecium and non Pharmacokinetics and Adverse Reactions Imipenem is not absorbed orally. The drug is hydrolyzed rapidly by a dipeptidase found in the brush border of the proximal renal tubule (Kropp et al., 1982). Because concentrations of active drug in urine were low, an inhibitor of the dehydropeptidase was synthesized. This compound is called cilastatin. A preparation has been developed that contains equal amounts of imipenem and cilastatin ( PRIMAXIN After the intravenous administration of 500 mg of imipenem (as PRIMAXIN ), peak concentrations in plasma
average 33 Nausea and vomiting are the most common adverse reactions (1% to 20%).
Seizures also have been noted in up to 1.5% of patients, especially when high
doses are given to patients with CNS lesions and to those with renal
insufficiency. Patients who are allergic to other Therapeutic Uses Imipenemcilastatin is effective for a wide variety of infections (Eron
et al., 1983), including urinary tract and lower respiratory
infections; intraabdominal and gynecological infections; and skin,
soft-tissue, bone, and joint infections. The drug combination appears to be
especially useful for the treatment of infections caused by
cephalosporin-resistant nosocomial bacteria, such as Citrobacter freundii
and Enterobacter spp. It would be prudent to use imipenem for empiric
treatment of serious infections in hospitalized patients who have recently
received other Meropenem Meropenem MERREM IV) is a dimethylcarbamoyl pyrolidinyl derivative of thienamycin. It does not require coadministration with cilastatin as it is not sensitive to renal dipeptidase. Its toxicity is similar to that of imipenem except that it may be less likely to cause seizures (0.5% of meropenem- and 1.5% of imipenem-treated patients seized). Its in vitro activity is similar to that of imipenem, with activity against some imipenem-resistant P. aeruginosa but less activity against gram-positive cocci. Clinical experience with meropenem demonstrates therapeutic equivalence with imipenem. Aztreonam Aztreonam
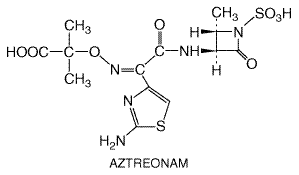
AZACTAM ) is a
monocyclic
Aztreonam interacts with penicillin-binding proteins of susceptible
microorganisms and induces the formation of long filamentous bacterial
structures. The compound is resistant to many of the The antimicrobial activity of aztreonam differs from those of other Aztreonam is administered either intramuscularly or intravenously.
Peak concentrations of aztreonam in plasma average nearly 50 Aztreonam generally is well tolerated. Interestingly, patients who are allergic to penicillins or cephalosporins appear not to react to aztreonam (Saxon et al., 1984). The usual dose of aztreonam for severe infections is 2 g every 6 to 8
hours. This should be reduced for patients with renal insufficiency.
Aztreonam has been used successfully for the therapy of a variety of
infections. One of its notable features is little allergic cross-reactivity
with |
![]() -Lactamase
Inhibitors
-Lactamase
Inhibitors
|
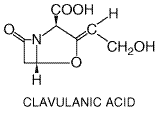
Certain molecules can bind to Clavulanic acid is produced by Streptomyces clavuligerus; its structural formula is as follows:
It has poor intrinsic antimicrobial activity, but it is a
'suicide' inhibitor (irreversible binder) of Amoxicillin plus clavulanate is effective in vitro and in
vivo for Sulbactam
is another Tazobactam
is a penicillanic acid sulfone The combination of piperacillin plus tazobactam does not increase the
activity of piperacillin against P. aeruginosa, as resistance is due
to either chromosomal |
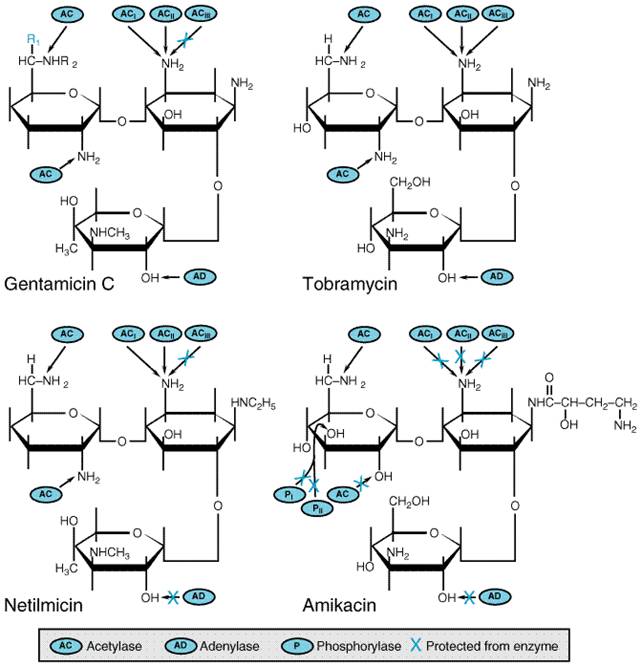
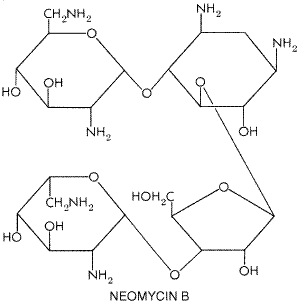
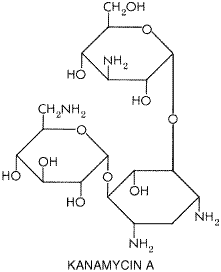
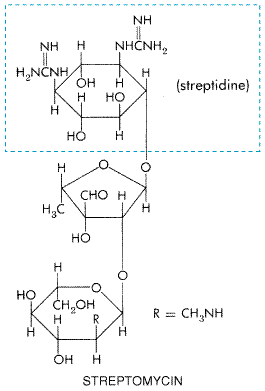
Chapter 46. Antimicrobial Agents: The Aminoglycosides
Overview
|
Aminoglycosides, which are aminoglycosidic aminocyclitols, are bactericidal inhibitors of protein synthesis. Although relatively toxic compared with other classes of antibiotics, they remain useful primarily in the treatment of infections caused by aerobic gram-negative bacteria. Streptomycin is an important agent for treatment of tuberculosis. This chapter covers the antibacterial spectrum, pharmacokinetics, and toxicity of this general class of drugs and the therapeutic uses of the individual agentsgentamicin, tobramycin, amikacin, netilmicin, kanamycin, streptomycin, and neomycin. |
Antimicrobial Agents: The Aminoglycosides: Introduction
|
Aminoglycosides contain amino sugars linked to an aminocyclitol ring by glycosidic bonds. They are polycations, and their polarity is in part responsible for pharmacokinetic properties shared by all members of the group. For example, none is absorbed adequately after oral administration, inadequate concentrations are found in cerebrospinal fluid, and all are excreted relatively rapidly by the normal kidney. The aminoglycosides are used primarily to treat infections caused by aerobic gram-negative bacteria; they interfere with protein synthesis in susceptible microorganisms. In contrast to most inhibitors of microbial protein synthesis, which are bacteriostatic, the aminoglycosides are bactericidal. Mutations affecting proteins in the bacterial ribosome, the target for these drugs, can confer marked resistance to their action. Resistance most commonly results from the acquisition of plasmids or transposon-encoding genes for aminoglycoside-metabolizing enzymes or from impaired transport of drug into the cell. There can be cross-resistance between some members of the class. Although they are widely used and important agents, serious toxicity is a major limitation to the usefulness of the aminoglycosides. The same spectrum of toxicity is shared by all members of the group. Most notable are nephrotoxicity and ototoxicity, which can involve both the auditory and vestibular functions of the eighth cranial nerve. History and Source The development of streptomycin was the result of a well-planned, scientific search for antibacterial substances. Stimulated by the discovery of penicillin, Waksman and coworkers examined a number of soil actinomycetes between 1939 and 1943. In 1943, a strain of Streptomyces griseus was isolated that elaborated a potent antimicrobial substance, streptomycin, which was shown to inhibit the growth of the tubercle bacillus and a number of aerobic gram-positive and gram-negative microorganisms. In less than two years, extensive bacteriological, chemical, and pharmacological investigations of streptomycin had been carried out, and its clinical usefulness was established (Waksman, 1949). However, streptomycin-resistant gram-negative bacilli and gram-positive cocci (enterococci) have emerged, limiting its clinical usefulness. It is now rarely used except for the treatment of certain types of streptococcal or enterococcal endocarditis, tularemia, and plague, and for treatment of tuberculosis. In 1949, Waksman and Lechevalier isolated a soil organism, Streptomyces fradiae, which produced a group of antibacterial substances that were named neomycin. One component, neomycin B, is still used topically or given orally for its local effect on bowel flora, because it causes severe renal toxicity and ototoxicity when administered parenterally. Kanamycin, an antibiotic elaborated by Streptomyces kanamyceticus, was first produced and isolated by Umezawa and coworkers at the Japanese National Institutes of Health in 1957. Because of toxicity and the emergence of resistant microorganisms, kanamycin has been replaced almost entirely by the newer aminoglycosides. Gentamicin
and netilmicin are both broad-spectrum antibiotics derived from
species of the actinomycete Micromonospora. The difference in spelling
(-micin) as compared with that of the other aminoglycoside antibiotics
(-mycin) reflects this difference in origin. Gentamicin was first
studied and described by Weinstein and coworkers in 1963. It has a broader
spectrum of activity than kanamycin and currently is used widely. Tobramycin
and amikacin were introduced into clinical practice in the 1970s.
Tobramycin is one of several components of an aminoglycoside complex
(nebramycin) that is produced by Streptomyces tenebrarius (Higgins and
Kastner, 1967). It is most similar in antimicrobial activity and toxicity to
gentamicin. In contrast to the other aminoglycosides, amikacin and netilmicin
are semisynthetic products. Amikacin, which is a derivative of kanamycin, was
described by Chemistry The aminoglycosides consist of two or more amino sugars joined in glycosidic linkage to a hexose nucleus, which is usually in a central position (seeFigure 461). This hexose, or aminocyclitol, is either streptidine (found in streptomycin) or 2-deoxystreptamine (all other available aminoglycosides). These compounds are thus aminoglycosidic aminocyclitols, although the simpler term aminoglycoside is commonly used to describe them. An additional drug, spectinomycin, is an aminocyclitol that does not contain amino sugars; it is discussed in Chapter 47: Antimicrobial Agents: Protein Synthesis Inhibitors and Miscellaneous Antibacterial Agents.
The aminoglycoside families are distinguished by the amino sugars attached to the aminocyclitol. In the neomycin family, which includes neomycin B and paromomycin, an aminoglycoside used orally for the treatment of intestinal parasitic infections, there are three amino sugars attached to the central 2-deoxystreptamine. The kanamycin and gentamicin families have only two such amino sugars. Neomycin B has the following structural formula:
In the kanamycin family, which includes kanamycins A and B, amikacin, and tobramycin, two amino sugars are linked to a centrally located 2-deoxystreptamine moiety; one of these is a 3-aminohexose (seeFigure 461). The structural formula of kanamycin A, which is the major component of the commercial product, is as follows:
Amikacin is a semisynthetic derivative prepared from kana mycin A by acylation of the 1-amino group of the 2-deoxy- streptamine moiety with 2-hydroxy-4-aminobutyric acid. The gentamicin family, which includes gentamicin C1, C1a, and C2, sisomicin, and netilmicin (the 1-N-ethyl derivative of sisomicin), contains a different 3-amino sugar (garosamine). Variations in methylation of the other amino sugar result in the different components of gentamicin (Figure 461). These modifications appear to have little effect on biological activity. Streptomycin and dihydrostreptomycin (the latter is no longer available because of excessive ototoxicity) differ from the other aminoglycoside antibiotics in that they contain streptidine rather than 2-deoxystreptamine, and their aminocyclitol is not in a central position. The structural formula of streptomycin is as follows:
Mechanism of Action The aminoglycoside antibiotics are rapidly bactericidal. Bacterial killing is concentration-dependent: the higher the concentration, the greater the rate at which bacteria are killed. A postantibiotic effect, that is, residual bactericidal activity persisting after the serum concentration has fallen below the minimum inhibitory concentration, also is characteristic of aminoglycoside antibiotics, and the duration of this effect is concentration-dependent. These properties probably account for the efficacy of once-daily dosing regimens of aminoglycosides. Although much is known about their capacity to inhibit protein synthesis and decrease the fidelity of translation of mRNA at the ribosome (Shannon and Phillips, 1982), the precise mechanism responsible for the rapidly lethal effect of aminoglycosides on bacteria is unknown. Aminoglycosides diffuse through aqueous channels formed by porin
proteins in the outer membrane of gram-negative bacteria to enter the
periplasmic space (Nakae and Nakae, 1982). Transport of aminoglycosides
across the cytoplasmic (inner) membrane depends on electron transport, in
part because of a requirement for a membrane electrical potential (interior
negative) to drive permeation of these antibiotics (Bryan and Kwan, 1983).
This phase of transport has been termed energy-dependent phase I. It
is rate-limiting and can be blocked or inhibited by divalent cations (e.g.,
Ca2+ and Mg2+), hyperosmolarity, a reduction in pH, and
anaerobiasis. The last two of these conditions impair the ability of the
bacteria to maintain the membrane potential, which is the driving force
necessary for transport. Thus, the antimicrobial activity of aminoglycosides
is reduced markedly in the anaerobic environment of an abscess, in
hyperosmolar acidic urine, and so forth. Once inside the cell,
aminoglycosides bind to polysomes and interfere with protein synthesis by
causing misreading and premature termination of translation of mRNA (seeFigure
462). The aberrant proteins produced may be inserted into the cell membrane,
leading to altered permeability and further stimulation of aminoglycoside
transport (Busse et al., 1992). This phase of aminoglycoside
transport, termed energy-dependent phase II (EDP2), is
poorly understood; however, it has been suggested that EDP2 is in
some way linked with disruption of the structure of the cytoplasmic membrane,
perhaps by the aberrant proteins. This concept is consistent with the
observed progression of the leakage of small ions, followed by larger
molecules and, eventually, by proteins from the bacterial cell prior to
aminoglycoside-induced death. This progressive disruption of the cell
envelope, as well as other vital cell processes, may help to explain the
lethal action of aminoglycosides (
The
primary intracellular site of action of the aminoglycosides is the 30 S
ribosomal subunit, which consists of 21 proteins and a single 16 S molecule
of RNA. At least three of these proteins and perhaps the 16 S ribosomal RNA
as well contribute to the streptomycin binding site, and alterations of these
molecules markedly affect the binding and subsequent action of streptomycin.
For example, a single amino acid substitution of asparagine for lysine at
position 42 of one ribosomal protein (S12) prevents binding of the
drug; the resultant mutant is totally resistant to streptomycin. Another
mutant, in which glutamine is the amino acid at this position, is dependent
on streptomycin, which is actually required for survival. The other
aminoglycosides also bind to the 30 S ribosomal subunit; however, they also
appear to bind to several sites on the 50 S ribosomal subunit ( Aminoglycosides disrupt the normal cycle of ribosomal function by interfering, at least in part, with the initiation of protein synthesis, leading to the accumulation of abnormal initiation complexes or 'streptomycin monosomes,' shown schematically in Figure 462B (Luzzatto et al., 1969). Another effect of the aminoglycosides is their capacity to induce misreading of the mRNA template, causing incorrect amino acids to be incorporated into the growing polypeptide chains (seeTai et al., 1978). The aminoglycosides vary in their capacity to cause misreading, and this property presumably depends on differences in their affinities for specific ribosomal proteins. Although there appears to be a strong correlation between bactericidal activity and the ability to induce misreading (Hummel and Bck, 1989), it remains to be established that this is the primary mechanism of aminoglycoside-induced cell death. Microbial Resistance to the Aminoglycosides Bacteria may be resistant to the antimicrobial activity of the aminoglycosides because of failure of permeation of the antibiotic, low affinity of the drug for the bacterial ribosome, or inactivation of the drug by microbial enzymes. Drug inactivation is by far the most important explanation for the acquired microbial resistance to aminoglycosides that is encountered in clinical practice. Penetration of drug through the pores in the outer membrane of gram-negative microorganisms into the periplasmic space may be retarded; resistance of this type is unimportant clinically. Once the aminoglycoside does reach the periplasmic space, it may be altered by microbial enzymes that phosphorylate, adenylate, or acetylate specific hydroxyl or amino groups (Figure 461). The genes for these enzymes are acquired primarily by conjugation and the transfer of DNA as plasmids and resistance transfer factors (Davies, 1994; seeChapter 43: Antimicrobial Agents: General Considerations). These plasmids have become widespread in nature (especially in hospital environments), and they code for a large number of enzymes (more than 20) that have markedly reduced the clinical usefulness of aminoglysides. Amikacin is less vulnerable to these inactivating enzymes because of protective molecular side chains (Figure 461); thus, this drug has a particularly important role in certain hospital settings. The metabolites of the aminoglycosides may compete with the unaltered drug for intracellular transport, but they are incapable of binding effectively to ribosomes and interfering with protein synthesis. Acquisition of aminoglycoside-inactivating enzymes by enterococci has
become a source of concern. In several centers, a significant percentage of
clinical isolates of these organisms (both Enterococcus faecalis and Enterococcus
faecium) are highly resistant to all aminoglycosides because of this
mechanism (Spera and Farber, 1992; Vemuri and Zervos, 1993). Because
different enzymes are responsible for inactivation of gentamicin and streptomycin,
a smal proportion of gentamicin-resistant strains of enterococci will be
susceptible to streptomycin. Resistance to gentamicin indicates resistance to
tobramycin, amikacin, kanamycin, and netilmicin, because the inactivating
enzyme is bifunctional and modifies all of these aminoglycosides ( Another common form of natural resistance to aminoglycosides is caused by failure of the drug to penetrate the cytoplasmic (inner) membrane. As mentioned above, the transport of aminoglycosides across the cytoplasmic membrane is an oxygen-dependent, active process. Strictly anaerobic bacteria are thus resistant to these drugs, since they lack the necessary transport system. Similarly, facultative bacteria become resistant when they are grown under anaerobic conditions (Mates et al., 1983). The significance of this transport defect for resistance to aminoglycosides among aerobic gram-negative bacilli is not known. Natural resistance to amikacin by Pseudomonas maltophilia and certain other microorganisms appears to have a similar basis, as does the low-level resistance of some gram-positive cocci to aminoglycosides. Resistance that results from alterations in ribosomal structure is relatively uncommon for aminoglycosides other than streptomycin. Single-step mutations in Escherichia coli that result in the substitution of an amino acid in a crucial ribosomal protein may prevent binding of the drug. Although such strains of E. coli are highly resistant to streptomycin, they are not widespread in nature. Similarly, only 5% of strains of Pseudomonas aeruginosa exhibit such ribosomal resistance to streptomycin. It has been estimated that approximately half of the streptomycin-resistant strains are ribosomally resistant (Eliopoulos et al., 1984). There is no synergistic effect of penicillin and streptomycin against these strains demonstrable in vitro. Because ribosomal resistance often is specific for streptomycin, these strains of enterococci generally are sensitive to a combination of penicillin and gentamicinin vitro. Antibacterial Activity of the Aminoglycosides The antibacterial activity of gentamicin, tobramycin, kanamycin, netilmicin, and amikacin is primarily directed against aerobic, gram-negative bacilli. Kanamycin, like streptomycin, has a more limited spectrum compared with other aminoglycosides, and in particular it should not be used to treat infections caused by Serratia or P. aeruginosa. As noted above, these antibiotics have little activity against anaerobic microorganisms or facultative bacteria under anaerobic conditions. Their action against most gram-positive bacteria is limited. Streptococcus pneumoniae and Streptococcus pyogenes are resistant, and, in fact, gentamicin has been added to blood-agar plates to aid in the isolation of these microorganisms from sputum and pharyngeal secretions. Although not active when used alone, either streptomycin or gentamicin in combination with a cell wallactive agent, such as a penicillin or vancomycin, is active against 'sensitive' strains of enterococci and streptococci at concentrations that can be achieved clinically. Such combinations result in a more rapid bactericidal effect than is produced by either drug alone (i.e., they are synergistic). Both gentamicin and tobramycin are active in vitro against more than 90% of strains of Staphylococcus aureus and 75% of strains of Staphylococcus epidermidis. The clinical efficacy of aminoglycosides alone in the treatment of serious staphylococcal infections has not been documented, and they should not be used. Gentamicin-resistant mutant strains of staphylococci emerge rapidly during exposure to the drug. Moreover, staphylococcal resistance that is mediated by conjugative plasmids that code for aminoglycoside-modifying enzymes is common among methicillin-resistant strains of staphylococci. The aerobic gram-negative bacilli vary in their susceptibility to the
aminoglycosides as shown in Table 461. 'Sensitive' microorganisms
are defined as those inhibited by concentrations that can be achieved
clinically in plasma without a high incidence of toxicity; when given at 8-
or 12-hour intervals, these therapeutic peak values range from 4 to 12 |
Absorption, Distribution, Dosing, and Elimination of the Aminoglycosides
|
Absorption The aminoglycosides are highly polar cations and therefore are very poorly absorbed from the gastrointestinal tract. Less than 1% of a dose is absorbed following either oral or rectal administration. The drugs are not inactivated in the intestine, and they are eliminated quantitatively in the feces. However, long-term oral or rectal administration may result in accumulation of aminoglycosides to toxic concentrations in patients with renal impairment. Absorption of gentamicin from the gastrointestinal tract may be increased by gastrointestinal disease (ulcers, inflammatory bowel disease; Breen et al., 1972). Instillation of these drugs into body cavities with serosal surfaces may result in rapid absorption and unexpected toxicity, i.e., neuromuscular blockade. Similarly, intoxication may occur when aminoglycosides are applied topically for long periods to large wounds, burns, or cutaneous ulcers, particularly if there is renal insufficiency. All of the aminoglycosides are absorbed rapidly from intramuscular sites of injection. Peak concentrations in plasma occur after 30 to 90 minutes and are similar to those observed 30 minutes after completion of an intravenous infusion of an equal dose over a 30-minute period. In critically ill patients, especially those in shock, absorption of drug may be reduced from intramuscular sites because of poor perfusion. Distribution Because of their polar nature, the aminoglycosides largely are excluded from most cells, from the central nervous system, and from the eye. Except for streptomycin, there is negligible binding of aminoglycosides to plasma albumin. The apparent volume of distribution of these drugs is 25% of lean body weight and approximates the volume of extracellular fluid (Barza et al., 1975). Concentrations of aminoglycosides in secretions and tissues are low. High concentrations are found only in the renal cortex and in the endolymph and perilymph of the inner ear; this may contribute to the nephrotoxicity and ototoxicity caused by these drugs. Concentrations in bile approach 30% of those found in plasma as a result of active hepatic secretion, but this represents a very minor excretory route for the aminoglycosides. Penetration into respiratory secretions is poor (Levy, 1986). Diffusion into pleural and synovial fluid is relatively slow, but concentrations that approximate those in the plasma may be achieved after repeated administration. Inflammation increases the penetration of aminoglycosides into peritoneal and pericardial cavities. Concentrations of aminoglycosides in cerebrospinal fluid (CSF) that are achievable with parenteral administration of drug usually are subtherapeutic. In experimental animals and human beings, concentrations in CSF are less than 10% of those in plasma in the absence of inflammation; this value may approach 25% when there is meningitis (Strausbaugh et al., 1977). The concentrations achieved are therefore inadequate for the treatment of gram-negative bacillary meningitis in adults. Intrathecal or intraventricular administration of aminoglycosides has been used to achieve therapeutic levels, but the availability of third-generation cephalosporins has now made this unnecessary in most cases. There is no proven benefit of either intrathecal or intraventricular injection of aminoglycosides to neonates with meningitis, perhaps because of the immaturity of the blood-brain barrier (McCracken et al., 1980). Penetration of aminoglycosides into ocular fluids is so poor that effective therapy of bacterial endophthalmitis requires periocular and intraocular injections of the drugs (Barza, 1978). Administration of aminoglycosides to women late in pregnancy may result in accumulation of drug in fetal plasma and amniotic fluid. Streptomycin can cause hearing loss in children born to women who receive the drug during pregnancy (Warkany, 1979), as can tobramycin. Insufficient data are available regarding the other aminoglycosides; it is thus recommended that they be used with caution in pregnancy and only for strong clinical indications in the absence of suitable alternatives. Dosing Recommended doses of individual aminoglycosides in the treatment of specific infections are given in later sections of this chapter. Traditionally, the total daily dose of aminoglycosides is administered as two or three equally divided doses. Administration of the total dose once daily, however, appears to be less toxic and just as effective (Verpooten et al., 1989; Gilbert, 1991; Prins et al., 1993; The International Antimicrobial Therapy Cooperative Group of the European Organization for Research and Treatment of Cancer, 1993; Charnas et al., 1997; Urban and Craig, 1997; Gilbert et al., 1998; Rybak et al., 1999). Toxicity results from accumulation of drug in the inner ear and kidney. The amount of drug that accumulates increases with higher plasma concentrations and longer periods of exposure. Elimination (or washout) of aminoglycoside from these organs occurs more slowly than from plasma and is retarded by high plasma concentrations (Tran Ba Huy et al., 1983), accounting for the association between toxicity and high plasma trough concentrations (Swan, 1997). Toxicity, then, can be considered as a threshold phenomenon, more likely to occur the longer the plasma concentration exceeds a relatively safe upper limit (e.g., above a recommended trough concentration) (Figure 463). A once-daily dosing regimen, despite the higher peak concentration, provides a longer period when concentrations are below the threshold for toxicity than does a multiple-dosing regimen (12 hours versus less than 3 hours total in the example shown in the figure), accounting for its lower toxicity. Aminoglycoside bactericidal activity, on the other hand, is directly related to the concentration achieved, because aminoglycosides have concentration-dependent killing and a concentration-dependent postantibiotic effect. This enhanced activity at higher concentrations probably accounts for the equivalent efficacy of a once-daily regimen compared to a multiple-dosing regimen despite the relatively prolonged periods of time that plasma concentrations are 'subtherapeutic,'i.e., below the minimum inhibitory concentration (MIC).
Numerous studies in a variety of clinical settings employing virtually every commonly used aminoglycoside have demonstrated that once-daily regimens are just as safe as or safer than multiple-dosing regimens and as efficacious (Barza et al., 1996; Deaney and Tate, 1996; Ferriols-Lisart and Alos-Alminana, 1996; Freeman and Strayer, 1996; Ali and Goetz, 1997; Bailey et al., 1997; Charnas et al., 1997; Freeman et al., 1997; Deamer, 1998). Once-daily dosing also costs less and is more easily administered. Administration of aminoglycosides as a single daily dose is for these reasons generally preferred with few exceptions. Exceptions are use in pregnancy, neonatal infections, and low-dose combination therapy of bacterial endocarditis, because data documenting equivalent safety and efficacy are inadequate. Once-daily dosing should also be avoided in patients with creatinine clearances less than 20 to 25 ml/min, because accumulation is likely to occur. Less frequent dosing (e.g., every 48 hours) is more appropriate for these patients. Whether once-daily or multiple-daily dosing is chosen, the dose must be adjusted for patients with creatinine clearances below 80 to 100 ml/min (Table 462). If it is anticipated that the patient will be treated with an aminoglycoside for more than three to four days, then plasma concentrations should be monitored to avoid accumulation of the drug. In addition, an aminoglycoside in general should not be used as a single agent except for urinary tract infections because of relatively poor tissue penetration and poorer outcome compared to combination regimens or other classes of antibiotics (Bodey et al., 1985; Leibovici et al., 1997). For twice-daily or three-times-daily dosing regimens, both trough and
peak plasma concentrations are determined. The trough sample is obtained just
prior to a dose, and the peak sample is obtained thirty minutes following
intramuscular injection or thirty minutes after an intravenous infusion given
over thirty minutes. The peak concentration is used to document that the dose
produces therapeutic concentrations, generally accepted to be 4 to 10 Monitoring of aminoglycoside plasma concentrations also is important
when using a once-daily dosing regimen, although peak concentrations are not
routinely determined (these will be three to four times higher than the peak
achieved with a multiple-daily-dosing regimen). Several approaches may be
used to determine that drug is being cleared and not accumulating. The
simplest method is to obtain a trough sample 24 hours after dosing and adjust
the dose to achieve the recommended plasma concentration, e.g., below
1 to 2 Elimination The aminoglycosides are excreted almost entirely by glomerular
filtration, and concentrations in the urine of 50 to 200 Following a single dose of an aminoglycoside, disappearance from the plasma exceeds renal excretion by 10% to 20%; however, after 1 to 2 days of therapy, nearly 100% of subsequent doses is eventually recovered in the urine. This lag period probably represents saturation of binding sites in tissues. The rate of elimination of drug from these sites is considerably longer than from plasma; the half-life for tissue-bound aminoglycoside has been estimated to range from 30 to 700 hours (Schentag and Jusko, 1977). For this reason, small amounts of aminoglycosides can be detected in the urine for 10 to 20 days after drug administration is discontinued. Aminoglycoside bound to renal tissue exhibits antibacterial activity and protects experimental animals against bacterial infections of the kidney, even when the drug no longer can be detected in serum (Bergeron et al., 1982). The concentration of aminoglycoside in plasma produced by the initial dose is dependent only on the volume of distribution of the drug. Since the elimination of aminoglycosides is almost entirely dependent on the kidney, a linear relationship exists between the concentration of creatinine in plasma and the half-life of all aminoglycosides in patients with moderately compromised renal function. In anephric patients, the half-life varies from 20 to 40 times that determined in normal individuals. Because the incidence of nephrotoxicity and ototoxicity is related to the concentration to which an aminoglycoside accumulates, it is critical to reduce the maintenance dosage of these drugs in patients with impaired renal function. The size of the individual dose, the interval between doses, or both, can be altered. There is no conclusive information on the best approach, and even the currently accepted therapeutic range has been questioned (McCormack and Jewesson, 1992). The most consistent plasma concentrations are achieved when the loading dose is given in milligrams per kilogram of body weight; and since aminoglycosides are minimally distributed in fatty tissue, the lean or expected body weight should be used. Methods for calculation of dosage are described in Appendix II. There are obvious difficulties in utilizing any of these approaches for ill patients with rapidly changing renal function (Lesar et al., 1982). In addition, even when known factors are taken into consideration, concentrations of aminoglycosides achieved in plasma after a given dose vary widely among patients (Barza et al., 1975). If the extracellular volume is expanded, the volume of distribution is increased and concentrations will be reduced. For unknown reasons, the clearances are increased, and the half-lives of the aminoglycosides are reduced in patients with cystic fibrosis; the volume of distribution is increased in patients with leukemia (Rosenthal et al., 1977; Spyker et al., 1978). Patients with anemia (hematocrit <25%) have a concentration in plasma that is higher than expected, probably because of a reduction in the number of binding sites on red blood cells (Siber et al., 1975). Determination of the concentration of drug in plasma is an essential guide to the proper administration of aminoglycosides. In patients with life-threatening systemic infections, aminoglycoside concentrations should be determined several times per week (more frequently if renal function is changing) and always should be determined within 24 hours after a change in dosage. Aminoglycosides are removed from the body by either hemodialysis or peritoneal dialysis. Approximately 50% of the administered dose is removed in 12 hours by hemodialysis, which has been used for the treatment of overdosage. As a general rule, a dose equal to half the loading dose administered after each hemodialysis should maintain the plasma concentration in the desired range; however, a number of variables make this a rough approximation at best. Continuous arteriovenous hemofiltration (CAVH) and continuous venovenous hemofiltration (CVVH) will result in aminoglycoside clearances approximately equivalent to 15 ml/min and 15 to 30 ml/min of creatinine clearance, respectively, depending on the flow rate. The amount of aminoglycoside removed can be replaced by administering approximately 15% to 30% of the maximum daily dose (Table 462) each day. Frequent monitoring of drug concentrations in plasma is again crucial. Peritoneal dialysis is less effective than hemodialysis in removing aminoglycosides. Clearance rates are approximately 5 to 10 ml per minute for the various drugs, but are highly variable. If a patient who requires dialysis has bacterial peritonitis, a therapeutic concentration of the aminoglycoside probably will not be achieved in the peritoneal fluid, since the ratio of the concentration in plasma to that in peritoneal fluid may be 10 to 1 (Smithivas et al., 1971). It is thus recommended that antibiotic be added to the dialysate to achieve concentrations equal to those desired in plasma. For intermittent dosing via peritoneal dialysate, 2 mg/kg of amikacin is added to the bag once a day. The corresponding dose for gentamicin, netilmicin, or tobramycin is 0.6 mg/kg. For continuous dosing, the dose of amikacin is 12 mg per liter (25 mg/l loading dose in the first bag) and the dose of gentamicin, netilmicin, or tobramycin is 4 mg per liter in each bag (8 mg/l loading dose). This should be preceded by administration of a loading dose, either parenterally or in dialysis fluid. Although excretion of aminoglycosides is similar in adults and children over 6 months of age, half-lives of the drugs may be significantly prolonged in the newborn. Newborn infants who weigh less than 2 kg have half-lives for aminoglycosides of 8 to 11 hours during the first week of life, while those who weigh over 2 kg eliminate these drugs with half-lives of about 5 hours (Yow, 1977). It is thus critically important to monitor concentrations of aminoglycosides during treatment of neonates (Philips et al., 1982). Aminoglycosides can be inactivated by various penicillins in vitro (Konishi et al., 1983) and in patients with end-stage renal failure (Blair et al., 1982), thus making dosage recommendations even more difficult. Amikacin appears to be the least affected by this interaction. |
Untoward Effects of the Aminoglycosides
|
All aminoglycosides have the potential to produce reversible and irreversible vestibular, cochlear, and renal toxicity. These side effects complicate the use of these compounds and make their proper administration difficult. Ototoxicity Both vestibular and auditory dysfunction can follow the administration of any of the aminoglycosides. Studies of both animals and human beings have documented progressive accumulation of these drugs in the perilymph and endolymph of the inner ear (Tran Ba Huy et al., 1983). Accumulation occurs predominantly when concentrations in plasma are high. Diffusion back into the bloodstream is slow; the half-lives of the aminoglycosides are five to six times longer in the otic fluids than in plasma. Back-diffusion is concentration-dependent and is facilitated at the trough concentration of drug in plasma. Ototoxicity is more likely to occur in patients with persistently elevated concentrations of drug in plasma. However, even a single dose of tobramycin has been reported to produce slight temporary cochlear dysfunction during periods when the concentration in plasma is at its peak (Wilson and Ramsden, 1977). The relationship of this observation to permanent loss of hearing is not known. Ototoxicity is largely irreversible and results from progressive destruction of vestibular or cochlear sensory cells, which are highly sensitive to damage by aminoglycosides (Brummett, 1983). Studies in guinea pigs exposed to large doses of gentamicin reveal degeneration of the type I sensory hair cells in the central part of the crista ampullaris (vestibular organ) and fusion of individual sensory hairs into giant hairs (Wersll et al., 1973). Similar studies with gentamicin and tobramycin also demonstrate loss of hair cells in the cochlea of the organ of Corti (Theopold, 1977). With increasing dosage and prolonged exposure, damage progresses from the base of the cochlea, where high-frequency sounds are processed, to the apex, which is necessary for the perception of low frequencies. While these histological changes correlate with the ability of the cochlea to generate an action potential in response to sound, the biochemical mechanism for ototoxicity is poorly understood. Early changes induced by aminoglycosides have been shown in experimental ototoxicity to be reversible by Ca2+. Once sensory cells are lost, however, regeneration does not occur; retrograde degeneration of the auditory nerve follows, resulting in irreversible hearing loss. It has been suggested that aminoglycosides interfere with the active transport system essential for the maintenance of the ionic balance of the endolymph (Neu and Bendush, 1976). This would lead to alteration in the normal concentrations of ions in the labyrinthine fluids, with impairment of electrical activity and nerve conduction. Eventually, the electrolyte changes, or perhaps the drugs themselves, damage the hair cells irreversibly. Interest also has centered on the interaction of aminoglycosides with membrane phospholipids, particularly phosphatidyl-inositol and its phosphorylated derivatives, which are the precursors of the intracellular second messengers inositol 1,4,5-trisphosphate and diacylglycerol. The degree of permanent dysfunction correlates with the number of destroyed or altered sensory hair cells and is thought to be related to sustained exposure to the drug. Repeated courses of aminoglycosides, each resulting in the loss of more cells, can lead to deafness. Since there is a decrease in the number of cells with age, older patients may be more susceptible to ototoxicity. Drugs such as ethacrynic acid and furosemide potentiate the ototoxic effects of the aminoglycosides in animals (Brummett, 1983); data implicating furosemide are less convincing in human beings (Moore et al., 1984a). Hearing loss also is more likely to develop in patients with preexisting auditory impairment following exposure to these agents. Although all aminoglycosides are capable of affecting both cochlear and vestibular function, some preferential toxicity is evident. Streptomycin and gentamicin produce predominantly vestibular effects, whereas amikacin, kanamycin, and neomycin primarily affect auditory function; tobramycin affects both equally. The incidence of ototoxicity is extremely difficult to determine. Data from audiometry suggest that the incidence may be as high as 25% (Moore et al., 1984a). The relative incidence appears to be equal for tobramycin, gentamicin, and amikacin. Initial studies in laboratory animals and human beings suggested that netilmicin is less ototoxic than other aminoglycosides (Lerner et al., 1983); however, the incidence of ototoxicity from netilmicin is not negligiblesuch complications developed in 10% of patients in one clinical trial of netilmicin (Trestman et al., 1978). The incidence of vestibular toxicity is particularly high in patients receiving streptomycin; nearly 20% of individuals who received 500 mg twice daily for 4 weeks for enterococcal endocarditis developed clinically detectable, irreversible vestibular damage (Wilson et al., 1984). In addition, up to 75% of patients who received 2 g of streptomycin for more than 60 days showed evidence of nystagmus or postural imbalance. It is recommended that patients receiving high doses and/or prolonged courses of aminoglycosides be monitored carefully for ototoxicity, since the initial symptoms may be reversible; however, deafness may occur several weeks after therapy is discontinued. Clinical Symptoms of Cochlear Toxicity A high-pitched tinnitus often is the first symptom of toxicity. If the drug is not discontinued, auditory impairment may develop after a few days. The tinnitus may persist for several days to 2 weeks after therapy is stopped. Since perception of sound in the high-frequency range (outside the conversational range) is lost first, the affected individual is not always aware of the difficulty, and it will not be detected unless careful audiometric examination is carried out. If the loss of hearing progresses, the lower sound ranges are affected, and conversation becomes difficult. Clinical Symptoms of Vestibular Toxicity Moderately intense headache lasting 1 or 2 days may precede the onset of labyrinthine dysfunction. This is immediately followed by an acute stage, in which nausea, vomiting, and difficulty with equilibrium develop and persist for 1 to 2 weeks. Vertigo in the upright position, inability to perceive termination of movement ('mental past pointing'), and difficulty in sitting or standing without visual cues are prominent symptoms. Drifting of the eyes at the end of a movement so that focusing and reading are difficult, positive Romberg test, and rarely pendular trunk movement and spontaneous nystagmus are outstanding signs. The acute stage ends suddenly and is followed by the appearance of manifestations consistent with chronic labyrinthitis, in which, although symptomless while in bed, the patient has difficulty when attempting to walk or make sudden movements; ataxia is the most prominent feature. The chronic phase persists for approximately 2 months; it is gradually superseded by a compensatory stage, in which symptoms are latent and appear only when the eyes are closed. Adaptation to the impairment of labyrinthine function is accomplished by the use of visual cues and deep proprioceptive sensation for determining movement and position. It is more adequate in the young than in the old, but may not be sufficient to permit the high degree of coordination required in many special trades. Recovery from this phase may require 12 to 18 months, and most patients have some permanent residual damage. Although there is no specific treatment for the vestibular deficiency, early discontinuation of the drug may permit recovery prior to irreversible damage of the hair cells. Nephrotoxicity Approximately 8% to 26% of patients who receive an aminoglycoside for
more than several days will develop mild renal impairment that is almost
always reversible (Smith et al., 1977, 1980). The toxicity results
from accumulation and retention of aminoglycoside in the proximal tubular
cells (Aronoff et al., 1983; Lietman and Smith, 1983). The initial
manifestation of damage at this site is excretion of enzymes of the renal
tubular brush border (Patel et al., 1975). After several days, there
is a defect in renal concentrating ability, mild proteinuria, and the
appearance of hyaline and granular casts. The glomerular filtration rate is
reduced after several additional days (Schentag et al., 1979). The
nonoliguric phase of renal insufficiency has been postulated to be due to the
effects of aminoglycosides on the distal portion of the nephron. They are
thought by some investigators to decrease the sensitivity of the
collecting-duct epithelium to endogenous antidiuretic hormone (Appel, 1982).
While severe acute tubular necrosis may occur rarely, the most common
significant finding is a mild rise in plasma creatinine (0.5 to 2.0 mg/dl; 40
to 175 Several variables appear to influence nephrotoxicity from aminoglycosides. Toxicity correlates with the total amount of drug administered. Consequently toxicity is more likely to be encountered with longer courses of therapy. Continuous infusion is more nephrotoxic in dogs and rats than is intermittent dosing (Reiner et al., 1978; Powell et al., 1983), and constant concentrations of drug in plasma above a critical level, which is manifest by elevated trough serum concentrations, correlate with toxicity in human beings (Keating et al., 1979). The nephrotoxic potential varies among individual aminoglycosides. The relative toxicity correlates with the concentration of drug found in the renal cortex in experimental animals. Neomycin, which concentrates to the greatest degree, is highly nephrotoxic in human beings and should not be administered systemically. Streptomycin does not concentrate in the renal cortex and is the least nephrotoxic. Most of the controversy has concerned the relative toxicities of gentamicin and tobramycin. Gentamicin is concentrated in the kidney to a greater degree than is tobramycin, but several controlled clinical trials have given different estimates of their relative nephrotoxicities (Smith et al., 1977, 1980; Fong et al., 1981; Keys et al., 1981). If differences between the renal toxicity of these two aminoglycosides do exist in human beings, they appear to be slight. Comparative studies with amikacin, sisomicin, and netilmicin are not conclusive. Other drugs, such as amphotericin B, vancomycin, cisplatin, and cyclosporine, may potentiate aminoglycoside-induced nephrotoxicity (Wood et al., 1986). Several studies suggest that cephalothin aggravates the nephrotoxicity produced by aminoglycosides (Klastersky et al., 1975; Wade et al., 1978). Furosemide enhances the nephrotoxicity of aminoglycosides in rats if there is concurrent fluid depletion (Mitchell et al., 1977). It has been suggested that the diuretic-induced loss of K+ might be responsible for this toxicity. Clinical studies have not proven conclusively that furosemide aggravates nephrotoxicity (Smith and Lietman, 1983); however, both volume depletion and wasting of K+ have been incriminated. Advanced age, liver disease, and septic shock have been suggested as risk factors for the development of nephrotoxicity from aminoglycosides, but data are not convincing (Moore et al., 1984b). It should be emphasized, however, that renal function is overestimated in the elderly patient from measurement of creatinine concentration in plasma, and overdosing will occur if this value is used as the only guide in this patient population. Whereas aminoglycosides consistently alter the structure and function
of renal proximal tubular cells, these effects usually are reversible. The
most important result of this toxicity may be reduced excretion of the drug,
which, in turn, predisposes ototoxicity. Monitoring drug concentrations in
plasma is useful, particularly during prolonged and/or high-dose therapy.
However, it never has been proven that toxicity can be prevented by avoiding
excessive peak or trough concentrations of aminoglycosides. In fact,
experience with once-daily dosing regimens strongly suggests that high peaks
(e.g., 25 The biochemical events leading to tubular cell damage and glomerular dysfunction are poorly understood, but they may involve perturbations of the structure of cellular membranes. Aminoglycosides inhibit various phospholipases, sphingomyelinases, and ATPases, and they alter the function of mitochondria and ribosomes (Silverblatt, 1982; Queener et al., 1983; Humes et al., 1984). Because of the ability of cationic aminoglycosides to interact with anionic phospholipids, these drugs may impair the generation of membrane-derived autacoids and intracellular second messengers such as prostaglandins, inositol phosphates, and diacylglycerol. Derangements of prostaglandin metabolism might explain the relationship between tubular damage and reduction in glomerular filtration rate. Others have observed morphological changes in glomerular endothelial cells (decreased number of endothelial fenestrations) in animals receiving aminoglycosides (Luft and Evan, 1980) and drug-induced reduction in the glomerular capillary ultrafiltration coefficient (Baylis et al., 1977). Ca2+ has been shown to inhibit the uptake and binding of aminoglycosides to the renal brush-border luminal membrane in vitro, and supplementary dietary Ca2+ attenuates experimental nephrotoxicity (Bennett et al., 1982). Aminoglycosides eventually are internalized by pinocytosis. Morphologically, there is clear evidence of accumulation of drug in liposomes, a means by which aminoglycosides are trapped, concentrated (up to 50 times the plasma concentration; Aronoff et al., 1983), and prepared for extrusion into the urine as multilamellar, phospholipid structures called myeloid bodies (Silverblatt, 1982). Neuromuscular Blockade An unusual toxic reaction of acute neuromuscular blockade and apnea has been attributed to the aminoglycosides. A review of 83 reports of prolonged muscular paralysis implicated neomycin as the most frequent cause (Pittinger et al., 1970). The order of decreasing potency for blockade is neomycin, kanamycin, amikacin, gentamicin, and tobramycin. In human beings, neuromuscular blockade generally has occurred after intrapleural or intraperitoneal instillation of large doses of an aminoglycoside; however, the reaction can follow intravenous, intramuscular, and even the oral administration of these agents (Holtzman, 1976). Most episodes have occurred in association with anesthesia or the administration of other neuromuscular blocking agents. Patients with myasthenia gravis are particularly susceptible to neuromuscular blockade by aminoglycosides. Animal studies indicate that the aminoglycosides inhibit prejunctional release of acetylcholine while also reducing postsynaptic sensitivity to the transmitter (Pittinger and Adamson, 1972; Sokoll and Gergis, 1981). Ca2+ overcomes the effect of the aminoglycoside at the neuromuscular junction, and the intravenous administration of a calcium salt is the preferred treatment for this toxicity (Singh et al., 1978). Inhibitors of cholinesterase (edrophonium, neostigmine) also have been used with varying degrees of success. Since physicians have become aware of this complication, it is now relatively uncommon. Other Effects on the Nervous System The administration of streptomycin in particular may produce dysfunction of the optic nerve. Scotomas, presenting as enlargement of the blind spot, have been associated with the drug. Among the less common toxic reactions to streptomycin is peripheral neuritis. This may be due either to accidental injection of a nerve during the course of parenteral therapy or to toxicity involving nerves remote from the site of antibiotic administration. Paresthesia, most commonly perioral but also present in other areas of the face or in the hands, occasionally follows the use of the antibiotic and usually appears within 30 to 60 minutes after injection of the drug. It may persist for several hours. Other Untoward Effects In general, the aminoglycosides have little allergenic potential; both anaphylaxis and rash are unusual. Rare hypersensitivity reactionsincluding skin rashes, eosinophilia, fever, blood dyscrasias, angioedema, exfoliative dermatitis, stomatitis, and anaphylactic shockhave been reported. Parenterally administered aminoglycosides are not associated with pseudomembranous colitis, probably because they do not disrupt the normal anaerobic flora. Other reactions that have been attributed to individual drugs are discussed below. |
Streptomycin
|
Streptomycin is used today for the treatment
of certain unusual infections, generally in combination with other
antimicrobial agents. It is in general less active than other members of the
class against aerobic gram-negative rods. It therefore has fallen into
disuse, and for a time the drug was unavailable in the Therapeutic Uses Bacterial Endocarditis Streptomycin and penicillin produce a synergistic bactericidal effect in
vitro and in animal models of infection against strains of enterococci,
group D streptococci, and the various oral streptococci of the viridans
group. Many authorities recommend a combination of such antibiotics (although
gentamicin has almost entirely replaced streptomycin) for treatment of
endocarditis caused by these microorganisms. Penicillin G alone is
ineffective in the therapy of enterococcal endocarditis, and either
streptomycin (500 mg twice daily) or gentamicin (1 mg/kg three times daily)
also must be given to ensure cure. Gentamicin is preferred when the strain is
resistant to streptomycin (MIC > 2000 Endocarditis caused by penicillin-sensitive streptococci (MIC < 0.1
Tularemia Patients with tularemia benefit dramatically from the administration of streptomycin (Evans et al., 1985). The best results are obtained when therapy is instituted early; however, chronicity does not exclude the possibility of complete cure. Most cases respond to the administration of 1 to 2 g (15 to 25 mg/kg) of streptomycin per day (in divided doses) for 7 to 10 days. The tetracyclines also are highly effective in tularemia and are preferred by some physicians for milder forms of the disease. Plague Streptomycin is one of the most effective agents for the treatment of all forms of plague. The tetracyclines and chloramphenicol also are beneficial in this disease. When streptomycin is used, a dose of 1 to 4 g per day may be given in two to four divided doses for 7 to 10 days. Tuberculosis In treatment of tuberculosis, streptomycin always should be used in combination with at least one or two other drugs to which the causative strain is susceptible. The dose for patients with normal renal function is 15 mg/kg per day as a single intramuscular injection for two to three months, then two or three times a week thereafter. |
Gentamicin
|
Gentamicin is an important agent for the treatment of many serious gram-negative bacillary infections. It is the aminoglycoside of first choice because of its low cost and its reliable activity against all but the most resistant gram-negative aerobes. However, emergence of resistant microorganisms in some hospitals has become a serious problem and may limit the future use of this agent. Therapeutic Uses of Gentamicin and Other Aminoglycosides Gentamicin, tobramycin, amikacin, and netilmicin can be used interchangeably for the treatment of most of the following infections and are therefore discussed together. For most indications, gentamicin is the preferred agent because of long experience with its use and its relatively low cost. The recommended intramuscular or intravenous dose of gentamicin
sulfate (GARAMYCIN) for
adults is a loading dose of 2 mg/kg, then 3 to 5 mg/kg per day, one-third
being given every 8 hours when administered as a multiple-daily-dosing
regimen. The once-daily dose is 5.1 mg/kg given over 30 to 60 minutes for
patients with normal renal function. Several dosage schedules have been
suggested for infants: 2 to 2.5 mg/kg every 8 hours has been found to be safe
for children up to 2 years of age; 5 mg/kg daily, divided into two equally
spaced injections, has been recommended for neonates with severe infections.
Peak plasma concentrations range from 4 to 10 A large variety of infections have been treated successfully with these aminoglycosides; however, due to their toxicities, prolonged use should be restricted to the therapy of life-threatening infections and those for which a less toxic agent is contraindicated or less effective. These antibiotics frequently are used (often in combination with a penicillin or a cephalosporin) for the therapy of proven or suspected serious gram-negative microbial infectionsespecially those due to P. aeruginosa, Enterobacter, Klebsiella, Serratia, and other species resistant to less toxic antibioticsurinary tract infections, bacteremia, infected burns, osteomyelitis, pneumonia, peritonitis, and otitis. Penicillins and aminoglycosides must never be mixed in the same bottle because the penicillin inactivates the aminoglycoside to a significant degree. Similar incompatibilities exist in vitro to different degrees between gentamicin and heparin, amphotericin B, and the various cephalosporins. Urinary Tract Infections Aminoglycosides usually are not indicated for the treatment of
uncomplicated urinary tract infections, although a single intramuscular dose
of gentamicin (5 mg/kg) has been effective in curing over 90% of
uncomplicated infections of the lower urinary tract (Varese et al.,
1980). In the seriously ill patient with pyelonephritis, an aminoglycoside
alone or in combination with a Pneumonia The frequency of pneumonia caused by various gram-negative bacilli is
increasing, especially in hospitalized patients, patients on respirators, and
those with impaired defenses (especially granulocytopenia). Selection of an
antibiotic depends on the sensitivity of the microorganism. The majority of
organisms that cause community-acquired pneumonia will be susceptible to
broad-spectrum Gentamicin- and tobramycin-resistant strains of Klebsiella, Enterobacter, Serratia, Proteus, and Pseudomonas have emerged in many hospitals, particularly in burn units and intensive care units, where these drugs are used extensively. Critically ill patients with tracheostomies and impaired host defenses and those with indwelling intravenous and urinary catheters frequently are colonized or infected by resistant bacteria. Aminoglycosides are ineffective for treatment of pneumonia due to anaerobes or S. pneumoniae, which are common causes of community-acquired pneumonia. They should not be considered as effective single-drug therapy for any aerobic gram-positive cocci (including S. aureus or streptococci), the microorganisms commonly responsible for suppurative pneumonia or lung abscess. Thus, gentamicin (or other aminoglycosides) should never be used as the sole agent to treat pneumonia acquired in the community or as the initial treatment for pneumonia acquired in the hospital (Kunin, 1977). Meningitis Availability of third-generation cephalosporins, especially cefotaxime
and ceftriaxone, has reduced the need for treatment with
aminoglycosides in most cases of meningitis, except for infections caused by
gram-negative organisms that are resistant to Peritonitis Patients who develop peritonitis as a result of peritoneal dialysis may benefit from therapy with an aminoglycoside. Since suboptimal intraperitoneal concentrations of the antibiotic may follow intramuscular or intravenous administration in patients undergoing dialysis, the procedure should be continued with fluids containing an appropriate concentration of the aminoglycoside. Gram-Positive Infections Although there are very few indications for the use of aminoglycosides
for gram-positive bacterial infections, at times it may be necessary and
lifesaving. In cases of enterococcal endocarditis, up to 50% of isolates of
enterococci are not killed by penicillin plus streptomycin; these strains,
however, are nearly always sensitive to penicillin plus gentamicin. This is
not revealed by testing for sensitivity to a standard dose of gentamicin, and
the breakpoint for resistance is defined as 500 Sepsis When a patient has granulocytopenia and infection (sepsis) with P. aeruginosa is suspected, the administration of an antipseudomonal penicillin in combination with gentamicin, tobramycin, amikacin, or netilmicin is recommended. Treatment of gram-negative bacillary sepsis, especially in neutropenic patients, has been improved by the use of such synergistic combinations (Klastersky, 1987). Topical Applications Gentamicin is very slowly absorbed when applied in an ointment, but
absorption may be more rapid when a cream is used topically. When the
antibiotic is applied to large areas of denuded body surface, as may be the
case in burned patients, plasma concentrations can reach 4 Untoward Effects The untoward effects of gentamicin are similar to those of other aminoglycosides. The most important and serious side effects of the use of gentamicin are nephrotoxicity and irreversible ototoxicity. Intrathecal or intraventricular administration may cause local inflammation and can result in radiculitis and other complications and therefore is rarely used (see above). |
Tobramycin
|
The antimicrobial activity and
pharmacokinetic properties of tobramycin (NEBCIN) are very similar to those of gentamicin.
Tobramycin may be given either intramuscularly or intravenously. Dosages and
serum concentrations are identical with those for gentamicin. Toxicity is
most common at minimal (trough) concentrations that exceed 2 Tobramycin ( TOBREX ) also is available in ophthalmic ointments and solutions. Therapeutic Uses Indications for the use of tobramycin are essentially identical with
those for gentamicin. The superior activity of tobramycin against P.
aeruginosa may make it desirable in the treatment of bacteremia,
osteomyelitis, and pneumonia caused by Pseudomonas species. It usually
should be used concurrently with an antipseudomonal In contrast to gentamicin, tobramycin shows poor activity in combination with penicillin against many strains of enterococci. Most strains of E. faecium are highly resistant (Moellering et al., 1979). Tobramycin is ineffective against mycobacteria (Gangadharam and Candler, 1977). Untoward Effects Tobramycin, like other aminoglycosides, causes both nephrotoxicity and ototoxicity, as discussed above. Studies in experimental animals suggest that tobramycin may be less toxic to hair cells in the cochlear and vestibular end organs and cause less renal tubular damage than does gentamicin. However, clinical data are less convincing. |
Amikacin
|
The spectrum of antimicrobial activity of amikacin ( AMIKIN ) is the broadest of the group, and because of its unique resistance to the aminoglycoside-inactivating enzymes, it has a special role in hospitals where gentamicin- and tobramycin-resistant microorganisms are prevalent. Amikacin is similar to kanamycin in dosage and pharmacokinetic properties. The recommended dose of amikacin is 15 mg/kg per day, as a single
daily dose or divided into two or three equal portions. The individual dose
or the interval between doses must be altered in patients with renal failure.
The drug is rapidly absorbed after intramuscular injection, and peak
concentrations in plasma approximate 20 Therapeutic Uses Amikacin has become the preferred agent for initial treatment of serious nosocomial gram-negative bacillary infections in hospitals where resistance to gentamicin and tobramycin has become a significant problem. Some hospitals have restricted its use to avoid emergence of resistant strains, although some suggest that this is not likely (Betts et al., 1984). Because of its unique resistance to aminoglycoside-inactivating
enzymes, amikacin is active against the vast majority of aerobic
gram-negative bacilli in both the community and the hospital. This includes
most strains of Serratia, Proteus, and P. aeruginosa. It is
active against nearly all strains of Klebsiella, Enterobacter, and E.
coli that are resistant to gentamicin and tobramycin. Most resistance to
amikacin is found among strains of Acinetobacter, Providencia, and Flavobacter
and strains of Pseudomonas other than P. aeruginosa. These are
all unusual pathogens. Like tobramycin, amikacin is less active than
gentamicin against enterococci and should not be used. Amikacin is not active
against the majority of gram-positive anaerobic bacteria. It is effective
against M. tuberculosis (99% of strains inhibited by 4 Untoward Effects Like the other aminoglycosides, amikacin causes both ototoxicity and nephrotoxicity. Auditory deficits are most commonly produced, as discussed above. |
Netilmicin
|
Netilmicin NETROMYCIN) is the latest of the aminoglycosides to be marketed. It is similar to gentamicin and tobramycin in its pharmacokinetic properties and dosage. Its antibacterial activity is broad against aerobic gram-negative bacilli. Like amikacin, it is not metabolized by the majority of the aminoglycoside-inactivating enzymes, and it may be active against certain bacteria that are resistant to gentamicin. The recommended dose of netilmicin for complicated urinary tract
infections in adults is 1.5 to 2 mg/kg every 12 hours. For other serious
systemic infections, a total daily dose of 4 to 6.5 mg/kg is administered as
a single dose or divided into two or three portions. Children should receive
3.0 to 7.5 mg/kg per day in two to three divided doses; neonates receive 4 to
6.5 mg/kg per day in two divided doses. The distribution and elimination of
netilmicin, gentamicin, and tobramycin are very similar. An intravenous
infusion of 2 mg/kg netilmicin, given over a 60-minute period, results in a
peak plasma concentration of approximately 11 Therapeutic Uses Netilmicin is a useful antibiotic for the treatment of serious infections due to susceptible Enterobacteriaceae and other aerobic gram-negative bacilli. It has been shown to be effective against certain gentamicin-resistant pathogens, except enterococci (Panwalker et al., 1978). Untoward Effects Like other aminoglycosides, netilmicin also may produce ototoxicity and nephrotoxicity. Although studies in animals have suggested that netilmicin may be less toxic (Luft et al., 1976), this remains to be proven in human beings (Trestman et al., 1978; Bock et al., 1980). |
Kanamycin
|
The use of kanamycin has declined markedly because its spectrum of activity is limited compared with other aminoglycosides, and it is among the most toxic. Kanamycin sulfate KANTREX) is available for injection and oral use. The parenteral dose for adults is 15 mg/kg per day (two to four equally divided and spaced doses), with a maximum of 1.5 g per day. Children may be given up to 15 mg/kg per day. Therapeutic Uses Kanamycin is all but obsolete, and there are few indications for its use. Kanamycin has been employed to treat tuberculosis in combination with other effective drugs. Because the therapy of this disease is protracted and involves the administration of large total doses of the drug, with the risk of ototoxicity and nephrotoxicity, kanamycin should be used only to treat patients who harbor microorganisms that are resistant to the more commonly used agents (seeChapter 48: Antimicrobial Agents: Drugs Used in the Chemotherapy of Tuberculosis, Mycobacterium avium Complex Disease, and Leprosy). Prophylactic Uses Kanamycin can be administered orally as adjunctive therapy in cases of hepatic coma. The rationale for such therapy is described under (seeNeomycin). The dose usually employed for these purposes is 4 to 6 g per day for 36 to 72 hours; quantities as large as 12 g per day (in divided doses) have been given. The effect on intestinal bacteria may not be sustained even when such large doses of kanamycin are administered. Untoward Effects The untoward effects of the oral administration of aminoglycosides are considered under Neomycin. |
Neomycin
|
Neomycin is a broad-spectrum antibiotic.
Susceptible microorganisms usually are inhibited by concentrations of 5 to 10
Neomycin sulfate MYCIFRADIN) is available for topical and oral administration. Neomycin and polymyxin B have been used for bladder irrigation in solutions containing 40 mg of neomycin and 200,000 units of polymyxin B per milliliter (NEOSPORIN G.U. IRRIGANT). One milliliter of this preparation is added to 1000 ml of 0.9% sodium chloride solution and is used for continuous irrigation of the urinary bladder through appropriate catheter systems. The goal is to prevent bacteriuria and bacteremia associated with the use of indwelling catheters. The bladder usually is irrigated at the rate of 1000 ml every 24 hours. Neomycin currently is available in many brands of creams, ointments, and other products both alone and in combination with polymyxin, bacitracin, other antibiotics, and a variety of corticosteroids. There is no evidence that these topical preparations shorten the time required for healing of wounds or that those containing a steroid are more effective. Therapeutic Uses Neomycin has been widely used for topical application in a variety of infections of the skin and mucous membranes caused by microorganisms susceptible to the drug. These include infections associated with burns, wounds, ulcers, and infected dermatoses. However, such treatment does not eradicate bacteria from the lesions. The oral administration of neomycin (usually in combination with erythromycin base) has been employed primarily for 'preparation' of the bowel for surgery. As an adjunct to the therapy of hepatic coma, a daily dose of 4 to 12 g (in divided doses) by mouth can be given without difficulty to patients, provided renal function is normal. Because severe renal insufficiency may develop in the late stages of hepatic failure, treatment with neomycin must be followed with the greatest care and stopped if evidence of ototoxicity or further injury to the kidney appears. Lactulose is a much less toxic agent for treatment of hepatic coma, and neomycin is used rarely for this condition. Absorption and Excretion Neomycin is poorly absorbed from the gastrointestinal tract and is
excreted by the kidney, as are the other aminoglycosides. An oral dose of 3 g
produces a peak plasma concentration of only 1 to 4 Untoward Effects Hypersensitivity reactions, primarily skin rashes, occur in 6% to 8% of patients when neomycin is applied topically. Individuals sensitive to this agent may develop cross-reactions when exposed to other aminoglycosides. The most important toxic effects of neomycin are renal damage and nerve deafness. These were most frequent when relatively large quantities of the antibiotic were used parenterally and are the reason the drug is no longer used in this way. Toxicity has even occurred in patients with normal renal function following topical application or irrigation of wounds with 0.5% neomycin solution. Neuromuscular blockade with respiratory paralysis also has occurred after irrigation of wounds or serosal cavities. The most important adverse effects resulting from the oral administration of neomycin are intestinal malabsorption and superinfection. Individuals treated with 4 to 6 g of the drug by mouth per day sometimes develop a spruelike syndrome with diarrhea, steatorrhea, and azotorrhea. Overgrowth of yeasts in the intestine also may occur; this is not associated with diarrhea or other symptoms in most cases. The oral administration of even large doses of neomycin usually has no effect on blood levels of prothrombin. For further information regarding particular infections for which the antimicrobial agents discussed in this chapter are useful, the reader is referred to the following chapters of Harrison's Principles of Internal Medicine, 16th ed. (McGraw-Hill, New York, 2005): diseases caused by gram-negative enteric bacilli and P. aeruginosa (Chapters 134 and 136); tularemia (Chapter 142); plague (Chapter 143); and tuberculosis (Chapter 150). |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 8969
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved