| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Introduction
Lebers hereditary optic neuropathy (LHON) is a mitochondrially inherited disease with male predominance. It is characterized by bilateral optic atrophy with loss of central vision due to degeneration of the retinal ganglion cells and optic nerve axons. More than 30 mitochondrial DNA (mtDNA) mutations have been associated with LHON. Mutations in complex I of the oxidative phosphorylation (OXPHOS) system at nucleotide positions ND4/11778, ND1/3460 and ND6/14484 are considered as high risk or so-called primary LHON mutations and are present in nearly 90% of LHON patients.
In general, pathology in LHON is limited to the optic system; however, in rare cases, complication by a Leigh-like encephalopathy, another mitochondrial disease which primarily affects the grey matter, or more frequently a multiple sclerosis-like syndrome may occur. The latter is mainly associated with mutation 11778 and extremely rarely with mutation 14484. The neurological characteristics, including MRI features and/or oligoclonal bands in CSF, of the multiple sclerosis-like syndrome associated with LHON are indistinguishable from those of multiple sclerosis in general.
There have been only a few neuropathological studies of LHON. These consistently show degeneration of the retinal ganglion layer and optic nerve with axonal loss, occasionally with mild inflammation. Others describe evidence of myelin splitting and reactive astrocytosis with vacuolar degeneration in the optic nerves. Additional findings include demyelination in the gracile columns of the spinal cord as well as some demyelination in peripheral nerves of the lower extremities.
Another early pathological analysis, carried out before genetic testing was introduced, reported diffuse fibrillary gliosis in the white matter of the cerebral hemispheres, and demyelinated zones lacking active degradation in the optic nerves, chiasma and optic tracts in addition to myelin loss with Marchi-positive lipid in the crossed and uncrossed pyramidal tracts, and central chromatolysis of neurons of the cervical and thoracic segments of the spinal cord. Although they could not demonstrate intraparenchymal inflammation or multiple sclerosis-like changes, diffuse mononuclear infiltration of the leptomeninges was mentioned.
In sum, despite repeated clinical reports, histopathological descriptions of brain white matter (WM) and optic nerve changes in LHON associated with multiple sclerosis-like syndrome are lacking. Neuropathological studies of the optic nerve also lack information about the kinetics of degeneration in LHON as these investigations are carried out with patients who lost vision many years prior to death. Here we demonstrate a case with WM disease associated with the rare T14484C LHON mutation which consists histopathologically of a spectrum of changes reminiscent of both multiple sclerosis and mitochondrial WM disease, as in KearnsSayre syndrome (KSS). Lesions include demyelination, vacuolation, cystic necrosis, plaques, and inflammatory cell infiltrates involving the optic nerve as well.
Neuropathology
After widespread sampling of formalin-fixed tissue, including brainstem, basal ganglia, cerebral cortical areas, cerebellum, gross hemispheric sections of five levels, spinal cord and dorsal roots, blocks were embedded in paraffin and routinely stained using haematoxylin and eosin (HE), Luxol fast blueperiodic acid Schiff (PAS), Woelcke myelin stain, Bielschowsky, van Gieson elastica and cresyl violet. Formalin-fixed dorsal roots of the cervical and lumbar level were embedded in epon and processed for semi-thin sections.
We used well characterized antibodies to evaluate inflammation and tissue damage (Table 1). To detect DNA fragmentation in situ (by terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling; TUNEL), a commercial kit was used. Immunohistochemistry for light microscopy and double immunolabelling for confocal laser microscopy were performed according to previously described protocols.
Discussion
The spectrum of neuropathological changes of the present case with T14484C LHON mutation includes axonal damage, active demyelination, partly extensive tissue destruction (cystic necrosis), inactive demyelinated areas and vacuolation of WM. Thus the features are reminiscent of both classical and atypical lesions of multiple sclerosis. Our observations suggest that the extensive and unselective tissue damage is mediated predominantly by T cells and activated macrophages/microglia. However, the low number of perivascular inflammatory infiltrates may be due to immunosuppressant treatment. The reason for the predominantly frontal
localization with relative sparing of other brain areas remains obscure.
Nonetheless, WM changes are not exclusively due to a multiple sclerosis-like demyelinative process. Vacuolation and diffuse myelin pallor, reminiscent of that seen in KSS, might also contribute to abnormalities picked up by MRI. Interestingly, vacuolation was also mentioned in the optic nerves in another study. However, previous pathological studies usually described end-stage changes, while we could demonstrate an active phase of the process. WM involvement without neurological manifestation, oligoclonal bands in CSF and response to steroid therapy may be associated with LHON. The presence of inflammatory cells within lesions of LHON is unusual since only rarely may mild inflammation be detected. In contrast, in our patient, introduction of corticosteroid intermittently improved visual and neurological function, suggesting an early immunological mechanism in addition to the primary degeneration of the optic nerve.
The mechanisms leading to inflammatory demyelination and tissue damage in LHON so far have not been determined. It may be due to a coincidental association of multiple sclerosis in a patient with LHON. In particular, reactive oxygen and nitrogen species, produced by activated macrophages in multiple sclerosis lesions, may impair mitochondrial function. This may be potentiated in the presence of a genetic defect of mitochondrial function. In this case, the mitochondrial dysfunction may aggravate or modify the pathogenesis of the lesions. However, similar mitochondrial DNA mutations were not found in an unselected multiple sclerosis population. Alternatively, tissue injury in LHON patients may by itself provoke an autoimmune response in genetically susceptible individuals. Molecular mimicry was implicated in the precipitation of an autoimmune process in mtDNA mutations. Alternatively, antigen or determinant spreading mediated through the liberation of autoantigen may induce the autoimmune response. The presence of another autoimmune disorder, Hashimoto thyroiditis, in our patient also supports this notion. It must be mentioned that we did not observe any signs of Hashimoto encephalitis. Vascular damage, angiopathy and Leigh disease-like neuropathology were also lacking.
In contrast to 11778, the 14484 LHON mutation is only exceptionally associated with multiple sclerosis-like disease, thus additional genetic or epigenetic factors might be suspected for the unusual clinical course in our case.
In conclusion, the various phenotypes of extraoptic LHON disease suggest that mtDNA mutations may affect the nervous system on a common metabolic basis and occasionally may aggravate or initiate autoimmune processes.
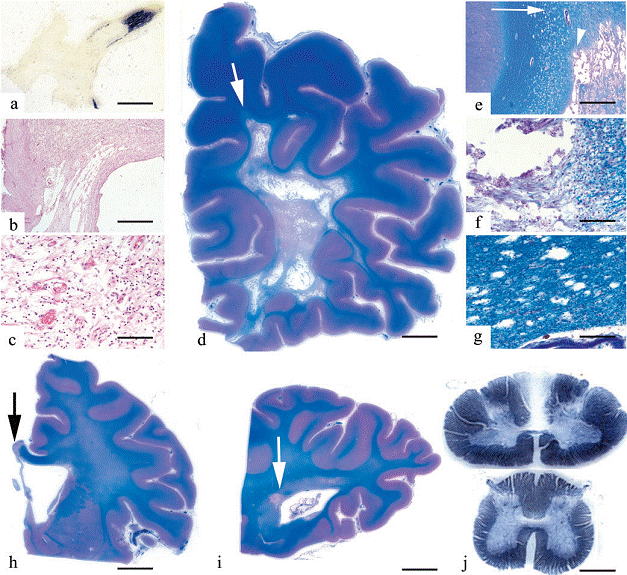
Fig. 2. Histopathological findings in LHON associated with white matter disease. Demyelination (A, Woelcke stain), cystic lesion
(B, H&E) and mononuclear infiltration (C, H&E) in the optic chiasma. There is a widespread cysticnecrotic white matter lesion in the frontal lobe (D). The arrow indicates the region enlarged in (E) demonstrating the edge of this with cystic destruction (F, indicated by an arrowhead in E) and vacuolation (G, indicated by an arrow in E). Plaques (indicated by arrows) in the corpus callosum (H) and around the occipital horn of the lateral ventricle (I), (DI; LuxolPAS staining). Degeneration of the gracile column in the cervical spinal cord (J, upper part), with sparing of the lumbar segment (J, lower part). Bars: A = 5 mm; B = 50 mm; C, F and G = 25 mm; D = 15 mm; E = 100 mm; H and I = 6 mm; J = 1.5 mm.
Neuropathology of white matter disease in Lebers hereditary optic neuropathy; Gbor G. Kovcs, Romana Hftberger, Katalin Majtnyi, Rita Horvth, Pter Barsi, Smuel Komoly, Hans Lassmann, Herbert Budka and Gbor Jakab]
DISCUSSION
In general, carriers are either blinded by the disease or are asymptomatic. This dichotomy seems to be true regardless of whether the individual is homoplasmic or heteroplasmic for the mutation. Although heteroplasmic populations exhibit a lower incidence of LHON, those who are affected are subject to the full spectrum of the disease. Speculations on the pathophysiology relating mitochondrial mutations to inefficient energy metabolism and oxidative stress in the development f LHON do not account for the lack of any clinical indings in asymptomatic carriers.
Mitochondrial dysfunction resulting from the 11778, 3460, and 14484 mutations have a role in the development of LHON. All three mutations affect the site of interaction of complex I with its natural quinone (CoQ) substrate. In a cybrid cellular model these mtDNA mutations induced a variable impairment of mitochondrial respiratory function. Consequentially there may develop a chronic increase of reactive oxygen species (ROS). Some recent publications have implicated the importance of ROS in LHON. Environmental (toxin exposure, diet, and exercise) and genetic factors have also been proposed as important in the pathogenesis of LHON. None of these theories can explain why asymptomatic carriers, who should also undergo some degree of the same pathological metabolic dysfunction, have absolutely no clinical signs or symptoms of visual impairment.
This study identifies dyschromatopsia in asymptomatic carriers of the 11778 LHON mutation. Verriest defined three types of acquired dyschromatopsias based on the FM-100. Type I (protan-like) defects are red/green defects with little or no loss of blue/yellow discrimination, frequently with moderate reduction in central acuity. Type II (deutan-like) defects are red/green defects and significant but less severe blue/yellow defects. Type III (tritan-like) defects are mild to moderate blue/yellow defects, lesser or absent red/green defects, and normal or mildly reduced visual acuity. Each congenital cone defect demonstrates a well defined axis of
colour confusion on FM-100 plots.
In acquired dyschromatopsias, colour errors tend to be more diffusely distributed, making FM-100 polar plots more difficult to interpret; leading to difficulties in determining whether different optic nerve defects result in characteristic dyschromatopsias. One previously well accepted generalisation was Kllners rule which stated that patients with macular disease tend to have blue-yellow defects, and patients with optic nerve disease tend to have red-green defects.
There are several exceptions to this rule. For example, the ONTT dyschromatopsia data reveals that optic neuritis patients initially present with a preponderance of blue-yellow defects, and months later demonstrate a preponderance of red-green defects. The results of our investigation also seem to violate Kllners rule, with the presence of blue-yellow defects in many asymptomatic carriers of LHON.
LHON selectively and severely degrades the small calibre fibres of the papillomacular bundle in affected individuals. If asymptomatic carriers of the mtDNA 11778 mutation exhibit any neural defect at all, it would probably be a subtle dysfunction corresponding to the same small calibre papillomacular fibres. These fibres subserve the foveal and perifoveal regions, with a greater area obviously devoted to the perifoveal region. The fovea itself is rich in red-green cones while the larger perifoveal region is rich in blue cones. Therefore, loss of the papillomacular fibres subserving the perifoveal region would be expected to decrease blue-yellow
discrimination and produce a tritanopsia, violating Kllners rule. Patients with retrobulbar optic neuritis exhibiting perifoveal visual field defects also exhibited a preponderance of tritanopsia while RBN patients with foveal field defects exhibited a preponderance of red-green defects. We suggest the observed tritan defects in our carrier population may serve as a sensitive marker for subclinical, papillomacular fibre dysfunction and eagerly await further analysis though the Cambridge Colour test.
It is also important to note the preponderance for deutanlike defects in the carrier group relative to the off pedigree controls. This finding also is consistent with the small calibre fibre dysfunction model. The fovea is considered to be primarily tritanopic, with sensitivity to red/green light constant throughout the central 2 of the visual field. The central fovea has a markedly reduced sensitivity to blue light, which increases in the perifoveal regions. Therefore, impairment of afferent channels serving the foveal region would prevent transmission of signals from red/green cones, whereas impairment of afferent channels from the perifoveal region would affect signals from blue cones. This red/green defect and significant but less severe blue/yellow defect is consistent with Verriests type II deutan-like dyschromatopsia and can explain the clinical findings of deutan dyschromatopsias in the carrier group.
The observed dyschromatopsias of the carriers are remarkable in that they are atypical of those commonly seen in other optic neuropathies, and may reflect a selective degeneration of primarily perifoveal as well as foveal fibres This study also serves to suggest that asymptomatic carriers of the mtDNA 11778 mutation may exhibit other subclinical, as of yet unidentified optic nerve dysfunctions.
[150-Colour vision defects in asymptomatic carriers of the Lebers hereditary optic neuropathy (LHON) mtDNA 11778 mutation from a large Brazilian LHON pedigree: a case-control study; P A Quiros, R J Torres, S Salomao, A Berezovsky, V Carelli, J Sherman, F Sadun, A De Negri, R Belfort, A A Sadun]
Lebers hereditary optic neuropathy (LHON) is a mitochondrial disease leading to bilateral loss of central vision and severe optic nerve atrophy. A subtype of LHON presents additional clinical and MRI aspects indistinguishable from those of multiple sclerosis (MS) (LHON-MS). In patients with LHON or LHON-MS, an assessment was made of (a) the severity of optic nerve damage, using MRI and magnetisation transfer imaging (MTI), and (b) the presence and extent of macroscopic and microscopic pathology in the brain and cervical cord, using MRI and MT ratio (MTR) and mean diffusivity (Dz ) histogram analysis.
Ten patients with LHON, four with LHON-MS, and 20 age and sex matched healthy controls were studied. For the optic nerve and the brain, dual-echo turbo spin echo (TSE), T1 weighted spin echo, andMTimages were obtained. For the brain, fast fluid attenuated inversion recovery (fast FLAIR) and diffusion weighted images were also obtained. For the cervical cord, fast short tau inversion recovery (STIR) and MT images were obtained. The volume and the average MTR value of both the optic nerves were measured. MTRand Dz histograms of the normal appearing brain tissue (NABT) and MTR histograms of the whole cervical cord tissue were created.
The mean values of optic nerve volumes and MTR were significantly lower in patients with LHON than in healthy controls. Mean NABT-MTR histogram peak height was significantly lower in patients with LHON than in controls, whereas no significant difference was found for any of the cervical cord MTR histogram derived measures. Average diffusivity (Dz ) was higher in patients with LHON than in controls. Optic nerve volume and MTR value and mean NABT-MTR were lower in patients with HON-MS than in those with LHON.
The severity of optic nerve pathology in LHON is measurable in vivo using MRI and MTI.MTR and Dz histogram analysis suggests that microscopic brain damage occurs in LHON and that it is more severe in the MS-like form of the disease.
Discussion
Lebers hereditary optic neuropathy is a familial disease with mitochondrial inheritance, causing severe visual loss and optic nerve atrophy. We investigated the characteristics of optic nerve damage in LHON, using MRI and MTI. As previous studies reported an association between LHON and clinical MRI or MRS findings suggesting a more widespread CNS involvement in this disease, we also assessed the presence and extent of microscopic pathology in the brain and cervical cord of patients with LHON using several MRI techniques.
We did not detect any macroscopic lesion in the optic nerve of patients with LHON. This
disagrees with a previous study, where, using a surface eye coil, a bilateral increase of the optic nerve signal was detected on T2 weighted images in eight patients with LHON. The discrepancy between these two studies might be the reflection of the different clinical characteristics of the enrolled patient populations. In the previous study, patients were scanned a few months after the onset of their visual disturbances, whereas our patients, at the time they underwent the MRI examination, had a disease duration of at least 1 year. The visual assessment of optic nerve atrophy on MRI can be difficult when there are bilateral abnormalities, such as in LHON. This is confirmed by the inability of a previous MRI study to detect changes in optic nerve size in patients with LHON, by contrast with the high frequency of unequivocal optic atrophy reported in postmortem studies. Using a quantitative approach, we detected a significant reduction of the optic nerve volumes in patients with LHON compared with those of healthy controls. This is consistent with the loss of axons and myelin sheaths, which has been described in LHON. The correlation between disease duration and optic nerve atrophy fits well with the notion of a progressive loss of optic nerve tissue in patients with LHON.
We also obtained MT scans of the optic nerves and found that MTR values were significantly reduced in patients with LHON. A postmortem study showed that MTR values are inversely correlated with the severity of myelin and axon loss and several in vivo studies showed that MTR changes are correlated with patients disability in several neurological conditions. Our results confirm the potential of MTI to quantify the degree of tissue loss and indicate that this technique might also be used in the in vivo assessment of tissue damage in optic nerve diseases. Clearly, the acquisition of MT images in the optic nerve is more challenging than in the brain, because of the small size and the mobility of the optic nerves. In addition, image postprocessing is also liable to limitations related to the inclusion of pixels with partial volume effects from the CSF. Nevertheless, as shown by this and other previous studies, it is possible to obtain reliable MTR measurements from the optic nerves. Interestingly, in patients with LHON, we did not find any significant correlation between disease duration and optic nerve MTR, by contrast with that found between disease duration and optic nerve atrophy. Although this remains speculative, one possible explanation of this discrepancy might be related to the higher sensitivity of MTI compared with optic nerve volume measurements in detecting subtle tissue changes.
No macroscopic T2 hyperintense or T1 hypointense lesions of the brain were found in patients with isolated LHON, even using fast FLAIR sequences, which are known to be more sensitive than spin echo sequences for the detection of brain white matter abnormalities. Consistently with other studies, patients with LHON-MS showed a pattern of brain MRI abnormalities which was indistinguishable from that of patients with MS. As it is unlikely that the association between LHON and MS would represent a coincidental occurrence, given their low prevalences, this prompts speculation about the role of immunological factors in the pathogenesis of LHON and that of mitochondrial genes in MS.911 However, given the limited number of patients in our report and the conflicting evidence derived from previous research, firm conclusions cannot be drawn.
Both MTR and Dz histogram derived measurements were compatible with the presence of microscopic pathology in the brain of patients with LHON. In detail, patients with LHON had significantly lower NABT-MTR histogram peak height than healthy controls. This MR quantity was not significantly different from that of patients with MS. As demonstrated by previous studies in MS and other CNS disorders, this finding indicates that the amount of truly normal brain tissue is reduced, due to the presence of damage not visible on T2 weighted scans. Diffusion weighted imaging provides a unique form of MRI contrast that enables the diffusional motion of water molecules to be measured in vivo and an increased Dz is the result of a net loss of barriers restricting water molecular motion.We found that the average Dz showed a significance trend towards increased values in patients with LHON compared with controls, but did not differ between patients with LHON and those with MS. All these findings might reflect a tissue loss and disorganisation in the optic tracts, secondary to neural fibre degeneration occurring in the retinal layer and in the prechiasmal tract of the optic nerve. However, they might also indicate that a diffuse and microscopic brain pathology is present in LHON. The second hypothesis fits with the notion of LHON as a genetic defect which should affect all the neural populations, as recently shown for other hereditary conditions, and raises again the issue of the pathophysiology of LHON and its relation with MS.
No macroscopic lesions were found in the cervical cord of patients with LHON, whereas
all patients with LHON-MS showed a pattern of cervical cord abnormalities indistinguishable from that of MS. Cervical cord MTR histogram derived measurements were also not significantly different between patients with LHON and healthy controls. These findings suggest that the cervical cord tissue is spared in patients with LHON and fit with the hypothesis that the microscopic brain damage in these patients might be due to a secondary degeneration of retrochiasmatic optic fibres.
[444-Magnetic resonance imaging, magnetisation transfer imaging, and diffusion weighted imaging correlates of optic nerve, brain, and cervical cord damage in Lebers hereditary optic neuropathy;M Inglese,M Rovaris, S Bianchi, L La Mantia, G L Mancardi, A Ghezzi, P Montagna, F Salvi, M Filippi]
Comment.
Leber hereditary optic neuropathy is a maternally inherited optic neuropathy that typically
affects males between the ages of 15 and 35 years. The classic funduscopic appearance is characterized by elevation and hyperemia of the optic disc, tortuosity of the retinal vasculature, and peripapillary telangiectatic microangiopathy. The discovery of several mitochondrial DNA mutations responsible for LHON has broadened the clinical spectrum of this disease. Many patients with genetically confirmed LHON do not report a family history of visual loss and do not have the classic fundus appearance. Some of these patients are women and others are outside the typical age range. The patient described in case 1 was young, had no family history of ophthalmologic disease, and never demonstrated the typical fundus features of LHON. His first neuro-ophthalmic evaluation occurred 2 months after his visual loss and the fundus features of LHON may have been observed had he been evaluated at the time of his visual loss. The patient described in case 2 was atypical in that he had no family history of ophthalmologic disease, a 4-year interval prior to involvement of the second eye, and the typical fundus features of LHON were never observed despite the fact that he had had a neuro-ophthalmologic evaluation 3 weeks after visual loss in his right eye.
Magnetic resonance images of the brain and orbits are typically normal in patients with LHON. However, increased signal from the retrobulbar optic nerve on T2-weighted fast spin echo and short time inversion recovery sequences has been described. The increased optic nerve signal may not occur until several months after the onset of visual loss. Optic nerve sheath distention has been demonstrated with orbital ultrasonography, computed tomography, and MRI in a patient with LHON.
Cranial MRI of our patients with LHON demonstrated enlargement of the optic chiasm in both cases and mild enlargement and enhancement of the proximal, intracranial optic nerves in case 1. The association of LHON with enlargement of the optic nerves and chiasm on MRI has not been reported. Although the classic fundus findings in LHON suggest pathology at the optic nerve head, chiasmal and perichiasmal pathology has been suggested. Weiner et al and Raaf and Bair have described bitemporal visual field defects that respect the vertical meridian in patients with LHON. Imachi and Nishizaki noted arachnoid thickening and adhesions around the chiasm during surgery in 120 patients with LHON.
Vaphiades and Newman described a patient with LHON who had enhancement of the retrobulbar optic nerves on MRI obtained several weeks after the onset of visual loss. To our knowledge, there are no other reported cases of optic nerve enhancement among patients with LHON, although many of the previous MRI studies did not obtain designated images of the orbits
with orbital fat suppression techniques following the administration of intravenous gadolinium.
These cases demonstrate that MRI may show enlargement of the optic nerves and chiasm and optic nerve enhancement in patients with LHON. These neuroimaging signs may mimic an optic pathway glioma or optic neuritis. Leber hereditary optic neuropathy should be considered as a possible cause of any unexplained case of unilateral or bilateral optic neuropathy.
[577-Chiasmal Enlargement and Optic Nerve Enhancement on Magnetic Resonance Imaging in Leber Hereditary Optic Neuropathy; Paul H. Phillips, Michael Vaphiades, Charles M. Glasier,
DISCUSSION
The increase in content of mtDNA with age has been considered to be a compensatory response for the decline in respiratory function. It is thought that as the respiratory function of tissue cells declines with age, the cells are mobilised to compensate for the reduced ATP synthesis by increasing the copy number of mtDNA. The energy deficit might be signalled to the nucleus, probably through a reactive oxygen species increase, which can activate the expression of nuclear DNA encoded transacting factors able to induce mitochondrial proliferation. Increased expression of mitochondrial transcription factor A and nuclear respiratory factor-1 were observed in aged human skeletal muscles. Over-proliferation of mitochondria and increased synthesis of mitochondrial respiratory enzymes were observed in a patient with mitochondrial myopathy. Increase in mitochondria and mtDNA was also found in human cells harbouring 4977 bp deleted mtDNA in response to oxidative stress.
Although LHON is known as a mitochondrial disease, the molecular mechanism of the disease is unknown. Variable functional defects of complex I in LHON have been reported. Brown et al reported an extensive biochemical analysis of the mitochondrial defects in lymphoblasts and trans-mitochondrial cybrids habouring LHON 3460, 11778 and 14484 mtDNAmutation. Respiration studies revealed that the maximal respiration rate reduced 2028%, 3036%, and 1015% in the cybrids with 3460, 11778, and 14484 mtDNA mutation, respectively. From this study, we found the content of mtDNA is increased in LHON patients and their asymptomatic maternal relatives harbouring 11778 mutation of mtDNA. However, the content of mtDNA did not correlate with the age of onset of the disease (Pearson correlation coefficient, p>0.05).We believe that the change is a compensatory response for respiratory chain defects of mitochondria harbouring the 11778 mutation.
However, mtDNA content does not increase in all pathological conditions. The content of mtDNA was found to be decreased in adult, but not fetal, pancreatic islets of diabetic rats compared with non-diabetic rats. The content of mtDNA was found to be increased in light smokers, but decreased in heavy smokers. These findings suggest that under some conditions, other factors, such as diabetes or smoking, may modulate the compensatory mechanism in the control of mtDNA replication. In this study,we did not find a modulatory factor that affects the compensatory effect in LHON patients. The compensation does not fade with time because the increased content did not correlate with the duration from the onset of visual loss to the time when the patient was examined (Pearson correlation coefficient, p>0.05).
Typical LHON is often seen in young males. The male to female ratio of affected individuals is about 4.2 to 1. Why males are predominantly affected is unknown. Our study did not show a statistically significant difference of mtDNA content between all males and females carrying 11778 mtDNA mutation.
The content of mtDNA between LHON patients and asymptomatic maternal relatives did not show a statistically significant difference. Both LHON patients and asymptomatic maternal relatives carried the mtDNA 11778 mutation. Since the ages of asymptomatic maternal relatives overlap with the LHON patients, some of them might develop LHON later. The increased mtDNA content in both groups cannot explain the pathogenesis of the onset of LHON.
[1027-Increase of mitochondrial DNA in blood cells of patients with Lebers hereditary optic neuropathy with 11778 mutation; M-Y Yen, C-S Chen, A-G Wang and Y-H Wei]
The pathogenesis of LHON remains largely unknown. The bioenergetic defect has little consistent support for a common enzymatic defect in complex I activity in LHON with the three pathogenic mutations. Heteroplasmy does not explain the clinical variations of patients. Whereas the possibility of nuclear gene involvement has been strongly suggested, linkage and X-inactivation analyses to demonstrate such a locus have been unsuccessful.
Deficiencies in respiratory chain function and reactive oxygen species (ROS) are believed to have pivotal roles in the pathogenesis of LHON. Human cells depend on mitochondrial oxidative phosphorylation to generate energy. Through defective respiration, mitochondria produce a large number of ROS, which may cause oxidative damage to cellular constituents, including membrane lipids, proteins, and DNA. Among the many types of modifications induced by ROS, 8-hydroxy-2-deoxyguanosine (8-OHdG) is one of the most abundant oxidative products of DNA. The 8-OHdG content in leukocyte DNA and in urine is a specific biomarker of DNA damage. In the present study, we measured the 8-OHdG content in leukocyte DNA from patients with LHON to determine whether oxidative stress is involved in LHON.
DISCUSSION
Oxidative DNA damage is thought to contribute to aging and to a host of degenerative diseases of aging, including cancer. DNA damage can occur intrinsically as a consequence of normal metabolism. The rate of oxidative DNA damage is directly related to the metabolic rate and is inversely related to the lifespan of the organism. Leukocyte DNA 8-OHdG levels are increased in cigarette smokers, patients with diabetes, and patients on chronic hemodialysis. The 8-OHdG content in cybrids increases as the proportion of mtDNA with the 4977 deletion increases. The present study indicated that the leukocyte DNA 8-OHdG content was significantly increased in patients with LHON. This is powerful evidence supporting the presence of oxidative stress in the pathogenesis of LHON.
Others have implicated oxidative stress in LHON. In in vitro studies, cybrid cell lines bearing the pathogenic LHON 11778 mutation were much more sensitive than the parental cell line to oxidative stress, which causes cell death in a Ca2+-dependent manner. Biochemical studies of LHON suggest that the cytotoxicity induced by the loss of complex-I activity was not from reductions in oxidative phosphorylation, but was due to increased production of ROS. Chronic overproduction of ROS may be an important consequence of the pathogenic mtDNA mutations. ROS production is increased in cybrids carrying the three primary mutations associated with LHON and different mutations in mtDNA result in a modified pattern of the antioxidant machinery.In animal studies, optic neuropathy induced by reductions in mitochondrial superoxide dismutase, the essential antioxidant that catalyzes the dismutation of superoxide radicals, was strikingly similar to the histopathology of LHON. Patients with LHON and asymptomatic carriers have a reduced α-tocopherol/lipid ratio in their plasma, which most probably reflects increased free radical generation and α -tocopherol consumption. In the present study, mtDNA damage was increased in patients with the 11778 mtDNA mutation, which may reflect the increased ROS production.
How does optic neuropathy in LHON occur? The pathogenic mtDNA mutations of complex-I genes result in defective respiration, inhibiting the electron transport chain, thereby generating ROS to levels beyond the capability of endogenous antioxidants present within the mitochondria. The selective vulnerability of the optic nerve in LHON, however, remains a mystery. Using a neuronal precursor cell line NT2 containing mitochondria bearing the 11778 and 3460 mutations, differentiation significantly reduced LHON cells (by 30%) compared with control subjects, indicating either a decreased proliferative potential or increased cell death of the LHON-NT2 cells. There are increased mitochondrial ROS observed in differentiated LHON-NT2 cells. The findings suggest that the LHON phenotype might be the result of an increase in mitochondrial ROS, which is caused by LHON mutations, possibly mediated through neuron-specific alterations in the complex-I structure. In summary, the present study indicated that the 8-OHdG content in leukocyte DNA was significantly increased in patients with LHON. This provides further evidence linking oxidative stress to the pathogenesis of LHON.
Increased 8-Hydroxy-2-Deoxyguanosine in Leukocyte DNA in Lebers Hereditary Optic Neuropathy; May-Yung Yen, Shu-Huei Kao, An-Guor Wang and Yau-Huei Wei]
DISCUSSION
LHON is a maternally inherited form of central vision loss, in which three prevalent pathogenic mtDNA mutations at positions 11778, 3460, and 14484 affecting different subunits of complex I cause RGC death and optic nerve atrophy. Cell death is painless and without inflammation, suggesting an apoptotic mechanism. Recently, the role of apoptosis in RGC degeneration has been tested extensively; Krishanamoorthy et al showed that in an immortalised rat RGC cell line, deprivation of trophic factors induced cellular death by apoptosis. Wein and Levin found that transection axotomy of the optic nerve in small animals induces retrograde axonal degeneration and cell death by apoptosis of RGCs. Activation of the apoptotic cascade in retinal neurones appears to occur via the major apoptotic pathway described for neurones of the central nervous system, including activation of caspases, (mainly caspases 9 and 3), c-jun kinase, and Bcl family proteins. In addition to these proteins, other molecules, such as tumour necrosis factor α and glutamate, have been shown to induce apoptosis in retinal neurones. Several authors have evaluated different aspects of apoptosis in tissue and cells from patients with LHON. Saadati et al compared the distinctive patterns of nerve fibre distribution and axonal dropout in LHON and other inherited disorders, such as optic nerve hypoplasia (ONH), and concluded that ONH is the result of an apoptotic process, whereas LHON is the result of a specific degenerative process. However, this postmortem study had two important limitations: only one nerve was observed and this was done 60 years after the onset of LHON. Mirabella and colleagues evaluated apoptosis in muscle biopsies of patients with different forms of mitochondrial encephaloneuromyopathies, and reported abnormalities in the process in all cases except a LHON specimen characterised by the absence of a detectable biochemical or morphological abnormality; however, only one case of LHON disease was examined in that study. Various results have recently been obtained; Danielson and colleagues were the first to discover that cells (osteosarcoma derived cybrid) with pathogenic LHON mutations were more sensitive to Fas dependent apoptosis than were control cells. Ghelli et al recently stressed the role of apoptosis in the same cell lines harbouring one of the three most frequent LHON pathogenic mutations. They documented that LHON cybrid cell death is apoptotic and saw increased release of cytochrome c into the cytosol, demonstrating mitochondrial involvement in the activation of the apoptotic cascade.
Other studies have provided direct proof that oxidative stress can damage mtDNA. In two recent papers, oxidative stress was engineered genetically in mice by targeted deletions in superoxide dismutase or the adenine nuclear transporter. Subsequent analysis showed a significant increase in mitochondrial rearrangements, associated with impaired mitochondrial function and morphology. Wong et al found that mtDNA mutations such as LHON mutations confer sensitivity to oxidative stress induced death. In a cell model with complex I impairment, Barrientos and Moraes identified a positive and quantitative correlation between apoptosis and free radical production. In 2002, Wong and colleagues and Ghelli et al, in two different experiments, showed that cybrids with LHON mutation have increased production of ROS. This suggested the reason for the cell specificity of the LHON degeneration phenotype for the first time, highlighting that degeneration of RGCs could be the result of an interaction between mtDNA mutation, complex I alteration, and increased ROS production. In an elegant experiment with mice in 2003, Qi and colleagues induced the reduction of mitochondrial superoxide dismutase and observed similar histopathological findings to those seen in the RGC of patients with LHON, confirming the key role of ROS in the pathogenesis of LHON.
Our study is the first one in which apoptosis induced by oxidative stress has been examined in cells from patients with LHON. Lymphocytes from patients with LHON treated with the oxidising agent dRib showed a significant increase in the percentage of apoptotic cells with respect to controls; no relation was evident between the percentage of apoptotic cells and the type of mtDNA mutation. The JC-1 test revealed depolarisation of the mitochondrial membrane potential in lymphocytes from patients with LHON, with a greater shift from redorange to green fluorescence after 48 hours of culture than in control cells. Our observation confirms that the apoptotic process induced by oxidative stress primarily involves the mitochondrial cascade. LHON cells showed a particular susceptibility to this inducer and confirmed the notion of a direct link between complex I (commonly altered in patients with LHON) and changes in mitochondrial
membrane permeability. Fontaine et al found that complex I may be part of the pore complex, strongly supporting this association. Finally, our data are in line with and complementary to those from LHON cybrid cells, confirming that the alteration of redox homeostasis renders the RGCs of patients with LHON vulnerable to apoptotic cell death. This factor could play a role in the different individual expression of genetic mutation and be a potential target in the development of new therapeutic strategies.
[1731-Cell response to oxidative stress induced apoptosis in patients with Lebers hereditary optic neuropathy; C Battisti, P Formichi, E Cardaioli, S Bianchi, P Mangiavacchi, S A Tripodi, P Tosi and A Federico]
Summary
Leber hereditary optic neuropathy (LHON) is a maternally inherited form of retinal ganglion cell degeneration leading to optic atrophy which is caused by point mutations in the mitochondrial genome (mtDNA). Three pathogenic mutations (positions 11778/ND4, 3460/ND1 and 14484/ND6) account for the majority of LHON cases and they affect genes that encode for different subunits of mitochondrial complex I. Excitotoxic injury to retinal ganglion cells and the optic nerve has been previously hypothesized, especially given the high susceptibility of this neural cell type to glutamate toxicity. Osteosarcoma-derived cytoplasmic hybrids (cybrids) generated from six unrelated LHON patients, two cell lines for each pathogenic mutation, were compared with cybrids obtained from three healthy controls. Molecular and biochemical analyses showed that excitatory amino acid transporter 1 (EAAT1)/GLAST is the most active glutamate transporter in this cellular model. The glutamate uptake maximal velocity was significantly reduced in all LHON cybrids compared with control cybrids. This reduction was correlated in a mutation-specific fashion with the degree of mitochondrial production of reactive oxygen species, which is enhanced in LHON cybrids. Our findings support the hypothesis that the genetically determined mitochondrial dysfunction in LHON patients leads to impaired activity of the EAAT1 glutamate transporter. This observation is particularly relevant since EAAT1 is the major means of glutamate removal in the inner retina and this prevents retinal ganglion cells being damaged as a result of excitotoxicity.
Discussion
Despite the well-established genetic aetiology of LHON, relatively little is known about the molecular mechanisms that link complex I mutations to retinal ganglion cell degeneration. Excitotoxicity has been hypothesized to be a concurring factor, based on the well-known sensitivity of retinal ganglion cells to injury mediated by over-activation of NMDA-type glutamate receptors. Nevertheless, no experimental evidence has so far been provided to document this hypothesis. The present study reports a novel finding in LHONresearch, demonstrating for the first time that LHON-associated mtDNA mutations negatively affect the activity of EAATs, in particular EAAT1 [glutainate-aspantate transporter (GLAST) in mice]. Our results suggest that the reduced activity of EAAT1, which varies among the different mtDNA mutations, might be the biochemical consequence of an oxidative modification of the transporter, caused by mitochondria-derived ROS. Thus, it is tempting to speculate that retinal Műller astroglial cells, which mostly rely on EAAT1 for glutamate clearance from the synaptic cleft, would remove glutamate less efficiently in LHON patients. This would imply a toxic rise of extracellular glutamate in the inner retina, which may contribute to the degenerative process affecting retinal ganglion cells.
We have investigated expression and function of glutamate transporters in cybrids obtained from six LHON patients and three healthy subjects. This cellular model is designed to study the cellular changes induced by mutant mtDNA dissected from the original nuclear background, given that only mitochondria from patients or controls are used to repopulate by cell fusion the perennial osteosarcoma-derived rho0 cell line (206), previously devoid from its own mtDNA. The resulting fusion cybrids express the exogenous mtDNA using the neutral nuclear background of the host parental osteosarcoma (143B.TKˉ) cell line, which was found to express naturally all three of the major EAATs. In particular, our data indicate that glutamate transport in 143B.TKˉ cells is mostly mediated by EAAT1. This finding is not surprising, since it has already been reported that EAAT1/GLAST is expressed and exerts a metabolic role in bone tissue whose osteosarcoma cell line represents a malignant variant. Interestingly, EAAT1/GLAST is the same transporter that accounts for most glutamate removal in the neuroretina by Műller astroglial cells. In addition, the observed glutamate transport Km and Vmax values in osteosarcoma cells are of the same order of magnitude of those described in Műller glia in culture. A direct link between EAAT1 function and excitotoxicity in retinal tissue was reported, showing that degenerative changes in the inner retinal layers after ischemic injury and glutamate toxicity were more severe in EAAT1-deficient mice compared with wildtype mice.
The cybrid cell system represents a convenient experimental model to study the effect of mtDNA point mutations, which affect complex I function, on EAAT1-mediated glutamate transport. The 3460/ND1 mutation, which consistently lowers complex I activity and is associated with a severe neuropathology, also resulted in a drastic decrease in glutamate transport. Furthermore, the 14484/ND6 mutation, which is associated with a benign visual prognosis and normal complex I activity, had the mildest effect. The 11778/ND4 mutation, which is the most common worldwide and is associated with the poorest visual outcome, showed an intermediate impairment in glutamate transport. Despite the degree that glutamate uptake impairment mirrors the known effect of single LHON mutations on complex I activity, it does not correlate exactly with the clinical severity of the disease, suggesting that other independent pathways might operate synergistically in the development of the overall clinical picture. The presence of the J haplogroup in the mtDNA background, which has been associated with the 11778/ND4 and 14484/ND6 mutations, did not seem to influence the glutamate uptake rate. We refer in particular to the direct comparison of HPE/HFF cybrid lines, carrying both the 11778/ND4 mutation but differing in their haplogroup, J and U, respectively.
EAAT1 expression did not show any change in LHON cybrids with respect to controls, suggesting that glutamate transporter molecules are synthesized correctly. Several recent studies showed that EAATs possess specific redox-sensing elements, consisting of cysteine residues, which regulate the transport rate by thiol-disulphide redox interconversion. The rate of glutamate uptake is maximal when the reactive cysteine residues are in the reduced state and minimal when they are in the oxidized state, without any change in the affinity for glutamate itself (Km). This regulatory mechanism probably reflects variation in transporter conformation. Hydrogen peroxide and xanthine oxidase activation generate ROS, which rapidly and irreversibly inhibit glutamate uptake in astrocyte cultures. Peroxynitrite, formed by the combination of superoxide and nitric oxide, potently inhibits glutamate uptake by purified or recombinant high affinity glutamate transporters reconstituted in liposomes. Lipids are especially vulnerable to oxidative stress and products of lipoperoxidation, such as acrolein and 4-hydroxy-2-nonenal, are able to reduce glutamate uptake. Thus, increased ROS generation from LHON mitochodria seems to represent a likely culprit for the observed inactivation of glutamate transporters. Preliminary studies on optic nerve and retinal histological specimens obtained from LHON patients indicate an increased staining for nitrotyrosine, possibly affecting the astroglial cellular component at the optic nerve head, and thus providing indirect evidence that the proposed pathway might be active in LHON.
In line with this hypothesis, we have shown that the impairment of glutamate uptake correlates strongly with mitochondrial ROS production in the single cybrid cell lines. Remarkably, an increase in hydroperoxide and mitochondrial-derived superoxide production with no changes in mitochondrial membrane potential were reported recently in neuronal-like LHON cybrids carrying the 11778/ND4 and 3460/ND1 mutations that were obtained using the neuronal precursor cell line Ntera-2/D1. On the contrary, no correlation was observed between the glutamate uptake rate and total ATP content. Energy production was assessed as total cellular ATP content, which is known to be almost unchanged in cybrids containing non-LHON mtDNA mutations when cultured in a complete medium. On the other hand, when assaying ATP synthesis specifically driven by complex I substrates, a drastic decrease was shown in LHON cybrids. As far as glutamate transporters are concerned, it is interesting to note that, even in mtDNA mutants, the subcellular concentration of ATP is preserved in the cytosolic and subplasma membrane compartmentsthose compartments that fuel the activity of glutamate transporterswhile being depleted in the mitochondrial matrix.
Lactate production has not yet been investigated in our cybrid cell lines. Although it is reasonable to speculate that LHON mutations might lead to lactic acid overproduction due to altered oxidative phosphorylation, this is unlikely to have biased glutamate uptake measures in this study, as the culture medium was changed frequently and the uptake experiments were conducted in a pH-controlled buffer. Rotenone, a selective inhibitor of complex I, was used as a
pharmacological tool to investigate the effect of altered complex I activity on glutamate transport.
Rotenone shares with LHON mutations the toxic properties of both inhibiting complex I activity and promoting the production of mitochondrial-derived ROS. In particular, one study showed that rotenone both reduces cell respiration and induces a dose-dependent oxidative damage in 143B.TKˉ cells within the same concentration range tested in the present study. The detrimental effect of both hydrogen peroxide and rotenone on EAAT1-mediated transport further confirms the sensitivity of glutamate transporters to increased ROS production and this occurs either as a result of external stimuli or from mitochondrial sources.
Although LHON mutations are usually homoplasmic and occur systemically, most patients report visual failure as the only symptom, raising the question of the specific vulnerability of the optic nerve. Conversely, it is noteworthy to recall that a number of patients with LHON have been described to have associated neurological abnormalities such as movement disorders, multiple sclerosis-like illness, polyneuropathy, epilepsy and dementia, indicating a potentially widespread neurodegenerative process, which might be the manifestation of an excitotoxic damage enhanced by unknown factors.
Multiple observations support the view that retinal ganglion cell death occurs preferentially via apoptosis under various physiological and pathological conditions, and this may apply to LHON. For example, surplus retinal ganglion cells die during development through an apoptotic pathway and optic nerve axotomy induces retinal ganglion cell apoptosis in animal models. In the acute phase of LHON, the absence of overt signs of inflammation at the examination of the fundus oculi has been suggested to argue against a necrotic type of cell death. Moreover, two recent studies showed respectively an increased sensitivity of LHON cybrids bearing the 11778/ND4 and 3460/ND6 mutations to Fas-induced apoptosis and the rapid induction of a mitochondrial-dependent apoptotic death in LHON cybrids carrying primary LHON mutations when cells were forced to use oxidative metabolism for ATP synthesis by replacing glucose with galactose in the medium.
There is increasing evidence that mitochondrial dysfunction and activation of NMDA channels may both contribute to neuronal apoptosis. Our findings indicate that interaction between mitochondrial dysfunction and excitotoxicity may also occur in LHON. The impaired glutamate uptake points to the possible relevant role of Műller astroglial cells, which express EAAT1 as their major glutamate transporter, in promoting excitotoxic injury to retinal ganglion cells. Although these findings need more in depth study, this working hypothesis might open new opportunities for the prevention or early treatment of LHON subjects and families with currently available anti-glutamatergic drugs.
Leber hereditary optic neuropathy mtDNA mutations disrupt glutamate transport in cybrid cell lines; Simone Beretta, Laura Mattavelli, Gessica Sala, Lucio Tremolizzo, Anthony H.V. Schapira, Andrea Martinuzzi, Valerio Carelli and Carlo Ferrarese]
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1644
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved