| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Therapeutic Gases: Oxygen, Carbon Dioxide, Nitric Oxide, and Helium
Oxygen
|
Oxygen is a fundamental requirement for animal existence. Hypoxia is a life-threatening condition in which oxygen delivery is inadequate to meet the metabolic demands of the tissues. Since oxygen delivery is the product of blood flow and oxygen content, hypoxia may result from alterations in tissue perfusion, decreased oxygen tension in the blood, or decreased oxygen carrying capacity. In addition, hypoxia may result from a problem in oxygen transport from the microvasculature to the cells or in utilization within the cell. Irrespective of cause, an inadequate supply of oxygen ultimately results in the cessation of aerobic metabolism and oxidative phosphorylation, depletion of high-energy compounds, cellular dysfunction, and death. History Soon after Priestley's discovery of oxygen in 1772 and Lavoisier's
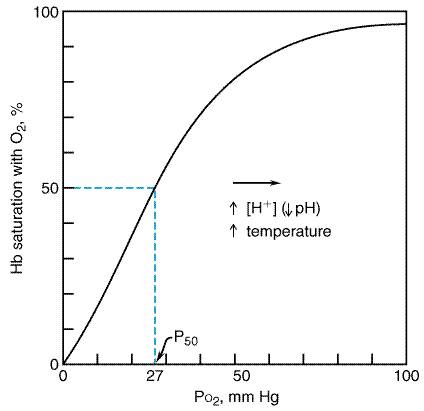
elucidation of its role in respiration, oxygen therapy was introduced in Normal Oxygenation Oxygen makes up 21% of air, which at sea level (1 atmosphere, 101 kPa) represents a partial pressure of 21 kPa (158 mmHg). While the fraction (percentage) of oxygen remains constant regardless of atmospheric pressure and altitude, the partial pressure of oxygen (PO ) decreases with lower atmospheric pressure. Since it is this partial pressure that drives the diffusion of oxygen, ascent to elevated altitude reduces the uptake and delivery of oxygen to the tissues. Conversely, increases in atmospheric pressure (hyperbaric therapy, or breathing at depth) increase the PO in inspired air and result in increased gas uptake. As the air is delivered to the distal airways and alveoli, the PO decreases by dilution with carbon dioxide and water vapor and by uptake into the blood. Under ideal conditions, when ventilation and perfusion are uniformly distributed, the alveolar PO will be approximately 14.6 kPa (110 mmHg). The corresponding alveolar partial pressures of water and carbon dioxide are 6.2 kPa (47 mmHg) and 5.3 kPa (40 mmHg), respectively. Under normal conditions, there is complete equilibration of alveolar gas and capillary blood, and the PO in end capillary blood is typically within a fraction of a kPa of that in the alveoli. Under conditions of disease, when the diffusion barrier for gas transport may be increased, or exercise, when high cardiac output reduces capillary transit time, full equilibration may not occur, and the alveolarend capillary PO gradient may be increased. The PO in arterial blood, however, is further reduced by venous admixture (shunt), the addition of mixed venous blood, which has a PO of approximately 5.3 kPa (40 mmHg). Together, the diffusional barrier, inhomogeneities of ventilation and perfusion, and the shunt fraction are the major causes of the alveolar-to-arterial oxygen gradient, which is normally 1.3 to 1.6 kPa (10 to 12 mmHg) when air is breathed and 4.0 to 6.6 kPa (30 to 50 mmHg) when 100% oxygen is breathed (Clark and Lambertsen, 1971). Oxygen is delivered to the tissue capillary beds by the circulation and again follows a gradient out of the blood and into cells. Tissue extraction of oxygen typically reduces the PO of venous blood by an additional 7.3 kPa (55 mmHg). The mean tissue PO is much lower than the value in the mixed venous blood because of substantial diffusional barriers and the consumption of oxygen in the tissues. Although the PO at the site of oxygen utilizationthe mitochondriais not known, oxidative phosphorylation can continue at a PO of only a few mm Hg (Robiolio et al., 1989). In the blood, oxygen is carried primarily in chemical combination with hemoglobin and to a small extent dissolved in solution. The quantity of oxygen combined with hemoglobin depends on the PO , as illustrated by the sigmoid-shaped oxyhemoglobin dissociation curve (Figure 161). Hemoglobin is about 98% saturated with oxygen when air is breathed under normal circumstances, and it binds 1.3 ml of oxygen per gram when fully saturated. The steep slope of this curve with falling PO facilitates unloading of oxygen from hemoglobin at the tissue level and reloading when desaturated, mixed venous blood arrives at the lung. Shifting of the curve to the right with increasing temperature, increasing PCO , and decreasing pH, as is found in metabolically active tissues, lowers the oxygen saturation for the same PO and thus delivers additional oxygen where and when it is most needed. However, the flattening of the curve with higher PO indicates that increasing blood PO by inspiring oxygen-enriched mixtures only minimally can increase the amount of oxygen carried by hemoglobin. Further increases in blood oxygen content can occur only by increasing the amount of oxygen dissolved in plasma. Because of the low solubility of oxygen (0.226 ml/liter per kPa or 0.03ml/liter per mm Hg at 37C), breathing 100% oxygen can increase the amount of oxygen in blood by only 15 ml per liter, less than one third of normal metabolic demands. However, if the inspired PO is increased to 3 atmospheres (304 kPa) in a hyperbaric chamber, the amount of dissolved oxygen is sufficient to meet normal metabolic demands even in the absence of hemoglobin (Table 161).
Oxygen Deprivation An understanding of the causes and effects of oxygen deficiency is necessary for the rational therapeutic use of the gas. Hypoxia is the term used to denote insufficient oxygenation of the tissues. Hypoxemia generally implies a failure of the respiratory system to oxygenate arterial blood. Pulmonary Mechanisms of Hypoxemia Classically there are five causes of hypoxemia: low inspired oxygen
fraction (FIO2), increased diffusion barrier,
hypoventilation, ventilation/perfusion ( Low FIO2 is a cause of hypoxemia only at high altitude or in the event of equipment failure, such as a gas blender malfunction or a mislabeled compressed-gas tank. An increase in the barrier to diffusion of oxygen within the lung is rarely a cause of hypoxemia in a resting subject, except in end-stage parenchymal lung disease. Both of these problems may be alleviated with administration of supplemental oxygen, the former by definition and the latter by increasing the gradient driving diffusion. Hypoventilation causes hypoxemia by reducing the alveolar PO in proportion to the build-up of CO2 in the alveoli. In essence, during hypoventilation there is decreased delivery of oxygen to the alveoli while its removal by the blood remains the same, causing its alveolar concentration to fall. The opposite occurs with carbon dioxide. This is described by the alveolar gas equation: PAO2= PIO2(PACO2/R), where PAO2 and PACO2 are the alveolar partial pressures of O2 and CO2, PIO2 the partial pressure of O2 in the inspired gas, and R the respiratory quotient. Under normal conditions, breathing room air at sea level (corrected for the partial pressure of water vapor), the PIO2 is about 20 kPa (150 mmHg), the PACO2 about 5.3 kPa (40 mmHg), R is 0.8, and thus the PAO2 is normally around 13.3 kPa (100 mmHg). It would require substantial hypoventilation, with the PACO2 rising to over 9.3 kPa (70 mmHg), to cause the PAO2 to fall below 7.8 kPa (60 mmHg). This cause of hypoxemia is readily prevented by administration of even small amounts of supplemental oxygen. Shunt and The deleterious effect of
Nonpulmonary Causes of Hypoxia In addition to failure of the respiratory system to adequately oxygenate the blood, there are a number of other factors that can contribute to hypoxia at the tissue level. These may be divided into categories of oxygen delivery and oxygen utilization. Oxygen delivery decreases globally when cardiac output falls or locally when regional blood flow is compromised, as from a vascular occlusion (stenosis, thrombosis, microvascular occlusion) or increased downstream pressure to flow (compartment syndrome, venous stasis, or venous hypertension). Decreased oxygen-carrying capacity of the blood will likewise decrease oxygen delivery, as occurs with anemia, carbon monoxide poisoning, or hemoglobinopathy. Finally, hypoxia may occur when transport of oxygen from the capillaries to the tissues is decreased (edema) or utilization of the oxygen by the cells impaired (cyanide toxicity). Multiple causes of hypoxia often coexist. A victim of smoke inhalation may have an airway obstruction as a result of thermal injury and reduced oxygen carrying capacity because of carbon monoxide poisoning and anemia. An organ with a marginal blood supply because of atherosclerosis may be seriously damaged if the PO of its arterial supply is decreased only slightly. Effects of Hypoxia Regardless of the cause, hypoxia ultimately results in the cessation of aerobic metabolism, exhaustion of high-energy intracellular stores, cellular dysfunction, and death. The time course of cellular demise depends upon the tissue's relative metabolic requirements, oxygen and energy stores, and anaerobic capacity. Survival times (the time from the onset of circulatory arrest to significant organ dysfunction) range from 1 minute in the cerebral cortex to around 5 minutes in the heart and 10 minutes in the kidneys and liver, with the potential for some degree of recovery if reperfused. Revival times (the duration of hypoxia beyond which recovery is no longer possible) are approximately 4 to 5 times longer. Less severe degrees of hypoxia have progressive physiological effects on different organ systems (Nunn, 1993b). Respiratory System Hypoxia stimulates the carotid and aortic baroreceptors to cause increases in both the rate and depth of ventilation. Minute volume almost doubles when normal individuals inspire gas with a PO of 6.6 kPa (50 mm Hg). Dyspnea is not always experienced with simple hypoxia but occurs when the respiratory minute volume approaches half the maximal breathing capacity; this may occur with minimum exertion in patients in whom maximal breathing capacity is reduced by lung disease. In general, little warning precedes the loss of consciousness resulting from hypoxia. Cardiovascular System Hypoxia causes reflex activation of the sympathetic nervous system via both autonomic and humoral mechanisms, resulting in tachycardia and increased cardiac output. Peripheral vascular resistance, however, decreases primarily via local autoregulatory mechanisms, with the net result that blood pressure is generally maintained unless hypoxia is prolonged or severe. In contrast to the systemic circulation, hypoxia causes pulmonary vasoconstriction and hypertension, an extension of the normal regional vascular response that matches perfusion with ventilation to optimize gas exchange in the lung (hypoxic pulmonary vasoconstriction). Central Nervous System (CNS) The CNS is least able to tolerate hypoxia. Hypoxia is manifest initially by decreased intellectual capacity and impaired judgment and psychomotor ability. This state progresses to confusion and restlessness and ultimately to stupor, coma, and death as the arterial PO decreases below 4 to 5.3 kPa (30 to 40 mm Hg). Victims often are unaware of this progression. Cellular and Metabolic Effects When the mitochondrial PO falls below about 0.13 kPa (1 mm Hg), anaerobic metabolism stops, and the less efficient anaerobic pathways of glycolysis become responsible for the production of cellular energy. End-products of anaerobic metabolism, such as lactic acid, may be released into the circulation in measurable quantities. Energy-dependent ion pumps slow and transmembrane ion gradients dissipate. Intracellular concentrations of Na+, Ca2+, and H+ increase, finally leading to cell death. The time course of cellular demise depends on the relative metabolic demands, O2 storage capacity, and anaerobic capacity of the individual organs. Restoration of perfusion and oxygenation prior to hypoxic cell death paradoxically can result in an accelerated form of cell injury (ischemia-reperfusion syndrome), thought to result from the generation of highly reactive oxygen free radicals (McCord, 1985). Adaptation to Hypoxia Long-term hypoxia results in adaptive physiological changes; these have been studied most thoroughly in persons exposed to high altitude. Adaptations include increased numbers of pulmonary alveoli, increased concentrations of hemoglobin in blood and myoglobin in muscle, and a decreased ventilatory response to hypoxia (Cruz et al., 1980). Short-term exposure to altitude produces similar adaptive changes. In susceptible individuals, however, acute exposure to high altitude may produce 'acute mountain sickness,' a syndrome characterized by headache, nausea, dyspnea, sleep disturbances, and impaired judgment, progressing to pulmonary and cerebral edema (Johnson and Rock, 1988). Mountain sickness is treated with supplemental oxygen, descent to lower altitude, or an increase in ambient pressure. Diuretics (carbonic anhydrase inhibitors) and steroids also may be helpful. The syndrome usually can be avoided by a slow ascent to altitude, permitting time for adaptation to occur. It has been noted that certain aspects of fetal and newborn physiology are strongly reminiscent of adaptation mechanisms found in hypoxia-tolerant animals (Mortola, 1999; Singer, 1999), including shifts in the oxyhemoglobin dissociation curve (fetal hemoglobin), reduction in metabolic rate and body temperature (hibernation-like mode), reduction in heart rate and circulatory redistribution (as in diving mammals), and redirection of energy utilization from growth to maintenance metabolism. These adaptations help account for the relative tolerance of the fetus and neonate to both chronic (uterine insufficiency) and short-term hypoxia. Oxygen Inhalation Physiological Effects of Oxygen Inhalation The primary use for inhalation of oxygen is to reverse or prevent the development of hypoxia; other consequences usually are minor. However, when oxygen is breathed in excessive amounts or for prolonged periods, secondary physiological changes and toxic effects can occur. Respiratory System Inhalation of oxygen at 1 atmosphere or above causes a small degree of respiratory depression in normal subjects, presumably as a result of loss of tonic chemoreceptor activity. However, ventilation typically increases within a few minutes of oxygen inhalation because of a paradoxical increase in the tension of carbon dioxide in tissues. This increase results from the increased concentration of oxyhemoglobin in venous blood, which causes less efficient removal of carbon dioxide from the tissues (Lambertsen et al., 1953; Plewes and Farhi, 1983). In a small number of patients whose respiratory center is depressed by long-term retention of carbon dioxide, injury, or drugs, ventilation is maintained largely by stimulation of carotid and aortic chemoreceptors, commonly referred to as the hypoxic drive. The provision of too much oxygen can depress this drive, resulting in respiratory acidosis. In these cases, supplemental oxygen should be titrated carefully to ensure adequate arterial saturation. If hypoventilation results, then mechanical ventilatory support with or without tracheal intubation should be provided. Expansion of poorly ventilated alveoli is maintained in part by the nitrogen content of alveolar gas; nitrogen is poorly soluble and thus remains in the airspaces while oxygen is absorbed. High oxygen concentrations delivered to poorly ventilated lung regions can promote absorption atelectasis, occasionally resulting in an increase in shunt and a paradoxical worsening of hypoxemia after a period of oxygen administration. Cardiovascular System Aside from reversing the effects of hypoxia, the physiological consequences of oxygen inhalation on the cardiovascular system are of little significance. Heart rate and cardiac output are slightly reduced when 100% oxygen is breathed; blood pressure changes little. While pulmonary arterial pressure changes little in normal subjects with oxygen inhalation, elevated pulmonary artery pressures in patients living at altitude who have chronic hypoxic pulmonary hypertension may reverse with oxygen therapy or return to sea level (Grover et al., 1966; Spievogel et al., 1969). In particular, in neonates with congenital heart disease and left-to-right shunting of cardiac output, oxygen supplementation must be carefully regulated because of the risk of further reducing pulmonary vascular resistance and increasing pulmonary blood flow. Metabolism Inhalation of 100% oxygen does not produce detectable changes in oxygen consumption, carbon dioxide production, respiratory quotient, or glucose utilization. Oxygen Administration Oxygen is supplied as a compressed gas in steel cylinders, and a
purity of 99% is referred to as 'medical grade.' Most hospitals
have oxygen piped from insulated liquid oxygen containers to areas of
frequent use. For safety, oxygen cylinders and piping are color-coded (green
in the Oxygen is delivered by inhalation except during extracorporeal circulation, when it is dissolved directly into the circulating blood. Only a closed delivery system, with an airtight seal to the patient's airway and complete separation of inspired from expired gases, can precisely control FIO2. In all other systems, the actual delivered FIO2 will depend upon the ventilatory pattern (rate, tidal volume, inspiratory:expiratory time ratio, and inspiratory flow) and delivery system characteristics. Low-Flow Systems Low-flow systems, in which the oxygen flow is lower than the inspiratory flow rate, have a limited ability to raise the FIO2 because they depend upon entrained room air to make up the balance of the inspired gas. The FIO2 of these systems is extremely sensitive to small changes in the ventilatory pattern. Devices such as face tents are used primarily for delivering humidified gases to patients and cannot be relied upon to provide predictable amounts of supplemental oxygen. Nasal cannulaesmall, flexible prongs that sit just inside each narisdeliver oxygen at 1 to 6 liters/minute. The nasopharynx acts as a reservoir for storing the oxygen, and patients may breathe through either the mouth or nose as long as the nasal passages remain patent. These devices typically deliver 24% to 28% FIO2 at 2 to 3 liters/minute. Up to 40% FIO2 is possible at higher flow rates, although this is poorly tolerated for more than brief periods because of mucosal drying. The simple face-mask, a clear plastic mask with side holes for clearance of expiratory gas and inspiratory air entrainment, is used when higher concentrations of oxygen delivered without tight control are desired. The maximum FIO2 of a facemask can be increased from around 60% at 6 to 15 liters/minute to greater than 85% by adding a 600- to 1000-ml reservoir bag. With this partial rebreathing mask, most of the inspired volume is drawn from the reservoir, avoiding dilution of the FIO2 by entrainment of room air. High-Flow Systems The most commonly used high-flow oxygen delivery device is the Venturi mask, which utilizes a specially designed mask insert to reliably entrain room air in a fixed ratio and thus provides a relatively constant FIO2 at relatively high flow rates. Typically, each insert is designed to operate at a specific oxygen flow rate, and different inserts are required to change the FIO2. Lower delivered FIO2 values use greater entrainment ratios, resulting in higher total (oxygen plus entrained air) flows to the patient, ranging from 80 liters/minute for 24% FIO2 to 40 liters/minute at 50% FIO2. While these flow rates are much higher than those obtained with low-flow devices, they still may be lower than the peak inspiratory flows for patients in respiratory distress, and thus the actual delivered oxygen concentration may be lower than the nominal value. Oxygen nebulizers, another type of Venturi device, provide patients with humidified oxygen at 35% to 100% FIO2 at high flow rates. Finally, oxygen blenders provide high inspired oxygen concentrations at very high flow rates. These devices mix high-pressure, compressed air and oxygen to achieve any concentration of oxygen from 21% to 100% at flow rates up to 100 liters/minute. These same blenders are used to provide control of FIO2 for ventilators, CPAP/BiPAP machines, oxygenators, and other devices with similar requirements. Again, despite the high flows, the delivery of high FIO2 to an individual patient also depends on maintaining a tight-fitting seal to the airway and/or the use of reservoirs to minimize entrainment of diluting room air. Monitoring of Oxygenation Monitoring and titration are required to meet the therapeutic goal of oxygen therapy and to avoid complications and side effects. Although cyanosis is a physical finding of substantial clinical importance, it is not an early, sensitive, or reliable index of oxygenation. Cyanosis appears when about 5 g/dl of deoxyhemoglobin is present in arterial blood (Lundsgaard and Van Slyke, 1923), representing an oxygen saturation of about 67% when a normal amount of hemoglobin (15 g/dl) is present. However, when anemia lowers the hemoglobin to 10 g/dl, then cyanosis does not appear until the arterial blood saturation has decreased to 50%. Invasive approaches for monitoring oxygenation include intermittent laboratory analysis of arterial or mixed venous blood gases and placement of intravascular cannulae capable of continuous measurement of oxygen tension. The latter method, which relies on fiberoptic oximetry, is used frequently for the continuous measurement of mixed venous hemoglobin saturation as an index of tissue extraction of oxygen, usually in critically ill patients. Noninvasive monitoring of arterial oxygen saturation now is widely available from transcutaneous pulse oximetry, in which oxygen saturation is measured from the differential absorption of light by oxyhemoglobin and deoxyhemoglobin and the arterial saturation determined from the pulsatile component of this signal. Application is simple and calibration not required. Because pulse oximetry measures hemoglobin saturation and not PO , it is not sensitive to increases in PO that exceed levels required to fully saturate the blood. However, pulse oximetry is very useful for monitoring the adequacy of oxygenation during procedures requiring sedation or anesthesia, rapid evaluation and monitoring of potentially compromised patients, and titrating oxygen therapy in situations where toxicity from oxygen or side effects of excess oxygen are of concern. Complications of Oxygen Therapy Administration of supplemental oxygen is not without potential complications. In addition to the potential to promote absorption atelectasis and depress ventilation, discussed above, high flows of dry oxygen can dry out and irritate mucosal surfaces of the airway and the eyes, as well as decrease mucociliary transport and clearance of secretions. Humidified oxygen thus should be used when therapy of greater than an hour's duration is required. Finally, any oxygen-enriched atmosphere constitutes a fire hazard, and appropriate precautions must be taken both in the operating room and for patients on oxygen at home. It is important to realize that hypoxemia still can occur despite the administration of supplemental oxygen. Furthermore, when supplemental oxygen is administered, desaturation occurs at a later time after airway obstruction or hypoventilation, potentially delaying the detection of these critical events. Therefore, whether or not oxygen is administered to a patient at risk for these problems, it is essential that both oxygen saturation and adequacy of ventilation be frequently assessed. Therapeutic Uses of Oxygen Correction of Hypoxia As stated above, the primary therapeutic use of oxygen is to correct hypoxia. However, hypoxia is most commonly a manifestation of an underlying disease, and administration of oxygen thus can be viewed as a symptomatic or temporizing therapy. Only rarely is hypoxia due to a primary deficiency in the inspired gas. Because of the many causes of hypoxia, supplementation of the inspired gas alone will not suffice to correct the problem. Efforts must be directed at correcting the cause of the hypoxia. For example, airway obstruction is unlikely to respond to an increase in inspired oxygen tension without relief of the obstruction. More importantly, while hypoxemia due to hypoventilation after a narcotic overdose can be improved with supplemental oxygen administration, the patient remains at risk for respiratory embarrassment if ventilation is not increased through stimulation, narcotic reversal, or mechanical ventilation. In general, the hypoxia that results from most pulmonary diseases can be alleviated at least partially by administration of oxygen, thereby allowing time for definitive therapy to reverse the primary process. Thus, administration of oxygen is a basic and important treatment to be used in all forms of hypoxia, with the understanding that the response will vary in a way that is generally predictable from knowledge of the underlying pathophysiological processes. Reduction of Partial Pressure of an Inert Gas. Since nitrogen constitutes some 79% of ambient air, it also is the predominant gas in most gas-filled spaces in the body. In certain situations, such as bowel distension from obstruction or ileus, intravascular air embolism, or pneumothorax, it is desirable to reduce the volume of these air-filled spaces. Since nitrogen is relatively insoluble, inhalation of high concentrations of oxygen (and thus low concentrations of nitrogen) rapidly lowers total body partial pressure of nitrogen and provides a substantial gradient for the removal of nitrogen from gas spaces. Administration of oxygen for air embolism is additionally beneficial, because it also helps to relieve the localized hypoxia distal to the embolic vascular obstruction. In the case of decompression sickness or bends, lowering of inert gas tension in blood and tissues by oxygen inhalation prior to or during a barometric decompression can reduce the degree of supersaturation that occurs after decompression so that bubbles do not form. If bubbles do form in either tissues or the vasculature, administration of oxygen is based on the same rationale as that described for gas embolism. Hyperbaric Oxygen Therapy Oxygen is administered at greater than atmospheric pressure for a number of conditions when 100% oxygen at a single atmosphere is insufficient (Buras, 2000; Shank and Muth, 2000; Myers, 2000). To achieve concentrations of greater than 1 atmosphere, a hyperbaric chamber must be used. These chambers range from small, single-person affairs to multiroom establishments, which may include complex medical equipment. Smaller, one-person chambers typically are pressurized with oxygen, while larger ones are filled with air. In the latter case, a patient must wear a mask to receive the oxygen at the increased pressure. The larger chambers are more suitable for critically ill patients who require ventilation, monitoring, and constant attendance. Any chamber must be built to withstand pressures which may range from 200 to 600 kPa (2 to 6 atmospheres), though inhaled oxygen tension that exceeds 300 kPa (3 atmospheres) rarely is used (see Oxygen Toxicity, below). Hyperbaric oxygen therapy has two components: increased hydrostatic pressure and increased oxygen pressure. Both factors are necessary for the treatment of decompression sickness and air embolism. The hydrostatic pressure reduces bubble volume, and the absence of inspired nitrogen increases the gradient for elimination of nitrogen and reduces hypoxia in downstream tissues. Increased oxygen pressure at the tissue level is the primary therapeutic goal for most of the other indications for hyperbaric oxygen. For example, even a small increase in PO in previously ischemic areas may enhance the bactericidal activity of leukocytes and increase angiogenesis. Thus, repetitive, brief exposure to hyperbaric oxygen is a useful adjunct in the treatment of chronic refractory osteomyelitis, osteoradionecrosis, or crush injury or for the recovery of compromised skin, tissue grafts, or flaps. Furthermore, increased oxygen tension can itself be bacteriostatic; the spread of infection with Clostridium perfringens and production of toxin by the bacteria are slowed when oxygen tensions exceed 33 kPa (250 mm Hg), justifying the early use of hyperbaric oxygen in clostridial myonecrosis (gas gangrene). Hyperbaric oxygen also is useful in selected instances of generalized hypoxia. In carbon monoxide poisoning, hemoglobin and myoglobin become unavailable for oxygen binding because of the high affinity of CO for these proteins. A high PO facilitates competition of oxygen with CO for binding sites, permitting the resumption of normal delivery of oxygen to the tissues. Hyperbaric oxygen decreases the incidence of neurological sequelae after CO intoxication; this effect may be independent of the ability of hyperbaric oxygen to speed the elimination of CO (Thom, 1989). However, a recent randomized study suggests that hyperbaric oxygen is not beneficial in carbon monoxide poisoning and might even be harmful (Scheinkestel et al., 1999). The occasional use of hyperbaric oxygen in cyanide poisoning has a similar rationale. Hyperbaric oxygen also may be useful in severe, short-term anemia, since sufficient oxygen can be dissolved in the plasma at 3 atmospheres to meet metabolic needs. However, such treatment must be limited, because oxygen toxicity is dependent on increased PO , not on the oxygen content of the blood. Hyperbaric oxygen therapy also has been used in such diverse conditions as multiple sclerosis, traumatic spinal cord injury, cerebrovascular accidents, bone grafts and fractures, and leprosy. However, data from well-controlled clinical trials are not sufficient to justify these uses, and the costs of hyperbaric therapy remain very high. Oxygen Toxicity Oxygen is used in cellular energy production and is crucial for cellular metabolism. However, oxygen also may have deleterious actions at the cellular level. Oxygen toxicity probably results from increased production of reactive agents such as superoxide anion, singlet oxygen, hydroxyl radical, and hydrogen peroxide (Turrens et al., 1982). These agents attack and damage biological membranes, and thus eventually result in damage to most cellular components. A variety of factors limit the toxicity of oxygen-derived, reactive agents. These factors include enzymes such as superoxide dismutase, glutathione peroxidase, and catalase, which scavenge toxic byproducts. In addition, there are reducing agents including iron, glutathione, and ascorbate. These factors, however, are insufficient to limit the destructive actions of oxygen when patients are exposed to high concentrations over a period of time. Tissues show differential sensitivity to oxygen toxicity, which is likely the result of differences in both their production of reactive compounds and protective mechanisms. Oxygen toxicity recently was reviewed by Carraway and Piantadosi (1999). Respiratory Tract The pulmonary system is usually the first to exhibit toxicity, a function of its being continuously exposed to the highest oxygen tensions in the body. Subtle changes in pulmonary function can occur after as little as 8 to 12 hours of exposure to 100% oxygen (Sackner et al., 1975). Increases in capillary permeability, which will increase the alveolar/arterial O2 gradient and ultimately lead to further hypoxemia, and decreased pulmonary function can be seen after only 18 hours of exposure (Davis et al., 1983; Clark, 1988). Serious injury and death, however, require much longer exposures. Pulmonary damage is directly related to the inspired oxygen tension, and concentrations of less than 0.5 atmosphere appear safe over long time periods. The capillary endothelium is the most sensitive tissue of the lung. Endothelial injury results in loss of surface area from interstitial edema and leaks into the alveoli (Crapo et al., 1980). Decreases of inspired oxygen concentrations remain the cornerstone of therapy for oxygen toxicity. Modest decreases in toxicity have been observed in animals treated with antioxidant enzymes (White et al., 1989). Tolerance also may play a role in protection from oxygen toxicity; animals exposed briefly to high oxygen tension are subsequently more resistant to toxicity (Kravetz et al., 1980; Coursin et al., 1987). Sensitivity in human beings also can be altered by preexposure to both high and low oxygen concentrations (Hendricks et al., 1977; Clark, 1988). These studies strongly suggest that changes in alveolar surfactant and cellular levels of antioxidant enzymes play a role in protection from oxygen toxicity. Retina Retrolental fibroplasia can occur when neonates are exposed to increased oxygen tensions (Betts et al., 1977). These changes can go on to cause blindness and are likely caused by angiogenesis (Kushner et al., 1977; Ashton, 1979). Incidence of this disorder has decreased with an improved appreciation of the issues and avoidance of excessive inspired oxygen concentrations. Adults do not seem to develop the disease. Central Nervous System CNS problems are rare, and toxicity occurs only under hyperbaric conditions where exposure exceeds 200 kPa (2 atmospheres). Symptoms include seizures and visual changes, which resolve when oxygen tensions are returned to normal. These problems are a further reason to replace oxygen with helium under hyperbaric conditions (see Helium). |
Carbon Dioxide
|
Transfer and Elimination of Carbon Dioxide Carbon dioxide is produced by the body's metabolism at approximately the same rate as oxygen consumption. At rest, this value is about 3 ml/kg per minute, but it may increase dramatically with heavy exercise. Carbon dioxide diffuses readily from the cells into the bloodstream, where it is carried partly as bicarbonate ion, partly in chemical combination with hemoglobin and plasma proteins, and partly in solution at a partial pressure of about 6 kPa (46 mmHg) in mixed venous blood. CO2 is transported to the lung, where it is normally exhaled at the same rate at which it is produced, leaving a partial pressure of about 5.2 kPa (40 mmHg) in the alveoli and in arterial blood. An increase in PCO results in a respiratory acidosis and may be due to decreased ventilation or the inhalation of CO2, while an increase in ventilation results in decreased PCO and a respiratory alkalosis. As carbon dioxide is freely diffusible, the changes in blood PCO and pH soon are reflected by intracellular changes in PCO and pH. Effects of Carbon Dioxide Alterations in PCO and pH have widespread effects in the body, particularly on respiration, circulation, and the CNS. More complete discussions of these and other effects are found in textbooks of physiology (see Nunn, 1993a). Respiration Carbon dioxide is a rapid, potent stimulus to ventilation in direct proportion to the inspired CO2. Inhalation of 10% carbon dioxide can produce minute volumes of 75 liters per minute in normal individuals. Carbon dioxide acts at multiple sites to stimulate ventilation. Respiratory integration areas in the brainstem are acted upon by impulses from medullary and peripheral arterial chemoreceptors. The mechanism by which carbon dioxide acts on these receptors probably involves changes in pH (Nattie, 1999; Drysdale et al., 1981). Elevated PCO causes bronchodilation, whereas hypocarbia causes constriction of airway smooth muscle; these responses may play a role in matching pulmonary ventilation and perfusion (Duane et al., 1979). Circulation The circulatory effects of carbon dioxide result from the combination of its direct local effects and its centrally mediated effects on the autonomic nervous system. The direct effect of carbon dioxide on the heart, diminished contractility, results from pH changes (van den Bos et al., 1979). The direct effect on systemic blood vessels results in vasodilation. Carbon dioxide causes widespread activation of the sympathetic nervous system and an increase in the plasma concentrations of epinephrine, norepinephrine, angiotensin, and other vasoactive peptides (Staszewska-Barczak and Dusting, 1981). The results of sympathetic nervous system activation are, in general, opposite to the local effects of carbon dioxide. The sympathetic effects consist of increases in cardiac contractility, heart rate, and vasoconstriction (see Chapter 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists). The balance of opposing local and sympathetic effects, therefore, determines the total circulatory response to carbon dioxide. The net effect of carbon dioxide inhalation is an increase in cardiac output, heart rate, and blood pressure. In blood vessels, however, the direct vasodilating actions of CO2 appear more important and total peripheral resistance decreases when the PCO is increased. Carbon dioxide also is a potent coronary vasodilator (Ely et al., 1982). Cardiac arrhythmias associated with increased PCO are due to the release of catecholamines. Hypocarbia results in opposite effects: decreased blood pressure and vasoconstriction in skin, intestine, brain, kidney, and heart. These actions are exploited clinically in the use of hyperventilation in the presence of intracranial hypertension. Central Nervous System Hypercarbia depresses the excitability of the cerebral cortex and increases the cutaneous pain threshold through a central action. This central depression has therapeutic importance. For example, in patients hypoventilating from narcotics or anesthetics, increasing PCO may result in further CNS depression, which in turn may worsen the respiratory depression. This positive feedback cycle can be deadly. The inhalation of high concentrations of carbon dioxide (about 50%) produces marked cortical and subcortical depression of a type similar to that produced by anesthetic agents. Under certain circumstances inspired CO2 (25% to 30%) can result in subcortical activation and seizures. Methods of Administration Carbon dioxide is marketed in gray metal cylinders as the pure gas or as carbon dioxide mixed with oxygen. It usually is administered at a concentration of 5% to 10% in combination with oxygen by means of a facemask. Another method for the temporary administration of carbon dioxide is by rebreathing, for example from an anesthesia breathing circuit when the soda lime canister is bypassed or from something as simple as a paper bag. A potential safety issue exists in that CO2 tanks containing oxygen are the same color as those that are 100% CO2. When tanks containing oxygen have been used inadvertently where a fire hazard exists (e.g., in the presence of electrocautery during laparoscopic surgery), explosions and fires have resulted. Therapeutic Uses Inhalation of carbon dioxide is used less commonly today than in the past because there are now more effective treatments for most indications. Inhalation of carbon dioxide has been used during anesthesia to increase the speed of induction and emergence from inhalational anesthesia by increasing minute ventilation and cerebral blood flow. However, this technique results in some degree of respiratory acidosis. Hypocarbia with its attendant respiratory alkalosis still has some uses in anesthesia. It constricts cerebral vessels, decreasing brain size slightly, and thus may facilitate the performance of neurosurgical operations. Although carbon dioxide stimulates respiration, it is not useful in situations where respiratory depression has resulted in hypercarbia or acidosis, since further depression results. A common use of CO2 is for insufflation during endoscopic procedures (e.g., laparoscopic surgery), because it is highly soluble and does not support combustion. Any inadvertent gas emboli are thus more easily dissolved and eliminated via the respiratory system. Recently CO2 has been utilized during open cardiac surgery, where it is used to flood the surgical field. Because of its density, CO2 displaces the air surrounding the open heart so that any gas bubbles trapped in the heart are CO2 rather than insoluble nitrogen (Nadolny and Svensson, 2000). For the same reasons, CO2 is used to debubble cardiopulmonary bypass and extracorporeal membrane oxygenation (ECMO) circuits. It also can be used to adjust pH during bypass procedures when a patient is cooled. |
Nitric Oxide
|
Nitric oxide (NO), a free radical gas long known as an air pollutant and a potentially toxic agent, particularly when further oxidized (see below), recently has been shown to be an endogenous cell-signaling molecule of great physiological importance. As knowledge of the important actions of NO have evolved, the use of NO as a therapeutic agent has grown in interest. Endogenous NO is produced from the amino acid L-arginine by a family of enzymes called NO synthases. NO is now recognized as a novel cell messenger implicated in a wide range of physiological and pathophysiological events in numerous cell types and processes, including the cardiovascular, immune, and nervous systems. In the vasculature, NO produced by endothelial cells is a primary determinant of resting vascular tone through basal release and causes vasodilation when synthesized in response to shear stress and to a variety of vasodilating agents. It also plays an active role in inhibiting platelet aggregation and adhesion. Impaired NO production has been implicated in disease states such as atherosclerosis, hypertension, cerebral and coronary vasospasm, and ischemia-reperfusion injury. In the immune system, NO serves as an important effector of macrophage-induced cytotoxicity, and its overproduction is an important mediator of inflammatory states. In neurons, NO serves multiple functions, acting as a mediator of long-term potentiation, of N-methyl-D-aspartate (NMDA)mediated cytotoxicity, and as the mediator of nonadrenergic noncholingeric neurotransmission; it also has been implicated in mediating central nociceptive pathways. The physiology and pathophysiology of endogenous NO have been extensively reviewed (Moncada and Palmer, 1991; Nathan, 1992; Ignarro et al., 1999). Therapeutic Use of NO Inhalation of NO gas has received considerable therapeutic attention due to its ability to dilate selectively the pulmonary vasculature with minimal systemic cardiovascular effects (Steudel et al., 1999). The lack of effect of inhaled NO on the systemic circulation is due to its strong binding and inactivation by oxyhemoglobin upon exposure to the pulmonary circulation. Ventilation-perfusion matching is preserved or improved by NO, because inhaled NO is distributed only to ventilated areas of the lung and dilates only those vessels directly adjacent to the ventilated alveoli. Thus, inhaled NO will decrease elevated pulmonary artery pressure and pulmonary vascular resistance and often improve oxygenation (Steudel et al., 1999; Haddad et al., 2000). Due to its selective pulmonary vasodilating action, inhaled NO is undergoing intensive study as a potential therapeutic agent for numerous diseases associated with increased pulmonary vascular resistance. Therapeutic trials of inhaled NO in a wide range of such conditions have confirmed its ability to decrease pulmonary vascular resistance and often increase oxygenation, but in all but a few cases these trials have yet to demonstrate long-term improvement in terms of morbidity or mortality (Dellinger, 1999; Cheifetz, 2000). Inhaled NO has been approved by the United States Food and Drug Administration only for use in newborns with persistent pulmonary hypotension. In this disease state, NO inhalation has been shown to reduce significantly the necessity for extracorporeal membrane oxygenation, although overall mortality has been unchanged (Kinsella et al., 1997; Roberts et al., 1997). Notably, numerous trials of inhaled NO in adult and pediatric acute respiratory distress syndrome have failed to demonstrate an impact on outcome (Dellinger, 1999; Cheifetz, 2000). Several small studies and case reports have suggested potential benefits of inhaled NO in a variety of conditions, including weaning from cardiopulmonary bypass in adult and congenital heart disease patients; primary pulmonary hypertension; pulmonary embolism; acute chest syndrome in sickle-cell patients; congenital diaphragmatic hernia; high-altitude pulmonary edema; and lung transplantation (Steudel et al., 1999; Haddad et al., 2000). Larger, prospective, randomized studies, however, have not yet been performed or have failed to confirm any changes in outcome. At the present time, outside of clinical investigation, therapeutic use and benefit of inhaled NO are limited to newborns with persistent pulmonary hypotension. Diagnostic Uses of NO Inhaled NO also is used in several diagnostic applications. Inhaled NO can be used during cardiac catheterization to evaluate safely and selectively the pulmonary vasodilating capacity of patients with heart failure and infants with congenital heart disease. Inhaled NO also is used to determine the diffusion capacity (DL) across the alveolar-capillary unit. NO is more effective than carbon dioxide in this regard because of its greater affinity for hemoglobin and its higher water solubility at body temperature (Steudel et al., 1999; Haddad et al., 2000). NO is produced from the nasal passages and from the lungs of normal human subjects and can be detected in exhaled gas. The measurement of exhaled NO has been investigated for its utility in assessment of respiratory tract diseases. Measurement of exhaled NO may prove to be useful in diagnosis of asthma and in respiratory tract infections (Haddad et al., 2000). Toxicity of NO Administered at low concentrations (0.1 to 50 parts per million), inhaled NO appears to be safe and without significant side effects. Pulmonary toxicity can occur with levels higher than 50 to 100 ppm. In the context of NO as an atmospheric pollutant, the Occupational Safety and Health Administration places the seven-hour exposure limit at 50 ppm. Part of the toxicity of NO may be related to its further oxidation to nitrogen dioxide (NO2) in the presence of high concentrations of oxygen. Even low concentrations of NO2 (2 ppm) have been shown to be highly toxic in animal models, with observed changes in lung histopathology, including loss of cilia, hypertrophy, and focal hyperplasia in the epithelium of terminal bronchioles. It is important, therefore, to keep NO2 formation during NO therapy at a low level. This can be achieved through appropriate filters and scavengers and the use of high-quality gas mixtures. Laboratory studies have suggested potential additional toxic effects of chronic low doses of inhaled NO, including surfactant inactivation and the formation of peroxynitrite by interaction with superoxide. The ability of NO to inhibit or alter the function of a number of iron- and heme-containing proteinsincluding cyclooxygenase, lipoxygenases, and oxidative cytochromesas well as its interactions with ADP-ribosylation suggest a need for further investigation of the toxic potential of NO under therapeutic conditions (Steudel et al., 1999; Haddad et al., 2000). The development of methemoglobinemia is a significant complication of inhaled NO at higher concentrations, and rare deaths have been reported with overdoses of NO. The blood content of methemoglobin, however, generally will not increase to toxic levels with appropriate use of inhaled NO. Methemoglobin concentrations should be intermittently monitored during NO inhalation (Steudel et al., 1999; Haddad et al., 2000). Inhaled NO can inhibit platelet function and has been shown to increase bleeding time in some clinical studies, although reports of bleeding complications are not apparent in the literature. In patients with impaired function of the left ventricle, NO has a potential to further impair left ventricular performance by dilating the pulmonary circulation and increasing the blood flow to the left ventricle, thereby increasing left atrial pressure and promoting pulmonary edema formation. Careful monitoring of cardiac output, left atrial pressure or pulmonary capillary wedge pressure is important in this situation (Steudel et al., 1999). Despite these concerns, there are limited reports of inhaled NO-related toxicity in humans. The most important requirements for safe NO inhalation therapy are outlined by Steudel et al. (1999) and include: (1) continuous measurement of NO and NO2 concentrations using either chemiluminescence or electrochemical analyzers; (2) frequent calibration of monitoring equipment; (3) intermittent analysis of blood methemoglobin levels; (4) the use of certified tanks of NO; and (5) administration of the lowest NO concentration required for therapeutic effect. Methods of Administration Courses of treatment of patients with inhaled NO are highly varied, extending from 0.1 to 40 ppm in dose and for periods of a few hours to several weeks in duration. The minimum effective inhaled NO concentration should be determined for each patient to minimize the chance for toxicity. Commercial NO systems are available that will accurately deliver inspired NO concentrations between 0.1 and 80 ppm and simultaneously measure NO and NO2 concentrations. A constant inspired concentration of NO is obtained by administering NO in nitrogen to the inspiratory limb of the ventilator circuit in either a pulse or continuous mode. While inhaled NO may be administered to spontaneously breathing patients via a closely fitting mask, it usually is delivered during mechanical ventilation. Nasal prong administration is being employed in therapeutic trials of home administration for treatment of primary pulmonary hypertension (Steudel et al., 1999; Haddad et al., 2000). Acute discontinuation of NO inhalation can lead to a rebound pulmonary artery hypertension with an increase in right-to-left intrapulmonary shunting and a decrease in oxygenation. To avoid this phenomenon, a graded decrease of inhaled NO concentration is important in the process of weaning a patient from inhaled NO (Steudel et al., 1999; Haddad et al., 2000). |
Helium
|
Helium is an inert gas whose low density, low solubility, and high thermal conductivity provide the basis for its medical and diagnostic use. Helium is produced by separation from liquefied natural gas and supplied in brown cylinders. Helium can be mixed with oxygen and administered by mask or tracheal tube. Under hyperbaric conditions, it can be substituted for the bulk of other gases, resulting in a mixture of much lower density that is easier to breathe. The primary uses of helium are in pulmonary function testing, the treatment of respiratory obstruction, during laser airway surgery, for diving at depth, and most recently, as a label in imaging studies. The determinations of residual lung volume, functional residual capacity, and related lung volumes require a highly diffusible, nontoxic gas that is insoluble (and thus does not leave the lung via the bloodstream), so that, by dilution, the lung volume can be measured. Helium is well suited to these needs and is much cheaper than alternatives. In these tests, a breath of a known concentration of helium is given and the concentration of helium then measured in the mixed expired gas, allowing calculation of the other pulmonary volumes. Pulmonary gas flow is normally laminar, but with increased flow rate or narrowed flow pathway a component becomes turbulent. Helium can be added to oxygen to treat this turbulence due to airway obstruction. The density of helium is substantially less than that of air, and flow rates under turbulent conditions are increased with lower-density gases. This results in decreased work of breathing with mixtures of helium and oxygen. However, several factors limit the usefulness of this approach. Oxygenation frequently is a principal problem in airway obstruction, and the need for increased inspired oxygen concentration may limit the amount of helium that may be used. Furthermore, the viscosity of helium is higher than that of air, and increased viscosity reduces laminar flow. Helium has high thermal conductivity, which makes it useful during laser surgery on the airway. This more rapid conduction of heat away from the point of contact of the laser beam reduces the spread of tissue damage and the likelihood that the ignition point of flammable materials in the airway will be reached. Its low density improves the flow through the small endotracheal tubes typically used in such procedures. Recently, laser-polarized helium has been used as an inhalational contrast agent for pulmonary magnetic resonance imaging. Optical pumping of nonradioactive helium increases the signal from the gas in the lung sufficiently to permit detailed imaging of the airways and inspired airflow patterns (Kauczor et al., 1998). Hyperbaric Applications The depth and duration of diving activity are limited by oxygen toxicity, inert gas (nitrogen) narcosis, and nitrogen supersaturation when decompressing. Oxygen toxicity is a problem with prolonged exposure to compressed air at 500 kPa (5 atmospheres) or more. This problem can be minimized by dilution of oxygen with helium, which lacks narcotic potential even at very high pressures and is quite insoluble in body tissues. This low solubility reduces the likelihood of bubble formation after decompression, which can therefore be achieved more rapidly. The low density of helium also reduces the work of breathing in the otherwise dense hyperbaric atmosphere. The lower heat capacity of helium also decreases respiratory heat loss, which can be significant when diving at depth. Acknowledgment The authors wish to acknowledge Drs. Roderic G. Eckenhoff and David E. Longnecker, authors of this chapter in the ninth edition of Goodman and Gilman's The Pharmacological Basis of Therapeutics, some of whose text we have retained in this edition. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 5948
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved