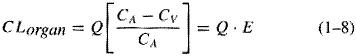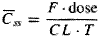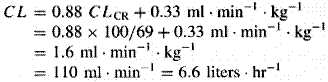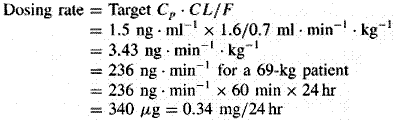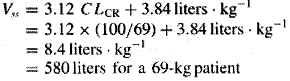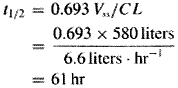| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination
Physicochemical Factors in Transfer of Drugs Across Membranes
|
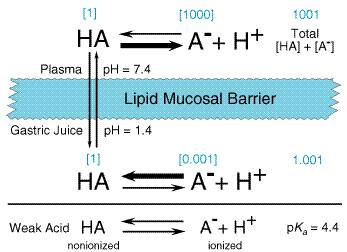
The absorption, distribution, metabolism, and excretion of a drug all involve its passage across cell membranes. Mechanisms by which drugs cross membranes and the physicochemical properties of molecules and membranes that influence this transfer are, therefore, important. The determining characteristics of a drug are its molecular size and shape, degree of ionization, relative lipid solubility of its ionized and nonionized forms, and its binding to tissue proteins. When a drug permeates a cell, it obviously must traverse the cellular plasma membrane. Other barriers to drug movement may be a single layer of cells (intestinal epithelium) or several layers of cells (skin). Despite such structural differences, the diffusion and transport of drugs across these various boundaries have many common characteristics, since drugs in general pass through cells rather than between them. The plasma membrane thus represents the common barrier. Cell Membranes The plasma membrane consists of a bilayer of amphipathic lipids, with their hydrocarbon chains oriented inward to form a continuous hydrophobic phase and their hydrophilic heads oriented outward. Individual lipid molecules in the bilayer vary according to the particular membrane and can move laterally, endowing the membrane with fluidity, flexibility, high electrical resistance, and relative impermeability to highly polar molecules. Membrane proteins embedded in the bilayer serve as receptors, ion channels, or transporters to elicit electrical or chemical signaling pathways and provide selective targets for drug actions. Most cell membranes are relatively permeable to water either by diffusion or by flow resulting from hydrostatic or osmotic differences across the membrane, and bulk flow of water can carry with it drug molecules. Such transport is the major mechanism by which drugs pass across most capillary endothelial membranes. However, proteins and drug molecules bound to them are too large and polar for this type of transport to occur; thus, transcapillary movement is limited to unbound drug. Paracellular transport through intercellular gaps is sufficiently large that passage across most capillaries is limited by blood flow and not by other factors (see below). As described later, this type of transport is an important factor in filtration across glomerular membranes in the kidney. Important exceptions exist in such capillary diffusion, however, since 'tight' intercellular junctions are present in specific tissues and paracellular transport in them is limited. Capillaries of the central nervous system (CNS) and a variety of epithelial tissues have tight junctions (see below). Although bulk flow of water can carry with it small, water-soluble substances, if the molecular mass of these compounds is greater than 100 to 200 daltons, such transport is limited. Accordingly, most large lipophilic drugs must pass through the cell membrane itself by one or more processes. Passive Membrane Transport Drugs cross membranes either by passive processes or by mechanisms involving the active participation of components of the membrane. In the former, the drug molecule usually penetrates by passive diffusion along a concentration gradient by virtue of its solubility in the lipid bilayer. Such transfer is directly proportional to the magnitude of the concentration gradient across the membrane, the lipid:water partition coefficient of the drug, and the cell surface area. The greater the partition coefficient, the higher is the concentration of drug in the membrane and the faster is its diffusion. After a steady state is attained, the concentration of the unbound drug is the same on both sides of the membrane if the drug is a nonelectrolyte. For ionic compounds, the steady-state concentrations will be dependent on differences in pH across the membrane, which may influence the state of ionization of the molecule on each side of the membrane and on the electrochemical gradient for the ion. Weak Electrolytes and Influence of pH Most drugs are weak acids or bases that are present in solution as both the nonionized and ionized species. The nonionized molecules are usually lipid-soluble and can diffuse across the cell membrane. In contrast, the ionized molecules are usually unable to penetrate the lipid membrane because of their low lipid solubility. Therefore, the transmembrane distribution of a weak electrolyte usually is determined by its pKa and the pH gradient across the membrane. The pKa is the pH at which half of the drug (weak electrolyte) is in its ionized form. To illustrate the effect of pH on distribution of drugs, the partitioning of a weak acid (pKa= 4.4) between plasma (pH = 7.4) and gastric juice (pH = 1.4) is depicted in Figure 12. It is assumed that the gastric mucosal membrane behaves as a simple lipid barrier that is permeable only to the lipid-soluble, nonionized form of the acid. The ratio of nonionized to ionized drug at each pH is readily calculated from the HendersonHasselbalch equation. Thus, in plasma, the ratio of nonionized to ionized drug is 1:1000; in gastric juice, the ratio is 1:0.001. These values are given in brackets in Figure 12. The total concentration ratio between the plasma and the gastric juice would therefore be 1000:1 if such a system came to a steady state. For a weak base with a pKa of 4.4, the ratio would be reversed, as would the thick horizontal arrows in Figure 12, which indicate the predominant species at each pH. Accordingly, at steady state, an acidic drug will accumulate on the more basic side of the membrane and a basic drug on the more acidic sidea phenomenon termed ion trapping. These considerations have obvious implications for the absorption and excretion of drugs, as discussed more specifically below. The establishment of concentration gradients of weak electrolytes across membranes with a pH gradient is a purely physical process and does not require an active transport system. All that is necessary is a membrane preferentially permeable to one form of the weak electrolyte and a pH gradient across the membrane. The establishment of the pH gradient is, however, an active process.
Carrier-Mediated Membrane Transport While passive diffusion through the bilayer is dominant in the disposition of most drugs, carrier-mediated mechanisms also can play an important role. Active transport is characterized by a requirement for energy, movement against an electrochemical gradient, saturability, selectivity, and competitive inhibition by cotransported compounds. The term facilitated diffusion describes a carrier-mediated transport process in which there is no input of energy and therefore enhanced movement of the involved substance is down an electrochemical gradient. Such mechanisms, which may be highly selective for a specific conformational structure of a drug, are involved in the transport of endogenous compounds whose rate of transport by passive diffusion otherwise would be too slow. In other cases, they function as a barrier system to protect cells from potentially toxic substances. The responsible transporter proteins often are expressed within cell membranes in a domain-specific fashion such that they mediate either drug uptake or efflux, and often such an arrangement facilitates vectorial transport across cells. Thus, in the liver, a number of basolaterally localized transporters with different substrate specificities are involved in the uptake of bile acids and amphipathic organic anions and cations into the hepatocyte, and a similar variety of ATP-dependent transporters in the canalicular membrane export such compounds into the bile. Analogous situations also are present in intestinal and renal tubular membranes. An important efflux transporter present at these sites and also in the capillary endothelium of brain capillaries is P-glycoprotein, which is encoded by the multidrug resistance-1 (MDR1) gene, important in resistance to cancer chemotherapeutic agents (Chapter 52: Antineoplastic Agents). P-glycoprotein localized in the enterocyte also limits the oral absorption of transported drugs since it exports the compound back into the intestinal tract subsequent to its absorption by passive diffusion. |
Drug Absorption, Bioavailability, and Routes of Administration
|
Absorption describes the rate at which a drug leaves its site of administration and the extent to which this occurs. However, the clinician is concerned primarily with a parameter designated as bioavailability, rather than absorption. Bioavailability is a term used to indicate the fractional extent to which a dose of drug reaches its site of action or a biological fluid from which the drug has access to its site of action. For example, a drug given orally must be absorbed first from the stomach and intestine, but this may be limited by the characteristics of the dosage form and/or the drug's physicochemical properties. In addition, drug then passes through the liver, where metabolism and/or biliary excretion may occur before it reaches the systemic circulation. Accordingly, a fraction of the administered and absorbed dose of drug will be inactivated or diverted before it can reach the general circulation and be distributed to its sites of action. If the metabolic or excretory capacity of the liver for the agent in question is large, bioavailability will be substantially reduced (the so-called first-pass effect). This decrease in availability is a function of the anatomical site from which absorption takes place; other anatomical, physiological, and pathological factors can influence bioavailability (see below), and the choice of the route of drug administration must be based on an understanding of these conditions. Oral (Enteral) versus Parenteral Administration Often there is a choice of the route by which a therapeutic agent may be given, and a knowledge of the advantages and disadvantages of the different routes of administration is then of primary importance. Some characteristics of the major routes employed for systemic drug effect are compared in Table 11. Oral ingestion is the most common method of drug administration. It also is the safest, most convenient, and most economical. Disadvantages to the oral route include limited absorption of some drugs because of their physical characteristics (e.g., water solubility), emesis as a result of irritation to the gastrointestinal mucosa, destruction of some drugs by digestive enzymes or low gastric pH, irregularities in absorption or propulsion in the presence of food or other drugs, and necessity for cooperation on the part of the patient. In addition, drugs in the gastrointestinal tract may be metabolized by the enzymes of the intestinal flora, mucosa, or the liver before they gain access to the general circulation. The parenteral injection of drugs has certain distinct advantages over oral administration. In some instances, parenteral administration is essential for the drug to be delivered in its active form. Availability is usually more rapid, extensive, and predictable than when a drug is given by mouth. The effective dose therefore can be more accurately delivered. In emergency therapy and when a patient is unconscious, uncooperative, or unable to retain anything given by mouth, parenteral therapy may be a necessity. The injection of drugs, however, has its disadvantages: asepsis must be maintained; pain may accompany the injection; it is sometimes difficult for patients to perform the injections themselves if self-medication is necessary; and there is the risk of inadvertent administration of a drug when it is not intended. Expense is another consideration. Oral Ingestion Absorption from the gastrointestinal tract is governed by factors such
as surface area for absorption, blood flow to the site of absorption, the
physical state of the drug (solution, suspension, or solid dosage form), its
water solubility, and concentration at the site of absorption. For drugs
given in solid form, the rate of dissolution may be the limiting factor in
their absorption, especially if they have low water solubility. Since most
drug absorption from the gastrointestinal tract occurs via passive
processes, absorption is favored when the drug is in the nonionized and more
lipophilic form. Based on the pH-partition concept presented in Figure 12,
it would be predicted that drugs that are weak acids would be better absorbed
from the stomach (pH 1 to 2) than from the upper intestine (pH 3 to 6), and vice
versa for weak bases. However, the epithelium of the stomach is lined
with a thick mucous layer, and its surface area is small; by contrast, the
villi of the upper intestine provide an extremely large surface area ( Drugs that are destroyed by gastric juice or that cause gastric irritation sometimes are administered in dosage forms with a coating that prevents dissolution in the acidic gastric contents. However, some enteric-coated preparations of a drug also may resist dissolution in the intestine, and very little of the drug may be absorbed. Controlled-Release Preparations The rate of absorption of a drug administered as a tablet or other solid oral-dosage form is partly dependent upon its rate of dissolution in the gastrointestinal fluids. This factor is the basis for the so-called controlled-release, extended-release, sustained-release, or prolonged-action pharmaceutical preparations that are designed to produce slow, uniform absorption of the drug for 8 hours or longer. Potential advantages of such preparations are reduction in the frequency of administration of the drug as compared with conventional dosage forms (possibly with improved compliance by the patient), maintenance of a therapeutic effect overnight, and decreased incidence and/or intensity of undesired effects by elimination of the peaks in drug concentration that often occur after administration of immediate-release dosage forms. Many controlled-release preparations fulfill these expectations. However, such products have some drawbacks. Generally, interpatient variability, in terms of the systemic concentration of the drug that is achieved, is greater for controlled-release than for immediate-release dosage forms. During repeated drug administration, trough drug concentrations resulting from controlled-release dosage forms may not be different from those observed with immediate-release preparations, although the time interval between trough concentrations is greater for a well-designed controlled-release product. It is possible that the dosage form may fail, and 'dose-dumping' with resultant toxicity can occur, since the total dose of drug ingested at one time may be several times the amount contained in the conventional preparation. Controlled-release dosage forms are most appropriate for drugs with short half-lives (less than 4 hours). So-called controlled-release dosage forms are sometimes developed for drugs with long half-lives (greater than 12 hours). These usually more expensive products should not be prescribed unless specific advantages have been demonstrated. Sublingual Administration Absorption from the oral mucosa has special significance for certain drugs, despite the fact that the surface area available is small. For example, nitroglycerin is effective when retained sublingually because it is nonionic and has a very high lipid solubility. Thus, the drug is absorbed very rapidly. Nitroglycerin also is very potent; relatively few molecules need to be absorbed to produce the therapeutic effect. Since venous drainage from the mouth is to the superior vena cava, the drug also is protected from rapid hepatic first-pass metabolism, which is sufficient to prevent the appearance of any active nitroglycerin in the systemic circulation if the sublingual tablet is swallowed. Rectal Administration The rectal route often is useful when oral ingestion is precluded because the patient is unconscious or when vomiting is presenta situation particularly relevant to young children. Approximately 50% of the drug that is absorbed from the rectum will bypass the liver; the potential for hepatic first-pass metabolism is thus less than that for an oral dose. However, rectal absorption often is irregular and incomplete, and many drugs cause irritation of the rectal mucosa. Parenteral Injection The major routes of parenteral administration are intravenous, subcutaneous, and intramuscular. Absorption from subcutaneous and intramuscular sites occurs by simple diffusion along the gradient from drug depot to plasma. The rate is limited by the area of the absorbing capillary membranes and by the solubility of the substance in the interstitial fluid. Relatively large aqueous channels in the endothelial membrane account for the indiscriminate diffusion of molecules regardless of their lipid solubility. Larger molecules, such as proteins, slowly gain access to the circulation by way of lymphatic channels. Drugs administered into the systemic circulation by any route, excluding the intraarterial route, are subject to possible first-pass elimination in the lung prior to distribution to the rest of the body. The lungs serve as a temporary storage site for a number of agents, especially drugs that are weak bases and are predominantly nonionized at the blood pH, apparently by their partition into lipid. The lungs also serve as a filter for particulate matter that may be given intravenously, and, of course, they provide a route of elimination for volatile substances. Intravenous Factors relevant to absorption are circumvented by intravenous injection of drugs in aqueous solution, because bioavailability is complete and rapid. Also, drug delivery is controlled and achieved with an accuracy and immediacy not possible by any other procedure. In some instances, as in the induction of surgical anesthesia, the dose of a drug is not predetermined but is adjusted to the response of the patient. Also, certain irritating solutions can be given only in this manner, since the blood vessel walls are relatively insensitive, and the drug, if injected slowly, is greatly diluted by the blood. As there are advantages to the use of this route of administration, so are there liabilities. Unfavorable reactions are likely to occur, since high concentrations of drug may be attained rapidly in both plasma and tissues. Because of this, it is advisable to intravenously administer a drug slowly by infusion rather than by rapid injection, and with close monitoring of the patient's response. Furthermore, once the drug is injected there is no retreat. Repeated intravenous injections are dependent upon the ability to maintain a patent vein. Drugs in an oily vehicle or those that precipitate blood constituents or hemolyze erythrocytes should not be given by this route. Subcutaneous Injection of a drug into a subcutaneous site often is used. It can be used only for drugs that are not irritating to tissue; otherwise, severe pain, necrosis, and tissue sloughing may occur. The rate of absorption following subcutaneous injection of a drug often is sufficiently constant and slow to provide a sustained effect. Moreover, it may be varied intentionally. For example, the rate of absorption of a suspension of insoluble insulin is slow compared with that of a soluble preparation of the hormone. The incorporation of a vasoconstrictor agent in a solution of a drug to be injected subcutaneously also retards absorption. Absorption of drugs implanted under the skin in a solid pellet form occurs slowly over a period of weeks or months; some hormones are effectively administered in this manner. Intramuscular Drugs in aqueous solution are absorbed quite rapidly after intramuscular injection, depending upon the rate of blood flow to the injection site. This may be modulated to some extent by local heating, massage, or exercise. For example, jogging may cause a precipitous drop in blood sugar when insulin is injected into the thigh, rather than into the arm or abdominal wall, since running markedly increases blood flow to the leg. Generally, the rate of absorption following injection of an aqueous preparation into the deltoid or vastus lateralis is faster than when the injection is made into the gluteus maximus. The rate is particularly slower for females after injection into the gluteus maximus. This has been attributed to the different distribution of subcutaneous fat in males and females, since fat is relatively poorly perfused. Very obese or emaciated patients may exhibit unusual patterns of absorption following intramuscular or subcutaneous injection. Very slow, constant absorption from the intramuscular site results if the drug is injected in solution in oil or suspended in various other repository vehicles. Antibiotics often are administered in this manner. Substances too irritating to be injected subcutaneously sometimes may be given intramuscularly. Intraarterial Occasionally a drug is injected directly into an artery to localize its effect in a particular tissue or organfor example, in the treatment of liver tumors and head/neck cancers. Diagnostic agents are sometimes administered by this route. Intraarterial injection requires great care and should be reserved for experts. The first-pass and cleansing effects of the lung are not available when drugs are given by this route. Intrathecal The bloodbrain barrier and the bloodcerebrospinal fluid barrier often preclude or slow the entrance of drugs into the CNS. Therefore, when local and rapid effects of drugs on the meninges or cerebrospinal axis are desired, as in spinal anesthesia or acute CNS infections, drugs are sometimes injected directly into the spinal subarachnoid space. Brain tumors also may be treated by direct intraventricular drug administration. Pulmonary Absorption Provided that they do not cause irritation, gaseous and volatile drugs may be inhaled and absorbed through the pulmonary epithelium and mucous membranes of the respiratory tract. Access to the circulation is rapid by this route, because the lung's surface area is large. The principles governing absorption and excretion of anesthetic and other therapeutic gases are discussed in Chapters 13: History and Principles of Anesthesiology, 14: General Anesthetics, and 16: Therapeutic Gases: Oxygen, Carbon Dioxide, Nitric Oxide, and Helium. In addition, solutions of drugs can be atomized and the fine droplets in air (aerosol) inhaled. Advantages are the almost instantaneous absorption of a drug into the blood, avoidance of hepatic first-pass loss, and, in the case of pulmonary disease, local application of the drug at the desired site of action. For example, drugs can be given in this manner for the treatment of bronchial asthma (seeChapter 28: Drugs Used in the Treatment of Asthma). Past disadvantages, such as poor ability to regulate the dose and cumbersomeness of the methods of administration, have to a large extent been overcome by technological advances, including metered-dose inhalers and more reliable aerolizers. Pulmonary absorption is an important route of entry of certain drugs of abuse and of toxic environmental substances of varied composition and physical states. Both local and systemic reactions to allergens may occur subsequent to inhalation. Topical Application Mucous Membranes Drugs are applied to the mucous membranes of the conjunctiva, nasopharynx, oropharynx, vagina, colon, urethra, and urinary bladder primarily for their local effects. Occasionally, as in the application of synthetic antidiuretic hormone to the nasal mucosa, systemic absorption is the goal. Absorption through mucous membranes occurs readily. In fact, local anesthetics applied for local effect sometimes may be absorbed so rapidly that they produce systemic toxicity. Skin Few drugs readily penetrate the intact skin. Absorption of those that do is dependent on the surface area over which they are applied and to their lipid solubility, since the epidermis behaves as a lipid barrier (seeChapter 65: Dermatological Pharmacology). The dermis, however, is freely permeable to many solutes; consequently, systemic absorption of drugs occurs much more readily through abraded, burned, or denuded skin. Inflammation and other conditions that increase cutaneous blood flow also enhance absorption. Toxic effects sometimes are produced by absorption through the skin of highly lipid-soluble substances (e.g., a lipid-soluble insecticide in an organic solvent). Absorption through the skin can be enhanced by suspending the drug in an oily vehicle and rubbing the resulting preparation into the skin. Because hydrated skin is more permeable than dry skin, the dosage form may be modified or an occlusive dressing may be used to facilitate absorption. Controlled-release topical patches are becoming increasingly available. A patch containing scopolamine, placed behind the ear where body temperature and blood flow enhance absorption, releases sufficient drug to the systemic circulation to protect the wearer from motion sickness. Transdermal estrogen replacement therapy yields low maintenance levels of estradiol while minimizing the high estrone metabolite levels observed following oral administration. Eye Topically applied ophthalmic drugs are used primarily for their local
effects (seeChapter 66: Ocular Pharmacology). Systemic absorption that
results from drainage through the nasolacrimal canal is usually undesirable.
In addition, drug that is absorbed after such drainage is not subject to
first-pass hepatic elimination. Unwanted systemic pharmacological effects may
occur for this reason when Bioequivalence Drugs are not administered as such; instead, they are formulated into drug dosage forms. Drug products are considered to be pharmaceutical equivalents if they contain the same active ingredients and are identical in strength or concentration, dosage form, and route of administration. Two pharmaceutically equivalent drug products are considered to be bioequivalent when the rates and extents of bioavailability of the active ingredient in the two products are not significantly different under suitable test conditions. In the past, dosage forms of a drug from different manufacturers and even different lots of preparations from a single manufacturer sometimes differed in their bioavailability. Such differences were seen primarily among oral dosage forms of poorly soluble, slowly absorbed drugs. They result from differences in crystal form, particle size, or other physical characteristics of the drug that are not rigidly controlled in formulation and manufacture of the preparations. These factors affect disintegration of the dosage form and dissolution of the drug and hence the rate and extent of drug absorption. The potential nonequivalence of different drug preparations has been a matter of concern. Strengthened regulatory requirements have resulted in few, if any, documented cases of nonequivalence between approved drug products. The significance of possible nonequivalence of drug preparations is further discussed in connection with drug nomenclature and the choice of drug name in writing prescription orders (seeAppendix I). |
Distribution of Drugs
|
Following absorption or administration into the systemic blood, a drug distributes into interstitial and intracellular fluids. This process reflects a number of physiological factors and the particular physicochemical properties of the individual drug. Cardiac output, regional blood flow, and tissue volume determine the rate of delivery and potential amount of drug distributed into tissues. Initially, liver, kidney, brain, and other well-perfused organs receive most of the drug, whereas delivery to muscle, most viscera, skin, and fat is slower. This second distribution phase may require minutes to several hours before the concentration of drug in tissue is in distribution equilibrium with that in blood. The second phase also involves a far larger fraction of body mass than does the initial phase and generally accounts for most of the extravascularly distributed drug. With exceptions such as the brain, diffusion of drug into the interstitial fluid occurs rapidly because of the highly permeable nature of the capillary endothelial membrane. Thus, tissue distribution is determined by the partitioning of drug between blood and the particular tissue. Lipid solubility is an important determinant of such uptake as is any pH gradient between intracellular and extracellular fluids for drugs that are either weak acids or bases. However, in general, ion trapping associated with the latter factor is not large, since the pH difference (7.0 versus 7.4) is small. The more important determinant of blood:tissue partitioning is the relative binding of drug to plasma proteins and tissue macromolecules. Plasma Proteins Many drugs are bound to plasma proteins, mostly to plasma albumin for
acidic drugs and to The fraction of total drug in plasma that is bound is determined by
the drug concentration, its affinity for the binding sites, and the number of
binding sites. Simple mass-action relationships determine the unbound and
bound concentrations (seeChapter 2: Pharmacodynamics: Mechanisms of
Drug Action and the Relationship Between Drug Concentration and Effect). At
low concentrations of drug (less than the plasma-protein binding dissociation
constant), the fraction bound is a function of the concentration of binding
sites and the dissociation constant. At high drug concentrations (greater
than the dissociation constant), the fraction bound is a function of the
number of binding sites and the drug concentration. Therefore, plasma binding
is a saturable and nonlinear process. For most drugs, however, the
therapeutic range of plasma concentrations is limited; thus, the extent of
binding and the unbound fraction is relatively constant. The percentage
values listed in Appendix II refer only to this situation unless otherwise
indicated. The extent of plasma binding also may be affected by
disease-related factors. For example, hypoalbuminemia secondary to severe
liver disease or the nephrotic syndrome results in reduced binding and an
increase in the unbound fraction. Also, conditions resulting in the acute
phase reaction response (cancer, arthritis, myocardial infarction, Crohn's
disease) lead to elevated levels of Because binding of drugs to plasma proteins is rather nonselective, many drugs with similar physicochemical characteristics can compete with each other and with endogenous substances for these binding sites. For example, displacement of unconjugated bilirubin from binding to albumin by the sulfonamides and other organic anions is known to increase the risk of bilirubin encephalopathy in the newborn. Concern for drug toxicities based on a similar competition between drugs for binding sites has, in the past, been overemphasized. Since drug responses, both efficacious and toxic, are a function of unbound concentrations, steady-state unbound concentrations will change only when either drug input (dosing rate) or clearance of unbound drug is changed [seeEquation (11) and discussion later in this chapter]. Thus, steady-state unbound concentrations are independent of the extent of protein binding. However, for narrow-therapeutic-index drugs, a transient change in unbound concentrations occurring immediately following the dose of a displacing drug could be of concern. A more common problem resulting from competition of drugs for plasma-protein binding sites is misinterpretation of measured concentrations of drugs in plasma, since most assays do not distinguish free drug from bound drug. Importantly, binding of a drug to plasma proteins limits its concentration in tissues and at its locus of action, since only unbound drug is in equilibrium across membranes. Accordingly, after distribution equilibrium is achieved, the concentration of active, unbound drug in intracellular water is the same as that in plasma except when carrier-mediated transport is involved. Binding also limits glomerular filtration of the drug, since this process does not immediately change the concentration of free drug in the plasma (water is also filtered). However, plasma-protein binding generally does not limit renal tubular secretion or biotransformation, since these processes lower the free drug concentration, and this is rapidly followed by dissociation of the drugprotein complex. Drug transport and metabolism also are limited by plasma binding except when these are especially efficient and drug clearance, calculated on the basis of unbound drug, exceeds organ plasma flow. In this situation, binding of the drug to plasma protein may be viewed as a transport mechanism that fosters drug elimination by delivering drug to sites for elimination. Tissue Binding Many drugs accumulate in tissues at higher concentrations than those in the extracellular fluids and blood. For example, during long-term administration of the antimalarial agent quinacrine, the concentration of drug in the liver may be several thousandfold higher than that in the blood. Such accumulation may be a result of active transport or, more commonly, binding. Tissue binding of drugs usually occurs with cellular constituents such as proteins, phospholipids, or nuclear proteins and generally is reversible. A large fraction of drug in the body may be bound in this fashion and serve as a reservoir that prolongs drug action in that same tissue or at a distant site reached through the circulation. Fat As a Reservoir Many lipid-soluble drugs are stored by physical solution in the neutral fat. In obese persons, the fat content of the body may be as high as 50%, and even in starvation it constitutes 10% of body weight; hence, fat can serve as an important reservoir for lipid-soluble drugs. For example, as much as 70% of the highly lipid-soluble barbiturate thiopental may be present in body fat 3 hours after administration. However, fat is a rather stable reservoir because it has a relatively low blood flow. Bone The tetracycline antibiotics (and other divalent-metal-ion chelating agents) and heavy metals may accumulate in bone by adsorption onto the bone-crystal surface and eventual incorporation into the crystal lattice. Bone can become a reservoir for the slow release of toxic agents such as lead or radium into the blood; their effects can thus persist long after exposure has ceased. Local destruction of the bone medulla also may lead to reduced blood flow and prolongation of the reservoir effect, since the toxic agent becomes sealed off from the circulation; this may further enhance the direct local damage to the bone. A vicious cycle results, whereby the greater the exposure to the toxic agent, the slower is its rate of elimination. Redistribution Termination of drug effect usually is by metabolism and excretion, but it also may result from redistribution of the drug from its site of action into other tissues or sites. Redistribution is a factor in terminating drug effect primarily when a highly lipid-soluble drug that acts on the brain or cardiovascular system is administered rapidly by intravenous injection or by inhalation. A good example of this is the use of the intravenous anesthetic thiopental, a highly lipid-soluble drug. Because blood flow to the brain is so high, the drug reaches its maximal concentration in brain within a minute after it is injected intravenously. After injection is concluded, the plasma concentration falls as thiopental diffuses into other tissues, such as muscle. The concentration of the drug in brain follows that of the plasma, because there is little binding of the drug to brain constituents. Thus, onset of anesthesia is rapid, but so is its termination. Both are directly related to the concentration of drug in the brain. Central Nervous System and Cerebrospinal Fluid The distribution of drugs into the CNS from the blood is unique, because functional barriers are present that restrict entry of drugs into this critical site. One reason for this is that the brain capillary endothelial cells have continuous tight junctions; therefore, drug penetration into the brain depends on transcellular rather than paracellular transport between cells. The unique characteristics of pericapillary glial cells also contribute to the bloodbrain barrier. At the choroid plexus, a similar bloodcerebrospinal fluid (CSF) barrier is present except that it is epithelial cells that are joined by tight junctions rather than endothelial cells. As a result, the lipid solubility of the nonionized and unbound species of the drug is an important determinant of its uptake by the brain; the more lipophilic it is, the more likely it is to cross the bloodbrain barrier. This situation often is used in drug design to alter brain distribution; for example, nonsedating antihistamines achieve far lower brain concentrations than do other agents in this class. Increasing evidence also indicates that drugs may penetrate into the CNS by specific uptake transporters normally involved in the transport of nutrients and endogenous compounds from blood into the brain and CSF. Recently, it has been discovered that another important factor in the functional bloodbrain barrier also involves membrane transporters which are, in this case, efflux carriers present in the brain capillary endothelial cell. P-glycoprotein is the most important of these and functions by a combination of not allowing drug to even translocate across the endothelial cell and also by exporting any drug that enters the brain by other means. Such transport may account for the brain, and other tissues where P-glycoprotein is similarly expressed (e.g., the testes), being pharmacological sanctuary sites where drug concentrations are below those necessary to achieve a desired effect even though blood levels are adequate. This situation apparently occurs with HIV protease inhibitors (Kim et al., 1998) and also with loperamidea potent, systemically active opioid that lacks any central effects characteristic of other opioids (seeChapter 23: Opioid Analgesics). Efflux transporters that actively secrete drug from the CSF into the blood also are present in the choroid plexus. Regardless of whether a drug is pumped out of the CNS by specific transporters or diffuses back into the blood, drugs also exit the CNS along with the bulk flow of CSF through the arachnoid villi. In general, the bloodbrain barrier's function is well maintained; however, meningeal and encephalic inflammation increase the local permeability. There also is the potential that the bloodbrain barrier may be advantageously modulated to enhance the treatment of infections or tumors in the brain. To date, however, such an approach has not been shown to be clinically useful. Placental Transfer of Drugs The potential transfer of drugs across the placenta is important, since drugs may cause congenital anomalies. Administered immediately before delivery, they also may have adverse effects on the neonate. Lipid solubility, extent of plasma binding, and degree of ionization of weak acids and bases are important general determinants, as previously discussed. The fetal plasma is slightly more acidic than that of the mother (pH 7.0 to 7.2 versus 7.4), so that ion-trapping of basic drugs occurs. As in the brain, P-glycoprotein is present in the placenta and functions as an export transporter to limit fetal exposure to potentially toxic agents. But the view that the placenta is an absolute barrier to drugs is inaccurate. A more appropriate approximation is that the fetus is to at least some extent exposed to essentially all drugs taken by the mother. |
Excretion of Drugs
|
Drugs are eliminated from the body either unchanged by the process of excretion or converted to metabolites. Excretory organs, the lung excluded, eliminate polar compounds more efficiently than substances with high lipid solubility. Lipid-soluble drugs thus are not readily eliminated until they are metabolized to more polar compounds. The kidney is the most important organ for excreting drugs and their metabolites. Substances excreted in the feces are mainly unabsorbed, orally ingested drugs or metabolites excreted either in the bile or secreted directly into the intestinal tract and, subsequently, not reabsorbed. Excretion of drugs in breast milk is important, not because of the amounts eliminated, but because the excreted drugs are potential sources of unwanted pharmacological effects in the nursing infant. Pulmonary excretion is important mainly for the elimination of anesthetic gases and vapors (seeChapters 13: History and Principles of Anesthesiology, 14: General Anesthetics, and 16: Therapeutic Gases: Oxygen, Carbon Dioxide, Nitric Oxide, and Helium); occasionally, small quantities of other drugs or metabolites are excreted by this route. Renal Excretion Excretion of drugs and metabolites in the urine involves three processes: glomerular filtration, active tubular secretion, and passive tubular reabsorption. Changes in overall renal function generally affect all three processes to a similar extent. Renal function is low compared to body size in neonates but rapidly matures within the first few months after birth. During adulthood there is a slow decline in renal function, about 1% per year, so that in the elderly a substantial degree of impairment is usually present. The amount of drug entering the tubular lumen by filtration is dependent on the glomerular filtration rate and the extent of plasma binding of the drug; only unbound drug is filtered. In the proximal renal tubule, active, carrier-mediated tubular secretion also may add drug to the tubular fluid. Transporters such as P-glycoprotein and the multidrug resistanceassociated protein-type 2 (MRP2) localized in the apical, brush-border membrane are largely responsible for the secretion of amphipathic anions and conjugated metabolites (such as glucuronides, sulfates, and glutathione adducts), respectively. Transport systems that are similar but more selective for organic cationic drugs (OCDs) are involved in the secretion of organic bases. Membrane transporters, mainly located in the distal renal tubule, also are responsible for any active reabsorption of drug from the tubular lumen back into the systemic circulation. However, most of such reabsorption occurs by nonionic diffusion. In the proximal and distal tubules, the nonionized forms of weak acids and bases undergo net passive reabsorption. The concentration gradient for back-diffusion is created by the reabsorption of water with Na+ and other inorganic ions. Since the tubular cells are less permeable to the ionized forms of weak electrolytes, passive reabsorption of these substances is pH-dependent. When the tubular urine is made more alkaline, weak acids are excreted more rapidly and to a greater extent, primarily because they are more ionized and passive reabsorption is decreased. When the tubular urine is made more acidic, the excretion of weak acids is reduced. Alkalinization and acidification of the urine have the opposite effects on the excretion of weak bases. In the treatment of drug poisoning, the excretion of some drugs can be hastened by appropriate alkalinization or acidification of the urine. Whether or not alteration of urine pH results in a significant change in drug elimination depends upon the extent and persistence of the pH change and the contribution of pH-dependent passive reabsorption to total drug elimination. The effect is greatest for weak acids and bases with pKa values in the range of urinary pH (5 to 8). However, alkalinization of urine can produce a fourfold to sixfold increase in excretion of a relatively strong acid such as salicylate when urinary pH is changed from 6.4 to 8.0. The fraction of nonionized drug would decrease from 1% to 0.04%. Biliary and Fecal Excretion Transport systems analogous to those in the kidney also are present in the canalicular membrane of the hepatocyte, and these actively secrete drugs and metabolites into bile. P-glycoprotein transports a plethora of amphipathic, lipid-soluble drugs, whereas MRP2 is mainly involved in the secretion of conjugated metabolites of drugs (glutathione conjugates, glucuronides, and some sulfates). MRP2 also is involved in the excretion of endogenous compounds, and the Dubin-Johnson syndrome is caused by a genetically determined absence of this transporter. Active biliary secretion of organic cations also involves transporters. Ultimately, drugs and metabolites present in bile are released into the intestinal tract during the digestive process. Because secretory transporters such as P-glycoprotein also are expressed on the apical membrane of enterocytes, direct secretion of drugs and metabolites may occur from the systemic circulation into the intestinal lumen. Subsequently, drugs and metabolites can be reabsorbed back into the body from the intestine which, in the case of conjugated metabolites like glucuronides, may require their enzymatic hydrolysis by the intestinal microflora. Such enterohepatic recycling, if extensive, may prolong significantly the presence of a drug and its effects within the body prior to elimination by other pathways. Excretion by Other Routes Excretion of drugs into sweat, saliva, and tears is quantitatively unimportant. Elimination by these routes is dependent mainly upon diffusion of the nonionized, lipid-soluble form of drugs through the epithelial cells of the glands and is pH-dependent. Drugs excreted in the saliva enter the mouth, where they are usually swallowed. The concentration of some drugs in saliva parallels that in plasma. Saliva therefore may be a useful biological fluid in which to determine drug concentrations when it is difficult or inconvenient to obtain blood. The same principles apply to excretion of drugs in breast milk. Since milk is more acidic than plasma, basic compounds may be slightly concentrated in this fluid, and the concentration of acidic compounds in the milk is lower than in plasma. Nonelectrolytes, such as ethanol and urea, readily enter breast milk and reach the same concentration as in plasma, independent of the pH of the milk. Although excretion into hair and skin also is quantitatively unimportant, sensitive methods of detection of drugs in these tissues have forensic significance. |
Metabolism of Drugs
|
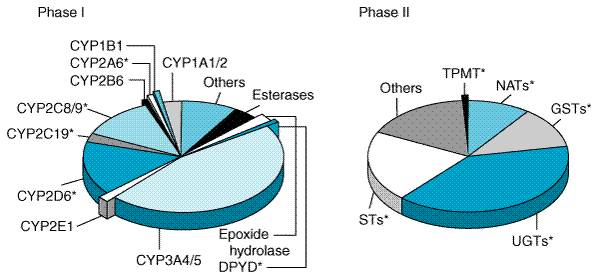
The lipophilic characteristics of drugs that promote their passage through biological membranes and subsequent access to their site of action hinder their excretion from the body. Renal excretion of unchanged drug plays only a modest role in the overall elimination of most therapeutic agents, since lipophilic compounds filtered through the glomerulus are largely reabsorbed back into the systemic circulation during passage through the renal tubules. The metabolism of drugs and other xenobiotics into more hydrophilic metabolites is therefore essential for the elimination of these compounds from the body and termination of their biological activity. In general, biotransformation reactions generate more polar, inactive metabolites that are readily excreted from the body. However, in some cases, metabolites with potent biological activity or toxic properties are generated. Many of the metabolic biotransformation reactions leading to inactive metabolites of drugs also generate biologically active metabolites of endogenous compounds. The following discussion focuses on the biotransformation of drugs but is generally applicable to the metabolism of all xenobiotics as well as a number of endogenous compounds, including steroids, vitamins, and fatty acids. Phase I and Phase II Metabolism Drug biotransformation reactions are classified as either phase I functionalization reactions or phase II biosynthetic (conjugation) reactions. Phase I reactions introduce or expose a functional group on the parent compound. Phase I reactions generally result in the loss of pharmacological activity, although there are examples of retention or enhancement of activity. In rare instances, metabolism is associated with an altered pharmacological activity. Prodrugs are pharmacologically inactive compounds, designed to maximize the amount of the active species that reaches its site of action. Inactive prodrugs are converted rapidly to biologically active metabolites, often by the hydrolysis of an ester or amide linkage. If not rapidly excreted into the urine, the products of phase I biotransformation reactions can then react with endogenous compounds to form a highly water-soluble conjugate. Phase II conjugation reactions lead to the formation of a covalent linkage between a functional group on the parent compound or phase I metabolite with endogenously derived glucuronic acid, sulfate, glutathione, amino acids, or acetate. These highly polar conjugates are generally inactive and are excreted rapidly in the urine and feces. An example of an active conjugate is the 6-glucuronide metabolite of morphine, which is a more potent analgesic than its parent compound. Site of Biotransformation The metabolic conversion of drugs generally is enzymatic in nature. The enzyme systems involved in the biotransformation of drugs are localized in the liver, although every tissue examined has some metabolic activity. Other organs with significant metabolic capacity include the gastrointestinal tract, kidneys, and lungs. Following nonparenteral administration of a drug, a significant portion of the dose may be metabolically inactivated in either the intestinal epithelium or the liver before it reaches the systemic circulation. This first-pass metabolism significantly limits the oral availability of highly metabolized drugs. Within a given cell, most drug-metabolizing activity is found in the endoplasmic reticulum and the cytosol, although drug biotransformations also can occur in the mitochondria, nuclear envelope, and plasma membrane. Upon homogenization and differential centrifugation of tissues, the endoplasmic reticulum breaks up, and fragments of the membrane form microvesicles, referred to as microsomes. The drug-metabolizing enzymes in the endoplasmic reticulum therefore often are classified as microsomal enzymes. The enzyme systems involved in phase I reactions are located primarily in the endoplasmic reticulum, while the phase II conjugation enzyme systems are mainly cytosolic. Often drugs biotransformed through a phase I reaction in the endoplasmic reticulum are conjugated at this same site or in the cytosolic fraction of the same cell. Cytochrome P450 Monooxygenase System The cytochrome P450 enzymes are a superfamily of heme-thiolate proteins widely distributed across all living kingdoms. The enzymes are involved in the metabolism of a plethora of chemically diverse, endogenous and exogenous compounds, including drugs, environmental chemicals, and other xenobiotics. Usually they function as a terminal oxidase in a multicomponent electron-transfer chain that introduces a single atom of molecular oxygen into the substrate with the other atom being incorporated into water. In microsomes, the electrons are supplied from NADPH via cytochrome P450 reductase, which is closely associated with cytochrome P450 in the lipid membrane of the smooth endoplasmic reticulum. Cytochrome P450 catalyzes many reactions, including aromatic and side-chain hydroxylation; N-, O- and S-dealkylation; N-oxidation; N-hydroxylation; sulfoxidation; deamination; dehalogenation; and desulfuration. Details and examples of cytochrome P450mediated metabolism are shown in Table 12. A number of reductive reactions also are catalyzed by these enzymes, generally under conditions of low oxygen tension. Of the approximately 1000 currently known cytochrome P450s, about 50 are functionally active in human beings. These are categorized into 17 families and many subfamilies according to the amino acidsequence similarities of the predicted proteins; the abbreviated term CYP is used for identification. Sequences that are greater than 40% identical belong to the same family, identified by an Arabic number; within a family, sequences greater than 55% identical are in the same subfamily, identified by a letter; and different individual isoforms within the subfamily are identified by an Arabic number. About 8 to 10 isoforms in the CYP1, CYP2, and CYP3 families primarily are involved in the majority of all drug metabolism reactions in human beings; members of the other families are important in the biosynthesis and degradation of steroids, fatty acids, vitamins, and other endogenous compounds. Each individual CYP isoform appears to have a characteristic substrate specificity based on structural features of the substrate; considerable overlap, however, often is present. As a result, two or more CYP isoforms and other drug-metabolizing enzymes often are involved in a drug's overall metabolism, leading to the formation of many primary and secondary metabolites. The various isoforms also have characteristic inhibition and induction profiles, as described later. Additionally, CYP-catalyzed metabolism is often regio- and stereoselective; the latter characteristic may be important if the administered drug is a racemate and the enantiomers have different pharmacological activities. The relative contributions of the various CYP isoforms in the metabolism of drugs is illustrated in Figure 13. CYP3A4 and CYP3A5, which are very similar isoforms, together are involved in the metabolism of about 50% of drugs; moreover, CYP3A is expressed in both the intestinal epithelium and the kidney. It is now recognized that metabolism by CYP3A during absorption through the intestinal enterocyte is a significant factor, along with hepatic first-pass metabolism, in the poor oral bioavailability of many drugs. Isoforms in the CYP2C family and CYP2D6 subfamily also are involved to a large extent in the metabolism of drugs. Although isoforms such as CYP1A1/2, CYP2A6, CYP2B1, and CYP2E1 are not involved to any major extent in the metabolism of therapeutic drugs, they do, however, catalyze the activation of many procarcinogenic environmental chemicals to the ultimate carcinogenic form. Accordingly, they are considered to be important in susceptibility to various cancers, such as tobacco smokingassociated lung cancer.
Other oxidative enzymes such as dehydrogenases and flavin-containing monooxygenases also are capable of catalyzing the metabolism of specific drugs, but, in general, such enzymes are of minor overall importance. Hydrolytic Enzymes The reactions of the major hydrolytic enzymes are illustrated in Table 12. A number of nonspecific esterases and amidases have been identified in the endoplasmic reticulum of human liver, intestine, and other tissues. The alcohol and amine groups exposed following hydrolysis of esters and amides are suitable substrates for conjugation reactions. Microsomal epoxide hydrolase is found in the endoplasmic reticulum of essentially all tissues and is in close proximity to the cytochrome P450 enzymes. Epoxide hydrolase generally is considered a detoxification enzyme, hydrolyzing highly reactive arene oxides generated from cytochrome P450 oxidation reactions to inactive, water-soluble transdihydrodiol metabolites. Protease and peptidase enzymes are widely distributed in many tissues and are involved in the biotransformation of polypeptide drugs. Delivery of such drugs across biological membranes requires the inhibition of these enzymes or the development of stable analogs. Conjugation Reactions Both an activated form of an endogenous compound and an appropriate transferase enzyme are necessary for the formation of a conjugated metabolite. In the case of glucuronidationthe most important conjugation reaction (Figure 13)uridine diphosphate glucuronosyltransferases (UGTs) catalyze the transfer of glucuronic acid to aromatic and aliphatic alcohols, carboxylic acids, amines, and free sulfhydryl groups of both exogenous and endogenous compounds to form O-, N-, and S-glucuronides, respectively. Glucuronidation also is important in the elimination of endogenous steroids, bilirubin, bile acids, and fat-soluble vitamins. The increased water solubility of a glucuronide conjugate promotes its elimination in the urine or bile. Unlike most phase II reactions, which are localized in the cytosol, UGTs are microsomal enzymes. This location facilitates direct access of phase I metabolites formed at the same site. In addition to the liver, UGTs also are found in the intestinal epithelium, kidney, and skin. About 15 human UGTs have been identified, and, based on amino acid similarity (>50% identity), two main families have been categorized. Members of the human UGT1A family are all encoded by a complex gene, and individual isoforms are produced by alternative splicing of 12 promoters/exon 1 with common exons 2 to 5 to produce multiple different proteins. By contrast, UGT2 contains only three subfamilies: 2A, 2B, and 2C. While it appears that individual UGTs have characteristic substrate specificities, there is considerable overlap, so that multiple isoforms may be responsible for formation of a particular glucuronide metabolite. Cytosolic sulfation also is an important conjugation reaction that involves the catalytic transfer by sulfotransferases (STs) of inorganic sulfur from activated 3'-phosphoadenosine-5'-phosphosulfate to the hydroxyl group of phenols and aliphatic alcohols. Therefore, drugs and primary metabolites with a hydroxyl group often form both glucuronide and sulfate metabolites. Two N-acetyltransferases (NAT1 and NAT2) are involved in the acetylation of amines, hydrazines, and sulfonamides. In contrast to most drug conjugates, acetylated metabolites often are less water-soluble than the parent drug, and this may result in crystalluria unless a high urine flow rate is maintained. Factors Affecting Drug Metabolism A hallmark of drug metabolism is a large interindividual variability that often results in marked differences in the extent of metabolism and, as a result, the drug's rate of elimination and other characteristics of its plasma concentrationtime profile. Such variability is a major reason why patients differ in their responses to a standard dose of a drug and it must be considered in optimizing a dosage regimen for a particular individual. A combination of genetic, environmental, and disease-state factors affect drug metabolism, with the relative contribution of each depending on the specific drug. Genetic Variation Advances in molecular biology have shown that genetic diversity is the rule rather than the exception with all proteins, including enzymes that catalyze drug-metabolism reactions. For an increasing number of such enzymes, allelic variants with different catalytic activities from that of the wild-type form have been identified. The differences involve a variety of molecular mechanisms leading to a complete lack of activity, a reduction in catalytic ability, or, in the case of gene duplication, enhanced activity. Furthermore, these traits are generally inherited in an autosomal, Mendelian recessive fashion and, if sufficiently prevalent, result in subpopulations with different drug-metabolizing abilities, i.e., genetic polymorphism. In addition, the frequency of specific allelic variants often varies according to the racial ancestry of the individual. It is possible to phenotype or genotype a person with respect to a particular genetic variant, and it is likely that such characterization will become increasingly useful in individualizing drug therapy, especially for drugs with a narrow therapeutic index. Accumulating evidence also suggests that individual susceptibility to diseases associated with environmental chemicals, such as cancer, may reflect genetic variability in drug-metabolizing enzymes. A number of genetic polymorphisms are present in several cytochrome
P450s that lead to altered drug metabolizing ability. The best characterized
of these is that associated with CYP2D6. About 70 single nucleotide
polymorphisms (SNPs) and other genetic variants of functional importance
have been identified in the CYP2D6 gene, many of which result in an inactive
enzyme while others reduce catalytic activity; gene duplication also occurs.
As a result, four phenotypic subpopulations of metabolizers exist: poor (PM),
intermediate (IM), extensive (EM), and ultrarapid (UM). Some of the variants
are relatively rare, whereas others are more common, and importantly, their
frequency varies according to racial background. For example, 5% to 10% of
Caucasians of European ancestry are PMs, whereas the frequency of this
homozygous phenotype in individuals of Southeast Asian origin is only about
1% to 2%. More than 65 commonly used drugs are metabolized by CYP2D6,
including tricyclic antidepressants, neuroleptic agents, selective serotonin
reuptake inhibitors, some antiarrhythmic agents, CYP2C9 catalyzes the metabolism of some 16 commonly used drugs, including that of warfarin and phenytoin, both of which have a narrow therapeutic index. The two most common allelic CYP2C9 variants have markedly reduced catalytic activity (5% to 12%) compared to the wild-type enzyme. As a consequence, patients who are heterozygous or homozygous for the mutant alleles require a lower anticoagulating dose of warfarin, especially the latter group, relative to homozygous, wild-type individuals. Also, initiating warfarin therapy is more difficult, and there is an increased risk of bleeding complications. Similarly, high plasma concentrations of phenytoin and associated adverse effects occur in patients with variant CYP2C9 alleles. Genetic polymorphism also occurs with CYP2C19, where 8 allelic variants have been identified that result in a catalytically inactive protein. About 3% of Caucasians are phenotypically PMs, whereas the frequency is far higher in Southeast Asians, 13% to 23%. Proton-pump inhibitors such as omeprazole and lansoprazole are among the 18 or so drugs importantly metabolized by CYP2C19 to an extent determined by the gene dose. The efficacy of the recommended 20-mg dose of omeprazole in combination with amoxicillin in eradicating Helicobacter pylori is markedly reduced in patients of the homozygous wild-type genotype compared with the 100% cure rate in homozygous PMs, reflecting differences in the drug's effect on gastric acid secretion. Although CYP3A activity shows marked interindividual variability (>10-fold), no significant functional polymorphisms have been found in the gene's coding region; it is, therefore, likely that unknown regulatory factors primarily determine such variability. Genetic variability also is present with dihydropyrimidine dehydrogenase (DPYD), which is a key enzyme in the metabolism of 5-fluorouracil. Accordingly, there is a marked risk of developing severe drug-induced toxicity in the 1% to 3% of cancer patients treated with this antimetabolite who have substantially reduced DPYD activity compared to the general population. A polymorphism in a conjugating drug-metabolizing enzyme, namely that in NAT2, was one of the first to be found to have a genetic basis some 50 years ago. This isoform is involved in the metabolism of about 16 common drugs including isoniazid, procainamide, dapsone, hydralazine, and caffeine. About 15 allelic variants have been identified, some of which are without functional effect, but others are associated with either reduced or absent catalytic activity. Considerable heterogeneity is present in the worldwide population frequency of these alleles, so that the slow-acetylator phenotype frequency is about 50% in American whites and blacks, 60% to 70% in North Europeans, but only 5% to 10% in Southeast Asians. It has been speculated that acetylator phenotype may be associated with environmental agentinduced disease such as bladder and colorectal cancer; however, definitive evidence is not yet available. Similarly, genetic variability in the catalytic activity of glutathione S-transferases may be linked to individual susceptibility to such diseases. Thiopurine methyltransferase (TPMT) is critically important in the metabolism of 6-mercaptopurine, the active metabolite of azathioprine. As a result, homozygotes for alleles encoding inactive TPMT (0.3% to 1% of the population) predictably exhibit severe pancytopenia if given standard doses of azathioprine; such patients typically can be treated with 10% to 15% of the usual dose. Environmental Determinants The activity of most drug-metabolizing enzymes may be modulated by exposure to certain exogenous compounds. In some instances, this may be a drug, which, if concomitantly administered with a second agent, results in a drug:drug interaction. Additionally, dietary micronutrients and other environmental factors can up- or down-regulate the enzymes, termed induction and inhibition, respectively. Such modulation is thought to be a major contributor to interindividual variability in the metabolism of many drugs. Inhibition of Drug Metabolism A consequence of inhibiting drug-metabolizing enzymes is an increase in the plasma concentration of parent drug and a reduction in that of metabolite, exaggerated and prolonged pharmacological effects, and an increased likelihood of drug-induced toxicity. These changes occur rapidly and with essentially no warning and are most critical for drugs that are extensively metabolized and have a narrow therapeutic index. Knowledge of the cytochrome-P450 isoforms that catalyze the main pathway of metabolism of a drug provides a basis for predicting and understanding inhibition, especially with regard to drug-drug interactions. This is because many inhibitors are more selective for some isoforms than others. Often, inhibition occurs because of competition between two or more substrates for the same active site of the enzyme, the extent of which depends on the relative concentrations of the substrates and their affinities for the enzyme. In certain instances, however, the enzyme may be irreversibly inactivated; for example, the substrate or a metabolite forms a tight complex with the heme iron of cytochrome P450 (cimetidine, ketoconazole) or the heme group may be destroyed (norethindrone, ethinylestradiol). A common mechanism of inhibition for some phase II enzymes is the depletion of necessary cofactors. Inhibition of the CYP3A-catalyzed mechanism is both common and important. Because of the high expression level of CYP3A in the intestinal epithelium and the fact that oral ingestion is the most common route of entry of drugs and environmental agents into the body, inhibition of the isoform's activity at this site is often particularly consequential, even if that in the liver is unaffected. This is because of the potential, large increase in bioavailability associated with the reduction in first-pass metabolism for drugs that usually exhibit this effect to a substantial extent. The antifungal agents ketoconazole and itraconazole, HIV protease inhibitors (especially ritonavir), macrolide antibiotics such as erythromycin and clarithromycin but not azithromycin, are all potent CYP3A inhibitors. Certain calcium channel blockers such as diltiazem, nicardipine, and verapamil also inhibit CYP3A, as does a constituent of grapefruit juice. Many inhibitors of CYP3A also reduce P-glycoprotein function, so that drug-drug interactions may involve a dual mechanism. Also, the disposition of drugs that are not significantly metabolized but are eliminated by P-glycoproteinmediated transport also may be affected by a CYP3A inhibitor. For example, the impaired excretion of digoxin by quinidine and a large number of other unrelated drugs is caused by inhibition of P-glycoprotein. With CYP2D6, quinidine and selective serotonin reuptake inhibitors are potent inhibitors that may produce phenocopying. On the other hand, other drugs are more general inhibitors of cytochrome P450catalyzed metabolism. For example amiodarone, cimetidine (but not ranitidine), paroxitene, and fluoxetine reduce the metabolic activity of several CYP isoforms. Phase I metabolic enzymes other than cytochrome P450 also may be inhibited by drug administration, as exemplified by the potent effect of valproic acid on microsomal epoxide hydrolase, and the inhibition of xanthine oxidase by allopurinol, which can result in life-threatening toxicity in patients concurrently receiving 6-mercaptopurine. Induction of Drug Metabolism Up-regulation of drug- metabolizing activity usually occurs by enhanced gene transcription following prolonged exposure to an inducing agent, although with CYP2E1 stabilization of the protein against degradation is the major mechanism. As a result, the consequences of induction take considerable time to be fully exhibited, c.f., inhibition of metabolism. Moreover, the consequences of induction are an increased rate of metabolism, enhanced oral first-pass metabolism and reduced bioavailability, and a corresponding decrease in the drug's plasma concentration, all factors that reduce drug exposure. By contrast, for drugs that are metabolized to an active or reactive metabolite, induction may be associated with increased drug effects or toxicity, respectively. In some cases, a drug can induce both the metabolism of other compounds and its own metabolism; such autoinduction occurs with the anticonvulsant carbamazepine. In many cases involving induction, the dosage of an affected drug must be increased to maintain the therapeutic effect. This is particularly the case when induction is extensive following administration of a highly effective inducer; in fact, women are advised to use an alternative to oral contraceptives for birth control during rifampin therapy because efficacy cannot be assured. The therapeutic risk associated with metabolic induction is most critical when administration of the inducing agent is stopped while maintaining the same dose of a drug that has been previously given. In this case, as the inducing effect wears off, plasma concentrations of the second drug will rise unless the dose is reduced, with an increase in the potential for adverse effects. Inducers generally are selective for certain CYP subfamilies and
isoforms, but at the same time, multiple other enzymes may be simultaneously
up-regulated through a common molecular mechanism. For example, polycyclic
aromatic hydrocarbons derived from environmental pollutants, cigarette smoke,
and charbroiled meats produce marked induction of the CYP1A subfamily of
enzymes both in the liver and extrahepatically. This involves activation of
the cytosolic arylhydrocarbon receptor (AhR), which interacts with another
regulatory protein, the AhR nuclear translocator (Arnt); the complex
functions as a transcription factor to up-regulate CYP1A expression. In
addition, the expression of phase II enzymes such as UGTs, GSTs, and
NAD(P)H:quinone oxidoreductase are simultaneously increased. A similar type
of receptor mechanism involving the pregnane X receptor (PXR) is involved in
the induction of CYP3A by a wide variety of diverse chemicals, including
drugs such as rifampin and rifabutin, barbiturates and other anticonvulsants,
some glucocorticoids, and even alternative medicines such as Disease Factors Since the liver is the major location of drug-metabolizing enzymes, dysfunction in this organ in patients with hepatitis, alcoholic liver disease, biliary cirrhosis, fatty liver, and hepatocarcinomas potentially can lead to impaired drug metabolism. In general, the severity of the liver damage determines the extent of reduced metabolism; unfortunately, common clinical tests of liver function are of little value in assessing this. Moreover, even in severe cirrhosis, the extent of impairment is only to about 30% to 50% of the activity in non-liver-diseased patients. However, with drugs that undergo substantial hepatic first-pass metabolism, oral bioavailability may be increased two- to fourfold in liver disease which, coupled with the prolonged presence of drugs in the body, increases the risk of exaggerated pharmacological responses and adverse effects. It appears that cytochrome-P450 isoforms are affected to a greater extent by liver disease than are those that catalyze phase II reactions such as glucuronosyltransferases. Severe cardiac failure and shock can result in both decreased perfusion of the liver and impaired metabolism. The best example of this is the almost twofold reduction in lidocaine metabolism in cardiac failure, which also is accompanied by a change in distribution to a similar extent. As a result, the loading and maintenance doses of lidocaine used to treat cardiac arrhythmias in such patients are substantially different from those used in patients without this condition. Age and Sex Functional cytochrome P450 isoforms and to a lesser degree phase II drug-metabolizing enzymes develop early in fetal development, but the levels, even at birth, are lower than those found postnatally. Both phase I and phase II enzymes begin to mature gradually following the first 2 to 4 weeks postpartum, although the pattern of development is variable for the different enzymes. Thus, newborns and infants are able to metabolize drugs relatively efficiently but generally at a slower rate than are adults. An exception to this is the impairment of bilirubin glucuronidation at birth, which contributes to the hyperbilirubinemia of newborns. Full maturity appears to occur in the second decade of life with a subsequent slow decline in function associated with aging. Unfortunately, few generalizations are possible regarding the extent or clinical importance of such age-related changes in an individual patient. This is particularly true for elderly patients who, because of multiple diseases, may be taking a large number of drugs, many of which may produce drug-drug interactions. In addition, increased sensitivity of target organs and impairment of physiological control mechanisms further complicate the use of drugs in the elderly population. Phase I drug-metabolizing enzymes appear to be affected to a greater extent than are those that catalyze phase II reactions. However, the changes are often modest relative to other causes of interindividual variability in metabolism. On the other hand, for drugs exhibiting a high first-pass effect, even a small reduction in metabolizing ability may significantly increase oral bioavailability. Drug use in the elderly, therefore, generally requires moderate reductions in drug dose and awareness of the possibility of exaggerated pharmacodynamic responsiveness. A number of examples indicate that drug treatment and/or responsiveness of men and women may be different for certain drugs. Some sex-related differences in drug-metabolizing activity, especially that catalyzed by CYP3A, also have been noted. However, such differences are minor and unimportant relative to other factors involved in interindividual variability in metabolism. One exception to this generalization is pregnancy, where induction of certain drug-metabolizing enzymes occurs in the second and third trimesters. As a result, drug dosage may have to be increased during this period and returned to its previous level postpartum. This situation is particularly important in the treatment of patients with seizures using phenytoin during their pregnancy. Many oral contraceptive agents also are potent irreversible inhibitors of CYP isoforms through a suicide-inactivation mechanism. |
Clinical Pharmacokinetics
|
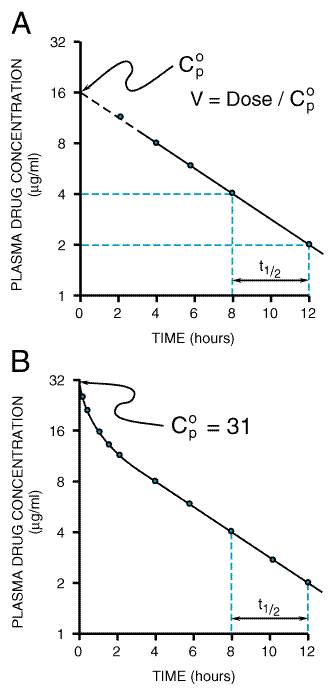
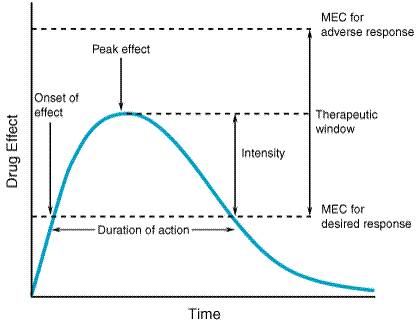
A fundamental hypothesis of clinical pharmacokinetics is that a relationship exists between the pharmacological effects of a drug and an accessible concentration of the drug (e.g., in blood or plasma). This hypothesis has been documented for many drugs, although for some drugs no clear or simple relationship has been found between pharmacological effect and concentration in plasma. In most cases, as depicted in Figure 11, the concentration of drug in the systemic circulation will be related to the concentration of drug at its sites of action. The pharmacological effect that results may be the clinical effect desired, a toxic effect, or, in some cases, an effect unrelated to therapeutic efficacy or toxicity. Clinical pharmacokinetics attempts to provide both a quantitative relationship between dose and effect and a framework with which to interpret measurements of concentrations of drugs in biological fluids. The importance of pharmacokinetics in patient care is based on the improvement in therapeutic efficacy that can be attained by application of its principles when dosage regimens are chosen and modified. The various physiological and pathophysiological variables that dictate adjustment of dosage in individual patients often do so as a result of modification of pharmacokinetic parameters. The four most important parameters are clearance, a measure of the body's efficiency in eliminating drug; volume of distribution, a measure of the apparent space in the body available to contain the drug; elimination half-life, a measure of the rate of removal of drug from the body; and bioavailability, the fraction of drug absorbed as such into the systemic circulation. Of lesser importance are the rates of availability and distribution of the agent. Clearance Clearance is the most important concept that needs to be considered when a rational regimen for long-term drug administration is to be designed. The clinician usually wants to maintain steady-state concentrations of a drug within a therapeutic window associated with therapeutic efficacy and a minimum of toxicity. Assuming complete bioavailability, the steady state will be achieved when the rate of drug elimination equals the rate of drug administration: Dosing rate =CL CSS (11) where CL is clearance from the systemic circulation and Css is the steady-state concentration of drug. Thus, if the desired steady-state concentration of drug in plasma or blood is known, the rate of clearance of drug by the patient will dictate the rate at which the drug should be administered. The concept of clearance is extremely useful in clinical pharmacokinetics, because its value for a particular drug usually is constant over the range of concentrations encountered clinically. This is true because systems for elimination of drugs such as metabolizing enzymes and transporters usually are not saturated, and thus the absolute rate of elimination of the drug is essentially a linear function of its concentration in plasma. A synonymous statement is that the elimination of most drugs follows first-order kineticsa constant fraction of drug in the body is eliminated per unit of time. If mechanisms for elimination of a given drug become saturated, the kinetics approach zero-ordera constant amount of drug is eliminated per unit of time. Under such a circumstance, clearance will vary with the concentration of drug, often according to the following equation: CL = vm/(Km+ C) (12) where Km represents the concentration at which half
of the maximal rate of elimination is reached (in units of mass/volume) and Principles of drug clearance are similar to those of renal physiology, where, for example, creatinine clearance is defined as the rate of elimination of creatinine in the urine relative to its concentration in plasma. At the simplest level, clearance of a drug is its rate of elimination by all routes normalized to the concentration of drug, C, in some biological fluid: CL= rate of elimination/C (13) Thus, when clearance is constant, the rate of drug elimination is directly proportional to drug concentration. It is important to note that clearance does not indicate how much drug is being removed but, rather, the volume of biological fluid such as blood or plasma from which drug would have to be completely removed to account for the elimination. Clearance is expressed as a volume per unit of time. Clearance usually is further defined as blood clearance (CLb), plasma clearance (CLp), or clearance based on the concentration of unbound drug (CLu), depending on the concentration measured (Cb, Cp, or Cu). Clearance by means of various organs of elimination is additive. Elimination of drug may occur as a result of processes that occur in the kidney, liver, and other organs. Division of the rate of elimination by each organ by a concentration of drug (e.g., plasma concentration) will yield the respective clearance by that organ. Added together, these separate clearances will equal systemic clearance: CLrenal+ CLhepatic+ CLother= CL (14) Other routes of elimination could include that in saliva or sweat, secretion into the intestinal tract, and metabolism at other sites. Systemic clearance may be determined at steady state by using Equation (11). For a single dose of a drug with complete bioavailability and first-order kinetics of elimination, systemic clearance may be determined from mass balance and the integration of Equation (13) over time. CL= Dose/AUC (15) where AUC is the total area under the curve that describes the concentration of drug in the systemic circulation as a function of time (from zero to infinity). Examples In Appendix II, the plasma clearance for cephalexin is reported as 4.3 ml min1 kg1, with 90% of the drug excreted unchanged in the urine. For a 70-kg man, the clearance from plasma would be 300 ml/minute, with renal clearance accounting for 90% of this elimination. In other words, the kidney is able to excrete cephalexin at a rate such that it is completely removed (cleared) from approximately 270 ml of plasma per minute. Because clearance usually is assumed to remain constant in a stable patient, the rate of elimination of cephalexin will depend on the concentration of drug in the plasma [Equation (13)]. Propranolol is cleared from the blood at a rate of 16 ml min1 kg1 (or 1120 ml/minute in a 70-kg man), almost exclusively by the liver. Thus, the liver is able to remove the amount of drug contained in 1120 ml of blood per minute. Even though the liver is the dominant organ for elimination, the plasma clearance of some drugs exceeds the rate of plasma (and blood) flow to this organ. Often this is because the drug partitions readily into red blood cells, and the rate of drug delivered to the eliminating organ is considerably higher than suspected from measurement of its concentration in plasma. The relationship between plasma and blood clearance at steady state is given by:
Clearance from the blood, therefore, may be estimated by dividing the plasma clearance by the drug's blood to plasma concentration ratio, obtained from knowledge of the hematocrit (H= 0.45) and the red cell to plasma concentration ratio. In most instances the blood clearance will be less than liver blood flow (1.5 liters/minute) or, if renal excretion also is involved, the sum of the two eliminating organs' blood flows. For example, the plasma clearance of tacrolimus, about 2 liters/minute, is more than twofold higher than the hepatic plasma flow rate and even exceeds the organ's blood flow, despite the fact that the liver is the predominant site of this drug's extensive metabolism. However, after taking into account the extensive distribution of tacrolimus into red cells, its clearance from the blood is only about 63 ml/minute, and it is actually a low- rather than high-clearance drug, as might be interpreted from the plasma clearance value. Sometimes, however, clearance from the blood by metabolism exceeds liver blood flow, and this indicates extrahepatic metabolism. In the case of esmolol (11.9 liters/minute), the blood clearance value is greater than cardiac output, because the drug is efficiently metabolized by esterases present in red blood cells. A further definition of clearance is useful for understanding the effects of pathological and physiological variables on drug elimination, particularly with respect to an individual organ. The rate of presentation of drug to the organ is the product of blood flow (Q) and the arterial drug concentration (CA), and the rate of exit of drug from the organ is the product of blood flow and the venous drug concentration (CV). The difference between these rates at steady state is the rate of drug elimination:
Division of Equation (17) by the concentration of drug entering the organ of elimination, CA, yields an expression for clearance of the drug by the organ in question:
The expression (CACV)/CA in Equation (18) can be referred to as the extraction ratio for the drug (E). Hepatic Clearance The concepts developed in Equation (18) have important implications for drugs that are eliminated by the liver. Consider a drug that is efficiently removed from the blood by hepatic processesmetabolism and/or excretion of drug into the bile. In this instance, the concentration of drug in the blood leaving the liver will be low, the extraction ratio will approach unity, and the clearance of the drug from blood will become limited by hepatic blood flow. Drugs that are cleared efficiently by the liver (e.g., drugs in Appendix II with systemic clearances greater than 6 ml min1 kg1, such as diltiazem, imipramine, lidocaine, morphine, and propranolol) are restricted in their rate of elimination, not by intrahepatic processes, but by the rate at which they can be transported in the blood to the liver. Additional complexities also have been considered. For example, the
equations presented above do not account for drug binding to components of
blood and tissues, nor do they permit an estimation of the intrinsic ability
of the liver to eliminate a drug in the absence of limitations imposed by
blood flow, termed intrinsic clearance. In biochemical terms and under
first-order conditions, intrinsic clearance is a measure of the ratio of the
MichaelisMenten kinetic parameters for the eliminating process, i.e.,
Renal Clearance Renal clearance of a drug results in its appearance as such in the urine; changes in the pharmacokinetic properties of drugs due to renal disease also may be explained in terms of clearance concepts. However, the complications that relate to filtration, active secretion, and reabsorption must be considered. The rate of filtration of a drug depends on the volume of fluid that is filtered in the glomerulus and the unbound concentration of drug in plasma, since drug bound to protein is not filtered. The rate of secretion of drug by the kidney will depend on the drug's intrinsic clearance by the transporters involved in active secretion as affected by the drug's binding to plasma proteins, the degree of saturation of these transporters, and the rate of delivery of the drug to the secretory site. In addition, processes involved in drug reabsorption from the tubular fluid must be considered. The influences of changes in protein binding, blood flow, and the number of functional nephrons are analogous to the examples given above for hepatic elimination. Distribution Volume of Distribution Volume is a second fundamental parameter that is useful in considering processes of drug disposition. The volume of distribution (V) relates the amount of drug in the body to the concentration of drug (C) in the blood or plasma, depending upon the fluid measured. This volume does not necessarily refer to an identifiable physiological volume, but merely to the fluid volume that would be required to contain all of the drug in the body at the same concentration as in the blood or plasma: V= amount of drug in body/C (19) A drug's volume of distribution, therefore, reflects the extent to
which it is present in extravascular tissues. The plasma volume of a typical
70-kg man is 3 liters, blood volume is about 5.5 liters, extracellular fluid
volume outside the plasma is 12 liters, and the volume of total body water is
approximately 42 liters. However, many drugs exhibit volumes of distribution
far in excess of these values. For example, if 500 The volume of distribution may vary widely depending on the relative degrees of binding to plasma and tissue proteins, the partition coefficient of the drug in fat, and so forth. As might be expected, the volume of distribution for a given drug can differ according to patient's age, gender, body composition, and presence of disease. Several volume terms commonly are used to describe drug distribution, and they have been derived in a number of ways. The volume of distribution defined in Equation (19) considers the body as a single homogeneous compartment. In this one-compartment model, all drug administration occurs directly into the central compartment and distribution of drug is instantaneous throughout the volume (V). Clearance of drug from this compartment occurs in a first-order fashion, as defined in Equation (13); that is, the amount of drug eliminated per unit of time depends on the amount (concentration) of drug in the body compartment. Figure 14A and Equation (110) describe the decline of plasma concentration with time for a drug introduced into this compartment.
C= (dose/V) exp(kt) (110) where k is the rate constant for elimination that reflects the fraction of drug removed from the compartment per unit of time. This rate constant is inversely related to the half-life of the drug (k= 0.693/t1/2). The idealized one-compartment model discussed above does not describe the entire time course of the plasma concentration. That is, certain tissue reservoirs can be distinguished from the central compartment, and the drug concentration appears to decay in a manner that can be described by multiple exponential terms (seeFigure 14B). Nevertheless, the one-compartment model is sufficient to apply to most clinical situations for most drugs. Rate of Drug Distribution The multiple exponential decay observed for a drug that is eliminated from the body with first-order kinetics results from differences in the rates at which the drug equilibrates with tissues. The rate of equilibration will depend upon the ratio of the perfusion of the tissue to the partition of drug into the tissue. In many cases, groups of tissues with similar perfusion/partition ratios all equilibrate at essentially the same rate, such that only one apparent phase of distribution (rapid initial fall of concentration, as in Figure 14B) is seen. It is as though the drug starts in a 'central' volume, which consists of plasma and tissue reservoirs that are in rapid equilibrium with it, and distributes to a 'final' volume, at which point concentrations in plasma decrease in a log-linear fashion with a rate constant of k (seeFigure 14B). If the pattern or ratio of blood flows to various tissues changes within an individual or differs among individuals, rates of drug distribution to tissues also will change. However, changes in blood flow also may cause some tissues that were originally in the 'central' volume to equilibrate sufficiently more slowly so as to appear only in the 'final' volume. This means that central volumes will appear to vary with disease states that cause altered regional blood flow. After an intravenous bolus dose, drug concentrations in plasma may be higher in individuals with poor perfusion (e.g., shock) than they would be if perfusion were better. These higher systemic concentrations may, in turn, cause higher concentrations (and greater effects) in tissues such as brain and heart, whose usually high perfusion has not been reduced by the altered hemodynamic state. Thus, the effect of a drug at various sites of action can be variable, depending on perfusion of these sites. Multicompartment Volume Terms Two different terms have been used to describe the volume of distribution for drugs that follow multiple exponential decay. The first, designated Varea, is calculated as the ratio of clearance to the rate of decline of concentration during the elimination (final) phase of the logarithmic concentration versustime curve:
The estimation of this parameter is straightforward, and the volume term may be determined after administration of a single dose of drug by intravenous or oral routes (where the dose used must be corrected for bioavailability). However, another multicompartment volume of distribution may be more useful, especially when the effect of disease states on pharmacokinetics is to be determined. The volume of distribution at steady state (Vss) represents the volume in which a drug would appear to be distributed during steady state if the drug existed throughout that volume at the same concentration as that in the measured fluid (plasma or blood). Following intravenous dosing, estimation of Vss is more complicated than Equation (111), but feasible (Benet and Galeazzi, 1979). It is more difficult to estimate Vss following oral dosing. Although Varea is a convenient and easily calculated parameter, it varies when the rate constant for drug elimination changes, even when there has been no change in the distribution space. This is because the terminal rate of decline of the concentration of drug in blood or plasma depends not only on clearance but also on the rates of distribution of drug between the 'central' and 'final' volumes. Vss does not suffer from this disadvantage. When using pharmacokinetics to make drug dosing decisions, the differences between Varea and Vss usually are not clinically significant. Nonetheless, both are quoted in the table of pharmacokinetic data in Appendix II, depending upon availability in the published literature. Half-Life The half-life (t1/2) is the time it takes for the plasma concentration or the amount of drug in the body to be reduced by 50%. For the simplest case, the one-compartment model (Figure 14A), half-life may be determined readily and used to make decisions about drug dosage. However, as indicated in Figure 14B, drug concentrations in plasma often follow a multiexponential pattern of decline; two or more half-life terms may thus be calculated. In the past, the half-life that was usually reported corresponded to the terminal log-linear phase of elimination. However, as greater analytical sensitivity has been achieved, the lower concentrations measured appeared to yield longer and longer terminal half-lives. For example, a terminal half-life of 53 hours is observed for gentamicin (versus the more clinically relevant 2- to 3-hour value in Appendix II), and biliary cycling is probably responsible for the 120-hour terminal value for indomethacin (as compared with the 2.4-hour half-life listed in Appendix II). The relevance of a particular half-life may be defined in terms of the fraction of the clearance and volume of distribution that is related to each half-life and whether plasma concentrations or amounts of drug in the body are best related to measures of response. The single half-life values given for each drug in Appendix II are chosen to represent the most clinically relevant half-life. Early studies of pharmacokinetic properties of drugs in disease were compromised by their reliance on half-life as the sole measure of alterations of drug disposition. It is now appreciated that half-life is a derived parameter that changes as a function of both clearance and volume of distribution. A useful approximate relationship between the clinically relevant half-life, clearance, and volume of distribution at steady state is given by: t Clearance is the measure of the body's ability to eliminate a drug; thus, as clearance decreases, due to a disease process, for example, half-life would be expected to increase. However, this reciprocal relationship is valid only when the disease does not change the volume of distribution. For example, the half-life of diazepam increases with increasing age; however, it is not clearance that changes as a function of age, but the volume of distribution (Klotz et al., 1975). Similarly, changes in protein binding of the drug may affect its clearance as well as its volume of distribution, leading to unpredictable changes in half-life as a function of disease. The half-life of tolbutamide, for example, decreases in patients with acute viral hepatitis, exactly the opposite from what one might expect. The disease alters the drug's protein binding in both plasma and tissues, causing no change in volume of distribution but an increase in clearance, because higher concentrations of unbound drug are present (Williams et al., 1977). Although it can be a poor index of drug elimination, half-life does provide a good indication of the time required to reach steady state after a dosage regimen is initiated or changed (i.e., four half-lives to reach approximately 94% of a new steady state), the time for a drug to be removed from the body, and a means to estimate the appropriate dosing interval (see below). Steady State Equation (11) indicates that a steady-state concentration eventually will be achieved when a drug is administered at a constant rate. At this point, drug elimination [the product of clearance and concentration; Equation (13)] will equal the rate of drug availability. This concept also extends to intermittent dosage (e.g., 250 mg of drug every 8 hours). During each interdose interval, the concentration of drug rises and falls. At steady state, the entire cycle is repeated identically in each interval. Equation (11) still applies for intermittent dosing, but it now describes the average drug concentration (Css) during an interdose interval. Steady-state dosing is illustrated in Figure 15.
Extent and Rate of Bioavailability Bioavailability It is important to distinguish between the rate and extent of drug absorption and the amount of drug that ultimately reaches the systemic circulation, as discussed above. The amount of the drug that reaches the systemic circulation depends not only on the administered dose but also on the fraction of the dose, F, which is absorbed and escapes any first-pass elimination. This fraction often is called bioavailability. Reasons for incomplete absorption have been discussed above. Also, as noted previously, if the drug is metabolized in the intestinal epithelium or the liver, or excreted in bile, some of the active drug absorbed from the gastrointestinal tract will be eliminated before it can reach the general circulation and be distributed to its sites of action. Knowing the extraction ratio (EH) for a drug across the liver [seeEquation (18)], it is possible to predict the maximum oral availability (Fmax), assuming hepatic elimination follows first-order processes: Fmax= 1 EH= 1 (CLhepatic/Qhepatic) Thus, if the hepatic blood clearance for the drug is large relative to hepatic blood flow, the extent of availability will be low when it is given orally (e.g., lidocaine). This reduction in availability is a function of the physiological site from which absorption takes place, and no modification of dosage form will improve the availability under conditions of linear kinetics. Incomplete absorption and/or intestinal metabolism following oral dosing will, in practice, reduce this predicted maximal value of F. When drugs are administered by a route that is subject to first-pass loss, the equations presented previously that contain the terms dose or dosing rate [Equations (11), (15), (110), and (111)] also must include the bioavailability term F, such that the available dose or dosing rate is used. For example, Equation (11) is modified to: F dosing rate =CL CSS (114) Rate of Absorption Although the rate of drug absorption does not, in general, influence the average steady-state concentration of the drug in plasma, it may still influence drug therapy. If a drug is absorbed rapidly (e.g., a dose given as an intravenous bolus) and has a small 'central' volume, the concentration of drug initially will be high. It will then fall as the drug is distributed to its 'final' (larger) volume (seeFigure 14B). If the same drug is absorbed more slowly (e.g., by slow infusion), it will be distributed while it is being given, and peak concentrations will be lower and will occur later. Controlled-release preparations are designed to provide a slow and sustained rate of absorption in order to produce a less fluctuating plasma concentrationtime profile during the dosage interval compared to more immediate-release formulations. A given drug may act to produce both desirable and undesirable effects at several sites in the body, and the rates of distribution of drug to these sites may not be the same. The relative intensities of these different effects of a drug may thus vary transiently when its rate of administration is changed. Nonlinear Pharmacokinetics Nonlinearity in pharmacokinetics (i.e., changes in such parameters as clearance, volume of distribution, and half-life as a function of dose or concentration of drug) usually is due to saturation of protein binding, hepatic metabolism, or active renal transport of the drug. Saturable Protein Binding As the molar concentration of drug increases, the unbound fraction eventually also must increase (as all binding sites become saturated). This usually occurs only when drug concentrations in plasma are in the range of tens to hundreds of micrograms per milliliter. For a drug that is metabolized by the liver with a low intrinsic clearance/extraction ratio, saturation of plasma-protein binding will cause both V and clearance to increase as drug concentrations increase; half-life may thus remain constant [seeEquation (112)]. For such a drug, Css will not increase linearly as the rate of drug administration is increased. For drugs that are cleared with high intrinsic clearances/extraction ratios, Css can remain linearly proportional to the rate of drug administration. In this case, hepatic clearance would not change, and the increase in V would increase the half-time of disappearance by reducing the fraction of the total drug in the body that is delivered to the liver per unit of time. Most drugs fall between these two extremes, and the effects of nonlinear protein binding may be difficult to predict. Saturable Elimination In this situation, the MichaelisMenten equation [Equation (12)] usually describes the nonlinearity. All active processes are undoubtedly saturable, but they will appear to be linear if values of drug concentrations encountered in practice are much less than Km. When they exceed Km, nonlinear kinetics is observed. The major consequences of saturation of metabolism or transport are the opposite of those for saturation of protein binding. When both conditions are present simultaneously, they may virtually cancel each others' effects, and surprisingly linear kinetics may result; this occurs over a certain range of concentrations for salicylic acid. Saturable metabolism causes oral first-pass metabolism to be less than expected (higher F), and there is a greater fractional increase in Css than the corresponding fractional increase in the rate of drug administration. The latter can be seen most easily by substituting Equation (12) into Equation (11) and solving for the steady-state concentration:
As the
dosing rate approaches the maximal elimination rate ( Phenytoin provides an example of a drug for which metabolism becomes
saturated in the therapeutic range of concentrations (seeAppendix II).
Km (5 to 10 mg per liter) is typically near the lower end
of the therapeutic range (10 to 20 mg per liter). For some individuals,
especially children, Km may be as low as 1 mg per liter.
If, for such an individual, the target concentration is 15 mg per liter, and
this is attained at a dosing rate of 300 mg per day, then, from Equation
(115), Design and Optimization of Dosage Regimens Following administration of a dose of drug, its effects usually show a characteristic temporal pattern (Figure 16). Onset of the effect is preceded by a lag period, after which the magnitude of the effect increases to a maximum and then declines; if a further dose is not administered, the effect eventually disappears. This time-course reflects changes in the drug's concentration as determined by the pharmacokinetics of its absorption, distribution, and elimination. Accordingly, the intensity of a drug's effect is related to its concentration above a minimum effective concentration, whereas the duration of this effect is a reflection of the length of time the drug level is above this value. These considerations, in general, apply to both desired and undesired (adverse) drug effects and, as a result, a therapeutic window exists reflecting a concentration range that provides efficacy without unacceptable toxicity. Similar considerations apply after multiple dosing associated with long-term therapy; therefore they determine the amount and frequency of drug administration to achieve an optimal therapeutic effect. In general, the lower limit of the therapeutic range appears to be approximately equal to the drug concentration that produces about half of the greatest possible therapeutic effect, and the upper limit of the therapeutic range is such that no more than 5% to 10% of patients will experience a toxic effect. For some drugs, this may mean that the upper limit of the range is no more than twice the lower limit. Of course, these figures can be highly variable, and some patients may benefit greatly from drug concentrations that exceed the therapeutic range, while others may suffer significant toxicity at much lower values.
For a
limited number of drugs, some effect of the drug is easily measured (e.g.,
blood pressure, blood glucose), and this can be used to optimize dosage,
using a trial-and-error approach. Even in this ideal case, certain
quantitative issues arise, such as how often to change dosage and by how
much. These usually can be settled with simple rules of thumb based on the
principles discussed (e.g., change dosage by no more than 50% and no
more often than every three to four half-lives). Alternatively, some drugs
have very little dose-related toxicity, and maximum efficacy is usually
desired. For these drugs, doses well in excess of the average required will
both ensure efficacy (if this is possible) and prolong drug action. Such a
'maximal dose' strategy typically is used for penicillins and most For many drugs, however, the effects are difficult to measure (or the drug is given for prophylaxis), toxicity and lack of efficacy are both potential dangers, and/or the therapeutic index is narrow. In these circumstances, doses must be titrated carefully, and drug dosage is limited by toxicity rather than efficacy. Thus, the therapeutic goal is to maintain steady-state drug levels within the therapeutic window. For most drugs, the actual concentrations associated with this desired range are not and need not be known. It is sufficient to understand that efficacy and toxicity are generally concentration-dependent, and how drug dosage and frequency of administration affect the drug level. However, for a small number of drugs, where there is a small (two- to threefold) difference between concentrations resulting in efficacy and toxicity (e.g., digoxin, theophylline, lidocaine, aminoglycosides, cyclosporine, and anticonvulsants), a plasma-concentration range associated with effective therapy has been defined. In this case, a target level strategy is reasonable, wherein a desired (target) steady-state concentration of the drug (usually in plasma) associated with efficacy and minimal toxicity is chosen, and a dosage is computed that is expected to achieve this value. Drug concentrations are subsequently measured, and dosage is adjusted if necessary to approximate the target more closely (see alsoChapter 3: Principles of Therapeutics). Maintenance Dose In most clinical situations, drugs are administered in a series of repetitive doses or as a continuous infusion to maintain a steady-state concentration of drug associated with the therapeutic window. Thus, calculation of the appropriate maintenance dosage is a primary goal. To maintain the chosen steady-state or target concentration, the rate of drug administration is adjusted such that the rate of input equals the rate of loss. This relationship was defined previously in Equations (11) and (114) and is expressed here in terms of the desired target concentration: Dosing rate = target Cp CL/F (116) If the clinician chooses the desired concentration of drug in plasma and knows the clearance and bioavailability for that drug in a particular patient, the appropriate dose and dosing interval can be calculated. Example Oral digoxin is to be used as a maintenance dose to gradually 'digitalize' a 69-kg patient with congestive heart failure. A steady-state plasma concentration of 1.5 ng/ml is selected as an appropriate target. Based on the fact that the patient's creatinine clearance (CLCR) is 100 ml/min, digoxin's clearance may be estimated from data in Appendix II.
Equation (116) is then used to calculate an appropriate dosing rate knowing that the oral bioavailability of digoxin is 70% (F= 0.7)
In practice, the dose rate would be rounded to the closest dosage size, either 0.375 mg/24 hr, which would result in a steady- state plasma concentration of 1.65 ng/ml (1.5 x 375/340), or 0.25 mg/24 hr, which would provide a value of 1.10 ng/ml (1.5 x Dosing Interval for Intermittent Dosage In general, marked fluctuations in drug concentrations between doses are not desirable. If absorption and distribution were instantaneous, fluctuation of drug concentrations between doses would be governed entirely by the drug's elimination half-life. If the dosing interval (T) was chosen to be equal to the half-life, then the total fluctuation would be twofold; this is often a tolerable variation. Pharmacodynamic considerations modify this. If a drug is relatively nontoxic, such that a concentration many times that necessary for therapy can be tolerated easily, the maximal dose strategy can be used, and the dosing interval can be much longer than the elimination half-life (for convenience). The half-life of amoxicillin is about 2 hours, but it is often given in large doses every 8 or 12 hours. For some drugs with a narrow therapeutic range, it may be important to estimate the maximal and minimal concentrations that will occur for a particular dosing interval. The minimal steady-state concentration Css, min may be reasonably determined by the use of Equation (117):
where k equals 0.693 divided by the clinically relevant plasma half-life and T is the dosing interval. The term exp(kT) is, in fact, the fraction of the last dose (corrected for bioavailability) that remains in the body at the end of a dosing interval. For drugs that follow multiexponential kinetics and that are administered orally, the estimation of the maximal steady-state concentration Css, max involves a complicated set of exponential constants for distribution and absorption. If these terms are ignored for multiple oral dosing, one may easily predict a maximal steady-state concentration by omitting the exp(kT) term in the numerator of Equation (117) [see Equation (118), below]. Because of the approximation, the predicted maximal concentration from Equation (118) will be greater than that actually observed. Example In the patient with congestive heart failure discussed above, an oral maintenance dosing of 0.375 mg/24 hr of digoxin was calculated to achieve an average plasma concentration of 1.65 ng/ml during the dosage interval. Digoxin has a narrow therapeutic index, and plasma levels between 0.8 and 2.0 ng/ml are usually associated with efficacy and minimal toxicity. It is, therefore, important to know what maximum and minimum plasma concentrations would be predicted with the above regimen. This first requires estimation of digoxin's volume of distribution based on data in Appendix II.
Combining this value with that of digoxin's clearance provides an estimate of digoxin's elimination half-life in the patient [Equations (11) through (112)].
Accordingly, the fractional rate constant of elimination is equal to 0.01136 hr1 (0.693/61 hr). Maximum and minimum digoxin plasma concentrations may then be predicted depending upon the dosage interval. If this were every 2 days:
Accordingly, the plasma concentrations would fluctuate about twofold, consistent with the similarity of the dosage interval to digoxin's half-life. Also, the peak concentration would be above the upper value of the therapeutic range, exposing the patient to possible adverse effects, and at the end of the dosing interval the concentration would be above but close to the lower limit. By using the same dosing rate but decreasing the frequency of dosing, a much smoother plasma concentration versus time profile would be obtained while still maintaining an average steady-state value of 1.65 ng/ml. For example, with a dose every 24 hours, the predicted maximum and minimum plasma concentrations would be 1.90 and 1.44 ng/ml, respectively, which are in the upper portion of the therapeutic window. By contrast, administering a more conservative dosing rate of 0.25 every 24 hours would produce peak and trough values of 1.26 and 0.96 ng/ml, respectively, that would be associated with a steady-state value of 1.10 ng/ml. Of course the clinician must balance the problem of compliance with regimens that involve frequent dosage against the problem of periods when the patient may be subjected to concentrations of the drug that could be too high or too low. Loading Dose The 'loading dose' is one or a series of doses that may be given at the onset of therapy with the aim of achieving the target concentration rapidly. The appropriate magnitude for the loading dose is: Loading dose = target Cp VSS/F (120) A loading dose may be desirable if the time required to attain steady state by the administration of drug at a constant rate (four elimination half-lives) is long relative to the temporal demands of the condition being treated. For example, the half-life of lidocaine is usually 1 to 2 hours. Arrhythmias encountered after myocardial infarction obviously may be life threatening, and one cannot wait 4 to 8 hours to achieve a therapeutic concentration of lidocaine by infusion of the drug at the rate required to attain this concentration. Hence, use of a loading dose of lidocaine in the coronary care unit is standard. The use of a loading dose also has significant disadvantages. First, the particularly sensitive individual may be exposed abruptly to a toxic concentration of a drug. Moreover, if the drug involved has a long half-life, it will take a long time for the concentration to fall if the level achieved was excessive. Loading doses tend to be large, and they are often given parenterally and rapidly; this can be particularly dangerous if toxic effects occur as a result of actions of the drug at sites that are in rapid equilibrium with plasma. This occurs because the loading dose calculated on the basis of Vss subsequent to drug distribution is initially constrained within the initial and smaller 'central' volume of distribution. It is, therefore, usually advisable to divide the loading dose into a number of smaller fractional doses that are administered over a period of time. Alternatively, the loading dose should be administered as a continuous intravenous infusion over a period of time. Ideally this should be given in an exponentially decreasing fashion to mirror the concomitant accumulation of the maintenance dose of the drug, and this is now technically feasible using computerized infusion pumps. Example 'Digitalization,' in the patient described above, is gradual if only a maintenance dose is administered (for at least 10 days based on a half-life of 61 hours). A more rapid response could be obtained (if deemed necessary by the physician; seeChapter 34: Pharmacological Treatment of Heart Failure) by using a loading dose strategy and Equation (120):
To avoid toxicity, this oral loading dose, which also could be intravenously administered, would be given as an initial 0.5-mg dose followed by a 0.25-mg dose at 6 to 8 hours later along with careful monitoring of the patient. It also would be prudent to give the final 0.25-mg fractional dose, if necessary, in two 0.125-mg divided doses separated by 6 to 8 hours to avoid overdigitalization, particularly if there were a plan to initiate an oral maintenance dose within 24 hours of beginning digoxin therapy. Individualizing Dosage A rational dosage regimen is based on knowledge of F, CL, Vss, and t1/2, and some information about rates of absorption and distribution of the drug. Recommended dosage regimens generally are designed for an 'average' patient; usual values for the important determining parameters and appropriate adjustments that may be necessitated by disease or other factors are presented in Appendix II. This 'one size fits all' approach, however, overlooks the considerable and unpredictable interpatient variability that usually is present in these pharmacokinetic parameters. For many drugs, one standard deviation in the values observed for F, CL, and Vss is about 20%, 50%, and 30%, respectively. This means that 95% of the time the Css that is achieved will be between 35% and 270% of the target; this is an unacceptably wide range for a drug with a low therapeutic index. Individualization of the dosage regimen to a particular patient is, therefore, critical for optimal therapy. The pharmacokinetic principles, described above, provide a basis for modifying the dosage regimen to obtain a desired degree of efficacy with a minimum of unacceptable adverse effects. In situations where the drug's plasma concentration can be measured and related to the therapeutic window, additional guidance for dosage modification is obtained. Such measurement and adjustment are appropriate for many drugs with low therapeutic indices (e.g., cardiac glycosides, antiarrhythmic agents, anticonvulsants, theophylline, and others). Therapeutic Drug Monitoring The major use of measured concentrations of drugs (at steady state) is to refine the estimate of CL/F for the patient being treated [using Equation (114) as rearranged below]: CL/F(patient) = dosing rate/CSS(measured) (121) The new estimate of CL/F can be used in Equation (116) to adjust the maintenance dose to achieve the desired target concentration. Certain practical details and pitfalls associated with therapeutic drug monitoring should be kept in mind. The first of these relates to the time of sampling for measurement of the drug concentration. If intermittent dosing is used, when during a dosing interval should samples be taken? It is necessary to distinguish between two possible uses of measured drug concentrations to understand the possible answers. A concentration of drug measured in a sample taken at virtually any time during the dosing interval will provide information that may aid in the assessment of drug toxicity. This is one type of therapeutic drug monitoring. It should be stressed, however, that such use of a measured concentration of drug is fraught with difficulties because of interindividual variability in sensitivity to the drug. When there is a question of toxicity, the drug concentration can be no more than just one of many items that serve to interpret the clinical situation. Changes in the effects of drugs may be delayed relative to changes in plasma concentration because of a slow rate of distribution or pharmacodynamic factors. Concentrations of digoxin, for example, regularly exceed 2 ng/ml (a potentially toxic value) shortly after an oral dose, yet these peak concentrations do not cause toxicity; indeed, they occur well before peak effects. Thus, concentrations of drugs in samples obtained shortly after administration can be uninformative or even misleading. When concentrations of drugs are used for purposes of adjusting dosage regimens, samples obtained shortly after administration of a dose are almost invariably misleading. The purpose of sampling during supposed steady state is to modify the estimate of CL/F and thus the choice of dosage. Early postabsorptive concentrations do not reflect clearance; they are determined primarily by the rate of absorption, the 'central' (rather than the steady-state) volume of distribution, and the rate of distribution, all of which are pharmacokinetic features of virtually no relevance in choosing the long-term maintenance dosage. When the goal of measurement is adjustment of dosage, the sample should be taken well after the previous doseas a rule of thumb just before the next planned dose, when the concentration is at its minimum. There is an exception to this approach: some drugs are nearly completely eliminated between doses and act only during the initial portion of each dosing interval. If it is questionable whether or not efficacious concentrations of such drugs are being achieved, a sample taken shortly after a dose may be helpful. On the other hand, if a concern is whether or not low clearance (as in renal failure) may cause accumulation of drug, concentrations measured just before the next dose will reveal such accumulation and are considerably more useful for this purpose than is knowledge of the maximal concentration. For such drugs, determination of both maximal and minimal concentrations is thus recommended. A second important aspect of the timing of sampling is its relationship to the beginning of the maintenance dosage regimen. When constant dosage is given, steady state is reached only after four half-lives have passed. If a sample is obtained too soon after dosage is begun, it will not accurately reflect this state and the drug's clearance. Yet, for toxic drugs, if sampling is delayed until steady state is ensured, the damage may have been done. Some simple guidelines can be offered. When it is important to maintain careful control of concentrations, the first sample should be taken after two half-lives (as calculated and expected for the patient), assuming no loading dose has been given. If the concentration already exceeds 90% of the eventual expected mean steady-state concentration, the dosage rate should be halved, another sample obtained in another two (supposed) half-lives, and the dosage halved again if this sample exceeds the target. If the first concentration is not too high, the initial rate of dosage is continued; even if the concentration is lower than expected, it is usually reasonable to await the attainment of steady state in another two estimated half-lives and then proceed to adjust dosage as described above. If dosage is intermittent, there is a third concern with the time at which samples are obtained for determination of drug concentrations. If the sample has been obtained just prior to the next dose, as recommended, concentration will be a minimal value, not the mean. However, as discussed above, the estimated mean concentration may be calculated by using Equation (114). If a drug follows first-order kinetics, the average, minimum, and maximum concentrations at steady state are linearly related to dose and dosing rate [seeEquations (114), (117), and (118)]. Therefore, the ratio between the measured and the desired concentrations can be used to adjust the dose, consistent with available dosage sizes:
In the previously described patient given 0.375 mg digoxin every 24 hours, for example, if the measured steady-state concentration was found to be 1.65 ng/ml rather than a desired level of 1.3 ng/ml, an appropriate, practical change in the dosage regimen would be to reduce the daily dose to 0.25 mg digoxin.
Compliance Ultimately, therapeutic success is dependent on the patient's actually taking the drug according to the prescribed dosage regimen'Drugs don't work if you don't take them.' Noncompliance with the dosing schedule is a major and often unappreciated reason for therapeutic failure, especially in the long-term treatment of disease using antihypertensive, antiretroviral, and anticonvulsant agents. When no special efforts are made to address this issue, only about 50% of patients follow the prescribed dosage regimen in a reasonably satisfactory fashion; approximately one-third only partly comply; and about 1 in 6 patients is essentially noncompliant. Missed doses are more common than too many doses. The number of drugs does not appear to be as important as the number of times a day doses must be remembered (Farmer, 1999). Reducing the number of required dosing occasions will improve adherence to a prescribed dosage regimen. Equally important is the need to involve patients in the responsibility for their own health, using a variety of strategies based on improved communication regarding the nature of the disease and the overall therapeutic plan. Acknowledgment The author wishes to acknowledge Drs. Leslie Z. Benet, Deanna L. Kroetz, and Lewis B. Sheiner, authors of this chapter in the ninth edition of Goodman and Gilman's The Pharmacological Basis of Therapeutics, some of whose text has been retained in this edition. |
Chapter 2. Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect
Overview
|
This chapter provides an introduction to the concept of receptors, the structural and functional families of receptors, and the interplay among the diverse signaling pathways activated by receptor occupancy. These introductory concepts are amplified in subsequent chapters detailing the structure and function of receptors for individual drug groups. The latter part of the chapter presents means for quantifying receptor activation by agonists and blockade by antagonists. The functional relevance of partial agonists and inverse antagonists also is described as a prelude to the intentional development of mechanistically diverse drugs via classical or new combinatorial strategies. |
Pharmacodynamics: Introduction
|
Pharmacodynamics can be defined as the study of the biochemical and physiological effects of drugs and their mechanisms of action. The objectives of the analysis of drug action are to delineate the chemical or physical interactions between drug and target cell and to characterize the full sequence and scope of actions of each drug. Such a complete analysis provides the basis for both the rational therapeutic use of a drug and the design of new and superior therapeutic agents. Basic research in pharmacodynamics also provides fundamental insights into biochemical and physiological regulation. |
Mechanisms of Drug Action
|
The effects of most drugs result from their interaction with macromolecular components of the organism. These interactions alter the function of the pertinent component and thereby initiate the biochemical and physiological changes that are characteristic of the response to the drug. The term receptor denotes the component of the organism with which the chemical agent was presumed to interact. The statement that the receptor for a drug can be any functional macromolecular component of the organism has several fundamental corollaries. One is that a drug potentially is capable of altering the rate at which any bodily function proceeds. Another is that drugs do not create effects, but instead modulate intrinsic physiological functions. Drug Receptors At least from a numerical standpoint, proteins form the most important class of drug receptors. Examples are the receptors for hormones, growth factors, and neurotransmitters, the enzymes of crucial metabolic or regulatory pathways (e.g., dihydrofolate reductase, acetylcholines-terase), proteins involved in transport processes (e.g., Na+,K+-ATPase), or structural proteins (e.g., tubulin). Specific binding properties of other cellular constituents also can be exploited. Thus, nucleic acids are important drug receptors, particularly for cancer chemotherapeutic agents. A particularly important group of drug receptors are proteins that normally serve as receptors for endogenous regulatory ligands (e.g., hormones, neurotransmitters). Many drugs act on such physiological receptors and are often particularly selective, because physiological receptors are specialized to recognize and respond to individual signaling molecules with great selectivity. Drugs that bind to physiological receptors and mimic the regulatory effects of the endogenous signaling compounds are termed agonists. Other drugs bind to receptors without regulatory effect, but their binding blocks the binding of the endogenous agonist. Such compounds, which may still produce useful effects by inhibiting the action of an agonist (e.g., by competition for agonist binding sites), are termed antagonists. Agents that are only partly as effective as agonists are termed partial agonists, and those that stabilize the receptor in its inactive conformation are termed inverse agonists. (See 'Quantitation of DrugReceptor Interactions and Elicited Effect.') The binding of drugs to receptors can involve all known types of interactionsionic, hydrogen bonding, hydrophobic, van der Waals, and covalent. In most interactions between drugs and receptors, it is likely that bonds of multiple types are important. If binding is covalent, the duration of drug action is frequently, but not necessarily, prolonged. Noncovalent interactions of high affinity also may appear to be essentially irreversible. StructureActivity Relationship and Drug Design Both the affinity of a drug for its receptor and its intrinsic activity are determined by its chemical structure. This relationship is frequently quite stringent. Relatively minor modifications in the drug molecule may result in major changes in pharmacological properties. Exploitation of structureactivity relationships has on many occasions led to the synthesis of valuable therapeutic agents. Because changes in molecular configuration need not alter all actions and effects of a drug equally, it is sometimes possible to develop a congener with a more favorable ratio of therapeutic to toxic effects, enhanced selectivity among different cells or tissues, or more acceptable secondary characteristics than those of the parent drug. Therapeutically useful antagonists of hormones or neurotransmitters have been developed by chemical modification of the structure of the physiological agonist. Minor modifications of structure also can have profound effects on the pharmacokinetic properties of drugs. Given adequate information about both the molecular structures and the pharmacological activities of a relatively large group of congeners, it should be possible to identify those chemical properties that are required for optimal action at the receptorsize, shape, the position and orientation of charged groups or hydrogen bond donors, and so on. Advances in computational chemistry, structural analysis of organic compounds, and the biochemical measurement of the primary actions of drugs at their receptors have enriched the quantitation of structureactivity relationships and its use in drug design (Kuntz, 1992; Schreiber, 1992). The importance of specific drugreceptor interactions can be evaluated further by analyzing the responsiveness of receptors that have been selectively mutated at individual amino acid residues. Such information is increasingly allowing the optimization or design of chemicals that can bind to a receptor with improved affinity, selectivity, or regulatory effect. Similar structure-based approaches also are used to improve pharmacokinetic properties of drugs (see Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination). Knowledge of the structures of receptors and of drug-receptor complexes, determined at atomic resolution by X-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy, is even more helpful in the design of ligands. Ironically, advances in molecular biology that contribute to structure-motivated drug design also have spawned powerful but entirely random searches for new drugs. In this approach, huge libraries of randomly synthesized chemicals are generated either by synthetic chemistry or by genetically engineered microbes. A library then is screened for pharmacologically active agents using mammalian cells or microorganisms that have been engineered to express the receptor of therapeutic interest and the associated biochemical machinery necessary for detection of the receptor's response. Active compounds initially discovered by such random screens then can be modified and improved using knowledge of their structurefunction relationships. Cellular Sites of Drug Action Because drugs act by altering the activities of their receptors, the sites at which a drug acts and the extent of its action are determined by the location and functional capacity of its receptors. Selective localization of drug action within the organism is therefore not necessarily dependent upon selective distribution of the drug. If a drug acts on a receptor that serves functions common to most cells, its effects will be widespread. If the function is a vital one, the drug may be particularly difficult or dangerous to use. Nevertheless, such a drug may be clinically important. Digitalis glycosides, important in the treatment of heart failure, are potent inhibitors of an ion transport process that is vital to most cells. As such, they can cause widespread toxicity, and their margin of safety is dangerously low. Other examples could be cited, particularly in the area of cancer chemotherapy. If a drug interacts with receptors that are unique to only a few types of differentiated cells, its effects are more specific. Hypothetically, the ideal drug would cause its therapeutic effect by such an action. Side effects would be minimized, but toxicity might not be. If the differentiated function were a vital one, this type of drug also could be very dangerous. Some of the most lethal chemical agents known (e.g., botulinum toxin) show such specificity. Note also that, even if the primary action of a drug is localized, the consequent physiological effects of the drug may be widespread. Receptors for Physiological Regulatory Molecules The term receptor has been used operationally to denote any cellular macromolecule to which a drug binds to initiate its effects. Among the most important drug receptors are cellular proteins, whose normal function is to act as receptors for endogenous regulatory ligandsparticularly hormones, growth factors, and neurotransmitters. The function of such physiological receptors consists of binding the appropriate ligand and, in response, propagating its regulatory signal in the target cell. Identification of the two functions of a receptor, ligand binding and message propagation, correctly suggests the existence of functional domains within the receptor: a ligand-binding domain and an effector domain. The structure and function of these domains often can be deduced from high-resolution structures of receptor proteins and/or by analysis of the behavior of intentionally mutated receptors. Increasingly, the mechanism of intramolecular coupling of ligand binding with functional activation also can be learned. The biological importance of these functional domains is further indicated by the evolution both of different receptors for diverse ligands that act by similar biochemical mechanisms and of multiple receptors for a single ligand that act by unrelated mechanisms. The regulatory actions of a receptor may be exerted directly on its cellular target(s), effector protein(s), or may be conveyed by intermediary cellular signaling molecules called transducers. The receptor, its cellular target, and any intermediary molecules are referred to as a receptor-effector system or signal transduction pathway. Frequently, the proximal cellular effector protein is not the ultimate physiological target, but rather is an enzyme or transport protein that creates, moves, or degrades a small molecule metabolite or ion known as a second messenger. Second messengers can diffuse through a cell and convey information to a wide variety of targets, which can respond simultaneously to the output of a single receptor. Receptors and their associated effector and transducer proteins also act as integrators of information as they coordinate signals from multiple ligands with each other and with the metabolic activities of the cell (see below). This integrative function is particularly evident when one considers that the different receptors for scores of chemically unrelated ligands utilize relatively few biochemical mechanisms to exert their regulatory functions, and that even these few pathways may share common signaling molecules. An important property of physiological receptors, which also makes them excellent targets for drugs, is that they act catalytically and hence are biochemical signal amplifiers. The catalytic nature of receptors is obvious when the receptor itself is an enzyme, but all known physiological receptors are formally catalysts. For example, when a single agonist molecule binds to a receptor that is an ion channel, many ions flow through the channel. Similarly, a single steroid hormone molecule binds to its receptor and initiates the transcription of many copies of specific mRNAs, which in turn can give rise to multiple copies of a single protein. A Framework for Considering Agonist Activity If two drugs bind to the same receptor at the same site, why can one be an agonist and the other an antagonist? This question, more broadly stated, is also central to understanding the dynamics of protein structure and proteinligand interactions. At a detailed level, the atomic interactions that allow a bound ligand to alter the structure of the protein to which it binds are increasingly well understood. High-resolution structures of liganded and unliganded proteins combined with increasingly accurate theoretical structural modeling and functional studies of mutant proteins are clarifying how we can think about induced conformational changes. From a functional perspective, however, the question of agonist action can be restated as: How does a receptor use the free energy of ligand binding to shift its conformation to the active state? A receptor, by definition, exists in at least two conformational states, active (a) and inactive (i).
These conformations might correspond to the open and closed states of an ion channel, the active and inactive states of a protein tyrosine kinase, or the productive versus nonproductive conformations of a receptor for coupling to G proteins. If these states are in equilibrium and the inactive state predominates in the absence of drug, then the basal signal output will be low. The extent to which the equilibrium is shifted toward the active state is determined by the relative affinity of the drug for the two conformations (Figure 21). A drug that has a higher affinity for the active conformation than for the inactive conformation will drive the equilibrium to the active state and thereby activate the receptor. Such a drug will be an agonist. A full agonist is sufficiently selective for the active conformation that, at a saturating concentration, it will drive the receptor essentially completely to the active state (see Figure 21). If a different but perhaps structurally similar compound binds to the same site on R but with only moderately greater affinity for Ra than for Ri, its effect will be less, even at saturating concentrations. A drug that displays such intermediate effectiveness is referred to as a partial agonist. Note that, in an absolute sense, all agonists are partial; selectivity for Ra over Ri cannot be total. A drug that binds with equal affinity to either conformation will not alter the activation equilibrium and will act as a competitive antagonist. Last, a drug with preferential affinity for Ri will actually produce an effect opposite to that of an agonist; examples of such inverse agonists do exist (see Chapters 11: 5-Hydroxytryptamine (Serotonin): Receptor Agonists and Antagonists and 17: Hypnotics and Sedatives). If the preexisting equilibrium for unliganded receptors lies far in the direction of Ri, negative antagonism may be difficult to observe and to distinguish from simple competitive antagonism.
Careful biochemical studies of receptordrug interactions, coupled with the analysis of receptors in which the intrinsic Ra/Ri equilibrium has been shifted by mutation, have supported this general model of drug action. The model is readily applicable to experimental data through the use of appropriate computer-assisted analysis and is frequently used as a guide to understanding drug action. Physiological Receptors: Structural and Functional Families The last two decades have witnessed both an explosion in our appreciation of the number of physiological receptors and, in parallel, the development of our understanding of the fundamental structural motifs and biochemical mechanisms that characterize them. Molecular cloning has identified both completely novel receptors (and their regulatory ligands) and numerous isoforms of previously known receptors. There now exist data banks devoted exclusively to structures of single classes of receptors. Members of various classes of receptors, transducers, and effector proteins have been purified, and their mechanisms of action are understood in considerable biochemical detail. Receptors, transducers, and effectors can be expressed via molecular genetic strategies and studied in cultured cells. Alternatively, they can be expressed in large amounts in cells of convenience (bacteria, yeast, etc.) to facilitate their purification. Receptors for physiological regulatory molecules can be assigned to a relatively few functional families whose members share both common mechanisms of action and similar molecular structures (Figure 22). For each receptor family, there is now at least a rudimentary understanding of the structures of ligand-binding domains and effector domains and of how agonist binding influences the regulatory activity of the receptor. The relatively small number of biochemical mechanisms and structural formats used for cellular signaling is fundamental to the ways in which target cells integrate signals from multiple receptors to produce additive, synergistic, or mutually inhibitory responses. Figure 21 provides a schematic diagram of various receptor families and their transducer and effector molecules.
Receptors as Enzymes: Receptor Protein Kinases The largest group of receptors with intrinsic enzymatic activity are cell surface protein kinases, which exert their regulatory effects by phosphorylating diverse effector proteins at the inner face of the plasma membrane. Phosphorylation can alter the biochemical activities of an effector or its interactions with other proteins. Most receptor protein kinases target tyrosine residues in their substrates; these include receptors for insulin, many cytokines and diverse peptides, and proteins that direct growth or differentiation. A few receptor protein kinases also phosphorylate serine or threonine residues. The structurally most simple receptor protein kinases are composed of an agonist-binding domain on the extracellular surface of the plasma membrane, a single membrane-spanning element, and a protein kinase domain on the inner membrane face. Many variations on this basic plan exist, including obligate oligomerization and the addition of multiple regulatory or protein-binding domains to the intracellular protein kinase domain. Another family of receptors that are functionally protein kinases contains a modification of the structure described above. Protein kinaseassociated receptors lack the intracellular enzymatic domains but, in response to agonists, bind and/or activate distinct protein kinases on the cytoplasmic face of the plasma membrane. Receptors of this group include several receptors for neurotrophic peptides and the multisubunit antigen receptors on T and B lymphocytes. The antigen receptors also involve tyrosine protein phosphatases in their cellular regulatory activity, and it is plausible that other receptors that apparently lack cytoplasmic effector domains may recruit still other effector proteins. Receptors with Other Enzymatic Activity The domain structure just described for cell surface protein kinases is varied in other receptors to utilize other signaling outputs. A family of protein tyrosine phosphatases has extracellular domains with a sequence reminiscent of cellular adhesion molecules. Although the extracellular ligands for many of these phosphatases are not yet known, the importance of their enzymatic activity has been demonstrated through genetic and biochemical experiments in multiple cell types. In the receptors for atrial natriuretic peptides and the peptide guanylin, the intracellular domain is not a protein kinase but a guanylyl cyclase, which synthesizes the second messenger, cyclic GMP. Receptors with guanylyl cyclase activity also serve as pheromone receptors in invertebrates. There may be other variations on this transmembrane topology. Ion Channels Receptors for several neurotransmitters form agonist-regulated, ion-selective channels in the plasma membrane, termed ligand-gated ion channels, which convey their signals by altering the cell's membrane potential or ionic composition. This group includes the nicotinic cholinergic receptor; the GABAA receptor for gamma-aminobutyric acid; and receptors for glutamate, aspartate, and glycine (see Chapters 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia, 12: Neurotransmission and the Central Nervous System, and 17: Hypnotics and Sedatives). They are all multisubunit proteins, with each subunit predicted to span the plasma membrane several times. Symmetrical association of the subunits allows each to form a segment of the channel wall and to cooperatively control channel opening and closing. G ProteinCoupled Receptors A large family of receptors use distinct heterotrimeric GTP-binding regulatory proteins, known as G proteins, as transducers to convey signals to their effector proteins. G proteincoupled receptors include those for many biogenic amines, eicosanoids, and other lipid signaling molecules as well as numerous peptide and protein ligands. G proteinregulated effectors include enzymes such as adenylyl cyclase and phospholipase C and plasma membrane ion channels selective for Ca2+ and K+ (Figure 22). Because of their number and physiological importance, G proteincoupled receptors are widely used targets for drugs; it has been estimated that about half of all nonantibiotic prescription drugs are directed toward these receptors. G proteincoupled receptors span the plasma membrane as a bundle of
seven alpha helices. Agonists bind to a cleft within the extracellular face
of the bundle or to a globular ligand-binding domain sometimes found at the
amino terminus. G proteins bind to the cytoplasmic face of the receptors.
Receptors in this family respond to agonists by promoting the binding of GTP
to the G protein. GTP activates the G protein and allows it in turn to
activate the effector protein. The G protein remains active until it
hydrolyzes bound GTP to GDP, which does not activate. G proteins are composed
of a GTP-binding A cell may express as many as twenty G proteincoupled receptors, each
with distinctive specificity for one or several of its half-dozen G proteins.
Each G Transcription Factors Receptors for steroid hormones, thyroid hormone, vitamin D, and the retinoids are soluble DNA-binding proteins that regulate the transcription of specific genes (Mangelsdorf et al., 1994). They are part of a larger family of transcription factors whose members may be regulated by phosphorylation, association with other cellular proteins, or by binding to metabolites or cellular regulatory ligands. These receptors act as dimerssome as homodimers and some as heterodimerswith homologous cellular proteins, but they may be regulated by higher-order oligomerization with other regulating molecules. They provide striking examples of conservation of structure and mechanism, in part because they are assembled as three largely independent domains. The region nearest the carboxyl terminus binds hormone and serves a negative regulatory role; that is, removal of this domain leaves a constitutively active fragment that may be nearly as effective in regulating transcription as is the intact hormone-liganded receptor. Hormone binding presumably also relieves this inhibitory constraint. The central region of the receptor mediates binding to specific sites on nuclear DNA to activate or inhibit transcription of the nearby gene. These regulatory sites in DNA are likewise receptor-specific: the sequence of a 'glucocorticoid-responsive element,' with only slight variation, is associated with each glucocorticoid-responsive gene. The function of the amino-terminal region of the receptor is less well defined, but its loss decreases the receptor's regulatory activity. The activity of each domain is largely independent, a phenomenon best demonstrated by the construction of chimeric receptors that reflect the hormone binding or DNA regulatory activity characteristic of the 'parent' receptor contributing that domain. Cytoplasmic Second Messengers Cyclic AMP Physiological signals also are integrated within the cell as a result of interactions between second-messenger pathways. Compared with the number of receptors and cytosolic signaling proteins, there are relatively few recognized cytoplasmic second messengers. However, their synthesis or release and degradation or excretion reflect the activities of many pathways. Well-studied second messengers include cyclic AMP and cyclic GMP, Ca2+, inositol phosphates, diacylglycerol, and nitric oxide; our list of this diverse group of molecules is still growing. Second messengers influence each other both directly, by altering the other's metabolism, and indirectly, by sharing intracellular targets. This superficially confusing pattern of regulatory pathways allows the cell to respond to agonists, singly or in combination, with an integrated array of cytoplasmic second messengers and responses. Cyclic AMP, the prototypical second messenger, remains a good example for understanding the regulation and function of most second messengers. It is synthesized by adenylyl cyclase under the control of many G proteincoupled receptors; stimulation is mediated by Gs and inhibition by Gi. There are at least ten tissue-specific adenylyl cyclase isozymes, each
with its unique pattern of regulatory responses. Several adenylyl cyclase
isozymes are either stimulated or inhibited by G protein In most cases, cyclic AMP functions by activating cyclic AMPdependent protein kinases, a relatively small group of closely related proteins. However, these protein kinases phosphorylate both final physiological targets (metabolic enzymes or transport proteins) and numerous protein kinases and other regulatory proteins. This latter group includes transcription factors that allow cyclic AMP to regulate gene expression in addition to more acute cellular events. In addition to activating a protein kinase, cyclic AMP also directly regulates the activity of plasma membrane cation channels, which are particularly important in olfactory neurons. Cyclic AMP signals are thus propagated throughout the biochemical behavior of the target cell. Calcium Intracellular Ca2+, another particularly well-studied second messenger, offers several interesting comparisons with cyclic AMP. The release of Ca2+ into the cytoplasm is mediated by diverse channels: plasma membrane channels regulated by G proteins, membrane potential, K+ or Ca2+ itself, and channels in specialized regions of endoplasmic reticulum that respond to the second messenger inositol trisphosphate (IP3), or, in muscle, to cell depolarization. Ca2+ is removed both by extrusion and by reuptake into the endoplasmic reticulum. Ca2+ propagates its signals through a much wider range of proteins than does cyclic AMP, including metabolic enzymes, protein kinases, and Ca2+-binding regulatory proteins that regulate still other ultimate and intermediary effectors. Regulation of Receptors Receptors not only initiate regulation of physiological and biochemical function but also are themselves subject to many regulatory and homeostatic controls. These controls include regulation of the synthesis and degradation of the receptor by multiple mechanisms, covalent modification, association with other regulatory proteins, and/or relocalization within the cell. Transducer and effector proteins similarly are regulated. Modulating inputs may come from other receptors, directly or indirectly, and receptors are almost always subject to feedback regulation by their own signaling outputs. Continued stimulation of cells with agonists generally results in a
state of desensitization (also referred to as refractoriness or
down-regulation), such that the effect that follows continued or
subsequent exposure to the same concentration of drug is diminished (Figure
23). This phenomenon is very important in therapeutic situations; an example
is attenuated response to the repeated use of
Feedback inhibition of signaling may be limited to output only from the stimulated receptor, a situation known as homologous desensitization. Attenuation also may extend to the action of all receptors that share a common signaling pathway, heterologous desensitization (Figure 23). Homologous desensitization indicates feedback directed to the receptor molecule itself (phosphorylation, proteolysis, decreased synthesis, etc.), whereas heterologous desensitization may involve inhibition or loss of one or more downstream proteins that participate in signaling from other receptors. Mechanisms involved in homologous and heterologous desensitization of specific receptors and signaling pathways are discussed in greater detail in later chapters related to individual receptor families. Predictably, supersensitivity to agonists also frequently follows
chronic reduction of receptor stimulation. Such situations can result, for
example, from the long-term administration of Diseases Resulting from Receptor Malfunction In addition to variability among individuals in their responses to drugs (see Chapter 3: Principles of Therapeutics), several definable diseases arise from disorders in receptors or receptoreffector systems. The loss of a receptor in a highly specialized signaling system may cause a relatively limited phenotypic disorder, such as the genetic deficiency of the androgen receptor in the testicular feminization syndrome (Griffin et al., 1995). Deficiencies of more widely used signaling systems have a broader spectrum of effects, as are seen in myasthenia gravis or some forms of insulin-resistant diabetes mellitus, which result from autoimmune depletion of nicotinic cholinergic receptors (see Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia) or insulin receptors (see Chapter 61: Insulin, Oral Hypoglycemic Agents, and the Pharmacology of the Endocrine Pancreas), respectively. A lesion in a component of a signaling pathway that is used by many receptors can cause a generalized endocrinopathy. Heterozygous deficiency of Gs, the G protein that activates adenylyl cyclase in all cells, causes multiple endocrine disorders (Spiegel and Weinstein, 1995). Homozygous deficiency in Gs presumably would be lethal. The expression of aberrant or ectopic receptors, effectors, or coupling proteins potentially can lead to supersensitivity, subsensitivity, or other untoward responses. Among the most interesting and significant events is the appearance of aberrant receptors as products of oncogenes, which transform otherwise normal cells into malignant cells. Virtually any type of signaling system may have oncogenic potential. The erbA oncogene product is an altered form of a receptor for thyroid hormone, constitutively active because of the loss of its ligand-binding domain (Mangelsdorf et al., 1994). The ros and erbB oncogene products are activated, uncontrolled forms of the receptors for insulin and epidermal growth factor, respectively, both known to enhance cellular proliferation (Yarden and Ulrich, 1988). The mas oncogene product (Young et al., 1986) is a G proteincoupled receptor, probably the receptor for a peptide hormone. Constitutive activation of G proteincoupled receptors due to subtle mutations in receptor structure has been shown to give rise to retinitis pigmentosa, precocious puberty, and malignant hyperthyroidism (reviewed in Clapham, 1993). G proteins can themselves be oncogenic when either overexpressed or constitutively activated by mutation (Lyons et al., 1990). Mutation of receptors can alter either acute responsiveness to drug
therapy or its continuing efficacy. For example, a mutation of Classification of Receptors and Drug Effects Traditionally, drug receptors have been identified and classified primarily on the basis of the effect and relative potency of selective agonists and antagonists. For example, the effects of acetylcholine that are mimicked by the alkaloid muscarine and that are selectively antagonized by atropine are termed muscarinic effects. Other effects of acetylcholine that are mimicked by nicotine are described as nicotinic effects. By extension, these two types of cholinergic effects are said to be mediated by muscarinic or nicotinic receptors. Although it frequently contributes little to delineation of the mechanism of drug action, such categorization provides a convenient basis for summarizing drug effects. A statement that a drug activates a specified type of receptor is a succinct summary of its spectrum of effects and of the agents that will regulate it. However, the accuracy of this statement may be altered when new receptors or receptor subtypes are identified or additional drug mechanisms or side effects are revealed. Significance of Receptor Subtypes As the diversity and selectivity of drugs increased, it became clear
that multiple subtypes of receptors exist within many previously defined
classes of receptors. Molecular cloning further accelerated discovery of
novel receptor subtypes, and their expression as recombinant proteins has
facilitated discovery of subtype-selective drugs. Distinct but related
receptors may, but need not, display distinctive patterns of selectivity
among agonist or antagonist li-gands. When selective ligands are not known,
the receptors are more commonly referred to as isoforms rather than as
subtypes. Receptor subtypes may display different mechanisms of signal
output. For example, M1- and M3-muscarinic receptors
activate Gq to initiate Ca2+ signaling, and M2-
and M4-muscarinic receptors activate Gi to activate
other signaling pathways. The distinction between classes and subtypes of
receptors is, however, often arbitrary and/or historical. The Pharmacological differences among receptor subtypes are exploited therapeutically through the development and use of receptor-selective drugs. Such drugs may be used to elicit different responses from a single tissue when receptor subtypes initiate different intracellular signals, or they may serve to differentially modulate different cells or tissues that express one or another receptor subtype. Increasing the selectivity of a drug among tissues or among responses elicited from a single tissue may determine whether the drug's therapeutic benefits outweigh its unwanted effects. The molecular biological search for novel receptors has moved well beyond the search for isoforms of known receptors toward the discovery of hundreds of genes for completely novel human receptors. Many of these receptors can be assigned to known families based on sequence, and their functions can be confirmed with appropriate ligands. However, many are 'orphans,' the designation give to receptors whose ligands are unknown. Discovery of the endogenous ligands and physiological functions of orphan receptors is widely hoped to lead to new drugs that can modulate currently intractable disease states. The discovery of numerous receptor isoforms raises the question of their importance to the organism, particularly when their signaling mechanisms and specificity for endogenous ligands are indistinguishable. Perhaps this multiplicity of genes facilitates the independent, cellspecific, and temporally controlled expression of receptors according to the developmental needs of the organism. Regardless of their mechanistic implications (or lack thereof), discovery of isoform-selective ligands may substantially improve our targeting of therapeutic drugs. Actions of Drugs Not Mediated by Receptors If one restricts the definition of receptors to macromolecules, then several drugs may be said not to act upon receptors as such. Some drugs specifically bind small molecules or ions that are normally or abnormally found in the body. One example is the therapeutic neutralization of gastric acid by a base (antacid). Another example is the use of mesna, a free radical scavenger rapidly eliminated by the kidneys, to bind to reactive metabolites associated with some cancer chemotherapeutic agents and thus minimize their untoward effects on the urinary tract (see Chapter 52: Antineoplastic Agents). Other agents act according to colligative effects without a requirement for highly specific chemical structure. For example, certain relatively benign compounds, such as mannitol, can be administered in quantities sufficient to increase the osmolarity of various body fluids and thereby cause appropriate changes in the distribution of water (see Chapter 29: Diuretics). Depending on the agent and route of administration, this effect can be exploited to promote diuresis, catharsis, expansion of circulating volume in the vascular compartment, or reduction of cerebral edema. Certain drugs that are structural analogs of normal biological chemicals may be incorporated into cellular components and thereby alter their function. This property has been termed a 'counterfeit incorporation mechanism,' and has been particularly useful with analogs of pyrimidines and purines that can be incorporated into nucleic acids; such drugs have clinical utility in antiviral and cancer chemotherapy (see Chapters 50 and 52). |
Quantitation of DrugReceptor Interactions and Elicited Effect
|
Receptor Pharmacology The major aim of receptor pharmacology is to understand and quantify the effects of chemicals (drugs) on biological systems. This is important in the therapeutic arena because drugs are nearly always used therapeutically in systems different from those in which they were discovered and tested. Biological systems interpret the effects of drugs in different ways, and these interpretations can be confusing. What is needed is a standard scale of drug activity that transcends biological systems and can be used to predict the effects of the drug in all systems. Receptor pharmacology strives to furnish the tools to accomplish this goal. The basic currency of receptor pharmacology is the doseresponse curve, a depiction of the observed effect of a drug as a function of its concentration in the receptor compartment. Figure 24A shows a typical doseresponse curve; it reaches a maximal asymptote value when the drug occupies all of the receptor sites. The range of concentrations needed to fully depict the doseresponse relationship usually is too wide to be useful in the format shown in Figure 24A. Most doseresponse curves are therefore plotted with the logarithm of the concentration as the x axis (see Figure 24B). Doseresponse curves have three basic properties: threshold, slope, and maximal asymptote; these parameters characterize and quantitate the activity of the drug.
In general, drugs can do two things to receptors: (1) bind to them and (2) possibly change their behavior toward the host cell system. The first function is governed by the chemical property of affinity, ruled by the chemical forces that cause the drug to associate with the receptor. The second is governed by a quantity referred to as efficacy. Efficacy is the information encoded in a drug's chemical structure that causes the receptor to change accordingly when the drug is bound. Historically, efficacy has been treated operationally as a proportionality constant that quantifies the extent of functional change imparted to a receptor upon binding a drug. Classical Receptor Theory Receptor occupancy theory, in which it is assumed that response emanates from a receptor occupied by a drug, has its basis in the law of mass action, with modifying constants added to accommodate experimental findings. Agonism was described by modification of this model by Arins (1954), Stephenson (1956), and Furchgott (1966). Stephenson introduced another important concept, stimulus, which is the initial effect of drug upon the receptor itself; stimulus is then processed by the systEM To yield the observable response. Antagonism was modeled by Gaddum (1937, 1957) and Schild (1957) to determine the affinity of antagonists. The basic components of drug receptormediated response are shown in Figure
25. Affinity is measured by the equilibrium dissociation constant of the
drugreceptor complex (denoted Kd); the fraction of
receptors occupied by the drug is determined by the concentration of drug and
Kd, as shown (see Figure 25). Intrinsic efficacy is
a proportionality constant (denoted
Transmission of Receptor Stimulus by the Target Tissue The activation of a receptor by a drug can be thought of as an initial signal that is then amplified by the cell. Different cells have different amplification properties; thus a weak receptor signal may produce no visible response in one cell type and a powerful signal in another. The amplification properties of the cell (referred to as the stimulus-response capability) control the observed outcome of drugreceptor interaction, as shown in Figure 26 for three hypothetical drugs and three different cell types. In cell I, which amplifies a stimulus relatively weakly, drug A produces a full tissue response and would be labeled a full agonist. Drug B produces a partial (submaximal) tissue response and would be a partial agonist. Drug C produces no response, but nevertheless occupies the receptor and therefore would antagonize the effects of either drug A or drug B; it would be referred to as an antagonist. When these same drugs are tested on cell II, which has a more efficiently coupled stimulus-response mechanism, drug A remains a full agonist, drug B is now also a full agonist, and drug C, which had insufficient efficacy to cause a physiological response in cell I, is now a partial agonist. The properties of these drugs have not changed; only the efficiency of the signaling system has changed. Thus, the labels for these drugs change as well. Drug B goes from a partial agonist to a full agonist and drug C goes from an antagonist to a partial agonist. This progression continues when these drugs are tested in cell III, which has even more efficient signaling machinery. Now all three drugs act as full agonists (Figure 26). This example illustrates the potential fallacy of classifying drugs on the basis of what they do rather than what they are. What drugs do depends on the receptor and its associated signaling proteins; classification by magnitude of physiological effect can be seriously misleading when drugs are tested in one cellular format for therapeutic use in another. The alternative is to classify drugs according to the magnitude of their two molecular properties: affinity for the receptor and efficacy once bound. By quantifying these system-independent properties, drug activity can be predicted in all systems as long as the identity of the receptor is known.
Quantifying Agonism Drugs have two observable properties in biological systems: potency and magnitude of effect (when a biological response is produced). Potency is controlled by four factors: two relate to the biological system containing the receptors (receptor density and efficiency of the stimulusresponse mechanisms of the tissue) and two relate to the interaction of drug with its receptor (affinity and efficacy). When the relative potency of two agonists of equal efficacy is measured in the same biological system, downstream signaling effects cancel and the comparison yields a relative measure of the affinity and efficacy of the two agonists (see Figure 27A). Thus, measuring agonist potency ratios is one method of measuring the capability of different agonists to induce a response in a test system and for predicting comparable activity in another. Another method of estimating agonist activity is to compare maximal asymptotes in systems where the agonists do not produce system maximal response (Figure 27B). The advantage of using maxima is that this property is solely dependent upon efficacy, whereas potency is a mixed function of both affinity and efficacy.
Quantifying Antagonism Characteristic patterns of antagonism are associated with certain mechanisms of blockade of receptors. One is simple competitive antagonism, whereby a drug that lacks intrinsic efficacy but retains affinity competes with the agonist for the binding site. The characteristic pattern of such antagonism is the concentration-dependent production of a parallel shift to the right of the agonist doseresponse curve with no change in the maximal asymptotic response (Figure 28A). The magnitude of the rightward shift of the curve depends only upon the concentration of the antagonist and its affinity for the receptor. The affinity of a competitive antagonist for its receptor can therefore be determined according to its concentration-dependent ability to shift the doseresponse curve for an agonist rightward, as first noted by Schild (1957). Note that a partial agonist similarly can compete with a 'full' agonist for binding to the receptor. However, increasing concentrations of a partial agonist will inhibit response to a finite level characteristic of the drug's intrinsic efficacy; a competitive antagonist will reduce the response to zero. Partial agonists thus can be used therapeutically to buffer a response by inhibiting untoward stimulation without totally abolishing the stimulus from the receptor.
An antagonist may dissociate so slowly from the receptor as to be essentially irreversible in its action. Under these circumstances, the maximal response to the agonist will be depressed at some antagonist concentrations (Figure 28B). Operationally, this is referred to as noncompetitive antagonism, although the molecular mechanism of action really cannot be unequivocally inferred from the effect. An irreversible antagonist competing for the same binding site as the agonist also can produce the pattern of antagonism shown in Figure 28B. Noncompetitive antagonism can be produced by another type of drug, referred to as an allosteric antagonist. This type of drug produces its effect by binding a site on the receptor distinct from that of the primary agonist and thereby changing the affinity of the receptor for the agonist (see Figure 28). In the case of an allosteric antagonist, the affinity of the receptor for the agonist is decreased by the antagonist (see Figure 28C). In contrast, some allosteric effects potentiate the effects of agonists (Figure 28D). Thus, in cases where the pathology may involve a failing agonist system (i.e., myasthenia gravis, Alzheimer's disease), an allosteric potentiator of the endogenous response would strengthen the signal and, importantly, preserve the natural pattern of response. By allowing both the overexpression of wild-type receptors and the creation (and discovery) of constitutively active mutant receptors, molecular genetic technology has facilitated the study of a novel class of functional antagonists, the inverse agonists. As discussed above, receptors spontaneously can adopt active conformations that produce a cellular response. The fraction of unoccupied receptors in the active conformation usually is too low to allow observation of their agonist-independent activity; but this activity can be observed readily, either when the receptor is expressed at heterologously high levels or when mutation shifts the conformational equilibrium toward the active form. In these situations, the tissue behaves as if there were an agonist present, and a conventional competitive antagonist has no effect. However, because inverse agonists selectively bind to the inactive form of the receptor and shift the conformational equilibrium toward the inactive state, these agents are capable of inhibiting agonist-independent or constitutive signaling. In systems that are not constitutively active, inverse agonists will behave exactly like competitive antagonists, which in part explains why the properties of inverse agonists and the number of such agents previously described as competitive antagonists were not appreciated until recently. It is not known to what extent constitutive receptor activity is a pathologically important phenomenon, and it is therefore unclear to what extent inverse agonism is a therapeutically relevant property. In some cases, however, the preferability of an inverse agonist over a competitive antagonist is obvious. For example, the human herpesvirus KSHV encodes a constitutively active chemokine receptor that generates a second messenger that drives cell growth and viral replication (Arvanitakis et al., 1997). Clearly, in such a case a conventional antagonist would not be useful as the chemokine agonist is not involved, and an inverse agonist would be the only viable intervention. |
Prospectus
|
The continuing identification and expansion of molecular families for receptors, especially at the advent of the human genome era, coupled with the enormous potential for generating new molecules with combinatorial chemistry or recombinant DNA strategies, forecast a new era of diversity and specificity in therapeutic intervention. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 8331
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved