| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Antimicrobial Agents: Antifungal Agents
Overview
|
Fungal infections traditionally have been divided into two distinct classes: systemic and superficial. Consequently, the major antifungal agents described in this chapter are discussed under two major headings, systemic and topical, although this distinction is becoming arbitrary. For example, the imidazole, triazole, and polyene antifungal agents may be used either systemically or topically, and, similarly, many superficial mycoses can be treated either systemically or topically. Amphotericin B, fluconazole, and itraconazole are discussed in considerable detail. Although Pneumocystis carinii, responsible for life-threatening pneumonia in immunocompromised patients, has been demonstrated to be a fungus and not a protozoan, its treatment is discussed in other chapters in this section, because currently employed agents in this setting are primarily antibacterial or antiprotozoal rather than antifungal. |
Antimicrobial Agents: Antifungal Agents: Introduction
|
Azole antifungal agents have dominated drug development and clinical use for nearly three decades. While the antifungal spectrum, physical properties, and pharmacology differ among compounds, azoles are remarkable as a drug class for their broad spectrum, oral bioavailability, and low toxicity. New compounds are still being developed, though the innovations are becoming more marginal, and resistance to azoles is slowly emerging in species that formerly were susceptible, particularly Candida albicans. Another area of antifungal drug development has been new, lipid formulations of amphotericin B. Compared with the original deoxycholate formulation (DOC), the lipid formulations have provided a major reduction in renal toxicity, with more variable reduction in infusion-related chills and fever. Oddly, none of the preparations has been evaluated prospectively in the treatment of established mycoses. Rather, they have been used to treat and are approved for use in patients who have failed or been unable to tolerate DOC, so-called salvage therapy. Although some formulations also have been studied as empiric therapy of neutropenic patients, efficacy has been extraordinarily difficult to assess in these trials. It remains unclear whether any lipid preparation is more effective in any mycosis than the DOC preparation given at full dosages, but the probable answer is no. Determination of blood concentrations of antifungal drugs remains a research procedure except in the case of flucytosine. The toxicity of flucytosine clearly depends on drug concentration; azotemia readily increases flucytosine to toxic levels. For the azoles and amphotericin B formulations, blood levels have not predicted either toxicity or efficacy. The advance of systemic antifungal agents into common medical practice has made it important to define their efficacy, toxicity, and interactions with other drugs. The wider choice of topical agents has complicated the selection process for the physician and, because of new over-the-counter azole drugs, for the patient as well. Table 491 lists the most common mycoses and the treatments of choice. |
Systemic Antifungal Agents
|
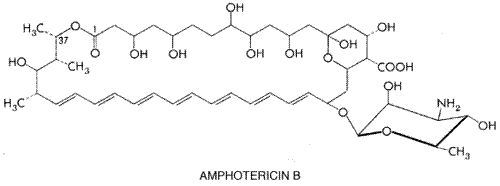
Amphotericin B History and Source Amphotericin B was discovered in 1956 by Gold and coworkers, who were studying a strain of Streptomyces nodosus, an aerobic actinomycete, obtained from the Orinoco River Valley of Venezuela. Chemistry Amphotericin B is one of a family of some 200 polyene macrolide antibiotics. Those studied to date share the characteristics of four to seven conjugated double bonds, an internal cyclic ester, poor aqueous solubility, substantial toxicity on parenteral administration, and a common mechanism of antifungal action. Amphotericin B (see below for structure) is a heptaene macrolide containing seven conjugated double bonds in the trans position and 3-amino-3,6-dideoxymannose (mycosamine) connected to the main ring by a glycosidic bond. The amphoteric behavior for which the drug is named derives from the presence of a carboxyl group on the main ring and a primary amino group on mycosamine; these groups confer aqueous solubility at extremes of pH. X-ray crystallography has shown the molecule to be rigid and rod-shaped, with the hydrophilic hydroxyl groups of the macrolide ring forming an opposing face to the lipophilic polyenic portion.
Drug Formulations Amphotericin B is insoluble in water but was formulated for
intravenous infusion by complexing it with the bile salt deoxycholate. The
complex is marketed as a lyophilized powder (FUNGIZONE) containing 50 mg of amphotericin
B, 41 mg of deoxycholate, and a small amount of sodium phosphate buffer. The
amphotericin Bdeoxycholate complex (DOC) forms a colloid in water, with
particles largely below 0.4 N-acyl and
O-acyl derivatives of amphotericin B form water-soluble salts, but
none is available commercially. The amphipathic nature of amphotericin B has
made it possible to create lipid formulations for intravenous infusion. Three
such formulations of amphotericin B are marketed in the A small unilamellar vesicle formulation of amphotericin B ( AMBISOME ) also is available. Amphotericin B (50 mg) is combined with 350 mg of lipid in an approximately 10% molar ratio. The lipid contains hydrogenated soy lecithin (phosphatidylcholine), cholesterol, and distearoylphosphatidylglycerol in a 10:5:4 molar ratio. The drug is supplied as a lyophilized powder, which is reconstituted with sterile water for injection and then the dose diluted with 5% dextrose solution. With complete dispersion, particle size is about 80 nm. Blood levels following intravenous infusion are almost equivalent to those obtained with DOC, and, because AMBISOME can be given at higher doses, blood levels have been achieved that exceed those obtained with DOC. Amphotericin B accumulation in the liver and spleen is higher with AMBISOME than with DOC (de Marie et al., 1994). In a series of 23 patients receiving 3 mg/kg daily for an average of 27 days, the average serum creatinine rise was only 34%, with nephrotoxicity leading to dose reduction in only one patient (Coker et al., 1993). Nephrotoxicity, hypokalemia, and infusion-related reactionssuch as fever, chills, hypoxia, hypotension, and hypertensionare less with AMBISOME than with either DOC or ABLC (see below), but they still occur (Walsh et al., 1999). Infusion-related pain in the back, abdomen, or chest occurs in occasional patients, usually with the first few doses (Johnson et al., 1998). Anaphylaxis has been reported. Most of the information about the efficacy of AMBISOME comes from open, noncomparative studies. Blinded, randomized comparisons of DOC and AMBISOME as empirical therapy or prophylaxis in febrile neutropenic patients are very helpful in comparing toxicity of the two drugs (Walsh et al., 1999; Prentice et al., 1997; Kelsey et al., 1999). By the primary endpoints of these studies, both preparations have been comparable. However, because the efficacy of DOC in these patients cannot be quantitated from historical data, the relative efficacy of AMBISOME is impossible to ascertain. Secondary analysis of mycoses emerging despite the use of AMBISOME or DOC in these trials has been limited by the rarity of this event and the tendency of investigators to reduce the DOC doses to subtherapeutic levels. AMBISOME is approved for empiric therapy of fever in the neutropenic host not responding to appropriate antibacterial agents as well as for salvage therapy of aspergillosis, cryptococcosis, and candidiasis. The recommended dose for empiric therapy is 3 mg/kg daily, and that for treatment of mycoses is 3 to 5 mg/kg intravenously. AMBISOME also is effective in visceral leishmaniasis at doses of 3 to 4 mg/kg daily. The drug is administered in 5% dextrose in water, with initial doses being infused over 2 hours. If well tolerated, infusion duration can be shortened to one hour. Although doses up to 10 mg/kg have been used in a few patients without causing death, 8 mg/kg is highly toxic in dogs (Bekersky et al., 1999). The third lipid formulation is amphotericin B lipid complex (ABLC, ABELCET ). This preparation of
dimyristoylphosphatidylcholine and dimyristoylphosphatidylglycerol in a 7:3
mixture with approximately 35 mol% amphotericin B forms ribbon-like sheets
which range in size from 1.6 to 11 DOC has been mixed with a 20% lipid emulsion (INTRALIPID) and infused intravenously. Whether amphotericin B aggregates or is bound to lipid in the infusate is unknown. Nephrotoxicity appears to be less than with DOC at 1 mg/kg, but there is a suggestion that both blood levels and efficacy are also lower (Chavanet et al., 1992). Intravenous infusion of DOC with lipid emulsion is not recommended. Rational use of the lipid formulations has been problematic for hospital pharmacies and physicians. Cost of the formulations is 20 to 50 times that of DOC, raising formidable cost-benefit issues. The person making the choice must weigh the uncertainties about relative efficacy of the lipid formulations against the gravity of potential nephrotoxicity of the formulation in individual patients. In some patients, the additive burden of amphotericin B nephrotoxicity can help precipitate advanced renal failure, with attendant morbidity and financial burden (Wingard et al., 1999). Antifungal Activity Amphotericin B has useful clinical activity against Candida spp., Cryptococcus neoformans, Blastomyces dermatitidis, Histoplasma capsulatum, Sporothrix schenckii, Coccidioides immitis, Paracoccidioides braziliensis, Aspergillus spp., Penicillium marneffei, and the agents of mucormycosis. Some isolates of Candida lusitaniae have appeared to be relatively resistant to amphotericin B. Amphotericin B has limited activity against the protozoa Leishmania braziliensis and Naegleria fowleri. The drug has no antibacterial activity. Mechanism of Action The antifungal activity of amphotericin B depends at least in part on its binding to a sterol moiety, primarily ergosterol, that is present in the membrane of sensitive fungi. By virtue of their interaction with the sterols of cell membranes, polyenes appear to form pores or channels. The result is an increase in the permeability of the membrane, allowing leakage of a variety of small molecules. Additional mechanisms of action may include oxidative damage to fungal cells, at least in vitro. Fungal Resistance Mutants selected in vitro for nystatin or amphotericin B resistance replace ergosterol with certain precursor sterols. The rarity of significant amphotericin B resistance arising during therapy has left it unclear whether or not ergosterol-deficient mutants retain sufficient pathogenicity to survive in deep tissue. Failure of amphotericin B to penetrate the fungal cell wall of some resistant species has been suggested by the greater susceptibility of protoplasts. Absorption, Distribution, and Excretion Absorption of all amphotericin B formulations from the gastrointestinal
tract is negligible. Repeated daily intravenous infusions to adults of 0.5
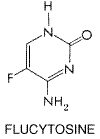
mg/kg of DOC result in concentrations in plasma of about 1.0 to 1.5 Therapeutic Uses The usual therapeutic dose of amphotericin B DOC is 0.5 to 0.6 mg/kg, administered in 5% glucose over 4 hours. Candida esophagitis in adults responds to 0.15 to 0.2 mg/kg daily. Rapidly progressive mucormycosis or invasive aspergillosis is treated with doses of 1.0 to 1.2 mg/kg daily until progression is arrested. Double-dose alternate day therapy may be more convenient but is not less toxic and is therefore rarely indicated. The infusion bottle need not be protected from light, as once recommended. Infusion intervals as short as one hour have been used, but, during the first 5 to 7 days of therapy, they lead to more febrile reactions. Intrathecal infusion of amphotericin B DOC is useful in patients with meningitis caused by Coccidioides. The drug can be injected into the CSF of the lumbar spine, cisterna magna, or lateral cerebral ventricle. Regardless of the site of injection, the treatment is begun with 0.05 to 0.1 mg and increased on a three-times-a-week schedule to 0.5 mg, as tolerance permits. Therapy is then continued on a twice-a-week schedule. Fever and headache are common reactions and may be decreased by intrathecal administration of 10 to 15 mg of hydrocortisone. Less common but more serious problems attend the use of intrathecal injections; the nature of the problem depends on the injection site chosen. Local injections of amphotericin B into a joint or peritoneal dialysate fluid commonly produce irritation and pain. Intraocular injection following pars plana vitrectomy has been used successfully for fungal endophthalmitis. Intravenous administration of amphotericin B is the treatment of choice for mucormycosis, invasive aspergillosis, extracutaneous sporotrichosis, cryptococcosis, fusariosis, alternariosis, trichosporonosis, and penicilliosis marneffei. Although imidazoles or triazoles are useful in many patients with blastomycosis, histoplasmosis, coccidioidomycosis, and paracoccidioidomycosis, amphotericin B is preferred when these mycoses are rapidly progressive, occur in an immunosuppressed host, or involve the central nervous system. Amphotericin B (DOC or AMBISOME ) also can be useful in selected patients with profound neutropenia and fever that is unresponsive to broad-spectrum antibacterial agents. Amphotericin B given once weekly has been used to prevent relapse in patients with AIDS who have been treated successfully for cryptococcosis or histoplasmosis. Bladder irrigation with 50 Untoward Effects The major acute reaction to intravenous amphotericin B DOC is fever and chills. Sometimes hyperpnea and respiratory stridor or modest hypotension may occur, but true bronchospasm or anaphylaxis is rare. Patients with preexisting cardiac or pulmonary disease may tolerate the metabolic demands of the reaction poorly and develop hypoxia or hypotension. A test dose of 1 mg may be considered for unstable patients; the patient should be observed for two hours prior to infusing the usual therapeutic dose. Although the reaction ends spontaneously in 30 to 45 minutes, meperidine may shorten it. Pretreatment with oral acetaminophen or use of intravenous hydrocortisone hemisuccinate, 0.7 mg/kg, at the start of the infusion decreases reactions. Febrile reactions abate with subsequent infusions. Infants, children, and patients receiving therapeutic doses of corticosteroids are less prone to reactions. Azotemia occurs in 80% of patients who receive amphotericin B for deep mycoses (Carlson and Condon, 1994). Toxicity is dose-dependent and transient and is increased by concurrent therapy with other nephrotoxic agents, such as aminoglycosides or cyclosporine. Although permanent histological damage to renal tubules occurs even during short courses, permanent functional deficits are uncommon in patients whose renal function was normal prior to treatment unless a total dose in excess of 3 to 4 g is given (to an adult). Renal tubular acidosis and renal wasting of K+ and Mg2+ also may be seen during and for several weeks after therapy. Supplemental K+ is required in one-third of patients on prolonged therapy. An increase in intrarenal vascular resistance is the major cause of nephrotoxicity in amphotericin Btreated rats (Tolins and Raij, 1988). In patients and experimental animals, loading with sodium chloride has decreased nephrotoxicity, even in the absence of water or salt deprivation. Administration of 1 liter of saline intravenously on the day that amphotericin B is to be given has been recommended for adults who are able to tolerate the Na+ load and who are not already receiving that amount in intravenous fluids (Branch, 1988). Hypochromic, normocytic anemia is usual; the average hematocrit declined to 27% in one study. Decreased production of erythropoietin is the probable mechanism. Patients with low plasma erythropoietin may respond to administration of recombinant erythropoietin. Anemia reverses slowly following therapy. Headache, nausea, vomiting, malaise, weight loss, and phlebitis at peripheral infusion sites are common side effects. Encephalopathy also has been attributed to amphotericin B (Balmaceda and Walker, 1994). Thrombocytopenia or mild leukopenia is observed rarely. Hepatotoxicity is not firmly established. Flucytosine Chemistry Flucytosine is a fluorinated pyrimidine related to fluorouracil and floxuridine. It is 5-fluorocytosine, the formula of which is as follows:
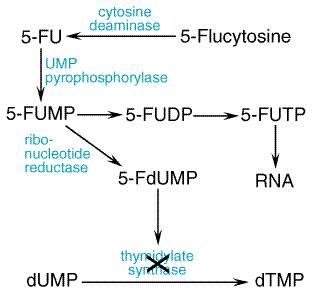
Antifungal Activity Flucytosine has clinically useful activity against Cryptococcus neoformans, Candida spp., and the agents of chromomycosis. Within these species, determination of susceptibility in vitro has been extremely dependent on the method employed, and susceptibility testing performed on isolates obtained prior to treatment has not correlated with clinical outcome. Mechanism of Action All susceptible fungi are capable of deaminating flucytosine to 5-fluorouracil, a potent antimetabolite (seeFigure 491). Fluorouracil is metabolized first to 5-fluorouridylic acid by the enzyme uridine monophosphate (UMP) pyrophosphorylase. It can then either be incorporated into RNA (via synthesis of 5-fluorouridine triphosphate) or be metabolized to 5-fluorodeoxyuridylic acid, a potent inhibitor of thymidylate synthetase. DNA synthesis is impaired as the ultimate result of this latter reaction. Mammalian cells do not convert flucytosine to fluorouracil. This fact is crucial for the selective action of this compound.
Fungal Resistance Drug resistance that arises during therapy (secondary resistance) is
an important cause of therapeutic failure when flucytosine is used alone for
cryptococcosis and candidiasis. In chromomycosis, resurgence of lesions after
an initial response has led to the presumption of secondary drug resistance.
In isolates of Cryptococcus and Candida species, secondary drug
resistance has been accompanied by a change in the minimal inhibitory
concentration from less than 2.5 Absorption, Distribution, and Excretion Flucytosine is absorbed rapidly and well from the gastrointestinal
tract. It is widely distributed in the body, with a volume of distribution that
approximates total body water. The drug is minimally bound to plasma
proteins. The peak plasma concentration in patients with normal renal
function is approximately 70 to 80 Flucytosine is present in CSF at a concentration about 65% to 90% of that simultaneously present in the plasma. The drug also appears to penetrate into the aqueous humor. Therapeutic Uses Flucytosine ( ANCOBON ) is given orally in amounts of 100 to 150 mg/kg per day, divided into 4 doses at 6-hour intervals. Dosage must be adjusted for decreased renal function. Flucytosine is used predominantly in combination with amphotericin B. Flucytosine had a possible, though not statistically significant, benefit and no added toxicity when added to 0.7 mg/kg of amphoterin B for the initial two weeks of therapy of cryptococcal meningitis in AIDS patients (van der Horst et al., 1997). An all-oral regimen of flucytosine plus fluconazole also has been advocated for therapy of AIDS patients with cyrptococcosis, but there is substantial gastrointestinal toxicity with the combination and no evidence that flucytosine adds benefit to the regimen. In cryptococcal meningitis of non-AIDS patients, in whom the goal is cure and not suppression, the role of flucytosine is more conjectural. Addition of flucytosine to six weeks or more of therapy with amphotericin B runs the risk of substantial bone marrow suppression or colitis if the flucytosine dose is not promptly adjusted downward as amphotericin Binduced azotemia occurs. More rapid culture conversion has been shown in cryptococcal meningitis of non-AIDS patients when flucytosine is added to an amphotericin B dose of 0.3 mg/kg daily but not when added to the more commonly used dosage of 0.7 mg/kg. Use of flucytosine in deep candidiasis has all but disappeared because of toxicity, absence of an intravenous formulation, and availability of other agents. Untoward Effects Flucytosine may depress the function of bone marrow and lead to the
development of leukopenia and thrombocytopenia; patients are more prone to
this complication if they have an underlying hematological disorder, are
being treated with radiation or drugs that injure the bone marrow, or have a
history of treatment with such agents. Other untoward effectsincluding rash,
nausea, vomiting, diarrhea, and severe enterocolitishave been noted. In
approximately 5% of patients, plasma levels of hepatic enzymes are elevated,
but this effect reverses when therapy is stopped. Toxicity is more frequent
in patients with AIDS or azotemia (including those who are receiving amphotericin
B concurrently) and when concentrations of the drug in plasma exceed 100 Imidazoles and Triazoles The azole antifungals include two broad classes, imidazoles and
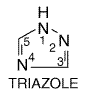
triazoles. Both classes share the same antifungal spectrum and mechanism of
action. The systemic triazoles are more slowly metabolized and have less
effect on human sterol synthesis than do the imidazoles. Because of these
advantages, new congeners under development are mostly triazoles, not
imidazoles. Of the drugs now on the market in the
Antifungal Activity Susceptibility testing with azole antifungals has not been useful in predicting which fungal species will respond to therapy. Although individual drugs have their own useful spectrum, azoles as a group have clinically useful activity against C. albicans, Candida tropicalis, Candida glabrata, C. neoformans, B. dermatitidis, H. capsulatum, C. immitis, Paracoccidioides brasiliensis, and ringworm fungi (dermatophytes). Aspergillus spp. and S. schenckii are intermediate in susceptibility. Candida krusei and the agents of mucormycosis appear to be resistant. These drugs do not appear to have any useful antibacterial or antiparasitic activity, with the possible exception of antiprotozoal effects against Leishmania major. Mechanism of Action At concentrations achieved during systemic use, the major effect of
imidazoles and triazoles on fungi is inhibition of sterol 14- Some azoles, such as clotrimazole, directly increase permeability of the fungal cytoplasmic membrane, but the concentrations required are likely only obtained with topical use. Azole resistance has emerged gradually during prolonged azole therapy
and has caused clinical failure in patients with far-advanced HIV infection
and oropharyngeal or esophageal candidiasis. The primary mechanism of
resistance in C. albicans is accumulation of mutations in ERG11, the
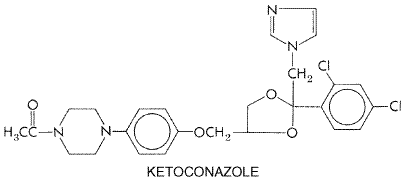
gene coding for the C14- Ketoconazole Ketoconazole, administered orally, has been replaced by itraconazole for the treatment of all mycoses except when the lower cost of ketoconazole outweighs the advantage of itraconazole. Itraconazole lacks ketoconazole's hepatotoxicity and corticosteroid suppression, while retaining most of ketoconazole's pharmacological properties and expanding the antifungal spectrum. Introduction of even newer triazoles likely will reduce further the usefulness of ketoconazole. The structural formula of ketoconazole is as follows:
Absorption, Distribution, and Excretion Oral absorption of ketoconazole varies among individuals. Since an
acidic environment is required for the dissolution of ketoconazole,
bioavailability is markedly depressed in patients taking H2-histamine
receptor blocking agents such as cimetidine or proton pump inhibitors.
Simultaneous administration of antacids also may impair absorption, as will didanosine
(an anti-HIV agent) products, which contain a buffer to neutralize gastric
acid and increase the absorption of didanosine. Ingestion of food has no
significant effect on the maximal concentration of the drug achieved in
plasma. After oral doses of 200, 400, and 800 mg, peak plasma concentrations
of ketoconazole are approximately 4, 8, and 20 Ketoconazole reaches keratinocytes efficiently, and its concentration in vaginal fluid approaches that in plasma. The concentration of ketoconazole in the CSF of patients with fungal meningitis is less than 1% of the total drug concentration in plasma. Induction of hepatic microsomal enzymes by rifampin, isoniazid, and possibly by phenytoin accelerates the metabolic clearance of ketoconazole, and concentrations of the antifungal agent may be reduced by more than 50%. Ketoconazole raises plasma concentrations of cyclosporine, midazolam, triazolam, indinavir, and phenytoin because the drugs are metabolized by the cytochrome P450 enzyme CYP3A4. The anticoagulant effect of warfarin also may be enhanced. Therapeutic Uses Ketoconazole ( NIZORAL ) is effective in blastomycosis, histoplasmosis, coccidioidomycosis, pseudallescheriasis, paracoccidioidomycosis, ringworm, tinea versicolor, chronic mucocutaneous candidiasis, Candida vulvovaginitis, and oral and esophageal candidiasis. Efficacy is poor in immunosuppressed patients and in meningitis. The usual adult dose is 400 mg taken once daily. Children are given 3.3 to 6.6 mg/kg daily. Duration of therapy is 5 days for Candida vulvovaginitis, 2 weeks for Candida esophagitis, and 6 to 12 months for deep mycoses. The slow response to therapy has made ketoconazole inappropriate for patients with severe or rapidly progressive mycoses. For all of the above indications, itraconazole has replaced ketoconazole for patients who can afford the more expensive, newer product. Untoward Effects The most common side effects of ketoconazole are dose-dependent nausea, anorexia, and vomiting, which occur in about 20% of patients receiving 400 mg daily. Administration of the drug with food, at bedtime, or in divided doses may improve tolerance. An allergic rash occurs in about 4% of ketoconazole-treated patients and pruritus without rash in about 2%. Hair loss has also been reported. Ketoconazole inhibits steroid biosynthesis in patients, as it does in
fungi, by inhibition of cytochrome P450dependent enzyme systems. Several
endocrinologic abnormalities thus may be evident. Approximately 10% of
females report menstrual irregularities. A variable number of males
experience gynecomastia and decreased libido and potency. At high doses,
azoospermia has been reported, but sterility has not been permanent. Doses of
ketoconazole as low as 400 mg can cause a transient drop in the plasma
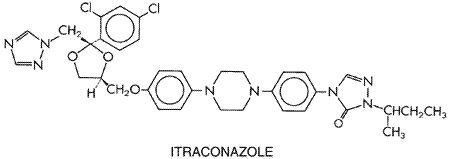
concentrations of free testosterone and estradiol C-17 Mild, asymptomatic elevation of aminotransferase activity in plasma is common, occurring in 5% to 10% of patients; these values revert to normal spontaneously. Symptomatic drug-induced hepatitis is rare but is potentially fatal. Hepatitis may occur after a few days of treatment, or it may be delayed for many months. The earliest symptoms are anorexia, malaise, nausea, and vomiting, with or without dull abdominal pain. Liver function tests usually mimic the pattern seen with hepatitis A, but a cholestatic or mixed picture can occur. Patients should be alerted to the symptoms and asked to return for liver function tests should this toxicity be suspected. Ketoconazole is teratogenic in animals, causing syndactyly in rats. Its use during pregnancy is not recommended, and because of secretion of the drug into breast milk, its use in nursing mothers also is unwise. Itraconazole This synthetic triazole is a 1:1:1:1 racemic mixture of four diastereoisomers (two enantiomeric pairs), each possessing three chiral centers. The structural formula is closely related to the imidazole, ketoconazole, as shown below:
Absorption, Distribution, and Excretion Itraconazole SPORONOX) is available as a capsule and
two solution formulations, one for oral and one for intravenous
administration. The capsule form of the drug is best absorbed in the fed
state, but the oral solution is better absorbed in the fasting state and
provides, under that condition, peak plasma concentrations that are more than
150% of those obtained with the capsule. Both the oral solution and intravenous
formulation are solubilized in a 40:1 weight ratio of hydroxypropyl- Drug Interactions Table 492 lists the known interactions of itraconazole with other drugs, but this list is still expanding. Many of the interactions can cause serious toxicity of the companion drug, as potentially fatal cardiac arrhythmias with cisapride, quinidine, or astemizole. Other interactions may decrease the itraconazole concentrations below therapeutic levels. Therapeutic Uses Itraconazole given as a capsule is the drug of choice for patients with indolent, nonmeningeal infections due to B. dermatitidis, H. capsulatum, P. brasiliensis, and C. immitis. This dosage form also is useful in therapy of indolent invasive aspergillosis outside the central nervous system, particularly after the infection has been stabilized with amphotericin B. The intravenous formulation is approved for the initial two weeks of therapy with blastomycosis, histoplasmosis, and indolent aspergillosis. The intravenous route would be most appropriate for patients unable to tolerate the oral formulation or unable to absorb it because of decreased gastric acid. Approximately half the patients with distal subungual onychomycosis respond well to itraconazole (Evans and Sigurgeirsson, 1999). Although not an approved use, itraconazole is often the best choice for treatment of pseudallescheriasis, an infection not responding to amphotericin B therapy, as well as cutaneous and extracutaneous sporotrichosis, tinea corporis, and extensive tinea versicolor. HIV-infected patients with disseminated histoplasmosis or P. marneffei infections have a decreased incidence of relapse if given prolonged itraconazole 'maintenance' therapy (Wheat et al., 1993; Supparatpinyo et al., 1998). It is as yet unclear whether patients responding to highly active antiretroviral therapy (HAART) will require less than lifelong therapy (seeChapter 51: Antiretroviral Agents: Antiretroviral Agents). Itraconazole is not recommended for maintenance therapy of cryptococcal meningitis in HIV-infected patients because of a high incidence of relapse. However, itraconazole prophylaxis in patients with advanced HIV infection yielded a decreased incidence of cryptococcosis as well as histoplasmosis. The incidence of mucosal candidiasis was not reduced (McKinsey et al., 1999). This study was done in the pre-HAART era, and itraconazole prophylaxis is not recommended. Long-term therapy has been used in non-HIV-infected patients with allergic bronchopulmonary aspergillosis to decrease the dose of corticosteroids and reduce attacks of acute bronchospasm (Salez et al., 1999). Itraconazole solution is effective and approved for use in oropharyngeal and esophageal candidiasis. Because the preparation has more gastrointestinal side effects than do fluconazole tablets, itraconazole solution usually is reserved for patients not responding to fluconazole (Saag et al., 1999). Unfortunately, these patients are often receiving protease inhibitors and other drugs that make itraconazole contraindicated. Itraconazole capsules and oral solution are not bioequivalent and should not be used interchangeably. Dosage In treating deep mycoses, two 100-mg capsules are given twice daily with food. Divided doses are said to increase the area under the curve (AUC) (seeChapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination) compared to once-daily dosing, even though the half-life is about 30 hours. For the first three days, 200 mg three times daily is used as a loading dose. For maintenance therapy of HIV-infected patients with disseminated histoplasmosis, 200 mg once daily is used. Onychomycosis can be treated with either 200 mg once daily for 12 weeks or as 200 mg twice daily for one week out of each month, so-called pulse therapy (Evans and Sigurgeirsson, 1999). Retention of active drug in the nail keratin permits intermittent treatment. Daily therapy is preferred by some authorities for infections likely to be more refractory but is twice the cost of pulse therapy. Once-daily terbinafine (250 mg) is slightly superior to pulse therapy with itraconazole (see below). Treatment is usually continued for three months. Intravenous itraconazole is reserved for seriously ill patients; it is given as an infusion over one hour of 200 mg twice daily for two days followed by 200 mg once daily for 12 days. Safety and efficacy of the intravenous regimen beyond 14 days is currently unknown. Itraconazole oral solution should be taken fasting in a dose of 100 mg in 10 ml once daily and swished vigorously in the mouth before swallowing to optimize any topical effect. Patients with fluconazole-resistant oropharyngeal or esophageal thrush are given 100 mg twice a day for 2 to 4 weeks. Untoward Effects Adverse effects of itraconazole therapy can occur as a result of interactions with many other drugs (see below). Itraconazole capsules, in the absence of interacting drugs, are well tolerated at 200 mg daily. Gastrointestinal distress occasionally prevents use of 400 mg per day. In a series of 189 patients receiving 50 to 400 mg per day, nausea and vomiting were recorded in 10%, hypertriglyceridemia in 9%, hypokalemia in 6%, increased serum aminotransferase in 5%, rash in 2%, and at least one side effect in 39%. Occasionally, hepatotoxicity or rash leads to drug discontinuation, but most adverse effects can be handled with dose reduction. Profound hypokalemia has been seen in patients receiving 600 mg or more daily (Sharkey et al., 1991) and in those who recently have received prolonged amphotericin B therapy. Doses of 300 mg twice daily have led to other side effects, including adrenal insufficiency, lower limb edema, hypertension, and, in one patient, rhabdomyolysis (Sharkey et al., 1991). Doses above 400 mg per day are not recommended for long-term use. Intravenous itraconazole has been well tolerated except for chemical
phlebitis. A dedicated catheter port is required. If other medications are
administered through the same port at a later time, the catheter should be
thoroughly flushed with normal saline. Although the volume of infusion is
small, 25 ml of itraconazole in excipient plus 50 ml of saline, infusion
durations less than one hour are not recommended. Toxicity of high plasma
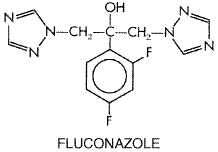
levels of hydroxypropyl- The oral solution of itraconazole is well tolerated but has all the adverse effects of itraconazole capsules. Anaphylaxis has been observed rarely, as well as severe rash, including StevensJohnson syndrome. Some patients complain of the taste, and gastrointestinal side effects are common, though compliance is generally unimpaired. Diarrhea, abdominal cramps, anorexia, and nausea are more common than with the capsules. Fluconazole Fluconazole is a fluorinated bistriazole. Its structure is as follows:
Absorption, Distribution, and Excretion Fluconazole is almost completely absorbed from the gastrointestinal
tract. Concentrations in plasma are essentially the same whether the drug is
given orally or intravenously, and bioavailability is not altered by food or
gastric acidity. Peak plasma concentrations are 4 to 8 Interactions Fluconazole significantly increases plasma concentrations of astemizole, cisapride, cyclosporine, rifampin, rifabutin, sulfonylureas (glipizide, tolbutamide, others), theophylline, tacrolimus, and warfarin. Patients who receive more than 400 mg daily or azotemic patients who have elevated fluconazole blood levels may experience drug interactions not otherwise seen. Rifampin decreases the fluconazole AUC by about 25%, an amount that ordinarily would not be significant. Drugs that decrease gastric acidity do not significantly lower fluconazole blood levels. Therapeutic Uses Candidiasis Fluconazole, 200 mg on the first day and then 100 mg daily for at least 2 weeks, is effective in oropharyngeal candidiasis. Esophageal candidiasis responds to 100 to 200 mg daily. This dose also has been used to decrease candiduria in high-risk patients (Sobel et al., 2000). A single dose of 150 mg is effective in vaginal candidiasis. A dose of 400 mg daily decreases the incidence of deep candidiasis in allogeneic bone marrow transplant recipients and is useful in treating candidemia of nonimmunosuppressed patients. Fluconazole is not known to be effective treatment for deep candidiasis in profoundly neutropenic patients. Based on resistance in vitro, Candida krusei would not be expected to respond to fluconazole or other azoles. Cryptococcosis Fluconazole, 400 mg daily, is used for the initial 8 weeks in cryptococcal meningitis of AIDS after the patient's clinical condition has been stabilized with intravenous amphotericin B. After the 8 weeks, the dose is decreased to 200 mg daily for life. It is still unclear whether or not patients with a sustained response to HAART can stop lifelong maintenance therapy. For AIDS patients with cryptococcal meningitis who are alert and oriented and have other favorable prognostic signs, initial therapy with 400 mg daily may be considered. The role of fluconazole for non-AIDS patients with cryptococcosis is not defined. Other Mycoses Fluconazole has become the drug of choice for treatment of coccidioidal meningitis because of much less morbidity than with intrathecal amphotericin B. In other forms of coccidioidomycosis, fluconazole appears roughly comparable to itraconazole. Fluconazole has activity against histoplasmosis, blastomycosis, sporotrichosis, and ringworm, but response is less than with equivalent doses of itraconazole. Fluconazole is not effective in the prevention or treatment of aspergillosis. As with other azoles, there is no activity in mucormycosis. Fluconazole ( DIFLUCAN ) is marketed in the Untoward Effects Nausea and vomiting may be seen at doses above 200 mg daily. Patients receiving 800 mg daily may require antiemetics and may need to be treated intravenously to prevent emesis, which reduces drug availability. Data provided in the package insert states that side effects in patients receiving more than 7 days of drug, irrespective of dose, include the following: nausea 3.7%, headache 1.9%, skin rash 1.8%, vomiting 1.7%, abdominal pain 1.7%, and diarrhea 1.5%. Reversible alopecia may occur with prolonged therapy at 400 mg daily. Rare cases of deaths due to hepatic failure (Jacobson et al., 1994) or StevensJohnson syndrome have been reported. Although the relationship between these deaths and fluconazole treatment is not completely clear in these cases, it is prudent to be alert to early symptoms of these disorders and to stop fluconazole if they occur. Fluconazole is teratogenic in rodents and has been associated with skeletal and cardiac deformities in three infants born to two women taking high doses during pregnancy (Pursley et al., 1996). The drug should be avoided during pregnancy. Pneumocandins Caspofungin acetate (CANCIDASE, MK-0991) is a water-soluble, semisynthetic lipopeptide derivative of
pneumocandin Bo. Drugs of this class, also referred to as
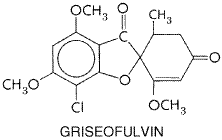
echinocandins, inhibit the formation of Griseofulvin Chemistry The structural formula of griseofulvin is as follows:
The drug is practically insoluble in water. Antifungal Activity Griseofulvin is fungistatic in vitro for various species of the dermatophytes Microsporum, Epidermophyton, and Trichophyton. The drug has no effect on bacteria or on other fungi. Resistance Although failure of ringworm lesions to improve is not rare, isolates from these patients usually are still susceptible to griseofulvinin vitro. Mechanism of Action A prominent morphological manifestation of the action of griseofulvin is the production of multinucleate cells as the drug inhibits fungal mitosis. An explanation for this phenomenon appears to come from studies of the effects on mammalian cells of higher concentrations of the antibiotic. Griseofulvin causes disruption of the mitotic spindle by interacting with polymerized microtubles. Although the effects of the drug are thus similar to those of colchicine and the vinca alkaloids, its binding sites on the microtubular protein are distinct. There is evidence that griseofulvin binds to a microtubule-associated protein in addition to its binding to tubulin. Absorption, Distribution, and Excretion The oral administration of a 0.5-g dose of griseofulvin produces peak
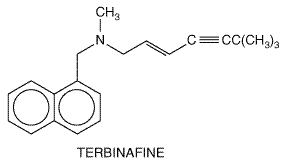
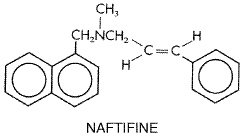
plasma concentrations of approximately 1 The drug is deposited in keratin precursor cells. The antibiotic present in such cells when they differentiate is tightly bound to, and persists in, keratin and makes this substance resistant to fungal invasion. For this reason, the new growth of hair or nails is the first to become free of disease. As the fungus-containing keratin is shed, it is replaced by normal tissue. Griseofulvin is detectable in the stratum corneum of the skin within 4 to 8 hours of oral administration. Sweat and transepidermal fluid loss play an important role in the transfer of the drug in the stratum corneum. Only a very small fraction of a dose of the drug is present in body fluids and tissues. Therapeutic Uses Mycotic disease of the skin, hair, and nails due to Microsporum, Trichophyton, or Epidermophyton responds to griseofulvin therapy. Infections that are readily treatable with this agent include infections of the hair (tinea capitis) caused by Microsporum canis, Microsporum audouini, Trichophyton schoenleinii, and Trichophyton verrucosum;'ringworm' of the glabrous skin; tinea cruris and tinea corporis caused by M. canis, Trichophyton rubrum, T. verrucosum, and Epidermophyton floccosum; and tinea of the hands (T. rubrum, Trichophyton mentagrophytes) and beard (Trichophyton species). Griseofulvin also is highly effective in 'athlete's foot' or epidermophytosis involving the skin and nails, the vesicular form of which is most commonly due to T. mentagrophytes and the hyperkeratotic type to T. rubrum. However, topical therapy is preferred (see below). T. rubrum and T. mentagrophytes infections may require higher-than-conventional doses. Since very high doses of griseofulvin are carcinogenic and teratogenic in laboratory animals, the drug should not be used to treat trivial infections that respond to topical therapy. The recommended daily dose of griseofulvin is 5 to 15 mg/kg for children and 500 mg to 1 g for adults. Doses of 1.5 to 2.0 g daily may be used for short periods in severe or extensive infections. Best results are obtained when the daily dose is divided and given at 6-hour intervals, although the drug often is given twice per day. Treatment must be continued until infected tissue is replaced by normal hair, skin, or nails, which requires 1 month for scalp and hair ringworm, 6 to 9 months for fingernails, and at least a year for toenails. Itraconazole or terbinafine is preferred for onychomycosis. Griseofulvin is not effective in treatment of subcutaneous or deep mycoses. Untoward Effects The incidence of serious reactions associated with the use of griseofulvin is very low. One of the minor effects is headache; it is sometimes severe and usually disappears as therapy is continued. The incidence of headache may be as high as 15%. Other nervous system manifestations include peripheral neuritis, lethargy, mental confusion, impairment of performance of routine tasks, fatigue, syncope, vertigo, blurred vision, transient macular edema, and augmentation of the effects of alcohol. Among the side effects involving the alimentary tract are nausea, vomiting, diarrhea, heartburn, flatulence, dry mouth, and angular stomatitis. Hepatotoxicity also has been observed. Hematologic effects include leukopenia, neutropenia, punctate basophilia, and monocytosis; these often disappear despite continuation of therapy. Blood studies should be carried out at least once a week during the first month of treatment or longer. Common renal effects include albuminuria and cylindruria without evidence of renal insufficiency. Reactions involving the skin are cold and warm urticaria, photosensitivity, lichen planus, erythema, erythema multiforme-like rashes, and vesicular and morbilliform eruptions. Serum sickness syndromes and severe angioedema develop rarely during treatment with griseofulvin. Estrogen-like effects have been observed in children. A moderate but inconsistent increase of fecal protoporphyrins has been noted when the drug is used for a long time. Griseofulvin induces hepatic microsomal enzymes, thus increasing the rate of metabolism of warfarin; adjustment of the dosage of the latter agent may be necessary in some patients. The drug may reduce the efficacy of some oral contraceptive agents, probably by a similar mechanism. Terbinafine Terbinafine is a synthetic allylamine, structurally similar to the topical agent naftifine. Its structural formula is shown below:
Terbinafine is well absorbed, but bioavailability is decreased to about 40% because of first-pass metabolism in the liver. Proteins bind more than 99% of the drug in plasma. Drug accumulates in skin, nails, and fat. The initial half-life is about 12 hours but extends to 200 to 400 hours at steady state. Drug can be found in plasma for 4 to 8 weeks after prolonged therapy (Balfour and Faulds, 1992). Terbinafine is not recommended in patients with marked azotemia or hepatic failure because terbinafine plasma levels are increased by unpredictable amounts. Rifampin decreases and cimetidine increases plasma terbinafine concentrations. The drug is well tolerated, with a low incidence of gastrointestinal distress, headache, or rash. Rarely, hepatotoxicity, severe neutropenia, StevensJohnson syndrome, or toxic epidermal necrolysis may occur. The drug is in pregnancy category B. Its mechanism of action is probably inhibition of fungal squalene epoxidase, blocking ergosterol biosynthesis. Terbinafine ( LAMISIL ), given as one 250-mg tablet daily, is at least as effective for onychomycosis of nails as 200 mg daily of itraconazole, and slightly more effective than pulse itraconazole therapy (see above) (Evans, 1999). Duration of treatment varies with the nail being treated but typically is 3 months. Although not approved for this use, terbinafine (250 mg daily) also is effective in ringworm elsewhere on the body. No pediatric formulation is available, so there is little experience with the drug in tinea capitis, usually a disease of children. The topical use of terbinafine is discussed in the following section. |
Topical Antifungal Agents
|
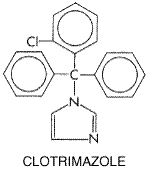
Topical treatment is useful in many superficial fungal infectionsthat is, those confined to the stratum corneum, squamous mucosa, or cornea. Such diseases include dermatophytosis (ringworm), candidiasis, tinea versicolor, piedra, tinea nigra, and fungal keratitis. Topical administration of antifungal agents is usually not successful for mycoses of the nails (onychomycosis) and hair (tinea capitis) and has no place in the treatment of subcutaneous mycoses, such as sporotrichosis and chromomycosis. The efficacy of topical agents in the superficial mycoses depends not only on the type of lesion and the mechanism of action of the drug but also on the viscosity, hydrophobicity, and acidity of the formulation. Regardless of formulation, penetration of topical drugs into hyperkeratotic lesions often is poor. Removal of thick, infected keratin is sometimes a useful adjunct to therapy; this is, for example, the principal mode of action of Whitfield's ointment (see below). A plethora of topical agents are available for the treatment of superficial mycoses. Many of the older drugsincluding gentian violet, carbol-fuchsin, acrisorcin, triacetin, sulfur, iodine, and aminacrineare now rarely indicated and are not discussed here (see previous editions of this textbook). Among the topical agents to be discussed, the preferred formulation for cutaneous application usually is a cream or solution. Ointments are messy and are too occlusive for macerated or fissured intertriginous lesions. The use of powders, whether applied by shake containers or aerosols, is largely confined to the feet and moist lesions of the groin and other intertriginous areas. The systemic agents that are used for the treatment of superficial mycoses are discussed in the first section of this chapter. Some of these agents are also administered topically; their uses are described here and also in Chapter 65: Dermatological Pharmacology. Imidazoles and Triazoles for Topical Use As discussed above, these closely related classes of drugs are synthetic antifungal agents that are used both topically and systemically. Indications for their topical use include ringworm, tinea versicolor, and mucocutaneous candidiasis. Resistance to imidazoles or triazoles is very rare among the fungi that cause ringworm. Selection of one of these agents for topical use should be based on cost and availability, since testing in vitro for fungal susceptibility to these drugs is not predictive of clinical responses. Cutaneous Application The preparations for cutaneous use described below are effective for tinea corporis, tinea pedis, tinea cruris, tinea versicolor, and cutaneous candidiasis. They should be applied twice a day for 3 to 6 weeks. Despite some activity in vitro against bacteria, this effect is not clinically useful. The cutaneous formulations are not suitable for oral, vaginal, or ocular use. Vaginal Application The vaginal creams, suppositories, and tablets are the preparations of choice for vaginal candidiasis. None is useful in trichomoniasis, despite some activity in vitro. They are all used once a day, preferably at bedtime to facilitate retention. Most vaginal creams are administered in 5-g amounts. Three vaginal formulationsclotrimazole tablets, miconazole suppositories, and terconazole creamcome in both low- and high-dose preparations. A shorter duration of therapy is recommended for the higher dose of each. Except for the 500-mg clotrimazole tablet and tioconazole ointment, which are given only once, these preparations are administered for 3 to 7 days. Approximately 3% to 10% of the vaginal dose is absorbed. Although some imidazoles are embryotoxic in rodents, no adverse effects on the human fetus have been attributed to the vaginal use of imidazoles or triazoles. The most common side effect is vaginal burning or itching. A male sexual partner may experience mild penile irritation. Cross-allergenicity among these compounds is assumed to exist, based on their structural similarities. Oral Use Use of the oral troche of clotrimazole is properly considered as topical therapy. The only indication for this 10-mg troche is oropharyngeal candidiasis. Antifungal activity is due entirely to the local concentration of the drug; there is no systemic effect. The patient should be told to suck on the troche until it dissolves. Clotrimazole Clotrimazole has the following structure:
Absorption of clotrimazole is less than 0.5% after application to the
intact skin; from the vagina, it is 3% to 10%. Fungicidal concentrations
remain in the vagina for as long as 3 days after application of the drug. The
small amount absorbed is metabolized in the liver and excreted in bile. In
adults, an oral dose of 200 mg per day will give rise initially to plasma
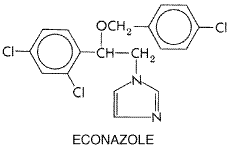
concentrations of 0.2 to 0.35 In a small fraction of recipients, clotrimazole on the skin may cause stinging, erythema, edema, vesication, desquamation, pruritus, and urticaria. When it is applied to the vagina, about 1.6% of recipients complain of a mild burning sensation and, rarely, of lower abdominal cramps, slight increase in urinary frequency, or skin rash. Occasionally, the sexual partner may experience penile or urethral irritation. By the oral route, clotrimazole can cause gastrointestinal irritation. In patients using troches, the incidence of this side effect is about 5%. Therapeutic Uses Clotrimazole is available as a 1% cream, lotion, and solution ( LOTRIMIN MYCELEX), 1% or 2% vaginal cream or vaginal tablets of 100, 200, or 500 mg (GYNELOTRIMIN MYCELEX G), and 10-mg troches (MYCELEX). On the skin, applications are made twice a day. For the vagina, the standard regimens are one 100-mg tablet once a day at bedtime for 7 days, one 200-mg tablet daily for 3 days, one 500-mg tablet inserted only once, or 5 g of cream once a day for 3 days (2% cream) or 7 days (1% cream). For nonpregnant females, two 100-mg tablets may be used once a day for 3 days. Troches are to be dissolved slowly in the mouth five times a day for 14 days. Clotrimazole has been reported to cure dermatophyte infections in 60% to 100% of cases. The cure rates in cutaneous candidiasis are 80% to 100%. In vulvovaginal candidiasis, the cure rate is usually above 80% when the 7-day regimen is used. A 3-day regimen of 200 mg once a day appears to be similarly effective, as does single-dose treatment (500 mg). Recurrences are common after all regimens. The cure rate with oral troches for oral and pharyngeal candidiasis may be as high as 100% in the immunocompetent host. Econazole Econazole, the deschloro derivative of miconazole, has the following structure:
Econazole readily penetrates the stratum corneum and is found in effective concentrations down to the mid-dermis. However, less than 1% of an applied dose appears to be absorbed into the blood. Approximately 3% of recipients have local erythema, burning, stinging, or itching. Econazole nitrate SPECTAZOLE ) is available as a water-miscible cream (1%) to be applied twice a day. Miconazole Miconazole is a very close chemical congener of econazole, with the following structure:
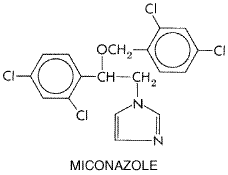
Miconazole readily penetrates the stratum corneum of the skin and persists there for more than 4 days after application. Less than 1% is absorbed into the blood. Absorption is no more than 1.3% from the vagina. Adverse effects from topical application to the vagina include burning, itching, or irritation in about 7% of recipients and, infrequently, pelvic cramps (0.2%), headache, hives, or skin rash. Irritation, burning, and maceration are rare after cutaneous application. Miconazole is considered safe for use during pregnancy, although some authors believe that its vaginal use should be avoided during the first trimester. Therapeutic Uses Miconazole nitrate is available as a dermatologic ointment, cream, solution, spray, powder, or lotion (MICATIN MONISTAT DERM, others). To avoid maceration, only the lotion should be applied to intertriginous areas. It is available as a 2% and 4% vaginal cream, as 100-mg suppositories (MONISTAT 7), to be applied high in the vagina at bedtime for 7 days, and as 200-mg vaginal suppositories (MONISTAT 3) for 3-day therapy. In the treatment of tinea pedis, tinea cruris, and tinea versicolor, the cure rate may be over 90%. In the treatment of vulvovaginal candidiasis, the mycologic cure rate at the end of 1 month is about 80% to 95%. Pruritus sometimes is relieved after a single application. Some vaginal infections caused by Candida glabrata also respond. Terconazole and Butoconazole Terconazole TERAZOL) is a ketal triazole with structural similarities to ketoconazole. Its structure is as follows:
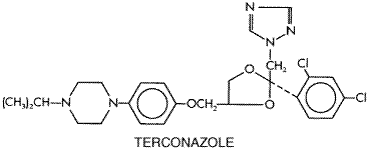
The mechanism of action of terconazole is similar to that of the imidazoles. The 80-mg vaginal suppository is inserted at bedtime for 3 days, while the 0.4% vaginal cream is used for 7 days and the 0.8% cream for 3 days. Clinical efficacy and patient acceptance of both preparations are at least as good as for clotrimazole in patients with vaginal candidiasis. Butoconazole is an imidazole quite comparable to clotrimazole. Its structural formula is as follows:
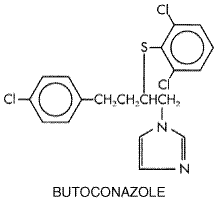
Butoconazole nitrate FEMSTAT 3) is available as a 2% vaginal cream; it is used at bedtime for 3 days in nonpregnant females. Because of the slower response during pregnancy, a 6-day course is recommended (during the second and third trimester). Tioconazole Tioconazole VAGISTAT 1) is an imidazole that is marketed for treatment of Candida vulvovaginitis. A single 4.6-g dose of ointment containing 6.5% drug is given at bedtime. Oxiconazole and Sulconazole Oxiconazole and sulconazole are two imidazole derivatives that are used for the topical treatment of infections caused by the common pathogenic dermatophytes. Oxiconazole nitrate ( OXISTAT ) is available as a cream and lotion; sulconazole nitrate ( EXELDERM ) is supplied as a solution and cream. Ciclopirox Olamine Ciclopirox olamine LOPROX ) has broad-spectrum antifungal activity. The chemical structure is as follows:
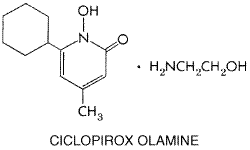
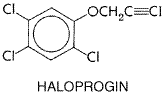
It is fungicidal to C. albicans, E. floccosum, M. canis, T. mentagrophytes, and T. rubrum. It also inhibits the growth of Malassezia furfur. After application to the skin, it penetrates through the epidermis into the dermis, but even under occlusion, less than 1.5% is absorbed into the systemic circulation. Since the half-life is 1.7 hours, no systemic accumulation occurs. The drug penetrates into hair follicles and sebaceous glands. It can sometimes cause hypersensitivity. It is available as a 1% cream and lotion for the treatment of cutaneous candidiasis and for tinea corporis, cruris, pedis, and versicolor. Cure rates in the dermatomycoses and candidal infections have been variously reported to be 81% to 94%. No topical toxicity has been noted. Haloprogin Haloprogin is a halogenated phenolic ether with the following structure:
It is fungicidal to various species of Epidermophyton, Pityrosporum, Microsporum, Trichophyton, and Candida. During treatment with this drug, irritation, pruritus, burning sensations, vesiculation, increased maceration, and 'sensitization' (or exacerbation of the lesion) occasionally occur, especially on the foot if occlusive footgear is worn. Haloprogin is poorly absorbed through the skin; it is converted to trichlorophenol in the body. The systemic toxicity from topical application appears to be low. Haloprogin HALOTEX) cream or solution is applied twice a day for 2 to 4 weeks. Its principal use is against tinea pedis, for which the cure rate is about 80%; it is thus approximately equal in efficacy to tolnaftate (see below). It also is used against tinea cruris, tinea corporis, tinea manuum, and tinea versicolor. Tolnaftate Tolnaftate is a thiocarbamate with the following structure:
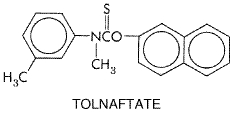
Tolnaftate is effective in the treatment of most cutaneous mycoses caused by T. rubrum, T. mentagrophytes, T. tonsurans, E. floccosum, M. canis, M. audouinii, Microsporum gypseum, and M. furfur, but it is ineffective against Candida. In tinea pedis, the cure rate is around 80%, compared with about 95% for miconazole. Toxic or allergic reactions to tolnaftate have not been reported. Tolnaftate AFTATE TINACTIN, others) is available in a 1% concentration as a cream, gel, powder, aerosol powder, and topical solution, or as a topical aerosol liquid. The preparations are applied locally twice a day. Pruritus is usually relieved in 24 to 72 hours. Involution of interdigital lesions caused by susceptible fungi is very often complete in 7 to 21 days. Naftifine Naftifine is an allylamine with the following structure:
Naftifine is representative of the allylamine class of synthetic agents that inhibit squalene-2,3-epoxidase and thus inhibit fungal biosynthesis of ergosterol. The drug has broad-spectrum fungicidal activity in vitro. Naftifine hydrochloride (NAFTIN) is available as a 1% cream or gel. It is effective for the topical treatment of tinea cruris and tinea corporis; twice-daily application is recommended. The drug is well tolerated, although local irritation has been observed in 3% of treated patients. Allergic contact dermatitis also has been reported. Naftifine also may be efficacious for cutaneous candidiasis and tinea versicolor, although the drug has not been approved for these uses. Terbinafine Terbinafine cream is applied twice daily and is effective in tinea corporis, tinea cruris, and tinea pedis. Terbinafine is less active against Candida species and Malassezia furfur, but the cream also can be used in cutaneous candidiasis and tinea versicolor. In European studies, oral terbinafine has appeared to be effective in treatment of ringworm and in some cases of onychomycosis. The systemic use of terbinafine is discussed above. Butenafine Butenafine hydrochloride MENTAX ) is a benzylamine derivative with a mechanism of action similar to that of terbinafine and naftifine. Its spectrum of antifungal activity and use also are similar to those of the allylamines. Polyene Antifungal Antibiotics Nystatin Nystatin was discovered in the New York State Health Laboratory and was named accordingly; it is a tetraene macrolide produced by Streptomyces noursei. Nystatin is structurally similar to amphotericin B and has the same mechanism of action. Nystatin is not absorbed from the gastrointestinal tract, skin, or vagina. A liposomal formulation (NYOTRAN) is in clinical trials for candidemia. Nystatin MYCOSTATIN NILSTAT, others) is useful only for candidiasis and is supplied in preparations intended for cutaneous, vaginal, or oral administration for this purpose. Infections of the nails and hyperkeratinized or crusted skin lesions do not respond. Topical preparations include ointments, creams, and powders, all of which contain 100,000 U/g. Powders are preferred for moist lesions and are applied two or three times a day. Creams or ointments are used twice daily. Combinations of nystatin with antibacterial agents or corticosteroids also are available. Allergic reactions to nystatin are very uncommon. Vaginal tablets containing 100,000 U of the drug are inserted once daily for 2 weeks. Although the tablets are well tolerated, imidazoles or triazoles are more effective agents for vaginal candidiasis. An oral suspension that contains 100,000 U of nystain per milliliter is given four times a day. Premature and low-birth-weight neonates should receive 1 ml of this preparation, infants 2 ml, and children or adults 4 to 6 ml per dose. Older children and adults should be instructed to swish the drug around the mouth and then swallow. If not otherwise instructed, the patient may expectorate the bitter liquid and fail to treat the infected mucosa in the posterior pharynx or esophagus. Nystatin suspension is usually effective for oral candidiasis of the immunocompetent host. Other than the bitter taste and occasional complaints of nausea, adverse effects are uncommon. Oral tablets containing 500,000 U have been used to decrease gastrointestinal colonization with Candida in the hope of preventing relapse of vaginal candidiasis or of protecting the neutropenic patient from gastrointestinal candidiasis. Careful studies have failed to document efficacy for these indications. Amphotericin B Topical amphotericin B (FUNGIZONE) also is used for cutaneous and mucocutaneous candidiasis. A lotion, cream, and ointment are marketed; these preparations all contain 3% amphotericin B and are applied to the lesion two to four times daily. The systemic use of amphotericin B is discussed above. Miscellaneous Antifungal Agents Undecylenic Acid Undecylenic acid is 10-undecenoic acid, an 11-carbon unsaturated compound. It is a yellow liquid with a characteristic rancid odor. It is primarily fungistatic, although fungicidal activity may be observed with long exposure to high concentrations of the agent. The drug is active against a variety of fungi, including those that cause ringworm. Undecylenic acid (DESENEX) is available in a foam, ointment, cream, powder, spray powder, soap, and liquid. Zinc undecylenate is marketed in combination with other ingredients. The zinc provides an astringent action that aids in the suppression of inflammation. Compound undecylenic acid ointment contains both undecylenic acid (about 5%) and zinc undecylenate (about 20%). Calcium undecylenate (CALDESENE CRUEX) is available as a powder. Undecylenic acid preparations are used in the treatment of various dermatomycoses, especially tinea pedis. Concentrations of the acid as high as 10%, as well as those of the acid and salt in the compound ointment, may be applied to the skin. The preparations as formulated are usually not irritating to tissue, and sensitization to them is uncommon. It is of undoubted benefit in retarding fungal growth in tinea pedis, but the infection frequently persists despite intensive treatment with preparations of the acid and the zinc salt. At best, the clinical 'cure' rate is about 50% and is thus much lower than that obtained with the imidazoles, haloprogin, or tolnaftate. The efficacy in the treatment of tinea capitis is marginal, and the drug is no longer used for that purpose. Undecylenic acid preparations also are approved for use in the treatment of diaper rash, tinea cruris, and other minor dermatologic conditions. Benzoic Acid and Salicylic Acid An ointment containing benzoic and salicylic acids is known as Whitfield's ointment. It combines the fungistatic action of benzoate with the keratolytic action of salicylate. It contains benzoic acid and salicylic acid in a ratio of 2 to 1 (usually 6% to 3%). It is used mainly in the treatment of tinea pedis. Since benzoic acid is only fungistatic, eradication of the infection occurs only after the infected stratum corneum is shed, and continuous medication is required for several weeks to months. The salicylic acid accelerates the desquamation. The ointment also is sometimes used to treat tinea capitis. Mild irritation may occur at the site of application. Propionic Acid and Caprylic Acid Propionic acid and sodium propionate are promoted for the treatment of the dermatomycoses. Both their low efficacy and exaggerated price make them irrational choices for treatment. They may be compounded together or with sodium caprylate or other agents. Sodium propionate is used in proprietary preparations in concentrations of 1% to 5%. Potassium Iodide A saturated solution of potassium iodide, containing 1 g/ml, is useful in treating cutaneous sporotrichosis. The drug has a bitter taste and causes nausea, bitter eructation, and excessive salivation. Patient acceptance is improved if the initial dosage is low, such as 10 drops in a small amount of water, taken three times daily. Drinking water or juice immediately following the dose lessens the bitter aftertaste. Dosage is increased gradually to 25 to 40 drops three times daily in children and to 40 to 50 drops three times a day in adults. Therapy is continued for at least 6 weeks and until the cutaneous lesions have flattened and any ulcerations have healed. Gradual enlargement of the salivary and lacrimal glands is usual, and adults may develop an acneiform rash over the cape area of the chest. These side effects go away after therapy is discontinued and are not a cause for drug discontinuation. Patients with true allergic rashes should be changed to itraconazole therapy. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3048
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved