| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Hypnotics and Sedatives
Overview
|
A wide variety of agents have the capacity to depress the function of the central nervous system (CNS) such that calming or drowsiness (sedation) is produced. Older sedative-hypnotic drugs depress the CNS in a dose-dependent fashion, progressively producing sedation, sleep, unconsciousness, surgical anesthesia, coma, and, ultimately, fatal depression of respiration and cardiovascular regulation. The CNS depressants that are addressed in this chapter include the benzodiazepines and barbiturates as well as sedative-hypnotic agents of diverse chemical structure (e.g., paraldehyde, chloral hydrate). Volatile anesthetics are discussed in Chapter 14: General Anesthetics. Benzodiazepines have only a limited capacity to produce profound and potentially fatal CNS depression. Although coma may be produced at very high doses, benzodiazepines cannot induce a state of surgical anesthesia by themselves and virtually are incapable of causing fatal respiratory depression or cardiovascular collapse unless other CNS depressants also are present. Because of this measure of safety, benzodiazepines and their newer analogs have largely replaced older agents for the treatment of insomnia or anxiety. Sedative-hypnotic drugs, particularly the benzodiazepines, also are used to produce sedation and amnesia before or during diagnostic or operative procedures, and some, notably certain barbiturates, are used at high doses to induce or maintain surgical anesthesia (see Chapter 14: General Anesthetics). A few barbiturates and benzodiazepines are used as antiepileptic agents (see Chapter 21: Drugs Effective in the Therapy of the Epilepsies), and a few benzodiazepines may be used as muscle relaxants (see Chapter 22: Treatment of Central Nervous System Degenerative Disorders). The role of the benzodiazepines and other agents in the pharmacotherapy of anxiety will be discussed in Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders. CNS depressants also include the aliphatic alcohols, particularly ethanol. Ethanol shares many pharmacological properties with the nonbenzodiazepine sedative-hypnotic drugs. However, its usefulness in the treatment of sleep disorders is limited, and often it may be more disruptive than beneficial. The pharmacology of ethanol is discussed in Chapter 18: Ethanol. Abuse of ethanol and other CNS depressants is discussed in Chapter 24: Drug Addiction and Drug Abuse. |
Hypnotics and Sedatives: Introduction
|
A sedative drug decreases activity, moderates excitement, and calms the recipient, whereas a hypnotic drug produces drowsiness and facilitates the onset and maintenance of a state of sleep that resembles natural sleep in its electroencephalographic characteristics and from which the recipient can be aroused easily. The latter effect sometimes is called hypnosis, but the sleep induced by hypnotic drugs does not resemble the artificially induced passive state of suggestibility also called hypnosis. The nonbenzodiazepine sedative-hypnotic drugs belong to a group of agents that depress the central nervous system (CNS) in a dose-dependent fashion, progressively producing calming or drowsiness (sedation), sleep (pharmacological hypnosis), unconsciousness, coma, surgical anesthesia, and fatal depression of respiration and cardiovascular regulation. They share these properties with a large number of chemicals, including general anesthetics (seeChapter 14: General Anesthetics) and aliphatic alcohols, most notably ethanol (seeChapter 18: Ethanol). Only two landmarks on the continuum of CNS depression produced by increasing concentrations of these agents can be defined with a reasonable degree of precision: surgical anesthesia, a state in which painful stimuli elicit no behavioral or autonomic response (seeChapter 13: History and Principles of Anesthesiology), and death, resulting from sufficient depression of medullary neurons to disrupt coordination of cardiovascular function and respiration. The 'end points' at lower concentrations of CNS depressants are defined with less precisionin terms of deficits in cognitive function (including attention to environmental stimuli) or motor skills (e.g., ataxia), or of the intensity of sensory stimuli needed to elicit some reflex or behavioral response. Other important indices of decreased activity of the CNS, such as analgesia and seizure suppression, do not necessarily fall along this continuum; they may not be present at subanesthetic concentrations of a CNS-depressant drug (e.g., a barbiturate), or they may be achieved with minimal sedation or other evidence of CNS depression (e.g., with low doses of opioids, phenytoin, ethosuximide). Sedation is a side effect of many drugs that are not general CNS depressants (e.g., antihistamines, neuroleptics). Although such agents can intensify the effects of CNS depressants, they usually produce more specific therapeutic effects at concentrations far lower than those causing substantial CNS depression. They cannot, for example, induce surgical anesthesia in the absence of other agents. The benzodiazepine sedative-hypnotics resemble such agents; although coma may occur at very high doses, neither surgical anesthesia nor fatal intoxication is produced by benzodiazepines in the absence of other drugs with CNS-depressant actions. Moreover, certain congeners can specifically antagonize the actions of benzodiazepines without eliciting significant effects in their absence. This constellation of properties sets the benzodiazepines apart from other sedative-hypnotic drugs and imparts a measure of safety that has resulted in benzodiazepines largely displacing older agents for the treatment of insomnia and anxiety. History Since antiquity, alcoholic beverages and potions containing laudanum and various herbals have been used to induce sleep. The first agent to be introduced specifically as a sedative and soon thereafter as a hypnotic was bromide, in the middle of the nineteenth century. Chloral hydrate, paraldehyde, urethane, and sulfonal came into use before the introduction of barbital in 1903 and phenobarbital in 1912. Their success spawned the synthesis and testing of over 2500 barbiturates, of which approximately 50 were distributed commercially. The barbiturates so dominated the stage that less than a dozen other sedative-hypnotics were successfully marketed before 1960. The partial separation of sedative-hypnotic-anesthetic from anticonvulsant properties embodied in phenobarbital led to searches for agents with more selective effects on the functions of the CNS. As a result, relatively nonsedative anticonvulsants, notably phenytoin and trimethadione, were developed in the late 1930s and early 1940s (seeChapter 21: Drugs Effective in the Therapy of the Epilepsies). The advent of chlorpromazine and meprobamate in the early 1950s, with their taming effects in animals, and the development of increasingly sophisticated methods for evaluating the behavioral effects of drugs set the stage in the 1950s for the synthesis of chlordiazepoxide by Sternbach and the discovery of its unique pattern of actions by Randall (seeSymposium, 1982). The introduction of chlordiazepoxide into clinical medicine in 1961 ushered in the era of benzodiazepines. Most of the benzodiazepines that have reached the marketplace were selected for high anxiolytic potency in relation to their depression of CNS function. However, all benzodiazepines possess sedative-hypnotic properties to varying degrees; these properties are extensively exploited clinically, especially to facilitate sleep. Mainly because of their remarkably low capacity to produce fatal CNS depression, the benzodiazepines have displaced the barbiturates as sedative-hypnotic agents. Over the past decade, it has become clear that all benzodiazepines in clinical use have the capacity to promote the binding of the major inhibitory neurotransmitter, gamma-aminobutyric acid (GABA), to the GABAA subtype of GABA receptors, which exist as multisubunit, ligand-gated chloride channels. Benzodiazepines enhance the GABA-induced ionic currents through these channels. Pharmacological investigations have provided evidence for heterogeneity among sites of binding and action of benzodiazepines, while biochemical and molecular biological investigations have revealed the numerous varieties of subunits that make up the GABA-gated chloride channels expressed in different neurons. Since receptor subunit composition appears to govern the interaction of various allosteric modulators with these channels, there has been a surge in efforts to find agents displaying a different mixture of benzodiazepine-like properties that may reflect selective actions on one or more subtypes of GABA receptors. One result of these efforts has been the introduction of zolpidem, one of several imidazopyridine compounds that appear to exert sedative-hypnotic actions by interacting with a subset of benzodiazepine binding sites. Zaleplon, a pyrazolopyrimidine, also has specificity for a subset of GABAA receptors. Investigation of compounds in many other chemical classes is in progress. |
Benzodiazepines
|
Although the benzodiazepines in clinical use exert qualitatively similar effects, important quantitative differences in their pharmacodynamic spectra and pharmacokinetic properties have led to varying patterns of therapeutic application. There is reason to believe that a number of distinct mechanisms of action contribute in varying degrees to the sedative-hypnotic, muscle-relaxant, anxiolytic, and anticonvulsant effects of the benzodiazepines. Recent findings provide evidence that specific subunits of GABAA receptor are responsible for specific pharmacological properties of benzodiazepines. While only the benzodiazepines used primarily for hypnosis will be discussed in detail, this chapter will describe the general properties of the group and the important differences among individual agents (see alsoChapters 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders and 21: Drugs Effective in the Therapy of the Epilepsies). Chemistry The structures of the benzodiazepines in use in the In addition to various benzodiazepine or imidazobenzodiazepine
derivatives, a large number of nonbenzodiazepine compounds have been
synthesized that compete with classic benzodiazepines or flumazenil for
binding at specific sites in the CNS (seeGardner et al., 1993).
These include representatives from the Pharmacological Properties Virtually all effects of the benzodiazepines result from actions of these drugs on the CNS. The most prominent of these effects are sedation, hypnosis, decreased anxiety, muscle relaxation, anterograde amnesia, and anticonvulsant activity. Only two effects of these drugs appear to result from actions on peripheral tissues: coronary vasodilation, seen after intravenous administration of therapeutic doses of certain benzodiazepines, and neuromuscular blockade, seen only with very high doses. A variety of benzodiazepine-like effects have been observed in vivo and in vitro and have been classified as full agonistic effects (i.e., faithfully mimicking agents such as diazepam with relatively low fractional occupancy of binding sites) or partial agonistic effects (i.e., producing less intense maximal effects and/or requiring relatively high fractional occupancy compared to agents such as diazepam). Some compounds produce effects opposite to those of diazepam in the absence of benzodiazepine-like agonists and have been termed inverse agonists; partial inverse agonists also have been recognized. The vast majority of effects of agonists and inverse agonists can be reversed or prevented by the benzodiazepine antagonist flumazenil, which competes with agonists and inverse agonists for binding to the benzodiazepine receptor. In addition, representatives from various classes of compounds behave like flumazenil and act only to block the effects of agonists or inverse agonists. Central Nervous System While benzodiazepines affect activity at all levels of the neuraxis, some structures are affected to a much greater extent than others. The benzodiazepines are not capable of producing the same degrees of neuronal depression as do barbiturates and volatile anesthetics. All of the benzodiazepines have very similar pharmacological profiles. Nevertheless, the drugs differ in selectivity, and the clinical usefulness of individual benzodiazepines thus varies considerably. As the dose of a benzodiazepine is increased, sedation progresses to hypnosis and then to stupor. The clinical literature often refers to the 'anesthetic' effects and uses of certain benzodiazepines, but the drugs do not cause a true general anesthesia, since awareness usually persists, and relaxation sufficient to allow surgery cannot be achieved. However, at 'preanesthetic' doses, there is amnesia for events subsequent to the administration of the drug; this may create the illusion of previous anesthesia. The recent discovery of a molecular basis for numerous benzodiazepine receptor subtypes (see below) has provided the rationale for attempts to separate the anxiolytic actions of these drugs from their sedative/hypnotic effects. However, distinguishing between these behaviors remains problematic. Measurements of anxiety and sedation are difficult in human beings, and the validity of animal models for anxiety and sedation is uncertain. The existence of multiple benzodiazepine receptors may partially explain the diversity of pharmacological responses in different species. Animal Models of Anxiety In animal models of anxiety, most attention has been focused on the ability of benzodiazepines to increase locomotor, feeding, or drinking behavior that has been suppressed by novel or aversive stimuli. For such tests, animal behaviors that previously had been rewarded by food or water are periodically punished by an electric shock. The time during which shocks are delivered is signaled by some auditory or visual cue, and untreated animals stop performing almost completely when the cue is perceived. The difference in behavioral responses during the punished and unpunished periods is eliminated by benzodiazepine agonists, usually at doses that do not reduce the rate of unpunished responses or produce other signs of impaired motor function. Similarly, rats placed in an unfamiliar environment exhibit markedly reduced exploratory behavior (neophobia), whereas animals treated with benzodiazepines do not. Opioid analgesics and neuroleptic (antipsychotic) drugs do not increase suppressed behaviors, and phenobarbital and meprobamate usually do so only at doses that also reduce spontaneous or unpunished behaviors or produce ataxia. The difference between the dose required to impair motor function and that necessary to increase punished behavior varies widely among the benzodiazepines and depends on the species and experimental protocol. Although such differences may have encouraged the marketing of some benzodiazepines as selective sedative-hypnotic agents, they have not predicted with any accuracy the magnitude of sedative effects among those benzodiazepines marketed as anxiolytic agents. Tolerance to Benzodiazepines Studies on tolerance in laboratory animals often are cited to support the belief that disinhibitory effects of benzodiazepines are separate from their sedative-ataxic effects. For example, tolerance to the depressant effects on rewarded or neutral behavior occurs after several days of treatment with benzodiazepines; the disinhibitory effects of the drugs on punished behavior are augmented initially and decline after 3 to 4 weeks (seeFile, 1985). Although most patients who chronically ingest benzodiazepines report that drowsiness wanes over a few days, tolerance to the impairment of some measures of psychomotor performance (e.g., visual tracking) usually is not observed. The development of tolerance to the anxiolytic effects of benzodiazepines is a subject of debate (Lader and File, 1987). However, many patients can maintain themselves on a fairly constant dose; increases or decreases in dosage appear to correspond to changes in problems or stresses. Nevertheless, some patients either do not reduce their dosage when stress is relieved or steadily escalate dosage. Such behavior may be associated with the development of drug dependence (seeWoods et al., 1987; DuPont, 1988). Some benzodiazepines induce muscle hypotonia without interfering with normal locomotion and can decrease rigidity in patients with cerebral palsy. However, in contrast to effects in animals, there is only a limited degree of selectivity in human beings. Clonazepam in nonsedative doses does cause muscle relaxation in patients, but diazepam and most other benzodiazepines do not. Tolerance occurs to both the muscle relaxant and ataxic effects of these drugs. Experimentally, benzodiazepines inhibit seizure activity induced by either pentylenetetrazol or picrotoxin, but strychnine- and maximal electroshock-induced seizures are suppressed only with doses that also severely impair locomotor activity. Clonazepam, nitrazepam, and nordazepam are among those compounds with more selective anticonvulsant activity than most other benzodiazepines. Benzodiazepines also suppress photic seizures in baboons and ethanol-withdrawal seizures in human beings. However, the development of tolerance to the anticonvulsant effects has limited the usefulness of benzodiazepines in the treatment of recurrent seizure disorders in human beings (seeChapter 21: Drugs Effective in the Therapy of the Epilepsies). Although analgesic effects of benzodiazepines have been observed in experimental animals, only transient analgesia is apparent in human patients after intravenous administration. Such effects actually may involve the production of amnesia. However, it is clear that benzodiazepines do not cause hyperalgesia, unlike the barbiturates. Effects on the Electroencephalogram (EEG) and Sleep Stages The effects of benzodiazepines on the waking EEG resemble those of other sedative-hypnotic drugs. Alpha activity is decreased, but there is an increase in low-voltage fast activity. Tolerance occurs to these effects. Most benzodiazepines decrease sleep latency, especially when first used, and diminish the number of awakenings and the time spent in stage 0 (a stage of wakefulness). Time in stage 1 (descending drowsiness) usually is decreased, and there is a prominent decrease in the time spent in slow-wave sleep (stages 3 and 4). Most benzodiazepines increase the time from onset of spindle sleep to the first burst of rapid-eye-movement (REM) sleep, and the time spent in REM sleep usually is shortened. However, the number of cycles of REM sleep usually is increased, mostly late in the sleep time. Zolpidem does not suppress REM sleep to the same extent as do benzodiazepines and thus may be superior to benzodiazepines for use as a hypnotic (Dujardin et al., 1998). Despite the shortening of stage 4 and REM sleep, the net effect of administration of benzodiazepines typically is an increase in total sleep time, largely because of an increase in time spent in stage 2 (which is the major fraction of non-REM sleep). The effect is greatest in subjects with shortest baseline total sleep time. In addition, despite the increase in the number of REM cycles, the number of shifts to lighter sleep stages (1 and 0) and the amount of body movement are diminished. The nocturnal peaks in the concentrations of growth hormone, prolactin, and luteinizing hormone in plasma are not affected. During chronic nocturnal use of benzodiazepines, the effects on the various stages of sleep usually decline within a few nights. When such use is discontinued, the pattern of drug-induced changes in sleep parameters may 'rebound,' and an increase in the amount and density of REM sleep may be especially prominent. However, if the dosage has not been excessive, patients usually will note only a shortening of sleep time rather than an exacerbation of insomnia. Although some differences in the patterns of effects exerted by the various benzodiazepines have been noted, their use usually imparts a sense of deep or refreshing sleep. It is uncertain to which effect on sleep parameters this feeling can be attributed. As a result, variations in the pharmacokinetic properties of individual benzodiazepines appear to be much more important determinants of the utility of the available drugs for their effects on sleep than are any potential differences in their pharmacodynamic properties. Molecular Targets for Benzodiazepine Actions in the CNS Benzodiazepines are believed to exert most of their effects by interacting with inhibitory neurotransmitter receptors directly activated by GABA. GABA receptors are membrane-bound proteins that can be divided into two major subtypes: GABAA and GABAB receptors. The ionotropic GABAA receptors are composed of five subunits that coassemble to form an integral chloride channel. GABAA receptors are responsible for most inhibitory neurotransmission in the CNS. In contrast, the metabotropic GABAB receptors, made up of single peptides with seven transmembrane domains, are coupled to their signal transduction mechanisms by G proteins. Benzodiazepines act at GABAA but not GABAB receptors by binding directly to a specific site that is distinct from that of GABA binding on the receptor/ion channel complex. Unlike barbiturates, benzodiazepines do not directly activate GABAA receptors but require GABA to express their effects; i.e., they only modulate the effects of GABA. Benzodiazepines and GABA analogs bind to their respective sites on brain membranes with nanomolar affinity. Benzodiazepines modulate GABA binding and GABA alters benzodiazepine binding in an allosteric fashion. Benzodiazepine-receptor ligands can act as agonists, antagonists, or inverse agonists at the benzodiazepine receptor site, depending on the compound. Agonists at the benzodiazepine receptor increase, while inverse agonists decrease, the amount of chloride current generated by GABAA-receptor activation. Benzodiazepine receptor agonists produce shifts of GABA concentration-response curves to the left, while inverse agonists shift the curves to the right. Both of these effects can be blocked by antagonists at the benzodiazepine-receptor site. In the absence of a benzodiazepine-receptor agonist or inverse agonist, a benzodiazepine-receptor antagonist does not affect GABAA-receptor function. One such antagonist, flumazenil, is used clinically to reverse the effects of high doses of benzodiazepines. The behavioral and electrophysiological effects of benzodiazepines also can be reduced or prevented by prior treatment with antagonists (e.g., bicuculline) at the GABA binding site. The strongest evidence that benzodiazepines act directly on GABAA
receptors comes from molecular cloning of cDNAs encoding subunits of the GABAA
receptor complex (Schofield et al., 1987; Pritchett et al.,
1989). When receptors formed of the appropriate subunits (see below)
are studied in an in vitro expression system, high-affinity
benzodiazepine binding sites are seen, as are GABA-activated chloride
conductances that are enhanced by benzodiazepine-receptor agonists. The
properties of the expressed receptors are generally similar to those of GABAA
receptors found in most CNS neurons. Each GABAA receptor is
believed to consist of a pentamer of homologous subunits. Thus far, 16
different subunits have been identified and classified into seven subunit
families: six Recent work is beginning to show which GABAA-receptor
subunits are responsible for particular effects of benzodiazepines in vivo.
The mutation to arginine of a histidine residue at position 101 of the GABAA
receptor GABAA-receptor subunits also may play roles in the proper targeting
of assembled receptors to their proper locations in synapses. The production
of GABAA Receptor-Mediated Electrical Events: In Vivo Properties The remarkable safety of the benzodiazepines is likely related to the fact that the production of their effects in vivo depends on the presynaptic release of GABA; in the absence of GABA, benzodiazepines have no effects on GABAA-receptor function. Although barbiturates also enhance the effects of GABA at low doses, they directly activate GABA receptors at higher doses, which can lead to profound CNS depression (see below). Further, the ability of benzodiazepines to release suppressed behaviors and to produce sedation can be ascribed in part to potentiation of GABA-ergic pathways that serve to regulate the firing of neurons containing various monoamines (seeChapter 12: Neurotransmission and the Central Nervous System). These neurons are known to promote behavioral arousal and are important mediators of the inhibitory effects of fear and punishment on behavior. Finally, inhibitory effects on muscular hypertonia or the spread of seizure activity can be rationalized by potentiation of inhibitory GABA-ergic circuits at various levels of the neuraxis. In most studies conducted in vivo or in situ, the local or systemic administration of benzodiazepines reduces the spontaneous or evoked electrical activity of major (large) neurons in all regions of the brain and spinal cord. The activity of these neurons is regulated in part by small inhibitory interneurons (predominantly GABA-ergic) arranged in both feedback and feedforward types of circuits. The magnitude of the effects produced by benzodiazepines can vary widely and depends on such factors as the types of inhibitory circuits that are operating, the sources and intensity of excitatory input, and the manner in which experimental manipulations are performed and assessed. For example, feedback circuits often involve powerful inhibitory synapses on the neuronal soma near the axon hillock, which are supplied predominantly by recurrent pathways. The synaptic or exogenous application of GABA to this region increases chloride conductance and can prevent neuronal discharge by shunting electrical currents that would otherwise depolarize the membrane of the initial segment. Accordingly, benzodiazepines markedly prolong the period following brief activation of recurrent GABA-ergic pathways during which neither spontaneous nor applied excitatory stimuli can evoke neuronal discharge; this effect is reversed by the GABAA-receptor antagonist bicuculline. Molecular Basis for Benzodiazepine Regulation of GABAA Receptor-Mediated Electrical Events Electrophysiological studies in vitro have shown that the enhancement of GABA-induced chloride currents by benzodiazepines results primarily from an increase in the frequency of bursts of openings of chloride channels produced by submaximal amounts of GABA (Twyman et al., 1989). Inhibitory synaptic transmission measured after stimulation of afferent fibers is potentiated by benzodiazepines at therapeutically relevant concentrations. Prolongation of spontaneous miniature inhibitory postsynaptic currents (IPSCs) by benzodiazepines also has been observed. Although sedative barbiturates also enhance such chloride currents, they do so by prolonging the duration of individual channel-opening events. Macroscopic measurements of GABAA receptor-mediated currents indicate that benzodiazepines shift the GABA concentration-response curve to the left without increasing the maximum current evoked with GABA. Taken together with the in vivo data, these findings are consistent with a model in which benzodiazepines exert their major actions by increasing the gain of inhibitory neurotransmission mediated by GABAA receptors. As noted above, certain experimental benzodiazepines and other structurally related compounds act as inverse agonists to reduce GABA-induced chloride currents, promote convulsions, and produce other in vivo effects opposite to those induced by the benzodiazepines in clinical use (seeGardner, 1988; Gardner et al., 1993). A few compounds, most notably flumazenil, can block the effects of both clinically used benzodiazepines and inverse agonists in vitro and in vivo but have no detectable actions by themselves. The conceptual advances brought about by molecular studies have strengthened the hypothesis that benzodiazepines act mainly at GABAA receptors. Moreover, molecular diversity helps clarify many previous observations that appeared to conflict with this hypothesis (for reviews, seeDe Lorey and Olsen, 1992; Doble and Martin, 1992; Sieghart, 1992; Ragan et al., 1993; and Symposium, 1992). Nonetheless, some observations are difficult to reconcile with the hypothesis that all effects of benzodiazepines are mediated via GABAA receptors. Low concentrations of benzodiazepines that are not blocked by bicuculline or picrotoxin induce depressant effects on hippocampal neurons (Polc, 1988). The induction of sleep in rats by benzodiazepines also is insensitive to bicuculline or picrotoxin but is prevented by flumazenil (seeMendelson, 1992). At higher concentrations, corresponding to those producing hypnosis and amnesia during preanesthetic medication (seeChapter 14: General Anesthetics) or those achieved during the treatment of status epilepticus (seeChapter 21: Drugs Effective in the Therapy of the Epilepsies), the actions of the benzodiazepines may involve the participation of a number of other mechanisms. These include inhibition of the uptake of adenosine and the resultant potentiation of the actions of this endogenous neuronal depressant (seePhillis and O'Regan, 1988), as well as the GABA-independent inhibition of Ca2+ currents, Ca2+-dependent release of neurotransmitter, and tetrodotoxin-sensitive Na+ channels (seeMacdonald and McLean, 1986). The macromolecular complex containing GABA-regulated chloride channels
also may be a site of action of general anesthetics, ethanol, inhaled drugs
of abuse, and certain metabolites of endogenous steroids (Mehta and Ticku,
1999; Beckstead et al., 2000). Among the latter, allopregnanolone (3 Respiration Hypnotic doses of benzodiazepines are without effect on respiration in normal subjects, but special care must be taken in the treatment of children (Kriel et al., 2000) and individuals with impaired hepatic function, such as alcoholics (Guglielminotti et al., 1999). At higher doses, such as those used for preanesthetic medication or for endoscopy, benzodiazepines slightly depress alveolar ventilation and cause respiratory acidosis as the result of a decrease in hypoxic rather than hypercapnic drive; these effects are exaggerated in patients with chronic obstructive pulmonary disease (COPD), and alveolar hypoxia and/or CO2 narcosis may result. These drugs can cause apnea during anesthesia or when given with opioids, and patients severely intoxicated with benzodiazepines usually require respiratory assistance only when they also have ingested another CNS-depressant drug, most commonly alcohol. By contrast, hypnotic doses of benzodiazepines may worsen sleep-related breathing disorders by adversely affecting the control of the upper airway muscles or by decreasing the ventilatory response to CO2 (see Guilleminault, in Symposium, 1990b). The latter effect may be sufficient to cause hypoventilation and hypoxemia in some patients with severe COPD, although benzodiazepines may improve sleep and sleep structure in some instances. In patients with obstructive sleep apnea (OSA), hypnotic doses of benzodiazepines may decrease muscular tone in the upper airway and exaggerate the impact of apneic episodes on alveolar hypoxia, pulmonary hypertension, and cardiac ventricular load. Many physicians consider the presence of OSA to be a contraindication for the use of alcohol or any sedative-hypnotic agent, including a benzodiazepine; caution also should be exercised in patients who snore regularly, because partial airway obstruction may be converted to OSA under the influence of these drugs. In addition, benzodiazepines may promote the appearance of episodes of apnea during REM sleep (associated with decreases in oxygen saturation) in patients recovering from a myocardial infarction (Guilleminault, in Symposium, 1990b); however, the potential impact of these drugs on survival of patients with cardiac disease has not been investigated as yet. Cardiovascular System The cardiovascular effects of benzodiazepines are minor in normal subjects except in severe intoxication; the adverse effects in patients with obstructive sleep disorders or cardiac disease were noted above. In preanesthetic doses, all benzodiazepines decrease blood pressure and increase heart rate. With midazolam, the effects appear to be secondary to a decrease in peripheral resistance, but with diazepam they are secondary to a decrease in left ventricular work and cardiac output. Diazepam increases coronary flow, possibly by an action to increase interstitial concentrations of adenosine, and the accumulation of this cardiodepressant metabolite also may explain the negative inotropic effects of the drug. In large doses, midazolam decreases considerably both cerebral blood flow and oxygen assimilation (Nugent et al., 1982). Gastrointestinal Tract Benzodiazepines are thought by some gastroenterologists to improve a variety of 'anxiety-related' gastrointestinal disorders. There is a paucity of evidence for direct actions. Benzodiazepines partially protect against stress ulcers in rats, and diazepam markedly decreases nocturnal gastric secretion in human beings. Absorption, Fate, and Excretion The physicochemical and pharmacokinetic properties of the benzodiazepines greatly affect their clinical utility. They all have high lipid: water distribution coefficients in the nonionized form; nevertheless, lipophilicity varies more than 50-fold according to the polarity and electronegativity of various substituents. All of the benzodiazepines essentially are completely absorbed, with the exception of clorazepate; this drug is rapidly decarboxylated in gastric juice to N-desmethyldiazepam (nordazepam), which subsequently is absorbed completely. Some benzodiazepines (e.g., prazepam and flurazepam) reach the systemic circulation only in the form of active metabolites. Drugs active at the benzodiazepine receptor may be divided into four categories based on their elimination half-lives: (1) ultra-short-acting benzodiazepines; (2) short-acting agents, with t1/2 less than 6 hours, including triazolam, the nonbenzodiazepine zolpidem (t1/2 approximately 2 hours), and zopiclone (t1/2 5 to 6 hours); (3) intermediate-acting agents, with t1/2 of 6 to 24 hours, including estazolam and temazepam; and (4) long-acting agents, with t1/2 greater than 24 hours, including flurazepam, diazepam, and quazepam. The benzodiazepines and their active metabolites bind to plasma proteins. The extent of binding correlates strongly with lipid solubility and ranges from about 70% for alprazolam to nearly 99% for diazepam. The concentration in the cerebrospinal fluid (CSF) is approximately equal to the concentration of free drug in plasma. While competition with other protein-bound drugs may occur, no clinically significant examples have been reported. The plasma concentrations of most benzodiazepines exhibit patterns that are consistent with two-compartment models (seeChapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination), but three-compartment models appear to be more appropriate for the compounds with the highest lipid solubility. Accordingly, there is rapid uptake of benzodiazepines into the brain and other highly perfused organs after intravenous administration (or oral administration of a rapidly absorbed compound); rapid uptake is followed by a phase of redistribution into tissues that are less well perfused, especially muscle and fat. Redistribution is most rapid for drugs with the highest lipid solubility. In the regimens used for nighttime sedation, the rate of redistribution sometimes can have a greater influence than the rate of biotransformation on the duration of CNS effects (Dettli, in Symposium, 1986a). The kinetics of redistribution of diazepam and other lipophilic benzodiazepines are complicated by enterohepatic circulation. The volumes of distribution of the benzodiazepines are large, and in many cases are increased in elderly patients (Swift and Stevenson, in Symposium, 1983). These drugs cross the placental barrier and are secreted into breast milk. The benzodiazepines are metabolized extensively by enzymes in the cytochrome P450 family, particularly CYP3A4 and CYP2C19. Some benzodiazepines, such as oxazepam, are conjugated directly and are not metabolized by these enzymes (seeTanaka, 1999). Erythromycin, clarithromycin, ritonavir, itraconazole, ketoconazole, nefazodone, and grapefruit juice are inhibitors of CYP3A4 and can affect the metabolism of benzodiazepines (Dresser et al., 2000). Because active metabolites of some benzodiazepines are biotransformed more slowly than are the parent compounds, the duration of action of many benzodiazepines bears little relationship to the half-time of elimination of the drug that has been administered. For example, the half-life of flurazepam in plasma is 2 to 3 hours, but that of a major active metabolite (N-desalkylflurazepam) is 50 hours or more. Conversely, the rate of biotransformation of those agents that are inactivated by the initial reaction is an important determinant of their duration of action; these agents include oxazepam, lorazepam, temazepam, triazolam, and midazolam. Metabolism of the benzodiazepines occurs in three major stages. These and the relationships between the drugs and their metabolites are shown in Table 172. For those benzodiazepines that bear a substituent at position 1 (or 2) of the diazepine ring, the initial and most rapid phase of metabolism involves modification and/or removal of the substituent. With the exception of triazolam, alprazolam, estazolam, and midazolam, which contain either a fused triazolo or imidazolo ring, the eventual products are N-desalkylated compounds; these are all biologically active. One such compound, nordazepam, is a major metabolite common to the biotransformation of diazepam, clorazepate, prazepam, and halazepam; it also is formed from demoxepam, an important metabolite of chlordiazepoxide. The second phase of metabolism involves hydroxylation at position 3 and also usually yields an active derivative (e.g., oxazepam from nordazepam). The rates of these reactions are usually very much slower than the first stage (half-times greater than 40 to 50 hours), such that appreciable accumulation of hydroxylated products with intact substituents at position 1 does not occur. There are two significant exceptions to this rule: (1) Small amounts of temazepine accumulate during the chronic administration of diazepam (not shown in Table 172) and (2) following the replacement of sulfur with oxygen in quazepam, most of the resultant 2-oxoquazepam is slowly hydroxylated at position 3 without removal of the N-alkyl group. However, only small amounts of the 3-hydroxyl derivative accumulate during the chronic administration of quazepam, because this compound is conjugated at an unusually rapid rate. By contrast, the N-desalkylflurazepam that is formed by the 'minor' metabolic pathway does accumulate during quazepam administration, and it contributes significantly to the overall clinical effect. The third major phase of metabolism is the conjugation of the
3-hydroxyl compounds, principally with glucuronic acid; the half-times of
these reactions are usually between 6 and 12 hours, and the products are
invariably inactive. Conjugation is the only major route of metabolism
available for oxazepam and lorazepam, and it is the preferred pathway for temazepam
because of the slower conversion of this compound to oxazepam. Triazolam and alprazolam
are metabolized principally by initial hydroxylation of the methyl group on
the fused triazolo ring; the absence of a chlorine residue in ring C of
alprazolam slows this reaction significantly. The products, sometimes
referred to as Midazolam is metabolized rapidly, primarily by hydroxylation of the
methyl group on the fused imidazo ring; only small amounts of 3-hydroxyl
compounds are formed. The The aromatic rings (A and C) of the benzodiazepines are hydroxylated to only a small extent. The only important metabolism at these sites is the reduction of the 7-nitro substituents of clonazepam, nitrazepam, and flunitrazepam; the half-lives of these reactions are usually 20 to 40 hours. The resulting amines are inactive and are acetylated to varying degrees before excretion. Because the benzodiazepines apparently do not significantly induce the synthesis of hepatic microsomal enzymes, their chronic administration usually does not result in the accelerated metabolism of other substances or of the benzodiazepines. Cimetidine and oral contraceptives inhibit N-dealkylation and 3-hydroxylation of benzodiazepines. Ethanol, isoniazid, and phenytoin are less effective in this regard. These reactions usually are reduced to a greater extent in elderly patients and in patients with chronic liver disease than are those involving conjugation. Ideally, a useful hypnotic agent would have a rapid onset of action when taken at bedtime, a sufficiently sustained action to facilitate sleep throughout the night, and no residual action by the following morning. Among those benzodiazepines that are commonly used as hypnotic agents, triazolam theoretically fits this description most closely. Because of the slow rate of elimination of desalkylflurazepam, flurazepam (or quazepam) might seem to be unsuitable for this purpose. However, in practice there appear to be some disadvantages to the use of agents that have a relatively rapid rate of disappearance; these disadvantages include the early-morning insomnia that is experienced by some patients and a greater likelihood of rebound insomnia upon discontinuance of use (seeGillin et al., 1989; Roehrs et al., in Symposium, 1990b; Roth and Roehrs, 1992). With careful selection of dosage, flurazepam and other benzodiazepines with slower rates of elimination than triazolam can be used effectively (seeVogel, 1992). The biotransformation and pharmacokinetic properties of the benzodiazepines have been reviewed by Greenblatt (1991), Greenblatt and Wright (1993), Greenblatt et al. (1983a,b, 1991), and Hilbert and Battista (1991). Untoward Effects At the time of peak concentration in plasma, hypnotic doses of benzodiazepines can be expected to cause varying degrees of lightheadedness, lassitude, increased reaction time, motor incoordination, impairment of mental and motor functions, confusion, and anterograde amnesia. Cognition appears to be affected less than motor performance. All of these effects can greatly impair driving and other psychomotor skills. Interaction with ethanol may be especially serious. When the drug is given at the intended time of sleep, the persistence of these effects during the waking hours is adverse. These residual effects are clearly dose-related and can be insidious, since most subjects underestimate the degree of their impairment. Residual daytime sleepiness also may be present as an adverse effect, even though successful drug therapy can reduce the daytime sleepiness resulting from chronic insomnia (seeDement, 1991). The intensity and incidence of CNS toxicity generally increase with age; both pharmacokinetic and pharmacodynamic factors are involved (seeMeyer, 1982; Swift et al., in Symposium, 1983; Monane, 1992). Other relatively common side effects of benzodiazepines are weakness, headache, blurred vision, vertigo, nausea and vomiting, epigastric distress, and diarrhea; joint pains, chest pains, and incontinence may occur in a few recipients. Anticonvulsant benzodiazepines sometimes actually increase the frequency of seizures in patients with epilepsy. The possible adverse effects of alterations in the sleep pattern are discussed at the end of this chapter. Adverse Psychological Effects Benzodiazepines may cause paradoxical effects. For example, flurazepam
may occasionally increase the incidence of nightmares, especially during the
first week of use, and sometimes causes garrulousness, anxiety, irritability,
tachycardia, and sweating. Amnesia, euphoria, restlessness, hallucinations,
and hypomanic behavior have been reported to occur during use of various
benzodiazepines. The release of bizarre uninhibited behavior has been noted
in some users, while hostility and rage may occur in others; collectively,
these are sometimes referred to as disinhibition or dyscontrol
reactions. Paranoia, depression, and suicidal ideation also occasionally may
accompany the use of these agents. The incidence of such paradoxical or
disinhibition reactions is rare and appears to be dose-related. Because of
reports of an increased incidence of confusion and abnormal behaviors, triazolam
has been banned in the Chronic benzodiazepine use poses a risk for development of dependence and abuse, but not to the same extent as seem with older sedatives and other recognized drugs of abuse (Ulemhuth et al., 1999). Abuse of benzodiazepines includes the use of flunitrazepam (ROHYPNOL) as a 'date-rape' drug (Woods and Winger, 1997). Mild dependence may develop in many patients who have taken therapeutic doses of benzodiazepines on a regular basis for prolonged periods. Withdrawal symptoms may include temporary intensification of the problems that originally prompted their use (e.g., insomnia, anxiety). Dysphoria, irritability, sweating, unpleasant dreams, tremors, anorexia, and faintness or dizziness also may occur, especially when withdrawal of the benzodiazepine occurs abruptly (Petursson, 1994). Hence, it is prudent to taper the dosage gradually when therapy is to be discontinued. During conventional treatment regimens, very few individuals increase their intake without instructions to do so, and very few manifest compulsive drug-seeking behavior upon discontinuation of a benzodiazepine. Patients who have histories of drug or alcohol abuse are most apt to use these agents inappropriately, and abuse of benzodiazepines usually occurs as part of a pattern of abuse of multiple drugs. In such individuals, benzodiazepines seldom are preferred to barbiturates or even alcohol, but they often are combined with those drugs to either accentuate their effect (e.g., alcohol, opiates) or reduce their toxicity (e.g., cocaine). The use of high doses of benzodiazepines over prolonged periods can lead to more severe symptoms after discontinuing the drug, including agitation, depression, panic, paranoia, myalgia, muscle twitches, and even convulsions and delirium. Dependence on benzodiazepines and their abuse have been reviewed by Woods et al. (1992) and in a report edited by DuPont (1988). In spite of the adverse effects reviewed above, the benzodiazepines are relatively safe drugs. Even huge doses are rarely fatal unless other drugs are taken concomitantly. Ethanol is a common contributor to deaths involving benzodiazepines, and true coma is uncommon in the absence of another CNS depressant. Although overdosage with a benzodiazepine rarely causes severe cardiovascular or respiratory depression, therapeutic doses can further compromise respiration in patients with COPD or obstructive sleep apnea (see discussion of effects in Respiration, above). A wide variety of allergic, hepatotoxic, and hematologic reactions to the benzodiazepines may occur, but the incidence is quite low; these reactions have been associated with the use of flurazepam and triazolam but not with temazepam. Large doses taken just prior to or during labor may cause hypothermia, hypotonia, and mild respiratory depression in the neonate. Abuse by the pregnant mother can result in a withdrawal syndrome in the newborn. Except for additive effects with other sedative or hypnotic drugs, reports of clinically important, pharmacodynamic interactions between benzodiazepines and other drugs have been infrequent. Ethanol increases both the rate of absorption of benzodiazepines and the associated CNS depression. Valproate and benzodiazepines in combination may cause psychotic episodes. Pharmacokinetic interactions are mentioned above. Therapeutic Uses The therapeutic uses and routes of administration of individual
benzodiazepines that currently are marketed in the The use of the benzodiazepines as hypnotics and sedatives is discussed later in this chapter (see alsoSymposium, 1990b; Teboul and Chouinard, 1991; Vogel, 1992; Dement, 1992; Walsh and Engelhardt, 1992; Maczaj, 1993). The use of benzodiazepines as antianxiety agents and anticonvulsants is discussed in Chapters 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders and 21: Drugs Effective in the Therapy of the Epilepsies, respectively, and their roles in preanesthetic medication and anesthesia are described in Chapters 13: History and Principles of Anesthesiology and 14: General Anesthetics. The utility of benzodiazepines as muscle relaxants is discussed in Chapter 22: Treatment of Central Nervous System Degenerative Disorders. Novel Benzodiazepine-Receptor Agonists Hypnotics in this class include zolpicone (not available in the
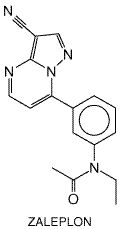
Zaleplon and zolpidem are effective in relieving sleep-onset insomnia. Both drugs have been approved by the FDA for use for up to 7 to 10 days at a time. There is evidence that both zaleplon and zolpidem have sustained hypnotic efficacy, without occurrence of rebound insomnia on abrupt discontinuation (Mitler, 2000; Walsh et al., 2000). Zaleplon and zolpidem have similar degrees of efficacy. Zolpidem has a half-life of about 2 hours, which is sufficient to cover most of a typical 8-hour sleep period, and is presently approved for bedtime use only. Zaleplon has a shorter half-life, about 1 hour, which offers up the possibility for safe dosing later in the night, within 4 hours of the anticipated rising time. As a result, zaleplon is approved for use immediately at bedtime or when the patient has difficulty falling asleep after bedtime. Because of its short half-life, zaleplon has not been shown to be different from placebo in measures of duration of sleep and number of awakenings. Zaleplon and zolpidem may differ in residual side effects; late-night administration of zolpidem has been associated with morning sedation, delayed reaction time, and anterograde amnesia, whereas zaleplon has had no more side effects than has placebo. The abuse potential of these drugs appears to be similar to that of benzodiazepines. Zaleplon Zaleplon (SONATA) is a nonbenzodiazepine and is a member of the pyrazolopyrimidine class of compounds. The structural formula is shown below.
Although its chemical structure is unrelated to that of
benzodiazepines, zaleplon preferentially binds to the benzodiazepine receptor
site on GABAA receptors containing the Zaleplon (usually administered in 5-, 10-, or 20-mg doses) has been studied in clinical trials on patients with chronic or transient insomnia (for a review, seeDooley and Plosker, 2000). Studies have focused on its effects in decreasing sleep latency. Zaleplon-treated subjects with either chronic or transient insomnia have experienced shorter periods of sleep latency than have placebo-treated subjects. Tolerance to zaleplon does not appear to occur, nor do rebound insomnia or withdrawal symptoms after stopping treatment. Zolpidem Zolpidem (AMBIEN) is a
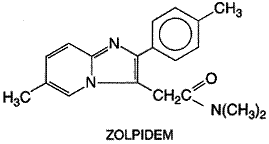
nonbenzodiazepine sedative-hypnotic drug that became available in the
Although the actions of zolpidem are due to agonist effects on benzodiazepine receptors and generally resemble those of benzodiazepines, it produces only weak anticonvulsant effects in experimental animals, and its relatively strong sedative actions appear to mask anxiolytic effects in various animal models of anxiety (seeLangtry and Benfield, 1990). Although the chronic administration of zolpidem to rodents produces neither tolerance to its sedative effects nor signs of withdrawal when the drug is discontinued and flumazenil is injected (Perrault et al., 1992), evidence of tolerance and physical dependence has been observed with chronic administration of zolpidem to baboons (Griffiths et al., 1992). Unlike the benzodiazepines, zolpidem has little effect on the stages of sleep in normal human subjects. The drug is as effective as benzodiazepines in shortening sleep latency and prolonging total sleep time in patients with insomnia. Following discontinuation of zolpidem, the beneficial effects on sleep have been reported to persist for up to one week (Herrmann et al., 1993), but mild rebound insomnia on the first night also has occurred (Anonymous, 1993). The development of tolerance and physical dependence has been seen only very rarely and under unusual circumstances (Cavallaro et al., 1993; Morselli, 1993). Indeed, zolpidem-induced improvement in sleep time of chronic insomniacs was found in one study to be sustained during as much as 6 months of treatment without signs of withdrawal or rebound after stopping the drug (Kummer et al., 1993). Nevertheless, zolpidem currently is approved only for the short-term treatment of insomnia despite the apparently benign consequences of its chronic administration. At therapeutic doses (10 to 20 mg; 5 to 10 mg in elderly patients), zolpidem infrequently produces residual daytime sedation or amnesia, and the incidence of other adverse effects (e.g., gastrointestinal complaints, dizziness) also is low. Like the benzodiazepines, large overdoses of zolpidem do not produce severe respiratory depression unless other agents (e.g., alcohol) also are ingested (Garnier et al., 1994). Hypnotic doses increase the hypoxia and hypercarbia of patients with obstructive sleep apnea. Zolpidem is absorbed readily from the gastrointestinal tract; first-pass hepatic metabolism results in an oral bioavailability of about 70%, but this value is lower when the drug is ingested with food because of slowed absorption and increased hepatic blood flow. Zolpidem is eliminated almost entirely by conversion to inactive products in the liver, largely through oxidation of the methyl groups on the phenyl and imidazopyridine rings to the corresponding carboxylic acids. Its half-life in plasma is approximately 2 hours in individuals with normal hepatic blood flow or function. This value may be increased twofold or more in those with cirrhosis, and it also tends to be greater in older patients; adjustment of dosage often is necessary in both categories of patients. Although little or no unchanged zolpidem is found in the urine, the elimination of the drug is slower in patients with chronic renal insufficiency, largely owing to an increase in its apparent volume of distribution. The properties of zolpidem and its therapeutic utility have been reviewed by Langtry and Benfield (1990) and by Hoehns and Perry (1993). Flumazenil: A Benzodiazepine-Receptor Antagonist Flumazenil ROMAZICON) is an imidazobenzodiazepine (seeTable
171) that behaves as a specific benzodiazepine antagonist. It is the first
such agent to undergo an extensive clinical trial, and it was released for
clinical use in 1991. As noted above, flumazenil binds with high affinity to
specific sites, where it competitively antagonizes the binding and allosteric
effects of benzodiazepines and other ligands. Both the electrophysiological
and behavioral effects of agonist or inverse-agonist benzodiazepines or Flumazenil is available only for intravenous administration. Although it is rapidly absorbed after oral administration, less than 25% of the drug reaches the systemic circulation as a result of extensive first-pass hepatic metabolism; effective oral doses are apt to cause headache and dizziness (Roncari et al., 1993). Upon intravenous administration, flumazenil is eliminated almost entirely by hepatic metabolism to inactive products with a half-life of about 1 hour; the duration of clinical effects is thus brief, and they usually persist for only 30 to 60 minutes. The primary indications for the use of flumazenil are the management of suspected benzodiazepine overdose and the reversal of sedative effects produced by benzodiazepines administered during either general anesthesia or diagnostic and/or therapeutic procedures. The administration of a series of small injections is preferred to a single bolus injection. A total of 1 mg of flumazenil given over 1 to 3 minutes usually is sufficient to abolish the effects of therapeutic doses of benzodiazepines; patients with suspected benzodiazepine overdose should respond adequately to a cumulative dose of 1 to 5 mg given over 2 to 10 minutes, and a lack of response to 5 mg of flumazenil strongly suggests that a benzodiazepine is not the major cause of sedation. Additional courses of treatment with flumazenil may be necessary within 20 to 30 minutes should sedation reappear. Flumazenil is not effective in single-drug overdoses with either barbiturates or tricyclic antidepressants. To the contrary, the administration of flumazenil may be associated with the onset of seizures under these circumstances; the risk of seizures is especially high in patients poisoned with tricyclic antidepressants (Spivey, 1992). Seizures or other withdrawal symptoms also may be precipitated in patients who had been taking benzodiazepines for protracted periods and in whom tolerance and/or dependence may have developed. The properties and therapeutic uses of flumazenil have been reviewed by Hoffman and Warren (1993). |
Barbiturates
|
The barbiturates enjoyed a long period of extensive use as sedative-hypnotic drugs; however, except for a few specialized uses, they have been largely replaced by the much safer benzodiazepines. A more detailed description of the barbiturates can be found in the fifth edition of this textbook. Chemistry Barbituric acid is 2,4,6-trioxohexahydropyrimidine. This compound lacks central-depressant activity, but the presence of alkyl or aryl groups at position 5 confers sedative-hypnotic and sometimes other activities. The general structural formula for the barbiturates and the structures of selected compounds are included in Table 174. The carbonyl group at position 2 takes on acidic character because of lactamlactim ('keto''enol') tautomerization, which is favored by its location between the two electronegative amido nitrogens. The lactim form is favored in alkaline solution, and salts result. Barbiturates in which the oxygen at C2 is replaced by sulfur are sometimes called thiobarbiturates. These compounds are more lipid-soluble than the corresponding oxybarbiturates. In general, structural changes that increase lipid solubility decrease duration of action, decrease latency to onset of activity, accelerate metabolic degradation, and often increase hypnotic potency. Pharmacological Properties The barbiturates reversibly depress the activity of all excitable tissues. The CNS is exquisitely sensitive, and, even when barbiturates are given in anesthetic concentrations, direct effects on peripheral excitable tissues are weak. However, serious deficits in cardiovascular and other peripheral functions occur in acute barbiturate intoxication. Central Nervous System The barbiturates can produce all degrees of depression of the CNS, ranging from mild sedation to general anesthesia. The use of barbiturates for general anesthesia is discussed in Chapter 14: General Anesthetics. Certain barbiturates, particularly those containing a 5-phenyl substituent (phenobarbital, mephobarbital), have selective anticonvulsant activity (seeChapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania). The antianxiety properties of the barbiturates are not equivalent to those exerted by the benzodiazepines, especially with respect to the degree of sedation that is produced. The barbiturates may have euphoriant effects. Except for the anticonvulsant activities of phenobarbital and its congeners, the barbiturates possess a low degree of selectivity and therapeutic index. Thus, it is not possible to achieve a desired effect without evidence of general depression of the CNS. Pain perception and reaction are relatively unimpaired until the moment of unconsciousness, and in small doses the barbiturates increase the reaction to painful stimuli. Hence, they cannot be relied upon to produce sedation or sleep in the presence of even moderate pain. In some individuals and in some circumstances, such as in the presence of pain, barbiturates cause overt excitement instead of sedation. The fact that such paradoxical excitement occurs with other CNS depressants suggests that it may result from depression of inhibitory centers. Effects on Stages of Sleep Hypnotic doses of barbiturates increase the total sleep time and alter the stages of sleep in a dose-dependent manner. Like the benzodiazepines, these drugs decrease sleep latency, the number of awakenings, and the durations of REM and slow-wave sleep. During repetitive nightly administration, some tolerance to the effects on sleep occurs within a few days, and the effect on total sleep time may be reduced by as much as 50% after 2 weeks of use. Discontinuation leads to rebound increases in all the parameters reported to be decreased by barbiturates. Tolerance Both pharmacodynamic (functional) and pharmacokinetic tolerance to barbiturates can occur. The former contributes more to the decreased effect than does the latter. With chronic administration of gradually increasing doses, pharmacodynamic tolerance continues to develop over a period of weeks to months, depending on the dosage schedule, whereas pharmacokinetic tolerance reaches its peak in a few days to a week. Tolerance to the effects on mood, sedation, and hypnosis occurs more readily and is greater than that to the anticonvulsant and lethal effects; thus, as tolerance increases, the therapeutic index decreases. Pharmacodynamic tolerance to barbiturates confers tolerance to all general CNS-depressant drugs, including ethanol. Abuse and Dependence Like other CNS-depressant drugs, barbiturates are abused, and some individuals develop a dependence upon them. These topics are discussed in Chapter 24: Drug Addiction and Drug Abuse. Sites and Mechanisms of Action on the CNS Barbiturates act throughout the CNS; nonanesthetic doses preferentially suppress polysynaptic responses. Facilitation is diminished, and inhibition usually is enhanced. The site of inhibition is either postsynaptic, as at cortical and cerebellar pyramidal cells and in the cuneate nucleus, substantia nigra, and thalamic relay neurons, or presynaptic, as in the spinal cord. Enhancement of inhibition occurs primarily at synapses where neurotransmission is mediated by GABA acting at GABAA receptors. The barbiturates exert several distinct effects on excitatory and
inhibitory synaptic transmission. For example, ()-pentobarbital potentiates
GABA-induced increases in chloride conductance and depresses
voltage-activated Ca2+ currents at similar concentrations (below
10 As noted earlier in this chapter, the mechanisms underlying the
actions of barbiturates on GABAA receptors appear to be distinct
from those of either GABA or the benzodiazepines, for reasons that include
the following: (1) Although barbiturates also enhance the binding of GABA to
GABAA receptors in a chloride-dependent and picrotoxin-sensitive
fashion, they promote (rather than displace) the binding of benzodiazepines.
(2) Barbiturates potentiate GABA-induced chloride currents by prolonging
periods during which bursts of channel opening occur rather than by
increasing the frequency of these bursts, as benzodiazepines do. (3) Only Subanesthetic concentrations of barbiturates also can reduce glutamate-induced depolarizations (seeChapter 12: Neurotransmission and the Central Nervous System; Macdonald and McLean, 1982); only the AMPA subtypes of glutamate receptors sensitive to kainate or quisqualate appear to be affected (Marszalec and Narahashi, 1993). Recombinant AMPA receptors also are blocked by barbiturates. At higher concentrations that produce anesthesia, pentobarbital suppresses high-frequency, repetitive firing of neurons, apparently as a result of inhibiting the function of voltage-dependent, tetrodotoxin-sensitive Na+ channels; in this case, however, both stereoisomers are about equally effective (Frenkel et al., 1990). At still higher concentrations, voltage-dependent K+ conductances are reduced. Taken together, the findings that barbiturates activate inhibitory GABAA receptors and inhibit excitatory AMPA receptors can explain the CNS-depressant effects of these agents. The mechanism of action of barbiturates has been reviewed by Saunders and Ho (1990). Peripheral Nervous Structures Barbiturates selectively depress transmission in autonomic ganglia and reduce nicotinic excitation by choline esters. This effect may account, at least in part, for the fall in blood pressure produced by intravenous oxybarbiturates and by severe barbiturate intoxication. At skeletal neuromuscular junctions, the blocking effects of both tubocurarine and decamethonium are enhanced during barbiturate anesthesia. These actions probably result from the capacity of barbiturates at hypnotic or anesthetic concentrations to inhibit the passage of current through nicotinic cholinergic receptors. Several distinct mechanisms appear to be involved, and little stereoselectivity is evident (Roth et al., 1989). Respiration Barbiturates depress both the respiratory drive and the mechanisms responsible for the rhythmic character of respiration. The neurogenic drive is diminished by hypnotic doses, but usually no more so than during natural sleep. However, neurogenic drive is essentially eliminated by a dose three times greater than that normally used to induce sleep. Such doses also suppress the hypoxic drive and, to a lesser extent, the chemoreceptor drive. At still higher doses, the powerful hypoxic drive also fails. However, the margin between the lighter planes of surgical anesthesia and dangerous respiratory depression is sufficient to permit the ultra-short-acting barbiturates to be used, with suitable precautions, as anesthetic agents. The barbiturates only slightly depress protective reflexes until the degree of intoxication is sufficient to produce severe respiratory depression. Coughing, sneezing, hiccoughing, and laryngospasm may occur when barbiturates are employed as intravenous anesthetic agents. Indeed, laryngospasm is one of the chief complications of barbiturate anesthesia. Cardiovascular System When given orally in sedative or hypnotic doses, the barbiturates do not produce significant overt cardiovascular effects except for a slight decrease in blood pressure and heart rate such as occurs in normal sleep. In general, the effects of thiopental anesthesia on the cardiovascular system are benign in comparison with those of the volatile anesthetic agents; there is usually either no change or a fall in mean arterial pressure. Apparently, a decrease in cardiac output usually is sufficient to offset an increase in total calculated peripheral resistance, which sometimes is accompanied by an increase in heart rate. Cardiovascular reflexes are obtunded by partial inhibition of ganglionic transmission. This is most evident in patients with congestive heart failure or hypovolemic shock whose reflexes already are operating maximally and in whom barbiturates can cause an exaggerated fall in blood pressure. Because barbiturates also impair reflex cardiovascular adjustments to inflation of the lung, positive-pressure respiration should be used cautiously and only when necessary to maintain adequate pulmonary ventilation in patients who are anesthetized or intoxicated with a barbiturate. Other cardiovascular changes often noted when thiopental and other intravenous thiobarbiturates are administered after conventional preanesthetic medication include a decrease in renal plasma flow and in cerebral blood flow, with a marked fall in CSF pressure. Although cardiac arrhythmias are observed only infrequently, intravenous anesthesia with barbiturates can increase the incidence of ventricular arrhythmias, especially when epinephrine and halothane are also present. Anesthetic concentrations of barbiturates have direct electrophysiological effects in the heart; in addition to depressing Na+ channels, they reduce the function of at least two types of K+ channels (Nattel et al., 1990; Pancrazio et al., 1993). However, direct depression of cardiac contractility occurs only when doses several times those required to cause anesthesia are administered, which probably contributes to the cardiovascular depression that accompanies acute barbiturate poisoning. Gastrointestinal Tract The oxybarbiturates tend to decrease the tonus of the gastrointestinal musculature and the amplitude of rhythmic contractions. The locus of action is partly peripheral and partly central, depending on the dose. A hypnotic dose does not significantly delay gastric emptying in human beings. The relief of various gastrointestinal symptoms by sedative doses is probably largely due to the central-depressant action. Liver The best-known effects of barbiturates on the liver are those on the microsomal drug-metabolizing system (seeChapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination). Acutely, the barbiturates combine with several species of cytochrome P450 and competitively interfere with the biotransformation of a number of other drugs as well as of endogenous substrates, such as steroids; other substrates may reciprocally inhibit barbiturate biotransformations. Drug interactions may result even when the other substances and barbiturates are oxidized by different microsomal enzyme systems. The chronic administration of barbiturates causes a marked increase in
the protein and lipid content of the hepatic smooth endoplasmic reticulum, as
well as in the activities of glucuronyl transferase and the oxidases
containing cytochrome P450. The inducing effect on these enzymes results in
an increased rate of metabolism of a number of drugs and endogenous
substances, including steroid hormones, cholesterol, bile salts, and vitamins
K and D. An increase in the rate of barbiturate metabolism also results,
which accounts for part of the tolerance to barbiturates. Many
sedative-hypnotics, various anesthetics, and ethanol also are metabolized by
and/or induce the microsomal enzymes, and some degree of cross-tolerance can
occur on this basis. Not all microsomal biotransformations of drugs and
endogenous substrates are affected to the same degree, but a convenient rule
of thumb is that, at maximal induction in human beings, the rates are
approximately doubled. The inducing effect is not limited to the microsomal
enzymes; for example, there is an increase in Kidney Severe oliguria or anuria may occur in acute barbiturate poisoning, largely as a result of the marked hypotension. Absorption, Fate, and Excretion For sedative-hypnotic use, the barbiturates usually are administered orally (seeTable 174). Such doses are rapidly and probably completely absorbed; sodium salts are absorbed more rapidly than the corresponding free acids, especially from liquid formulations. The onset of action varies from 10 to 60 minutes, depending on the agent and the formulation, and is delayed by the presence of food in the stomach. When necessary, intramuscular injections of solutions of the sodium salts should be placed deeply into large muscles in order to avoid the pain and possible necrosis that can result at more superficial sites. With some agents, special preparations are available for rectal administration. The intravenous route is usually reserved for the management of status epilepticus (phenobarbital sodium) or for the induction and/or maintenance of general anesthesia (e.g., thiopental, methohexital). Barbiturates are distributed widely and readily cross the placenta. The highly lipid-soluble barbiturates, led by those used to induce anesthesia, undergo redistribution after intravenous injection. Uptake into less vascular tissues, especially muscle and fat, leads to a decline in the concentration of barbiturate in the plasma and brain. With thiopental and methohexital, this results in the awakening of patients within 5 to 15 minutes of the injection of the usual anesthetic doses. With the exception of the less lipid-soluble aprobarbital and phenobarbital, nearly complete metabolism and/or conjugation of barbiturates in the liver precedes their renal excretion. The oxidation of radicals at C5 is the most important biotransformation responsible for termination of biological activity. Oxidation results in the formation of alcohols, ketones, phenols, or carboxylic acids, which may appear in the urine as such or as glucuronic acid conjugates. In some instances (e.g., phenobarbital), N-glycosylation is an important metabolic pathway. Other biotransformations include N-hydroxylation, desulfuration of thiobarbiturates to oxybarbiturates, opening of the barbituric acid ring, and N-dealkylation of N-alkylbarbiturates to active metabolites (e.g., mephobarbital to phenobarbital). About 25% of phenobarbital and nearly all of aprobarbital are excreted unchanged in the urine. Their renal excretion can be greatly increased by osmotic diuresis and/or alkalinization of the urine. The metabolic elimination of barbiturates is more rapid in young people than in the elderly and infants, and half-lives are increased during pregnancy, partly because of the expanded volume of distribution. Chronic liver disease, especially cirrhosis, often increases the half-life of the biotransformable barbiturates. Repeated administration, especially of phenobarbital, shortens the half-life of barbiturates that are metabolized as a result of the induction of microsomal enzymes (see above). The data on half-lives given in Table 174 show that none of the barbiturates used for hypnosis in the United States appears to have an elimination half-life that is short enough for elimination to be virtually complete in 24 hours. However, the relationship between duration of action and half-time of elimination is complicated in part by the fact that enantiomers of optically active barbiturates often differ in both biological potencies and rates of biotransformation. Nevertheless, all of these barbiturates will accumulate during repetitive administration unless appropriate adjustments in dosage are made. Furthermore, the persistence of the drug in plasma during the day favors the development of tolerance and abuse. Untoward Effects After Effects Drowsiness may last for only a few hours after a hypnotic dose of barbiturate, but residual depression of the CNS sometimes is evident the following day. Even in the absence of overt evidence of residual depression, subtle distortions of mood and impairment of judgment and fine motor skills may be demonstrable. For example, a 200-mg dose of secobarbital has been shown to impair performance of driving or flying skills for 10 to 22 hours. Residual effects also may take the form of vertigo, nausea, vomiting, or diarrhea, or sometimes may be manifested as overt excitement. The user may awaken slightly intoxicated and feel euphoric and energetic; later, as the demands of daytime activities challenge possibly impaired faculties, the user may display irritability and temper. Paradoxical Excitement In some persons, barbiturates repeatedly produce excitement rather than depression, and the patient may appear to be inebriated. This type of idiosyncrasy is relatively common among geriatric and debilitated patients and occurs most frequently with phenobarbital and N-methylbarbiturates. Pain Barbiturates have been prescribed for localized or diffuse myalgic, neuralgic, or arthritic pain but often do not effectively treat these symptoms, especially in psychoneurotic patients with insomnia. Barbiturates may cause restlessness, excitement, and even delirium when given in the presence of pain and may make a patient's perception of pain worse. Hypersensitivity Allergic reactions occur especially in persons who tend to have asthma, urticaria, angioedema, and similar conditions. Hypersensitivity reactions in this category include localized swellings, particularly of the eyelids, cheeks, or lips, and erythematous dermatitis. Rarely, exfoliative dermatitis may be caused by phenobarbital and can prove fatal; the skin eruption may be associated with fever, delirium, and marked degenerative changes in the liver and other parenchymatous organs. Drug Interactions Barbiturates combine with other CNS depressants to cause severe depression; ethanol is the most frequent offender, and interactions with antihistamines are also common. Isoniazid, methylphenidate, and monoamine oxidase inhibitors also increase the CNS-depressant effects. Barbiturates competitively inhibit the metabolism of certain other drugs; however, the greatest number of drug interactions results from induction of hepatic microsomal enzymes and the accelerated disappearance of many drugs and endogenous substances. The metabolism of vitamins D and K are accelerated, which may hamper bone mineralization and lower Ca2+ absorption in patients taking phenobarbital and may be responsible for the reported instances of coagulation defects in neonates whose mothers had been taking phenobarbital. Hepatic enzyme induction enhances metabolism of endogenous steroid hormones, which may cause endocrine disturbances, as well as of oral contraceptives, which may result in unwanted pregnancy. Barbiturates also induce the hepatic generation of toxic metabolites of chlorocarbon anesthetics and carbon tetrachloride and consequently promote lipid peroxidation, which facilitates the periportal necrosis of the liver caused by these agents. Other Untoward Effects Because barbiturates enhance porphyrin synthesis, they are absolutely contraindicated in patients with acute intermittent porphyria or porphyria variegata. In hypnotic doses, the effects of barbiturates on the control of respiration are minor; however, in the presence of pulmonary insufficiency, serious respiratory depression may occur, and the drugs are thus contraindicated. Rapid intravenous injection of a barbiturate may cause cardiovascular collapse before anesthesia ensues, so that the CNS signs of depth of anesthesia may fail to give an adequate warning of impending toxicity. Blood pressure can fall to shock levels; even slow intravenous injection of barbiturates often produces apnea and occasionally laryngospasm, coughing, and other respiratory difficulties. Barbiturate Poisoning The incidence of barbiturate poisoning has declined markedly in recent years, largely as a result of the decline in the use of these drugs as sedative-hypnotic agents. Nevertheless, poisoning with barbiturates is a significant clinical problem; death occurs in a few percent of cases. Most of the cases are the result of deliberate attempts at suicide, but some are from accidental poisonings in children or in drug abusers. The lethal dose of barbiturate varies with many factors, but severe poisoning is likely to occur when more than ten times the full hypnotic dose has been ingested at once. If alcohol or other depressant drugs are also present, the concentrations that can cause death are lower. In severe intoxication, the patient is comatose; respiration is affected early. Breathing may be either slow or else rapid and shallow. Superficial observation of respiration may be misleading with regard to actual minute volume and to the degree of respiratory acidosis and cerebral hypoxia. Eventually, blood pressure falls owing to the effect of the drug and of hypoxia on medullary vasomotor centers; depression of cardiac contractility and sympathetic ganglia also contribute. Pulmonary complications (atelectasis, edema, and bronchopneumonia) and renal failure are likely to be the fatal complications of severe barbiturate poisoning. The optimal treatment of acute barbiturate intoxication is based on general supportive measures. Hemodialysis or hemoperfusion is only rarely necessary, and the use of CNS stimulants increases the rate of mortality. The present treatment is applicable in most respects to poisoning by any CNS depressant. Constant attention must be given to the maintenance of a patent airway and adequate ventilation and to the prevention of pneumonia; oxygen should be administered. After precautions to avoid aspiration, gastric lavage should be considered if fewer than 24 hours have elapsed since ingestion, since the barbiturate can reduce gastrointestinal motility. After lavage, the administration of activated charcoal and a cathartic such as sorbitol may shorten the half-life of the less lipid-soluble agents such as phenobarbital. If renal and cardiac function are satisfactory and the patient is hydrated, forced diuresis and alkalinization of the urine will hasten the excretion of aprobarbital and phenobarbital. Measures to prevent or treat atelectasis should be taken, and mechanical ventilation should be initiated when indicated. In severe acute barbiturate intoxication, circulatory collapse is a major threat. Often the patient is admitted to the hospital with severe hypotension or shock, and dehydration is often severe. Hypovolemia must be corrected, and, if necessary, the blood pressure can be supported with dopamine. Acute renal failure consequent to shock and hypoxia accounts for perhaps one-sixth of the deaths. In the event of renal failure, hemodialysis should be instituted. Intoxication by barbiturates and its management have been reviewed by Gary and Tresznewsky (1983). Therapeutic Uses The use of barbiturates as sedative-hypnotic drugs has declined enormously because they lack specificity of effect in the CNS, they have a lower therapeutic index than do the benzodiazepines, tolerance occurs more frequently than with benzodiazepines, the liability for abuse is greater, and the number of drug interactions is considerable. The major uses of individual barbiturates are listed in Table 174. Like the benzodiazepines, selection of particular barbiturates for a given therapeutic indication is based primarily on pharmocokinetic considerations. CNS Uses Although barbiturates largely have been replaced by benzodiazepines and other compounds for daytime sedation, phenobarbital and butabarbital are still available as 'sedatives' in a host of combinations of questionable efficacy for the treatment of functional gastrointestinal disorders and asthma. They also are included in analgesic combinations, possibly counterproductively. Barbiturates, especially butabarbital and phenobarbital, are sometimes used to antagonize unwanted CNS-stimulant effects of various drugs, such as ephedrine, dextroamphetamine, and theophylline, although a preferred approach is adjustment of dosage or substitution of alternative therapy for the primary agents. Phenobarbital still is a widely used, and probably the only effective, treatment for hypnosedative withdrawal (Martin et al., 1979). Barbiturates are still employed in the emergency treatment of convulsions, such as occur in tetanus, eclampsia, status epilepticus, cerebral hemorrhage, and poisoning by convulsant drugs; however, benzodiazepines generally are superior in these uses. Phenobarbital sodium is most frequently used because of its anticonvulsant efficacy; however, even when administered intravenously, 15 minutes or more may be required for it to attain peak concentrations in the brain. The ultrashort- and short-acting barbiturates have a low ratio of anticonvulsant to hypnotic action, and these drugs or inhalational anesthetic agents are employed only when general anesthesia must be used to control seizures refractory to other measures. Diazepam usually is chosen for the emergency treatment of seizures. The use of barbiturates in the symptomatic therapy of epilepsy is discussed in Chapter 21: Drugs Effective in the Therapy of the Epilepsies. Ultrashort-acting agents such as thiopental or methohexital continue to be employed as intravenous anesthetics (Chapter 14: General Anesthetics). In children, the rectal administration of methohexital sometimes is used for the induction of anesthesia or for sedation during imaging procedures (Manuli and Davies, 1993). Short- and ultrashort-acting barbiturates occasionally are used as adjuncts to other agents in the production of obstetrical anesthesia. Several studies have failed to affirm gross depression of respiration in full-term infants, but premature infants clearly are more susceptible. Since evaluation of the effects on the fetus and neonate is difficult, it is prudent to avoid the use of barbiturates in obstetrics. The barbiturates are employed as diagnostic and therapeutic aids in psychiatry; these uses sometimes are referred to as narcoanalysis and narcotherapy, respectively. In low concentrations, amobarbital has been administered directly into the carotid artery prior to neurosurgery as a means of identifying the dominant cerebral hemisphere for speech. The use of this procedure has been expanded to include a more extensive neuropsychological evaluation of patients with medically intractable seizure disorders who may benefit from surgical therapy (seeSmith and Riskin, 1991). Anesthetic doses of barbiturates attenuate cerebral edema resulting from surgery, head injury, or cerebral ischemia, and they may decrease infarct size and increase survival. General anesthetics do not provide protection. The procedure is not without serious danger, however, and the ultimate benefit to the patient has been questioned (seeShapiro, 1985; Smith and Riskin, 1991). Hepatic Metabolic Uses Because hepatic glucuronyl transferase and the bilirubin-binding Y protein are increased by the barbiturates, phenobarbital has been used successfully to treat hyperbilirubinemia and kernicterus in the neonate. The nondepressant barbiturate phetharbital (N-phenylbarbital) works equally well. Phenobarbital may improve the hepatic transport of bilirubin in patients with hemolytic jaundice. |
Miscellaneous Sedative-Hypnotic Drugs
|
Over the years, many drugs with diverse structures have been used for their sedative-hypnotic properties, including paraldehyde (introduced before the barbiturates), chloral hydrate, ethchlorvynol, glutethimide, methyprylon, ethinamate, and meprobamate (introduced just before the benzodiazepines). With the exception of meprobamate, the pharmacological actions of these drugs generally resemble those of the barbiturates: They all are general CNS depressants that can produce profound hypnosis with little or no analgesia; their effects on the stages of sleep are similar to those of the barbiturates; their therapeutic index is limited, and acute intoxication, which produces respiratory depression and hypotension, is managed similarly to barbiturate poisoning; their chronic use can result in tolerance and physical dependence; and the syndrome following chronic use can be severe and life threatening. The properties of meprobamate bear some resemblance to those of the benzodiazepines, but the drug has a distinctly higher potential for abuse and has less selective antianxiety effects. The clinical use of these agents has decreased markedly, and deservedly so. Nevertheless, some of them are useful in certain settings, particularly in hospitalized patients. The chemical structures and major pharmacological properties of paraldehyde, ethchlorvynol, chloral hydrate, and meprobamate are presented in Table 175. Further information on glutethimide, methyprylon, and ethinamate can be found in previous editions of this book. Paraldehyde Paraldehyde is a polymer of acetaldehyde, but it perhaps is best regarded as a polyether of cyclic structure. It has a strong aromatic odor and a disagreeable taste. Orally, it is irritating to the throat and stomach, and it is not administered parenterally because of its injurious effects on tissues. When given rectally as a retention enema, the drug is diluted with olive oil. Oral paraldehyde is rapidly absorbed and widely distributed; sleep usually ensues in 10 to 15 minutes after hypnotic doses. About 70% to 80% of a dose is metabolized in the liver, probably by depolymerization to acetaldehyde and subsequent oxidation to acetic acid, which is ultimately converted to carbon dioxide and water; most of the remainder is exhaled, producing a strong, characteristic smell to the breath. Commonly observed consequences of poisoning with the drug include acidosis, bleeding gastritis, and fatty changes in the liver and kidney with toxic hepatitis and nephrosis. The clinical uses of paraldehyde include the treatment of abstinence phenomena (especially delirium tremens in hospitalized patients) and other psychiatric states characterized by excitement. Paraldehyde also has been used for the treatments of convulsions (including status epilepticus) in children. Individuals who become addicted to paraldehyde may have become acquainted with the drug during treatment of their alcoholism and then, surprisingly in view of its disagreeable taste and odor, prefer it to alcohol. Chloral Hydrate Chloral hydrate is formed by adding one molecule of water to the carbonyl group of chloral (2,2,2-trichloroacetaldehyde). In addition to its hypnotic use, the drug has been employed in the past for the production of sedation in children undergoing diagnostic, dental, or other potentially uncomfortable procedures. Chloral hydrate is rapidly reduced to the active compound, trichloroethanol (CCl3CH2OH), largely by alcohol dehydrogenase in the liver; significant amounts of chloral hydrate are not found in the blood after its oral administration. Therefore, its pharmacological effects probably are caused by trichloroethanol. Indeed, the latter compound can exert barbiturate-like effects on GABAA receptor channels in vitro (Lovinger et al., 1993). Trichloroethanol is mainly conjugated with glucuronic acid, and the product (urochloralic acid) is excreted mostly into the urine. Chloral hydrate is irritating to the skin and mucous membranes. These irritant actions give rise to an unpleasant taste, epigastric distress, nausea, and occasional vomiting, all of which are particularly likely to occur if the drug is insufficiently diluted or if it is taken on an empty stomach. Undesirable CNS effects include light-headedness, malaise, ataxia, and nightmares. 'Hangover' also may occur, although it is less common than with most barbiturates and some benzodiazepines. Rarely, patients exhibit idiosyncratic reactions to chloral hydrate and may be disoriented and incoherent and show paranoid behavior. Acute poisoning by chloral hydrate may cause icterus. Individuals using chloral hydrate chronically may exhibit sudden, acute intoxication, which can be fatal; this situation results either from an overdose or from a failure of the detoxification mechanism owing to hepatic damage; parenchymatous renal injury also may occur. Sudden withdrawal from the habitual use of chloral hydrate may result in delirium and seizures, with a high frequency of death when untreated. Ethchlorvynol In addition to pharmacological actions that are very similar to those of barbiturates, ethchlorvynol has anticonvulsant and muscle relaxant properties. Ethchlorvynol is rapidly absorbed and widely distributed following oral administration. Two-compartment kinetics is manifest, with a distribution half-life of about 1 to 3 hours and an elimination half-life of 10 to 20 hours. As a result, the duration of action of the drug is relatively short, and early morning awakening may occur after its administration at bedtime. Approximately 90% of the drug eventually is destroyed in the liver. Ethchlorvynol is used as a short-term hypnotic for the management of insomnia. The most common side effects caused by ethchlorvynol are mint-like aftertaste, dizziness, nausea, vomiting, hypotension, and facial numbness. Mild 'hangover' also is relatively common. An occasional patient responds with profound hypnosis, muscular weakness, and syncope unrelated to marked hypotension. Idiosyncratic responses range from mild stimulation to marked excitement and hysteria. Hypersensitivity reactions include urticaria, rare but sometimes fatal thrombocytopenia, and occasionally cholestatic jaundice. Acute intoxication resembles that produced by barbiturates, except for more severe respiratory depression and a relative bradycardia. Ethchlorvynol may enhance the hepatic metabolism of other drugs such as oral anticoagulants, and it is contraindicated in patients with intermittent porphyria. Meprobamate Meprobamate is a bis-carbamate ester; it was introduced as an
antianxiety agent in 1955, and this remains its only approved use in the The pharmacological properties of meprobamate resemble those of the benzodiazepines in a number of ways. Like the benzodiazepines, meprobamate can release suppressed behaviors in experimental animals at doses that cause little impairment of locomotor activity, and, although it can cause widespread depression of the CNS, it cannot produce anesthesia. Unlike the benzodiazepines, ingestion of large doses of meprobamate alone may cause severe or even fatal respiratory depression, hypotension, shock, and heart failure. Meprobamate appears to have a mild analgesic effect in patients with musculoskeletal pain, and it enhances the analgesic effects of other drugs. Meprobamate is well absorbed when administered orally. Nevertheless, an important aspect of intoxication with meprobamate is the formation of gastric bezoars consisting of undissolved meprobamate tablets; hence, treatment may require endoscopy, with mechanical removal of the bezoar. Most of the drug is metabolized in the liver, mainly to a side-chain hydroxy derivative and a glucuronide; the kinetics of elimination may be dependent on the dose. The half-life of meprobamate may be prolonged during its chronic administration, even though the drug can induce some hepatic microsomal enzymes. The major unwanted effects of the usual sedative doses of meprobamate are drowsiness and ataxia; larger doses produce considerable impairment of learning and motor coordination and prolongation of reaction time. Like the benzodiazepines, meprobamate enhances the CNS depression produced by other drugs. The abuse of meprobamate has continued despite a substantial decrease in the clinical use of the drug. Carisoprodol (SOMA), a skeletal muscle relaxant whose active metabolite is meprobamate, also has abuse potential and has become a popular 'street drug' (Reeves et al., 1999). Meprobamate is preferred to the benzodiazepines by subjects with a history of drug abuse. After long-term medication, abrupt discontinuation evokes a withdrawal syndrome usually characterized by anxiety, insomnia, tremors, and, frequently, hallucinations; generalized seizures occur in about 10% of cases. The intensity of symptoms depends on the dosage ingested. Others Etomidate AMIDATE) is used in the Clomethiazole has sedative, muscle relaxant, and anticonvulsant properties. It is
used outside the Nonprescription Hypnotic Drugs An advisory review panel of the FDA has recommended that, except for certain antihistamines (doxylamine, diphenhydramine, and pyrilamine), all putative active ingredients be eliminated from nonprescription sleep aids. Despite the prominent sedative side effects encountered during their use in the treatment of allergic diseases (seeChapter 25: Histamine, Bradykinin, and Their Antagonists), these antihistamines are not consistently effective in the treatment of sleep disorders. Contributing factors may include the rapid development of tolerance, paradoxical stimulation, and the inadequacy of the doses that currently are approved. Nevertheless, these doses sometimes produce prominent residual daytime CNS depression. For example, the elimination half-lives of doxylamine and diphenhydramine are about 9 hours. |
Management of Insomnia
|
Insomnia is one of the most common complaints in general medical practice and its treatment is predicated upon proper diagnosis. A variety of pharmacological agents are available for the treatment of insomnia. The 'perfect' hypnotic would allow sleep to occur, with normal sleep architecture, rather than produce a pharmacologically altered sleep pattern. It would not cause next-day effects, either of rebound anxiety or continued sedation. It would not interact with other medications. It could be used chronically without causing dependence or rebound insomnia on discontinuation. Regular moderate exercise meets these criteria, but often is not effective by itself, and patients with significant cardiorespiratory disease may not be able to exercise. However, even small amounts of exercise often are effective in promoting sleep. Although the precise function of sleep is not known, adequate sleep improves the quality of daytime wakefulness, and hypnotics should be used judiciously to avoid its impairment. Controversy in the management of insomnia revolves around two issues:
pharmacological versus nonpharmacological treatment and the use of
short-acting versus long-acting hypnotics. Benzodiazepine hypnotics
have been prescribed less commonly over the past decade. The British tend to
take a conservative attitude toward prescribing benzodiazepines, for either
anxiety or insomnia ( Two aspects of the management of insomnia traditionally have been underappreciated. They are a search for specific medical causes and the use of nonpharmacological treatments. In addition to appropriate pharmacological treatment, the management of insomnia should correct identifiable causes, address inadequate sleep hygiene, eliminate performance anxiety related to falling asleep, provide entrainment of the biological clock so that maximum sleepiness occurs at the hour of attempted sleep, and suppress the use of alcohol and over-the-counter sleep medications (Nino-Murcia, 1992). Categories of Insomnia The National Institute of Mental Health Consensus Development Conference (1984) divided insomnia into three categories:
Insomnia Accompanying Major Psychiatric Illnesses The insomnia caused by major psychiatric illnesses often responds to specific pharmacological treatment for that illness. For example, in major depressive episodes with insomnia, even such medications as the selective serotonin-reuptake inhibitors, which may cause insomnia as a side effect, usually will result in improved sleep as they treat the depressive syndrome. In patients whose depression is responding to the serotonin-reuptake inhibitor but who have persistent insomnia as a side effect of the medication, judicious use of evening trazodone may improve sleep (Nierenberg et al., 1994) as well as augment the antidepressant effect of the reuptake inhibitor. However, the patient should be monitored for priapism, orthostatic hypotension, and arrhythmias. Adequate control of anxiety in patients with anxiety disorders often
produces adequate resolution of the accompanying insomnia. Sedative use in
the anxiety disorders is decreasing because of a growing appreciation of the
effectiveness of other agents, such as Insomnia Accompanying Other Medical Illnesses For long-term insomnia due to other medical illnesses, adequate treatment of the underlying disorder, such as congestive heart failure, asthma, or chronic obstructive pulmonary disease, may resolve the insomnia. Adequate pain management in conditions of chronic pain, including terminal cancer pain, will treat both the pain and the insomnia and may make hypnotics unnecessary. Many patients simply manage their sleep poorly. Adequate attention to sleep hygiene, including reduced caffeine intake, avoidance of alcohol, adequate exercise, and regular sleep and wake times often will reduce the insomnia. Conditioned (Learned) Insomnia In those who have no major psychiatric or other medical illness and in whom attention to sleep hygiene is ineffective, attention should be directed to conditioned (learned) insomnia. These patients have associated the bedroom with activities consistent with wakefulness rather than sleep. In such patients, the bed should be used only for sex and sleep. All other activities associated with waking, even such quiescent activities as reading and watching television, should be done outside of the bedroom. Some patients complain of poor sleep but have been shown to have no objective polysomnographic evidence of insomnia. They are difficult to treat. Some patients are simply consitutional short sleepers, who do not need the typical seven to eight hours of sleep per day to function. If daytime wakefulness, mood, and functioning are unimpaired, no treatment is necessary. Some patients with sleep apnea may ask for sleeping pills because they do not feel rested in the morning. Hypnotic agents usually are contraindicated in such patients. These individuals benefit from all-night sleep studies for proper evaluation and recommendations for appropriate treatment. Long-Term Insomnia Nonpharmacological treatments are important for all patients with long-term insomnia. These include education about sleep hygiene, adequate exercise (where possible), relaxation training, and behavioral modification approaches, such as sleep restriction and stimulus control therapy. In sleep restriction therapy, the patient keeps a diary of the amount of time spent in bed and then chooses a time in bed of 30 to 60 minutes less than this time. This induces a mild sleep debt, which aids sleep onset. In stimulus control therapy, the patient is instructed to go to bed only when sleepy, to use the bedroom only for sleep and sex, to get up and leave the bedroom if sleep does not occur within 15 to 20 minutes, to return to bed again only when sleepy, to arise at the same time each morning regardless of sleep quality the preceding night, and to avoid daytime naps. Nonpharmacological treatments for insomnia have been found to be particularly effective in reducing sleep-onset latency and time awake after sleep onset (Morin et al., 1994). Side effects of hypnotic agents may limit their usefulness for insomnia management. The use of hypnotics for long-term insomnia is problematic for many reasons. Long-term hypnotic use leads to a decrease in effectiveness and may produce rebound insomnia upon discontinuance. Almost all hypnotics change sleep architecture. The barbiturates reduce REM sleep; the benzodiazepines reduce slow-wave non-REM sleep and, to a lesser extent, REM sleep. While the significance of these findings is still unclear, there is an emerging consensus that slow-wave sleep is particularly important for physical restorative processes. REM sleep may aid in the consolidation of learning. The blockade of slow-wave sleep by benzodiazepines may help to account for their diminishing effectiveness over the long term, and it also may explain their effectiveness in blocking sleep terrors, a disorder of arousal from slow-wave sleep. Benzodiazepines produce cognitive changes. Long-acting agents can cause next-day confusion, with a concomitant increase in falls, while shorter-acting agents can produce rebound next-day anxiety. Paradoxically, the acute amnestic effects of benzodiazepines may be responsible for the patient's subsequent report of restful sleep. Triazolam has been postulated to induce cognitive changes that blur the subjective distinction between waking and sleeping (Mendelson, 1993). Anterograde amnesia may be more common with triazolam. While the performance-disruptive effects of alcohol and diphenhydramine are reduced after napping, those of triazolam are not (Roehrs et al., 1993). Benzodiazepines may worsen sleep apnea. Some hypersomnia patients do not feel refreshed after a night's sleep and so may ask for sleeping pills to improve the quality of their sleep. The consensus is that hypnotics should not be given to the patients with sleep apnea, especially of the obstructive type, because these agents decrease upper airway muscle tone while also decreasing the arousal response to hypoxia (Robinson and Zwillich, 1989). Insomnia in Older Patients The elderly, like the very young, tend to sleep in a polyphasic (multiple sleep episodes per day) pattern, rather than the monophasic pattern characteristic of younger adults. They may have single or multiple daytime naps in addition to nighttime sleep. This pattern makes assessment of adequate sleep time difficult. Anyone who naps regularly will have shortened nighttime sleep without evidence of impaired daytime wakefulness, regardless of age. This pattern is exemplified in 'siesta' cultures and probably is adaptive. Changes in the pharmacokinetic profiles of hypnotic agents occur in the elderly because of reduced body water, reduced renal function, and increased body fat, leading to a longer half-life for benzodiazepines. A dose that produces pleasant sleep and adequate daytime wakefulness during week 1 of administration may produce daytime confusion and amnesia by week 3 as the level continues to rise, particularly with long-acting hypnotics. For example, the benzodiazepine diazepam is highly lipid soluble and is excreted by the kidney. Because of the increase in body fat and the decrease in renal excretion that typically occurs from age 20 to 80, the half-life of the drug may increase fourfold over this span. Elderly people who are living full lives with relatively unimpaired daytime wakefulness may complain of insomnia because they are not sleeping as long as they did when they were younger. Injudicious use of hypnotics in these individuals can produce daytime cognitive impairment and so impair overall quality of life. Once an older patient has been taking benzodiazepines for an extended period, whether for daytime anxiety or nighttime sedation, terminating administration of the drug can be a long, involved process. It may be warranted to leave the patient on the medication, with adequate attention to daytime side effects. Management of Patients Following Long-Term Treatment with Hypnotic Agents Patients who have been taking hypnotics for many months or even years represent a special problem group (Fleming, 1993). If a benzodiazepine has been used regularly for more than 2 weeks, it should be tapered rather than discontinued abruptly. In some patients on hypnotics with a short half-life, it is easier to switch first to a hypnotic with a long half-life and then taper. In a study of nine patients in whom the nonbenzodiazepine agent zopiclone was abruptly substituted for a benzodiazepine agent for 1 month and then itself abruptly terminated, improved sleep was reported during the zopiclone treatment, and withdrawal effects were absent on discontinuation of zopiclone (Shapiro et al., 1993). The onset of withdrawal symptoms from medications with a long half-life may be delayed. Consequently, the patient should be warned about the symptoms associated with withdrawal effects. Prescribing Guidelines for the Management of Insomnia Hypnotics that act at benzodiazepine receptors, including the benzodiazepine hypnotics as well as the newer agents zolpidem, zopiclone, and zaleplon are preferred to barbiturates because they have a greater therapeutic index, are less toxic in overdose, have smaller effects on sleep architecture, and have less abuse potential. Compounds with a shorter half-life are favored in patients with sleep-onset insomnia but without significant daytime anxiety who need to function at full effectiveness all day. These compounds also are appropriate for the elderly, because of a decreased risk of falls and respiratory depression. However, the patient and physician should be aware that early morning awakening, rebound daytime anxiety, and amnestic episodes also may occur. These undesirable side effects are more common at higher doses of the benzodiazepines. Benzodiazepines with a longer half-life are favored for patients who have significant daytime anxiety and who may be able to tolerate next-day sedation but would be impaired further by rebound daytime anxiety. These benzodiazepines also are appropriate for patients receiving treatment for major depressive episodes, because the short-acting agents can worsen early morning awakening. However, longer-acting benzodiazepines can be associated with next-day cognitive impairment or delayed daytime cognitive impairment (after 2 to 4 weeks of treatment) as a result of drug accumulation with repeated administration. Older agents such as barbiturates, glutethimide, and meprobamate should be avoided for the management of insomnia. They have high abuse potential and are dangerous in overdose. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1579
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved