| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Drugs Used in the Chemotherapy of Helminthiasis
Overview
|
Helminthiasis, or infection with parasitic worms, affects over two billion people worldwide. In tropical regions, where prevalence is greatest, simultaneous infection with more than one type of helminth is common. Moreover, human beings can spread these pathogens to previously uninvolved populations through travel, migration, and military operations. Worms pathogenic for human beings are Metazoa, classified into roundworms (nematodes) and two types of flatworms, flukes (trematodes) and tapeworms (cestodes). These biologically diverse eukaryotes vary with respect to life cycle, bodily structure, development, physiology, localization within the host, and susceptibility to chemotherapy. Immature forms invade human beings via the skin or gastrointestinal tract and evolve into well-differentiated adult worms that have characteristic tissue distributions. With few exceptions, such as Strongyloides and Echinococcus, these organisms cannot complete their life cycles, i.e., replicate themselves, within the human host. Therefore, the extent of exposure to these parasites dictates the severity of infection, and reduction in the number of adult organisms by chemotherapy is sustained unless reinfection occurs. The prevalence of parasitic helminths typically displays a negative binomial distribution within an infected population such that relatively few persons carry heavy parasite burdens. Without treatment, those individuals are most likely to become ill and to perpetuate infection within their community. Anthelmintics are drugs that act either locally to expel worms from the gastrointestinal tract or systemically to eradicate adult helminths or developmental forms that invade organs and tissues. Due to discovery and development of anthelmintics, particularly for veterinary applications, physicians now have effective and, in some cases, broad-spectrum agents that will cure or control most human infections caused by either flukes or intestinal helminths. But cysticercosis, echinococcosis, filariasis, and trichinosis are examples of systemic infections caused by tissue-dwelling helminths that at best respond only partially to currently available drugs. Because metazoan parasites are generally long-lived and have relatively complex life cycles, acquired resistance to anthelmintics in human beings has yet to become a major factor limiting clinical efficacy. However, based on extensive use of anthelmintics, such as the benzimidazoles in veterinary medicine, the potential for drug resistance among human helminths cannot be discounted. This chapter begins with brief descriptions of recommended chemotherapy for common human infections caused by parasitic helminths. The following section features properties of selected antihelmintic drugs in alphabetical order, and the chapter concludes with a prospectus about the chemotherapy of helminthiasis. |
Treatment of Helminth Infections
|
Nematodes (Roundworms) Ascaris lumbricoides Ascaris lumbricoides, known as the 'roundworm,' parasitizes more than 1.4
billion people worldwide. Although ascariasis is not uncommon in temperate
climates, it may affect from 70% to 90% of persons in some tropical regions.
In the More effective, less toxic compounds largely have replaced the older
ascaricides. Mebendazole, pyrantel pamoate, and albendazole are
preferred agents. Piperazine also is effective but is used less often
because of occasional neurotoxicity and hypersensitivity reactions. Cure with
any of these drugs can be achieved in nearly 100% of cases, and all infected
persons should be treated. Mebendazole and albendazole are preferred for
therapy of asymptomatic to moderate ascariasis. Both of these benzimidazoles
are cidal and have a broad spectrum of activity against mixed infections with
other gastrointestinal nematodes. Albendazole is useful against infections
with some systemic nematodes and some cestodes as well (see Table
VII1). Both compounds should be used with caution to treat heavy Ascaris
infections, alone or with hookworms. In rare instances, hyperactive ascarids
may migrate to unusual loci and cause serious complications such as
appendicitis, occlusion of the common bile duct, intestinal obstruction, and
intestinal perforation with peritonitis. Therapy with noncidal agents such as
pyrantel or piperazine is preferred for heavy Ascaris infections;
pyrantel also is useful for mixed infections with hookworms. Surgery still
may be required despite the use of these agents. Pyrantel and piperazine are
considered safe for use during pregnancy, whereas the bendimidazoles should
be avoided during the first trimester because of their teratogenic potential.
Pyrantel and albendazole are considered 'investigational' drugs for
treatment of ascariasis in the Hookworm Necator americanus, Ancylostoma duodenale. These closely related hookworm species
infest about 1.3 billion people, chiefly between the latitudes 30S and 40N.
N. americanus predominates in the Treatment of hookworm disease involves two related objectives. The first is to restore the blood values to normal, and the second is to expel the intestinal parasites. Proper diet and treatment with iron usually are sufficient for the first objective, but blood transfusion is required occasionally. Albendazole and mebendazole are now agents of first choice against both A. duodenale and N. americanus, and both benzimidazoles have the advantage of effectiveness against other roundworms when there is multiple infection. However, use of these cidal drugs in heavy mixed infections with Ascaris may risk unwanted ascarid activation and migration (see'Ascaris lumbricoides,' above). Therapy with pyrantel, which just paralyzes ascarids, may be safe under these circumstances. Topical or oral thiabendazole is the drug of choice for treating cutaneous larva migrans or 'creeping eruption,' which is due most commonly to penetration of the skin of human beings by larvae of the dog hookworm, A. braziliense. Trichuris trichiura Trichuris (whipworm) infection affects nearly one billion people throughout the world, especially children in warm, humid climates, where it frequently is found along with Ascaris and hookworms. The infection is acquired by eating food contaminated with parasite eggs. The adult worms usually do not cause problems except in heavily infected young children, who may exhibit abdominal cramping, diarrhea, and some degree of anemia. Rarely, worms lodge in the appendix or penetrate the bowel wall to cause peritonitis. Mebendazole and albendazole are considered the safest agents and the most effective for treatment of whipworm, alone or together with Ascaris and hookworm infections. Pyrantel pamoate is ineffective against Trichuris. Strongyloides stercoralis S. stercoralis, sometimes called the threadworm or dwarf threadworm, is exceptional
among helminths because it can replicate and cause cycles of larval
reinfection within the human host. The organism infects more than 200 million
people worldwide, most frequently in the tropics and other hot, humid
locales. In the Enterobius vermicularis Enterobius,
the pinworm, causes the most common helminthic infection in the Pyrantel pamoate, mebendazole, and albendazole are highly effective. Single oral doses of each should be repeated after 2 weeks. When their use is combined with rigid standards of personal hygiene, a very high proportion of cures can be obtained. Treatment is simple and almost devoid of side effects. The benzimidazoles should not be used during pregnancy because of their teratogenic potential. Daily doses of piperazine for 1 week also are effective but less convenient. Trichinella spiralis The trichina worm is ubiquitous, regardless of climate, and can live
outside its hosts. It is found in Albendazole and mebendazole appear to be effective against the intestinal forms of T. spiralis that are present early in infection. The efficacy of these agents or any anthelmintic agent on larvae that have migrated to muscle is questionable. Glucocorticoids may be of considerable value in controlling the acute and dangerous manifestations of established infection. Filariae Adult worms that cause human filariasis dwell either in the lymphatic
system (Wurchereria bancrofti, Brugia malayi, Brugia timori) or other
tissues (Loa loa, Onchocerca volvulus, Mansonella species). Spread by
the bites of infected mosquitoes, lymphatic filariasis affects nearly 90
million people, about 90% of them infected with W. bancrofti and most
of the rest with B. malayi. W. bancrofti is especially a risk in
central Africa, Diethylcarbamazine and ivermectin are the primary compounds used to treat lymphatic filariasis. Population-based, yearly chemotherapy with a single oral dose of either diethylcarbamazine or ivermectin together with an oral dose of albendazole may be more effective in markedly reducing the microfilaremia and prevalence of this infection (Ottesen et al., 1999). Ivermectin is the safer primary agent for areas where bancroftian filariasis may coexist with either loiasis or onchocerciasis; diethylcarbamazine can be used with albendazole elsewhere. The best results are achieved in W. bancrofti and B. malayi infections if chemotherapy is started early, before obstructive lesions of the lymphatics have occurred. Even in late cases, some improvement may result. In long-standing elephantiasis, surgical measures are required to improve lymph drainage and remove redundant tissue. Diethylcarbamazine currently is the best single drug for the treatment of loiasis, but it is advisable to start with a small initial dose to diminish host reactions that result from destruction of microfilariae. Glucocorticoids often are required to control acute reactions. In rare instances, serious cerebral reactions occur in the treatment of loiasis, probably due to destruction of microfilariae in the brain. If headache is severe and there is other evidence of an adult L. loa near the orbit, extra caution for initial dosing is advised. Ivermectin is the best single drug for control and treatment of onchocerciasis. Diethylcarbamazine no longer is recommended. Both agents kill only microfilariae of O. volvulus, but ivermectin produces far milder systemic reactions and few if any ocular complications. With diethylcarbamazine, such reactions are likely to be severe, particularly in cases where there are lesions of the eye. Although suramin (see Chapter 41: Drugs Used in the Chemotherapy of Protozoal Infections: Amebiasis, Giardiasis, Trichomoniasis, Trypanosomiasis, Leishmaniasis, and Other Protozoal Infections) is lethal to adult O. volvulus, treatment with this relatively toxic agent probably is unwarranted. Less toxic macrofilaricides are needed. Dracunculus medinensis Known as the guinea, dragon, or There is no suitable anthelmintic that acts directly against D. medinensis. Traditional treatment for this disabling condition is to draw the live adult female worm out day by day by rolling it onto a small piece of wood. This procedure risks severe infection if rupture occurs. Metronidazole, 250 mg given 3 times a day for 10 days, can provide symptomatic and functional relief by facilitating removal of the worm through indirect suppression of host inflammatory responses. Strategies such as filtering drinking water and reducing contact of infected individuals with water have caused marked reductions in the transmission and prevalence of dracunculiasis in most endemic regions. Cestodes (Flatworms) Taenia saginata Human beings are the definitive hosts for Taenia saginata,
known as the beef tapeworm. This most common form of tapeworm usually is
detected after passage of proglottids from the intestine. It is cosmopolitan,
occurring most commonly in sub-Saharan Africa and the Praziquantel is the drug of choice for treatment of infection by T. saginata, although niclosamide also is used because it is cheap and available. Both are very effective, simple to administer, and comparatively free from side effects. Assessment of cure can be difficult because the worm (segments as well as scolex) is usually passed in a partially digested state. If parasitological diagnosis is uncertain, praziquantel is the preferred drug because of the danger of cysticercosis (see below). Taenia solium Taenia solium, or pork tapeworm, also has a cosmopolitan distribution; immigrant
populations are a common source of infection in the Diphyllobothrium latum Diphyllobothrium latum, the fish tapeworm, is found most commonly in rivers and lakes of the
northern hemisphere. In Hymenolepis nana Hymenolepis nana, the dwarf tapeworm, is the smallest and most common tapeworm
parasitizing human beings. Infection with this cestode is cosmopolitan, more
prevalent in tropical than temperate climates, and most common among
institutionalized children, including those in the southern Echinococcus Species Human beings serve as one of several intermediate hosts for larval forms of Echinococcus species that cause 'cystic' (E. granulosus) and 'alveolar' (E. multilocoularis and E. vogeli) hydatid disease. Dogs are definitive hosts for these tapeworms. Parasite eggs from canine stools are a major worldwide cause of disease in associated livestock, e.g., sheep and goats. E. granulosus produces unilocular, slowly growing cysts, most often in liver and lung, whereas E. multilocularis creates multilocular invasive cysts predominantly in the same organs. Removal of the cysts by surgery is the preferred treatment, but leakage from ruptured cysts may spread disease to other organs. Prolonged regimens of albendazole, either alone or as an adjunct to surgery, are reportedly of some benefit. However, some patients are not cured despite multiple courses of therapy. Treatment of infected dogs with praziquantel eradicates adult worms and interrupts transmission of these infections. Trematodes (Flukes) Schistosoma haematobium, Schistosoma mansoni, Schistosoma japonicum These are the main species of blood flukes that cause human
schistosomiasis; less common species are Schistosoma intercalatum and Schistosoma
mekongi. The infection affects about 200 million people, and more than
600 million are considered at risk. Schistosomiasis is widely distributed
over the South American continent and certain Caribbean islands (S.
mansoni), much of Africa and the Arabian Peninsula (S. mansoni and
S. haematobium), and Praziquantel
is the drug of choice for treating all species of schistosomes that infect
human beings. The drug is safe and effective when it is given in single or
divided oral doses on the same day. These properties make praziquantel
especially suitable for population-based chemotherapy. Although not effective
clinically against S. haematobium and S. japonicum, oxamniquine
is effective for treatment of S. mansoni infections, particularly in Paragonimus westermani and Other Paragonimus Species Called lung flukes, a number of Paragonimus species, of which P.
westermani is the most common, are pathogenic for human beings and
carnivores. Found in the Clonorchis sinensis, Opisthorchis viverrini, Opisthorchis felineus These closely related trematodes exist in the Fasciola hepatica Human beings are only accidentally infected with F. hepatica, the large liver fluke that exists worldwide and primarily affects herbivorous ruminants such as cattle and sheep. Eating contaminated freshwater plants such as watercress initiates the infection. Migratory larvae penetrate the intestine, invade the liver from the peritoneum, and eventually reside in the biliary tract. The acute illness is characterized by fever, urticaria, and abdominal symptoms, whereas chronic infection resembles that caused by other hepatic flukes. However, F. hepatica infection is not associated with cholangiocarcinoma. Unlike infections with other flukes, fascioliasis does not usually respond to praziquantel. The current recommended drug is bithionol, 30 to 50 mg/kg given on alternate days for 10 to 15 doses. Bithionol may be obtained from the United States Centers for Disease Control and Prevention Drug Service. Based on limited trials, triclabendazole, a narrow-spectrum benzimidazole used in veterinary medicine, shows more promise for treating fascioliasis (see Arjona et al., 1995). Fasciolopsis buski, Heterophyes heterophyes, Metagonimus yokogawai, Nanophyetus salmincola Obtained by eating contaminated water chestnuts and other caltrops in |
Anthelmintic Drugs
|
Benzimidazoles History The discovery by Brown and coworkers (1961) that thiabendazole possessed potent activity against gastrointestinal nematodes sparked development of the benzimidazoles as broad-spectrum anthelmintic agents against parasites of both veterinary and human medical importance. Of the hundreds of derivatives tested, those most therapeutically useful have modifications at the 2 and/or 5 positions of the benzimidazole ring system (see Townsend and Wise, 1990). Three compounds, thiabendazole, mebendazole, and albendazole, have been used extensively for the treatment of human helminthiasis. The chemical structures of these drugs are shown in Table 421. Thiabendazole, which contains a thiazole ring at position 2, is active against a wide range of nematodes that infect the gastrointestinal tract. However, its clinical use against these organisms has declined markedly because of thiabendazole's toxicity relative to that of other equally effective drugs. Mebendazole, the prototype benzimidazole carbamate, was introduced for the treatment of intestinal roundworm infections as a result of research carried out by Brugmans and collaborators (1971). Albendazole is a newer benzimidazole carbamate that is used worldwide, primarily against a variety of intestinal and tissue nematodes but also against larval forms of certain cestodes (de Silva et al., 1997; Venkatesan, 1998). As to the latter, albendazole has become the drug of choice for treating cysticercosis (see Sotelo and Jung, 1998; Garcia and Del Brutto, 2000) and the drug of choice for cystic hydatid disease (see Horton, 1997). However, albendazole is not effective against F. hepatica. When used yearly in conjunction with either ivermectin or diethylcarbamazine, single doses of albendazole have shown considerable promise for global control of lymphatic filariasis and related tissue filarial infections (see Ottesen et al., 1999). Anthelmintic Action The benzimidazoles, exemplified by mebendazole and albendazole, are versatile anthelmintic agents, particularly against gastrointestinal nematodes, where their action is not dictated by the systemic drug concentration. Appropriate doses of mebendazole and albendazole are highly effective in ascariasis, intestinal capillariasis, enterobiasis, trichuriasis, and hookworm (Ancylostoma duodenale and Necator americanus) infection as single or mixed infections. These drugs are active against both larval and adult stages of nematodes that cause these infections, and they are ovicidal for Ascaris and Trichuris. Immobilization and death of susceptible gastrointestinal parasites occur slowly, and their clearance from the gastrointestinal tract may not be complete until a few days after treatment. Albendazole is superior to mebendazole in curing hookworm infections in children (de Silva et al., 1997; Bennett and Guyatt, 2000). Moreover, albendazole is more effective than mebendazole against strongyloidiasis (Liu and Weller, 1993), cystic hydatid disease caused by Echinococcus granulosus (Horton, 1997; Davis et al., 1989), and neurocysticercosis caused by larval forms of Taenia solium (Evans et al., 1997; Garcia and Del Brutto, 2000). The benzimidazoles probably are active against the intestinal stages of Trichinella spiralis in human beings but probably do not affect the larval stages in tissues. Albendazole is highly effective against the migrating forms of dog and cat hookworms that cause cutaneous larval migrans, although thiabendazole can be used topically for this purpose. Regimens in which albendazole with either ivermectin or diethylcarbamazine are given as single annual doses show great promise for controlling lymphatic filariasis occurring either alone or together with other filarial infections (see Ottesen et al., 1999). Such combined therapy has the additional benefit of reducing intestinal roundworm infestations in school-aged children (Albonico et al., 1999). Certain microsporidial species that cause intestinal infections in people with AIDS respond partially (Enterocytozoon bieneusi) or completely (Encephalitozoon intestintalis and related Encephalitozoon species) to albendazole; albendazole's sulfoxide metabolite appears to be especially effective against these parasites in vitro (see Katiyar and Edlind, 1997). Albendazole also has some efficacy against anaerobic protozoa such as Trichomonas vaginalis and Giardia lamblia (Ottesen et al., 1999). While benzimidazoles have antifungal activity, their clinical use against human mycoses is limited. Benzimidazoles produce many biochemical changes in susceptible
nematodes, e.g., inhibition of mitochondrial fumarate reductase,
reduced glucose transport, and uncoupling of oxidative phosphorylation (see
Lacey, 1988). There is strong evidence, however, that the primary action of
these drugs is to inhibit microtubule polymerization by binding to Absorption, Fate, and Excretion Benzimidazoles have only limited solubility in water; consequently,
minor differences in solubility tend to have a major effect on absorption. Thiabendazole
is absorbed rapidly after oral ingestion and reaches peak concentrations in
plasma after about 1 hour. Most of the drug is excreted in the urine within
24 hours as 5-hydroxythiabendazole, conjugated either as the glucuronide or
as the sulfate. In contrast, tablet formulations of mebendazole are poorly
and erratically absorbed, and concentrations of the drug in plasma are low
and do not reflect the dosage taken (Witassek et al., 1981). The low
systemic bioavailability (22%) of mebendazole results from a combination of
poor absorption and rapid first-pass hepatic metabolism. Mebendazole is about
95% bound to plasma proteins and is extensively metabolized. Two major
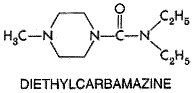
metabolites, methyl-5-( Albendazole is variably and erratically absorbed after oral administration; absorption is enhanced by the presence of fatty foods and possibly by bile salts as well. After a 400-mg oral dose, albendazole cannot be detected in plasma, because the drug is rapidly metabolized in the liver and possibly in the intestine as well, to albendazole sulfoxide, which has potent anthelmintic activity (Marriner et al., 1986; Moroni et al., 1995; Redondo et al., 1999). Both the (+) and () enantiomers of albendazole sulfoxide are formed, but in human beings the (+) enantiomer reaches much higher peak concentrations in plasma and is cleared much more slowly than the () form (Delatour et al., 1991; Marques et al., 1999). Total sulfoxide attains peak plasma concentrations of about 300 ng/ml, but with wide interindividual variation. Albendazole sulfoxide is about 70% bound to plasma proteins and has a highly variable plasma half-life ranging from about 4 to 15 hours (Delatour et al., 1991; Jung et al., 1992; Marques et al., 1999). It is well distributed into various tissues, including hydatid cysts, where it reaches a concentration of about one-fifth that in plasma (Marriner et al., 1986; Morris et al., 1987). This probably explains why albendazole is more effective than mebendazole for treating hydatid cyst disease. Formation of albendazole sulfoxide is catalyzed by both microsomal flavin monooxygenase and isoforms of cytochrome P450 in the liver and possibly also in the intestine (Redondo et al., 1999). Hepatic flavin monooxygenase activity appears associated with (+) albendazole sulfoxide formation, whereas cytochromes P450 preferentially produce the () sulfoxide metabolite (Delatour et al., 1991). Both sulfoxide derivatives are oxidized further to the nonchiral sulfone metabolite of albendazole, which is pharmacologically inactive; this reaction favors the () sulfoxide and probably becomes rate limiting in determining the clearance and plasma half-life of the bioactive (+) sulfoxide metabolite (Delatour et al., 1991). Induction of enzymes involved in sulfone formation from the (+) sulfoxide could account for some of the wide variation noted in plasma half-lives of albendazole sulfoxide. Indeed, in animal models, benzimidizoles can induce their own metabolism (for example, see Gleizes et al., 1991). Albendazole metabolites are excreted mainly in the urine. Therapeutic Uses The introduction of thiabendazole (MINTEZOL) was a major advance in the therapy of cutaneous larva migrans (creeping eruption) and S. stercoralis infection. The majority of patients experience marked relief of symptoms of creeping eruption, and a high percentage of cures are achieved after topical treatment with 15% thiabendazole in a water-soluble cream base applied to the affected area two or three times per day for 5 days (Davies et al., 1993). A common regimen for therapy of strongyloidiasis is 25 mg/kg of thiabendazole given twice daily after meals for 2 days; the total daily dose should not exceed 3 grams. This schedule should be extended for 5 to 7 days for treatment of disseminated strongyloidiasis or until the parasites have been eliminated. The thiabendazole regimen has been replaced largely by a single dose of ivermectin for treatment of intestinal strongyloidiasis (see'Ivermectin,' below), but as yet thiabendazole is the only drug with established efficacy against disseminated strongyloidiasis (Gann et al., 1994; Liu and Weller, 1998). Thiabendazole given at a dosage of 25 mg/kg twice per day for 7 days may produce some benefit for early trichinosis. The drug, however, has no effect on migrating or muscle-stage larvae. Thiabendazole also is effective in gastrointestinal nematode infections, but because of its toxicity, it should no longer be used for those infections. Mebendazole VERMOX) is highly effective against gastrointestinal nematode infections and is particularly valuable for the treatment of mixed infections. Mebendazole is always taken orally, and the same dosage schedule applies to adults and children more than 2 years of age. For control of enterobiasis, a single 100-mg tablet is taken; a second should be given after 2 weeks. For control of ascariasis, trichuriasis, and hookworm infection, the recommended regimen is 100 mg of mebendazole taken in the morning and evening for 3 consecutive days. If the patient is not cured 3 weeks after treatment, a second course should be given. The 3-day mebendazole regimen is more effective than single doses of either mebendazole (500 mg) or albendazole (400 mg), which also have been used successfully to control these mixed infections. A 500-mg dose of mebendazole may be slightly more effective against trichuriasis than a 400-mg dose of albendazole, but the single dose of albendazole appears superior to mebendazole against hookworms (de Silva et al., 1997; Bennett and Guyatt, 2000). Infections with Capillaria philippinensis are more resistant to treatment with mebendazole; 400 mg of the drug has been given per day in two divided doses for at least 20 days (Cross, 1992). Mebendazole has been used to treat cystic hydatid disease, although surgery should be performed first, and chemotherapy with albendazole is now considered to be superior (see Horton, 1997). Like mebendazole, albendazole (ALBENZA) provides safe and highly effective therapy against infections with gastrointestinal nematodes, including mixed infections of Ascaris, Trichuria, and hookworms. For treatment of enterobiasis, ascariasis, trichuriasis, and hookworm infections, albendazole is taken as a single oral 400-mg dose by adults and children more than 2 years of age. Cure rates for light to moderate Ascaris infections are typically over 97%, although heavy infections may require therapy for 2 to 3 days. A 400-mg dose of albendazole appears to be superior to a 500-mg dose of mebendazole for curing hookworm infections and reducing egg counts (Sacko et al., 1999; Bennett and Guyatt, 2000). Given at a dose of 400 mg daily for 3 days, albendazole exhibits highly variable efficacy against strongyloidiasis; both thiabendazole and ivermectin are more effective in treating this infection. Albendazole is the drug of choice for treating people with cystic hydatid disease due to E. granulosus. While the drug provides only a modest cure rate when used alone, it produces superior results when used before and after either surgery to remove cysts or aspiration/injection of cysts with protoscolicidal agents (see Horton, 1997; Schantz, 1999). A typical dosage regimen for adults is 400 mg given twice a day for 28 days, the course to be repeated as necessary. However, albendazole at 10 to 12 mg/kg per day has been given continuously for 3 to 6 months without producing serious or irreversible toxicity (Franchi et al., 1999). While still the best drug available, albendazole appears to be just marginally effective against alveolar echinococcosis caused by E. multilocularis (see Venkatesan, 1998). Albendazole also is the preferred treatment of neurocysticercosis caused by larval forms of T. solium (see Evans et al., 1997; Sotelo and Jung, 1998; Garcia and Del Brutto, 2000). The recommended dosage is 400 mg given twice a day for adults with therapy continued for 3 to 28 days, depending on the number, type, and location of the cysts. Glucocorticoids usually are given for several days prior to the start of albendazole therapy to reduce the incidence of side effects resulting from inflammatory reactions to dead and dying cysticerci. Such pretreatment also increases plasma levels of albendazole sulfoxide. Albendazole, 400 mg per day, has shown efficacy for therapy of microsporidial intestinal infections in patients with AIDS; Encephalitozoon intestinalis responds well to this drug, whereas the related Enterocytozoon bieneusi seems to be more refractory (see Venkatesan, 1999). A notable recent advance is the use of albendazole together with either diethylcarbamazine or ivermectin in programs directed toward controlling lymphatic filariasis (reviewed by Ottesen et al., 1999). Albendazole itself has delayed microfilaricidal activity but only slight macrofilarcidal activity against W. bancrofti. But a single dose of albendazole (400 to 600 mg) given alone with a single dose of either diethylcarbamazine (6 mg/kg) or ivermectin (0.2 to 0.4 mg/kg) markedly decreases the microfilaremia in W. bancrofti infection for well over a year. By annual dosing for 4 to 6 years, the strategy is to maintain the microfilaremia at such low levels that transmission cannot occur. The period of therapy is estimated to correspond to the duration of fecundity of adult worms. Albendazole is given with diethylcarbamazine to treat lymphatic filariasis in most parts of the world. However, to avoid serious reactions to dying microfilariae, the albendazole/ivermectin combination should be used in locations where filariasis coexists with either onchocerciasis or loiasis. Toxicity, Side Effects, Precautions, and Contraindications The clinical utility of thiabendazole is compromised by its toxicity. Side effects frequently encountered with therapeutic doses are anorexia, nausea, vomiting, and dizziness. Less frequently, diarrhea, weariness, drowsiness, giddiness, and headache occur. Occasional fever, rashes, erythema multiforme, hallucinations, sensory disturbances, and StevensJohnson syndrome have been reported. Angioneurotic edema, shock, tinnitus, convulsions, and intrahepatic cholestasis are rare complications of therapy. Some patients excrete a metabolite that imparts an odor to the urine much like that occurring after ingestion of asparagus. Crystalluria without hematuria has been reported on occasion; it promptly subsides with discontinuation of therapy. Transient leukopenia has been noted in a few patients on thiabendazole therapy. There are no absolute contraindications to the use of thiabendazole. Because central nervous system (CNS) side effects occur frequently, activities requiring mental alertness should be avoided during therapy. Thiabendazole has hepatotoxic potential, so it should be used with caution in patients with hepatic disease or decreased hepatic function. The effects of thiabendazole in pregnant women have not been studied adequately, so it should be used in pregnancy only when the potential benefit justifies the potential risk. Unlike thiabendazole, mebendazole does not cause significant systemic toxicity in routine clinical use, even in the presence of anemia and malnutrition. This probably results from its low systemic bioavailability. Transient symptoms of abdominal pain, distention, and diarrhea have occurred in cases of massive infestation and expulsion of gastrointestinal worms. Rare side effects in patients treated with high doses of mebendazole include allergic reactions, alopecia, reversible neutropenia, agranulocytosis, and hypospermia. Reversible elevation of serum transaminases is not uncommon in this population. Mebendazole is a potent embryotoxin and teratogen in laboratory animals; effects may occur in pregnant rats at single oral doses as low as 10 mg/kg. Thus, despite a lack of evidence for teratogenicity in human beings, it is advised that mebendazole not be given to pregnant women or to children less than 2 years of age. It should not be used in patients who have experienced allergic reactions to the agent. Like mebendazole, albendazole produces few side effects when used for short-term therapy of gastrointestinal helminthiasis, even in patients with heavy worm burdens. Transient abdominal pain, diarrhea, nausea, dizziness, and headache occur on occasion. Even when used for long-term therapy of cystic hydatid disease and neurocysticercosis, albendazole is well tolerated by most patients. The most common side effect is an increase in serum aminotransferase activity; in rare instances, jaundice or chemical cholestasis may be noted, but enzyme activities return to normal after therapy is completed. Routine liver function should be monitored during protracted albendazole therapy, and the drug is not recommended for patients with hepatic cirrhosis (Davis et al., 1989). Especially if not pretreated with glucocorticoids, some patients with neurocysticercosis may experience serious neurological sequelae that depend on the location of inflamed cysts with dying cysticerci. Other side effects reported during extended therapy include gastrointestinal pain, severe headaches, fever, fatigue, loss of hair, leukopenia, and thrombocytopenia. Albendazole is teratogenic and embryotoxic in animals. Thus, it should not be given to pregnant women. The safety of albendazole in children less than 2 years of age has not been established. The benzimidazoles as a group display remarkably few clinically significant interactions with other drugs. The most versatile of this family, albendazole, probably induces its own metabolism, and plasma levels of its sulfoxide metabolites can be increased by coadministration of glucocorticoids and possibly praziquantel. Caution is advised when using high doses of albendazole together with general inhibitors of hepatic cytochromes P450. Diethylcarbamazine History Over 1500 cases of filariasis in American military personnel during World War II stimulated the search for effective filaricides. The most promising group of antifilarial compounds to emerge were piperazine derivatives, of which diethylcarbamazine is the most important (Hawking, 1979; Mackenzie and Kron, 1985). Diethylcarbamazine is a first-line agent for control and treatment of lymphatic filariasis and for therapy of tropical pulmonary eosinophilia caused by W. bancrofti and B. malayi (see Ottesen and Ramachandran, 1995). Although this agent is effective against onchocerciasis and loiasis, it can cause serious reactions to affected microfilariae in both infections. For this reason, ivermectin has replaced diethylcarbamazine for therapy of onchocerciasis. Despite its toxicity, diethylcarbamazine remains the best drug available to treat loiasis. Programs that feature annual single doses of both dietnylcarbamazine and albendazole show considerable promise for the control of lymphatic filariasis in geographic regions where onchocerciasis and loiasis are not endemic (see Ottesen et al., 1999). Chemistry Diethylcarbamazine (HETRAZAN) is formulated as the water-soluble citrate salt containing 51% by
weight of the active base. Because the compound is tasteless, odorless, and
stable to heat, it also can be taken in the form of fortified table salt
containing 0.2% to 0.4% by weight of the base. The drug is available outside
the
Anthelmintic Action Microfilarial forms of susceptible filarial species are most affected by diethylcarbamazine, which elicits rapid disappearance of these developmental forms of W. bancrofti, B. malayi, and L. loa from human blood. The drug causes microfilariae of O. volvulus to disappear from skin but does not kill microfilariae in nodules that contain the adult (female) worms. It does not affect the microfilariae of W. bancrofti in a hydrocele, despite penetration into the fluid. The mechanism of action of diethylcarbamazine on susceptible microfilariae is not well understood (see Martin et al., 1997; de Silva et al., 1997). The drug exerts little effect in vitro, whereas it acts rapidly in vivo. One possibility is that diethylcarbamazine may perturb arachidonic acid metabolism in both microfilariae and host endothelial cells with resulting vasoconstriction and aggregation of host platelets and granulocytes around membrane-damaged parasites. The compound does not appear to activate adaptic immune responses in the host, but innate immune response(s) could be involved (Maizels and Denham, 1992; Maizels et al., 1993). There is evidence that diethylcarbamazine kills worms of adult L. loa and probably adult W. bancrofti and B. malayi as well. However, it has little action against adult O. volvulus. The mechanism of filaricidal action of diethylcarbamazine against adult worms is unknown (see Hawking, 1979). Some studies suggest that diethylcarbamazine compromises intracellular processing and transport of certain macromolecules to the plasma membrane (Spiro et al., 1986). The drug also may affect specific immune and inflammatory responses of the host by as yet undefined mechanisms (see Mackenzie and Kron, 1985; Martin et al., 1997). Absorption, Fate, and Excretion Diethylcarbamazine is absorbed rapidly from the gastrointestinal tract. Peak plasma levels occur within 1 to 2 hours after a single oral dose, and the plasma half-life of this base varies from 2 to 10 hours, depending on the urinary pH. Metabolism is both rapid and extensive (Faulkner and Smith, 1972). A major metabolite, diethylcarbamazine-N-oxide, is bioactive. Diethylcarbamazine is excreted by both urinary and extraurinary routes; over 50% of an oral dose appears in acidic urine as the unchanged drug, but this value is decreased when the urine is alkaline (Edwards et al., 1981). Indeed, alkalinizing the urine can elevate plasma levels, prolong the plasma half-life, and increase both the therapeutic effect and toxicity of diethylcarbamazine (Awadzi et al., 1986). Therefore, dosage reduction may be required for people with renal dysfunction or sustained alkaline urine. Therapeutic Uses Dosages of diethylcarbamazine citrate used to prevent or treat filarial infections have evolved empirically and vary according to local experience. Recommended regimens differ according to whether the drug is used for population-based chemotherapy, control of filarial disease, or prophylaxis against infection. W. bancrofti, B. malayi, and B. timori For mass treatment with the objective of reducing microfilaremia to subinfective levels for mosquitoes, the recent introduction of diethylcarbamazine into table salt (0.2% to 0.4% by weight of the base) has markedly reduced the prevalence, severity, and transmission of lymphatic filariasis in many endemic areas (Gelband, 1994). Moreover, single 6-mg/kg doses given orally every 6 to 12 months have proven just as effective as former prolonged daily dosage regimens. A major discovery was that diethylcarbamazine given annually as a single oral dose of 6 mg/kg was most effective in reducing microfilaremia when given along with a single dose of another antifilarial agent (see Ottesen et al., 1999). Initial studies were done with ivermectin as the partner drug, but more recent evidence indicates that 400 mg of albendazole taken orally in addition to diethylcarbamazine is an even more appropriate choice for mass chemotherapy (see'Benzimidazoles,' above). Adverse reactions to microfilarial destruction, greater after the oral diethylcarbamazine tablet than the table salt preparation, usually are well tolerated. However, mass chemotherapy with diethylcarbamazine should not be used in regions where onchocerciasis or loiasis coexist because, even in the table salt formulation, this drug may induce especially severe reactions related to parasite burden in these infections. Diethylcarbamazine, given in doses of 2 mg/kg three times daily for 14 days, causes rapid disappearance of symptoms of tropical eosinophilia, the pulmonary inflammatory response typical of infection with W. bancrofti or B. malayi. After taking a small test dose of 50 mg, asymptomatic individuals with microfilaremia should be treated with 'standard' courses of diethylcarbamazine, e.g., 6 mg/kg in 3 divided doses per day for 14 to 21 days; the 21-day period is customary for B. malayi. Such therapy should minimize further lymphatic and renal damage. It is largely ineffective against more advanced complications of lymphatic filariasis, such as lymphangitis and chronic obstructive lymphedema (elephantiasis), that respond better to antibiotics and maintenance of good local hygiene, respectively. A monthly dose of 50 mg of diethylcarbamazine is effective for prophylaxis against lymphatic filariasis. O. volvulus and L. loa Diethylcarbamazine no longer is recommended for initial treatment of onchocerciasis, because it causes severe reactions related to microfilarial destruction (see below). Such reactions are far less severe in response to ivermectin, the drug now preferred for this infection. Diethylcarbamazine, despite its drawbacks, remains the best available drug for therapy of loiasis. Treatment is initiated with test doses of 1 mg/kg daily for 2 to 3 days, escalating to maximally tolerated daily doses of 8 to 10 mg/kg for a total of 2 to 3 weeks. Low test doses are used, often accompanied by pretreatment with glucocorticoids or antihistamines, to minimize reactions to dying microfilariae and adult worms; these reactions consist of severe allergic reactions and, occasionally, meningoencephalitis and coma from invasion of the CNS by microfilariae. Repeated courses of treatment with diethylcarbamazine, separated by 3 to 4 weeks, may be required to cure loiasis, and doses of 300 mg weekly have proven to be effective for prophylaxis against this infection. Ivermectin does not provide a good alternative to diethylcarbamazine for treatment of loiasis, but albendazole may prove to be useful in patients who either fail therapy with diethylcarbamazine or who cannot take the drug (Klion et al., 1999). Diethylcarbamazine is clinically effective against microfilariae and adult worms of Dipetalonema streptocerca, but filariasis due to Mansonella perstans, M. ozzardi, or Dirofilaria immitis responds minimally to this agent. Although therapy with diethylcarbamazine, 6 mg/kg daily in divided doses for 7 to 10 days, has been recommended for treatment of toxocariasis, the drug is considered experimental for this indication. Toxicity and Side Effects Unless a daily dose of 8 to 10 mg/kg is exceeded, direct toxic reactions to diethylcarbamazine are rarely severe and usually disappear within a few days despite continuation of therapy. These reactions include anorexia, nausea, headache, and, at high doses, vomiting. Major adverse effects result directly or indirectly from the host response to destruction of parasites, primarily microfilariae. Reactions are especially severe in patients heavily infected with O. volvulus. They usually are less serious in B. malayi or L. loa infections and usually are mild in bancroftian filariasis, but the drug occasionally induces retinal hemorrhages and severe encephalitis in patients heavily infected with L. loa. In patients with onchocerciasis, there usually is a typical reaction (termed a Mazzotti reaction) that occurs within a few hours after the first oral dose. This consists of intense itching and skin rashes, enlargement and tenderness of the lymph nodes, sometimes a fine papular rash, fever, tachycardia, arthralgia, and headache. These symptoms persist for 3 to 7 days and then subside, after which quite high doses can be tolerated. Ocular complications include limbitis, punctate keratitis, uveitis, and atrophy of the retinal pigment epithelium (Rivas-Alcala et al., 1981; Dominguez-Vazquez et al., 1983). In patients with bancroftian or brugian filariasis, nodular swellings may occur along the course of the lymphatics, and there is often an accompanying lymphadenitis. This reaction also subsides within a few days. Almost all patients receiving therapy exhibit a leukocytosis, first evident on the second day, reaching its peak on the fourth or fifth day, and gradually subsiding over a period of a few weeks. Reversible proteinuria may occur, and the eosinophilia so frequently observed in patients with filariasis can be intensified by therapy with diethylcarbamazine. Delayed reactions to more mature dying filarial forms include lymphangitis, swelling and lymphoid abscesses in bancroftian and brugian filariasis, and small skin weals in loiasis. Diethylcarbamazine appears to be safe for use during pregnancy. Precautions and Contraindications Population-based chemotherapy with diethylcarbamazine should be avoided in areas where onchocerciasis or loiasis is endemic, even though the drug can be used to protect foreign travelers from these infections. Pretreatment with glucocorticoids and antihistamines often is undertaken to minimize indirect reactions to diethylcarbamazine that result from dying microfilariae. Dosage reduction must be considered for patients with impaired renal function or persistent alkaline urine. Ivermectin History In the mid-1970s, a survey of natural products revealed that a
fermentation broth of the soil actinomycete Streptomyces avermitilis
ameliorated infection with Nematospiroides dubius in mice (Burg et
al., 1979; Egerton et al., 1979; Miller et al., 1979).
Isolation of the anthelmintic components from cultures of this organism led
to discovery of the avermectins, a novel class of 16-membered lactones
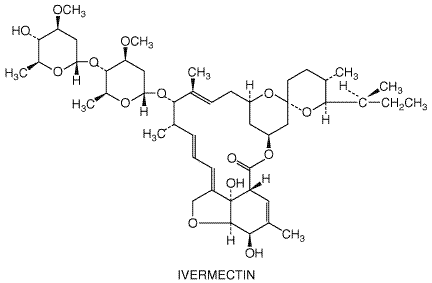
(see The milbemycins are macrocyclic lactone analogs of the avermectins. Some of these compounds have antiparasitic activity similar to that of the avermectins and probably act by similar mechanisms (Fisher and Mrozik, 1992; Arena et al., 1995). The chemical structure of ivermectin is shown below.
Antiparasitic Activity and Resistance Several reviews have focused on the antiparasitic action of and resistance to the avermectins and related milbemycins (see Cully et al., 1996; Sangster, 1996; Martin et al., 1997; Sangster and Gill, 1999). Ivermectin is effective and highly potent against at least some developmental stages of many parasitic nematodes and insects that affect animals and human beings. The drug immobilizes affected organisms by inducing a tonic paralysis of the musculature. Work with the free-living nematode Caenorhabditis elegans indicates that avermectins act on a group of glutamate-gated Cl channels to produce this effect. Found only in invertebrates, these channels and two of their cloned subunits have been expressed and characterized in Xenopus laevis oocytes. There is close correlation among activation and potentiation by avermectins and milbemycin D of glutamate-sensitive Cl current, nematicidal activity, and membrane binding affinity in this system (see Arena et al., 1995; Cully et al., 1996). Moreover, glutamate-gated Cl channels are expressed in the pharyngeal muscle cells of these worms, consistent with the marked and potent inhibitory effect of avermectins on the feeding behavior of the organisms (see Sangster and Gill, 1997). The basis for resistance or relative unresponsiveness to avermectin action shown by different nematodes, especially those species parasitizing livestock, is complex. Several different avermectin-'resistant' developmental and physiological phenotypes have been described, but definitive relationships among these phenotypes and native avermectin receptor subtypes, locations, numbers, and binding affinities require clarification (see Sangster and Gill, 1999; Hejmadi et al., 2000). Alterations in genes encoding ATP-dependent P-glycoprotein transporters that bind avermectins and in those encoding putative components of the glutamate-gated Cl channel have been associated with the development of resistance in Haemonchus contortus (Xu et al., 1998; Blackhall et al., 1998). A large increase in low-affinity glutamate binding has been detected in ivermectin-resistant nematodes, but how this relates to drug resistance is unclear (Hejmadi et al., 2000). Glutamate-gated Cl channels probably serve as one site of ivermectin action in insects and crustaceans too (Duce and Scott, 1985; Scott and Duce, 1985; Zufall et al., 1989). Avermectins also bind with high affinity to GABA-gated and other ligand-gated Cl channels in nematodes such as ascaris and in insects, but the physiological consequences are less well defined. Lack of high-affinity avermectin receptors in cestodes and trematodes may explain why these helminths are not sensitive to ivermectin (Shoop et al., 1995). Avermectins do interact with gamma-aminobutyric acid (GABA) receptors in vertebrate (mammalian) brain, but their affinity for invertebrate receptors is about 100-fold greater (see Schaeffer and Haines, 1989). In human beings infected with Onchocerca volvulus, ivermectin causes a rapid, marked decrease in microfilarial counts in the skin and ocular tissues that lasts for 6 to 12 months (Greene et al., 1987; Newland et al., 1988). The drug has little discernible effect on adult parasites, but affects developing larvae and blocks egress of microfilariae from the uterus of adult female worms (Awadzi et al., 1985; Court et al., 1985). By reducing microfilariae in the skin, ivermectin decreases transmission to the Simulium black fly vector (Cupp et al., 1986, 1989). Ivermectin also is effective against microfilaria but not against adult worms of W. bancrofti, B. malayi, L. loa, and M. ozzardi (see de Silva et al., 1997). The drug exhibits excellent efficacy in human beings against A. lumbricoides, S. stercoralis, and cutaneous larva migrans. Other gastrointestinal nematodes are either partially affected (T. trichuria and E. vermicularis) or unresponsive (N. americanus and A. duodenale) (Naquira et al., 1989; de Silva et al., 1997). Absorption, Fate, and Excretion In human beings, peak levels of ivermectin in plasma are achieved within 4 to 5 hours after oral administration. The long terminal half-life of about 57 hours in adults primarily reflects a low systemic clearance (about 1 to 2 liters/hour) and a large apparent volume of distribution (see Appendix II). Ivermectin is about 93% bound to plasma proteins (Klotz et al., 1990). The drug is extensively converted by hepatic CYP3A4 to at least 10 metabolites, mostly hydroxylated and demethylated derivatives (Zeng et al., 1998). Virtually no ivermectin appears in human urine in either unchanged or conjugated form (Krishna and Klotz, 1993). In animals, ivermectin is recovered in feces, nearly all as unchanged drug, and the highest tissue concentrations occur in liver and fat. Extremely low levels are found in brain, even though ivermectin would be expected to penetrate the blood-brain barrier on the basis of its lipid solubility. Studies in transgenic mice suggest, however, that a P-glycoprotein efflux pump in the bloodbrain barrier prevents ivermectin from entering the CNS (Schinkel et al., 1994). This and the limited affinity of ivermectin for CNS receptors may explain the paucity of CNS side effects and the relative safety of this drug in human beings. Therapeutic Uses Onchocerciasis As reviewed by Lymphatic Filariasis Initial studies indicate that single annual doses of ivermectin (400 Infections with Intestinal Nematodes The finding that a single dose of 150 to 200 Other Indications Although ivermectin has activity against microfilaria but not against
adult worms of L. loa and M. ozzardi, it is not used clinically
for treating infections with these parasitic worms. Taken as a single 150 to
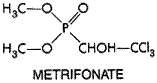
200 Toxicity, Side Effects, and Precautions Ivermectin is well tolerated by uninfected human beings and other mammals. In animals, signs of CNS toxicity, including lethargy, ataxia, mydriasis, tremors, and eventually death, occur only at very high doses; dogs, particularly collie breeds, are especially vulnerable (Campbell and Benz, 1984). In human beings, ivermectin toxicity nearly always results from Mazzotti-like reactions to dying microfilariae; the intensity and nature of these reactions relate to the microfilarial burden and the duration and type of filarial infection. After treatment of O. volvulus infections with ivermectin, these side effects usually are limited to mild itching and swollen, tender lymph nodes, which arise in 5% to 35% of people, last just a few days, and are relieved by aspirin and antihistamine drugs (see Goa et al., 1991). Rarely, more severe reactions occur that include high fever, tachycardia, hypotension, prostration, dizziness, headache, myalgia, arthralgia, diarrhea, and facial and peripheral edema; these may respond to glucocorticoid therapy. Ivermectin induces milder side effects than does diethylcarbamazine and, unlike the latter, seldom exacerbates lesions of ocular tissues in onchocerciasis. The drug can cause rare but serious side effects including marked disability and encephalopathies in patients coinfected with heavy burdens of L. loa microfilaria (Gardon et al., 1997). There is little evidence that ivermectin is teratogenic or carcinogenic. Because of its effects on gamma-aminobutyric acid receptors in the CNS, ivermectin is contraindicated in conditions associated with an impaired blood-brain barrier, e.g., African trypanosomiasis and meningitis. Caution also is advised about coadministration of ivermectin with other agents that depress CNS activity. Possible adverse interactions of ivermectin with other drugs that are extensively metabolized by hepatic CYP3A4 have yet to be evaluated. Ivermectin has not yet been approved for use in children less than 5 years old and in pregnant women, but both populations undoubtedly have been exposed to the drug in mass treatment programs. Lactating women taking the drug secrete low levels in their milk; the consequences for nursing infants are unknown. Metrifonate Metrifonate (trichlorfon; BILARCIL) is an organophosphorous compound used first as an insecticide and later as an anthelmintic, especially for treatment of schistosomiasis hematobium. Metrifonate has the following chemical structure:
Metrifonate is a prodrug; it is converted nonenzymatically at physiological pH to dichlorvos (2,2-dichlorovinyl dimethyl phosphate, DDVP), a potent cholinesterase inhibitor (Hinz et al., 1996). However, inhibition of cholinesterase alone is unlikely to explain the antischistosomal properties of metrifonate (Bloom, 1981). In vitro, dichlorvos is about equally potent as a inhibitor of both S. mansoni and S. haematobium acetylcholinesterases, yet metrifonate is effective clinically only against infections with S. hematobium. The molecular basis for this species-selective effect is not understood. Peak concentrations in plasma of metrifonate (30 Because of its low cost, effectiveness, and ready acceptance, metrifonate
is used as an alternative to praziquantel for treatment of urinary
schistosomiasis caused by S. haematobium. The usual dose is 7.5 to 10
mg/kg, given orally three times at intervals of 2 weeks. Recently, metrifonate
has been investigated for the treatment of central cholinergic deficits
associated with Alzheimer's disease (see Williams, 1999). Metrifonate
is not an approved drug in the At therapeutic doses, metrifonate inhibits the activities of plasma cholinesterase and erythrocyte acetylcholinesterase; both recover to almost normal levels within weeks of stopping treatment. Despite these changes, metrifonate is well tolerated. Side effects such as mild vertigo, lassitude, nausea, and colic are dose-related and occur infrequently. Treated individuals should be free from recent exposure to insecticides that might add to the anticholinesterase effect. They also should not receive depolarizing neuromuscular blocking agents for at least 48 hours after treatment. Niclosamide Niclosamide NICLOCIDE), a halogenated salicylanilide
derivative, was introduced in the 1960s for human use as a taeniacide. This
compound is considered as a second-choice drug to praziquantel for treating
human intestinal infections with T. saginata, D. latum, H. nana, and
most other cestodes because it was cheap, effective, and readily available in
many parts of the world. However, therapy with niclosamide poses a risk to
people infected with T. solium, because ova released from drug-damaged
gravid worms develop into larvae that can cause cysticercosis, a dangerous
infection that responds poorly to chemotherapy. Niclosamide is no longer
approved for use in the Oxamniquine Oxamniquine VANSIL) is a
2-aminomethyltetrahydroquinoline derivative that is used as a second-choice
drug to praziquantel for the treatment of schistosomiasis. Most strains of S.
mansoni are highly susceptible to oxamniquine, but S. haematobium
and S. japonicum are virtually unaffected by therapeutic doses.
Because of a low incidence of mild side effects together with normally high
efficacy after a single oral dose, oxamniquine continues to be used in S.
mansoni control programs, especially in Piperazine The discovery of the anthelmintic properties of piperazine usually is
credited to Fayard (1949), but these were first observed by Boismare, a
Piperazine is highly effective against both A. lumbricoides and E. vermicularis. The predominant effect of piperazine on Ascaris is to cause a flaccid paralysis that results in expulsion of the worm by peristalsis. Affected worms recover if incubated in drug-free medium. Piperazine acts as a GABA agonist. By increasing chloride ion conductance of Ascaris muscle membrane, the drug produces hyperpolarization and reduced excitability that leads to muscle relaxation and flaccid paralysis (Martin, 1985). The basis for its selectivity of action is not clear, but studies like those done on ivermectin (Arena et al., 1995) could help resolve this issue. Piperazine is rapidly absorbed after an oral dose. About 20% is excreted unchanged in the urine (Fletcher et al., 1982). Piperazine citrate MULTIFUGE),
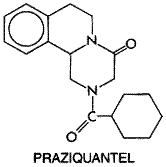
the form available in the In enterobiasis, single daily doses of 65 mg/kg, with a maximum of 2.5 grams, given for 7 days, will result in 95% to 100% cure. Treatment of enterobiasis is complicated by the readiness with which reinfection may occur. Thus, a second course of therapy should be given to ambulatory patients 1 to 2 weeks after the first. Many authorities advocate the simultaneous treatment of the entire household with piperazine in lieu of diagnosing each member by anal swabs. There is a large difference between effective therapeutic and overtly toxic doses of piperazine. Occasional gastrointestinal upset, transient neurological effects, and urticarial reactions have attended its use. Piperazine has been used without ill effect during pregnancy. Lethal doses cause convulsions and respiratory depression. Piperazine is contraindicated in patients with a history of epilepsy. Neurotoxic effects occur in individuals with renal dysfunction, because urinary excretion is the main route of elimination of the drug. Praziquantel History Praziquantel BILTRICIDE, DISTOCIDE) is a pyrazinoisoquinoline derivative developed after this class of compounds was discovered to have anthelmintic activity in 1972. In animals and human beings, infections with many different cestodes and trematodes respond favorably to this agent, whereas nematodes are unaffected (see Symposium, 1981; Andrews, 1985). Praziquantel has the following chemical structure:
The () isomer is responsible for most of the drug's anthelmintic activity. Anthelmintic Action After rapid and reversible uptake, praziquantel has two major effects on adult schistosomes. At the lowest effective concentrations, it causes increased muscular activity, followed by contraction and spastic paralysis. Affected worms detach from blood vessel walls, resulting in a rapid shift from the mesenteric veins to the liver. At slightly higher therapeutic concentrations, praziquantel causes tegumental damage, which exposes a number of tegumental antigens (see Redman et al., 1996). Comparisons of stage-specific susceptibility of S. mansoni to praziquantel in vitro and in vivo indicate that the clinical efficacy of this drug correlates better with tegumental action (Xiao et al., 1985). Studies in laboratory animals have shown that praziquantel is less effective against S. mansoni and S. japonicum in immunosuppressed mice (see Fallon et al., 1996). Whether or not host immune status is important for clinical efficacy of praziquantel in human beings is not known. The tegument of schistosomes seems to be the primary site of action of praziquantel. The drug causes an influx of Ca2+ across the tegument, and the effect is blocked in Ca2+-free medium. A number of praziquantel-sensitive sites have been suggested as possible targets, but the precise molecular mechanism of action remains elusive (see Redman et al., 1996). Praziquantel also produces a variety of biochemical changes, but most appear to be secondary to its primary tegumental action (see Andrews, 1985). Nearly all available information about the action of praziquantel has come from studies on schistosomes. Although it generally is assumed that the anthelmintic action of praziquantel against other trematodes and cestodes is the same as for schistosomes, direct evidence is lacking. Absorption, Fate, and Excretion Praziquantel is readily absorbed after oral administration, so that maximal levels in human plasma occur in 1 to 2 hours. The pharmacokinetics of praziquantel are dose-related. Extensive first-pass metabolism to many inactive hydroxylated and conjugated products limits the bioavailability of this drug and results in plasma concentrations of metabolites at least 100-fold higher than that of praziquantel. The drug is about 80% bound to plasma proteins. Its plasma half-life is 0.8 to 3.0 hours, depending on the dose, compared with 4 to 6 hours for its metabolites, but this may be prolonged in patients with severe liver disease, including those with hepatosplenic schistosomiasis. About 70% of an oral dose of praziquantel is recovered as metabolites in the urine within 24 hours; most of the remainder is metabolized in the liver and eliminated in the bile. Therapeutic Uses Praziquantel is approved in the Praziquantel is the drug of choice for treating schistosomiasis caused
by all Schistosoma species that infect human beings. Although dosage
regimens vary, a single oral dose of 40 mg/kg or three doses of 20 mg/kg
each, given 4 to 6 hours apart, generally produce cure rates of 70% to 95%
and consistently high reductions (over 85%) in egg counts. Strains of S.
mansoni and S. japonicum resistant to praziquantel have been
selected in laboratory studies (see Fallon et al., 1996).
Moreover, decreased clinical efficacy of praziquantel against infections with
S. mansoni has been reported in two human populations, one in a focal
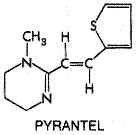
area of high transmission in northern Three doses of 25 mg/kg taken 4 to 8 hours apart on the same day result in high rates of cure for infections with either the liver flukes, C. sinensis and O. viverrini, or the intestinal flukes, Fasciolopsis buski, Heterophyes heterophyes, and Metagonimus yokogawi. The same three-dose regimen used for two days is highly effective against infections with the lung fluke, Paragonimus westermani. Infections with Fasciola hepatica are unresponsive to high doses, even though praziquantel penetrates this trematode. The reason for the insensitivity of F. hepatica to praziquantel is unknown. Low doses of praziquantel can be used successfully to treat intestinal infections with adult cestodes, for example, a single oral dose of 25 mg/kg for Hymenolepis nana and 10 to 20 mg/kg for D. latum, T. saginata, or T. solium. Retreatment after 7 to 10 days is advisable for individuals heavily infected with H. nana. While albendazole is preferred for therapy of human cysticercosis, the tissue infection with intermediate cyst larvae of T. solium, prolonged high-dose therapy with praziquantel remains an alternative treatment (see Evans et al., 1997). Neither the 'cystic' nor 'alveolar' hydatid diseases caused by larval stages of Echinococcus tapeworms respond to praziquantel; here too, albendazole is effective (see Horton, 1997; Schantz, 1999). Toxicity, Precautions, and Interactions Abdominal discomfort, particularly pain and nausea, headache, dizziness, and drowsiness may occur shortly after taking praziquantel; these direct effects are transient and dose-related. Indirect effects such as fever, pruritus, urticaria, rashes, arthralgia, and myalgia are noted occasionally. Such side effects and increases in eosinophilia often relate to parasite burden. In neurocysticercosis, inflammatory reactions to praziquantel may produce meningismus, seizures, mental changes, and cerebrospinal fluid pleocytosis. These effects usually are delayed in onset, last 2 to 3 days, and respond to appropriate symptomatic therapy such as analgesics and anticonvulsants. Praziquantel is considered safe in children over 4 years of age, who probably tolerate the drug better than do adults. Low levels of the drug appear in the maternal milk, but there is no evidence that this compound is mutagenic or carcinogenic. High doses of praziquantel do increase abortion rates in rats, so the drug probably is best avoided during human pregnancy. The bioavailability of praziquantel is reduced by inducers of hepatic cytochromes P450 such as carbamazepine and phenobarbital; predictably, coadministration of the cytochrome P450 inhibitor cimetidine has the opposite effect (Bittencourt et al., 1992; Dachman et al., 1994). Dexamethasone reduces the bioavailability of praziquantel, but the mechanism is not understood. Under certain conditions, praziquantel may increase the bioavailability of albendazole (Homeida et al., 1994). Praziquantel is contraindicated in ocular cysticercosis, because the host response can produce irreversible damage to the eye. Shortly after taking the drug, driving, operating machinery, and other tasks requiring mental alertness should be avoided. The half-life of praziquantel can be prolonged in patients with severe hepatic disease; dosage adjustment in such patients may be required (Mandour et al., 1990). Pyrantel Pamoate Pyrantel pamoate ANTIMINTH, others) was introduced first into veterinary practice as a broad-spectrum anthelmintic directed against pinworm, roundworm, and hookworm infections (Austin et al., 1966). Its effectiveness and lack of toxicity led to its trial against related intestinal helminths in human beings (Bumbalo et al., 1969). Oxantel pamoate, an m-oxyphenol analog of pyrantel, is effective for single-dose treatment of trichuriasis. Pyrantel is employed as the pamoate salt. It has the following chemical structure:
Pyrantel and its analogs are depolarizing neuromuscular blocking agents. They open nonselective cation channels and induce marked, persistent activation of nicotinic acetylcholine receptors, which results in spastic paralysis of the worm (Robertson et al., 1994). Pyrantel also inhibits cholinesterases. It causes a slowly developing contracture of preparations of Ascaris at 1% of the concentration of acetylcholine required to produce the same effect. In single muscle cells of this helminth, pyrantel causes depolarization and increased spike-discharge frequency, accompanied by increase in tension. Pyrantel is effective against hookworm, pinworm, and roundworm; unlike its analog oxantel, it is ineffective against T. trichiura. Pyrantel pamoate is poorly absorbed from the gastrointestinal tract, a property that contributes to its selective action on gastrointestinal nematodes. Less than 15% is excreted in the urine as parent drug and metabolites. The major proportion of an administered dose is recovered in the feces. Pyrantel pamoate is an alternative to mebendazole in the treatment of ascariasis and enterobiasis. High cure rates have been achieved after a single oral dose of 11 mg/kg, to a maximum of 1 gram. Pyrantel also is effective against hookworm infections caused by A. duodenale and N. americanus, although repeated doses are needed to cure heavy infections with N. americanus. The drug should be used in combination with oxantel for mixed infections with T. trichiura. In the case of pinworm, it is wise to repeat the treatment after an interval of 2 weeks. When given parenterally, pyrantel can produce complete neuromuscular blockade in animals; if given orally, toxic effects are produced only by very large doses. Transient and mild gastrointestinal symptoms are occasionally observed in human beings, as are headache, dizziness, rash, and fever. Pyrantel pamoate has not been studied in pregnant women. Thus, its use in pregnant patients and children less than 2 years of age is not recommended. Because pyrantel pamoate and piperazine are mutually antagonistic with respect to their neuromuscular effects on parasites, the two should not be used together. |
Prospectus
|
Eradication of helminthiasis is highly unlikely because of its close association with human poverty. Although these infections are both common and cosmopolitan, their subtle clinical course encourages neglect until disease becomes obvious. Helminthiasis flourishes especially in warm environments marked by inadequate sanitation, parasitized reservoirs and vectors, and contaminated food and water sources. Affluence, however, does not protect against these infections; young or debilitated individuals are particularly vulnerable regardless of socioeconomic status. Chemotherapy now provides the single most efficient, practical, and inexpensive strategy to control helminth infections. Moreover, it still will be needed even if effective vaccines become available. To achieve optimal results, chemotherapy must be combined with other measures such as improved sanitation and vector and reservoir control. Even for chemotherapy itself to succeed, there must be a felt need for the drug regimen, a suitable delivery infrastructure for it, and appropriate follow-up to ensure compliance. Periodic treatment with effective agents can reduce the parasite burden to the point where transmission is affected, because few helminth species multiply within the human host. A major challenge is to develop better drugs against systemic infections with helminths that respond inadequately to current compounds, i.e., the filariases, echinococcosis, fascioliasis, trichinosis, toxocariasis, and cysticercosis. Discovery of agents effective against all developmental stages of parasitic helminths, e.g., adult filarial worms instead of just microfilariae, would constitute a major advance in this context. That most effective anthelmintics have been in use for years invites eventual development of resistance to these drugs. Although the emergence of drug-resistant helminths has not yet become a problem for treatment of infections in human beings, resistance to benzimidazoles and avermectins is well established in animal infections. The specter of drug resistance mandates the search for improved anthelmintics, even for therapy of infections that now respond adequately to available drugs. Because of lack of economic incentives, scientific knowledge relevant to drug discovery is likely to outpace drug development in this field. Broad applications to helminths of modern techniques such as genome sequencing projects and proteomics together with other approaches should accelerate identification and characterization of drug receptors and increase understanding of mechanisms of drug resistance. Understanding the relationship between avermectin resistance of helminths in vivo to their cloned avermectin-modulated, glutamate-gated Cl channel 'receptors' is a case in point. Rapid automatic screening of compounds for reactivity with essential parasite macromolecules in vitro also is feasible, so components with anthelmintic activity can be identified readily in large libraries of structurally diverse synthetic compounds. Barring development of novel drugs, what other pharmacological measures might be taken to control helminth infections of human beings? One strategy would be to improve the use of available anthelmintics. Indeed, introduction of diethylcarbamazine into table salt already has modulated lymphatic filariasis, and periodic use of anthelmintics such as praziquantel for schistosomiasis and either diethylcarbamazine or ivermectin for lymphatic filariasis has reduced the parasite burden, rate of transmission, and severity of disease in these infections. Empirical dosage regimens must undergo continuing clinical evaluation; this process already has resulted in the reduction of doses of diethylcarbamazine and ivermectin used to control the filariases. Concomitant use of current drugs with different mechanisms of action against the same organism represents a major advance in anthelmintic chemotherapy. This concept is aptly illustrated by the periodic use of albendazole together with either diethylcarbamazine or ivermectin for successful control of lymphatic filariasis. Human populations coinfected with different classes of helminths should benefit from periodic therapy with two or more broad-spectrum drugs, each with a different anthelmintic profile. This approach already has had success, i.e., albendazole and ivermectin, used together to control lymphatic filariasis, are highly effective against intestinal nematodes. Praziquantel, ivermectin, and albendazole together have been advocated for periodic treatment of populations infected with several species of helminths. At far less cost than required for development of novel drugs, profitable drugs used for veterinary purposes or even approved for other clinical indications in human beings could improve the control of human helminthiasis. Indeed, the avermectins and benzimidazoles were developed initially for veterinary use, and both ivermectin and albendazole have been donated by their respective manufacturers for control of human helminth infections. Limited studies have shown that the veterinary benzimidazole, triclabendazole, is probably the most effective known drug against fascioliasis in human beings. Moxidectin, a veterinary drug related to the avermectins, shows macrofilaricidal activity against onchocerciasis and lymphatic filariasis and is now ready for evaluation in human beings. A veterinary formulation of ivermectin administered subcutaneously to bypass hepatic metabolism might achieve higher plasma levels and greater efficacy against certain human systemic infections with marginally drug-responsive helminths. Moreover, animal models of helminthic infections provide a powerful approach to explore mechanisms of anthelmintic drug resistance. Ultimately, society will be forced to pay the high cost of developing novel anthelmintic drugs for human use. The technology is either available now or soon will be. Both drugs and vaccines may be needed to combat helminth infections that now exact a heavy toll on human health and productivity. For further discussion of helminth infections, see Chapters 192,
193, and 200204 in Harrison's Principles of Internal Medicine, 16th
ed., |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 2386
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved