| CATEGORII DOCUMENTE |
| Astronomie | Biofizica | Biologie | Botanica | Carti | Chimie | Copii |
| Educatie civica | Fabule ghicitori | Fizica | Gramatica | Joc | Literatura romana | Logica |
| Matematica | Poezii | Psihologie psihiatrie | Sociologie |
Spectroscopia de rezonanta magnetica nucleara
(1H-RMN si 13C-RMN) aplicata in stabilirea structurii compusilor organici
1. INTRODUCERE. GENERALITATI.
Dintre toate metodele fizice, rezonanta magnetica nucleara (RMN) este aceea care ofera cea mai bogata si completa informatie structurala asupra compusilor organici. Spre deosebire de spectroscopia IR, in RMN practic toate semnalele sunt interpretabile relativ usor, iar spre deosebire de spectroscopia electronica metoda RMN ofera mult mai multe informatii. In timp ce spectroscopia IR sau cele de masa sunt prea bogate in informatii, deci greu interpretabile, iar cele UV-VIZ prea sarace, spectrele RMN, atat cele 1H cat si cele 13C, contin exact informatia necesara, care poate fi pusa in legatura directa cu formula structurara a substantei.
Dezvoltata prin analogie cu rezonanta electronica de spin, rezonanta magnetica nucleara de inalta rezolutie, aplicata initial pentru studiul protonilor si extinsa ulterior pentru o serie de alti nuclizi: 13C, 19F, 31P, 17O etc., a devenit in prezent cea mai importanta metoda de studiu a structurii, configuratiei compusilor organici.
Magnetismul nuclear
Intocmai ca si electronul, protonul efectueaza o miscare rapida de rotatie in jurul axei sale, miscare numita spin nuclear ". Miscarii de rotatie a protonului (sarcina electrica) I se asociaza moment magnetic de spin.
Desi lipsit de sarcina, neutronul prezinta de asemenea un moment magnetic de spin. Acest fapt neasteptat se poate explica prin existenta unei structuri interne comportand sarcini electrice fractionare (quark).
In cazul unor nuclee compuse din mai multi protoni si neutroni, are loc o compensare spinilor particulelor elementare, spre exterior manifestandu-se un moment magnetic rezultant.
Magnetismul nuclear este caracterizat prin numarul cuantic de spin nuclear, I, care, spre deosebire de cel al electronului, poate avea valori diferite pentru nuclee diferit, in conformitate cu urmatoarele reguli:
a) nuclizii continand un numar par de protoni si un numar par de neutroni au numarul cuantic de spin nuclear egal cu zero (I=0). La acesti nuclizi momentele magnetice de spin ale protonilor si neutronilor se compenseaza separat; ne-aparand un moment magnetic de spin nuclear (μI=0). Exista 165 asemenea nuclizi stabili.
b) nuclizii continand un numar impar
fie de protoni fie de neutroni au ca valori I
numere fractionare: ![]() . Exista 110 asemenea nuclizi stabili,
impartiti aproape egal in nuclizi par-impari si impar-pari
(dupa valorile lui Z si lui N).
. Exista 110 asemenea nuclizi stabili,
impartiti aproape egal in nuclizi par-impari si impar-pari
(dupa valorile lui Z si lui N).
c) nuclizii in care atat protonii cat si neutronii sunt prezentati in numar impar au valori I intregi: I=1;2;3 (exista numai 6 asemenea nuclizi stabili: 2D, 6Li, 10B, 14N, 50V, 180Ta).
Cele de mai
sus se mai pot exprima si in felul urmator: nuclizi cu numar de
masa, A= Z N, impar au totdeauna spinul nuclear ![]() in timp de nuclizii cu A
par au spinul nul cand Z este par si numarul intreg diferit de zero
cand Z este impar.
in timp de nuclizii cu A
par au spinul nul cand Z este par si numarul intreg diferit de zero
cand Z este impar.
Se deci
ca nucleele izotopilor aceluiasi element pot avea comportari
magnetice foarte diferite. De exemplu, nuclizii 16O si 18O
nu poseda moment magnetic de spin (I=0)
in timp ce 17O are ![]() . (Pentru alte marimi I, v. tabelul 1).
. (Pentru alte marimi I, v. tabelul 1).
Unitatea de
moment magnetic nuclear este asa-numitul magneton nuclear, μn, (analog cu magnetonul
Bhor-Procopiu al electronului) definit prin relatia 1: ![]()
in care e reprezinta sarcina elementara, h - constanta lui Planck, mp - masa protonului iar c - viteza luminii.
Intre valoarea teoretica a momentului magnetic, calculata din impulsul de rotatie si valoarea reala exista o nepotrivire, datorita unei repartitii diferite a sarcinii electrice in raport cu masa. Aceasta face necesara introducerea asa-numitului factor giromagnetic nuclear, gn, reprezentand raportul intre valoarea reala si ce teoretica a momentului magnetic. Momentul magnetic nuclear μI va avea marimea:
![]()
(citeste μI este egal cu gn
![]() unitati
μn). Valorile g
pentru cativa nuclizi importanti in chimia organica sunt date in
tabelul 1.
unitati
μn). Valorile g
pentru cativa nuclizi importanti in chimia organica sunt date in
tabelul 1.
Orientare nucleelor in camp magnetic exterior
Momentul magnetic foarte mic al nucleelor se
poate pune in evidenta prin interactiune sa cuantificata,
cu un camp magnetic exterior. Vectorul momentului magnetic al nucleului se
orienteaza in raport cu directia campului exterior. Orientarea se
cuantifica, in sensul ca proiectia momentului magnetic nuclear pe
directia campului poate avea avea numai anumite valori. In general
momentul magnetic al nucleului poate adopta (2I+1) orientari. Pentru nucleele cele mai des
intalnite 1H, 13C, 19F, 31P, spinul
nuclear I are valoarea![]() , astfel incat rezulta numai 2 orientari posibile
(fig. 1.a), una aproximativ in sensul campului (facand cu aceasta un unghi
de
, astfel incat rezulta numai 2 orientari posibile
(fig. 1.a), una aproximativ in sensul campului (facand cu aceasta un unghi
de ![]() ) numita si orientare
paralela si alta aproximativ opusa sensului campului
(facand un unghi de 126s cu aceasta) numita si orientare antiparalela. Pentru a se
putea mentine la aceasta inclinatie constanta vectorul
momentului magnetic nuclear executa o miscare de precizie de (Larmor)
de frecventa proportionala cu intensitatea H a campului magnetic exterior (fig. 1.b).
) numita si orientare
paralela si alta aproximativ opusa sensului campului
(facand un unghi de 126s cu aceasta) numita si orientare antiparalela. Pentru a se
putea mentine la aceasta inclinatie constanta vectorul
momentului magnetic nuclear executa o miscare de precizie de (Larmor)
de frecventa proportionala cu intensitatea H a campului magnetic exterior (fig. 1.b).
In cele doua orientari posibile ale nuclizilor care au I=1 apare o diferenta de energie ΔE, data de relatia 3:
![]()
Spre deosebire de nivelele energetice din spectroscopia optica, in acest caz diferenta de energie dintre cele doua stari este influentabila din exterior prin valoarea intensitatii H a campului magnetic exterior (fig. 1.c)
Fenomenul de rezonanta magnetica nucleara
In mod asemanator spectroscopiei optice trecerea de la nivelul energetic inferior (orientarea paralela pe cel superior (orientarea antiparalela) se poate realiza prin absorbtia unei cuante de radiatie electromagnetica de energie egala cu ΔE:
![]()
Spectroscopia RMN se bazeaza tocmai pe acest fenomen de trecere de la un nivel energetic pe altul (simultan cu inversarea spinului in raport cu campul magnetic exterior) atunci cand nucleul situat in camp magnetic este iradiat cu o sursa de radiatii electromagnetice de frecventa adecvata v. Tranzitia din orientarea paralela in cea antiparalela este insotita de absorbtia de energie electromagnetica.
Ecuatia 4 este relatia fundamentala de rezonanta (egalitate a energiei radiatiei absorbite, hv, cu diferenta de energie ΔE a starilor nucleului) magnetica nucleara; cu ajutorul ei se poate determina marimea frecventei de rezonanta pentru diferiti nuclizi (v. tabelul 1).
Factorul de sensibilitate relativa fata proton, indicat in tabelul 1, este dat pentru acelasi camp magnetic exterior si acelasi numar de nuclee. In penultima coloana este indicat factorul care tine seama doar de caracteristicile magnetice nucleare (moment magnetic nuclear, raport giromagnetic), in timp ce ultima coloana s-a luat in consideratie abundenta izotopica naturala.
Factorii de sensibilitate arata ca toti nuclizii din tabel dau semnale de intensitate relativa mai mica decat protonul; intensitatea relativa este doar putin mai mica la 19F, dar la alti nuclizi ca de ex. 13C, la abundenta naturala, ea este atat de redusa incat pentru 13C-RMN trebuie folosite tehnici diferite (transformata Fourier, a se vedea mai departe). In aceasta ordine de ideii merita mentionat ca radioizotopul hidrogenului tritiul , are factorul de sensibilitate relativa 1,21, deci mai mare ca al protonului. De aici decurge importanta si sensibilitatea determinarilor de 3H-RMN, care se aplica in practica pe scara din ce in ce mai larga (in special in determinarile de distributie a 3H in molecule marcate).
Caracteristicile magnetice ale catorva nuclizi stabili (Tabelul 1)
|
Nuclidul |
Abundenta naturala (%) |
I |
Gn |
Frecventa de rezonanta (MHz) |
Factorul de sensibilitate relativa in raport cu 1H. |
||
|
la 14092 Oe |
la 23490 Oe | ||||||
|
H |
9,98 x 10-1 |
||||||
|
H |
1,6 x 10-6 |
||||||
|
H |
1,34 x 10-1 |
||||||
|
C |
1,76 x 10-4 |
||||||
|
N |
9,96 x 10-4 |
||||||
|
O |
1,1 x 10-5 |
||||||
|
F |
8,3 x 10-1 |
||||||
|
P |
7,0 x 10-2 |
||||||
|
* fara considerarea abundentei izotopice naturale ** cu considerarea abundentei izotopice naturale |
|||||||
Momentele magnetice nucleare, μI , fiind extrem de mici, diferentele de energie dintre cele doua nivele (orientari) sunt si ele extrem de mici si corespunzator acestora frecventele absorbite sunt de ordinul zecilor de megahertzi (la campuri uzuale de 10000 - 25000 Oe[2]) in functie de valoarea gn a nuclidului (v. tabelul 1). Aceste frecvente de absorbtie corespund frecventelor de precizie Larmor a nuclidului respectiv in campul H dat (tabelul 2).
Populatiile celor doua nivele, fundamental, N0, si excitat, N1, sunt corelate prin ecuatia lui Boltzmann) (5):
![]()
unde T este temperatura in K, ∆E diferenta de energie a nivelelor, iar ![]() erg grd este constanta
Boltzmann.
erg grd este constanta
Boltzmann.
Din cauza valorilor extrem de mici ale diferentei ∆E (tabelul 2) populatiile nivelelor sunt foarte apropiate: de exemplu pentru 1H la 15000 Oe, la o populatie N0=1000000 corespunde N1=999,993 protoni. De aici decurge necesitatea atingerii unei mari sensibilitati a tehnicii RMN care trebuie sa poata sesiza tranzitia catorva spini dintr-un milion.
Din cele de mai sus rezulta ca spectroscopia RMN are un principiu fundamental comun cu spectroscopia IR sau UV si anume aparitia unor tranzitii intre nivele diferite de energie, caracteristice sistemului (atomi sau moleculara). Diferentele de energie dintre nivele, frecventele si respectiv lungimile de unda ale radiatiilor excitante au insa valori mult diferite de la un tip de spectroscopie la altul (v. tabelul 2).
|
Marime |
Spectroscopie |
Electronica UV-VIZ |
Vibrationala IR |
H-RMN (la 23490 Oe) |
C-RMN (la 23490 Oe) |
|
λ(μm) |
3 x 106 |
1,193 x 107 |
|||
|
ν(Hz) |
De la 1,5 x 1015 la 3,75 x 1014 |
De la 1,2 x 1014 la 1,2 x 1013 |
100 x 106 |
25,15 x 106 (25,15 MHz) |
|
|
|
9,53 x 10-6 |
2,40 x 10-6 |
|||
Prin absorbtia de energie radianta se tinde catre egalizarea populatiei celor doua nivele. Revenirea la echilibru initial se realizeaza prin fenomene de relaxare, neradiative, in care se cedeaza energia absorbita (de exemplu sub forma de caldura), Daca relaxarea se face rapid, proba continua sa absoarba energie electromagnetica dar daca relaxarea este lenta, populatiile se egalizeaza usor si absorbtia de energie (semnalul) dispare, aparand saturatia semnalului RMN.
2. PRINCIPIUL APARATULUI
Dupa cum rezulta din relatia 4, pentru a putea avea loc inversarea orientarii momentului magnetic nuclear, la o anumita frecventa v a radiatie electromagnetice, intensitatea H a campului magnetic exterior trebuie sa aiba o valoare bine precizata. In mod asemanator, pentru o intensitate de camp magnetic data, radiofrecventa trebuie sa prezinte o anumita valoare. Instalatia experimentala RMN trebuie sa realizeze acest acord intre camp si frecventa, permitand in acelasi timp declararea absorbtiei (extrem de mici) de energiei care are loc in momentul "rezonantei".
Realizarea experientelor de RMN se poate face fie in aparate de baleiaj in camp, lucrand la frecventa fixa, cum se practica de obicei, fie in aparate cu baleiaj de frecventa, la camp magnetic fix.
Schita de principiu, mult simplificata a unui spectrometru RMN cu baleiaj in camp este prezentata in fig. 2.
Fig. 2. Schita de principiu a unui spectrometru RMN:
1,1' - polii electromagnetului; 2 - generatorul de radiofrecventa; 3 - tubul de proba; 4 - bobina de radiofrecventa; 5 - detector-amplificator; 6 - inregistrator de semnal; 7 - bobina generatoare a campului de baza; 8 - sursa de curent continuu stabilizat; 9 - bobina de variatie a campului magnetic; 10 - generator de curent tip "dinte de ferastrau".
Proba se introduce in fiola 3 plasata in campul magnetic omogen dat de electromagnetul 1,1' (sau de un magnet permanent). In scopul uniformizarii pozitiei tuturor protonilor in raport cu campul magnetic, proba se roteste in jurul axei verticale cu circa 20-30 rotatii/secunda. Generatorul 2 produce un camp de radiofrecventa (cu frecventa constanta v) din care o parte trece prin bobina 4 iradiind proba iar cealalta parte se transmite detectorului amplificator Majoritatea aparatelor 1H-RMN lucreaza cu frecvente de 60; 80; 90 sau 100 MHz, desi in prezent exista aparate de rezolutie mai mare, lucrand la 220 MHz sau la 360 MHz (care insa necesita folosirea supraconductorilor, deci racire cu heliu lichid). Generatorul 10 produce un curent continuu de intensitate crescatoare liniar in timp (curent tip "dinte de fierastrau"). Trecand prin bobina 9 acest curent face sa creasca intensitatea H a campului. La atingerea valorii critice, corespunzatoare rezonantei (v. relatia 4.), se produce o inversare a spinilor nucleari, bobina 4 absorbind un surplus de energie. In acest moment In acest timp la amplificator ajunge o energie micsorata, luand nastere un "semnal " care dupa o amplificare corespunzatoare este inregistrat de inregistratorul 6.
Daca in proba se afla mai multe probe de nuclizi diferind prin valorile gn si I, conditia de rezonanta va fi satisfacuta pe rand, la diferite campuri magnetice, aparand cate un semnal la fiecare specie de nuclizi. Acest tip de experiment RMN nu prezinta nici un interes pentru chimia organica, unde in general se cunoaste de la inceput tipul de nuclid existent in proba.
In functie de structura si de anturajul chimic, unul si acelasi tip de nuclid (de exemplu H) poate prezenta foarte mici diferente in valorile (aparente) ale lui gn. Datorita efectului de ecranare (v. mai departe) diferit al electronilor de legatura si datorita orientarii diferite a spinilor nucleelor vecine, campul local H la nivelul nuclidului respectiv poate prezenta foarte mici diferente fata de valoarea campului exterior.
Deci, in realitate , nu gn ci H prezinta mici abateri de la valoarea campului exterior.
Impingand rezolutia spre o limita extrema, prin realizarea unui camp magnetic intens, de o perfecta omogenitate in spatiu si constanta in timp, precum si realizarea unei frecvente perfect stabile, se pot sesiza mici diferente (aparente) de valori gn. Astfel se pot obtine pentru acelasi nuclid (de ex. 1H) o serie de semnale apropiate, rezultand un spectru RMN extrem de bogat in informatii asupra structurii compusului investigat.
Iradierea in pulsuri cu transformata Fourier (PFT) pentru 1H-RMN si 13C-RMN.
S-a aratat mai sus (tabelul 1) ca sensibilitatea
relativa a nuclidului 13C in spectrele 13C-RMN este
a zecea mia parte din cea a protonului lucrand la abundenta naturala
a 13C. Pentru a obtine spectre 13C-RMN, calea
obisnuita cu iradiere continua ("continuous wave "; CW) chiar cu magneti supraconductori (ce
permit frecvente de 220; 360 sau 400 MHz pentru protoni) nu duce la
rezultate satisfacatoare. O metoda care a fost un anumit timp,
consta in acumularea mai multor (n)
spectre in memoria unui calculator electronic legat ( on line") cu spectrometrul RMN; datorita caracterului aleator al zgomotului de fond, raportul semnal/zgomot se
imbunatateste cu ![]() . De exemplu, pentru a mari de 100 de ori intensitatea
semnalului in raport cu zgomotul, trebuie acumulate 10000 de spectre (ceea ce
la o durata de 1-5 minute pentru parcurgerea unui spectru, duce la timpi
foarte lungi).
. De exemplu, pentru a mari de 100 de ori intensitatea
semnalului in raport cu zgomotul, trebuie acumulate 10000 de spectre (ceea ce
la o durata de 1-5 minute pentru parcurgerea unui spectru, duce la timpi
foarte lungi).
Unica metoda care a putut rezolva in conditii optime
aceasta problema consta in tehnica iradierii in pulsuri si
prelucrarea informatiei prin transformarea Fourier (tehnica pulse Fourier transform"
PFT). In esenta ridicarea spectrului consta
in iradierea probei cu un puls intens care acopera un domeniu de
radiofrecventa (nu cu o singura radiofrecventa care
masoara in 1 - 5 minute domeniul deplasarilor chimice, ca in
tehnica CW). Daca frecventa semnalului este F Hz si daca
pulsul are o durata de t secunde, rezultatul este echivalent cu iradierea
simultana a probei cu toate radiofrecventele din domeniul ![]() , deci alegand un puls de aproximativ 0,1 secunde se pot
excita toate nucleele din proba in acelasi moment. Informatia
obtinuta consta in inregistrarea momentului cum se produce
relaxarea nucleara in timpul de 0,5 - 1 secunda cand nu se mai
iradiaza proba cu radiofrecvente externe. Aceasta
informatie (dezexcitarea prin inductie libera) se poate traduce
intr-un spectru RMN normal, folosind transformarea Fourier cu ajutorul unui
calculator montat on line. In fig. 3
este prezentat aspectul dezexcitarii prin inductie libera in
cazul ciclohexenei alaturi de spectrul uzual 13C-RMN de tip CW.
, deci alegand un puls de aproximativ 0,1 secunde se pot
excita toate nucleele din proba in acelasi moment. Informatia
obtinuta consta in inregistrarea momentului cum se produce
relaxarea nucleara in timpul de 0,5 - 1 secunda cand nu se mai
iradiaza proba cu radiofrecvente externe. Aceasta
informatie (dezexcitarea prin inductie libera) se poate traduce
intr-un spectru RMN normal, folosind transformarea Fourier cu ajutorul unui
calculator montat on line. In fig. 3
este prezentat aspectul dezexcitarii prin inductie libera in
cazul ciclohexenei alaturi de spectrul uzual 13C-RMN de tip CW.
Datorita perioadei de 0,5 - 1 secunde, cat dureaza obtinerea si prelucrarea unui spectru, in cateva minute se pot inregistra sute de pulsuri, ceea ce duce la un raport semnal/zgomot mult mai ridicat in cazul spectrelor PFT decat in al celor CW.
Exista insa o serie de consecinte ale aplicarii metodei PFT, de care trebuie sa se tina seama:
1) concentratia substantei in proba trebuie astfel aleasa incat raportul intre intensitatea celui mai intens semnal (acesta provine de obicei din solvent) si al celui mai redus semnal sa nu depaseasca 2000 (la calculatoarele uzuale cu 12 biti ).
2) relaxarea nucleelor nu se produce cu viteze egale, deci in 0,5 - 1 secunda nuclee nu vor ajunge sa se relaxeze, semnalul lor fiind deci mai aproape de saturatie decat al celor care s-au relaxat complet. In consecinta, semnalele nucleelor care se relaxeaza rapid vor avea intensitati mai mari si deci integrala nu mai masoara numarul relativ de nuclee, decat daca acestea au timpul de relaxare egal.
In cazul spectrelor 1H-RMN, tehnica PFT da o mare crestere a rezolutiei, dar nu este absolut necesara. Spectrele 13C-RMN se realizeaza in prezent cu aparate ce folosesc PFT (si care sunt mai costisitoare decat cele CW.
Timpul de relaxare al unui nucleu se compune din timpul de relaxare spin-retea (T1). Prezenta unor atomi de hidrogen legati de carbon scurteaza timpul de relaxare T1; ca urmare semnalele carbonilor cuaternari sau carbonilici in spectrele 13C-RMN au intotdeauna intensitati reduse. Prezenta impuritatilor paramagnetice, de exemplu oxigen dizolvat, scurteaza de asemenea timpul de relaxare; acest efect este uneori util, de exemplu adaosul de acetilacetonat cromic, paramagnetic, mareste intensitatea semnalelor carbonilor cuaternari sau carbonilici.
3. TEHNICA DE LUCRU
In spectroscopie RMN influenta diferitilor factori experimentali asupra aspectului spectrului este mai importanta decat in cazul spectroscopiei IR sau UV-VIZ.
Dintre acesti factori se amintesc cei mai importanti.
Rotirea probei in campul magnetic supune toti nucleii situati intr-un anumit moment pe un cerc orizontal de sectiune a tubului de proba aceluiasi camp magnetic mediu. Daca rotirea probei se face prea lent uniformizarea amintita nu este perfecta si semnalul apare aplatizat. Rotatia probei se manifesta in spectru prin aparitia unor sateliti de rotatie" (benzi foarte slabe situate simetric, deoparte si de alta a semnalului la diferente de frecventa egale cu frecventa de rotatie a probei). La viteze prea mici de rotatie intensitatea satelitilor de rotatie creste mult, putand ingreuna interpretarea spectrului (fig. 4). Vitezele prea mari de rotatie nu sunt de asemenea recomandate intrucat scad rezolutia datorita formarii unor vartejuri in proba.
Viteza de variatie a campului magnetic (in cazul baleiajului de camp) determina forma benzilor de absorbtie. La viteze mari de variatie (1 Hz/s) sfarsitul semnalului de rezonanta este in sfarsit de leganari caracteristice, a caror intensitate descreste rapid (Fig. 4.). Aceasta forma a semnalului RMN al unui compus etalon este un indiciu al omogenitatii campului si ea este testata de regula inainte de inceperea inregistrarii spectrului. La viteze mici de variatie a campului magnetic leganarile amintite dispar (Fig. 4.).
Realizarea unei sensibilitati ridicate in inregistrarile RMN este conditionata de o valoare ridicata a raportului semnal/zgomot de font. Pentru obtinerea unor sensibilitati mari se lucreaza cu probe relativ concentrate (la zgomot de fond egal intensitatea semnalelor creste prin cresterea numarului de molecule absorbante); se incepe inregistrarea dupa un anumit timp de la introducerea probei in aparat (omogenizarea temperaturii in proba, diminuarea zgomotelor de fond mari initiale) sau se utilizeaza dispozitive de acumulare a spectrelor , prin parcurgerea lor repetata.
Probele spectrale RMN se pregatesc de regula sub forma lichida sau de solutie. In aceste cazuri se obtin spectre de inalta rezolutie, caci moleculele se misca liber schimbandu-si orientarea cu frecvente mai ridicate decat frecventa radiatiei radio absorbite.
Pentru o proba obisnuita sunt necesare 50 - 100 mg substanta care se dizolva in solventi adecvati (v. anexa 1), realizandu-se solutii de concentratie de la 10 - 15%. In cazul unor probe foarte mici se poate lucra in microcuve (volum de solutie cca. 15μl) sau se pot utiliza anexele de acumulare ale spectrelor. La substante lichide de mica vascozitate se poate lucra si fara solvent (adica la concentratii de 100%).
Tehnica de lucru H-RMN (protonica)
In cazul spectrometriei RMN protonice se utilizeaza drept solventi compusi ce nu contin protoni, cum ar fi: CCl4; CS2; CDCl3; (D3C)2CO; C6D6; (D3C)2SO; D2O (v. anexa 1). In anumite cazuri se poate folosi H2SO4 sau alti acizi (F3C - COOH) la care semnalul protonilor acizi este pozitionat de obicei departe de semnalele utile ale compusilor organici. In cazuri speciale se pot utiliza si solventi protici, cu conditia de a se interpreta doar regiunile din spectru in care solventul nu da absorbtii directe sau benzi satelit.
Cu majoritatea aparatelor RMN se poate lucra pe un domeniu larg de temperatura (de la -185s pana la +250s ) fapt important pentru studiul unor fenomene dinamice (v. mai departe) dar si din punct de vedere al solubilitatii probelor.
Tehnica de lucru 13C-RMN
Pentru spectroscopia 13C-RMN se folosesc solventi anorganici ca: H2O; D2O; H2SO4 sau solventi organici ce contin doar 1 - 2 tipuri de atomi de carbon diferiti, de obicei tot solventii deuterati ca: CDCl3; CD2Cl2; C6D6; (D3C)2CO; (D3C)2SO; F3C - COOH. Intrucat toate aparatele moderne folosesc pentru ancorare frecventa nucleelor de deuteriu, daca solventul ales nu este deuterat miscibil. Semnalul de referinta este de obicei cel al tetrametilsilanului, standard intern" (v. mai jos) utilizat si in spectroscopia 1H-RMN.
4. SPECTRUL RMN. DEPLASAREA CHIMICA
Spectrul RMN este curba absorbtiei de energie radiata (radiatii electromagnetice) de catre nucleele probei, in functie de campul magnetic aplicat (in cazul baleiajului de camp) sau in functie de frecventa (in cazul baleiajului de frecventa).
In general, valoarea aparenta a lui gn pentru un nuclid oarecare este dependenta de anturajul chimic, determinat de locul nucleului in molecula (se considera moleculele in solutie, in rotatie rapida). Domeniul de valori in care se situeaza gn este functie de natura nuclidului respectiv si de natura invelisului electronic. In cazul 1H-RMN variatiile lui gn pentru diferite categorii de protoni din molecule organice nu depasesc decat exceptional 10 milionimi din valoarea absoluta.
In momentul plasarii probei in campul magnetic, circulatia electronilor de legatura este modificata, in sensul ca, in conformitate cu principiul actiunii si reactiunii, ei incep sa se miste astfel incat sa genereze un camp magnetic opus ca sens campului exterior H. Apare astfel un efect de ecranare diamagnetica, care diminueaza campul exterior ce patrunde pana la nucleul respectiv. Aceasta ecranare este desigur foarte mica, ea atingand la protoni maximum 10 milionimi din valoarea absoluta a campului. In mod evident ecranarea diamagnetica este proportionala cu denumirea de electroni din jurul nucleului respectiv, densitatea conditionata de anturajul chimic, de efectele atragatoare sau respingatoare de electroni etc. Din cele de mai sus rezulta ca valoarea critica a campului ce conditioneaza rezonanta va fi atinsa la nivelul nucleului abia atunci cand campul exterior a depasit valoarea H. La cresterea (desigur cu valori foarte mici ) a campului exterior, vor aparea pe rand semnale pentru diferite tipuri de nuclee, in functie de gradul lor de "ecranare". Esenta metodei RMN si sensibilitatea ei constau tocmai sesizarea si diferentierea intre nuclizi de acelasi fel (de exemplu protoni) avand ecranari diferite. Intr-un spectru RMN (fig. 5) nucleele mai puternic ecranate dau semnale la camp mai mare (respectiv frecventa mai mica in cazul baleiajului de frecventa) comparativ cu nucleele mai putin ecranate.
Fig. Spectrul RMN.
Diferenta valorilor campurilor magnetice de rezonanta (la baleiajul de camp) sau a frecventelor de rezonanta (la baleiajul de frecventa) in functie de vecinatatile structurale se numeste deplasare chimica. Deplasarea chimica indica deci pozitia din spectrul a semnalului fiecarui nucleu. Ea poate fi definita in mod absolut prin exprimarea marimii campului sau frecventei. Acest mod de exprimare nu este utilizat in practica din cauza ca el ar fi incomod si ar depinde de aparat. Se recurge de aceea la exprimarea deplasarii chimice ca diferenta relativa de ecranare a nucleului in raport cu un standard. Exprimarea se face fie in diferente de camp magnetic la frecvente fixa, fie in diferente de frecvente la camp magnetic fix.
Dupa cum s-a amintit si mai sus standardul uzual este de cele mai multe ori, atat pentru 1H-RMN cat ti pentru 13C-RMN, tetrametilsilanul (CH3)4Si (prescurtat TMS), care are un semnal unic (al celor patru atomi de carbon, respectiv al celor 12 protoni echivalenti) ce apare la extremitatea de camp magnetic maxim a spectrului (nuclee foarte ecranate) (fig. 5). TMS se dizolva de obicei in proba (standard intern) fiind inert din punct de vedere al reactivitatii chimice si foarte volatil, el se poate indeparta usor odata cu solventul, permitand recuperarea probei practic pure.
Deplasarea chimica se exprima in valori δ (mai rar si numai pentru 1H si in valori τ) definite de relatiile 6 si 7:
![]()
![]()
Se observa ca marimile adimensionale δ si
τ se exprima in parti pe milion si ele reflecta
sensibilitatea metodei. Astfel, pe o hartie uzuala de spectre 1H-RMN,
pe o lungime de 50 cm se gasesc de regula valori δ intre 0
si 8 ppm (mai rar intre 0 - 10 ppm).Inregistrarea intregului camp (sau
frecventa ar necesita ![]() hartie. Cu toate
acestea, practic semnalele tuturor protonilor din chimia organica se pot
inregistra pe hartie de 50 cm corespunzatoare la 10 ppm din camp (sau din
frecventa).
hartie. Cu toate
acestea, practic semnalele tuturor protonilor din chimia organica se pot
inregistra pe hartie de 50 cm corespunzatoare la 10 ppm din camp (sau din
frecventa).
Totusi, adeseori grupe de protoni neechivalenti chimic (de ex. protonii din catenele saturate lungi) apar suprapuse ducand la ingreunarea interpretarii spectrelor.
In schimb, in cazul spectrelor 13C-RMN deplasarile chimice au valori mult diferite (δ13C=0-220 ppm, in general). Din aceasta cauza arareori apar suprapuneri de carboni neechivalenti. Fig. 6. ilustreaza graitor aceste diferente pentru 3 - metilheptan, unde fiecare gupa CH, CH2 sau CH3 este neechivalenta.
Aceasta deosebire face ca metoda 13C-RMN sa fie net superioara rezonantei magnetice protonice in probleme de atributii structurale.
Ecranarea fiecarui nucleu, adica frecventa, respectiv campul la rezonanta, depinde de frecventa sau campul aparatului (v. relatia 5). Unitatea adimensionala δ (sau τ), rezultata prin divizarea diferentelor de frecventa (sau camp) frecventa (sau campul) de lucru are avantajul important de a nu mai depinde de frecventa (respectiv campul aparatului). Cu alte cuvinte, valoarea δ (ppm) este aceiasi, indiferent daca se lucreaza cu un aparat de 60 MHz sau de100 MHz precum si daca se exprima δ (sau τ) in functie de camp (H) sau de frecventa (v) (v. relatia 6.).
Uneori, pozitia semnalului se exprima in hertzi, prin diferenta dintre frecventa protonului si a standardului (pe hartia spectrala sunt gradate de obicei si diferentele de frecvente in Hz):
![]()
Valorile deplasarilor chimice exprimate astfel sunt dependente de frecventa de lucru, adica de tipul aparatului. Transformarea acestor marimi in marimi δ se face tinand seama de relatia de definitie (6):
![]()
Pentru un aparat de 60 MHz, relatia 9 devine:
![]()
Deci, pentru δ=1 ppm corespunde o diferenta de frecventa 60 Hz la un aparat de 60 MHz, de 100 Hz la un aparat de 100 MHz. De aici rezulta avantajul lucrului cu aparate de frecvente nominale mari care raspandesc semnalele pe un domeniu mai larg de frecvente, permitand o rezolutie mai buna.
Pentru spectrele 13C-RMN, spectrul se intinde de obicei pe 200 ppm, iar valorile δ trebuie citite cu o precizie de 0,002 ppm. Evident, o hartie gradata nu poate permite acest lucru, dar toate aparatele actuale folosesc transformata Fourier si ca atare au atasat un calculator care tipareste toate semnalele semnificative dand deplasarile chimice (in Hz si ppm, ultimele cu o precizie de o sutime de ppm) precum si intensitatile semnalelor, fara a mai fi nevoie sa se traseze si sa se masoare curba integrala. In exemplele din partea aplicativa a acestei lucrari se ilustreaza mai multe asemenea spectre.
FACTORI CARE INFLUENTEAZA DEPLASAREA CHIMICA IN 1H-RMN
Ecranarea unui anumit nucleu poate fi redata de un parametru δ care masoara diminuarea campului magnetic exterior si se traduce in deplasarile chimice. Campul H la care rezoneaza nucleul respectiv este dat de relatia 11:
![]()
Ecranarea δ este suma efectelor mai multor factori, interni sau externi:
![]()
unde δ1 este ecranarea locala diamagnetica, δ2 - ecranarea locala paramagnetica, δ3 - ecranarea indepartata iar δ4 - ecranarea datorita solventului. Termenul local" se refera la ecranari datorite electronilor ce apartin protonului studiat.
Ecranarea locala diamagnetica este o functie a densitatii electronice reale in jurul nucleului studiat. Influente majore in acest sens aduc efectele inductive si conjugative. De exemplu, derivatii monohalogenati ai metanului valorile deplasarilor chimice δ pentru protonii meticlici au urmatoarele valori:
CH3 - F CH3 - Cl CH3 - Br CH3 - I CH3 - H
3,06 2,69 2,16 0,23
Se observa ca substituentul electronegativ, saracind in electroni legaturile C - H, dezecraneaza protonii respectivi. Acestia vor rezona la camp mai mic, deci la valori δ mai mari cu cat efectul inductiv al substituentului sau electronegativitatea sa sunt mai puternice (corelatia valorilor δ cu electronegativitatea halogenilor este in acest caz perfect liniara).
Introducerea mai multor substituenti electronegativi duce la cresterea corespunzatoare a valorilor δ; dupa cum rezulta din exemplele urmatoare:
CH3 - Cl CH2Cl2 CHCl3
δ (ppm) 3,0 5,33 7,24
Influenta efectelor de conjugare rezulta clar din urmatoarele exemple:
H2C = CH2; δCH2=5,28
H2C = CH - CH = O H2C+ - CH = CH - O-; δCH2 = 6 - 6.3
H2C = CH - NR2 H2C - CH =
Scaderea densitatii electronice la grupa CH2 din acroleina ca urmare a efectului de conjugare corespunde cu dezecranarea respectivi si deci cu o valoare mai mare decat in etena. Dimpotriva, cresterea densitatii electronice la grupa CH2 din enamine substituite ca urmare a efectului de conjugare de sens opus celui din acroleina conduce la ecranari manifestate prin valori mai mici ca in etena
Starea de hibridizare a atomului de carbon de care sunt legati protonii studiati influenteaza marimea deplasarii chimice. Cu cat atomul de carbon are un component de tip s mai important in starea sa de hibridizare, ecranarea protonilor va fi mai mica, intrucat atomul de carbon atrage mai puternic electronii de tip s. Astfel, derivatii de etan (sp3) au deplasari chimice cu 4 - 5 ppm mai mici decat derivatii de etena (sp2).
Ecranare locala paramagnetica, dependenta de influenta naturii legaturilor chimice asupra circulatiei electronice, are importanta mica in cazul protonului, dar este puternica in cazul 13C.
Ecranarea indepartata reflecta efectul circulatiei electronice de la atomi mai departati asupra nucleului studiat. In aceasta categorie se incadreaza importantele efecte observate la moleculele cu anizotropie diamagnetica a legaturilor. De exemplu, molecula de acetilena situata intr-un camp magnetic exterior poate adopta doua orientari limita (fig. 7)
In orientare (a) nu apare nici o circulatie diamagnetica electronica. Dimpotriva, in orientare (b) circulatia diamagnetica a electronilor din tripla legatura produce un camp magnetic local, ΔH, care in zona protonilor acetilenici se opune campului magnetic exterior. Ca urmare, pentru a se atinge conditia de rezonanta a acestor protoni este necesar campul H0 ΔH sau, altfel spus, protonii acetilenici sunt puternic ecranati. Valoarea observata, δ=1.80 ppm, reprezinta o medie a tuturor orientarilor posibile intre (a) si (b) pe care le adopta in mod rapid molecula. Prin studierea a numeroase tipuri de molecule s-a demonstrat existenta unor zone spatiale de ecranare sau de dezecranare, situate in vecinatatea unor grupe functionale caracteristice. Cele mai importante exemple de acest fel sunt prezentate in figura 7.
Un alt exemplu foarte interesant il constituie molecula de benzen figura 8. Circulatia electronilor π aromatici situati deasupra si dedesubtul planului atomilor de carbon si hidrogen da nastere unui camp magnetic local ΔH. Acest camp este de acelasi sens cu campul exterior H0 in zona protonilor benzenului (in planul inelului) si este de sens opus cu campul exterior in regiunile situate deasupra si dedesubtul inelului aromatic.
Ca urmare, protonii aromatici situati in planul inelului vor rezona la un camp mai mic (H - H), deci vor avea valori δ mari (7.25 ppm ) in timp ce protonii situati deasupra si dedesubtul inelului aromatic vor fi puternic ecranati. In fig. 8 sunt prezentate si alte exemple demonstrand efectele de ecranare si dezecranare datorite circulatiei electronice in moleculele aromatice (curent de inel). Aceste dezecranari generate de curentul de inel sunt considerate in prezent dovezi clare ale caracterului aromatic.
Ecranare produsa de solvent poate conduce in anumite cazuri la variatii de pana la 1 ppm in pozitia unei benzi spectrale 1H-RMN. Principalele efecte care intervin in aceste deplasari sunt datorate anizotropiei diamagnetice a solventului, polaritatii si polarizabilitatii solventului, interactiunilor Van der Waals sau formarii unor asociatii moleculare, de exemplu prin legaturi de hidrogen. De regula, semnalul protonilor care participa la legaturi de hidrogen apare la valori δ mai mari decat cel al protonilor neasociati. Astfel, protonul grupei -COOH, inclus in legaturi de hidrogen de un semnal la valori δ>10 ppm. Acest fenomen este important in atributii structurale: de exemplu semnalul protonului OH dintr-un alcool poate fi deplasat daca apare intr-o zona cu alte semnale complexe spre camp mai mare (δ mai mic) prin diluarea probei sau prin incalzirea acesteia (operatii care diminueaza numarul legaturilor de hidrogen). Se aminteste si posibilitatea indentificarii pe aceasta cale a tipului legaturilor de hidrogen: cele intramoleculare nu se desfac prin diluare ci numai prin incalzire, in timp ce legaturile intermoleculare se desfac si prin diluare si prin incalzire.
INTESITATEA SEMNALELOR 1H-RMN.
Spre deosebire de spectrele IR sau UV-VIZ, ude coeficientii de extinctie depind mult sau foarte mult de natura substantei (de exemplu grupa CO are valori ε diferite in IR pentru cetone, esteri, acizi, etc.), in rezonanta magnetica nucleara 1H-RMN intensitatea semnalului unui proton este aceiasi indiferent de natura acestuia. Marimea caracteristica pentru intensitatea semnalului RMN este aria de sub curba. Aparatele RMN au posibilitatea de a trasa nu numai spectrul ci si curbele integrale (fig. 9)
Valoarea absoluta a acestor integrale (suprafete, respectiv
inaltimi lA, lB etc.) nu are semnificatie.
Raportul ariilor integrale, exprimat prin raportul inaltimii curbelor
integrale, este insa egal cu raportul numarului de nuclee
responsabile de aparitia fiecarui semnal: ![]()
Relatia (13) este de cea mai mare utilitate la aflarea numarului de nuclee de un anumit tip in determinarile de structura.
CUPLAJUL SPIN-SPIN
Generalitati
Semnalul oricarui nucleu dintr-o molecula este influentat in mod caracteristic de nucleele vecine cu el (dar neechivalente cu el). Astfel, se considera cazul a doi protoni vecini A si X, avand valori δ diferite (fig. 10)
In momentul inregistrarii semnalului primului proton (A) spinul protonului vecin (X) poate fi orientat fie paralel" fie "antiparalel" cu campul. Intrucat s-a vazut mai inainte ca populatiile celor doua nivele sunt practic egale, urmeaza ca se poate considera proba ca fiind formata din doua specii diferite intr-una din specii (care reprezinta 50% din total) protonul X are spinul paralel iar in a doua specie (de asemenea in proportie de 50%) acelasi proton are spinul "antiparalel" cu campul exterior. Ca urmare, pentru protonul A vor aparea in spectru doua semnale, egale, situate la o oarecare diferenta de camp (de frecventa), corespunzatoare diferentei de "ecranare" ce rezulta pentru cele doua specii "diferite", mentionate. Reciproc, in mod corespunzator semnalul protonului X va fi "scindat" de catre spinul protonului A (scindarea spin-spin) in doua semnale egale, separate prin aceiasi diferenta de camp (sau de frecventa) ca si semnalele protonului A. Diferenta de frecventa, exprimata in Hz (se masoara direct pe spectrul) se numeste constanta de cuplaj si se noteaza cu J.
Constanta de cuplaj fiind un efect al campului magnetic intramolecular este independenta de intensitatea campului exterior, respectiv de frecventa aparatului.
Spectrele in care semnalele protonilor se afla separate prin diferente de frecventa Δv >10J se numesc spectre de ordinul intai si ele permit de regula o analiza uzuala.
Spectrele la care Δv < 10J sunt numite spectre de ordinul doi si ele se interpreteaza mult mai dificil.
In interpretarea spectrelor de ordinul intai se tine seama de urmatoarele reguli:
a) protonii echivalenti chimic si magnetic nu se cupleaza intre ei;
b) prin interactiunea unui proton A sau a unui grup de protoni echivalenti A cu n protoni, cu constante de cuplaj diferite, in spectru vor aparea 2n linii, egale ca intensitate (dar diferit distantate).
c) prin interactiunea unui proton A sau a unui grup de protoni echivalenti A cu un numar de n protoni, cu aceiasi constanta de cuplaj, din cauza echidistantei semnalelor o parte din acestea se vor suprapune, astfel ca rezulta un numar mai mic de linii. Numarul de linii este (n 1) iar intensitatile vor fi proportionale, in ordine, cu coeficientii dezvoltarii binomului (a 1)n. De exemplu, pentru n=3: (a+1)3=1a3+3a2+3a+1, deci cele 4 semnale vor fi in raportul 1 Determinarea intensitatii liniilor poate fi facuta si grafic cu ajutorul triunghiului Pascal (v. schema alaturata) in care liniile exterioare au intotdeauna valori unitare, iar valorile liniilor interioare se determina prin adunarea valorilor liniilor superioare din care provin.
d) prin interactiunea unui (unor) proton (i) A cu mai multe grupe diferite de protoni, de exemplu An Mm Xp se obtine pentru protonii A un semnal din (m 1) (p linii. Intensitatea acestora corespunde coeficientilor binomului (a+1)m.(b+1)p.
Pentru o mai buna intelegere a acestor reguli in figura 11 este prezentata detaliat scindarea semnalelor protonilor din grupa etil: protonii echivalenti, HA, din grupa CH2, sesizeaza starile de spin ale protonilor vecini, metilici, care scindeaza semnalul in 4 linii de intensitati 1:3:3:1.
Pe de alta parte, protonii echivalenti HX din CH3 sesizeaza starile de spin ale protonilor vecini CH2, scindandu-si semnalul in trei linii de intensitati 1:2:1. Ariile integrale ale semnalelor CH2 si CH3 se afla in raport 2:3.
Constanta de cuplaj JCH2 - CH3 masurata la semnalul grupei CH2 este egala cu constanta JCH3 - CH2 masurata la semnalul grupei CH3; aceasta este dovada ca protonii celor doua grupe sunt cuplati intre ei.
Cuplajele se transmit prin intermediul electronilor de legatura, de aceea protonii despartiti de un numar mare de legaturi, chiar vecini in spatiu, nu se cupleaza reciproc. Cuplajele protonilor de tipul H - C - H se numesc cuplaje geminale, cele de tipul H - C - C - H se numesc cuplaje vicinale, iar cele de tipul H - (C)n>0 - H se numesc cuplaje la distanta.
In analiza spectrelor RMN este foarte important a se diferentia multiplicitatile datorite cuplajelor de multiplicitatile aparente, datorite numai diferentelor de deplasare chimica. In asemenea diferentieri se tine seama de: a) intensitatile relative ale liniilor, datorite grupelor de protoni echivalenti (rapoarte de numere intregi), b) dependenta deplasarilor de campul magnetic si independenta constantelor de cuplaj de acest camp, c) variatia (relativ mica) a deplasarilor chimice in functie de natura solventului.
In acelasi scop este de mare importanta recunoasterea protonilor echivalenti (chimic si magnetic).
Nucleele echivalente chimic sunt nucleele iterschimbabile printr-o operatie de simetrie. Nucleele echivalente chimic au aceiasi valoare δ (ppm) in toate conditiile achirale.
De exemplu, protonii HA si HB sau protonii HC si HD di compusul ciclopropanic 1 sunt echivalenti chimic intrucat sunt interschimbabili prin rotatia 180˚ un jurul unei axe ce trece prin carbonul metilenic C1 si jumatatea legaturii C2 - C3.
Protonii echivalenti chimic care sunt interschimbabili prin celelalte operatii de simetrie mentionate (in afara de rotatia in jurul unei axe), se numesc enantiotopici; inlocuirea H cu D in aceste cazuri conduce la obtinerea unei perechi de enantiomeri (de ex. in C6H5 - O - CH2 - R).
Un alt exemplu uzual il constituie echivalenta chimica a protonilor grupei CH3 din compusi de tipul 2; in aceste cazuri echivalenta apare ca urmare a rotatiei foarte rapide in jurul legaturii C - C.
Deplasarea chimica a fiecaruia din cei trei protoni a grupei CH3 reprezinta media deplasarilor chimice corespunzatoare celor trei conformatii reprezentate in formule. In lipsa notatiilor distinctive, HB(A,C) cele trei conformatii ar fi identice. Pe de alta parte, daca nu ar exista rotatie libera C - C, fiecare proton ar fi situat in fiecare conformer in imprejurimi diferite si ar avea deci alta valoare δ.
Dimpotriva, in compusul chiral 3 protonii metilenici HA si HB nu sunt echivalenti chimic ei nu pot fi interschimbati prin operatii de simetrie (molecula nu are axa, plan sau centru de simetrie); pe de alta parte, desi exista o rotatie rapida in jurul legaturii C - C, protonii HA si HB nu devin interschimbabili prin aceasta operatie. In acest caz, chiar in lipsa notatiei HA(B) conformatiile moleculei nu sunt identice:
Protonii de acest fel, neinterschimbabili, se numesc protoni diastereotopici; ei nu sunt echivalenti chimic in nici o imprejurare. Inlocuirea unui H cu D in aceste cazuri conduce la obtinerea unei perechi de diastereoizomeri.
Nucleele echivalente magnetic indica, pe langa aceiasi deplasare chimica, o interactiune egala cu toate celelalte nuclee din sistemul de spini[5]. De multe ori doi sau mai multi protoni echivalenti chimic interactioneza cu constante de cuplaj diferite cu un al treilea nucleu si astfel nu sunt echivalenti magnetic. Pentru a se determina echivalenta magnetica este necesara examinarea geometriei sistemului; In cazul unor relatii geometrice identice intre nucleul investigat si fiecare din ceilalti protoni din sistem nuceele sunt echivalente magnetic. Asadar, doua nuclee sunt echivalente magnetic daca ele sunt dispuse simetric in raport cu fiecare nucleu din orice alt set al aceluiasi sistem de spini.
De exemplu, in butadiena (4) protonii HA si HA sunt echivalenti chimic, dar nu sunt echivalenti magnetic cu HC (caci JAC este un cuplaj vinilic trans, iar JA C este un cuplaj alilic, diferit ca valoare de JAC).
In mod asemanator, protonii HA si HA' din 5, deci si echivalenti chimic, nu sunt echivalenti magnetic, avand cuplaje diferite cu HX sau cu HX'.
Protonii echivalenti chimic ai grupei CH3 sunt si echivalenti magnetic datorita aceleiasi rotatii rapide C - C care mediaza si toate interactiunile cu grupele CH2 vecine. Protonii acestor grupe CH2 sunt de asemenea echivalenti magnetic (daca nu apar alte cuplaje suplimentare).
Cuplarea protonilor A si X, prezenta mai sus (fig. 10), este caracteristica pentru interactiunea de ordinul I a doi protoni neechivalenti, foarte diferiti intre ei. (simbolizati de acea prin litere de la extremitatile alfabetului). In cazul interactiunii de ordinul II a doi protoni neechivalenti, dar relativ asemanatori, simbolizati cu literele vecine din alfabet. A si B (de exemplu protonii aromatici din benzeni paradisubstituiti, spectrul RMN (fig. 12) consta de asemenea din doua dublete; se observa insa ca intensitatealiniilor exterioare este din ce in ce mai mica (fata de cea a liniilor interioare) pe masura scaderii raportului Δv/J, adica pe masura indepartarii de la spectrul de ordinul intai (AX). Cazul extrem, a doi protoni echivalenti, A2, conduce la disparitia totala a liniilor exterioare si la furnizarea celor doua linii interioare.
Astfel, sistemul AX2 genereaza un spectru caracteristic de ordinul intai cu aspect de triplet-dublet. Interactiunea a trei protoni neechivalenti, mult diferiti intre ei, AMX (de exemplu protonii din etilenele monosubstituite) conduce la un spectru de trei cuartete cu intensitati totale 1:1:1, fiecare cuartet avand liniile de intensitati egale (1:1:1:1). Cele trei constante de cuplaj, diferite, se pot identifica usor in spectru (fiecare dintre ele aparand la ambii protoni in interactiune, JAM la protonii A si M; JMX la protonii M si X iar JAX la protonii A si X). Interactiunea a trei protoni de tip ABX (ca in cazul stirenului) conduce la spectre de trei cuartete, neicadrabile insa in grupa spectrelor de ordinul intai.
Uneori cuplajele se pot identifica relativ usor (fig. 13), acordand atentie intercalarii sau chiar suprapunerii liniilor spectrale in zona protonilor A si B.
Cazul extrem al cuplarii a trei protoni foarte asemanatori, ABC (de exemplu protonii din 1, 2, 4 - triclorbenzen sau din furanii 2 - monosubstituiti) conduce la spectru complex, de ordinul doi format din 15 linii (uneori o parte din acestea sunt suprapuse). De asemenea, sistemul de trei protoni de tip AB2 (A2B) da nastere unor spectre complexe, de ordinul doi, continand intre 5 si 9 linii (in functie de raportul Δv J) a caror intensitate creste din ambele sensuri spre centru ( efect de acoperis", fig. 13). Exemple de astfel de spectre sunt cele ale unor derivati 1, 2, 3 - trisubstituiti ai benzenului; 9, 10 - dihidorfenanttrenii 9 - substituiti etc.
Interactiunea unui proton cu alti trei protoni echivalenti (de tip AX3), precum si interactiunea a doi protoni echivalenti cu alti doi protoni echivalenti intre ei (de tip A2X2) conduc la spectre caracteristice de ordinul intai (fig. 14) care se intalnesc deseori la compusii organici uzuali. Spectrul de tip A2X3 al grupei etil (de ex. clorura de etil) a fost discutat mai sus (fig. 11.).
FACTORI CARE INFLUENTEAZA CONSTANTELE DE CUPLAJ 1H-RMN
Influenta diferitelor efecte de vecinatate asupra constantelor de cuplaj a fost studiata in detaliu pentru cele mai variate tipuri de sisteme de spini. In acest paragraf se mentioneaza succint principalii factori, insistandu-se in special asupra sensului in care acestia modifica marimea constantelor de cuplaj. In tabele de la sfarsitul acestui capitol sunt date valori concrete pentru numeroase constante de cuplaj din diferite sisteme de spini ce apar in molecule organice.
In compusii cu conformatia mobila, de ex. in catene saturate cu rotatie libera in jurul legaturilor C-C, nu apar scindari de tip geminal. Asemenea scindari se pot manifesta insa la compusii cilici substituiti, unde nu mai exista rotatie in jurul legaturilor C-C.
In sisteme de tipul R - CH - X, constanta de cuplaj geminal intre protonii metilici descreste odata cu cresterea electronegativitatii substituientului X. De ex. Jgem. 12,4 Hz in metan si Jgem. 9,6 Hz in CH3F (marimi determinate prin marcj cu deuteruiu).
Variatia constantelor de cuplaj in functie de unghiul θ format intre atomii de hidrogen este prezentata grafic in fig. 15a. In practica apar abateri de la valorile teoretice prezentate in figura 15, ca urmare a interventiei unor alti factori (de exemplu electronegativitatea substituentilor, includerea sistemului intr-un inel ciclopropanic etc).
In sistemele aciclice, cu rotatie libera, in care grupa CH2 este vecina cu una sau mai multe legaturi π, constanta de cuplaj geminal indica o crestere fixa de 1,9 Hz pentru fiecare legatura π adiacenta. De exemplu, in metan, Jgem.= 12,4 Hz, in tolen (1 legatura π adiacenta) Jgem.= 14,5 Hz iar in acetonitril (2 legaturi π adiacente) Jgem.= 16,2 Hz. Pentru sistemele ciclice continand legaturi π adiacente cu grupa CH2 s-a demonstrat existenta unei contributii de tip π" negativa (se scade din constanta de cuplaj JCH2 a sistemului corespunzator fara legaturi π). Aceasta contributie este dependenta intr-un mod mai complicat de unghiul diedru φ format intre orbitalul π al carbonului α si o legatura CH a grupei CH2. Astfel, ea are valori de 3 - 4,5 Hz pentru φ=0-60˚ si valori mici, de 1 - 2 Hz, pentru φ=60-180˚.
Constantele de cuplaj vicinal in spectrele 1H-RMN
In catene saturate cu rotatie libera in jurul legaturilor C - C, constantele de cuplaj vicinal au valori de cca. 7 Hz. Acesta reprezinta o valoare medie pentru diferitele unghiuri diedre H - C - C -H care variaza continuu in cursul rotatie libere (v. mai jos ec. Karplus). In compusii unde rotatia libera lipseste sau are o rotatie mai joasa decat a radiatiei radio absorbite, constantele de cuplaj vicinale indica o dependenta importanta de unghiul diedru dintre protonii cuplati. Aceasta dependenta se exprima prin ec. Karplus (14, 15), reprezentate grafic in figura 15b.
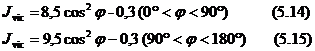
Din relatiile lui Karplus rezulta constante de cuplaj maxime cand protonii vicinali sunt coplanari (trans sau cis) si constantele de cuplaj) apropiate de zero cand protonii vicinali formeaza un unghi diedru de cca. 90˚.
Pentru protonii vicinali din etilene s+a dedus (pe baza unor ecuatii de tip Karplus) ca Jcis= 7 - 10 Hz (φ=0˚) si Jtrans= 12 - 18 Hz (φ=180˚). Practic, independent de substituientii dublei legaturi etilice Jcis<Jtans.
Penttru sisteme de tipul CH3A - CHBX - s-a demonstart o dependenta de tip liniar a constantei de cuplaj vicinal JAB in functie de electronegativitatea lui X; cu cat aceasta este mei mare, constanta de cuplaj JAB scade.
In sisteme de acelasi tip, ciclice, s-a dovedit dependenta constantei de cuplaj JAB de orientare (axiala sau ecuatoriala) a substituentului X. De exemplu, in ciclohexanol 7 (X=OH), sau in alti derivati asemanatori, JAB=2,5 Hz in izomerul axial (cu toate ca unghiul diedru este φ=60˚ in ambele cazuri).
Un alt factor important de care depind constantele de cuplaj vicinale
este marimea unghiului (unghiurilor) α si α formate de
legaturile H - C si C - C ale sistemului cuplat HA si
HB. Odata cu cresterea unghiurilor α si α'
se constata o descrestere a constantei de cuplaj JAB
creste odata cu scaderea lungimii legaturii C - C. De
exemplu, ![]() in etena (lC-C=1,33
A) si JAB = 8 Hz in
benzen (lC-C=1,39 A).
in etena (lC-C=1,33
A) si JAB = 8 Hz in
benzen (lC-C=1,39 A).
Constantele de cuplaj ale protonilor separati prin patru sau mai multe legaturi au in general valori mici care diminueaza puternic cu cresterea numarului de legaturi dintre protonii analizati. La distante mai mari de 4 - 5 legaturi nu se observa cuplaje, decat in cazuri speciale, la sisteme puternic nesaturate. Cateva cazuri de cuplare indepartata sunt insa de mare importanta in analiza structurala: cuplajele alilice; homoalilice, cuplajele protonilor aromatici si ale celor acetilenici:
Alte exemple caracteristice sunt prezentate in tabelele de la sfarsitul capitolului.
9. DUBLA REZONANTA. DECUPLAREA DE SPIN IN SPECTRE 1H-RMN SI 13C-RMN.
Spectre 1H-RMN. Decuplarea homonucleara.
Daca la inregistrarea spectrului 1H-RMN al unor protoni de tip HA se iradiaza protonii vecini HB (cu care protonii A sunt cuplati) cu o frecventa radio egala cu frecventa de rezonanta a protonilor B, atunci tranzitiile spinilor protonilor B vor fi mult stimulate, durata vietii efective a nucleelor in fiecare stare de spin va fi mult diminuata, iar populatiile nivelelor se vor egaliza. Protonii A sunt supusi astfel din parte protonilor B unor influente de sensuri opuse ce se schimba foarte rapid; aceasta influenta este sesizata doar ca valoare medie, in acest caz nula. Cu alte cuvinte, protonii B nu mai exercita nici o influenta asupra campului local mediu al protonilor A. Urmeaza ca din spectrul 1H-RMN vor dispare cuplajele pe care evidentiau semnalele protonilor cuplati magnetic cu protonul iradiat ( decuplare de spin homonucleara iar spectrul se va simplifica, permitand stabilirea prezentei si marimii acestor constante de cuplaj. Prin astfel de iradieri succesive se pot simplifica pe rand diferite regiuni ale unor spectre complexe, care devin mai usor interpretabile.
In ultimul timp se aplica si tehnici spectrale de dubla rezonanta ENDOR ( spin tickling") asupra carora nu se poate insista aici.
Spectre 13C-RMN. Decuplarea de spin heteronucleara. Efect Overhauser nuclear.
In cazul spectrelor 13C-RMN, trebuie avut in vedere ca lucrand cu abundentele nucleare (98,9% 12C cu I=0 si 1,1% 13C) cuplajele C - C sunt practic invizibile, caci probabilitatea aparitiei a doi atomi 13C vecini este neglijabila. Acest fapt poate fi considerat o fericita potrivire a abundentelor naturale, caci daca nuclidul 13C cu I diferit de zero ar fi fost predominant, cuplajele C - C din orice catena ar fi facut imposibil de interpretat spectrele 13C-RMN. Pe de alta parte, o abundenta si mai mica a izotopului 13C ar fi scazut sensibilitatea metodei sub limita admisibila.
Raman de luat in consideratie cuplajele 1H - 13C, care complica in realitate spectrele 13C-RMN, dar care sunt de obicei total eliminate prin ceea ce se numeste decuplare de spin heteronucleara prin zgomot". Cuplajel 1JCH pentru legaturile covalente C - H sunt de ordinul 100 - 200 Hz. De multe ori apar si cuplaje mai mici, datorita substantelor mai mari dintre atomi: H - C - 13C(2JCH) sau H - C - C - 13C(3JCH); acestea, pe deoparte complica spectrele, iar pe de alta parte, multiplicand liniile, diminueaza intensitatile.
Tehnica uzuala de decuplare heteronucleara consta in iradierea concomitenta, pe deoparte cu frecventa corespunzatoare excitarii nucleelor 13C, si pe de alta parte cu o frecventa modulata de decuplare, corespunzatoare protonilor: v(1H) la 24000 Oe, cu o largime de modulare de 1000 Hz. Efectul este echivalent cu iradierea simultana a tuturor protonilor cu frecventele lor corespunzatoare.
In spectru se observa pe deoparte o intensificare a liniilor, prin gruparea semnalelor datorite aceluiasi carbon, pe de alta parte o intensificare suplimentara considerabila (pana la 30%) datorita efectului Overhauser nuclear (EON). In esenta, acest efect consta in influentarea favorabila a populatiei spinilor carbonului prin decuplarea acestuia de protonii cu care fusese cuplat. Un exemplu de decuplare heteronucleara prin zgomot este ilustrat in fig. 16.
Odata cu decuplarea heteronucleara se pierde insa informatia structurala privind numarul protonilor legati de fiecare carbon. Din fericire exista posibilitatea de regasire a acestei informatii, efectuandu-se la dorinta, dupa ce se inregistreaza spectrul normal decuplat, un spectru partial decuplat (off resonance) care mentine doar o mica parte din cuplajele protonilor direct legati (o fractiune din 1JCH) eliminand celelalte cuplaje (2JCH; 3JCH etc.). In asemenea spectre, carbonii grupelor metil apar sub forma de cuadrupleti cu intensitati 1:3:3:1, carbonii grupelor CH2 sub forma de tripleti cu intensitati 1:2:1, carbonii grupelor CH sub forma de dubleti cu intensitati 1:1 iar carbonii ce nu sunt legati de atomi de hidrogen apar nescindati. Aceste date se noteaza uneori pe spectrele 13C-RMN publicate, sub forma: q(CH3): t(CH2): d(CH), si s(C), notata alaturi de fiecare semnal (q-cuartet; t-triplet; d-dublet; s-singlet).
Un exemplu comparativ de decuplare prin zgomot si de tip off resonance este prezentat in figura 17.
O importanta deosebita o prezinta spectroscopia 13C-RMN pentru studiul compusilor aromatici (in special policiclici) cu schelet carbociclic sau heterociclic. La asemenea compusi, cuplajele multiple JH-H nu permit elucidarea spectrelor 1H-RMN; de aceea spectrele 13C-RMN raman singurele care aduc informatii structurale. Desigur, in asemenea cazuri se cere o atributie initiala a semnalelor in sistemele aromatice de baza, dar aceasta problema este de regula deja rezolvata iar rezultatele se gasesc in literatura chimica din ultimii 15ani.
Un exemplu de diferentiere clara a atomilor de carbon aromatici prin valorile δ 13C este sarea de pirilin 13:
In scopul atribuirii semnalelor 13C-RMN pot fi utilizate si alte metode cum ar fi: a)marcarea cu 13C-RMN imbogatit, prin sinteza, in pozitii specifice si observarea semnalelor care cresc in intensitate; b) deuterarea, prin sinteza, cu marcarea unor pozitii specifice sau prin schimb izotopic al legaturii C-H.
Trebuie mentionat ca intrucat deuteriul are spin nuclear (I=1) semnalul CDCl3 in spectrul 13C-RMN este un triplet cu intensitatile 1:1:1 (scindarea in 2I 1 linii ; in mod asemanator, atomii de carbon din grupele CD3 ale unor solventi uzuali RMN ca D3C - SO - CD3 sau D3C - CO - CD3 apar ca hepteti cu intensitati 1:3:6:7:6:3:1.
In spectrele 13C-RMN decuplate heteronuclear prin zgomot de cuplaje cu protoni, cuplajele cu deuteroni raman neafectate; din cauza scindarii liniilor corespunzatoare carbonilor ce sunt deuterati si din cauza absentei efectului Overhauser nuclear, semnalele grupelor CD, CD2 sau CD3 se disting imediat in raport cu aceleasi grupe nedeuterate, prin intensitatea mult mai mica a semnalelor carbonilor deuterati: In urma deuterarii, semnalele carbonilor ce poarta deuteriu dispar practic complet, iar cele ale carbonilor invecinati cu acestia sunt diminuate apreciate datorita cuplajelor la distanta 2JCD; 3HCD etc.
Un avantaj considerabil al spectrelor 13C-RMN fata de spectrele 1H-RMN il constituie faptul ca doar primele pot identifica grupele lipsite de hidrogen cum sunt grupa cetonica, nitril, ester carboxilic. Prezenta acestor grupe poate fi confirmata de spectrele IR, dar se stie ca uneori intensitatea absorbtiei IR pentru grupa CN este atat de mica incat doar spectrele 13C-RMN raman in aceste cazuri un diagnostic structural sigur.
Cele spuse anterior cu privire la neechivalenta magnetica a protonilor grupelor CH2 in spectrele 1H-RMN ale computilor organici se aplica la fel spectrelor 13C-RMN ale compusilor organici care contin grupe C(CH3)2. In cazul spectrelor 13C-RMN insa, datorita diferentelor mult mai mari de deplasare chimica in raport cu spectrele 1H-RMN se intampla adesea ca protoni magnetic neechivalenti sa dea semnale confundate (degenerare accidentala).
STUDIUL FENOMENELOR DEPENDENTE DE TIMP (SPECTROSCOPIE RMN DINAMICA)
In capitolul 1 s-a vazut ca protonul supus actiunii unui camp magnetic exterior se comporta ca un giroscop, axa sa de rotatie (inclinata cu α=54˚ fata de axa campului magnetic exterior) efectuand o miscare de precesie ("precesie Larmor") in jurul axei campului magnetic exterior (fig. 1c).
La frecventele uzuale de lucru (60 - 100 MHz) fenomenul RMN poate fi considerat un fenomen macroscopic. In absenta campului de radio frecventa nucleele echivalente din proba efectueaza miscari de precesie Larmor cu aceiasi frecventa, dar in toate fazele posibile. Aplicarea frecventei radio conduce la coordonarea precesiilor tuturor protonilor echivalenti din proba. Numai dupa aceasta fazare protonii incep sa-si schimbe spinul, dand semnale RMN. Fenomenul de coordonare a precesiilor dureaza (la frecventele uzuale RMN) timpi de sutimi sau miimi de secunda. Metoda RMN nu poate percepe fenomenele care se produc intr-un timp mai scurt decat aceasta coordonare. De exemplu, un echilibru tautomer foarte rapid nu poate fi sesizat in sensul ca nu se vor distinge semnalele diferite, corespunzatoare celor doua forma tautomere, ci se va obtine un singur semnal, corespunzator unei ecranari medii.
Prin gasirea, de la caz la caz, a conditiilor favorabile (solvent, temperatura) la care viteza unor asemenea echilibre tautomere devine acceptabil de mica pentru masuratori RMN, in spectru vor aparea semnale caracteristice ambelor forme. Unele echilibre tautomere lente pot fi studiate chiar la temperatura camerei (de ex. acetil-acetona).
Ciclohexanul, 14, prezinta la temperatura camerei un spectru 1H-RMN format dintr-un singur semnal corespunzator la 12 protoni echivalenti chimic (datorita trecerilor foarte rapide ale protonilor ecuatoriali in protoni axiali prin inversari de conformatie):
Daca insa se lucreaza la temperaturi foarte scazute (-100˚) inversiile conformationale devin mult mai lente, fenomenul incadrandu-se in "scala de timp RMN"; ca urmare, in spectrul RMN se vor sesiza si diferentia (prin valori δ caracteristice) protonii ecuatoriali δ=1,6 ppm de cei axiali δ=1,1 ppm.
Fenomenele dependente de timp influenteaza si cuplajele. De exemplu, in spectrul obisnuit al etanolului, protonul OH nu este cuplat cu protonii vecini al grupei CH2. Aceasta "decuplare" se poate explica printr-un schimb foarte rapid al protonului hidroxilic intre diferite molecule sub influenta urmelor catalitice de acizi sau baze existente practic in orice proba. Protonul respectiv apartine pentru timpi foarte scurti multor molecule din proba si el sesizeaza toate aranjamentele posibile ale spinilor grupei vecine CH2. Intrucat schimbul protonic amintit este extrem de rapid, aranjamentele spinilor CH2 dau un efect mediat, observandu-se o singura linie RMN. In etanolul purificat in mod special, in care schimbul de protoni este incetinit, semnalul OH apare insa ca un triplet.
SPECTRE RMN IN PREZENTA REACTIVILOR DE DEPLASARE CHIMICA.
La inregistrarea spectrului RMN al unor compusi posedand electroni neparticipanti, in prezenta unor complecsi continand metale tranzitionale (paramagnetice) cu care complexeaza, semnalele protonilor probei sunt deplasate fata de pozitia lor normala. Aceasta modalitate de influenta reciproca intre electroni si spinul nuclear este cunoscuta sub denumirea de interactiune de pseudocontact.
Cel mai uzual compus utilizat in acest scop este tris (2,2,6,6 - tetrametilheptan - 3, 5-dionato) europiul, numit si tris (dipivaloil - metanato) europiu (prescurtat Eu(DPM)3) care produce deplasari spre campuri mai joase ale protonilor.
Compusul analog de praseodim, Pr(DPM)3 produce deplasari spre campuri mai inalte.
Complexarea moleculelor probei cu derivatii amintiti ai lantanidelor (si cu inca multi altii, utilizati pe scara larga in ultimul timp) este un fenomen dinamic rapid. Fiecare molecula este complexata un anumit timp, apoi necomplexata un alt timp, deplasarile chimice fiind valori mediate ale celor doua forme. Timpul cat molecule probei sunt complexate depinde de raportul molar proba Eu(DPM)3. Ca urmare, se pot obtine deplasari variabile, liniare, pentru acelasi proton pe scara δ in functie de raportul amintit (fig. 18). Deplasarea este cu atat mai puternica cu cat protonul respectiv este mai apropiat in spatiu de atomul de europiu si ea depinde si de unghiuri, dupa formula McConnell-Robertson (16):
![]()
in care ΔδI este deplasarea chimica indusa de europiu asupra protonului Hi, θI este unghiul H.Eu.X (X fiind atomul ce complexeaza europiul, de regula oxigen), rI este distanta H.Eu, iar k o constanta.
Avantajul important al utilizarii acestor complecsi de lantanide, numiti reactivi de deplasare chimica, este acela ca permite imprastierea semnalelor unui spectru prea complicat, usurand mult interpretarea sa. In plus, cuplajele complicate de ordinul de tip ABC se transforma in cuplaje de ordinul intai de tip AMX. Pentru molecule de mare complexitate se pot utiliza succesiv reactivi cu europiu si praseodim, putandu-se astfel deplasa semnalele pe un domeniu mult mai larg. De asemenea trebuie mentionat ca deplasarea chimica necunoscuta a unor protoni dintr-un spectru complex, cu semnale suprapuse, poate fi extrapolata din diagrame de tipul celei din fig. 18.
Tabelul 12.1
12 Tabelul de date spectrale 1H-RMN si 13C-RMN
H-RMN. Deplasari chimice. Alcani substituiti. (δ ppm fata de TMS).
|
Substituentul |
Metil - CH3 |
Etil - CH2 - CH3 |
n-Propil - CH2 - CH2 - CH3 |
izo-Propil - CH - CH3 |
t-Butil - CH3 |
|
- H - CH=CH2 - C ≡ CH - C6H5 - Cl - Br - I - OH - O - Alchil - O - C6H5 - O - COCH3 - NH2 - NHCOCH3 - NO2 - CHO - COC6H5 - COOH - COOCH3 |
0,86 1,00 1,15 1,24 1,33 1,66 1,88 1,18 1,15 1,38 1,21 1,10 1,12 1,58 1,13 1,18 1,16 1,12 |
1,33 0,91 1,81 1,06 1,88 1,03 1,56 0,97 |
0,91 |
Tabelul 12.2
H-RMN. Regulile Shoolery pentru calculul aditiv al deplasarilor chimice ale protonilor alifatici metilenici (CH2XY) si metinici (CHXYZ)[7]
(δ ppm fata de TMS)
|
Substituentul |
Incrementul ai |
Substituentul |
Incrementul aI |
|
H CH3 CH2 - Alchil C=C C ≡ C - R COOR NR2 CN COR |
I C6H5 Br OR Cl OH OCOR F |
Tabelul 12.3
Tabelul 12.4
H-RMN. Deplasari chimice alcani halogenati.
|
Compusul |
δ ppm fata de TMS, pentru X= |
|||
|
F |
Cl |
Br |
I |
|
|
CH3X CH2X2 CHX3 X - CH2 - CH3 XCH2CH2X | ||||
Tabelul 12.5
H-RMN. Constanta de cuplaj. Alcani (J, Hz)
Jgeminal=8..18 Hz
|
Compusul |
Jgem |
|
CH4 CH3Cl CH2Cl2 CH3OH CH3 - C6H5 CH3CN |
![]()
Tritiul, izotop radioactiv artificial, 3H , are spinul nuclear 1/2 si frecventa de rezonanta 96MHz la campul de 21 100 Oe
Aparatele moderne de RMN mentin automat la rezonanta o proba cu acelasi fel de nuclee sau cu nuclee diferite ( ancorare homo- respectiv heteronucleara pastrandu-se astfel absolut constant raportul camp frecventa.
Prin sistem de spini se inteleg seturile de nuclee, care pot fi cuplate intre ele dar care sunt izolate de alte nuclee (nu se cupleaza cu nici un alt nucleu din afara sistemului). Setul de nuclee cuprinde doua sau mai multe nuclee cu valori 8 egale.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 4571
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved