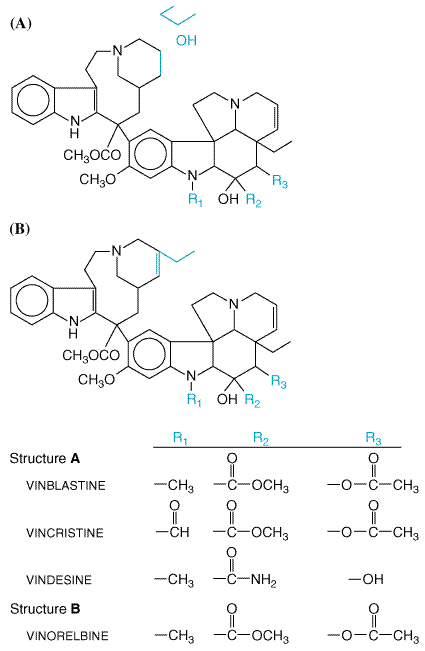
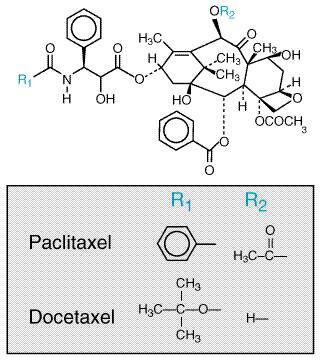
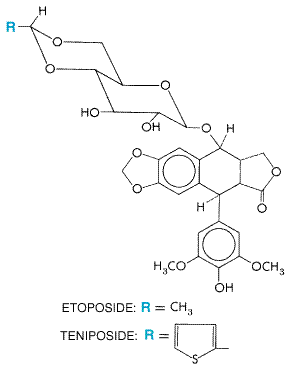
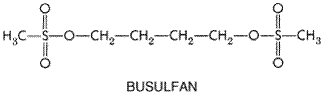| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Antineoplastic Agents
Alkylating Agents
|
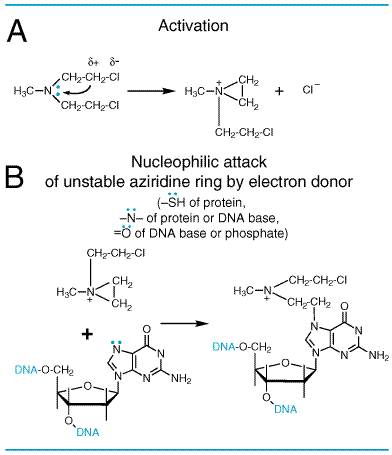
History Although synthesized in 1854, the vesicant properties of sulfur mustard were not described until 1887. During World War I, medical attention was first focused on the vesicant action of sulfur mustard on the skin, eyes, and respiratory tract. It was appreciated later, however, that serious systemic toxicity also follows exposure. In 1919, Krumbhaar and Krumbhaar made the pertinent observation that the poisoning caused by sulfur mustard is characterized by leukopenia and, in cases that came to autopsy, by aplasia of the bone marrow, dissolution of lymphoid tissue, and ulceration of the gastrointestinal tract. In the interval between World Wars I and II, extensive studies of the biological and chemical actions of the nitrogen mustards were conducted. The marked cytotoxic action on lymphoid tissue prompted Gilman, Goodman, and T.F. Dougherty to study the effect of nitrogen mustards on transplanted lymphosarcoma in mice, and in 1942 clinical studies were initiated. This launched the era of modern cancer chemotherapy (Gilman, 1963). In their early phases, all these investigations were conducted under secrecy restrictions imposed by the use of classified chemical-warfare agents. At the termination of World War II, however, the nitrogen mustards were declassified; a general review was presented by Gilman and Philips (1946). A more recent review is provided by Ludlum and Tong (1985). Thousands of variants of the basic chemical structure of the nitrogen mustards have been prepared, but only a few of these agents have proven more useful than the original compound in specific clinical circumstances (see below). At present five major types of alkylating agents are used in the chemotherapy of neoplastic diseases: (1) the nitrogen mustards, (2) the ethylenimines, (3) the alkyl sulfonates, (4) the nitrosoureas, and (5) the triazenes. Chemistry The chemotherapeutic alkylating agents have in common the property of becoming strong electrophiles through the formation of carbonium ion intermediates or of transition complexes with the target molecules. These reactions result in the formation of covalent linkages by alkylation of various nucleophilic moieties such as phosphate, amino, sulfhydryl, hydroxyl, carboxyl, and imidazole groups. The chemotherapeutic and cytotoxic effects are directly related to the alkylation of DNA. The 7 nitrogen atom of guanine is particularly susceptible to the formation of a covalent bond with bifunctional alkylating agents and may well represent the key target that determines their biological effects. It must be appreciated, however, that other atoms in the purine and pyrimidine bases of DNAparticularly, the 1 and 3 nitrogens of adenine, the 3 nitrogen of cytosine, and the 6 oxygen of guaninealso may be alkylated, as will be the phosphate atoms of the DNA chains and amino and sulfhydryl groups of proteins. To illustrate the actions of alkylating agents, possible consequences of the reaction of mechlorethamine (nitrogen mustard) with guanine residues in DNA chains are shown in Figure 521. First, one 2-chloroethyl side chain undergoes a first-order (SN1) intramolecular cyclization, with release of Cl and formation of a highly reactive ethyleniminium intermediate (Figure 521A). By this reaction, the tertiary amine is converted to an unstable quaternary ammonium compound, which can react avidly, through formation of a carbonium ion or transition complex intermediate, with a variety of sites that possess high electron density. This reaction proceeds as a second-order (SN2) nucleophilic substitution. Alkylation of the 7 nitrogen of guanine residues in DNA (Figure 521B), a highly favored reaction, may exert several effects of considerable biological importance. Normally, guanine residues in DNA exist predominantly as the keto tautomer and readily make WatsonCrick base pairs by hydrogen bonding with cytosine residues. However, when the 7 nitrogen of guanine is alkylated (to become a quaternary ammonium nitrogen), the guanine residue is more acidic and the enol tautomer is favored. The modified guanine can mispair with thymine residues during DNA synthesis, leading to the substitution of an adeninethymine base pair for a guaninecytosine base pair. Second, alkylation of the 7 nitrogen labilizes the imidazole ring, making possible the opening of the imidazole ring or depurination by excision of guanine residues. Either of these seriously damages the DNA molecule and must be repaired. Third, with bifunctional alkylating agents, such as nitrogen mustard, the second 2-chloroethyl side chain can undergo a similar cyclization reaction and alkylate a second guanine residue or another nucleophilic moiety, resulting in the cross-linking of two nucleic acid chains or the linking of a nucleic acid to a protein, alterations that would cause a major disruption in nucleic acid function. Any of these effects could adequately explain both the mutagenic and the cytotoxic effects of alkylating agents. However, cytotoxicity of bifunctional alkylators correlates very closely with interstrand cross-linkage of DNA (Garcia et al., 1988).
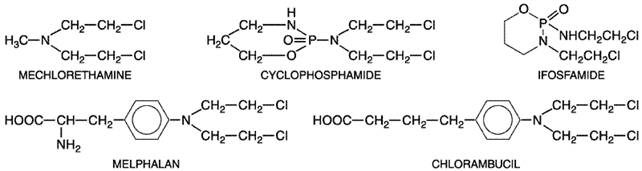
The ultimate cause of cell death related to DNA damage is not known. Specific cellular responses include cell-cycle arrest, DNA repair, and apoptosis, a specific form of nuclear fragmentation termed programmed cell death (Fisher, 1994). The p53 gene product senses DNA damage and initiates apoptosis in response to DNA alkylation. Mutations of p53 lead to alkylating-agent resistance (Kastan, 1999). All nitrogen mustards are chemically unstable but vary greatly in their degree of instability. Therefore, the specific chemical properties of each member of this class of drugs must be considered individually in therapeutic applications. For example, mechlorethamine is very unstable, and it reacts almost completely in the body within a few minutes of its administration. By contrast, agents such as chlorambucil are sufficiently stable to permit oral administration. Cyclophosphamide requires biochemical activation by the cytochrome P450 system of the liver before its cytotoxicity becomes evident. The ethylenimine derivatives such as chlorambucil and melphalan react by an SN2 reaction; since the opening of the ethylenimine intermediate is acid-catalyzed, they are more reactive at acidic pH. StructureActivity Relationship The alkylating agents used in chemotherapy encompass a diverse group of chemicals that have in common the capacity to contribute, under physiological conditions, alkyl groups to biologically vital macromolecules such as DNA. In most instances, physical and chemical parameters, such as lipophilicity, capacity to cross biological membranes, acid dissociation constants, stability in aqueous solution, and sites of macromolecular attack, determine drug activity in vivo. With several of the most valuable agents (e.g., cyclophosphamide and the nitrosoureas), the active alkylating moieties are generated in vivo after complex metabolic reactions. The nitrogen mustards may be regarded as nitrogen analogs of sulfur mustard. The biological activity of both types of compounds is based upon the presence of the bis-(2-chloroethyl) grouping. While mechlorethamine has been widely used in the past, various structural modifications have resulted in compounds with greater selectivity and stability and therefore less toxicity. Bis-(2-chloroethyl) groups have been linked to amino acids (phenylalanine), substituted phenyl groups (aminophenyl butyric acid, as in chlorambucil), pyrimidine bases (uracil), and other chemical entities in an effort to make a more stable and orally available form. Although none of these modifications has produced an agent highly selective for malignant cells, some have unique pharmacological properties and are more useful clinically than is mechlorethamine. Their structures are shown in Figure 522.
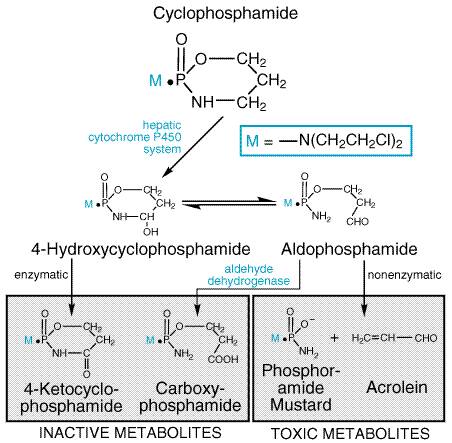
The addition of substituted phenyl groups has produced a series of relatively stable derivatives that retain the ability to form reactive charged intermediates; the electron-withdrawing capacity of the aromatic ring greatly reduces the rate of cyclization and carbonium ion formation, and these compounds therefore can reach distant sites in the body before reacting with components of blood and other tissues. Chlorambucil and melphalan are the most successful examples of such aromatic mustards. These compounds can be administered orally if desired. A classical example of the role of host metabolism in the activation of an alkylating agent is seen with cyclophosphamidenow the most widely used agent of this class. The design of this molecule was based on two considerations. First, if a cyclic phosphamide group replaced the N-methyl of mechlorethamine, the compound might be relatively inert, presumably because the bis-(2-chloroethyl) group of the molecule could not ionize until the cyclic phosphamide was cleaved at the phosphorusnitrogen linkage. Second, it was hoped that neoplastic tissues might possess high phosphatase or phosphamidase activity capable of accomplishing this cleavage, thus resulting in the selective production of an activated nitrogen mustard in the malignant cells. In accord with these predictions, the parent cyclophosphamide displays only weak cytotoxic, mutagenic, or alkylating activity in vitro and is relatively stable in aqueous solution. However, when administered to experimental animals or patients bearing susceptible tumors, it causes marked chemotherapeutic effects, as well as mutagenicity and carcinogenicity. The postulated role for phosphatases or phosphamidases in the mechanism of action of cyclophosphamide has proven incorrect. Rather, the drug undergoes metabolic activation (hydroxylation) by the cytochrome P450 mixed-function oxidase system of the liver (Figure 523), with subsequent transport of the activated intermediate to sites of action, as discussed below. The selectivity of cyclophosphamide against certain malignant tissues may result in part from the capacity of normal tissues, such as liver, to protect themselves against cytotoxicity by further degrading the activated intermediates via aldehyde dehydrogenase and other pathways.
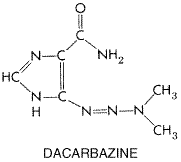
Ifosfamide is an oxazaphosphorine, similar to cyclophosphamide. Cyclophosphamide has two chloroethyl groups on the exocyclic nitrogen atom, whereas one of the two chloroethyl groups of ifosfamide is on the cyclic phosphamide nitrogen of the oxazaphosphorine ring. Like cyclophosphamide, ifosfamide is activated in the liver by hydroxylation. However, the activation of ifosfamide proceeds more slowly, with greater production of dechlorinated metabolites and chloroacetaldehyde. These differences in metabolism likely account for the higher doses of ifosfamide required for equitoxic effects and the possible differences in antitumor spectrum of the two agents. Although initially considered an antimetabolite, the triazene derivative 5-(3,3-dimethyl-1-triazeno)-imidazole-4-carboxamide, usually referred to as dacarbazine or DTIC, functions through alkylation. Its structural formula is shown below:
Dacarbazine requires initial activation by the cytochrome P450 system
of the liver through an N-demethylation reaction. In the target cell,
spontaneous cleavage of the metabolite yields an alkylating moiety,
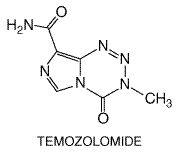
diazomethane. A related triazene, temozolomide undergoes spontaneous
activation, and has significant activity against gliomas and melanoma in
human beings (Agarwala and
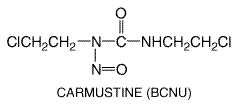
The nitrosoureas, which include compounds such as 1,3-bis-(2-chloroethyl)-1-nitrosourea (carmustine, BCNU), 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea (lomustine, CCNU), and its methyl derivative (semustine, methyl-CCNU), as well as the antibiotic streptozocin (streptozotocin), exert their cytotoxicity through the spontaneous breakdown to alkylating and carbamoylating moieties. The structural formula of carmustine is as follows:
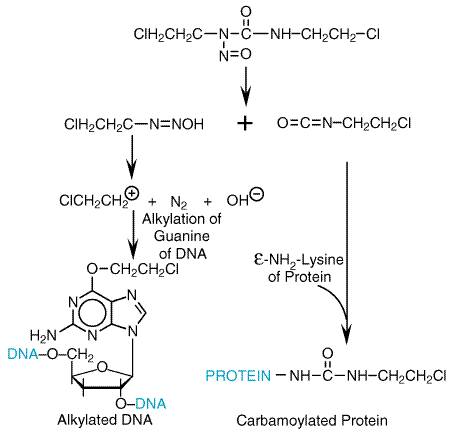
The antineoplastic nitrosoureas have in common the capacity to undergo spontaneous, nonenzymatic degradation with the formation of the 2-chloroethyl carbonium ion (from CNU compounds). This strong electrophile can alkylate a variety of substances; guanine, cytidine, and adenine adducts have been identified (Ludlum, 1990). Displacement of the halogen atom can then lead to interstrand or intrastrand cross-linking of the DNA. The formation of the cross-links after the initial alkylation reaction is relatively slow and can be interrupted by the DNA repair enzyme guanine O6-alkyl transferase (Dolan et al., 1990). The same enzyme, when overexpressed in gliomas, produces resistance to nitrosoureas and various methylating agents, including DTIC, temozolomide, and procarbazine. As with the nitrogen mustards, it is generally agreed that interstrand cross-linking is associated with the cytotoxicity of nitrosoureas (Hemminki and Ludlum, 1984). In addition to the generation of carbonium ions, the spontaneous degradation of BCNU, CCNU, and methyl-CCNU liberates organic isocyanates that attach carbamoyl groups to lysine residues of proteins, a reaction that apparently can inactivate certain DNA repair enzymes. The reactions of the nitrosoureas with macromolecules are shown in Figure 524.
Since the formation of the ethyleniminium ion constitutes the initial reaction of the nitrogen mustards, it is not surprising that stable ethylenimine derivatives have antitumor activity. Several compounds of this type, including triethylenemelamine (TEM) and triethylene thiophosphoramide (thiotepa), have been used clinically. In standard doses, thiotepa produces little toxicity other than myelosuppression and is thus increasingly used for high-dose chemotherapy regimens. Altretamine (hexamethylmelamine; HMM) is mentioned here because of its chemical similarity to TEM. The methylmelamines are N-demethylated by hepatic microsomes, with the release of formaldehyde, and there is a relationship between the degree of the demethylation and their activity against murine tumors. Altretamine requires microsomal activation to display cytotoxicity (Friedman, 2001). Several interesting compounds have emerged from a large group of esters of alkanesulfonic acids. One of these, busulfan, is of value in the treatment of chronic granulocytic leukemia and in high-dose chemotherapy; its structural formula is as follows:
Busulfan is a member of a series of symmetrical bis-substituted methanesulfonic acid esters in which the length of a bridge of methylene varies from 2 to 10. The compounds of intermediate length (n= 4 or 5) possess the highest activities and therapeutic indices. Cross-linked guanine residues have been identified in DNA incubated in vitro with busulfan (Tong and Ludlum, 1980). Pharmacological Actions The pharmacological actions of the various groups of alkylating agents are considered together in the following discussion. Although there are many similarities, some notable differences also are evident. Cytotoxic Actions The most important pharmacological actions of the alkylating agents are those that disturb DNA synthesis and cell division. The capacity of these drugs to interfere with DNA integrity and function in rapidly proliferating tissues provides the basis for their therapeutic applications and for many of their toxic properties. Whereas certain alkylating agents may have damaging effects on tissues with normally low mitotic indicesfor example, liver, kidney, and mature lymphocytesthey are most cytotoxic to rapidly proliferating tissues in which a large proportion of the cells are in division. These compounds may readily alkylate nondividing cells, but cytotoxicity is markedly enhanced if DNA is damaged in cells programmed to divide. Thus, DNA alkylation itself may not be a lethal event if DNA repair enzymes can correct the lesions in DNA prior to the next cellular division. In contrast to many other antineoplastic agents, the effects of the alkylating drugs, although dependent on proliferation, are not cell-cyclespecific, and the drugs may act on cells at any stage of the cycle. However, the toxicity is usually expressed when the cell enters the S phase and progression through the cycle is blocked. While not strictly cell-cyclespecific, quantitative differences may be detected when nitrogen mustards are applied to synchronized cells at different phases of the cycle. Cells appear more sensitive in late G1 or S than in G2, mitosis, or early G1. Polynucleotides are more susceptible to alkylation in the unpaired state than in the helical form; during replication of DNA, portions of the molecule are unpaired. The actual mechanism(s) of cell death related to DNA alkylation are not well understood. There is evidence that, in normal cells of the bone marrow and intestinal epithelium, DNA damage activates a checkpoint dependent on the presence of a normal p53 gene. Cells thus blocked in the G1/S interface either repair DNA alkylation or undergo apoptosis. Malignant cells with mutant or absent p53 fail to suspend cell-cycle progression and do not undergo apoptosis (Fisher, 1994). The great preponderance of evidence indicates that the primary target of pharmacological doses of alkylating agents is DNA, as illustrated in Figure 521. A crucial distinction that must be emphasized is between the bifunctional agents, in which cytotoxic effects predominate, and the monofunctional methylating agents (procarbazine, temozolomide), which, although cytotoxic, have greater capacity for mutagenesis and carcinogenesis. This suggests that the cross-linking of DNA strands represents a much greater threat to cellular survival than do other effects, such as single-base alkylation and the resulting depurination and chain scission. On the other hand, the latter reactions may cause permanent modifications in DNA structure and sequence that are compatible with continued life of the cell and are transmissible to subsequent generations; such modifications may result in mutagenesis or carcinogenesis. The remarkable DNA repair systems found in most cells likely play an important but as yet poorly defined role in the relative resistance of nonproliferating tissues, the selectivity of action against particular cell types, and acquired resistance to alkylating agents. Although alkylation of a single strand of DNA often may be repaired with relative ease, interstrand cross-linkages, such as those produced by the bifunctional alkylating agents, require more complex mechanisms for repair. Many of the cross-links formed in DNA by these agents at low doses also may be corrected; higher doses cause extensive cross-linkage, and DNA breakdown occurs. Specific repair enzymes for removing alkyl groups from the O-6 of guanine (guanine O6-alkyl transferase) and the N-3 of adenine and N-7 of guanine (3-methyladenine-DNA glycosylase) have been identified (Matijasevic et al., 1993). The presence of sufficient levels of guanine O6-alkyl transferase protects cells from cytotoxic effects of nitrosoureas and methylating agents (Pegg, 1990) and confers drug resistance. Detailed information is lacking on mechanisms of cellular uptake of alkylating agents. Mechlorethamine appears to enter murine tumor cells by means of an active transport system, the natural substrate of which is choline. Melphalan, an analog of phenylalanine, is taken up by at least two active transport systems that normally react with leucine and other neutral amino acids. The highly lipophilic drugs, including nitrosoureas, carmustine, and lomustine, diffuse into cells passively. Mechanisms of Resistance to Alkylating Agents Acquired resistance to alkylating agents is a common event, and the acquisition of resistance to one alkylating agent often but not always imparts cross-resistance to others; thus, there are at least theoretical reasons to combine alkylating agents in high-dose therapy. While definitive information on the biochemical mechanisms of clinical resistance is lacking, specific biochemical changes have been implicated in the development of such resistance by tumor cells. Among these changes are (1) decreased permeation of actively transported drugs (mechlorethamine and melphalan); (2) increased production of nucleophilic substances, principally thiols such as glutathione, that can conjugate with and detoxify electrophilic intermediates; (3) increased activity of the DNA repair enzymes, such as the guanine O6-alkyl transferase, that repair nitrosourea-produced alkylation; and (4) increased rates of metabolism of the activated forms of cyclophosphamide to its inactive keto and carboxy metabolites by aldehyde dehydrogenase (see Figure 523; Tew et al., 2001). To reverse cellular changes that lead to resistance, strategies have been devised and appear to be effective in selected experimental tumors. These include the use of compounds that deplete glutathione, such as L-buthionine-sulfoximine; sulfhydryl compounds, such as WR-2721, that selectively detoxify alkylating species in normal cells and thereby prevent toxicity; compounds such as O6-benzylguanine that inactivate the guanine O6-alkyl transferase DNA repair enzyme; and compounds such as ethacrynic acid that inhibit the enzymes (glutathione transferases) that conjugate thiols with alkylating agents. While each of these modalities has experimental evidence to support its use, the clinical efficacy has not yet been proven for these strategies. Of these, O6-benzylguanine has advanced to phase II trials used in conjunction with carmustine (BCNU) or procarbazine against malignant gliomas (Schilsky et al., 2000). Toxicities of Alkylating Agents The alkylating agents differ in their patterns of antitumor activity and in the sites and severity of their side effects. Most cause dose-limiting toxicity to bone marrow elements and, to a lesser extent, intestinal mucosa. Most alkylating agents, including nitrogen mustard, melphalan, chlorambucil, cyclophosphamide, and ifosfamide, produce an acute myelosuppression, with a nadir of the peripheral blood granulocyte count at 6 to 10 days and recovery in 14 to 21 days. Cyclophosphamide has lesser effects on peripheral blood platelet counts than do the other agents. Busulfan suppresses all blood elements, particularly stem cells, and may produce a prolonged and cumulative myelosuppression lasting months. For this reason, it is used as a preparative regimen in allogenic bone marrow transplantation. BCNU and other chloroethylnitrosoureas cause delayed and prolonged suppression of both platelets and granulocytes, reaching a nadir 4 to 6 weeks after drug administration and reversing slowly thereafter. Both cellular and humoral immunity are suppressed by alkylating agents, which have been used to treat various autoimmune diseases. Immunosuppression is reversible at doses used in most anticancer protocols. In addition to effects on the hematopoietic system, alkylating agents are highly toxic to dividing mucosal cells, leading to oral mucosal ulceration and intestinal denudation. The mucosal effects are particularly significant in high-dose chemotherapy protocols associated with bone marrow reconstitution, as they predispose to bacterial sepsis arising from the gastrointestinal tract. In these protocols, melphalan and thiotepa have the advantage of causing less mucosal damage than the other agents. In high-dose protocols, a number of toxicities not seen at conventional doses become dose-limiting. They are listed in Table 521. While mucosal and bone marrow toxicities occur predictably with conventional doses of these drugs, other organ toxicities, although less common, can be irreversible and at times lethal. All alkylating agents have caused pulmonary fibrosis, and in high-dose regimens, endothelial damage that may precipitate venoocclusive disease of the liver; the nitrosoureas, after multiple cycles of therapy, may lead to renal failure; ifosfamide in high-dose regimens frequently causes a central neurotoxicity, with seizures, coma, and at times death; and all such agents are leukemogenic, particularly procarbazine (a methylating agent) and the nitrosoureas. Cyclophosphamide and ifosfamide release a nephrotoxic and urotoxic metabolite, acrolein, which causes a severe hemorrhagic cystitis, a side effect that in high-dose regimens can be prevented by coadministration of the sulfhydryl-releasing agent mesna (2-mercaptoethanesulfonate). Mesna, when administered with the offending agent at 60% of the drug dosage, conjugates toxic metabolites in urine. The more unstable alkylating agents (particularly nitrogen mustard and the nitrosoureas) have strong vesicant properties, damage veins with repeated use, and, if extravasated, produce ulceration. Topical application of nitrogen mustard is an effective treatment for cutaneous neoplasms such as mycosis fungoides. Most alkylating agents cause alopecia. Central nervous system (CNS) toxicity is manifest in the form of nausea and vomiting, particularly after intravenous administration of nitrogen mustard or BCNU. Ifosfamide is the most neurotoxic of this class of agents, producing altered mental status, coma, generalized seizures, and paralysis. These side effects have been linked to the release of chloroacetaldehyde from the phosphate-linked chloroethyl side chain of ifosfamide. High-dose busulfan may cause seizures; in addition, it accelerates the clearance of phenytoin, an antiseizure medication (see Chapter 21: Drugs Effective in the Therapy of the Epilepsies). As a class of drugs, the alkylating agents are highly leukemogenic. Acute nonlymphocytic leukemia, often associated with partial or total deletions of chromosome 5 or 7, peaks in incidence about four years after therapy and may affect up to 5% of patients treated on regimens containing alkylating drugs (Levine and Bloomfield, 1992). Melphalan, the nitrosoureas, and the methylating agent procarbazine have the greatest propensity to cause leukemia, while cyclophosphamide is less potent in this regard. Finally, all alkylating agents have toxic effects on the male and female reproductive systems, causing an often permanent amenorrhea, particularly in perimenopausal women, and an irreversible azoospermia in men. Nitrogen Mustards The chemistry and the pharmacological actions of the alkylating agents as a group, and of the nitrogen mustards, have been presented above. Only the unique pharmacological characteristics of the individual agents are considered below. Mechlorethamine Mechlorethamine, the first nitrogen mustard to be introduced into clinical medicine, is the most reactive of the drugs in this class. Absorption and Fate Severe local reactions of exposed tissues necessitate intravenous injection of mechlorethamine for most clinical uses. In either water or body fluids, at rates affected markedly by pH, mechlorethamine rapidly undergoes chemical transformation and combines with either water or nucleophilic molecules of cells, so that the parent drug has an extremely short mean residence time in the body. Therapeutic Uses Mechlorethamine HCl MUSTARGEN) is used primarily in the combination chemotherapy regimen MOPP [mechlorethamine, ONCOVIN (vincristine), procarbazine, and prednisone] in patients with Hodgkin's disease (DeVita et al., 1972). It is given by intravenous bolus administration in doses of 6 mg/m2 on days 1 and 8 of the 28-day cycles of each course of treatment. It has been largely replaced in other regimens by cyclophosphamide, melphalan, and other, more stable, alkylating agents. Clinical Toxicity The major acute toxic manifestations of mechlorethamine are nausea, vomiting, and lacrimation as well as myelosuppression. Leukopenia and thrombocytopenia limit the amount of drug that can be given in a single course. Like other alkylating agents, nitrogen mustard blocks reproductive function and may produce menstrual irregularities or premature menopause in women and oligospermia in men. Since fetal abnormalities can be induced, this drug as well as other alkylating agents should not be used in the first trimester of pregnancy and should be used with caution in later stages of pregnancy. Breast-feeding should be terminated before therapy with mechlorethamine is initiated. Local reactions to extravasation of mechlorethamine into the subcutaneous tissue result in a severe, brawny, tender induration that may persist for a long time. If the local reaction is unusually severe, a slough may result. If it is obvious that extravasation has occurred, the involved area should be promptly infiltrated with a sterile isotonic solution of sodium thiosulfate (1/6 M); an ice compress then should be applied intermittently for 6 to 12 hours. Thiosulfate provides an ion that reacts avidly with the nitrogen mustard and thereby protects tissue constituents. Cyclophosphamide Pharmacological and Cytotoxic Actions Although the general cytotoxic action of this drug is similar to that of other alkylating agents, there are notable differences. Thrombocytopenia is less severe, while alopecia is marked. There are no severe acute or delayed central nervous system (CNS) manifestations either in conventional doses or in high-dose regimens. Nausea and vomiting, however, may occur. The drug is not a vesicant, and there is no local irritation. Absorption, Fate, and Excretion Cyclophosphamide is well absorbed orally. As mentioned above, the drug is activated by the hepatic cytochrome P450 system (see Figure 523). Cyclophosphamide is first converted to 4-hydroxycyclophosphamide, which is in a steady state with the acyclic tautomer aldophosphamide. In vitro studies with human liver microsomes and cloned P450 isoenzymes have shown that cyclophosphamide is activated by the CYP2B group of P450 isoenzymes, while a closely related oxazaphosphorine, ifosfamide, is hydroxylated by the CYP3A system (Chang et al., 1993). This difference may account for the somewhat different patterns of antitumor activity, the slower activation of ifosfamide in vivo, and the interpatient variability in toxicity of these two closely related molecules. 4-Hydroxycyclophosphamide may be oxidized further by aldehyde oxidase either in liver or in tumor tissue and perhaps by other enzymes, yielding the metabolites carboxyphosphamide and 4-ketocyclophosphamide, neither of which possesses significant biological activity. It appears that hepatic damage is minimized by these secondary reactions, whereas significant amounts of the active metabolites, such as 4-hydroxycyclophosphamide and its tautomer, aldophosphamide, are transported to the target sites by the circulatory system. In tumor cells, the aldophosphamide cleaves spontaneously, generating stoichiometric amounts of phosphoramide mustard and acrolein. The former is believed to be responsible for antitumor effects. The latter compound may be responsible for the hemorrhagic cystitis seen during therapy with cyclophosphamide. Cystitis can be reduced in intensity or prevented by the parenteral administration of mesna (MESNEX), a sulfhydryl compound that reacts readily with acrolein in the acid environment of the urinary tract (Tew et al., 2001). Pretreatment with P450 inducers such as phenobarbital enhances the rate of drug activation but does not alter toxicity or therapeutic activity in human beings. Urinary and fecal recovery of unchanged cyclophosphamide is minimal after intravenous administration. Maximal concentrations in plasma are achieved 1 hour after oral administration, and the half-life in plasma is about 7 hours. Therapeutic Uses Cyclophosphamide CYTOXAN, NEOSAR) is administered orally or intravenously. Recommended doses vary widely, and published protocols for the dosage of cyclophosphamide and other chemotherapeutic agents and for the method and sequence of administration should be consulted. As a single agent, a daily dose of 100 mg/m2 orally for 14 days has been recommended for patients with more susceptible neoplasms, such as lymphomas and chronic leukemias. A higher dosage of 500 mg/m2 intravenously every 3 to 4 weeks in combination with other drugs often is employed in the treatment of breast cancer and lymphomas. The leukocyte count generally serves as a guide to dosage adjustments in prolonged therapy. An absolute neutrophil count between 500 and 1000 cells per cubic millimeter is recommended as the desired target. In regimens associated with bone marrow or peripheral stem cell rescue, cyclophosphamide may be given in doses of 5 to 7 g/m2 over a 3-day period. Gastrointestinal ulceration, cystitis (counteracted by mesna and diuresis), and, less commonly, pulmonary, renal, hepatic, and cardiac toxicities may occur after high-dose therapy. The clinical spectrum of activity for cyclophosphamide is very broad. It is an essential component of many effective drug combinations for non-Hodgkin's lymphomas. Complete remissions and presumed cures have been reported when cyclophosphamide was given as a single agent for Burkitt's lymphoma. It is frequently used in combination with methotrexate (or doxorubicin) and fluorouracil as adjuvant therapy after surgery for carcinoma of the breast. Notable advantages of this drug are the availability of the oral route of administration and the possibility of giving fractionated doses over prolonged periods. For these reasons it possesses a versatility of action that allows an intermediate range of use, between that of the highly reactive intravenous mechlorethamine and that of oral chlorambucil. Beneficial results have been obtained in multiple myeloma; chronic lymphocytic leukemia; carcinomas of the lung, breast, cervix, and ovary; and neuroblastoma, retinoblastoma, and other neoplasms of childhood. Because of its potent immunosuppressive properties, cyclophosphamide has received considerable attention for the control of organ rejection after transplantation and in nonneoplastic disorders associated with altered immune reactivity, including Wegener's granulomatosis, rheumatoid arthritis, and the nephrotic syndrome in children. Caution is advised when the drug is considered for use in these conditions, not only because of its acute toxic effects but also because of its potential for inducing sterility, teratogenic effects, and leukemia. Clinical Toxicity Nausea and vomiting, myelosuppression with platelet sparing, and alopecia are common to virtually all regimens using cyclophosphamide. Mucosal ulcerations and, less frequently, interstitial pulmonary fibrosis also may result from cyclophosphamide treatment. Extravasation of the drug into subcutaneous tissues does not produce local reactions, and thrombophlebitis does not complicate intravenous administration. The occurrence of sterile hemorrhagic cystitis has been reported in 5% to 10% of patients. As noted above, this has been attributed to chemical irritation produced by acrolein. Its incidence is significantly reduced by coadministration of mesna (Brock and Pohl, 1986). For routine clinical use, ample fluid intake is recommended. Administration of the drug should be interrupted at the first indication of dysuria or hematuria. The syndrome of inappropriate secretion of antidiuretic hormone (ADH) has been observed in patients receiving cyclophosphamide, usually at doses higher than 50 mg/kg (DeFronzo et al., 1973). It is important to be aware of the possibility of water intoxication, since these patients usually are vigorously hydrated. Ifosfamide Ifosfamide, an analog of cyclophosphamide, also is activated by ring hydroxylation in the liver. Severe urinary tract toxicity limited the use of ifosfamide when it was first introduced in the early 1970s. However, adequate hydration and coadministration of mesna now permit effective use of ifosfamide. Therapeutic Uses Ifosfamide currently is approved for use in combination with other drugs for germ cell testicular cancer and is widely used to treat pediatric and adult sarcomas. Clinical trials also have shown ifosfamide to be active against carcinomas of the cervix and lung and against lymphomas. It is a common component of high-dose chemotherapy regimens with bone marrow or stem cell rescue; in these regimens, in total doses of 12 to 14 g/m2, it may cause severe neurological toxicity, including coma and death. This toxicity is thought to result from a metabolite, chloracetaldehyde (Colvin, 1982). In addition to hemorrhagic cystitis, ifosfamide causes nausea, vomiting, anorexia, leukopenia, nephrotoxicity, and CNS disturbances (especially somnolence or confusion) (see Brade et al., 1987). Ifosfamide IFEX) is infused intravenously over at least 30 minutes at a dose of 1.2 g/m2 per day for 5 days. Intravenous mesna is given as bolus injections in a dosage equal to 20% of the ifosfamide dosage concomitantly and again 4 and 8 hours later, for a total mesna dose of 60% of the ifosfamide dose. Alternatively, mesna may be given in a single dose equal to the ifosfamide dose concomitantly. Patients also should receive at least 2 liters of oral or intravenous fluid daily. Treatment cycles are usually repeated every 3 to 4 weeks. Pharmacokinetics Ifosfamide has a half-life in plasma of approximately 15 hours after doses of 3.8 to 5.0 g/m2 and a somewhat shorter half-life at lower doses. Toxicity Ifosfamide has virtually the same toxicity profile as does cyclophosphamide, with perhaps greater platelet suppression, neurotoxicity, and, in the absence of mesna, urothelial damage. Melphalan Pharmacological and Cytotoxic Actions The general pharmacological and cytotoxic actions of melphalan, the phenylalanine derivative of nitrogen mustard, are similar to those of other nitrogen mustards. The drug is not a vesicant. Absorption, Fate, and Excretion When given orally, melphalan is absorbed in an incomplete and variable manner, and 20% to 50% of the drug is recovered in the stool. The drug has a half-life in plasma of approximately 45 to 90 minutes, and 10% to 15% of an administered dose is excreted unchanged in the urine (Alberts et al., 1979b). Therapeutic Uses The usual oral melphalan (ALKERAN) dose for multiple myeloma is 6 mg daily for a period of 2 to 3 weeks, during which time the blood count should be carefully observed. A rest period of up to 4 weeks should then intervene. When the leukocyte and platelet counts are rising, maintenance therapy, ordinarily 2 to 4 mg daily, is begun. It usually is necessary to maintain a significant degree of bone marrow depression (total leukocyte count in the range of 2500 to 3500 cells per cubic millimeter) in order to achieve optimal results. The usual intravenous dose is 16 mg/m2 infused over 15 to 20 minutes. Doses are repeated at 2-week intervals for four doses and then at 4-week intervals based on response and tolerance. Dosage adjustments should be considered based on blood cell counts and in patients with renal impairment. Although the general spectrum of action of melphalan seems to resemble that of other nitrogen mustards, the advantages of administration by the oral route have made the drug useful in the treatment of multiple myeloma. Clinical Toxicity The clinical toxicity of melphalan is mostly hematological and is similar to that of other alkylating agents. Nausea and vomiting are infrequent. Alopecia does not occur at standard doses, and changes in renal or hepatic function have not been observed. Chlorambucil Pharmacological and Cytotoxic Actions The cytotoxic effects of chlorambucil on the bone marrow, lymphoid organs, and epithelial tissues are similar to those observed with the nitrogen mustards. Although CNS side effects can occur, these have been observed only with large doses. Nausea and vomiting may result from single oral doses of 20 mg or more. Absorption, Fate, and Excretion Oral absorption of chlorambucil is adequate and reliable. The drug has a half-life in plasma of approximately 1.5 hours, and it is almost completely metabolized (Alberts et al., 1979a). Therapeutic Uses The standard initial daily dosage of chlorambucil (LEUKERAN) is 0.1 to 0.2 mg/kg, continued for at least 3 to 6 weeks. The total daily dose, usually 4 to 10 mg, is given at one time. With a fall in the peripheral total leukocyte count or clinical improvement, the dosage is reduced; maintenance therapy (usually 2 mg daily) is feasible and may be required, depending on the nature of the disease. Other dosage schedules also are used. At the recommended dosages, chlorambucil is the slowest-acting nitrogen mustard in clinical use. It is a standard agent for patients with chronic lymphocytic leukemia and primary (Waldenstrm's) macroglobulinemia. Clinical Toxicity In chronic lymphocytic leukemia, chlorambucil may be given orally for months or years, achieving its effects gradually and often without toxicity to a precariously compromised bone marrow. Clinical improvement comparable to that with melphalan or cyclophosphamide has been observed in some patients with plasma cell myeloma. Beneficial results also have been reported in disorders with altered immune reactivity, such as vasculitis associated with rheumatoid arthritis and autoimmune hemolytic anemia with cold agglutinins. Although it is possible to induce marked hypoplasia of the bone marrow with excessive doses of chlorambucil administered over long periods, its myelosuppressive action is usually moderate, gradual, and rapidly reversible. Gastrointestinal discomfort, azoospermia, amenorrhea, pulmonary fibrosis, seizures, dermatitis, and hepatotoxicity may be rarely encountered. A marked increase in the incidence of leukemia and other tumors has been noted in a large controlled study of its use for the treatment of polycythemia vera by the National Polycythemia Vera Study Group, as well as in patients with breast cancer receiving long-term adjuvant chemotherapy (Lerner, 1978). Ethylenimines and Methylmelamines Triethylenemelamine (TEM), Thiotepa (Triethylene Thiophosphoramide), and Altretamine (Hexamethylmelamine; HMM) Pharmacological and Cytotoxic Effects Although nitrogen mustards have largely replaced ethylenimines in general clinical practice, this class of agents continues to have specific use. Thiotepa (THIOPLEX) is active as an intravesicular agent in bladder cancer and is used as a component of experimental high-dose chemotherapy regimens (Kletzel et al., 1992), and altretamine (HEXALEN), formerly known as hexamethylmelamine, is used in patients with advanced ovarian cancer after failure of first-line therapies. Both thiotepa and its primary metabolite, triethylenephosphoramide (TEPA), to which it is rapidly converted by hepatic mixed-function oxygenases (Ng and Waxman, 1991), are capable of forming DNA cross-links. The aziridine rings open after protonation of the ring-nitrogen, leading to a reactive molecule. Absorption, Fate, and Excretion TEPA becomes the predominant form of the drug present in plasma within 5 minutes of thiotepa administration. The parent compound has a plasma half-life of 1.2 to 2 hours, as compared to a half-life of 3 to 24 hours for TEPA. Thiotepa pharmacokinetics are essentially the same in children as in adults at conventional doses (up to 80 mg/m2), and drug and metabolite half-lives are unchanged in children receiving high-dose therapy of 300 mg/m2 per day for 3 days (Kletzel et al., 1992). Less than 10% of the administered drug appears in urine as the parent drug or the primary metabolite. The remainder is metabolized, interacts with biological molecules, or undergoes spontaneous chemical degradation. Clinical Toxicities The toxicities of thiotepa are essentially the same as those of the other alkylating agents, namely myelosuppression and, to a lesser extent, mucositis. Myelosuppression tends to develop somewhat later than with cyclophosphamide, with leukopenic nadirs at 2 weeks and platelet nadirs at 3 weeks. Alkyl Sulfonates Busulfan Pharmacological and Cytotoxic Actions Busulfan is unique in that, in conventional doses, it exerts few pharmacological actions other than myelosuppression. At low doses, selective depression of granulocytopoiesis is evident, leading to its primary use in the chronic phase of chronic myelogenous leukemia (CML). However, platelets and erythroid elements also may be suppressed as the dosage is raised, and in some patients a severe and prolonged pancytopenia results. In low doses, cytotoxic action does not appear to extend to either the lymphoid tissues or the gastrointestinal epithelium. In high-dose regimens, new toxicities, including pulmonary fibrosis and venoocclusive disease of the liver, become apparent. Absorption, Fate, and Excretion Busulfan is well absorbed after oral administration in doses of 2 to 6 mg/day, and it disappears from the blood with a half-life of 2 to 3 hours. Almost all of the drug is excreted in the urine as methanesulfonic acid. In high doses, children under 18 years of age clear the drug faster than do adults, and tolerate higher doses (Vassal et al., 1993). Therapeutic Uses In treating chronic granulocytic leukemia, the initial oral dose of busulfan
(MYLERAN,
BUSULFEX) varies with
the total leukocyte count and the severity of the disease; daily doses from 2
to 8 mg are recommended to initiate therapy and are adjusted appropriately to
subsequent hematological and clinical responses, with the aim of reduction of
the total leukocyte count to The beneficial effects of busulfan in chronic granulocytic leukemia are well established, and clinical remissions may be expected in 85% to 90% of patients after the initial course of therapy, but the drug has largely been replaced by interferon-alfa and hydroxyurea. In CML, reduction of the leukocyte count is noted during the second or third week, and regression of splenomegaly follows. Beneficial results have been reported in other myeloproliferative disorders, including polycythemia vera and myelofibrosis with myeloid metaplasia. High doses of busulfan (640 mg/m2) have been used effectively in combination with high doses of cyclophosphamide to prepare patients with acute myelogenous leukemia for bone marrow transplantation (Santos et al., 1983). High-dose regimens are given in multiple doses over 3 to 4 days to reduce the incidence of acute CNS toxicities, including tonic-clonic seizures, which may occur several hours after each dose. As mentioned earlier, busulfan induces the metabolism of phenytoin. Clinical Toxicity The major toxic effects of busulfan are related to its myelosuppressive properties, and prolonged thrombocytopenia may be a hazard. Occasional instances of nausea, vomiting, diarrhea, impotence, sterility, amenorrhea, and fetal malformation have been reported. The drug is leukemogenic. In the initial phase of chronic granulocytic leukemia treatment, hyperuricemia, resulting from extensive purine catabolism accompanying the rapid cellular destruction, and renal damage from precipitation of urates have been noted. The concurrent use of allopurinol is recommended to avoid this complication. A number of unusual complications have been observed in patients receiving busulfan, but their relation to the drug is poorly understood; these include a syndrome resembling Addison's disease (but without steroid deficiency), cataracts, gynecomastia, cheilosis, glossitis, anhidrosis, and pulmonary fibrosis (Tew et al., 2001). Nitrosoureas The nitrosoureas have an important role in the treatment of brain tumors and gastrointestinal neoplasms. They appear to function as bifunctional alkylating agents but differ in both pharmacological and toxicological properties from conventional nitrogen mustards. Carmustine (BCNU) and lomustine (CCNU) have attracted special interest because of their high lipophilicity and, thus, their capacity to cross the bloodbrain barrier, an important property in the treatment of brain tumors. Unfortunately, with the exception of streptozocin, the nitrosoureas used in the clinic to date cause profound, cumulative myelosuppression that restricts their therapeutic value. In addition, long-term treatment with the nitrosoureas, especially semustine (methyl-CCNU), has resulted in renal failure. As with other alkylating agents, the nitrosoureas are highly carcinogenic and mutagenic. Streptozocin, originally discovered as an antibiotic, is of special
interest. This compound has a methylnitrosourea (MNU) moiety attached to the
2 carbon of glucose. It has a high affinity for Carmustine (BCNU) Pharmacological and Cytotoxic Actions Carmustine's major action is its alkylation of DNA at the O6-guanine position. It kills cells in all phases of the cell cycle. This drug characteristically causes an unusually delayed myelosuppression, with a nadir of the leukocyte and platelet counts at 4 to 6 weeks. In high doses with bone marrow rescue, it produces hepatic venoocclusive disease, pulmonary fibrosis, renal failure, and secondary leukemia (Tew et al., 2001). Absorption, Fate, and Excretion Carmustine is unstable in aqueous solution and in body fluids. After intravenous infusion, it disappears from the plasma with a highly variable half-life of from 15 to 90 minutes or longer (see Levin et al., 1978). Approximately 30% to 80% of the drug appears in the urine within 24 hours as degradation products. The entry of alkylating metabolites into the cerebrospinal fluid (CSF) is rapid, and their concentrations in the CSF are 15% to 30% of the concurrent plasma values (Oliverio, 1976). Therapeutic Uses Carmustine BICNU) usually is administered intravenously at doses of 150 to 200 mg/m2, given by infusion over 1 to 2 hours, and it is not repeated for 6 weeks. When used in combination with other chemotherapeutic agents, the dose is usually reduced by 25% to 50%. The spectrum of activity of carmustine is similar to that of other alkylating agents, with significant responses observed in Hodgkin's disease and a lower response rate in other lymphomas and myeloma. Because of its ability to cross the bloodbrain barrier, carmustine is used as a component of multimodality treatment of malignant astrocytomas and metastatic tumors of the brain. Beneficial responses have been reported in patients with melanoma and gastrointestinal tumors. Streptozocin This naturally occurring nitrosourea is an antibiotic derived from Streptomyces acromogenes. It has been particularly useful in treating functional, malignant pancreatic islet cell tumors. It affects cells in all stages of the mammalian cell cycle. Absorption, Fate, and Excretion Streptozocin is administered parenterally. After intravenous infusions
of 200 to 1600 mg/m2, peak concentrations in the plasma are 30 to
40 Therapeutic Uses Streptozocin ZANOSAR) is administered intravenously, 500 mg/m2 once daily for 5 days; this course is repeated every 6 weeks. Alternatively, 1000 mg/m2 can be given weekly for 2 weeks, and the weekly dose can then be increased to a maximum of 1500 mg/m2. Streptozocin has been used primarily in patients with metastatic pancreatic islet cell carcinoma, and beneficial responses are translated into a significant increase in 1-year survival rate and a doubling of median survival time for the responders. Clinical Toxicity Nausea is a frequent side effect. Renal or hepatic toxicity occurs in approximately two-thirds of cases; although usually reversible, renal toxicity is dose-related and cumulative and may be fatal, and proximal tubular damage is the most important toxic effect. Serial determinations of urinary protein are most valuable in detecting early renal effects. Streptozocin should not be given with other nephrotoxic drugs. Hematological toxicityanemia, leukopenia, or thrombocytopeniaoccurs in 20% of patients. Triazenes Dacarbazine (DTIC) Dacarbazine functions as a methylating agent after metabolic activation in the liver. Its active metabolite is a monomethyl triazino derivative, the same metabolite formed spontaneously by its analog, temozolomide. It kills cells in all phases of the cell cycle. Dacarbazine resistance has been ascribed to the repair of methylated guanine bases in DNA by guanine O6-alkyl transferase. Absorption, Fate, and Excretion Dacarbazine is administered intravenously; after an initial rapid phase of disappearance (t1/2 of about 20 minutes), the drug is removed from plasma with a terminal half-life of about 5 hours (Loo et al., 1976). The half-life is prolonged in the presence of hepatic or renal disease. Almost one-half of the compound is excreted intact in the urine by tubular secretion. Elevated urinary concentrations of 5-aminoimidazole-4-carboxamide (AIC) are derived from the catabolism of dacarbazine, rather than by inhibition of de novo purine biosynthesis. Concentrations of dacarbazine in CSF are approximately 14% of those in plasma (Friedman, 2001). Therapeutic Uses Dacarbazine DTIC-DOME) is administered intravenously. The recommended regimen for malignant melanoma is to give 3.5 mg/kg per day, intravenously, for a 10-day period; this is repeated every 28 days. Alternatively, 250 mg/m2 can be given daily for 5 days and repeated every 3 weeks. Extravasation of the drug may cause tissue damage and severe pain. At present, dacarbazine is employed in combination regimens for the treatment of malignant melanoma, Hodgkin's disease, and adult sarcomas. Temozolomide (TEMODAL), the spontaneously activated analog, has shown activity in patients with malignant gliomas (Newlands et al., 1992; Agarwala and Kirkwood, 2000). Clinical Toxicity The toxicity of both DTIC and temozolomide includes nausea and vomiting in more than 90% of patients; this usually develops 1 to 3 hours after treatment and may last up to 12 hours. Myelosuppression, with both leukopenia and thrombocytopenia, is usually mild to moderate. A flulike syndrome, consisting of chills, fever, malaise, and myalgias, may occur during treatment with DTIC. Hepatotoxicity, alopecia, facial flushing, neurotoxicity, and dermatological reactions also have been reported. |
Antimetabolites
|
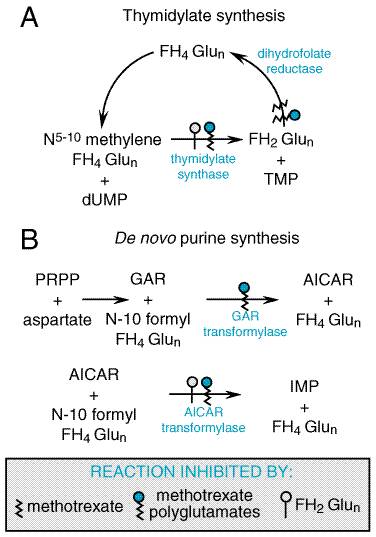
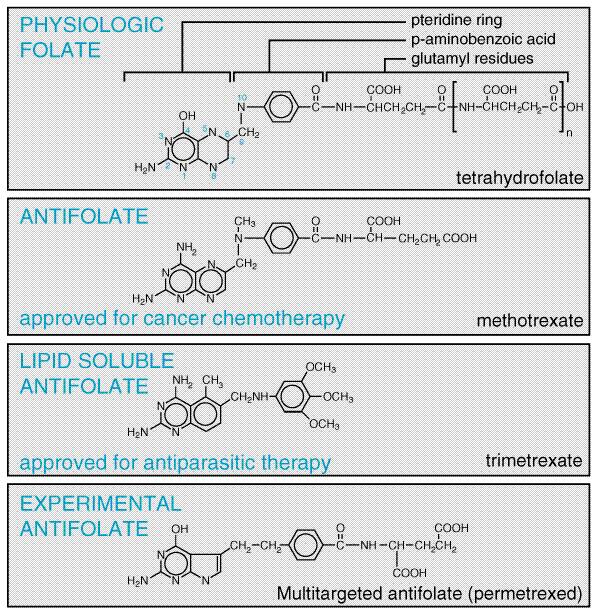
Folic Acid Analogs Methotrexate Antifolates occupy a special place in antineoplastic chemotherapy, in that they produced the first striking, although temporary, remissions in leukemia (Farber et al., 1948) and the first cure of a solid tumor, choriocarcinoma (Hertz, 1963). The consistent cure of choriocarcinoma by methotrexate provided great impetus to investigations into the chemotherapy of cancer. Interest in folate antagonists further increased with the introduction of high-dose regimens with 'rescue' of host toxicity by the reduced folate, leucovorin (folinic acid, citrovorum factor). These methods extend the usefulness of methotrexate to tumors such as osteogenic sarcoma that do not respond to lower doses. Recognition that methotrexate, an inhibitor of dihydrofolate reductase, also directly inhibits the folate-dependent enzymes of de novo purine and thymidylate synthesis focused attention on the development of antifolate analogs that specifically target these other folate-dependent enzyme targets of methotrexate (see Figure 525). Replacement of the 5, 8, and/or 10 nitrogens of the pteridine ring of folate, as well as various side-chain substitutions, has generated a series of new inhibitors that preserve the common folate potential to form long-lived, intracellular polyglutamates. These new agents, however, have greater capacity for transport into tumor cells (Messmann and Allegra, 2001), and exert their primary inhibitory effect on thymidylate synthesis (raltitrexed, TOMUDEX), purine biosynthesis (lometrexol) or both [the multitargeted antifolate permefrexed (MTA)] (Calvete et al., 1994; Beardsley et al., 1986; Chen et al., 1999).
Aside from its antineoplastic activity, methotrexate also has been used with benefit in the therapy of the common skin disease psoriasis (McDonald, 1981; see Chapter 65: Dermatological Pharmacology). Additionally, methotrexate inhibits cell-mediated immune reactions and is employed as an immunosuppressive agent, for example, in allogeneic bone marrow and organ transplantation and for the treatment of dermatomyositis, rheumatoid arthritis, Wegener's granulomatosis, and Crohn's disease (Messmann and Allegra, 2001; Feagan et al., 1995; see Chapter 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants). StructureActivity Relationship Folic acid is an essential dietary factor from which is derived a series of tetrahydrofolate cofactors that provide single carbon groups for the synthesis of precursors of DNA (thymidylate and purines) and RNA (purines). A detailed description of the biological functions and therapeutic applications of folic acid appears in Chapter 54: Hematopoietic Agents: Growth Factors, Minerals, and Vitamins. The enzyme dihydrofolate reductase (DHFR) is the primary site of action of most folate analogs studied to date (see Figures 525 and 526). Inhibition of DHFR leads to toxic effects through partial depletion of the tetrahydrofolate cofactors that are required for the synthesis of purines and thymidylate (Messmann and Allegra, 2001) and through direct inhibition of the folate-dependent enzymes of purine and thymidylate metabolism by the polyglutamates of methotrexate and the dihydrofolate polyglutamates that accumulate with DHFR inhibition (Figure 525) (Allegra et al., 1986, 1987b). Inhibitors of DHFR differ in their relative potency for blocking enzyme from different species. Agents have been identified that have little effect on the human enzyme but have strong activity against bacterial and parasitic infections (see discussions of trimethoprim, Chapter 44: Antimicrobial Agents: Sulfonamides, Trimethoprim-Sulfamethoxazole, Quinolones, and Agents for Urinary Tract Infections; pyrimethamine, Chapter 40: Drugs Used in the Chemotherapy of Protozoal Infections: Malaria). By contrast, methotrexate is an effective inhibitor of DHFR in all species investigated. Crystallographic studies have revealed the atomic basis for the high affinity of methotrexate for DHFR (Kraut and Matthews, 1987; Schweitzer et al., 1989; Bystroff and Kraut, 1991; Blakley and Sorrentino, 1998) and the species specificity of the various DHFR inhibitors (Matthews et al., 1985; Stone and Morrison, 1986).
Because folic acid and many of its analogs are very polar, they cross the bloodbrain barrier poorly and require specific transport mechanisms to enter mammalian cells (Elwood, 1989; Dixon et al., 1994). Two inward folate transport systems are found on mammalian cells: (1) a folate receptor, which has high affinity for folic acid but lesser ability to transport methotrexate and other analogs (Elwood, 1989); and (2) the reduced folate transporter, the major transit protein for methotrexate, raltitrexed, and most analogs (Westerhof et al., 1995). Once in the cell, additional glutamyl residues are added to the molecule by the enzyme folylpolyglutamate synthetase (Cichowicz and Shane, 1987). Intracellular methotrexate polyglutamates have been identified with up to six glutamyl residues. Since these higher polyglutamates cross cellular membranes poorly, if at all, this serves as a mechanism of entrapment and may account for the prolonged retention of methotrexate in tumors and normal tissues such as liver. Polyglutamylated folates and analogs have substantially greater affinity than the monoglutamate form for folate-dependent enzymes that are required for purine and thymidylate synthesis, but not for DHFR. Novel folate antagonists have been identified that exploit differences between the folate influx system in certain tumors and that in normal tissues (e.g., bone marrow). The analog 10-ethyl,10-deaza aminopterin (edatrexate) is transported into some tumor cells much more efficiently than into normal tissues and is an excellent inhibitor of DHFR. This promising compound is undergoing clinical evaluation (Grant et al., 1993). In efforts to bypass the obligatory membrane transport system and facilitate penetration of the bloodbrain barrier, lipid-soluble folate antagonists also have been synthesized. Trimetrexate (Figure 526) was one of the first to be tested for clinical activity. The analog was found to have modest antitumor activity, primarily in combination with leucovorin (5-formyl tetrahydrofolate) rescue. However, it has proven to be beneficial in the treatment of Pneumocystis carinii pneumonia (Allegra et al., 1987a). The other important new folate analog, MTA or pemetrexed (ALTIMA) (Figure 526), is a tetrahydrofolate analog. It readily converts to polyglutamates that inhibit thymidylate and purine biosynthesis, as well as dihydrofolate reductase. In early trials, it has shown activity against colon cancer, mesothelioma, and non-small cell lung cancer (Rusthoven et al., 1999). Mechanism of Action To function as a cofactor in one-carbon transfer reactions, folate
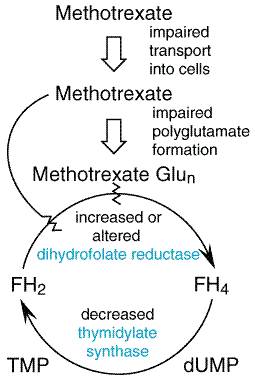
must first be reduced by DHFR to tetrahydrofolate (FH4).
Single-carbon fragments are added enzymatically to FH4 in various
configurations and may then be transferred in specific synthetic reactions.
In a key metabolic event catalyzed by thymidylate synthase (Figure 525),
2'-deoxyuridylate (dUMP) is converted to thymidylate, an essential component
of DNA. In this reaction, a one-carbon group is transferred to dUMP from
5,10-methylene FH4, and the reduced folate cofactor is oxidized to
dihydrofolate (FH2). To function again as a cofactor, FH2
must be reduced to FH4 by DHFR. Inhibitors, such as methotrexate,
with a high affinity for DHFR (Ki As with most antimetabolites, methotrexate is only partially selective for tumor cells and is toxic to all rapidly dividing normal cells, such as those of the intestinal epithelium and bone marrow. Folate antagonists kill cells during the S phase of the cell cycle and are most effective when cells are in the logarithmic phase of growth. Mechanisms of Resistance to Antifolates In experimental systems, a vast array of biochemical mechanisms of acquired resistance to methotrexate have been demonstrated (Figure 527) affecting each known step in methotrexate action, including: (1) impaired transport of methotrexate into cells (Assaraf and Schimke, 1987; Trippett et al., 1992); (2) production of altered forms of DHFR that have decreased affinity for the inhibitor (Srimatkandada et al., 1989); (3) increased concentrations of intracellular DHFR through gene amplification or altered gene regulation (Pauletti et al., 1990; Matherley et al., 1997); (4) decreased ability to synthesize methotrexate polyglutamates (Li et al., 1992); and (5) decreased thymidylate synthase activity (Curt et al., 1985). DHFR levels in leukemic cells increase within 24 hours after treatment of patients with methotrexate; this likely reflects induction of new enzyme synthesis. Recent investigations have demonstrated that the intracellular level of DHFR protein is controlled at the level of mRNA translational efficiency through an autoregulatory mechanism whereby the DHFR protein may bind to and control the translational efficiency of its own messenger RNA (Chu et al., 1993). Over longer periods of treatment, tumor cell populations emerge that contain markedly increased levels of DHFR. These cells contain multiple gene copies of DHFR either in mitotically unstable double-minute chromosomes or in stable, homogeneously staining regions or amplisomes of the tumor cell chromosomes. First identified as an explanation for resistance to methotrexate (Schimke et al., 1978), gene amplification has since been implicated in the resistance to many antitumor agents, including fluorouracil and pentostatin (2'-deoxycoformycin) (Stark and Wahl, 1984). Evidence supports the conclusion that DHFR gene amplification is clinically significant in patients with lung cancer (Curt et al., 1983) and leukemia (Goker et al., 1995).
To overcome resistance, high doses of methotrexate with leucovorin rescue may permit entry of drug into transport-defective cells and may permit the intracellular accumulation of methotrexate in concentrations that inactivate high levels of DHFR. General Toxicity and Cytotoxic Action The primary toxic effects of methotrexate and other folate antagonists used in cancer chemotherapy are exerted against rapidly dividing cells of the bone marrow and gastrointestinal epithelium. Mucositis, myelosuppression, and thrombocytopenia reach their maximum in 5 to 10 days after drug administration andexcept in instances of altered drug excretionreverse rapidly thereafter. In addition to its acute toxicities, methotrexate can cause pneumonitis, characterized by patchy inflammatory infiltrates that rapidly regress upon discontinuation of drug. In some cases, patients can be rechallenged with drug without toxicity. The etiology is not clearly allergic. A second toxicity of particular significance in its chronic administration in patients with psoriasis or rheumatoid arthritis is hepatic fibrosis and cirrhosis. Increased hepatic portal fibrosis is detected with higher frequency than in control patients after 6 months or longer of continuous oral methotrexate treatment of psoriasis. Its presence should lead to discontinuation of methotrexate. Acute, reversible elevation of hepatic enzymes is detected in serum after high-dose administration but is rarely associated with permanent changes. Folic acid antagonists are toxic to developing embryos. In preliminary trials, methotrexate has been highly effective when used with the prostaglandin analog, misoprostol, in inducing abortion in first trimester pregnancy (Hausknecht, 1995). Absorption, Fate, and Excretion Methotrexate is readily absorbed from the gastrointestinal tract at
doses of less than 25 mg/m2, but larger doses are absorbed
incompletely and are routinely administered intravenously. Peak
concentrations in the plasma of 1 to 10 Approximately 50% of methotrexate is bound to plasma proteins and may be displaced from plasma albumin by a number of drugs, including sulfonamides, salicylates, tetracycline, chloramphenicol, and phenytoin; caution should be used if these are given concomitantly. Of the drug absorbed, about 90% is excreted unchanged in the urine within 48 hours, mostly within the first 8 to 12 hours. A small amount of methotrexate also is excreted in the stool, probably through the biliary tract. Metabolism of methotrexate in human beings is usually minimal. After high doses, however, metabolites do accumulate; these include 7-hydroxy-methotrexate, which is potentially nephrotoxic (Messmann and Allegra, 2001). Renal excretion of methotrexate occurs through a combination of glomerular filtration and active tubular secretion. Therefore, the concurrent use of drugs that reduce renal blood flow (e.g., nonsteroidal antiinflammatory agents), that are nephrotoxic (e.g., cisplatin), or that are weak organic acids (e.g., aspirin or piperacillin) can delay drug excretion and lead to severe myelosuppression (Stoller et al., 1977; Iven and Brasch, 1988; Thyss et al., 1986). Particular caution must be exercised in treating patients with renal insufficiency, and the dose should be adjusted in these patients in proportion to decreases in renal function. Methotrexate is retained in the form of polyglutamates for long periodsfor example, for weeks in the kidneys and for several months in the liver. There also is evidence for enterohepatic recirculation. It is important to emphasize that concentrations of methotrexate in cerebrospinal fluid are only 3% of those in the systemic circulation at steady state; hence, neoplastic cells in the CNS are probably not killed by standard dosage regimens. When high doses of methotrexate are given (>1.5 g/m2), followed by leucovorin rescue (see below), cytotoxic concentrations of methotrexate may be attained in the CNS. Therapeutic Uses Methotrexate (methotrexate sodium; amethopterin; FOLEX, MEXATE, RHEUMATREX, others) has been used in the treatment of severe, disabling psoriasis in doses of 2.5 mg orally for 5 days, followed by a rest period of at least 2 days, or 10 to 25 mg intravenously weekly. An initial parenteral test dose of 5 to 10 mg is recommended to detect any possible idiosyncrasy. It also is used intermittently at low dosage to induce remission in refractory rheumatoid arthritis (Hoffmeister, 1983). Complete awareness of the pharmacology and toxic potential of methotrexate is a prerequisite for its use in these nonneoplastic disorders (Weinstein, 1977). Methotrexate is a useful drug in the management of acute lymphoblastic leukemia in children. It is of great value in remission induction and consolidation, used in high doses, and in the maintenance of remissions in leukemia. For maintenance therapy, it is administered intermittently at doses of 30 mg/m2 intramuscularly weekly in two divided doses or in 2-day 'pulses' of 175 to 525 mg/m2 at monthly intervals. Outcome of treatment in children correlates inversely with the rate of drug clearance. During methotrexate infusion, high steady-state levels are associated with a lower leukemia relapse rate (Borsi and Moe, 1987). Methotrexate is of very limited value in the types of leukemia seen in adults, except for treatment and prevention of leukemic meningitis. The intrathecal administration of methotrexate has been employed for treatment or prophylaxis of meningeal leukemia or lymphoma and for treatment of meningeal carcinomatosis. This route of administration achieves high concentrations of methotrexate in the CSF and is effective also in patients whose systemic disease has become resistant to methotrexate, since the leukemic cells in the CNS beyond the bloodbrain barrier have survived in a pharmacological sanctuary and may retain their original degree of sensitivity to the drug. The recommended intrathecal dose in all patients over 3 years of age is 12 mg (Bleyer, 1978). The dose is repeated every 4 days until malignant cells are no longer evident in the CSF. Leucovorin may be administered to counteract the toxicity of methotrexate that escapes into the systemic circulation, although this is generally not necessary. Since methotrexate administered into the lumbar space distributes poorly over the cerebral convexities, the drug may be more effectively distributed through the use of an intraventricular Ommaya reservoir. The use of 1-mg doses of methotrexate at intervals of 12 to 24 hours yields an effective regimen with reduced neurotoxicity. Methotrexate is of established value in choriocarcinoma and related trophoblastic tumors of women; cure is achieved in approximately 75% of advanced cases treated sequentially with methotrexate and dactinomycin, and in over 90% when early diagnosis is made. In the treatment of choriocarcinoma with methotrexate, 1 mg/kg is administered intramuscularly every other day for four doses, alternating with leucovorin (0.1 mg/kg every other day). Courses are repeated at 3-week intervals, toxicity permitting, and urinary gonadotropin titers are used as a guide for persistence of disease. Beneficial effects also are observed in patients with osteosarcoma and
mycosis fungoides and when methotrexate is used as part of the combination
therapy of Burkitt's and other non-Hodgkin's lymphomas and carcinomas of the
breast, head and neck, ovary, and bladder. High-dose methotrexate, with leucovorin
rescue, can cause substantial tumor regression in osteosarcoma and in
combination therapy of leukemias and non-Hodgkin's lymphomas. A 6- to 72-hour
infusion of relatively large amounts of methotrexate may be employed
intermittently (from 250 mg to 7.5 g/m2 or more), but only when
leucovorin rescue is used. Such regimens produce cytotoxic concentrations of
drug in the cerebrospinal fluid (CSF) and protect against leukemic
meningitis. A typical regimen includes the infusion of methotrexate for 6
hours followed by leucovorin at a dose of 15 mg/m2 every 6 hours
for seven doses, with the goal of rescuing normal cells and thereby
preventing toxicity. Other dosage regimens also are used. The administration
of methotrexate in high dosage has the potential for serious toxicity and
should be performed only by experienced chemotherapists who are able to
monitor concentrations of methotrexate in plasma. If methotrexate values
measured 48 hours after drug administration are 1 Clinical Toxicities As previously stated, the primary toxicities of methotrexate affect the bone marrow and the intestinal epithelium. Such patients may be at risk for spontaneous hemorrhage or life-threatening infection, and they may require prophylactic transfusion of platelets and broad-spectrum antibiotics if febrile. Side effects usually disappear within 2 weeks, but prolonged suppression of the bone marrow may occur in patients with compromised renal function who have delayed excretion of the drug. The dosage of methotrexate must be reduced in proportion to any reduction in creatinine clearance. Additional toxicities of methotrexate include alopecia, dermatitis, interstitial pneumonitis, nephrotoxicity, defective oogenesis or spermatogenesis, abortion, and teratogenesis. Hepatic dysfunction is usually reversible but sometimes leads to cirrhosis after long-term continuous treatment, as in patients with psoriasis. Intrathecal administration of methotrexate often causes meningismus and an inflammatory response in the CSF. Seizures, coma, and death may occur rarely. Leucovorin does not reverse neurotoxicity. Pyrimidine Analogs This class of agents encompasses a diverse and interesting group of drugs that have in common the capacity to inhibit the biosynthesis of pyrimidine nucleotides or to mimic these natural metabolites to such an extent that the analogs interfere with the synthesis or function of nucleic acids. Analogs of deoxycytidine and thymidine have been synthesized as inhibitors of DNA synthesis, and an analog of uracil, 5-fluorouracil, effectively inhibits both RNA function and/or processing and synthesis of thymidylate (see Figure 528). Drugs in this group have been employed in the treatment of diverse afflictions, including neoplastic diseases, psoriasis, and infections caused by fungi and DNA-containing viruses. The pathways for metabolic activation and degradation of these compounds during systemic administration present opportunities for the development of synergistic combination therapies with other clinically effective drugs.
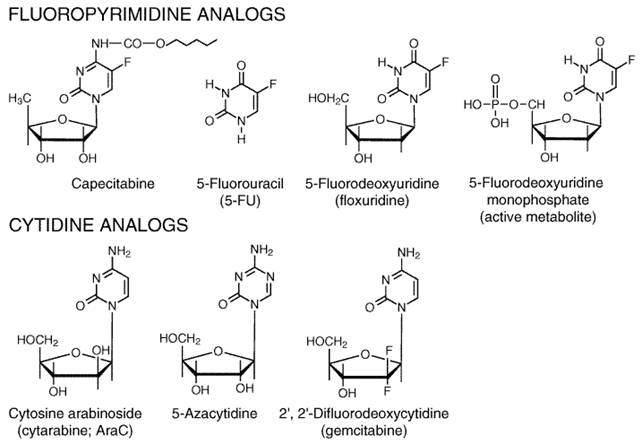
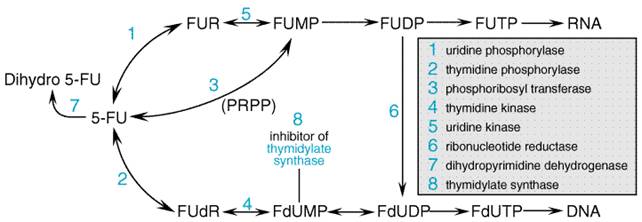
General Mechanism of Action The best-characterized agents in this class are the halogenated pyrimidines, a group that includes fluorouracil (5-fluorouracil, or 5-FU), floxuridine (5-fluoro-2'-deoxyuridine, or 5-FUdR), and idoxuridine (5-iodode-oxyuridine; see Chapter 50: Antimicrobial Agents: Antiviral Agents (Nonretroviral)). If one compares the van der Waals radii of the various 5-position substituents, the dimension of the fluorine atom resembles that of hydrogen, whereas the bromine and iodine atoms are larger and close in size to the methyl group. Thus, idoxuridine behaves as an analog of thymidine, and its primary biological action results from its phosphorylation and ultimate incorporation into DNA in place of thymidylate. In 5-FU, the smaller fluorine at position 5 allows the molecule to mimic uracil biochemically. However, the fluorinecarbon bond is much tighter than that of CH and prevents the methylation of the 5 position of 5-FU by thymidylate synthase. Instead, in the presence of the physiological cofactor 5,10-methylene tetrahydrofolate, the fluoropyrimidine locks the enzyme in an inhibited state. Thus, substitution of a halogen atom of the correct dimensions can produce a molecule that sufficiently resembles a natural pyrimidine to interact with enzymes of pyrimidine metabolism but at the same time interferes drastically with certain other aspects of pyrimidine action. A number of 5-FU analogs have reached the clinic. The most important of these is capecitabine (N4-pentoxycarbonyl-5'-deoxy-5-fluorocytidine), a drug with proven activity against colon and breast cancers. This orally administered agent is converted to 5'-deoxy-5-fluorocytidine by carboxylesterase activity in liver and other normal and malignant tissues. From that point, it is converted to 5'-deoxy-fluorodeoxyuridine by cytidine deaminase. The final step in its activation occurs when thymidine phosphorylase cleaves off the 5'-deoxy sugar, leaving intracellular 5-FU. Tumors with elevated thymidine phosphorylase activity seem particularly susceptible to this drug (Ishikawa et al., 1998). Nucleotides in RNA and DNA contain ribose and 2'-deoxyribose,
respectively. Among the various modifications of the sugar moiety that have
been attempted, the replacement of the ribose of cytidine with arabinose has
yielded a useful chemotherapeutic agent, cytarabine (AraC). As may be
seen in Figure 528, the hydroxyl group in this molecule is attached to the
2'-carbon in the Two other cytidine analogs have received extensive clinical evaluation. 5-Azacytidine, an inhibitor of DNA methylation as well as a cytidine antimetabolite, becomes incorporated predominantly into RNA and has antileukemic as well as differentiating actions in vitro. A newer analog, 2',2'-difluorodeoxycytidine (gemcitabine), becomes incorporated into DNA and inhibits the elongation of nascent DNA strands (see Figure 528). It has promising activity in various human solid tumors, including pancreatic, lung, and ovarian cancer. Fluorouracil and Floxuridine (Fluorodeoxyuridine) Mechanism of Action 5-FU requires enzymatic conversion to the nucleotide (ribosylation and phosphorylation) in order to exert its cytotoxic activity (Figure 529). Several routes are available for the formation of the 5'-monophosphate nucleotide (F-UMP) in animal cells. 5-FU may be converted to fluorouridine by uridine phosphorylase and then to F-UMP by uridine kinase, or it may react directly with 5-phosphoribosyl-1-pyrophosphate (PRPP), in a reaction catalyzed by the enzyme orotate phosphoribosyl transferase, to form F-UMP. Many metabolic pathways are available to F-UMP, including incorporation into RNA. A reaction sequence crucial for antineoplastic activity involves reduction of the diphosphate nucleotide by the enzyme ribonucleotide diphosphate reductase to the deoxynucleotide level and the eventual formation of 5-fluoro-2'-deoxyuridine-5'-phosphate (F-dUMP). 5-FU also may be converted directly to the deoxyriboside 5-FUdR by the enzyme thymidine phosphorylase and further to F-dUMP, a potent inhibitor of thymidylate synthesis, by thymidine kinase. This complex metabolic pathway for the generation of F-dUMP may be bypassed through use of the deoxyribonucleoside of fluorouracilfloxuridine (fluorodeoxyuridine, FUdR)which is converted directly to F-dUMP by thymidine kinase.
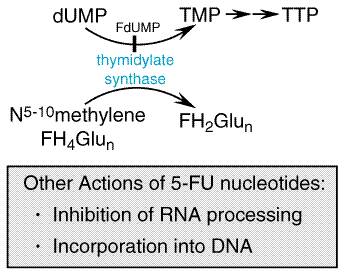
The interaction between F-dUMP and the enzyme thymidylate synthase leads to deletion of TTP, a necessary constituent of DNA (Figure 5210). The folate cofactor, 5,10-methylenetetra-hydrofolate, and F-dUMP form a covalently bound ternary complex with the enzyme. This inhibitory complex resembles the transition state formed during the normal enzymatic reaction when dUMP is converted to thymidylate. Although the physiological complex progresses to the synthesis of thymidylate by transfer of the methylene group and two hydrogen atoms from folate to dUMP, this reaction is blocked in the inhibitory complex by the stability of the fluorine carbon bond on F-dUMP; sustained inhibition of the enzyme results (Santi et al., 1974).
5-FU also is incorporated into both RNA and DNA. In 5-FUtreated cells, both F-dUTP and dUTP (the substrate that accumulates behind the blocked thymidylate synthase reaction) incorporate into DNA in place of the depleted physiological TTP. The significance of the incorporation of F-dUTP and dUTP into DNA is unclear (Canman et al., 1993). Presumably, the incorporation of deoxyuridylate and/or fluorodeoxyuridylate into DNA would call into action the excisionrepair process. This process may result in DNA strand breakage because DNA repair requires TTP, but this substrate is lacking as a result of thymidylate synthase inhibition (Mauro et al., 1993). 5-FU incorporation into RNA also causes toxicity as the result of major effects on both the processing and functions of RNA (Armstrong, 1989; Danenberg et al., 1990). A number of biochemical mechanisms have been identified that are associated with resistance to the cytotoxic effects of 5-FU or floxuridine. These mechanisms include loss or decreased activity of the enzymes necessary for activation of 5-FU, decreased pyrimidine monophosphate kinase (which decreases incorporation into RNA), amplification of thymidylate synthase (Washtein, 1982), and altered thymidylate synthase that is not inhibited by F-dUMP (Barbour et al., 1990). Both experimental studies and clinical trials support the position that the response to 5-FU correlates significantly with low levels of the degradative enzymes, dihydrouracil dehydrogenase and thymidine phosphorylase, and a low level of expression of the target enzyme, thymidylate synthase (van Triest et al., 2000). Recent investigations have demonstrated that the level of thymidylate synthase is finely controlled by an autoregulatory feedback mechanism wherein the thymidylate synthase protein interacts with and controls the translational efficiency of its own messenger RNA. This mechanism provides for the rapid modulation of the level of thymidylate synthase necessary for cellular division and also may be an important mechanism by which malignant cells become rapidly insensitive to the effects of 5-fluorouracil (Chu et al., 1991; Swain et al., 1989). Some malignant cells appear to have insufficient concentrations of 5,10-methylene tetrahydrofolate and, thus, cannot form maximal levels of the inhibited ternary complex with thymidylate synthase. Addition of exogenous folate in the form of 5-formyl-tetrahydrofolate (leucovorin) increases formation of the complex in both laboratory and clinical experiments and has enhanced responses to 5-FU in clinical trials (Ullman et al., 1978; Grogan et al., 1993). Except for inadequate intracellular folate pools, it is not established which (if any) of the other mechanisms is associated with clinical resistance to 5-FU and its derivatives (Grem et al., 1987). In addition to leucovorin, a number of other agents have been combined with 5-FU in attempts to enhance the cytotoxic activity through biochemical modulation. These agents, along with their proposed mechanisms of interaction, are shown in Table 52-2. The most clinically interesting combinations with 5-FU include methotrexate, interferon, leucovorin, or cisplatin, all of which are currently under investigation to define their ultimate clinical roles. Agents that inhibit early steps in pyrimidine biosynthesis, such as PALA (N-phosphonoacetyl-L-aspartate), an inhibitor of aspartate transcarbamylase, provide synergistic interaction with 5-FU in experimental systems, but these combinations have no proven clinical value (Grem et al., 1988). Methotrexate, by inhibiting purine synthesis and increasing cellular pools of PRPP, enhances the activation of 5-FU and increases antitumor activity of 5-FU when given prior to but not following 5-FU. In clinical trials, the combination of cisplatin and 5-FU has yielded impressive responses in tumors of the upper aerodigestive tract, but the molecular basis of their interaction is not well understood (Grem, 2001). Absorption, Fate, and Excretion 5-FU and floxuridine are administered parenterally, since absorption
after ingestion of the drugs is unpredictable and incomplete. Metabolic
degradation occurs in many tissues, particularly the liver. Floxuridine is
converted by thymidine or deoxyuridine phosphorylases into 5-FU. 5-FU is
inactivated by reduction of the pyrimidine ring; this reaction is carried out
by dihydropyrimidine dehydrogenase (DPD), which is found in liver, intestinal
mucosa, tumor cells, and other tissues. Inherited deficiency of this enzyme
leads to greatly increased sensitivity to the drug (Lu et al., 1993; Milano
et al., 1999). The rare individual who totally lacks this enzyme may
experience profound drug toxicity following conventional doses of the drug.
DPD deficiency can be detected either by enzymatic or molecular assays using
peripheral white blood cells, or by determining the plasma ratio of 5-FU to
its metabolite, 5-fluoro-5,6-dihydrouracil, which is ultimately degraded to Rapid intravenous administration of 5-FU produces plasma
concentrations of 0.1 to 1.0 mM; plasma clearance is rapid (t1/2
10 to 20 minutes). Urinary excretion of a single dose of 5-FU given intravenously
amounts to only 5% to 10% in 24 hours. Although the liver contains high
concentrations of DPD, dosage does not have to be modified in patients with
hepatic dysfunction, presumably because of degradation of the drug at
extrahepatic sites or by vast excess of this enzyme in the liver. Given by
continuous intravenous infusion for 24 to 120 hours, 5-FU achieves plasma
concentrations in the range of 0.5 to 8.0 Capecitabine is well absorbed orally, yielding high plasma concentrations of 5'-deoxy-fluorodeoxyuridine (5'-dFdU), which disappears with a half-life of about 1 hour. 5-FU levels are less than 10% of those of 5'-dFdU. Liver dysfunction delays the conversion of the parent compound to 5'-dFdU and 5-FU, but there is no consistent effect on toxicity (Twelves et al., 1999). Therapeutic Uses 5-Fluorouracil Accumulated experience with 5-FU (ADRUCIL) indicates that the drug produces partial responses in 10% to 20% of patients with metastatic carcinomas of the breast and the gastrointestinal tract; beneficial effects also have been reported in carcinoma of the ovary, cervix, urinary bladder, prostate, pancreas, and oropharyngeal areas. For average-risk patients in good nutritional status with adequate hematopoietic function, the weekly dosage regimen employs 750 mg/m2 alone or 500 to 600 mg/m2 with leucovorin once each week for 6 of 8 weeks. Other regimens use daily doses of 500 mg/m2 for 5 days, repeated in monthly cycles. When used with leucovorin, daily doses of 5-FU must be reduced to 375 to 425 mg/m2 for 5 days because of mucositis and diarrhea. It also has been given as a continuous infusion for up to 21 days (300 mg/m2 per day), or as a biweekly 48-hour continuous infusion (de Gramont et al., 1998). Floxuridine (FUdR) FUdR (fluorodeoxyuridine; FUDR) is used primarily by continuous infusion into the hepatic artery for treatment of metastatic carcinoma of the colon or following resection of hepatic metastases (Kemeny et al., 1999); the response rate to such infusion is 40% to 50%, or double that observed with intravenous administration. Intrahepatic arterial infusion for 14 to 21 days may be used with minimal systemic toxicity. However, there is a significant risk of biliary sclerosis if this route is used for multiple cycles of therapy (Kemeny et al., 1987; Hohn et al., 1986). Continuous infusion of floxuridine into the arterial blood supply of tumors at other sites, such as in the head and neck region, may provide beneficial clinical effects. With any of these regimens, treatment should be discontinued at the earliest manifestation of toxicity (usually stomatitis or diarrhea) because the maximal effects of bone marrow suppression and gut toxicity will not be evident until days 7 to 14. Capecitabine (XELODA Capecitabine is approved by the United States Food and Drug Administration (FDA) for the treatment of metastatic breast cancer in patients who have not responded to a regimen of paclitaxel and an anthracycline antibiotic (see below). The recommended dose is 2500 mg/m2 daily, given orally in two divided doses with food, for 2 weeks followed by a rest period of 1 week. This cycle is then repeated two more times. Combination Therapy Higher response rates are seen when 5-FU is used in combination with other agents, such as cyclophosphamide and methotrexate (breast cancer), cisplatin (head and neck cancer), and with leucovorin in colon cancer (see Table 52-2). The use of 5-FU in combination regimens has improved survival in the adjuvant treatment for breast cancer (Early Breast Cancer Trialists Collaborative Group, 1988) and, with leucovorin, for colorectal cancer (Wolmark et al., 1993). 5-FU is a potent radiation sensitizer and is being used with concurrent radiotherapy for primary therapy of locally advanced tumors of the head and neck, esophagus, lung, and rectum. 5-FU is used widely with very favorable results for the topical treatment of premalignant keratoses of the skin and multiple superficial basal cell carcinomas. It also is effective in severe recalcitrant psoriasis (Alper et al., 1985). Clinical Toxicities The clinical manifestations of toxicity caused by 5-FU and floxuridine are similar and may be difficult to anticipate because of their delayed appearance. The earliest untoward symptoms during a course of therapy are anorexia and nausea; these are followed by stomatitis and diarrhea, which constitute reliable warning signs that a sufficient dose has been administered. Mucosal ulcerations occur throughout the gastrointestinal tract and may lead to fulminant diarrhea, shock, and death, particularly in patients who are receiving continuous infusions of 5-FU or in those receiving 5-FU with leucovorin. The major toxic effects of bolus-dose regimens result from the myelosuppressive action of these drugs. The nadir of leukopenia is usually between days 9 and 14 after the first injection of drug. Thrombocytopenia and anemia also may occur. Loss of hair, occasionally progressing to total alopecia, nail changes, dermatitis, and increased pigmentation and atrophy of the skin may be encountered. Neurological manifestations, including an acute cerebellar syndrome, have been reported, and myelopathy has been observed after the intrathecal administration of 5-FU. Cardiac toxicity, particularly acute chest pain with evidence of ischemia in the electrocardiogram, also may occur. The low therapeutic indices of these agents emphasize the need for very skillful supervision by physicians familiar with the action of the fluorinated pyrimidines and the possible hazards of chemotherapy. Capecitabine causes much the same spectrum of toxicities as 5-FU (diarrhea, myelosuppression), but also a progressive hand-foot syndrome consisting of erythema, desquamation, pain, and sensitivity to touch of the palms and soles. Cytarabine (Cytosine Arabinoside; AraC) Cytarabine (1- Mechanism of Action This compound is an analog of 2'-deoxycytidine with the 2'-hydroxyl in a position trans to the 3'-hydroxyl of the sugar, as shown in Figure 528. The 2'-hydroxyl causes steric hindrance to the rotation of the pyrimidine base around the nucleosidic bond. The bases of polyarabinonucleotides cannot stack normally, as do the bases of polydeoxynucleotides. AraC penetrates cells by a carrier-mediated process shared by physiological nucleosides. As with most purine and pyrimidine antimetabolites, cytarabine must be 'activated' by conversion to the 5'-monophosphate nucleotide (AraCMP), a reaction catalyzed by deoxycytidine kinase. AraCMP can then react with appropriate nucleotide kinases to form the diphosphate and triphosphate nucleotides (AraCDP and AraCTP). AraC competes with the physiological substrate deoxycytidine 5'-triphosphate (dCTP) for incorporation into DNA by DNA polymerases. The incorporated AraCMP residue is a potent inhibitor of DNA polymerase. The effects of AraC on DNA polymerase activity extend not only to DNA chain elongation during semiconservative DNA replication, but also to DNA repair. There is a significant relationship between inhibition of DNA synthesis and the total amount of AraC incorporated into DNA. Thus, incorporation of about five molecules of AraC per 104 bases of DNA decreases cellular clonogenicity by about 50%. AraC also causes an unusual reiteration of DNA segments, thus increasing the possibility of recombination, crossover, and gene amplification. In addition, AraC is converted intracellularly to AraCDP-choline, an analog of the physiological CDP-choline, which inhibits the synthesis of membrane glycoproteins and glycolipids. Furthermore, AraCMP inhibits the transfer of galactose, N-acetylglucosamine, and sialic acid to cell-surface glycoproteins, and AraCTP inhibits the synthesis of CMP-acetylneuraminic acid. Thus, AraC may alter membrane structure, antigenicity, and function. AraC induces terminal differentiation of leukemic cells in tissue culture, an effect that is accompanied by decreased c-myc oncogene expression (Bianchi Scarra et al., 1986). These changes in morphology and oncogene expression occur at concentrations above the threshold for cytotoxicity and may simply represent terminal injury of cells. However, molecular analysis of bone marrow specimens from some leukemic patients in remission after AraC therapy has revealed persistence of leukemic markers, suggesting that differentiation may have occurred. The precise mechanism of cellular death caused by AraC is not fully
understood. Fragmentation of DNA is observed in AraC-treated cells, and there
is cytological and biochemical evidence for apoptosis in both tumor and
normal tissues (Smets, 1994). A complex system of interacting transduction
signals ultimately determines whether or not a cell exposed to a cytotoxic
agent is destined to die. Exposure of leukemic cells to AraC stimulates the
formation of ceramide, a potent inducer of apoptosis. On the other hand, an
increase in protein kinase C (PKC) activity is observed in leukemic cells in
response to AraC in vitro. Because PKC activation is known to oppose
apoptosis in hematopoietic cells, the lethal actions of AraC may depend, at
least partially, on its relative effects on the PKC and sphingomyelin
pathways (Strum et al., 1994). Transcriptional regulation of gene
expression may be another key mechanism through which malignant cell growth
and differentiation are regulated. The induction of some transcription
factors, such as AP-1 (a dimer of jun-fos or jun-jun proteins) and NF- In addition to biochemical factors that determine response, cell kinetic properties exert an important influence on the results of AraC treatment. It is likely that continued inhibition of DNA synthesis for a duration equivalent to at least one cell cycle is necessary to expose cells during the S, or DNA-synthetic, phase of the cycle. This mechanism may thus be important when AraC is administered by continuous prolonged infusion. A number of investigations have indicated that the optimal interval between bolus doses of AraC is about 8 to 12 hours. This interval may be determined by the need to maintain intracellular concentrations of AraCTP at inhibitory levels for at least one cell cycle. The mean cycle time of acute myelocytic leukemia cells is 1 to 2 days. Typical schedules for administration of AraC employ bolus doses every 12 hours for 5 to 7 days or continuous infusion for 7 days. Mechanisms of Resistance to Cytarabine Response to AraC is strongly influenced by the relative activities of anabolic and catabolic enzymes that determine the proportion of drug converted to AraCTP. The rate-limiting enzyme is deoxycytidine kinase, which produces AraCMP. An important degradative enzyme is cytidine deaminase, which deaminates AraC to a nontoxic metabolite, arauridine. Cytidine deaminase is found in high activity in many tissues, including some human tumors. A second degradative enzyme, dCMP deaminase, converts AraCMP to the inactive metabolite, AraUMP. Relationships have been shown between the synthesis and retention of AraCTP in leukemic cells and the duration of complete remission in patients with acute myeloblastic leukemia (Preisler et al., 1985). The ability of cells to transport AraC also appears to be an important determinant of the clinical response (Wiley et al., 1985). Because drug concentration in plasma rapidly falls below the level needed to saturate transport and activation processes, clinicians have employed high-dose regimens (2 to 3 g/m2 every 12 hours for 6 doses) to achieve 20- to 50-fold higher serum levels with improved results in remission induction (Bishop et al., 1996) and consolidation (Mayer et al., 1994) for acute nonlymphocytic leukemia (ANLL). Several biochemical mechanisms have been identified in AraC-resistant subpopulations in various murine and human tumor cell lines. Most commonly encountered is deficiency of deoxycytidine kinase (Flasshove et al., 1994). Another mechanism of resistance is marked expansion of the dCTP pool due to increased CTP synthase activity (Garcia-Carbonero et al., 2001). The increased concentrations of intracellular dCTP presumably can block the actions of AraCTP on DNA synthesis. Other mechanisms include increased cytidine deaminase activity and reduced affinity of DNA polymerase for AraCTP. Finally, while specific steps in AraC activation and degradation exert a strong influence on its ultimate action, the cellular response to AraC-mediated DNA damage also governs whether or not the genotoxic insult results in cell death. For example, overexpression of Bcl-2 and Bcl-xL in leukemic blasts have been associated with in vitro resistance to AraC-mediated apoptosis (Ibrado et al., 1996). Phosphorylation of DNA-damage response factors or transcription factors also may influence the cellular response to AraC toxic insult. Recent studies have shown that phosphorylation of Bcl-2 or the AP-1 transcription factor is associated with AraC resistance in human myeloid leukemia cell lines in vitro (Kolla and Studzinski, 1994). At the clinical level, AraC resistance is poorly understood. Absorption, Fate, and Excretion Due to the presence of high concentrations of cytidine deaminase in
the gastrointestinal mucosa and liver, only about 20% of the drug reaches the
circulation after oral AraC administration; thus, the drug is not given
orally. Peak concentrations of 2 to 50 Therapeutic Uses Two standard dosage schedules are recommended for administration of cytarabine (CYTOSARU, TARABINE PFS): (1) rapid intravenous injection of 100 mg/m2 every 12 hours for 5 to 7 days; or (2) continuous intravenous infusion of 100 to 200 mg/m2 daily for 5 to 7 days. In general, children seem to tolerate higher doses than do adults. Intrathecal doses of 30 mg/m2 every 4 days have been used to treat meningeal leukemia. The intrathecal administration of 50 mg of the liposomal formulation of cytarabine (DEPOCYT) every 2 weeks probably is at least as effective. In both pediatric and adult patients with acute nonlymphocytic leukemia, high-dose cytarabine (2 to 3 g/m2 administered over 2 hours every 12 hours for 6 doses) has greater efficacy but greater neurotoxicity, especially in elderly patients (Mayer et al., 1994). Cytarabine is indicated for induction of remission in acute leukemia in children and adults. When used alone, remission rates of 20% to 40% have been reported. The drug is particularly useful in acute nonlymphocytic leukemia in adults. Cytarabine is most effective when used with other agents, particularly anthracyclines or mitoxantrone. The drug also is used in combination therapy for aggressive presentations of non-Hodgkin's lymphomas in adults and in children and for treatment of relapses of acute lymphocytic leukemia in both age groups. Clinical Toxicities Cytarabine is primarily a potent myelosuppressive agent capable of producing severe leukopenia, thrombocytopenia, and anemia with striking megaloblastic changes. Other toxic manifestations include gastrointestinal disturbances, stomatitis, conjunctivitis, mild and reversible hepatic dysfunction, pneumonitis, fever, and dermatitis. Seizures and other manifestations of neurotoxicity may occur after intrathecal administration or when high doses (particularly greater than 3 g/m2) are administered intravenously to patients older than 40 and/or patients with poor renal function or abnormal alkaline phosphatase activity (Rubin et al., 1992). Gemcitabine Gemcitabine (2,2 difluorodeoxycytidine; dFdC) is the most important antimetabolite to enter the clinic in recent years and is part of the first-line regimen for patients with metastatic pancreatic cancer and non-small cell lung cancer. The drug was selected for development on the basis of its impressive activity against murine solid tumors and human xenografts in nude mice (Hertel et al., 1990). Mechanism of Action Gemcitabine retains many of the principal features of cytarabine. Influx of gemcitabine through the cell membrane occurs via active nucleoside transporters (Mackey et al., 1998). Intracellularly, deoxycytidine kinase phosphorylates gemcitabine to produce difluorodeoxycytidine monophosphate (Heinemann et al., 1988), from which point it is converted to difluorodeoxycytidine di- and triphosphate (dFdCDP, dFdCTP). While its metabolism to triphosphate status and its effects on DNA in general mimic those of cytarabine, there are differences in kinetics of inhibition, additional sites of action, incorporation in DNA, and spectrum of activity (Iwasaki et al., 1997). Unlike that of cytarabine, the cytotoxicity of gemcitabine is not confined to the S phase of the cell cycle, and the drug is equally effective against confluent cells and cells in log-phase growth. The cytotoxic activity may be a result of several actions on DNA synthesis: dFdCTP competes with dCTP as a weak inhibitor of DNA polymerase; dFdCDP is a potent inhibitor of ribonucleotide reductase, resulting in depletion of deoxyribonucleotide pools necessary for DNA synthesis; and dFdCTP is incorporated into DNA and after the incorporation of one more nucleotide leads to DNA strand termination (Heinemann et al., 1990; Huang et al., 1991). This 'extra' nucleotide may be important in hiding the dFdCTP from DNA repair enzymes, as incorporation of dFdCTP into DNA appears to be resistant to the normal mechanisms of DNA repair. The ability of cells to incorporate dFdCTP into DNA is critical for gemcitabine-induced apoptosis (Huang et al., 1995). Absorption, Fate, and Elimination Gemcitabine is administered as an intravenous infusion. The
pharmacokinetics of the parent compound are largely determined by
deamination, and the predominant elimination product is difluorodeoxyuridine
(dFdU). Gemcitabine has a t1/2 of 40 to 90 minutes,
depending on the age and gender of subjects. There is a biphasic elimination
of dFdU, which has a t1/2 Similar to that of cytarabine, conversion of gemcitabine to dFdCTP by
deoxycytidine kinase is saturated at infusion rates of approximately 10 mg/m2
per minute, which produce plasma drug concentrations in the range of 15 to
20 The activity of dFdCTP on DNA repair mechanisms may allow for increased cytotoxicity of other chemotherapeutic agents, particularly platinum compounds. Preclinical studies of tumor cell lines show that cisplatin-DNA adducts are enhanced in the presence of gemcitabine, presumably through suppression of nuclear excision repair (van Moorsel et al., 1999). Therapeutic Uses The standard dosing schedule for gemcitabine (GEMZAR) is a 30-minute intravenous infusion of 1000 to 1200 mg/m2 on days 1, 8, and 15 every 28 days. Besides pancreatic cancer and nonsmall cell lung cancer, activity has been noted in patients with transitional cell carcinoma, cervical cancer, ovarian cancer, and breast cancer. Clinical Toxicities The principal toxicity of gemcitabine is myelosuppression. In general, the longer duration infusions lead to greater myelosuppression. Nonhematologic toxicities including a flu-like syndrome, asthenia, and mild elevation in liver transaminases may occur in 40% or more of patients. Although severe nonhematologic toxicities are rare, interstitial pneumonitis may occur and is responsive to steroids. Rarely, hemolytic-uremic syndrome has been reported (Aapro et al., 1998). Purine Analogs Since the pioneering studies of Hitchings and associates, begun in 1942, many analogs of natural purine bases, nucleosides, and nucleotides have been examined in a wide variety of biological and biochemical systems. These extensive investigations have led to the development of several drugs, of use not only in the treatment of malignant diseases (mercaptopurine, thioguanine) but also for immunosuppressive (azathioprine) (Schwartz, 2000) and antiviral (acyclovir, ganciclovir, vidarabine, zidovudine) therapy (see Figure 5211). The hypoxanthine analog allopurinol, a potent inhibitor of xanthine oxidase, is an important by-product of this effort (see Chapter 27: Analgesic-Antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout). A promising development has been the discovery of powerful inhibitors of adenosine deaminase, for example, erythro-9-(2-hydroxy-3-nonyl)-adenine (EHNA) and pentostatin (2'-deoxycoformycin). Recent studies have confirmed that pentostatin has clinical activity against hairy cell and chronic lymphocytic leukemias and lymphomas. In experimental systems, these inhibitors of adenosine deaminase have produced marked synergistic effects in combination with various analogs of adenosine, such as vidarabine (arabinosyladenine; AraA); they also show promise as immunosuppressive agents. Two drugs that are resistant to deamination by adenosine deaminase, fludarabine phosphate and cladribine, have outstanding activity in several types of leukemias and lymphomas (Beutler, 1992; Cheson, 1992; Piro, 1992; Calabresi and Schein 1993; Chabner et al., 2001).
StructureActivity Relationship Mercaptopurine and thioguanine, both established clinical agents for
the therapy of human leukemias, are analogs of the natural purines
hypoxanthine and guanine, in which the keto group on carbon 6 of the purine
ring is replaced by a sulfur atom. Substitution in this position by chlorine
or selenium also yields antineoplastic compounds. Cytotoxicity also is
observed with the Many attempts have been made to modify the structures of such analogs to improve their therapeutic indices or selectivity. Azathioprine (see Figure 5211) was developed to decrease the rate of inactivation of 6-mercaptopurine by enzymatic S-methylation, nonenzymatic oxidation, or conversion to thiourate by xanthine oxidase. Azathioprine can react with sulfhydryl compounds such as glutathione (apparently nonenzymatically) and thus serves as a prodrug, permitting the slow liberation of mercaptopurine in tissues. Superior immunosuppressive activity is achieved in comparison with mercaptopurine (Elion, 1967). An important development has been the discovery of potent inhibitors of adenosine deaminase such as pentostatin (2'-deoxycoformycin; Ki= 2.5 pM) and erythro-9-(2-hydroxy-3-nonyl)-adenine (EHNA; Ki= 2 nM). Pentostatin (see Figure 5211) is a natural product derived from Streptomyces. Structurally, the drug resembles the transition state of adenosine as it is hydrolyzed by adenosine deaminase. As a result, the drug has an affinity for the enzyme that is 107-fold greater than that of the natural substrate. The enzymeinhibitor complex is very stable and dissociates with a t of about 25 to 30 hours (Agarwal et al., 1977; Agarwal, 1982). Thus, pentostatin blocks not only the deamination of natural nucleosides but also that of many analogs used in chemotherapy. Mechanism of Action Although animal tissues have nucleoside kinases that are capable of converting adenosine or the 2'-deoxyribonucleosides of guanine, hypoxanthine, adenine, and many of their analogs to the corresponding 5'-monophosphates, similar reactions do not occur with inosine, guanosine, or their analogs. The latter compounds must first undergo phosphorolysis by purine nucleoside phosphorylase, which is present in high activity in many human tissues. The liberated bases then may be converted to the corresponding nucleotides by hypoxanthine-guanine phosphoribosyltransferase (HGPRT). Similarly, 2'-deoxyguanosine, 2'-deoxyinosine, and many related analogs may react with purine nucleoside phosphorylase, and the product of this reactiona purine base or analogthen may be converted to the corresponding ribonucleoside 5'-monophosphate. Both thioguanine and mercaptopurine are excellent substrates for HGPRT and are converted to the ribonucleotides 6-thioguanosine-5'-phosphate (6-thioGMP) and 6-thioinosine-5'-phosphate (T-IMP), respectively. Because T-IMP is a poor substrate for guanylyl kinase, the enzyme that converts GMP to GDP, T-IMP accumulates intracellularly. Careful studies have demonstrated, however, that mercaptopurine can be incorporated into cellular DNA in the form of thioguanine deoxyribo-nucleotide, indicating that slow reactions catalyzed by enzymes of guanine metabolism can operate. The accumulation of T-IMP may inhibit several vital metabolic reactions, such as the conversion of inosinate (IMP) to adenylosuccinate (AMPS) and then to adenosine-5'-phosphate (AMP) and the oxidation of IMP to xanthylate (XMP) by inosinate dehydrogenase. These reactions are crucial steps in the conversion of IMP to adenine and guanine nucleotides. On the other hand, in cells incubated with thioguanine, 6-thioGMP first accumulates; it is a poor but definite substrate for guanylyl kinase. Thus, there is slow conversion to 6-thioGDP and 6-thioGTP and entry of thioguanine nucleotides into the nucleic acids of the cell. In addition, the concentrations of 6-thioGMP achieved are sufficient to cause progressive and irreversible inhibition of inosinate dehydrogenase, presumably through the formation of disulfide bonds. Furthermore, both 6-thioGMP and T-IMP, as well as a number of other 5'-monophosphate derivatives of purine nucleoside analogs, can cause 'pseudofeedback inhibition' of the first committed step in the de novo pathway of purine biosynthesis, the reaction of glutamine and PRPP to form ribosylamine-5-phosphate. This enzyme is a major control point in the biosynthesis of purine nucleotides, and its activity is regulated by the intracellular concentrations of 5'-mononucleotides (natural, as well as analogs). The synthesis of PRPP also is powerfully inhibited by ADP and ATP or related analogs. Mercaptopurine also inhibits 6-phosphofructo-2-kinase, an essential enzyme in the glycolytic pathway (Mojena et al., 1992). 6-Methymercaptopurine ribonucleoside (MMPR) is produced from mercaptopurine in an S-methylation reaction catalyzed by 6-thiopurine methytransferase. The nucleotides of MMPR are potent inhibitors of de novo purine biosynthesis. MMPR has been shown to have antiangiogenic activity in vivo (Presta et al., 1999). Despite extensive investigations, it is still not possible to assess precisely the role of incorporation of thioguanine or mercaptopurine into cellular DNA in the production of either the therapeutic or toxic effects of these drugs (Bo et al., 1999; Marathias et al., 1999). The incidence of pregnancy-related complications, however, was significantly increased when fathers used mercaptopurine within 3 months of conception (Rajapakse et al., 2000). These compounds can cause marked inhibition of the coordinated induction of various enzymes required for DNA synthesis, as well as potentially critical alterations in the synthesis of polyadenylate-containing RNA (Carrico and Sartorelli, 1977; Giverhaug et al., 1999). Other studies indicate that disruption of the synthesis of membrane glycoproteins may be caused by brief exposure to 6-thioguanine. These effects, which are potentially lethal to cellular survival, are likely mediated by depletion of guanosine diphosphate sugars. In view of these diverse biochemical actions, which involve vital systems such as purine biosynthesis, nucleotide interconversions, DNA and RNA synthesis, chromosomal replication, and glycoprotein synthesis, it is not possible to pinpoint a single biochemical event as the cause of thiopurine cytotoxicity. It seems likely that this class of drugs acts by multiple mechanisms (Hortelano and Bosca, 1997). Recently, a thiopurine analog, 6-methylmercaptopurine riboside also has been shown to inhibit angiogenesis (Presta et al., 1999). Of many adenosine analogs studied experimentally, vidarabine
(arabinosyladenine, AraA) is used for the treatment of herpetic infections (see
Chapter 50: Antimicrobial Agents: Antiviral Agents (Nonretroviral)), but it
has failed to produce useful antitumor activity due to its rapid deamination.
A deamination-resistant analog, 2-fluoro-9- As mentioned above, pentostatin is a potent inhibitor of adenosine deaminase. The relationship between this effect and drug-induced cytotoxicity, however, is not clear. Alterations of the usual intracellular concentrations of adenosine-containing compounds appear to cause feedback inhibition of S-adenosyl-homocysteine hydrolase; as a result, cellular methylation reactions are impaired. The drug interferes with the synthesis of nicotinamide adenine dinucleotide. The nucleoside triphosphate analog of pentostatin can be incorporated into DNA, resulting in strand breakage (Siaw and Coleman, 1984; Johnston et al., 1986; Begleiter et al., 1987; Calabresi et al., 1993; Chabner et al., 2001). Genetic deficiency of adenosine deaminase is associated with malfunction of both T and B lymphocytes, with little effect on other normal tissues (Giblett et al., 1972). Thus, animals treated with pentostatin display marked immunosuppression. Severe and sometimes fatal opportunistic infections also have been associated with the clinical use of pentostatin. Treatment with pentostatin alone has induced remissions in T lymphocyterelated diseases, such as T-cell leukemia and mycosis fungoides. These initial trials were predicated on the observation that malignant T cells have high levels of adenosine deaminase. However, encouraging results also have been obtained in B-cell disease; 25% of patients with refractory chronic lymphocytic leukemia have responded to the drug, as have 90% of patients with hairy cell leukemia. Another purine analog that has shown potent activity in hairy cell leukemia is 2-chlorodeoxyadenosine (2-CdA, cladribine). Cladribine is resistant to adenosine deaminase, and after intracellular phosphorylation by deoxycytidine kinase, it is incorporated into DNA. Because of its extremely high effectiveness and lower toxicity, cladribine often is preferred to pentostatin in hairy cell leukemia. It also is active in other leukemias and lymphoma (see Symposium, 1984; Tritsch, 1985; Beutler, 1992; Kay et al., 1992; Estey et al., 1992; Saven and Piro, 1992; Hoffman et al., 1994; Tallman et al., 1995; Dearden et al., 1999; Chabner et al., 2001). Mechanisms of Resistance to the Purine Antimetabolites As with other tumor-inhibiting antimetabolites, acquired resistance is a major obstacle to the successful use of the purine analogs. The most commonly encountered mechanism observed in vitro is deficiency or complete lack of the enzyme HGPRT. In addition, resistance can result from decreases in the affinity of this enzyme for its substrates. Cells that are resistant because of these mechanisms usually show cross-resistance to analogs such as mercaptopurine, thioguanine, and 8-azaguanine. Another mechanism of resistance identified in cells from leukemic patients is an increase in particulate alkaline phosphatase activity. Other mechanisms include (1) decreased drug transport, (2) increased rates of degradation of the drugs or their intracellular 'activated' analogs, (3) alteration in allosteric inhibition of ribosylamine 5-phosphate synthase, (4) altered DNA-repair efficiency, (5) loss or alterations of the enzymes adenine phosphoribosyltransferase or adenosine kinase (for adenine or adenosine analogs and deoxycytidine kinase for fludarabine phosphate), and (6) increased activity of multidrug resistance protein 5 (Wijnholds et al., 2000). However, the most important determinants of resistance to these drugs in the clinical setting remain uncertain (for review, see Brockman, 1974). Mercaptopurine The introduction of mercaptopurine by Elion and coworkers represents a landmark in the history of antineoplastic and immunosuppressive therapy. Mercaptopurine and its derivative, azathioprine, remain among the most important and most clinically useful drugs of the class (Relling et al., 1999a,b; Mahoney et al., 1998). Mercaptopurine is used principally in the maintenance therapy of acute lymphocytic leukemia. The structureactivity relationship and the mechanism of action and of drug resistance are discussed above. The structural formula of mercaptopurine is presented in Figure 5211. Absorption, Fate, and Excretion Absorption of mercaptopurine is incomplete after oral ingestion and
bioavailability is reduced by first-pass metabolism by the liver. Oral bioavailability
is only 5% to 37%, with great interpatient variability. Measurements of drug
concentrations in plasma may be necessary to optimize therapy with oral
mercaptopurine. Bioavailability is increased when mercaptopurine is combined
with high-dose methotrexate (Innocenti et al., 1996). After an
intravenous dose, the half-life of the drug in plasma is relatively short
(about 50 minutes in adults), due to uptake by cells, renal excretion, and
rapid metabolic degradation. Restricted brain distribution of mercaptopurine
is related to the efficient efflux transport system in the bloodbrain
barrier (Deguchi et al., 2000). In addition to the HGPRT-catalyzed
anabolism of mercaptopurine, there are two other pathways for its metabolism.
The first involves methylation of the sulfhydryl group and subsequent
oxidation of the methylated derivatives. Expression of the enzyme thiopurine
methyltransferase reflects the inheritance of polymorphic alleles (Yates et
al., 1997; Iyer, 1999; Relling et al., 1999b); up to 15% of the
population of the An attempt to modify the metabolic inactivation of mercaptopurine by xanthine oxidase led to the development of allopurinol. This analog of hypoxanthine is a powerful inhibitor of xanthine oxidase and not only blocks the conversion of mercaptopurine to 6-thiouric acid but also interferes with the production of uric acid from hypoxanthine and xanthine (see Chapter 27: Analgesic-Antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout). Because of its ability to interfere with the enzymatic oxidation of mercaptopurine and related derivatives, allopurinol increases the exposure of cells to the action of these compounds. Although it greatly potentiates the antineoplastic action of mercaptopurine in tumor-bearing mice, allopurinol increases the toxicity as well, and there is no apparent improvement in the therapeutic index (Zinner and Klastersky, 1985). Therapeutic Uses The initial average daily oral dose of mercaptopurine (6-mercaptopurine;PURINETHOL) is 2.5 mg/kg. Starting doses usually range from 100 to 200 mg per day; with hematological and clinical improvement, the dose is diminished to an appropriate multiple of 25 mg and, in general, maintenance therapy of 1.5 to 2.5 mg/kg per day is continued. If beneficial effects have not been noted after 4 weeks, the daily dose may be increased gradually up to 5 mg/kg until evidence of toxicity is encountered. The total dose required to produce depression of the bone marrow in patients with nonhematological malignancies is about 45 mg/kg and may range from 18 to 106 mg/kg. Hyperuricemia with hyperuricosuria may occur during treatment; the accumulation of uric acid presumably reflects the destruction of cells with release of purines that are oxidized by xanthine oxidase, as well as an inhibition of the conversion of inosinic acid to precursors of nucleic acids. This circumstance may be an indication for the use of allopurinol. Special caution must be employed if mercaptopurine or its imidazolyl derivative, azathioprine, is used with allopurinol, for reasons presented above. Patients treated simultaneously with both drugs should receive approximately 25% of the usual dose of mercaptopurine. In the early studies with mercaptopurine, bone marrow remissions were described in more than 40% of children with acute leukemia. In adults with acute leukemia, the results have been much less impressive, but occasional remissions have been obtained. The drug has contributed to the treatment of lymphoblastic leukemia more by maintaining than by inducing remissions. Cross-resistance does not occur between mercaptopurine and other classes of antileukemic agents. In the treatment of chronic granulocytic leukemia, maintenance therapy with mercaptopurine can be useful, but more effective agents are available. Mercaptopurine has not been of value in chronic lymphocytic leukemia, Hodgkin's disease and related lymphomas, and a wide variety of carcinomas, even at unusually high doses. Although active as an immunosuppressive agent, it has been superseded by its imidazolyl derivative, azathioprine. Clinical Toxicities The principal toxic effect of mercaptopurine is bone marrow depression, although, in general, this develops more gradually than with folic acid antagonists; accordingly, thrombocytopenia, granulocytopenia, or anemia may not be encountered for several weeks. When depression of normal bone marrow elements occurs, cessation of therapy with the drug usually results in prompt recovery. Anorexia, nausea, or vomiting is seen in approximately 25% of adults, but stomatitis and diarrhea are rare; manifestations of gastrointestinal effects are less frequent in children than in adults. The occurrence of jaundice in about one-third of adult patients treated with mercaptopurine has been reported; although the pathogenesis of this manifestation is obscure, it usually clears upon discontinuation of therapy. Its appearance has been associated with bile stasis and hepatic necrosis. Dermatological manifestations have been reported. The long-term complications associated with the use of mercaptopurine and its derivative, azathioprine, for immunosuppressive therapy are discussed by Schein and Winokur (1975), Kirschner (1998), and Korelitz et al. (1999). Teratogenic effects during the first trimester are associated with chronic mercaptopurine treatment, and acute myelogenous leukemia has been reported after prolonged use of mercaptopurine for Crohn's disease (Heizer and Peterson, 1998). Azathioprine Azathioprine, a derivative of 6-mercaptopurine, is used as an immunosuppressive agent; its structural formula is shown in Figure 5211. The rationale that led to its synthesis and its mechanism of action and metabolic degradation have been discussed above. Additional information is presented in Chapter 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants. Thioguanine The synthesis of thioguanine was first described by Elion and Hitchings in 1955. It is of particular value in the treatment of acute granulocytic leukemia when given with cytarabine. The structural formula of thioguanine is shown in Figure 5211, and its mechanism of action is discussed above. Absorption, Fate, and Excretion Absorption of thioguanine is incomplete and erratic, and concentrations of the drug in plasma may vary more than tenfold after oral administration. Peak concentrations in the blood are reached 2 to 4 hours after ingestion. When thioguanine is administered to human beings, the S-methylation product, 2-amino-6-methylthiopurine, rather than free thioguanine appears in the urine; inorganic sulfate and, after continuous intravenous infusion, 8-hydroxy-thioguanine also are major urinary metabolites (Kitchen et al., 1999). Lesser amounts of 6-thiouric acid are formed, suggesting that deamination catalyzed by the enzyme guanase does not have a major role in the metabolic inactivation of thioguanine. Since 6-thioxanthine, the deamination product of thioguanine, is inactive, thioguanine may be administered concurrently with allopurinol without reduction in dosage, unlike mercaptopurine and azathioprine. Therapeutic Uses Thioguanine (6-thioguanine, TG) is available for oral administration. The average daily dose is 2 mg/kg. If there is no clinical improvement or toxicity after 4 weeks, the dosage may be increased cautiously to 3 mg/kg daily. Clinically, thioguanine has been used in the treatment of acute leukemia and, in conjunction with cytarabine, is one of the most effective agents for induction of remissions in acute granulocytic leukemia; it has not been useful in the treatment of patients with solid tumors. This compound has been used as an immunosuppressive agent, particularly in patients with nephrosis or with collagen-vascular disorders. Clinical Toxicities Toxic manifestations include bone marrow depression and gastrointestinal effects, although the latter may be less pronounced than with mercaptopurine. Hepatotoxicity also is lower for thioguanine than for mercaptopurine. Fludarabine Phosphate A fluorinated deamination-resistant nucleotide analog of the antiviral
agent vidarabine (9- The structural formulas of fludarabine phosphate and a related adenosine analog, cladribine, are shown below:
Absorption, Fate, and Excretion Fludarabine phosphate is administered intravenously and is rapidly converted to fludarabine in the plasma. The terminal half-life of fludarabine is approximately 10 hours. The compound is primarily eliminated by renal excretion, and approximately 23% appears in the urine as fludarabine because of its relative resistance to deamination by adenosine deaminase. Therapeutic Uses Fludarabine phosphate FLUDARA) is available for intravenous use. The recommended dose of fludarabine phosphate is 20 to 30 mg/m2 daily for 5 days. The drug is administered intravenously by infusion during a period of 30 minutes to 2 hours. Dosage may need to be reduced in renal impairment. Treatment may be repeated every 4 weeks, and gradual improvement, at these doses, usually occurs during a period of two to three cycles. Fludarabine phosphate is used primarily for the treatment of patients with chronic lymphocytic leukemia (CLL), although experience is accumulating that suggests effectiveness in B-cell lymphomas refractory to standard therapy. Activity also has been seen with indolent non-Hodgkin's lymphoma, promyelocytic leukemia, cutaneous T-cell lymphoma, and Waldentrm's macroglobulinemia (Chun et al., 1991). In CLL patients previously refractory to a regimen containing a standard alkylating agent, response rates of 32% to 48% have been reported. In patients with previously untreated low-grade lymphomas, fludarabine phosphate in combination with cyclophosphamide has resulted in almost 90% complete responses (Hochster et al., 1994). In combination with the anthracycline, mitoxantrone, and dexamethasone, fludarabine phosphate gives response rates greater than 90% against indolent non-Hodgkin's lymphoma (McLaughlin et al., 1996; Emmanouilides et al., 1998). Clinical Toxicities Toxic manifestations include myelosuppression, nausea and vomiting, and chills and fever, as well as malaise, anorexia, and weakness. Lymphopenia and thrombocytopenia are dose-limiting and possibly cumulative (Malspeis et al., 1990). CD4-positive T cells are depleted with therapy (O'Brien et al., 1993). Opportunistic infections (Cheson, 1995) and tumor lysis syndrome have been reported. Peripheral neuropathy may occur at standard doses (Cheson et al., 1999), as well as altered mental status, seizures, optic neuritis, and coma, usually at higher doses. Neurotoxicity is seen more frequently and with increased severity in older patients. In combination with pentostatin, severe or even fatal pulmonary toxicity has been encountered. Because a significant fraction of drug (about one-quarter) is eliminated in the urine, patients with compromised renal function should be treated with caution. Initial doses should be reduced in proportion to serum creatinine levels. Pentostatin (2'-Deoxycoformycin) Pentostatin is a transition-state analog of the intermediate stage in
the adenosine deaminase ( The structural formula of pentostatin (2'-deoxycoformycin) is shown in Figure 5211. Absorption, Fate, and Excretion Pentostatin is administered intravenously, and a single dose of 4 mg/m2 has been reported to have a distribution half-life of 11 minutes and a mean terminal half-life of 5.7 hours. The drug is eliminated almost entirely by renal excretion. Appropriate reduction of dosage is recommended in patients with renal impairment as measured by reduced creatinine clearance. Therapeutic Uses Pentostatin NIPENT) is available for intravenous use. The recommended dosage is 4 mg/m2 administered every other week. After hydration with 500 to 1000 ml of 5%dextrose in half-normal saline, the drug is administered by rapid intravenous injection or by infusion during a period of up to 30 minutes, followed by an additional 500 ml of fluids. Extravasation does not produce cellulitis, vesication, or tissue necrosis. Pentostatin is extremely effective in producing complete remissions in hairy cell leukemia. Complete responses of 58% and partial responses of 28% have been reported even in patients who were refractory to interferon-alfa. Activity also is seen against CLL, CML, promyelocytic leukemia, cutaneous T-cell lymphoma, non-Hodgkin's lymphoma, and Langerhans-cell histiocytosis (Dillman, 1994; Cortes et al., 1997). Pentostatin has no significant activity against solid tumors or multiple myeloma. Clinical Toxicities Toxic manifestations include myelosuppression, gastrointestinal symptoms, skin rashes, and abnormal liver function studies at standard (4 mg/m2) doses. Depletion of normal T cells occurs at these doses, and neutropenic fever and opportunistic infections have been reported (Steis et al., 1991). Immunosuppression may persist for several years after discontinuation of pentostatin therapy (Kraut et al., 1990). At higher doses (10 mg/m2), major renal and neurological complications are encountered. The use of pentostatin in combination with fludarabine phosphate may result in severe or even fatal pulmonary toxicity. Cladribine An adenosine deaminase-resistant purine analog, cladribine (2-chlorodeoxyadenosine; 2-CdA) has demonstrated potent activity in hairy cell leukemia, chronic lymphocytic leukemia, and low-grade lymphomas (Estey et al., 1992; Kay et al., 1992; Beutler, 1992; Dearden et al., 1999; Tondini et al., 2000). It appears to be safe and moderately effective in patients with progressive multiple sclerosis (Rice et al., 2000). After intracellular phosphorylation by deoxycytidine kinase and conversion to cladribine triphosphate, it is incorporated into DNA. It produces DNA strand breaks and NAD and ATP depletion, as well as apoptosis in some cell lines (Piro, 1992; Beutler, 1992). Although the precise mechanism of action of cladribine is not fully understood, the drug does not require cell division to be cytotoxic. The structural formula of cladribine is shown above with that of fludarabine-5-phosphate. Absorption, Fate, and Excretion Cladribine is not well absorbed orally (55% 17%) and is administered intravenously (Liliemark et al., 1992). The drug is excreted primarily by the kidneys, with plasma half-lives of 35 minutes and 6.7 hours (Liliemark and Juliusson, 1991). Cladribine crosses the bloodbrain barrier and reaches CSF concentrations of about 25% of those seen in plasma. In patients with meningeal involvement, however, CSF concentrations can exceed those in plasma. Therapeutic Uses Cladribine LEUSTATIN) is available in an injectable dosage form. The recommended dose is a single course of 0.09 mg/kg per day for 7 days by continuous intravenous infusion. Cladribine is considered the drug of choice in hairy cell leukemia because of its high level of effectiveness and low profile of toxicity. Complete responses have been reported in 80% of patients, and partial responses in the rest after a single course of therapy (Saven and Piro, 1992; Dearden et al., 1999). The drug also is active in chronic lymphocytic leukemia, acute myelogenous leukemia, especially in pediatric patients, low-grade lymphomas, Langerhans-cell histiocytosis, cutaneous T-cell lymphomas, including mycosis fungoides and the Szary syndrome, and Waldenstrm's macroglobulinemia (Piro et al., 1988; Piro, 1992; Santana et al., 1992; Kuzel et al., 1992; Kay et al., 1992; Saven et al., 1992; Dimopoulos et al., 1993; Robak et al., 1999; Tondini et al., 2000; Saven and Burian, 1999). Clinical Toxicities The major dose-limiting toxicity of cladribine is myelosuppression, although cumulative thrombocytopenia may occur with repeated courses. Opportunistic infections are common and are correlated with decreased CD4+ cell counts. Other toxic effects include nausea, infections, high fever, headache, fatigue, skin rashes, and tumor lysis syndrome. Neurological and immunosuppressive adverse effects are less evident than with pentostatin at clinically active doses, perhaps because cladribine is not an inhibitor of adenosine deaminase. |
Natural Products
|
Antimitotic Drugs Vinca Alkaloids History The beneficial properties of the Madagascar periwinkle plant, Catharanthus roseus (formerly called Vinca rosea), a species of myrtle, have been described in medicinal folklore in various parts of the world. While exploring claims that extracts of the periwinkle might have beneficial effects in diabetes mellitus, Noble and coworkers (1958) observed granulocytopenia and bone marrow suppression in rats, effects that led to purification of an active alkaloid. Other investigations, by Johnson and associates, demonstrated activity of certain alkaloidal fractions against an acute lymphocytic neoplasm in mice. Fractionation of these extracts yielded four active dimeric alkaloids: vinblastine, vincristine, vinleurosine, and vinrosidine. Two of these, vinblastine and vincristine, are important clinical agents for treatment of leukemias, lymphomas, and testicular cancer. Another agent, vinorelbine, has important activity against lung cancer and breast cancer (see Budman, 1997). Chemistry The vinca alkaloid antimitotic agents are asymmetrical dimeric compounds; the structures of vinblastine, vincristine, vindesine, and vinorelbine are shown opposite. StructureActivity Relationship Minor differences in structure result in notable differences in toxicity and antitumor spectra among the vinca alkaloids. A number of related dimeric alkaloids are without biological activity. Removal of the acetyl group at C4 of one portion of vinblastine destroys its antileukemic activity, as does acetylation of the hydroxyl groups. Either hydrogenation of the double bond or reductive formation of carbinols reduces or destroys activity of these compounds. Mechanism of Action The vinca alkaloids are cell-cyclespecific agents and, in common with other drugs such as colchicine, podophyllotoxin, and taxanes, block cells in mitosis. The biological activities of these drugs can be explained by their ability to bind specifically to tubulin and to block the ability of the protein to polymerize into microtubules. When cells are incubated with vinblastine, dissolution of the microtubules occurs, and highly regular crystals are formed that contain 1 mol of bound vinblastine per mol of tubulin. Through disruption of the microtubules of the mitotic apparatus, cell division is arrested in metaphase. In the absence of an intact mitotic spindle, the chromosomes may disperse throughout the cytoplasm (exploded mitosis) or may clump in unusual groupings, such as balls or stars. The inability to segregate chromosomes correctly during mitosis presumably leads to cell death. Both normal and malignant cells exposed to vinca alkaloids undergo changes characteristic of apoptosis (Smets, 1994).
In addition to their key role in the formation of mitotic spindles, microtubules are involved in other cellular functions such as movement, phagocytosis, and axonal transport. Side effects of the vinca alkaloids, such as their neurotoxicity, may be due to disruption of these functions. Drug Resistance Despite their structural similarity, cross-resistance among the individual vinca alkaloids is not absolute. For over a decade, however, attention has been drawn to the phenomenon of multidrug resistance, in which tumor cells become cross-resistant to a wide range of chemically dissimilar agents after exposure to a single (natural product) drug. Such multidrug-resistant tumor cells display cross-resistance to vinca alkaloids, the epipodophyllotoxins, anthracyclines, and taxanes. Chromosomal abnormalities consistent with gene amplification have been observed in resistant cells in culture, and the cells contain markedly increased levels of the P-glycoprotein, a membrane efflux pump that transports drugs from the cells (see Endicott and Ling, 1989). Ca2+channel blockers, such as verapamil, can reverse resistance of this type. Other membrane transporters such as the multidrug resistance-associated protein (Kuss et al., 1994) may mediate multidrug resistance; still other forms of resistance to vinca alkaloids involve mutations in tubulin that prevent the effective binding of the inhibitors to their target. Cytotoxic Actions Both vincristine and vinblastine, as well as the analog vinorelbine, have potent and selective antitumor effects, although their actions on normal tissue differ significantly. Vincristine is a standard component of regimens for treating pediatric leukemias and solid tumors and is frequently used in adult lymphoma treatment. Vinblastine is employed primarily in treating testicular carcinomas and lymphomas and as second-line therapy of various solid tumors. Vinorelbine has activity against nonsmall cell lung cancer and breast cancer, and its range of usefulness is expanding as new trials mature. The limited myelosuppressive action of vincristine makes it a valuable component of a number of combination therapy regimens for leukemia and lymphoma, while the lack of neurotoxicity of vinblastine is a decided advantage in relapsed lymphomas or in combination with cisplatin against testicular cancer. Vinorelbine, which causes a mild neurotoxicity as well as myelosuppression, has an intermediate toxicity profile. Myelosuppression The nadir of leukopenia following vinblastine or vinorelbine occurs 7 to 10 days following drug administration. Vincristine in standard doses, 1.4 to 2 mg/m2, causes little reduction of formed elements in the blood. All three agents cause hair loss and local cellulitis if extravasated. A syndrome of inappropriate secretion of antidiuretic hormone occurs rarely after vincristine administration. Neurological Toxicity While all three derivatives may cause neurotoxic symptoms, vincristine has predictable cumulative effects. Numbness and tingling of the extremities and loss of deep tendon reflexes constitute the most common and earliest signs and are followed by motor weakness. The sensory changes do not usually warrant an immediate reduction in drug dose, but loss of motor function should lead to a reevaluation of the therapeutic plan and, under most circumstances, discontinuation of the drug. Rarely, patients may experience vocal cord paralysis or loss of extraocular muscle function. High-dose vincristine causes severe constipation or obstipation. Inadvertent intrathecal vincristine administration produces devastating and invariably fatal central neurotoxicity, with seizures and irreversible coma (Williams et al., 1983). Absorption, Fate, and Excretion All three agents are extensively metabolized by the liver, and the conjugates and metabolites are excreted in the bile (Zhou and Rahmani, 1992; Robieux et al., 1996). Only a small fraction of a dose (less than 15%) is found in the urine unchanged. In patients with hepatic dysfunction (bilirubin greater than 3 mg/dl), a 75% reduction in dose of any of the vinca alkaloids is advisable, although firm guidelines for dose adjustment have not been established. The pharmacokinetics of each of the three drugs are similar, with elimination half-lives of 1 and 20 hours for vincristine, 3 and 23 hours for vinblastine, and 1 and 45 hours for vinorelbine (Marquet et al., 1992). Vinblastine Therapeutic Uses Vinblastine sulfate VELBAN) is given intravenously; special precautions must be taken against subcutaneous extravasation, since this may cause painful irritation and ulceration. The drug should not be injected into an extremity with impaired circulation. After a single dose of 0.3 mg/kg of body weight, myelosuppression reaches its maximum in 7 to 10 days. If a moderate level of leukopenia (approximately 3000 cells per cubic millimeter) is not attained, the weekly dose may be increased gradually by increments of 0.05 mg/kg of body weight. In regimens designed to cure testicular cancer, vinblastine is used in doses of 0.3 mg/kg every 3 weeks irrespective of blood cell counts or toxicity. The most important clinical use of vinblastine is with bleomycin and cisplatin (see below) in the curative therapy of metastatic testicular tumors (Williams and Einhorn, 1985), although it has been significantly supplanted by etoposide in this disease. Beneficial responses have been reported in various lymphomas, particularly Hodgkin's disease, where significant improvement may be noted in 50% to 90% of cases. The effectiveness of vinblastine in a high proportion of lymphomas is not diminished when the disease is refractory to alkylating agents. It also is active in Kaposi's sarcoma, neuroblastoma, and LettererSiwe disease (histiocytosis X), as well as in carcinoma of the breast and choriocarcinoma. Clinical Toxicities The nadir of the leukopenia that follows the administration of vinblastine usually occurs within 7 to 10 days, after which recovery ensues within 7 days. Other toxic effects of vinblastine include neurological manifestations as described above. Gastrointestinal disturbances, including nausea, vomiting, anorexia, and diarrhea, may be encountered. The syndrome of inappropriate secretion of antidiuretic hormone has been reported. Loss of hair, mucositis of the mouth, and dermatitis occur infrequently. Extravasation during injection may lead to cellulitis and phlebitis. Local injection of hyaluronidase and application of moderate heat to the area may be of help by dispersing the drug. Vincristine Therapeutic Uses Vincristine sulfate ONCOVIN, VINCASAR PFS, others) used together with corticosteroids is presently the treatment of choice to induce remissions in childhood leukemia; common dosages for these drugs are vincristine, intravenously, 2 mg/m2 of body surface area, weekly, and prednisone, orally, 40 mg/m2, daily. Adult patients with Hodgkin's disease or non-Hodgkin's lymphomas usually receive vincristine as part of a complex protocol. When used in the MOPP regimen (see below), the recommended dose of vincristine is 1.4 mg/m2. Vincristine seems to be tolerated better by children than by adults, who may experience severe, progressive neurological toxicity. Administration of the drug more frequently than every 7 days or at higher doses seems to increase the toxic manifestations without proportional improvement in the response rate. Maintenance therapy with vincristine is not recommended in children with leukemia. Precautions also should be used to avoid extravasation during intravenous administration of vincristine. Despite their structural similarity, vincristine has a spectrum of clinical activity that differs significantly from that of vinblastine, although vincristine is effective in Hodgkin's disease and other lymphomas. It appears to be somewhat less beneficial than vinblastine when used alone in Hodgkin's disease, but when used with mechlorethamine, prednisone, and procarbazine (the so-called MOPP regimen), it was a part of the first curative treatment for the advanced stages (III and IV) of this disease (DeVita, 1981). In large-cell non-Hodgkin's lymphomas, vincristine remains an important agent, particularly when used in the CHOP regimen with cyclophosphamide, doxorubicin, and prednisone. As mentioned previously, vincristine is more useful for remission induction in lymphocytic leukemia. Vincristine also is a standard component of a number of regimens used to treat pediatric solid tumors such as Wilms' tumor, neuroblastoma, and rhabdomyosarcoma. Clinical Toxicities The clinical toxicity of vincristine is mostly neurological, as described above. The more severe neurological manifestations may be avoided or reversed by either suspending therapy or reducing the dosage upon occurrence of motor dysfunction. Severe constipation, sometimes resulting in colicky abdominal pain and obstruction, may be prevented by a prophylactic program of laxatives and hydrophilic agents and is usually a problem only with doses above 2 mg/m2. Alopecia occurs in about 20% of patients given vincristine; however, it is always reversible, frequently without cessation of therapy. Although less common than with vinblastine, leukopenia may occur with vincristine, and thrombocytopenia, anemia, polyuria, dysuria, fever, and gastrointestinal symptoms have been reported occasionally. The syndrome of hyponatremia associated with high urinary concentration of Na+ and inappropriate secretion of antidiuretic hormone occasionally has been observed during vincristine therapy. In view of the rapid action of the vinca alkaloids, it is advisable to prevent hyperuricemia by the administration of allopurinol. Vinorelbine Vinorelbine NAVELBINE) is administered in normal saline as an intravenous infusion of 30 mg/m2 given over 6 to 10 minutes. A lower dose (20 to 25 mg/m2) may be required for patients who have received prior chemotherapy. It is initially given every week until progression of disease or dose-limiting toxicity when used alone. When used with cisplatin for the treatment of non-small cell lung carcinoma, it is given every 3 weeks. Its primary toxicity is granulocytopenia, with only modest thrombocytopenia and less neurotoxicity than other vinca alkaloids. It may cause allergic reactions and mild, reversible changes in liver enzymes. In experimental studies, it has been given in an oral capsule, but bioavailability is only 30% to 40% (Fumoleau et al., 1993). Paclitaxel This compound, first isolated from the bark of the Western yew tree in 1971 (Wani et al., 1971), exhibits unique pharmacological actions as an inhibitor of mitosis, differing from the vinca alkaloids and colchicine derivatives in that it promotes rather than inhibits microtubule formation. The drug has a central role in the combination therapy of cisplatin-refractory ovarian, breast, lung, esophagus, bladder, and head and neck cancers (Rowinsky et al., 1993; Rowinsky and Donehower, 1995). The optimal dose, schedule, and use in drug combinations are incompletely understood. Chemistry Paclitaxel is a diterpenoid compound that contains a complex taxane ring as its nucleus (Figure 5212). The side chain linked to the taxane ring at carbon 13 is essential for its antitumor activity. Modification of the side chain has led to identification of a more potent analog, docetaxel (TAXOTERE) (Figure 5212), which has clinical activity against breast and ovarian cancers. Originally purified as the parent molecule from yew bark, paclitaxel can now be obtained for commercial purposes by semisynthesis from 10-desacetylbaccatin, a precursor found in yew leaves. It also has been successfully synthesized (Nicolaou et al., 1994) in a complex series of reactions. The molecule has very limited solubility and must be administered in a vehicle of 50% ethanol and 50% polyethoxylated castor oil (Cremophor EL), a formation likely responsible for a high rate of hypersensitivity reactions in patients not protected with both a histamine H1-receptor antagonist such as diphenhydramine, an H2-receptor antagonist such as cimetidine (see Chapter 25: Histamine, Bradykinin, and Their Antagonists), and a corticosteroid such as dexamethasone (see Chapter 60: Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones).
Mechanism of Action Interest in paclitaxel was stimulated by the finding that the drug
possessed the unique ability to promote microtubule formation at cold
temperatures and in the absence of GTP. It binds specifically to the In cultured tumor cells, resistance to paclitaxel is associated in
some lines with increased expression of the mdr-1 gene and its product,
the P-glycoprotein; other resistant cells have Absorption, Fate, and Excretion Paclitaxel is administered as a 3-hour or 24-hour infusion every 3 weeks, or as a weekly 1-hour infusion. Longer infusions (96 hours) have yielded significant response rates in breast cancer patients in preliminary trials (Wilson et al., 1994), but this form of treatment has serious practical limitations. The drug undergoes extensive P450-mediated hepatic metabolism (isoenzymes CYP3A4 and CYP2C8), and less than 10% of a dose is excreted in the urine intact. The primary metabolite identified thus far is 6-OH paclitaxel, but multiple additional products are found in urine and plasma (Cresteil et al., 1994). Paclitaxel clearance is saturable and decreases with increasing dose
or dose rate (Table 52-3). In studies of 96-hour infusion of 35 mg/m2
per day, the presence of hepatic metastases greater than 2 cm in diameter
decreased clearance and led to high drug levels in plasma and greater
myelosuppression. Paclitaxel disappears from the plasma compartment with
half-lives of approximately 0.2, 2, and 20 hours. The critical plasma
concentration for inhibiting bone marrow elements depends on duration of
exposure but likely lies in the range of 0.01 to 0.1 While precise guidelines for dose reduction in patients with abnormal hepatic function have not been established, 50% to 75% doses should be used in the presence of hepatic metastases greater than 2 cm in size or in patients with abnormal serum bilirubin (Donehower, 2001). Drugs that induce CYP3A4, such as phenytoin or phenobarbital, or those that inhibit the same cytochrome, such as antifungal imidazoles, may significantly alter drug clearance and toxicity. Therapeutic Uses Paclitaxel TAXOL) has undergone testing in patients with metastatic ovarian and breast cancers; it has significant activity as a component of primary combination therapy regimens and in adjuvant therapy of breast cancer (McGuire et al., 1996; Seidman, 1998). Response rates in relapsed patients range from 20% to 30%, depending on the treatment history and the regimen employed. Clinical trials indicate significant response rates in lung, head and neck, esophageal, and bladder carcinomas as well (Redman et al., 1998). The optimal schedule of paclitaxel administration, alone or in combination with other drugs, has not been defined. Clinical Toxicities Paclitaxel exerts its primary toxic effects on the bone marrow. Neutropenia usually occurs 8 to 11 days after a dose and reverses rapidly by days 15 to 21. Used with filgrastim (G-CSF), doses as high as 250 mg/m2 over 24 hours are well tolerated, and peripheral neuropathy becomes dose-limiting (Kohn et al., 1994). Many patients experience myalgias for several days after receiving paclitaxel. In high-dose schedules, a stockingglove sensory neuropathy can be disabling, particularly in patients with underlying diabetic or alcoholic neuropathy. Mucositis is prominent in 72- or 96-hour infusions and in the weekly schedule. Hypersensitivity reactions occurred in patients receiving paclitaxel infusions of short duration (1 to 6 hours) but have largely been averted by pretreatment with dexamethasone, diphenhydramine, and histamine H2-receptor antagonists, as noted above. Premedication is not necessary with 96-hour infusion. Many patients experience asymptomatic bradycardia, and occasional episodes of silent ventricular tachycardia also occur and resolve spontaneously during 3- or 24-hour infusions. Epipodophyllotoxins Podophyllotoxin, extracted from the mandrake plant (may-apple; Podophyllum peltatum), was used as a folk remedy by the American Indians and early colonists for its emetic, cathartic, and anthelmintic effects. Two semisynthetic glycosides of the active principle, podophyllotoxin, have been developed that show significant therapeutic activity in several human neoplasms, including pediatric leukemia, small cell carcinomas of the lung, testicular tumors, Hodgkin's disease, and large cell lymphomas. These derivatives are referred to as etoposide (VP-16-213) and teniposide (VM-26). Although podophyllotoxin binds to tubulin at a site distinct from that for interaction with the vinca alkaloids, etoposide and teniposide have no effect on microtubular structure or function at usual concentrations (for reviews of the epipodophyllotoxins, see Hande, 1998; and Pommier et al., 2001). Chemistry The chemical structures of etoposide and teniposide are shown below. They have been selected from many derivatives of podophyllotoxin that have been synthesized during the past 20 years.
Mechanism of Action Etoposide and teniposide are similar in their actions and in the spectrum of human tumors affected. Unlike podophyllotoxin, they do not arrest cells in mitosis; rather, these compounds form a ternary complex with topoisomerase II and DNA. This complex results in double-stranded DNA breaks, but the strand passage and resealing of the break that normally follow topoisomerase binding to DNA are inhibited by the drug. The enzyme remains bound to the free end of the broken DNA strand, leading to an accumulation of DNA breaks and cell death (Pommier et al., 2001). Cells in the S and G2 phases of the cell cycle are most sensitive to etoposide and teniposide. Resistant cells demonstrate either amplification of the mdr-1 gene that encodes the P-glycoprotein drug efflux transporter, mutation or decreased expression of topoisomerase II, or mutations of the p53 tumor suppressor gene, a required component of the apoptosis, or cell-death, pathway (Lowe et al., 1993). Etoposide Absorption, Fate, and Excretion Oral administration of etoposide results in variable absorption that
averages about 50% of the drug. After intravenous injection, peak plasma
concentrations of 30 Therapeutic Uses The intravenous dose of etoposide (VEPESID, TOPOSAR, ETOPOPHOS) for testicular cancer in combination therapy is 50 to 100 mg/m2 for 5 days, or 100 mg/m2 on alternate days for three doses. For small cell carcinoma of the lung, the dose in combination therapy is 50 to 120 mg/m2 per day intravenously for 3 days or 50 mg per day orally for 21 days. Cycles of therapy are usually repeated every 3 to 4 weeks. The drug should be administered slowly during a 30- to 60-minute infusion to avoid hypotension and bronchospasm, which likely result from the additives used to dissolve etoposide, a relatively insoluble compound. A disturbing complication of etoposide therapy has emerged in long-term follow-up of patients with childhood acute lymphoblastic leukemia, who develop an unusual form of acute nonlymphocytic leukemia with a translocation in chromosome 11 at 11q23. At this locus is found a gene(s) (the MLL or mixed-lineage leukemia gene) that regulates the proliferation of pluripotent stem cells. The leukemic cells have the cytological appearance of acute monocytic or monomyelocytic leukemia. Another distinguishing feature of etoposide-related leukemia is the short time interval between end of treatment and leukemia (1 to 3 years), as compared to the 4- to 5-year interval for secondary leukemias related to alkylating agents, and the absence of a myelodysplastic period preceding leukemia (Levine and Bloomfield, 1992; Pui et al., 1995; Sandler et al., 1997; Smith et al., 1999). Patients receiving weekly or twice-weekly doses of etoposide, with cumulative doses above 2000 mg/m2, seem to be at higher risk of leukemia. Etoposide is used primarily for treatment of testicular tumors, in combination with bleomycin and cisplatin, and in combination with cisplatin and ifosfamide for small cell carcinoma of the lung (Nemati et al., 2000). It also is active against non-Hodgkin's lymphomas, acute nonlymphocytic leukemia, and Kaposi's sarcoma associated with acquired immunodeficiency syndrome (AIDS) (Chao et al., 2000; Tung et al., 2000). Etoposide has a favorable toxicity profile for dose escalation in that its primary toxicity is myelosuppression. In combination with ifosfamide and carboplatin, it is frequently used for high-dose chemotherapy in total doses of 1500 to 2000 mg/m2 (Sobecks et al., 2000; Donato et al., 2000; Josting et al., 2000). Clinical Toxicities The dose-limiting toxicity of etoposide is leukopenia, with a nadir at 10 to 14 days and recovery by 3 weeks. Thrombocytopenia occurs less often and usually is not severe. Nausea, vomiting, stomatitis, and diarrhea occur in approximately 15% of patients treated intravenously and in about 55% of patients who receive the drug orally. Alopecia is common but reversible. Fever, phlebitis, dermatitis, and allergic reactions including anaphylaxis have been observed. Hepatic toxicity is particularly evident after high-dose treatment. For both etoposide and teniposide, toxicity is increased in patients with decreased serum albumin, an effect related to decreased protein binding of the drug (Stewart et al., 1991). Teniposide Teniposide VUMON) is administered intravenously. It has a multiphasic pattern of clearance from plasma. After distribution, half-lives of 4 hours and 10 to 40 hours are observed. Approximately 45% of the drug is excreted in the urine but, in contrast to etoposide, as much as 80% is recovered as metabolites. Anticonvulsants such as phenytoin increase the hepatic metabolism of teniposide and reduce systemic exposure (Baker et al., 1992). Dosage need not be reduced for patients with impaired renal function (Sinkule et al., 1984; Pommier et al., 2001). Less than 1% of the drug crosses the bloodbrain barrier. However, teniposide has produced responses in small cell and non-small cell lung cancer metastases in the brain (Postmus et al., 1995; Boogerd et al., 1999). Teniposide is available for treatment of refractory acute lymphoblastic leukemia in children and appears to be synergistic with cytarabine. It is administered by intravenous infusion in doses that range from 50 mg/m2 per day for 5 days to 165 mg/m2 per day twice weekly. The clinical spectrum of activity includes acute leukemia in children, particularly monocytic leukemia in infants, as well as glioblastoma, neuroblastoma, and brain metastases from small cell carcinomas of the lung (Odom and Gordon, 1984; Postmus et al., 1995; Boogerd et al., 1999). Myelosuppression, nausea, and vomiting are its primary toxic effects. Camptothecin Analogs The camptothecins are a promising new class of antineoplastic agents that target the nuclear enzyme topoisomerase I. The first compound in this class, camptothecin, was isolated from the Chinese tree Camptotheca acuminata in 1966. Despite the significant antitumor activity observed with the parent compound in preclinical models and in early clinical trials, further clinical development was compromised by severe and unpredictable toxicity, principally myelosuppression and hemorrhagic cystitis. Improved understanding of the mechanism of action and clinical pharmacology of these agents during the 1980s, led to the development of more soluble and less toxic analogs. Irinotecan and topotecan are currently the most widely used camptothecin analogs in the clinical setting, with established activity in colorectal, ovarian, and small cell lung cancer (for review, see Takimoto and Arbuck, 2001). Chemistry All camptothecins contain a basic five-ring structure, with a chiral center at C-20 of the terminal lactone ring (Figure 5213). The naturally occurring (S)-isomer is 10- to 100-times more active against topoisomerase I than is the (R)-isomer. Substitutions at C-9 and C-10 can enhance water solubility and increase topoisomerase I inhibition. Topotecan [(S)-9-dimethylaminoethyl-10-hydroxycamptothecin hydrochloride] is a semisynthetic camptothecin analog. It incorporates a basic dimethyl-amino side chain at C-9 which increases its water solubility. Irinotecan (7-ethyl-10-[4-(1-piperidino)-1-piperidino]car-bonyloxycamptothecin, or CPT-11) differs from topotecan in that it is a prodrug. Its bulky piperidino side chain at position C-10, is cleaved by a carboxylesterase-converting enzyme to form the biologically active metabolite, SN-38. SN-38 is 1000-fold more potent than irinotecan in inhibiting topoisomerase I.
Although an intact lactone ring is necessary for camptothecin's activity, it is unstable in aqueous solutions at neutral or basic pH. The lactone ring undergoes a rapid, reversible, nonenzymatic hydrolysis to form the carboxylate, which has greater water solubility but is 10-fold less potent than the lactone. In the absence of proteins, the lactone hydrolysis of different camptothecin analogs occurs at about the same rate, and at physiological pH the carboxylate form predominates. However, the equilibrium ratio in plasma between the carboxylate and lactone species of different topoisomerase I inhibitors is greatly dependent upon their relative degree of albumin binding. For camptothecin, for instance, the carboxylate form binds to serum albumin with a 200-fold greater affinity, and it is the predominant form found in blood. In contrast, for SN-38, the lactone form is the one that preferentially binds to serum albumin, thus shifting the equilibrium in the opposite direction. Mechanism of Action The DNA topoisomerases are nuclear enzymes that reduce supercoiled DNA torsional stress, allowing selected regions of DNA to become sufficiently untangled and relaxed to permit essential cellular processes to occur, such as DNA replication, recombination, repair, and transcription. Two classes of topoisomerase (I and II) are known to mediate DNA strand breakage and resealing, and both have become the target of cancer chemotherapies. Camptothecin analogs inhibit the function of topoisomerase I, while a number of different chemical entities (anthracyclines, epipodophyllotoxins, acridines) inhibit topoisomerase II. Topoisomerase I binds covalently to double-stranded DNA through a reversible trans-esterification reaction. This reaction yields an intermediate complex in which the tyrosine of the enzyme is bound to the 3-phosphate end of the DNA strand, creating a single-strand DNA break. This 'cleavable complex' allows for relaxation of the DNA torsional strain, either by passage of the intact single-strand through the nick, or by free rotation of the DNA about the noncleaved strand. Once the DNA torsional strain has been relieved, the topoisomerase I reseals the cleavage and dissociates from the newly relaxed double helix. The camptothecins bind to and stabilize the normally transient DNA-topoisomerase I cleavable complex (Hsiang et al., 1985). Although the initial cleavage action of topoisomerase I is not affected, the religation step is inhibited, leading to the accumulation of single-stranded breaks in DNA. This DNA damage, by itself, is not toxic to the cell, because upon drug removal, religation of DNA occurs. However, the collision of a DNA replication fork with this cleaved strand of DNA causes an irreversible double-strand DNA break, ultimately leading to cell death (Tsao et al., 1993). Since ongoing DNA synthesis is necessary for cytotoxicity, camptothecins are S-phase specific drugs. This has important clinical implications, because S-phase-specific cytotoxic agents generally require prolonged exposures of tumor cells to drug concentrations above a minimum threshold in order to optimize therapeutic efficacy. In fact, preclinical studies of low-dose, protracted administration of camptothecin analogs have shown less toxicity, and equal or greater antitumor activity than shorter, more intense courses. The precise sequence of events that lead from drug-induced DNA damage to cell death has not been fully elucidated. In vitro studies have shown that camptothecin-induced DNA damage abolishes the activation of p34cdc2/cyclin B complex, leading to cell-cycle arrest at the G2 phase (Tsao et al., 1992). It also has been observed that treatment with camptothecins can induce the transcription of c-fos and c-jun early-response genes, and this occurs in association with internucleosomal DNA fragmentation, a characteristic of programmed cell death (Kharbanda et al., 1991). Camptothecins also can induce the differentiation of human leukemia cells. Finally, camptothecin-induced cytotoxicity also has been observed in cells not actively synthesizing DNA. Replication-independent mechanisms of cytotoxicity may involve the induction of serine proteases and endonucleases. Mechanisms of Resistance A variety of mechanisms of resistance to topoisomerase Itargeted agents have been characterized in vitro, although little is known about their significance in the clinical setting. Decreased intracellular drug accumulation has been observed in several cell lines resistant to camptothecin analogs. Several topoisomerase I inhibitors have been shown to be substrates for P-glycoprotein, a cell membrane transporter that carries different cytotoxic drugs and toxins out of the cell and confers multidrug resistance to tumor cells. However, multidrug resistance (MDR) gene-expressing cell lines are only 9- to 12-fold more resistant to topotecan or SN-38 than are their parental wild-type counterparts, a much smaller degree of resistance than the 200-fold change in sensitivity for classic P-glycoprotein substrates such as etoposide or doxorubicin. Other reports have associated topotecan or irinotecan resistance and the multidrug resistance-associated protein (MRP) class of transporters. Another energy-dependent pump (MRP-3) confers resistance to mitoxantrone (an anthracenedione), but also to topotecan, irinotecan, and SN-38, but not to the parent compound, camptothecin (Miyake et al., 1999). Drug metabolism may play an important role in the resistance to the prodrug irinotecan. Cell lines that lack carboxylesterase activity, and therefore are unable to convert irinotecan to SN-38, demonstrate resistance to this camptothecin analog (van Ark-Otte et al., 1998). Camptothecin resistance also may result from decreased expression or mutation of topoisomerase I. Although a good correlation has been found in certain tumor cell lines between sensitivity to camptothecin analogs and topoisomerase I levels (Sugimoto et al., 1990), clinical studies have not confirmed this association. Chromosomal deletions or hypermethylation of the topoisomerase I gene are possible mechanisms of decreased topoisomerase I expression in resistant cells. A transient down-regulation of topoisomerase I has been demonstrated following prolonged exposure to camptothecins in vitro and in vivo. Moreover, an association between the degree of topoisomerase I down-regulation in peripheral blood mononuclear cells and the area under the plasma concentration-time curve (AUC) or neutrophil nadir has been observed in ovarian cancer patients treated with a 21-day continuous intravenous infusion of topotecan (Hochster et al., 1999). Mutations leading to reduced topoisomerase I enzyme catalytic activity or DNA binding affinity also have been described in vitro in association with camptothecin resistance (Tamura et al., 1991). In addition, some posttranscriptional events, such as enzyme phosphorylation (Pommier et al., 1990) or poly-ADP ribosylation (Kasid et al., 1989), may have a significant impact on the activity of topoisomerase I and on its susceptibility to inhibition. Finally, an observation of potential clinical interest is that exposure of cells to topoisomerase Itargeted agents leads to increased expression of topoisomerase II, providing a rationale for sequential therapy with topoisomerase I and II inhibitors. Very little is known about how the cell deals with the stabilized DNA-topoisomerase complexes. As cleavable complexes normally are present in the untreated cell, the drug-enzyme-DNA complex may not be recognized easily by cellular repair processes. However, an enzyme with specific tyrosyl-DNA phosphodiesterase activity may be involved in the disassembly of topoisomerase IDNA complexes (Yang et al., 1996). Entry into S phase is required to kill tumor cells exposed to camptothecins. Drugs that abolish the G1-S checkpoint enhance lethality of camptothecins (Shao et al., 1997). The fact that cell cycle arrest in G2 has been correlated with drug resistance to topoisomerase I-targeted drugs in colon cancer and leukemia cell lines in vitro suggests the possibility that enhanced DNA repair activity can lead to camptothecin resistance. The role of p53 in mediating cell death due to camptothecins is unclear. These drugs induce p53 expression, but cells without functional p53 also can undergo apoptosis following exposure to camptothecins. Absorption, Fate, and Excretion Topotecan Topotecan is administered intravenously, and is rapidly cleared from the plasma. Only 20% to 35% of the total drug in plasma is found to be in the active lactone form. The terminal half-life of topotecan lactone ranges between 2 and 3.5 hours, which is relatively short compared with other camptothecins. Bioavailability of topotecan following oral administration is about 30% to 40%. Unlike other topoisomerase I inhibitors, plasma-protein binding of topotecan is low (7% to 35%), a finding that may explain the higher CNS penetration compared with other camptothecins (29% to 42%). Few data are available concerning topotecan metabolism. Three novel metabolites, N-desmethyl topotecan, topotecan-O-glucuronide, and N-desmethyl topotecan-O-glucuronide, which are found at low concentrations in plasma, urine, and bile, have been characterized (Rosing et al., 1998). Elimination of the lactone form is thought to result mainly from the rapid hydrolysis to the carboxylate species followed by renal excretion. Between 25% and 70% of the administered dose is excreted in the urine within 24 hours, and doses should be reduced in proportion to reductions in creatinine clearance. Irinotecan Both the lactone and the open-ring carboxylate species of irinotecan and SN-38 can be measured in plasma shortly after an intravenous infusion. However, the overall AUC ratio of SN-38 to irinotecan is only about 4%. Compared to other camptothecin derivatives, a relatively large fraction of both irinotecan and SN-38 in plasma is present as the biologically active lactone form. Another potential advantage of this camptothecin analog are the longer plasma terminal half-lives of both irinotecan and its active metabolite, SN-38 (both about 10 hours). The oral bioavailability of irinotecan is low (8%). However, the molar SN-38 to irinotecan AUC ratio is threefold higher after oral administration than after identical intravenous doses. A potential explanation for this observation is extensive first-pass metabolism, with significant conversion of irinotecan to SN-38 by carboxylesterase present in the intestine and liver. Plasma-protein binding is 43% or higher for irinotecan and 92% to 96% for SN-38. CSF penetration of SN-38 in humans has not been characterized yet, although in rhesus monkeys it is only 14%, significantly lower than that observed for topotecan. Irinotecan is converted to its active metabolite, SN-38, by the carboxylesterase-converting enzyme. Variations in this enzyme's activity in tumor cells or in normal host tissues may be important in determining irinotecan antitumor effect and/or toxicity. SN-38, although much more potent than the parent drug, represents only a small fraction of the total irinotecan metabolized. Other irinotecan metabolites have been characterized, including 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino] carbonyloxycamptothecin (APC), and 7-ethyl-10-(4-amino-1-piperidino) carbonyloxycamptothecin (NPC), both of which are poor inhibitors of topoisomerase I. CYP3A seems to be responsible for the production of these 2 metabolites (Haaz et al., 1998). Since this enzyme is involved in the metabolism of a large number of commonly used drugs, drug interactions may have significant impact on irinotecan pharmacokinetics. Glucuronidation is the major metabolic route of SN-38. The uridine
diphosphate glucuronosyl transferase (UGT), particularly the UGT1A1 isoform,
converts SN-38 to its glucuronidated derivative (Iyer et al., 1998).
Both SN-38 and its glucuronide are excreted in the bile. The extent of SN-38
glucuronidation has been inversely correlated with the risk of severe
diarrhea after irinotecan therapy. UGT1A1 also glucuronidates bilirubin.
Polymorphisms of this enzyme are associated with familial hyperbilirubinemia
syndromes such as CriglerNajjar (CN) syndrome and Gilbert syndrome. CN
syndromes are rare (1 in a million births), but Gilbert syndrome occurs in up
to 15% of the general population, and results in a mild hyperbilirubinemia
that may be clinically silent. The existence of UGT enzyme polymorphisms may
have a major impact on the clinical use of irinotecan. A positive correlation
has been found between baseline serum unconjugated bilirubin concentration
and both severity of neutropenia and the AUC of irinotecan and SN-38 in
patients treated with this camptothecin analog. Moreover, severe irinotecan
toxicity has been observed in cancer patients with Gilbert syndrome,
presumably due to decreased glucuronidation of SN-38. Biliary excretion
appears to be the primary elimination route of irinotecan, SN-38, and their
glucuronides, although urinary excretion also contributes significantly (14%
to 37%). The presence of bacterial Therapeutic Uses Topotecan Topotecan HYCAMTIN) is given as a 30-minute infusion of 1.5 mg/m2 per day for 5 consecutive days every 3 weeks. A variety of prolonged infusion schedules also have been tested. In experimental studies, topotecan has been administered orally, but this route is still investigational. Since a significant fraction of the topotecan administered is excreted in the urine, severe toxicities have been observed in patients with decreased creatinine clearance (O'Reilly et al., 1996). Therefore, the dose of topotecan should be reduced to 0.75 mg/m2 per day in patients with moderate renal dysfunction (creatinine clearance 20 to 40 ml/minute), and topotecan should not be administered to patients with severe renal impairment (creatinine clearance <20 ml/minute). Topotecan clearance and toxicity are not significantly altered in patients with hepatic dysfunction, and therefore, no dose reduction is necessary in these patients. Topotecan is active in previously treated patients with ovarian (ten Bokkel Huinink et al., 1997) or small cell lung cancer (von Pawel et al., 1999). Its significant hematological toxicity, though, has limited its use in combination with other active agents in these diseases (e.g., cisplatin). Promising antitumor activity also has been observed in hematological malignancies, particularly in chronic myelomonocytic leukemia and in myelodysplastic syndromes. Irinotecan Approved dosage schedules of irinotecan (CAMPTOSAR) in the United States include: 125 mg/m2 as a 90-minute infusion administered weekly for 4 out of 6 weeks; 350 mg/m2 given every 3 weeks; 100 mg/m2 every week; or 150 mg/m2 every other week. Prolonged irinotecan infusions and oral administration also have been explored. Irinotecan has significant clinical activity in patients with advanced colorectal cancer. It is now the treatment of choice in combination with fluoropyrimidines for advanced colorectal cancer in patients who have not received chemotherapy previously (Douillard et al., 2000) or as a single agent following failure on a 5-fluorouracil regimen (Cunningham et al., 1998). Encouraging results from different phase II studies suggest that irinotecan may have an increasing role in the treatment of other solid tumors, including small cell and nonsmall cell lung cancer, cervical cancer, ovarian cancer, and gastric cancer. Clinical Toxicities Topotecan The dose-limiting toxicity with all schedules is neutropenia, with or without thrombocytopenia. The incidence of severe neutropenia at the recommended phase II dose of 1.5 mg/m2 daily for 5 days every 3 weeks may be as high as 81%, with a 26% incidence of febrile neutropenia. Dose intensity with the 21-day continuous infusion exceeds that achieved with other schedules, but this is associated with a higher incidence of thrombocytopenia and cumulative anemia. In patients with hematological malignancies, gastrointestinal side effects such as mucositis and diarrhea become dose-limiting. Other less common and generally mild topotecan-related toxicities include nausea and vomiting, elevated liver transaminases, fever, fatigue, and rash. Irinotecan The dose-limiting toxicity with all schedules is delayed diarrhea, with or without neutropenia. In the initial studies, up to 35% of the patients experienced severe diarrhea. Adoption of the intensive loperamide (see Chapter 39: Agents Used for Diarrhea, Constipation, and Inflammatory Bowel Disease; Agents Used for Biliary and Pancreatic Disease) regimen (4 mg of loperamide starting at the onset of any loose stool beginning more than a few hours after receiving therapy, followed by 2 mg every 2 hours) has effectively reduced this incidence by more than half. However, once severe diarrhea does occur, standard doses of antidiarrheal agents tend to be ineffective, although the diarrhea episode generally resolves within a week and, unless associated with fever and neutropenia, is rarely fatal. The second most common irinotecan-associated toxicity is myelosuppression. Severe neutropenia occurs in 14% to 47% of the patients treated with the every-3-week schedule, and is less frequently encountered among patients treated with the weekly schedule. Febrile neutropenia is observed in 3% of the patients, and may be fatal, particularly when associated with concomitant diarrhea. A cholinergic syndrome resulting from the inhibition of acetylcholinesterase activity by irinotecan may occur within the first 24 hours after irinotecan administration. Symptoms include acute diarrhea, diaphoresis, hypersalivation, abdominal cramps, visual accommodation disturbances, lacrimation, rhi-norrhea, and less often, asymptomatic bradycardia. These effects are short lasting and respond within minutes to atropine. Atropine may be prophylactically administered to patients who have previously experienced a cholinergic reaction, prior to the administration of additional cycles of irinotecan. Other common and generally manageable toxicities include nausea and vomiting, fatigue, vasodilation or skin flushing, mucositis, liver transaminases elevation, and alopecia. Finally, there have been case reports of dyspnea and interstitial pneumonitis associated with irinotecan therapy in Japanese patients with lung cancer (Fukuoka et al., 1992). Antibiotics Dactinomycin (Actinomycin D) History The first crystalline antibiotic agent to be isolated from a culture broth of a species of Streptomyces was actinomycin A (Waksman and Woodruff, 1940). Many related antibiotics, including actinomycin D, subsequently have been obtained (Waksman Conference on Actinomycins, 1974). Dactinomycin has beneficial effects in the treatment of a number of tumors, particularly certain neoplasms of childhood and choriocarcinoma. Chemistry and StructureActivity Relationship The actinomycins are chromopeptides, and most of them contain the same chromophore, the planar phenoxazone actinocin, which is responsible for the yellow-red color of the compounds. The differences among naturally occurring actinomycins are confined to the peptide side chains, and the variations are in the structure of the constituent amino acids. By varying the amino acid content of the growth medium, it is possible to alter the types of actinomycins produced and the biological activity of the molecule (Crooke, 1983). The chemical structure of dactinomycin is as follows:
Mechanism of Action The capacity of actinomycins to bind with double-helical DNA is responsible for their biological activity and cytotoxicity. X-ray studies of a crystalline complex between dactinomycin and deoxyguanosine permitted formulation of a model that appears to explain the binding of the drug to DNA (Sobell, 1973). The planar phenoxazone ring intercalates between adjacent guaninecytosine base pairs of DNA, where the guanine moieties are on opposite strands of the DNA, while the polypeptide chains extend along the minor groove of the helix. The summation of these interactions provides great stability to the dactinomycinDNA complex, and, as a result of the binding of dactinomycin, the transcription of DNA by RNA polymerase is blocked. The DNA-dependent RNA polymerases are much more sensitive to the effects of dactinomycin than are the DNA polymerases. In addition, dactinomycin causes single-strand breaks in DNA, possibly through a free-radical intermediate or as a result of the action of topoisomerase II (see Waksman Conference on Actinomycins, 1974; Goldberg et al., 1977). Cytotoxic Action Dactinomycin inhibits rapidly proliferating cells of normal and neoplastic origin and, on a molar basis, is among the most potent antitumor agents known. Atrophy of thymus, spleen, and other lymphatic tissues occurs in experimental animals. The drug may produce alopecia, and, when extravasated subcutaneously, causes marked local inflammation. Erythema, sometimes progressing to necrosis, has been noted in areas of the skin exposed to x-radiation before, during, or after administration of dactinomycin. Absorption, Fate, and Excretion Dactinomycin is much less potent when given orally than when administered by parenteral injection. The drug is excreted both in bile and in the urine and disappears from plasma with a terminal half-life of 36 hours. Metabolism of the drug is minimal. Dactinomycin does not cross the bloodbrain barrier. Therapeutic Uses Dactinomycin
(actinomycin D;COSMEGEN) is supplied for intravenous use. The usual daily dose is 10 to 15 The most important clinical use of dactinomycin is in the treatment of
rhabdomyosarcoma and Wilms' tumor in children, where it is curative in
combination with primary surgery, radiotherapy, and other drugs, particularly
vincristine and cyclophosphamide (Pinkel and Howarth, 1985). Antineoplastic
activity has been noted in Clinical Toxicities Toxic manifestations include anorexia, nausea, and vomiting, usually beginning a few hours after administration. Hematopoietic suppression with pancytopenia may occur in the first week after completion of therapy. Proctitis, diarrhea, glossitis, cheilitis, and ulcerations of the oral mucosa are common; dermatological manifestations include alopecia, as well as erythema, desquamation, and increased inflammation and pigmentation in areas previously or concomitantly subjected to x-radiation. Severe injury may occur as a result of local toxic extravasation. Daunorubicin, Doxorubicin, and Idarubicin These anthracycline antibiotics and their derivatives are among the most important antitumor agents. They are produced by the fungus Streptococcus peucetius var. caesius. Idarubicin is a synthetic derivative. Although they differ only slightly in chemical structure, daunorubicin and idarubicin have been used primarily in the acute leukemias, whereas doxorubicin displays broader activity against human neoplasms, including a variety of solid tumors. The clinical value of these agents is limited by an unusual and often irreversible cardiomyopathy, the occurrence of which is related to the total dose of the drug. In a search for agents with high antitumor activity but reduced cardiac toxicity, hundreds of anthracycline derivatives and related compounds have been prepared. Several of these have shown promise in clinical studies, including idarubicin for leukemia, epirubicin for solid-tumor chemotherapy, and mitoxantrone for prostate cancer, leukemia, and high-dose chemotherapy. Mitoxantrone, an anthracenedione, has significantly less cardiotoxicity than do the anthracyclines (see Arlin et al., 1990; Feldman et al., 1993; Berman et al., 1991; Wiernik et al., 1992; Launchbury and Habboubi, 1993). Chemistry The anthracycline antibiotics have tetracycline ring structures with an unusual sugar, daunosamine, attached by glycosidic linkage. Cytotoxic agents of this class all have quinone and hydroquinone moieties on adjacent rings that permit them to function as electron-accepting and -donating agents. Although there are marked differences in the clinical use of daunorubicin and doxorubicin, their chemical structures differ only by a single hydroxyl group on C-14. Idarubicin is 4-demethoxydaunorubicin, a synthetic derivative of daunorubicin without the methoxy group on C-4 of the aglycone ring. The chemical structures of doxorubicin, daunorubicin, epirubicin, and idarubicin are as follows:
Mechanism of Action A number of important biochemical effects have been described for the anthracyclines and anthracenediones, any one or all of which could have a role in the therapeutic and toxic effects of such drugs. These compounds can intercalate with DNA. Many functions of DNA are affected, including DNA and RNA synthesis. Single- and double-strand breaks occur, as does sister chromatid exchange. Thus, the anthracyclines are both mutagenic and carcinogenic. Scission of DNA is believed to be mediated by drug binding to DNA and topoisomerase II, an action that prevents the resealing of DNA breaks created by the enzyme (Tewey et al., 1984). Anthracyclines, by virtue of their quinone groups, also generate free radicals in solution and in both normal and malignant tissues (Gewirtz, 1999; Ikeda et al., 1999). The anthracyclines react with cytochrome P450 reductase in the presence of reduced nicotinamide adenine dinucleotide phosphate (NADPH) to form semiquinone radical intermediates, which in turn can react with oxygen to produce superoxide anion radicals. These can generate both hydrogen peroxide and hydroxyl radicals (OH), which attack DNA (Serrano et al., 1999) and oxidize DNA bases. The production of free radicals is significantly stimulated by the interaction of doxorubicin with iron (Myers, 1988). In addition, intramolecular electron-transfer reactions of the semiquinone intermediates result in the generation of lipid peroxides, nitric oxide, and other destructive radicals. Enzymatic defenses such as superoxide dismutase and catalase are believed to have an important role in protecting cells against the toxicity of the anthracyclines, and these defenses can be augmented by exogenous antioxidants such as alpha tocopherol or by an iron chelator, dexrazoxane (formerly called ICRF-187), which protects against cardiac toxicity (Speyer et al., 1988; Swain et al., 1997). The anthracyclines also can interact with cell membranes, producing lipid peroxides and altering their functions; this may play an important part in both the antitumor actions and the cardiac toxicity caused by these drugs (Tritton et al., 1978). Exposure of cells to anthracyclines leads to apoptosis; mediators of this process include the p53 DNA-damage sensor and activated caspases (proteases), although ceramide, a lipid breakdown product, and the fas receptor-ligand system also have been implicated in selected tumor cells (Friesen et al., 1996; Jaffrezou et al., 1996). As discussed above, the phenomenon of pleiotropic drug resistance is observed in tumor cell populations exposed to anthracyclines. This appears to result from acceleration of the efflux of anthracyclines and other agents from the cell. The P-glycoprotein, synthesized in high quantity as a result of gene amplification, has been implicated (Endicott and Ling, 1989). Anthracyclines also are exported from tumor cells by members of the MRP transporter family (Doyle et al., 1998). Other biochemical changes in resistant cells include increased glutathione peroxidase activity (Sinha et al., 1989) and decreased activity of topoisomerase II (Deffie et al., 1989; Jarvinen et al., 1998). Absorption, Fate, and Excretion Daunorubicin, doxorubicin, epirubicin, and idarubicin usually are administered intravenously and are cleared by hepatic metabolism and biliary excretion. The disappearance curve for doxorubicin is multiphasic, with elimination half-lives of 3 hours and about 30 hours. Idarubicin has a half-life of about 15 hours, and its active metabolite, idarubicinol, has a half-life of about 40 hours. There is rapid uptake of the drugs in the heart, kidneys, lungs, liver, and spleen. They do not cross the bloodbrain barrier. Daunorubicin and doxorubicin are eliminated by metabolic conversion to a variety of less active or inactive products. Idarubicin is primarily metabolized to idarubicinol, which accumulates in plasma and resembles the parent compound in activity. Daunorubicin and doxorubicin are converted to their alcohols, to aglycones, and to other derivatives. Precise guidelines for reduction of dosage in patients with impaired hepatic function have not been defined. Clearance is delayed in the presence of hepatic dysfunction, and at least a 50% initial reduction in dose should be considered in patients with abnormal serum bilirubin levels (Twelves et al., 1998). Idarubicin Hydrochloride (IDAMYCIN The recommended dosage for idarubicin is 12 mg/m2 daily for 3 days by intravenous injection in combination with cytarabine. Slow injection with care over 10 to 15 minutes is recommended to avoid extravasation, as with other anthracyclines. Daunorubicin Therapeutic Uses Daunorubicin hydrochloride (daunomycin, rubidomycin; CERUBIDINE) is available for intravenous use. The recommended dosage is 30 to 60 mg/m2 daily for 3 days. The agent is administered with appropriate care to prevent extravasation, since severe local vesicant action may result. A daunorubicin citrate liposomal product (DAUNOXOME) is indicated for the treatment of AIDS-related Kaposi's sarcoma. It is given in a dose of 40 mg/m2 infused over 60 minutes and repeated every 2 weeks. Patients should be advised that the drug may impart a red color to the urine. Daunorubicin is very useful in the treatment of acute lymphocytic and acute myelogenous leukemias. It is among the most active drugs for treatment of AML in adults and, given with cytarabine, either it or idarubicin is the treatment of choice in these conditions. Clinical Toxicities The toxic manifestations of daunorubicin as well as idarubicin include bone marrow depression, stomatitis, alopecia, gastrointestinal disturbances, and dermatological manifestations. Cardiac toxicity is a peculiar adverse effect observed with these agents. It is characterized by tachycardia, arrhythmias, dyspnea, hypotension, pericardial effusion, and congestive failure poorly responsive to digitalis (see below). Doxorubicin Therapeutic Uses Doxorubicin hydrochloride ADRIAMYCIN, RUBEX) is available for intravenous use. The recommended dose is 60 to 75 mg/m2, administered as a single rapid intravenous infusion that is repeated after 21 days. Care should be taken to avoid extravasation, since severe local vesicant action and tissue necrosis may result. A doxorubicin liposomal product (DOXIL) is available for treatment of AIDS-related Kaposi's sarcoma. It is given intravenously in a dose of 20 mg/m2 over 30 minutes and repeated every 3 weeks. As for daunorubicin, patients should be advised that the drug may impart a red color to the urine. Doxorubicin is effective in acute leukemias and malignant lymphomas;
however, in contrast to daunorubicin, it also is active in a number of solid
tumors, particularly breast cancer. Used concurrently with cyclophosphamide, vincristine,
procarbazine, and other agents, it is an important ingredient for the successful
treatment of Hodgkin's disease and non-Hodgkin's lymphomas. It is a valuable
component of various regimens of chemotherapy for carcinoma of the breast and
small cell carcinoma of the lung. The drug also is particularly beneficial in
a wide range of pediatric and adult sarcomas, including osteogenic, Clinical Toxicities The toxic manifestations of doxorubicin are similar to those of daunorubicin. Myelosuppression is a major dose-limiting complication, with leukopenia usually reaching a nadir during the second week of therapy and recovering by the fourth week; thrombocytopenia and anemia follow a similar pattern but usually are less pronounced. Stomatitis, gastrointestinal disturbances, and alopecia are common but reversible. Erythematous streaking near the site of infusion ('ADRIAMYCIN flare') is a benign local allergic reaction and should not be confused with extravasation. Facial flushing, conjunctivitis, and lacrimation may occur rarely. The drug may produce severe local toxicity in irradiated tissues (e.g., the skin, heart, lung, esophagus, and gastrointestinal mucosa). Such reactions may occur even when the two therapies are not administered concomitantly. Cardiomyopathy is a unique characteristic of the anthracycline antibiotics. Two types of cardiomyopathies may occur. (1) An acute form is characterized by abnormal electrocardiographic changes, including STT-wave alterations and arrhythmias. This is brief and rarely a serious problem. Cineangiographic studies have shown an acute, reversible reduction in ejection fraction 24 hours after a single dose. An exaggerated manifestation of acute myocardial damage, the 'pericarditismyocarditis syndrome,' may be characterized by severe disturbances in impulse conduction and frank congestive heart failure, often associated with pericardial effusion. (2) Chronic, cumulative dose-related toxicity (usually at or above total doses of 550 mg/m2) is manifested by congestive heart failure that is unresponsive to digitalis. The mortality rate is in excess of 50%. Total dosage of doxorubicin as low as 250 mg/m2 can cause myocardial toxicity, as demonstrated by subendocardial biopsies. Nonspecific alterations, including a decrease in the number of myocardial fibrils, mitochondrial changes, and cellular degeneration, are visible by electron microscopy. The most promising noninvasive technique used to detect the early development of drug-induced congestive heart failure is radionuclide cineangiography. Although no completely practical and reliable predictive tests are available, the frequency of serious cardiomyopathy is 1% to 10% at total doses below 450 mg/m2. The risk increases markedly (to >20% of patients) at total doses higher than 550 mg/m2, and this total dosage should be exceeded only under exceptional circumstances or with the concomitant use of dexrazoxane (ZINECARD), a cardioprotective intracellular chelating agent. (Speyer et al., 1988; Swain et al., 1997). Cardiac irradiation or administration of high doses of cyclophosphamide or another anthracycline may increase the risk of cardiotoxicity. Late-onset cardiac toxicity, with onset of congestive heart failure years after treatment, may occur in both pediatric and adult populations (Lipschultz et al., 1991). Newer Analogs of Doxorubicin Valrubicin VALSTAR) was approved in 1998 for intravesical therapy of BCG-refractory urinary bladder carcinoma in situ in patients for whom immediate cystectomy would be associated with unacceptable morbidity or mortality. Epirubicin (4'-epidoxorubicin, ELLENCE) was approved by the FDA in 1999 as a component of adjuvant therapy following resection of early lymph-node-positive breast cancer. A related anthracenedione, mitoxantrone, has been approved for use in acute nonlymphocytic leukemias. Its structural formula is as follows:
Mitoxantrone has limited ability to produce quinone-type free radicals and causes less cardiac toxicity than does doxorubicin. Mitoxantrone exerts its antitumor action by stimulating the formation of strand breaks in DNA; this is mediated by topoisomerase II; it also intercalates with DNA. Its range of antitumor activity is confined to leukemias, breast cancer, and prostate cancer (Shenkenberg and Von Hoff, 1986). Mitoxantrone produces acute myelosuppression, cardiac toxicity, and mucositis as its major toxicities; the drug causes less nausea and vomiting and alopecia than does doxorubicin. It also is used as a component of experimental high-dose chemotherapy regimens, with uncertain efficacy. Mitoxantrone (NOVANTRONE) is supplied for intravenous infusion. To induce remission in acute nonlymphocytic leukemia in adults, the drug is given in a daily dose of 12 mg/m2 for 3 days as a component of a regimen that also includes cytosine arabinoside. Mitoxantrone also is used in advanced hormone-resistant prostate cancer in a dose of 12 to 14 mg/m2 every 21 days. In 2000, mitoxantrone was approved by the FDA for the treatment of late-stage, secondary progressive multiple sclerosis. Bleomycins The bleomycins are an important group of DNA-cleaving antibiotics discovered by Umezawa and colleagues as fermentation products of Streptococcus verticillus. The drug currently employed clinically is a mixture of the two copper-chelating peptides, bleomycins A2 and B2. The bleomycins differ only in their terminal amine (see below), which can be altered by adding various amines to the fermentation medium. Bleomycins have attracted interest both because of their significant antitumor activity against squamous carcinomas of the cervix, head and neck, and lungs, and against lymphomas and testicular tumors. They are minimally myelo- and immunosuppressive but cause unusual cutaneous and pulmonary side effects. Because their toxicities do not overlap with those of other drugs, and because of their unique mechanism of action, the bleomycins have won an important role in combination chemotherapy. Chemistry The bleomycins are water-soluble, basic glycopeptides. The structures
of bleomycin A2 and B2 are shown in Figure 5214. The
core of the bleomycin molecule is a complex metal-binding structure
containing a pyrimidine chromophore linked to propionamide, a
Mechanism of Action Although the bleomycins have a number of interesting biochemical properties, their cytotoxic action results from their ability to cause oxidative damage to the deoxyribose of thymidylate and other nucleotides leading to single- and double-stranded breaks in DNA. Studies in vitro indicate that bleomycin causes accumulation of cells in the G2 phase of the cell cycle, and many of these cells display chromosomal aberrations, including chromatid breaks, gaps, and fragments, as well as translocations (Twentyman, 1983). Bleomycin causes scission of DNA by interacting with O2 and Fe2+. In the presence of O2 and a reducing agent, such as dithiothreitol, the metaldrug complex becomes activated and functions mechanistically as a ferrous oxidase, transferring electrons from Fe2+ to molecular oxygen to produce activated species of oxygen (Burger et al., 1986; Burger, 1998). It also has been shown that metallobleomycin complexes can be activated by reaction with the flavin enzyme, NADPH-cytochrome P450 reductase. Bleomycin binds to DNA through its amino-terminal peptide, and the activated complex generates free radicals that are responsible for scission of the deoxyribose backbone of the DNA chain (see Grollman et al., 1985). Bleomycin is degraded by a specific hydrolase found in various normal tissues, including liver; hydrolase activity is low in skin and lung (Sebti et al., 1987; Brmme et al., 1996). Some bleomycin-resistant cells contain high levels of hydrolase activity (Sebti et al., 1991). In other resistant cell lines different mechanisms, such as enhanced capacity to repair DNA, may lead to resistance (Zuckerman et al., 1986). In experimental models, resistance to bleomycin has been attributed to decreased uptake, cleavage by the hydrolase, repair of strand breaks, or drug inactivation by thiols. Resistance of human tumors is poorly understood. Absorption, Fate, and Excretion Bleomycin is administered parenterally or instilled into the bladder for local treatment of bladder cancer (Bracken et al., 1977). After intravenous infusion, relatively high drug concentrations are detected in the skin and lungs of experimental animals, and these organs become major sites of toxicity. Having a high molecular mass, bleomycin crosses the bloodbrain barrier poorly. After intravenous administration of a bolus dose of 15 units/m2, peak concentrations of 1 to 5 mU/ml are achieved in plasma. The half-time for elimination is approximately 3 hours. The average steady-state concentration of bleomycin in plasma of patients receiving continuous intravenous infusions of 30 units daily for 4 to 5 days is approximately 0.15 mU/ml. About two-thirds of the drug is normally excreted in the urine, probably by glomerular filtration. Concentrations in plasma are greatly elevated if usual doses are given to patients with renal impairment, and such patients are at high risk of developing pulmonary toxicity. Doses of bleomycin should be reduced in the presence of severe renal failure (see Dalgleish et al., 1984). Therapeutic Uses Bleomycin sulfate BLENOXANE) is available for injection. The recommended dose of bleomycin is 10 to 20 units/m2 given weekly or twice weekly by the intravenous or intramuscular route. It also may be administered as a subcutaneous injection or as an intrapleural or intracystic instillation. Total courses exceeding 250 units should be given with great caution because of a marked increase in pulmonary toxicity above this total dose. However, pulmonary toxicity may occur at lower doses (see below). Bleomycin is highly effective against germ cell tumors of the testis and ovary. In testicular cancer it is curative when used with cisplatin and vinblastine or cisplatin and etoposide (Williams and Einhorn, 1985), and it is highly active when used with cisplatin and other agents in combination therapy of squamous carcinomas of the head and neck, esophagus, and genitourinary tract. It often is used as a component of combination therapy of Hodgkin's and non-Hodgkin's lymphomas. Clinical Toxicities Because bleomycin causes little myelosuppression, it has significant advantages in combination with other cytotoxic drugs. However, it does cause significant cutaneous toxicity, including hyperpigmentation, hyperkeratosis, erythema, and even ulceration. These changes may begin with tenderness and swelling of the distal digits and progress to erythematous, ulcerating lesions over the elbows, knuckles, and other pressure areas. Skin changes often leave a residual hyperpigmentation at these points and may recur when patients are treated with other antineoplastic drugs. The most serious adverse reaction to bleomycin is pulmonary toxicity,
which begins with a dry cough, fine rales, and diffuse basilar infiltrates on
x-ray and may progress to life-threatening pulmonary fibrosis. Radiologic
changes may be indistinguishable from interstitial infection or tumor, but
may progress to dense fibrosis, cavitation, atelectasis or lobar collapse, or
even apparent consolidation. Approximately 5% to 10% of patients receiving
bleomycin develop clinically apparent pulmonary toxicity, and about 1% die of
this complication. Most who recover experience a significant improvement in
pulmonary function, but fibrosis may be irreversible (Van Barneveld et al.,
1987). Pulmonary function tests are not of predictive value for detecting
early onset of this complication. The CO diffusion capacity declines in
patients receiving doses above 250 U. The risk is related to total dose, with
a significant increase above total doses of 250 U and in patients over 70
years of age and in those with underlying pulmonary disease; single doses of
30 U/m2 or more also are associated with an increased risk of
pulmonary toxicity. Administration of high inspired oxygen concentrations
during anesthesia or respiratory therapy may aggravate or precipitate
pulmonary toxicity in patients previously treated with the drug. There is no
known specific therapy for bleomycin lung injury except for standard
symptomatic management and pulmonary care. Steroids are of uncertain benefit.
The etiology of bleomycin pulmonary toxicity has been the subject of intense
investigation in rodent models. These studies implicate cytokine [tumor
growth factor beta (TGF- Other toxic reactions to bleomycin include hyperthermia, headache, nausea, and vomiting, as well as a peculiar, acute fulminant reaction observed in patients with lymphomas. This is characterized by profound hyperthermia, hypotension, and sustained cardiorespiratory collapse; it does not appear to be a classical anaphylactic reaction and possibly may be related to release of an endogenous pyrogen. Because this reaction has occurred in approximately 1% of patients with lymphomas and has resulted in deaths, it is recommended that patients with lymphomas receive a 1-unit test dose of bleomycin, followed by a 1-hour period of observation, before administration of the drug on standard dosage schedules. Unexplained exacerbations of rheumatoid arthritis also have been reported during bleomycin therapy. Raynaud's phenomenon and coronary artery disease have been reported in patients with testicular tumors treated with bleomycin in combination with other chemotherapeutic agents. Mitomycin This antibiotic was isolated from Streptococcus caespitosus by Wakaki and associates in 1958. Mitomycin contains an aziridine group and a quinone group in its structure, as well as a mitosane ring, and each of these participates in the alkylation reactions with DNA. Its structural formula is as follows:
Mechanism of Action After intracellular enzymatic or spontaneous chemical reduction of the quinone and loss of the methoxy group, mitomycin becomes a bifunctional or trifunctional alkylating agent (Verweij et al., 2001). Reduction occurs preferentially in hypoxic cells in some experimental systems. The drug inhibits DNA synthesis and cross-links DNA at the N6 position of adenine and at the O6 and N7 positions of guanine. In addition, single-strand breakage of DNA and chromosomal breaks are caused by mitomycin. Mitomycin is a potent radiosensitizer and is teratogenic and carcinogenic in rodents. Resistance has been ascribed to deficient activation, intracellular inactivation of the reduced quinone, and P-glyco-proteinmediated drug efflux (Dorr, 1988; Crooke and Bradner, 1976). Absorption, Fate, and Excretion Mitomycin is absorbed inconsistently from the gastrointestinal tract
and is therefore administered intravenously. It disappears rapidly from the
blood after injection, with a half-life of 25 to 90 minutes. Peak
concentrations in plasma are 0.4 Therapeutic Uses Mitomycin (mitomycin-C; MUTAMYCIN) is administered by intravenous infusion; extravasation may result in severe local injury. The usual dose (6 to 10 mg/m2) may be administered intravenously as a single bolus infusion every 6 weeks and is usually given as part of a combination regimen for treatment of carcinoma of the colon or stomach. Dosage is modified based on hematological recovery. Mitomycin also may be used by direct instillation into the bladder to treat superficial carcinomas (Boccardo et al., 1994). Mitomycin is used primarily in combination with 5-FU, cisplatin, or doxorubicin in carcinomas of the cervix, stomach, breast, bladder, head and neck, and lung. It is a potent radiation sensitizer and continues to attract interest in clinical trials with concurrent irradiation in the above diseases. Clinical Toxicities The major toxic effect is myelosuppression, characterized by marked leukopenia and thrombocytopenia; after higher doses, the nadirs may be delayed and cumulative, with recovery only after 6 to 8 weeks of pancytopenia. Nausea, vomiting, diarrhea, stomatitis, dermatitis, fever, and malaise also are observed. A hemolytic-uremic syndrome represents the most dangerous toxic manifestation of mitomycin and is believed to result from drug-induced endothelial damage. Patients who have received more than 50 mg/m2 total dose may acutely develop hemolysis, neurological abnormalities, interstitial pneumonia, and glomerular damage resulting in renal failure. The incidence of renal failure increases to 28% in patients who receive total doses of 70 mg/m2 or higher (Valavaara and Nordman, 1985). There is no effective treatment for the disorder; blood transfusion may cause pulmonary edema. Mitomycin causes interstitial pulmonary fibrosis, and total doses above 30 mg/m2 have infrequently led to congestive heart failure (Verweij et al., 1988). It also may potentiate the cardiotoxicity of doxorubicin when used in conjunction with this drug (Bachur et al., 1978). Enzymes L-Asparaginase History In 1953, Kidd reported that guinea pig serum had antileukemic activity and identified L-asparaginase as the source of this activity (Kidd, 1953). Fifteen years later, the enzyme was introduced into cancer chemotherapy in an effort to exploit a distinct, qualitative difference between normal and malignant cells (Broome, 1981). Mechanism of Action Most normal tissues synthesize L-asparagine in amounts sufficient for protein synthesis. Certain neoplastic tissues, however, including acute lymphoblastic leukemic cells, require an exogenous source of this amino acid. L-Asparaginase, by catalyzing the hydrolysis of circulating asparagine to aspartic acid and ammonia, deprives these cells of the asparagine necessary for protein synthesis, leading to cell death. L-Asparaginase is commonly used in combination with methotrexate, doxorubicin, vincristine, and prednisone for the treatment of acute lymphoblastic leukemia. The sequence of drug administration in these combinations may be critical; for example, synergistic cytotoxicity results when methotrexate precedes the enzyme, but the reverse sequence leads to abrogation of methotrexate cytotoxicity. The latter outcome is a consequence of the inhibition of protein synthesis by L-asparaginase, an effect that stops the progression of cells through the cell cycle and negates the effect of methotrexate, a drug that exerts its greatest effect during the DNA synthetic phase of the cell cycle (Capizzi and Handschumacher, 1982). L-Asparaginase produces cell death through activation of apoptosis. Resistance arises through induction of the capacity of tumor cells to synthesize L-asparagine. Absorption, Fate, and Excretion L-Asparaginase is given parenterally. The rate of clearance from plasma varies considerably with different preparations. After intravenous administration, L-asparaginase has a clearance rate from plasma of 0.035 ml/minute per kg. Its apparent volume of distribution is only 55 ml/kg, approximately the volume of plasma in human beings. Its average half-life is 30 hours (Asselin et al., 1993), the Merck and Kyawa Hacco preparations having a somewhat longer plasma half-life than the Bayer preparation. An Erwinia preparation (see below), used in patients hypersensitive to the enzyme from Escherichia coli, has a shorter half-life of 16 hours and thus requires administration of higher doses. Pegaspargase (see below) is cleared much less rapidly (plasma half-life is 14.9 days) (Ho et al., 1986). Therapeutic Uses E. coli produces two L-asparaginase isozymes, only one of which (EC-2) has antileukemic activity. The purified E. coli enzyme (ELSPAR) is available for clinical use and has a molecular weight of 130,000 daltons. It consists of four equivalent subunits (see Patterson, 1975). Also available for use in patients hypersensitive to the native E. coli enzyme are the enzyme from Erwinia chrysanthemi (Minton et al., 1986) and a form of the E. coli enzyme modified by conjugation to polyethylene glycol (pegaspargase, ONCASPAR). The E. coli enzyme is administered intravenously or intramuscularly in a variety of regimens and schedules. Typically, 5000 to 10,000 U per m2 are given every other or every third day for 2 to 4 weeks, or single doses of up to 25,000 U per week. L-Asparagine levels in plasma fall immediately with drug administration and remain undetectable for 1 week after a single large dose. Recovery of L-asparagine levels occurs when enzyme levels fall below 0.03 U/ml in plasma. Because of its longer half-life, pegaspargase is given in doses of 2500 U/m2 every other week. Intermittent dosage regimens have an increased risk of anaphylaxis. In hypersensitive patients, circulating antibodies lead to immediate inactivation of the enzyme and L-asparaginase levels rapidly become unmeasurable after drug administration. L-Asparaginase is a useful component of regimens for treatment of acute lymphoblastic leukemia and other lymphoid malignancies. Clinical Toxicity Few objective responses have occurred in extensive trials in solid tumors. L-Asparaginase has minimal effects on bone marrow and gastrointestinal mucosa. Its most serious toxicities result from its antigenicity as a foreign protein and its inhibition of protein synthesis. Hypersensitivity reactions occur in 5% to 20% of patients and may be fatal. These reactions are heralded by the appearance of circulating neutralizing antibody in some, but not all, hypersensitive patients. In these patients, pegaspargase is a safe alternative, and the Erwinia enzyme may be used with caution (Ho et al., 1986; Keating et al., 1993). Other toxicities result from inhibition of protein synthesis in normal tissues and include hyperglycemia due to insulin deficiency, clotting abnormalities due to deficient clotting factors, and hypoalbuminemia. The clotting problems may take the form of spontaneous thrombosis related to deficient factor S, factor C, or antithrombin III, or, less frequently, hemorrhagic episodes. Thrombosis of cortical sinus vessels frequently goes unrecognized. Magnetic resonance imaging studies should be considered in patients treated with L-asparaginase who present with seizures, headache, or altered mental status (Bushara and Rust, 1997). There is evidence that the majority of L-asparaginaseinduced thromboses occur in patients with underlying inherited disorders of coagulation, such as Factor V Leiden, elevated serum homocysteine, protein C or S deficiency, AT III deficiency, or the 620210A variant of prothrombin (Nowak-Gottl et al., 1999). Intracranial hemorrhage in the first week of L-asparaginase treatment is an infrequent but often devastating complication. L-Asparaginase suppresses immune function as well. In addition to these side effects, coma may result rarely and has been attributed to ammonia toxicity resulting from L-asparagine hydrolysis. Pancreatitis also has been observed; its cause is uncertain. |
Miscellaneous Agents
|
Platinum Coordination Complexes The platinum coordination complexes were first identified by Rosenberg
and coworkers as cytotoxic agents in 1965. They observed that a current
delivered between platinum electrodes produced inhibition of E. coli
proliferation. The inhibitory effects on bacterial replication were later
ascribed to the formation of inorganic platinum-containing compounds in the
presence of ammonium and chloride ions (Rosenberg et al., 1965, 1967).
cis-Diamminedichloro-platinum (II) (cisplatin) was the most
active of these substances in experimental tumor systems and has proven to be
of great clinical value ( Chemistry cis-Diamminedichloroplatinum (II) (cisplatin) is a divalent inorganic water-soluble, platinum-containing complex. Other platinum complexes, some of which lack cross-resistance with cisplatin in preclinical tests, are currently in clinical trials; these include tetraplatin, ormiplatin, iproplatin, and oxaliplatin (Kelland, 1993). In each case, the coordination of di- or tetravalent platinum with various organic adducts reduces its renal toxicity and stabilizes the metal ion, but none of the complexes has unique clinical effects as an antitumor agent at this point in development. In carboplatin, platinum is incorporated into a more complex carbon-containing molecule. The structural formulas of cisplatin and carboplatin are as follows:
Mechanism of Action Cisplatin appears to enter cells by diffusion. The chloride atoms may be displaced directly by reaction with nucleophiles such as thiols; replacement of chloride by water yields a positively charged molecule and is probably responsible for formation of the activated species of the drug, which then reacts with nucleic acids and proteins. Aquation is favored at low concentrations of chloride. High concentrations of the anion stabilize the drug, explaining the effectiveness of chloride diuresis in preventing nephrotoxicity (see below). Hydrolysis of carboplatin removes the bidentate cyclobutanedicarboxylato group; this activation reaction occurs more slowly with carboplatin than with cisplatin. The platinum complexes can react with DNA, forming both intrastrand and interstrand cross-links. The N7 of guanine is very reactive, and platinum cross-links between adjacent guanines on the same DNA strand; guanineadenine cross-links also readily form. The formation of interstrand cross-links is a slower process and occurs to a lesser extent. DNA adducts formed by cisplatin inhibit DNA replication and transcription and lead to breaks and miscoding. Although no conclusive association between platinum-DNA adduct formation and efficacy has been documented, the ability of patients to form and sustain platinum adducts appears to be an important predictor of clinical response. When quantifying the effect of platinum adduct formation, it is difficult to measure the relative importance of pharmacogenetic factors and environmental exposures common to tumor and normal tissues, as well as the contribution of concomitantly administered chemotherapies. Nevertheless, preclinical data suggest that the formation of the platinum-adenosine-to-guanosine adduct may be the most critical adduct in terms of cytotoxicity (Reed et al., 1986; Parker et al., 1991b; Comess et al., 1992; Fichtinger-Schepman et al., 1995; Welters et al., 1999). The specificity of cisplatin with regard to phase of the cell cycle appears to differ among cell types, although the effects on cross-linking are most pronounced during the S phase. Cisplatin is mutagenic, teratogenic, and carcinogenic. Secondary leukemias after cisplatin have been reported, and the use of cisplatin- or carboplatin-based chemotherapy for women with ovarian cancer is associated with a fourfold increased risk of developing secondary leukemia (Jeha et al., 1992; Travis et al., 1999). The causes of tumor cell resistance to cisplatin and its analogs are incompletely understood. The various analogs differ in their degree of cross-resistance with cisplatin in experimental tumor systems. Carboplatin tends to share cross-resistance in most experimental tumors, while oxaliplatin and the tetravalent analogs do not, a finding that has led to interest in their clinical evaluation. A number of factors influence cisplatin sensitivity in experimental cells, including intracellular drug accumulation, intracellular levels of glutathione and other sulfhydryls such as metallothionein that bind to and inactivate the drug (Meijer et al., 1990), and rates of repair of DNA adducts (Parker et al., 1991a). The cisplatin adduct with DNA produces a bend in the helix, a change that is recognized by specific proteins of the high-mobility group (Huang et al., 1994), which are believed to inhibit the repair process. Repair of cisplatin-DNA adducts occurs through the nucleotide excision repair pathway, designated NER (Dabholkar et al., 1994; Reed, 1998; de Laat et al., 1999). Through a series of enzymatic steps, NER recognizes and excises the affected base, inserts a new base, and religates the affected strand. Inhibition of NER may increase sensitivity to cisplatin. Resistance to cisplatin appears to be mediated to some extent through the mismatch repair (MMR) proteins. Defects or deficiencies in MMR proteins, particularly hMLH1 or hMSH6, may be important in the recognition of platinum-DNA adducts and in initiating apoptosis. Loss of MMR has been associated with resistance to cisplatin in vitro (Vaisman et al., 1998). In addition, MMR proteins also may be involved in mediating apoptosis. Through an hMLH1-dependent event, cisplatin induces overexpression of p73, a member of the p53 family, as well as c-ABL tyrosine kinase and consequently activates apoptosis (Gong et al., 1999). In response to cisplatin exposure, induction of apoptosis is not seen in those cells deficient in MMR and in those cells unable to upregulate c-ABL tyrosine kinase. Cisplatin Absorption, Fate, and Excretion After rapid intravenous administration of usual doses, the drug has an initial elimination half-life in plasma of 25 to 50 minutes; concentrations of total drug, bound and unbound, fall thereafter, with a half-life of 24 hours or longer. More than 90% of the platinum in the blood is covalently bound to plasma proteins. High concentrations of cisplatin are found in the kidney, liver, intestine, and testes, but there is poor penetration into the CNS. Only a small portion of the drug is excreted by the kidney during the first 6 hours; by 24 hours up to 25% is excreted, and by 5 days up to 43% of the administered dose is recovered in the urine. When given by infusion instead of rapid injection, the plasma half-life is shorter and the amount of drug excreted is greater. Biliary or intestinal excretion of cisplatin appears to be minimal (Bajorin et al., 1986). Therapeutic Uses Cisplatin PLATINOL-AQ) is available for intravenous dosing. The usual intravenous dose of cisplatin is 20 mg/m2 per day for 5 days or 100 mg/m2, given once every 4 weeks. Doses as high as 40 mg/m2 daily for 5 consecutive days have been used alone or together with cyclophosphamide for the treatment of patients with advanced ovarian cancer, but result in greater renal, hearing, and neurological toxicity (Ozols et al., 1984). To prevent renal toxicity, hydration of the patient by the infusion of 1 to 2 liters of normal saline prior to treatment is recommended. The appropriate amount of cisplatin is then diluted in a solution of dextrose and saline and administered intravenously over a period of 6 to 8 hours. Since aluminum reacts with and inactivates cisplatin, it is important not to use needles or other equipment that contains aluminum when preparing or administering the drug. Combination chemotherapy with cisplatin, bleomycin, etoposide, and vinblastine is curative for 85% of patients with advanced testicular cancer (Williams and Einhorn, 1985; Einhorn, 1986). The drug also is beneficial in carcinoma of the ovary, particularly when used with paclitaxel, cyclophosphamide, or doxorubicin (Durant and Omura, 1985). Cisplatin consistently produces responses in cancers of the bladder, head and neck, and endometrium; small cell carcinoma of the lung; and some neoplasms of childhood. Interestingly, the drug also sensitizes cells to the cytotoxic effects of radiation therapy (see Pearson and Raghavan, 1985). Clinical Toxicities Cisplatin-induced nephrotoxicity has been largely abrogated by the routine use of hydration and diuresis. However, ototoxicity caused by cisplatin is unaffected by diuresis and is manifested by tinnitus and hearing loss in the high-frequency range (4000 to 8000 Hz). The ototoxicity can be unilateral or bilateral, tends to be more frequent and severe with repeated doses, and may be more pronounced in children. Marked nausea and vomiting occur in almost all patients and usually can be controlled with ondansetron or high-dose corticosteroids. At higher doses or after multiple cycles of treatment, cisplatin causes peripheral neuropathy, which may worsen after discontinuation of the drug. Mild-to-moderate myelosuppression may occur, with transient leukopenia, thrombocytopenia, and anemia. Electrolyte disturbances, including hypomagnesemia, hypocalcemia, hypokalemia, and hypophosphatemia, are common. Hypocalcemia and hypomagnesemia secondary to renal electrolyte wasting have been observed and may produce tetany. Routine measurement of Mg2+ concentrations in plasma is recommended. Hyperuricemia, seizures, hemolytic anemia, and cardiac abnormalities have been reported. Anaphylactic-like reactions, characterized by facial edema, bronchoconstriction, tachycardia, and hypotension, may occur within minutes after administration and should be treated by intravenous injection of epinephrine and with corticosteroids or antihistamines. Carboplatin The mechanism of action and spectrum of clinical activity of carboplatin (CBDCA, JM-8) are similar to those of cisplatin (see above). However, there are significant differences in the chemical, pharmacokinetic, and toxicological properties of the two drugs (see Von Hoff, 1987; Muggia, 1989; Ozols, 1989). Carboplatin is less reactive than cisplatin, and the drug is not bound to plasma proteins to a significant extent. As a result, there are no appreciable quantities of low-molecular-weight platinum-containing species (other than carboplatin itself) in plasma, and most of the drug is eliminated in the urine as such, with a half-life of about 2 hours. Platinum from the drug does become irreversibly bound to plasma proteins, and this fraction of the metal disappears slowly (half-life of 5 days or more). Carboplatin is relatively well tolerated clinically. There is less nausea, neurotoxicity, ototoxicity, and nephrotoxicity than with cisplatin. Instead, the dose-limiting toxicity is myelosuppression, primarily evident as thrombocytopenia. Carboplatin and cisplatin appear to be equally effective in the treatment of suboptimally debulked ovarian cancer, nonsmall cell lung cancer, and extensive stage small cell lung cancer; however, carboplatin may be less effective than cisplatin in germ cell, head and neck, and esophageal cancers (Go and Adjei, 1999). Carboplatin is an effective alternative for patients with responsive tumors who are unable to tolerate cisplatin because of impaired renal function, refractory nausea, significant hearing impairment, or neuropathy. In addition, it may be used in high-dose therapy with bone marrow or peripheral stem cell rescue. The dose of carboplatin should be adjusted in proportion to the reduction in creatinine clearance for patients with a creatinine clearance below 60 mg/ml (Van Echo et al., 1989). Calvert et al. (1989) have proposed the following formula for calculation of dose: Dose (mg) = AUC x (GFR + 25) (521) where the target AUC (area under the plasma concentrationtime curve) is in the range of 5 to 7 mg/ml per minute for acceptable toxicity in patients receiving single-agent carboplatin. (GFR = glomerular filtration rate; see Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination.) Carboplatin PARAPLATIN) is administered as an intravenous infusion over at least 15 minutes. The usual dose is 360 mg/m2, given once every 28 days. Carboplatin currently is approved for use in combination with paclitaxel or cyclophosphamide in patients with advanced ovarian cancer. It also has been shown to be effective in a number of tumors including lung cancer, bladder cancer, and head and neck cancer. Oxaliplatin Oxaliplatin or trans-1-diaminocyclohexane oxala-toplatinum has a diaminocyclohexane (DACH) carrier ligand that endows it with unique properties, thus allowing it to escape recognition by NER and MMR proteins. Its structure is as follows:
Unlike cisplatin or carboplatin, oxaliplatin is equally effective in MMR-proficient and -deficient cell lines and tumor xenografts (Fink et al., 1996; Fink et al., 1997). In particular, hMLH1 defects that produce cisplatin resistance in vitro have no effect on oxaliplatin cytotoxicity (Vaisman et al., 1998). Furthermore, the upregulation of c-ABL tyrosine kinase seen with cisplatin in the presence of functional MMR proteins is not observed after the administration of oxaliplatin (Nehme et al., 1999). In contrast to cisplatin and carboplatin, oxaliplatin has a very large
volume of distribution (Graham et al., 2000). The pharmacokinetics of
oxaliplatin are triexponential with short initial Like cisplatin, oxaliplatin shows a wide range of antitumor activity
and is active in ovarian cancer, germ-cell cancer, and cervical cancer.
Unlike cisplatin, oxaliplatin in combination with 5-fluorouracil is active in
colorectal cancer, perhaps due to its MMR-independent effects. In combination
with 5-fluorouracil, it is approved for treatment of patients with advanced
colorectal cancer in Europe, Asia, and Hydroxyurea Hydroxyurea originally was synthesized by Dresler and Stein in 1869, but its potential biological significance was not recognized until 1928, when leukopenia and megaloblastic anemia were observed in experimental animals treated with this compound. In the 1950s, the drug was evaluated in a large number of experimental murine tumor models and was found to have broad antitumor activity against both leukemia and solid-tumor models. Clinical trials with hydroxyurea began in the 1960s. Since then, this drug has continued to be of interest to both clinical and laboratory investigators, as it has a number of unique and surprisingly diverse biological effects that have led to exploration of its clinical utility in a wide range of malignant and nonmalignant diseases. Its use has been encouraged by the facts that the drug can be administered orally and that its toxicity in most patients is very modest. Comprehensive descriptions of the pharmacology of hydroxyurea have been published (Navarra and Preziosi, 1999; Paz-Ares and Donehower, 2001). The structural formula of hydroxyurea is as follows:
Cytotoxic Action Hydroxyurea is representative of a group of compounds that have as
their primary site of action the enzyme ribonucleoside diphosphate reductase.
A striking correlation has been observed between the relative growth rate of
a series of rat hepatomas and the activity of ribonucleoside diphosphate
reductase. This enzyme, which catalyzes the reductive conversion of
ribonucleotides to deoxyribonucleotides, is a crucial and probably
rate-limiting step in the biosynthesis of DNA, and it represents a logical
target for the design of chemotherapeutic agents. Hydroxyurea destroys a
tyrosyl free radical that is formed in the catalytic center of the enzyme.
The drug is specific for the S phase of the cell cycle, in which
concentrations of the target reductase are maximal, and it causes cells to
arrest at the G1S interface. Since cells are highly sensitive to
irradiation in the G1 phase of the cycle, combinations of
hydroxyurea and irradiation cause synergistic toxicity (Schilsky et al.,
1992). Hydroxyurea also may potentiate the antiproliferative effects of
DNA-damaging agents such as cisplatin, alkylating agents, or topoisomerase II
inhibitors. Even more interesting are the modulator effects of hydroxyurea on
antimetabolite drugs, particularly nucleotide analogs. The decrease of the
intracellular deoxyribonucleotide pools after hydroxyurea exposure
facilitates the incorporation of drugs such as AraC, gemcitabine, or fludarabine
into DNA. This type of interaction has implications for the anti-HIV effect
of hydroxyurea. The inhibition of cellular ribonucleoside diphosphate reductase
favors the incorporation of an increased proportion of the nucleoside reverse
transcriptase inhibitors into the viral DNA (Lori et al., 1994).
Hydroxyurea also has been shown to be converted in vivo to nitric
oxide, to induce the expression of a number of genes [for TNF, interleukin-6
(IL-6), The principal mechanism by which cells achieve resistance to hydroxyurea is through elevation in cellular ribonucleoside diphosphate reductase activity. Several different molecular mechanisms may contribute to the increased ribonucleoside reductase activity in hydroxyurea-resistant cells, including gene amplification and increased translational efficiency. It has been suggested that some examples of resistance to hydroxyurea may be the result of the production of a ribonucleoside diphosphate reductase with decreased sensitivity to inhibition by hydroxyurea. Absorption, Fate, and Excretion The oral bioavailability of hydroxyurea is excellent (80% to 100%), and comparable plasma concentrations are seen after oral or intravenous dosing (Rodriguez et al., 1998). Peak plasma concentrations are reached 1.0 to 1.5 hours after oral doses of 15 to 80 mg/kg. Hydroxyurea disappears from plasma with a half-life from 3.5 to 4.5 hours. The drug readily crosses the bloodbrain barrier, and it appears in significant quantities in human breast milk. From 40% to 80% of the drug is recovered in the urine within 12 hours after either intravenous or oral administration. Although precise guidelines are not available, it seems prudent to modify doses for patients with abnormal renal function until individual tolerance can be assessed. Data from several experimental animal systems suggest that metabolism of hydroxyurea does occur, but the extent and significance of metabolism of the drug in human beings have not been established. Therapeutic Uses Two dosage schedules for hydroxyurea (HYDREA), alone or in combination with other drugs, are most commonly used in a variety of clinical situations: (1) intermittent therapy with 80 mg/kg administered orally as a single dose every third day or (2) continuous therapy with 20 to 30 mg/kg administered as a single daily dose. Dosage should be adjusted according to the number of leukocytes in the peripheral blood. Treatment is typically continued for a period of 6 weeks in malignant diseases to determine its effectiveness; if satisfactory antineoplastic results are obtained, therapy can be continued indefinitely, although leukocyte counts at weekly intervals are advisable. The principal use of hydroxyurea has been as a mye-losuppressive agent in the myeloproliferative syndromes, particularly chronic granulocytic leukemia, polycythemia vera, and essential thrombocytosis. Currently, hydroxyurea is prescribed for patients with myeloproliferative syndromes who are not candidates for interferon treatment, or the drug is given in combination with interferon during the induction phase of therapy (Silver et al., 1999). Hydroxyurea cannot be considered to be standard therapy either as a single agent or as part of the standard chemotherapy regimen for any solid tumor, although it has produced anecdotal, temporary remissions in patients with advanced cancers (e.g., head and neck or genitourinary carcinomas, melanoma). Hydroxyurea has been incorporated into several schedules with concurrent irradiation, as it is able to synchronize cells into a radiation-sensitive phase of the cell cycle. This combination has shown promise in several diseases, including cervical carcinoma, primary brain tumors, head and neck cancer, and nonsmall cell lung cancer, although it has not been proven to be superior to regimens including cisplatin and irradiation. Hydroxyurea has been approved by the FDA for the treatment of adult
patients with sickle cell disease. The drug reduces the number of painful crises,
the frequency of acute chest syndrome and hospitalization, and the need for
blood transfusion (Charache et al., 1995). Hydroxyurea appears to be
effective in children with sickle cell disease and in patients with sickle
cell Hydroxyurea may serve as an important paradigm for agents that contribute to inhibition of HIV replication by a mechanism other than the one that targets a viral enzyme or a structural protein (Lori, 1999). Currently available clinical data reveal that hydroxyurea has little activity as a single agent but produces pronounced inhibition of HIV replication when combined with didanosine or with didanosine plus stavudine in nonheavily pretreated patients (see Chapter 51: Antiretroviral Agents: Antiretroviral Agents). Importantly, hydroxyurea appears to maintain the activity of the nucleoside reverse transcriptase inhibitors even in the presence of genotypic mutations of the HIV characteristically associated with resistance to the drugs. Clinical Toxicity Hematopoietic depressioninvolving leukopenia, megaloblastic anemia, and occasionally thrombocy-topeniais the major toxic effect; recovery of the bone marrow usually is prompt if the drug is discontinued for a few days. Other adverse reactions include gastrointestinal disturbances and mild dermatological reactions; more rarely, stomatitis, alopecia, and neurological manifestations have been encountered. Inflammation and increased pigmentation may occur in areas previously exposed to radiation. Hydroxyurea may increase the risk of secondary leukemia in patients with myeloproliferative disorders and should be used with caution in nonmalignant diseases. Hydroxyurea is a potent teratogen in all animal species tested and should not be used in women with childbearing potential. Procarbazine The methylhydrazine derivatives were synthesized among a large number of substituted hydrazines in a search for inhibitors of monoamine neurotransmitters. Several compounds in this series (Bollag, 1963) were discovered to have anticancer activity, but only procarbazine, an agent useful in Hodgkin's disease, has won a place in clinical chemotherapy. Its structural formula is as follows:
Cytotoxic Action Procarbazine must undergo metabolic activation to generate the proximal cytotoxic reactants, which methylate DNA. The activation pathways are complex and not fully understood. The first step involves oxidation of the hydrazine function with formation of the azo analog. This can occur spontaneously in neutral solution by reaction with molecular oxygen and also can occur enzymatically by reaction with the cytochrome P450 system of the liver. Further oxidations can generate the methylazoxy and benzylazoxy intermediates. It is postulated that the methylazoxy compound can react further to liberate an entity resembling diazomethane, a potent methylating reagent. Free-radical intermediates also may be involved in cytotoxicity. Activated procarbazine can produce chromosomal damage, including chromatid breaks and translocations. These effects are consistent with its mutagenic and carcinogenic actions. Exposure to procarbazine leads to inhibition of DNA, RNA, and protein synthesis in vivo. Resistance to procarbazine develops rapidly when it is used as a single agent. One mechanism results from the increased ability to repair methylation of guanine via guanine-O6-alkyl transferase (Souliotis et al., 1990). Absorption, Fate, and Excretion Procarbazine is absorbed almost completely from the gastrointestinal tract. After parenteral administration, the drug is readily equilibrated between the plasma and the CSF. It is rapidly metabolized in human beings, and its half-life in the blood after intravenous injection is approximately 7 minutes. Oxidation of procarbazine produces the corresponding azo compound and hydrogen peroxide. Further metabolism, presumably in the liver, yields azoxy derivatives that circulate in the bloodstream and have potent cytotoxic activity (Erikson et al., 1989). Induction of microsomal enzymes by phenobarbital and other agents enhances the rate of conversion of procarbazine to its active metabolites; the potential for drug interaction thus exists when procarbazine is administered with other agents that are metabolized by microsomal enzymes. From 25% to 70% of an oral or parenteral dose given to human beings is recovered from the urine during the first 24 hours after administration; less than 5% is excreted as the unchanged compound, and the rest is mostly in the form of a metabolite, N-isopropylterephthalamic acid (Friedman, 2001). Therapeutic Uses The recommended dose of procarbazine (MATULANE) for adults is 100 mg/m2 daily for 10 to 14 days in combination regimens. The drug rarely is used alone. Procarbazine primarily is used in the combination therapy of Hodgkin's disease. It is given with mechlorethamine, vincristine, and prednisone (the MOPP regimen) (DeVita, 1981). Of primary importance, procarbazine lacks cross-resistance with other mustard-type alkylating agents. Procarbazine also has demonstrated activity against brain tumors, small cell carcinoma of the lung, non-Hodgkin's lymphomas, myeloma, and melanoma. Clinical Toxicity Most common toxic effects include leukopenia and thrombocytopenia, which begin during the second week of therapy and reverse within 2 weeks off treatment. Gastrointestinal symptoms such as mild nausea and vomiting occur in most patients; gastrointestinal symptoms and neurological and dermatological manifestations have been noted in 5% to 10% of cases. Disturbances in behavior also have been reported. Because of augmentation of sedative effects, the concomitant use of CNS depressants should be avoided. The ingestion of alcohol by patients receiving procarbazine may cause intense warmth and reddening of the face, as well as other effects resembling the acetaldehyde syndrome produced by disulfiram (see Chapter 18: Ethanol). Since procarbazine is a weak monoamine oxidase inhibitor, hypertensive reactions may result from its use concurrently with sympathomimetic agents, tricyclic antidepressants, or foods with high tyramine content. Procarbazine is highly carcinogenic, mutagenic, and teratogenic, and its use in MOPP therapy is associated with a 5% to 10% risk of acute leukemia; the greatest risk is for patients who also receive radiation therapy (Tucker et al., 1988). Procarbazine is also a potent immunosuppressive agent, and it causes infertility, particularly in males. Mitotane The principal application of mitotane (o,p'-DDD), a compound chemically similar to the insecticides DDT and DDD, is in the treatment of neoplasms derived from the adrenal cortex. In studies of the toxicology of related insecticides in dogs, it was noted that the adrenal cortex was severely damaged, an effect caused by the presence of the o,p' isomer of DDD, whose structural formula is as follows:
Cytotoxic Action The mechanism of action of mitotane has not been elucidated, but its relatively selective attack on adrenocortical cells, normal or neoplastic, is well established. Thus, administration of the drug causes a rapid reduction in the levels of adrenocorticosteroids and their metabolites in blood and urine, a response that is useful both in guiding dosage and in following the course of hyperadrenocorticism (Cushing's syndrome) resulting from an adrenal tumor or adrenal hyperplasia. Damage to the liver, kidneys, or bone marrow has not been encountered. Absorption, Fate, and Excretion Clinical studies indicate that approximately 40% of mitotane is
absorbed after oral administration. After daily doses of 5 to 15 g,
concentrations of 10 to 90 Therapeutic Uses Mitotane LYSODREN) is administered in initial daily oral doses of 2 to 6 g, usually given in three or four divided portions, but the maximal tolerated dose may vary from 2 to 16 g per day. Treatment should be continued for at least 3 months; if beneficial effects are observed, therapy should be maintained indefinitely. Spironolactone should not be administered concomitantly, since it interferes with the adrenal suppression produced by mitotane (Wortsman and Soler, 1977). Treatment with mitotane is indicated for the palliation of inoperable adrenocortical carcinoma. Hutter and Kayhoe (1966) reported on treatment in 138 patients, and 115 were studied by Lubitz and associates (1973). Clinical effectiveness was reported in 34% to 54% of these cases. Apparent cures have been reported in some patients with metastatic disease (Becker and Schumacher, 1975; Ostuni and Roginsky, 1975). Clinical Toxicity Although the administration of mitotane produces anorexia and nausea in approximately 80% of patients, somnolence and lethargy in about 34%, and dermatitis in 15% to 20%, these effects do not contraindicate the use of the drug at lower doses. Since this drug damages the adrenal cortex, administration of adrenocorticosteroids is indicated, particularly in patients with evidence of adrenal insufficiency, shock, or severe trauma (Hogan et al., 1978). |
Hormones and Related Agents
|
Adrenocorticosteroids The pharmacology, major therapeutic uses, and toxic effects of the glucocorticoids are discussed in Chapter 60: Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones. Only the applications of the hormones in the treatment of neoplastic disease are considered here. Because of their lympholytic effects and their ability to suppress mitosis in lymphocytes, the greatest value of these steroids as cytotoxic agents is in the treatment of acute leukemia in children and malignant lymphoma in children and adults. In acute lymphoblastic or undifferentiated leukemia of child-hood, glucocorticoids may produce prompt clinical improvement and objective hematological remissions in up to 30% of children. Although these responses frequently are characterized by complete disappearance of all detectable leukemic cells from the peripheral blood and bone marrow, the duration of remission is brief. Remissions occur more rapidly with glucocorticoids than with antimetabolites, and there is no evidence of cross-resistance to unrelated agents. For these reasons, therapy is initiated with prednisone and vincristine often followed by an anthracycline, or methotrexate, and L-asparaginase. Glucocorticoids are a valuable component of curative regimens for Hodgkin's disease and non-Hodgkin's lymphoma, as well as for treatment of multiple myeloma and chronic lymphocytic leukemia (CLL). Glucocorticoids are extremely helpful in controlling autoimmune hemolytic anemia and thrombocytopenia associated with CLL. The glucocorticoids, particularly dexamethasone, are used in conjunction with x-ray therapy to reduce edema related to tumors in critical areas such as the superior mediastinum, brain, and spinal cord. Doses of 4 to 6 mg every 6 hours have dramatic effects in restoring neurological function in patients with cerebral metastases, but these effects are temporary. Acute changes in dexamethasone dosage can lead to a rapid recrudescence of symptoms. Dexamethasone should not be discontinued abruptly in patients receiving radiotherapy or chemotherapy for brain metastases. Gradual tapering of the dosage may be undertaken if a clinical response to definitive antitumor therapy has been achieved. The antitumor effects of glucocorticoids are mediated by their binding to a specific cytoplasmic receptor, which, when activated, induces a program of gene expression that leads to apoptosis. Several glucocorticoids are available and at appropriate dosages exert similar effects (see Chapter 60: Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones). Prednisone, for example, is usually administered orally in doses as high as 60 to 100 mg, or even higher, for the first few days and gradually reduced to levels of 20 to 40 mg per day. A continuous attempt should be made to establish the lowest possible dosage required to control the manifestations of the disease. These agents, when used chronically, exert a wide range of side effects, including glucose intolerance, immunosuppression, osteoporosis, gastrointestinal ulceration, and psychosis (see Chapter 60: Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones). Aminoglutethimide and Other Aromatase Inhibitors Aminoglutethimide Originally developed as an anticonvulsant, aminoglutethimide subsequently was found to inhibit the synthesis of adrenocortical steroids (see Chapter 60: Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones). Aminoglutethimide inhibits the conversion of cholesterol to pregnenolone, the first step in the synthesis of cortisol. Inhibition of cortisol synthesis, however, results in a compensatory rise in the secretion of adrenocorticotropic hormone (ACTH) sufficient to overcome the adrenal blockade. Administration of dexamethasone does not prevent the increase in ACTH secretion because aminoglutethimide accelerates the metabolism of dexamethasone. Since the metabolism of hydrocortisone (cortisol) is not affected by aminoglutethimide, this combination produces reliable inhibition of the synthesis of cortisol (Santen et al., 1980). Amino-glutethimide has been used to treat patients with adrenocortical carcinoma and Cushing's syndrome or metastatic, hormone-dependent breast cancer refractory to other hormonal approaches. Although aminoglutethimide effectively blocks the secretion of cortisol, the production of other adrenal steroidssuch as testosterone, dihydrotestosterone, androstenedione, progesterone, and 17-hydroxyprogesteroneis only partially inhibited. In certain tissues, including fat, muscle, and liver, androstenedione is converted by aromatization to estrone and estradiol. In postmenopausal and castrated women, the adrenal gland does not produce estrogens, but it is the most important source of precursors of estrogens. By inhibiting cytochrome P450dependent hydroxylation reactions that are necessary for aromatization reactions, aminoglutethimide is a potent inhibitor of the conversion of androgens to estrogens in extraadrenal tissues. Patients treated with aminoglutethimide and hydrocortisone thus experience a lowering of plasma and urinary concentrations of estradiol that is equivalent to that observed in patients treated by surgical adrenalectomy (Santen et al., 1982). Therapeutic Uses When it is used to treat patients with metastatic breast cancer, aminoglutethimide (CYTADREN) is administered orally at a dose of 125 mg twice daily in combination with 20 mg of hydrocortisone for 2 weeks, then increasing to 250 mg twice a day together with 40 mg of hydrocortisone in divided doses. The largest dose of hydrocortisone, 20 mg, is given at night. After 2 weeks of aminoglutethimide therapy, corticosteroid synthesis recovers spontaneously, and hydrocortisone supplementation may be discontinued. When used to control Cushing's syndrome, aminoglutethimide is given in the same dosage but without hydrocortisone. In these patients, plasma concentrations of hydrocortisone should be monitored, and the dose of aminoglutethimide is titrated as necessary (up to 2 g per day) to achieve suppression of adrenal function. In some patients, significant inhibition of adrenal function occurs at doses of 250 to 500 mg daily, and toxicity is thus reduced. The major indication for the use of aminoglutethimide is to produce inhibition of aromatase activity (the conversion of androgens to estrogens) in patients with advanced carcinoma of the breast when the tumor contains estrogen receptors. Its primary role in this setting has been supplanted by tamoxifen, with aminoglutethimide considered as either a second- or a third-line endocrine maneuver. If women are selected for therapy without regard to the status of estrogen receptors in the tumor, the response rate is 37%; patients whose tumor cells contain estrogen receptors experience a 50% response rate. Skin, soft tissue, and bone lesions respond more frequently than do lesions at other sites of metastasis. Such treatment is equal or superior to surgical adrenalectomy or hypophysectomy. Clinical Toxicity Early toxic effects of aminoglutethimide include lethargy, visual blurring, drowsiness, and ataxia. These symptoms usually resolve after 4 to 6 weeks of treatment. A pruritic, maculopapular rash usually appears 10 days after treatment is initiated and resolves after approximately 5 days without withdrawal of the drug. Since the adrenal gland recovers normal secretory activity and the response to stress 36 hours after hydrocortisone is withdrawn, it is not necessary to taper the administration of this agent. Steroidal and Imidazole Aromatase Inhibitors Two newer classes of inhibitors of aromatase, the enzyme that converts androgens to estrogens, have found a useful role in breast cancer treatment; these include the steroidal androstenedione analogs formestane and exemestane and the imidazole inhibitors anastrozole, vorozole, and letrozole (see also Chapter 58: Estrogens and Progestins). The imidazoles have become the dominant aromatase inhibitors in clinical use because of their oral route of administration, their greater effectiveness in lowering serum estrogen levels, and their favorable toxicity profile (Ellis and Swain, 2001). Formestane, which has not been approved for use in the Exemestane
(AROMASIN) is a more potent, orally
administered analog that lowers estrogen levels more effectively than does formestane.
It is FDA-approved for use in the The imidazole aromatase inhibitors have the advantage of oral
administration, rapid onset of action, total suppression of estrogen levels
below limits of detection, no androgenic side effects, and clearance by
hepatic metabolism (no dose adjustment needed for renal dysfunction). Anastrozole
and letrozole are FDA-approved for use in the Progestins Progestational agents (see Chapter 58: Estrogens and Progestins) are useful as second-line hormonal therapy for metastatic hormone-dependent breast cancer and in the management of endometrial carcinoma previously treated by surgery and radiotherapy. In addition, progestins stimulate appetite and restore a sense of well-being in cachectic patients with advanced stages of cancer and AIDS. While progesterone itself is poorly absorbed when given orally and must be used with an oil carrier when given intramuscularly, there are synthetic progesterone preparations. Hydroxyprogesterone caproate usually is administered intramuscularly in doses of 1000 mg one or more times weekly; medroxyprogesterone acetate (DEPO-PROVERA) can be administered intramuscularly in doses of 400 to 1000 mg weekly. An alternative and more commonly used oral agent is megestrol acetate (MEGACE; 40 to 320 mg daily, in divided doses). Beneficial effects have been observed in approximately one-third of patients with endometrial cancer. The response of breast cancer to megestrol is predicted by both the presence of hormonal receptors and the evidence of response to a prior hormonal treatment. Progestin therapy in breast cancer appears to be dose-dependent, with patients demonstrating second responses following escalation of megestrol to 1600 mg/day. Responses to progestational agents also have been reported in metastatic carcinomas of the prostate and kidney. Estrogens and Androgens Discussions of the pharmacology of the estrogens and androgens appear in Chapters 58: Estrogens and Progestins and 59: Androgens. Their use in the treatment of certain neoplastic diseases is discussed here. They are of value in this connection because certain organs that are often the primary sites of growth, notably the prostate and the mammary gland, are dependent upon hormones for their growth, function, and morphological integrity. Carcinomas arising from these organs often retain some of the hormonal responsiveness of their normal counterparts for varying periods of time. By changing the hormonal environment of such tumors it is possible to alter the course of the neoplastic process. Androgen-Control Therapy of Prostatic Carcinoma The development of antiandrogenic therapy for prostatic carcinoma is largely the contribution of Huggins and associates (1941). Although the hormonal treatment of metastatic prostate carcinoma is palliative, life expectancy is increased and thousands of patients have enjoyed its benefit. Localized prostate cancer is curable with surgery or radiation therapy. However, when distant metastases are already present, hormonal therapy becomes the primary treatment. Standard approaches to achieve reduction in the concentrations of endogenous androgens or inhibition of their effects include bilateral orchiectomy, antiandrogens, or most commonly, the administration of gonadotropin-releasing hormone (GnRH) agonists with or without antiandrogens (see below). Subjective and objective improvements rapidly follow the institution of androgen-control therapy of prostatic carcinoma in the majority of patients with metastatic disease, and these benefits last an average of one year. From the patient's point of view, the most gratifying is relief of bone pain. This is associated with an increase in appetite, weight gain, and a feeling of well-being. Objectively, there are regressions of the primary tumor and soft tissue metastases, but neoplastic cells do not disappear completely. The concentration of prostate-specific antigen (PSA) in plasma is a useful marker of response. Eventually prostatic tumors become insensitive to androgen deprivation through loss or mutation of the androgen receptor, which in some patients recognizes androgen antagonists such as flutamide (see below and Chapter 59: Androgens) as agonists. In such cases, withdrawal of the antagonist may lead to a response. Estrogens and Androgens in the Treatment of Mammary Carcinoma Because of the paucity of side effects and the equivalence of response, the use of antiestrogens such as tamoxifen largely has replaced treatment with estrogens or androgens as the initial approach to the hormonal therapy of breast cancer. Although the choice of regimen for the treatment of carcinoma of the breast is largely empirical, progress in endocrinology has led to the development of methods that are very useful for the selection of patients likely to respond. Tissues responsive to estrogens contain receptors for the hormones that can be detected by either ligand-binding techniques or monoclonal antibodies. Carcinomas that lack specific estrogen-binding capacity rarely respond to hormonal therapy. The tumors that contain receptors for either estrogen or progesterone have a 50% or greater response rate to hormonal therapy and, furthermore, have a better overall prognosis independent of the type of therapy. The onset of action of the hormones is slow. It often is necessary to continue therapy for 8 to 12 weeks before a decision can be reached as to effectiveness. If a favorable response is obtained, hormonal treatment should be continued until an exacerbation of symptoms occurs. Withdrawal of the hormone at this time is followed by remission of disease in 30% of cases. The duration of an induced remission averages about 1 year; however, some patients may receive benefit for years. Antiestrogens Tamoxifen The introduction of effective and nontoxic antiestrogen agents that block the actions of estrogen has been a relatively recent event (see Chapter 58: Estrogens and Progestins). However, these agents (principally the selective estrogen receptor modulator tamoxifen) have become first-line therapy for the hormonal treatment of breast cancer, both for adjuvant treatment and for the therapy of metastatic disease. Most recently, tamoxifen has shown effectiveness in reducing breast cancer incidence in women at high risk of developing breast cancer as the result of heredity, age greater than 60, or history of prior benign breast disease (Fisher et al., 1998). Mechanism of Action Tamoxifen is a competitive inhibitor of estradiol binding to the
estrogen receptor (ER). When bound to the ER, tamoxifen induces a change in
the three-dimensional shape of the receptor, inhibiting its binding to the
estrogen-responsive element (ERE) on DNA. Under normal physiological
conditions, estrogen stimulation increases tumor cell production of
transforming growth factor Absorption, Fate, and Excretion Tamoxifen is readily absorbed following oral administration, with peak concentrations measurable after 3 to 7 hours and steady-state levels reached at 4 to 6 weeks (Jordan, 1982). The drug is metabolized predominantly to N-desmethyltamoxifen and to 4-hydroxytamoxifen, a more potent metabolite. Both of these metabolites can be further converted to 4-hydroxy-N-desmethyltamoxifen, which retains high affinity for the ER. The parent drug has a terminal half-life of 7 days, while the half-lives of N-desmethyltamoxifen and 4-hydroxytamoxifen are significantly longer. After enterohepatic circulation, glucuronides and other metabolites are excreted in the stool; excretion in the urine is minimal. Therapeutic Uses Tamoxifen citrate NOLVADEX) is
marketed for oral administration. The usual dose prescribed in the Tamoxifen is the endocrine treatment of choice for postmenopausal women with estrogen-receptor positive (ER+) metastatic breast cancer or following primary tumor therapy in the adjuvant setting, where it is frequently used following chemotherapy. Tamoxifen also is used in premenopausal women with ER+ tumors; although response rates appear to be equal to those in postmenopausal patients, other alternatives such as oophorectomy or gonadotropin-releasing hormone analogs (leuprolide, goserelin) have the advantage of eliminating ovarian estrogen production. The combined use of tamoxifen and a gonadotropin-releasing hormone analog (to reduce high estrogen levels resulting from tamoxifen effects on the gonadal-pituitary axis) has had a better response rate and improved overall survival than has either drug alone (Klijn et al., 2000). Tamoxifen has been used alone as an adjuvant therapy for ER+ women at risk for recurrence following initial diagnosis and treatment of primary breast cancer. Both the NOLVADEX Adjuvant Trial Organization (NATO) study, which compared 2 years of tamoxifen treatment to observation (Baum, 1988), and the Scottish trial, which compared 5 years of tamoxifen to observation (Breast Cancer Trials Committee, 1987), indicate an overall survival advantage for the patients receiving tamoxifen. Five years of adjuvant therapy with tamoxifen yields superior results compared to 1 or 2 years of therapy (Early Breast Cancer Trialists' Collaborative Group, 1998). Tamoxifen and a related antiestrogen, raloxifene (EVISTA; see Chapter 58: Estrogens
and Progestins), have shown striking effectiveness in initial trials for
preventing breast cancer in high-risk women ( Clinical Toxicity The most frequent adverse reactions to tamoxifen include hot flashes, nausea, and vomiting. These may occur in as many as 25% of patients and are rarely sufficiently severe to require discontinuation of therapy. Menstrual irregularities, vaginal bleeding and discharge, pruritus vulvae, and dermatitis occur frequently, depending on the menopausal state of the patient. There is increasing concern about the potential of tamoxifen for causing endometrial cancer. The incidence of this tumor appears to be at least twofold higher in women who received 20 mg per day for 2 years or longer than in untreated controls. Patients receiving tamoxifen should have at least yearly pelvic examinations and should report symptoms or signs such as pelvic discomfort or vaginal bleeding (Fisher, 1994). Tamoxifen increases the risk of thromboembolic events. Like estrogen, tamoxifen is a hepatic carcinogen in animals, although increases in primary hepatocellular carcinoma have not been reported in patients on the drug. Tamoxifen causes retinal deposits, decreased visual acuity, and cataracts in occasional patients, although the frequency of these changes is uncertain (Longstaff et al., 1989). The estrogenic effect of tamoxifen also has potentially salubrious effects beyond its potential to prevent the recurrence or development of breast cancer. Tamoxifen may slow the development of osteoporosis in postmenopausal women (Fornander et al., 1990). In addition, like certain estrogens, tamoxifen lowers total serum cholesterol, LDL cholesterol, and lipoproteins and raises apolipoprotein AI levels, potentially decreasing the risk of myocardial infarction (Love et al., 1994). Gonadotropin-Releasing Hormone Analogs Gonadotropin-releasing hormone (GnRH) analogs came into use in the
1980s, and provided a medical form of castration for prostate carcinoma and
an additional hormonal manipulation for breast cancer (see Chapter 56:
Pituitary Hormones and Their Hypothalamic Releasing Factors). The analogs of
the GnRH peptideleuprolide (LUPRON), goserelin (ZOLADEX), triptorelin (TRELSTAR DEPOT), and buserelin (SUPREFACT; not available in the Antiandrogens Antiandrogens are competitive inhibitors that prevent the natural ligands of the androgen receptor from binding to the receptor. These compounds, therefore, have activity on their own against prostate cancer. They also are effective in preventing the flare reaction induced by the testosterone surge that can occur with GnRH monotherapy. Theoretically, antiandrogen therapy, in combination with a GnRH agonist, leads to a more complete androgen blockade by additionally inhibiting the biological effects of androgens produced in the adrenal glands. Clinical data, however, do not support the systematic use of complete androgen blockade, which is associated with more side effects than medical or surgical castration alone, but comparable efficacy (Eisenberger et al., 1998; Prostate Cancer Trialists' Collaborative Group, 2000). Antiandrogen monotherapy is not indicated either as routine, first-line treatment for patients with advanced disease. The antiandrogens typically are divided structurally and mechanistically into steroidal and nonsteroidal antiandrogens (NSAAs) (Reid et al., 1999). The steroidal agents have some partial agonist activity at the androgen receptor, whereas the NSAAs do not have significant agonist activity at the wild-type androgen receptor. The steroidal antiandrogens with which there is the most experience are cyproterone acetate (ANDROCUR) and megestrol acetate (MEGACE) (see above and Chapter 59: Androgens). The steroidal antiandrogens are weak partial agonists and competitive inhibitors of the androgen receptor in target tissues. In addition, they have progestational agonist properties at the level of the pituitary that reduce LH secretion. Consequently, LH-stimulated testosterone production decreases. The loss of libido, decreased sexual potency, and low testosterone levels produced by steroidal antiandrogens are among the major distinctions between the steroidal antiandrogens and NSAAs. The NSAAs in common usage are flutamide (EULEXIN), nilutamide (NILANDRON), and bicalutamide (CASODEX). These anilid derivatives inhibit the translocation of the androgen receptor to the nucleus from the cytoplasm of target cells. This appears to be the only mechanism by which NSAAs exert their antiproliferative effect in prostate cancer patients. In fact, the blockade of testosterone binding in the CNS interrupts the negative feedback of testosterone on gonadotropin secretion (Knuth et al., 1984). As a consequence, and in contrast to steroidal antiandrogens, testosterone levels increase with the use of NSAA monotherapy and the loss of sexual desire and loss of potency are less pronounced. Flutamide was the first androgen receptor antagonist to achieve
widespread use. It is metabolized to Biological Response Modifiers Biological response modifiers include agents or approaches that affect the patient's biological response to a neoplasm beneficially. Included are agents that act indirectly to mediate their antitumor effects (e.g., by enhancing the immunological response to neoplastic cells) or directly on the tumor cells (e.g., differentiating agents). Recombinant DNA technology has greatly facilitated the identification and production of a number of human proteins with potent effects on the function and growth of both normal and neoplastic cells. Proteins that are currently in clinical trials include the interferons (see Chapters 50: Antimicrobial Agents: Antiviral Agents (Nonretroviral) and 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants), interleukins (see Chapter 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants), hematopoietic growth factors (see Chapter 54: Hematopoietic Agents: Growth Factors, Minerals, and Vitamins) such as erythropoietin, filgrastim [granulocyte colony-stimulating factor (G-CSF)], and sargramostim [granulocyte/macrophage colony-stimulating factor (GM-CSF)] (see below), tumor necrosis factor (TNF), and monoclonal antibodies such as trastuzumab and rituximab (see below). Several of these agents now have been approved for clinical use because of their activity in specific diseases. For example, interferon-alfa is approved for use in hairy cell leukemia, condylomata acuminata, and Kaposi's sarcoma associated with AIDS; interleukin-2 (IL-2) for kidney cancer; filgrastim for prophylaxis against cancer treatmentinduced neutropenia; and sargramostim for rescue from graft failure or to speed graft recovery in patients undergoing autologous bone marrow transplantation. Other biological agents have been approved for the treatment of nonmalignant disease, including interferon-beta for multiple sclerosis and interferon-gamma for chronic granulomatous disease (see Chapter 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants), herceptin for breast cancer, and rituximab for B-cell lymphomas. Interleukin-2 (IL-2) The isolation of a cytokine initially named T-cell growth factor, subsequently renamed IL-2, allowed the first attempts to treat cancer by producing lymphocytes specifically cytolytic for the malignant cell (Morgan et al., 1976). IL-2 is not directly cytotoxic; rather, it induces and expands a T-cell response cytolytic for tumor cells. Clinical trials have studied the antitumor activity of IL-2 both as a single agent and with adoptive cellular therapy using IL-2stimulated autologous lymphocytes obtained by leukapheresis, termed lymphokine-activated killer (LAK) cells. Randomized trials have not shown that the addition of LAK cells to the treatment regimen improves overall response rates (Rosenberg et al., 1989). Later studies in adoptive cellular therapy have used expanded populations of lymphocytes obtained from tumor biopsies and expanded in vitro, so-called tumor-infiltrating lymphocytes (TIL cells) (Rosenberg et al., 1994). Because the half-life of IL-2 in human beings is short (t The toxicities of IL-2 are likely related to the activation and expansion of lytic lymphocytes in organs and within vessels, resulting in inflammation and vascular leak, and to the secondary release of other cytokines, such as tumor necrosis factor and interferon, by activated cells. When given at maximally tolerated doses of 600,000 U/kg every 8 hours for up to 5 days, IL-2 causes hypotension, arrhythmias, peripheral edema, prerenal azotemia, elevated liver function tests, anemia, thrombocytopenia, nausea, vomiting, diarrhea, confusion, and fever (Rosenberg et al., 1989). Reproducible antitumor activity has been reported in advanced malignant melanoma and renal cell cancer, where response rates (partial and complete) are seen in 20% to 30% of patients. Complete responses, seen in approximately 5% to 10% of all patients, appear to be durable, with some patients now free of disease beyond 5 years of treatment. IL-2 currently is being studied in the treatment of acute myelogenous leukemia, where it is capable of inducing remission in relapsed patients (Meloni et al., 1994). In some studies where IL-2 is given following bone marrow transplantation, it appears that IL-2 can lengthen the remission duration as compared to historical controls (Fefer et al., 1993). Randomized trials are in progress to test this hypothesis prospectively. Granulocyte Colony-Stimulating Factor (Filgrastim) Filgrastim NEUPOGEN) is a commercially available granulocyte colony-stimulating factor (G-CSF). Filgrastim is approved for clinical use in the prophylaxis of chemotherapy-induced neutropenia. It was initially isolated and cloned from a human bladder cancer cell line (Souza et al., 1986). In vitro, G-CSF was found not only to expand the population of neutrophil granulocyte precursors, but also to augment granulocyte function by enhancing chemotaxis and antibody-dependent cellular cytotoxicity. Its effects are confined to the granulocyte lineage. It also enhances the mobilization of stem cells in the peripheral blood following cytotoxic chemotherapy. In normal volunteers or cancer patients not receiving other treatment,
administration of filgrastim leads to an initial reduction of circulating
neutrophils within 1 hour, followed by a dose-dependent (1 to 60 The recommended dose of filgrastim is 5 Granulocyte/Macrophage Colony-Stimulating Factor (Sargramostim) Sargramostim LEUKINE) is a commercially available recombinant granulocyte/macrophage colony-stimulating factor (GM-CSF) produced in a yeast expression system. Sargramostim is approved to rescue bone marrow graft failure or speed graft recovery in patients undergoing autologous bone marrow transplantation and to shorten time to neutrophil recovery after induction chemotherapy in patients with acute myelogenous leukemia. Human GM-CSF was initially purified from a T-cell leukemia-infected lymphoblastoid cell line and cloned in 1985 (Wong et al., 1985). The in vitro effects of GM-CSF are more protean than those of G-CSF, as the agent is active earlier in the differentiation pathway of the pleuripotential stem cell. Macrophages, neutrophils, and eosinophils all respond to GM-CSF with proliferation and enhanced antibody-dependent cellular cytotoxicity (Lopez et al., 1986). The glycosylation of GM-CSF is variable and dependent on whether the preparation is derived from yeast (glycosylated) or bacterial (nonglycosylated) cells. In normal volunteers or cancer patients not receiving other treatment, GM-CSF causes a dose-dependent increase in neutrophils as well as increases in eosinophils and macrophages. In the setting of autologous bone marrow transplantation, GM-CSF accelerates the recovery of neutrophils in the peripheral blood while decreasing the need for antibiotics to treat febrile neutropenia (Nemunaitis et al., 1991). The toxicity of GM-CSF preparations appears to be dependent, at least
in part, on whether or not the molecule is glycosylated. The glycosylated
product commonly causes fever, bone pain, and myalgias. The nonglycosylated
product has similar toxicities but additionally can cause pericarditis and a
first-dose phenomenon of flushing, hypotension, hypoxia, and tachycardia.
These symptoms diminish as the patient continues through a treatment cycle
but recur at the beginning of each subsequent cycle. The commonly recommended
dose of sargramostim is 250 Monoclonal Antibodies in Cancer Therapy For the past two decades, since the discovery of the method for fusing mouse myeloma cells with B lymphocytes, it has been possible to produce a single species of antibody that recognizes a specific antigen. The mouse antibody can now be 'humanized'; that is, the domains not responsible for antigen recognition can be converted to human type to prevent a human-antimouse neutralizing response. Such antibodies now are used for immune suppression, anticoagulation, and, more recently, for cancer treatment. Two such antibodies, trastuzumab (HERCEPTIN) and rituximab (RITUXAN), have won a role in treating breast cancer and B-cell lymphomas, respectively (Hainsworth, 2000). The mechanism by which these antibodies kill cells is unresolved. There is no doubt that binding to specific cell-surface antigens is required as a first step. Trastuzumab binds to the her-2/neu growth factor receptor, a member of the epidermal growth factor receptor family. Whether its cytotoxic/cytostatic effect is related to T-cellmediated recognition of the bound antibody or to its inhibitory effect on the growth factor pathway has not been clarified. About one-quarter of breast cancers are positive for her-2/neu antigen, and their tumors lie at the more aggressive end of the spectrum of breast cancers. Herceptin demonstrated only modest activity against metastatic breast cancer in its initial trials, but when used with either paclitaxel or doxorubicin, it markedly enhanced the rate of response to these cytotoxic drugs and improved survival of the patients so treated (Norton et al., 1999). Like all monoclonal antibodies (including the humanized types), acute hypersensitivity reactions may occur, including hypotension, flushing, bronchoconstriction, and rash. In addition, trastuzumab administration has resulted in ventricular dysfunction and congestive heart failure. When combined with doxorubicin, trastuzumab enhances the cardiac toxicity of the anthracyclines; in rare individuals with underlying intrinsic or metastatic disease in the lungs, it has caused pulmonary infiltrates, hypoxia, and fatal pulmonary insufficiency; it should be used with caution in these cases. Trastuzumab usually is used in conjunction with paclitaxel, docetaxel, or venorelbine in treating breast cancer. The recommended loading dose of trastuzumab is 4 mg/kg as a 90-minute intravenous infusion, and the maintenance dose is 2 mg/kg per week, which can be given as a 30-minute infusion if the loading dose was well tolerated. Rituximab recognizes the CD20 antigen found on the surface of virtually all B-lymphocyte tumors. It has significant activity as a single agent in the treatment of indolent or follicular B-cell lymphomas, producing a response rate of 40% to 50% in patients who have relapsed or who have become refractory to standard chemotherapy (Maloney, 1998). In an effort to enhance its activity, the antibody has been combined with standard chemotherapy in primary-treatment regimens for aggressive forms of B-cell lymphoma. An alternative approach has been to label the anti-CD20 antibody with iodine 131 (131I). The resulting antibody, tositumomab (BEXXAR), has potent myelosuppressive effects but, when used with bone-marrow replacement, produces a high rate of complete response in drug-resistant patients. Standard doses of rituximab are 375 mg/m2 given weekly for 4 doses by intravenous infusion. Its acute toxicity is much the same as trastuzumab's and is related to hypersensitivity, but it does not cause the cardiac effects associated with trastuzumab administration. Uncommonly, patients develop neutropenia after multiple doses. |
Prospectus
|
While the current therapy for cancer depends primarily on the use of surgery, irradiation, and chemotherapy, the evolution in understanding the biology of malignant transformation and differences in the control of normal and malignant cell proliferation has provided a myriad of new possible targets for cancer treatment. Central to this understanding has been the elucidation of events in the cell cycle that monitor the integrity of DNA, check progression through the cell cycle when nutrients or growth factors are lacking, and direct the cell to undergo apoptosis (programmed cell death) when either intrinsic or extrinsic factors are unfavorable for survival. As might have been anticipated, a malfunction in the machinery that controls normal cell proliferation can lead to consequences that favor malignant transformation: a loss of cell-cycle checkpoints such as mutation or deletion of the p53 and p16 oncogenes, an increase in genes that protect cells from apoptosis (such as the bcl-2 gene that is translocated in nodular lymphomas), and an increase in expression of the D cyclin (the prad oncogene) that promotes cell entry into DNA synthesis. Not only do these changes promote cell proliferation, they also increase the frequency with which mutant and drug-resistant cells escape normal surveillance mechanisms and apoptosis. Loss of apoptotic pathway(s) in itself predisposes to resistance to radiation therapy and drugs. Thus, a major effort now is under way to identify compounds that restore apoptosis and cell-cycle checkpoints. The replacement of a missing function, such as that resulting from mutation of p53, represents an exceedingly difficult challenge, in that one seeks a small molecule that replaces the function of a complex protein or, alternatively, a targeted gene therapy approach to introduce wild-type p53 into affected tissues (see Chapter 5: Gene Therapy). Other directions for cancer drug discovery and development have emerged from tumor biology research and include differentiation inducers and inhibitors of angiogenesis and metastasis (Kerbel, 2000). The field of differentiation induction has received a significant impetus from the discovery of effectiveness of all-trans-retinoic acid in the treatment of acute promyelocytic leukemia. Although not curative as a single agent in this disease, all-trans-retinoic acid induces a remission in drug-refractory disease and does so without the period of marrow hypoplasia characteristic of cytotoxic drugs. Hormonal agents, planar-polar chemicals, and various retinoids and vitamin D analogs are being tested as differentiation drugs in established cancer and in preventing progression of premalignant disease. As genetic testing becomes increasingly able to identify individuals at high risk of developing cancer, the emphasis in cancer drug development will inevitably shift to the discovery of preventive or differentiating agents. The discovery of the important role of angiogenesis in allowing malignant cells to establish a generous blood supply also has led to current trials of inhibitors of endothelial cell proliferation, including low doses of cytotoxic agents as well as experimental drugs such as monoclonal antibodies to endothelial growth factors and their receptors and low-molecular-weight inhibitors of the receptors, and antiangiogenic peptides such as endostatin and angiostatin. The reader is referred to Kaelin (1999) and the series of articles that follow his introduction for a discussion of new strategies for drug discovery. Special note should be made of imatinib (STI-571, GLEEVEC), an inhibitor of the bcr-abl tyrosine kinase found in chronic myelocytic leukemia; this drug has shown remarkable remission-inducing activity as a single agent (Drucker and Lydon, 2000) and recently was approved by the FDA for clinical use. In addition to these targeted drug discovery efforts, new efforts are in clinical trial exploring the possibility that the immune system can be harnessed to treat cancer. These approaches include tumor-specific vaccines directed against unique antigens, such as oncogene products or the products of translocated genes; monoclonal antibodies armed with toxins or radioisotopes (see Kawakami et al., 1994); and genetically manipulated components of the immune system. Monoclonal antibodies directed against cell-surface antigens have become important new tools for producing antitumor responses as single agents (rituximab in B-cell lymphomas) or for enhancing response to chemotherapy (trastuzumab in breast cancer). Other antibodies directed against epithelial cancers, such as C225 (an anti-epidermal growth factor-receptor antibody), and 17.1 (directed against colon cancer antigens), are showing promising activity in early clinical trials. Completion of the sequencing of the human genome provides the basis for a more detailed understanding of the specific genetic mutations involved in various human cancers. Although initially this understanding will provide more specific tumor markers useful in detection and diagnosis, eventually it is anticipated that more targeted therapeutic agents will be devised. Future editions of this text undoubtedly will contain a much different spectrum of effective anticancer drugs, and there is reason to be quite optimistic about the prospects for a much more effective and specific collection of weapons for treating this group of fatal diseases. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 7627
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved