| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Most of us do not suffer any harmful effects from our defective genes because we carry two copies of nearly all genes, one derived from our mother and the other from our father. The only exceptions to this rule are the genes found on the male sex chromosomes. Males have one X and one Y chromosome, the former from the mother and the latter from the father, so each cell has only one copy of the genes on these chromosomes. In the majority of cases, one normal gene is sufficient to avoid all the symptoms of disease. If the potentially harmful gene is recessive, then its normal counterpart will carry out all the tasks assigned to both. Only if we inherit from our parents two copies of the same recessive gene will a disease develop.
On the other hand, if the gene is dominant, it alone can produce the disease, even if its counterpart is normal. Clearly only the children of a parent with the disease can be affected, and then on average only half the children will be affected. Huntington's chorea, a severe disease of the nervous system, which becomes apparent only in adulthood, is an example of a dominant genetic disease. Finally, there are the X chromosome-linked genetic diseases. As males have only one copy of the genes from this chromosome, there are no others available to fulfil the defective gene's function. Examples of such diseases are Duchenne muscular dystrophy and, perhaps most well known of all, haemophilia.
Queen
Not all defective genes necessarily produce detrimental effects, since the environment in which the gene operates is also of importance. A classic example of a genetic disease having a beneficial effect on survival is illustrated by the relationship between sickle-cell anaemia and malaria. Only individuals having two copies of the sickle-cell gene, which produces a defective blood protein, suffer from the disease. Those with one sickle-cell gene and one normal gene are unaffected and, more importantly, are able to resist infection by malarial parasites. The clear advantage, in this case, of having one defective gene explains why this gene is common in populations in those areas of the world where malaria is endemic.
Gene Therapy
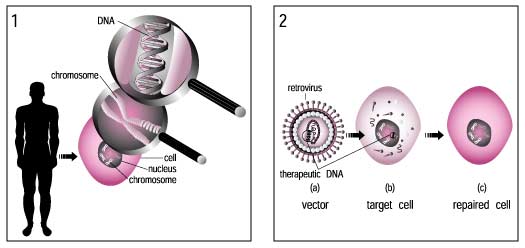
Much attention has been focused on the so-called genetic metabolic diseases in which a defective gene causes an enzyme to be either absent or ineffective in catalyzing a particular metabolic reaction effectively. A potential approach to the treatment of genetic disorders in man is gene therapy. This is a technique whereby the absent or faulty gene is replaced by a working gene, so that the body can make the correct enzyme or protein and consequently eliminate the root cause of the disease.
If gene therapy does become practicable, the biggest impact would be on the treatment of diseases where the normal gene needs to be introduced into only one organ. One such disease is phenylketonuria (PKU). PKU affects about one in 12,000 white children, and if not treated early can result in severe mental retardation. The disease is caused by a defect in a gene producing a liver enzyme. If detected early enough, the child can be placed on a special diet for their first few years, but this is very unpleasant and can lead to many problems within the family.
The types of gene therapy described thus far all have one factor in common: that is, that the tissues being treated are somatic. In contrast to this is the replacement of defective genes in the germ line cells. Gene therapy in germ line cells has the potential to affect not only the individual being treated, but also his or her children as well. Germ line therapy would change the genetic pool of the entire human species, and future generations would have to live with that change. In addition to these ethical problems, a number of technical difficulties would make it unlikely that germ line therapy would be tried on humans in the near future.
Before treatment for a genetic disease can begin, an accurate diagnosis of the genetic defect needs to be made. It is here that biotechnology is also likely to have a great impact in the near future. Genetic engineering research has produced a powerful tool for pinpointing specific diseases rapidly and accurately. Already, the genes for Duchenne muscular dystrophy, cystic fibrosis, and retinoblastoma have been identified, and more such information is emerging all the time.
Gene therapy is a technique for correcting defective genes responsible for disease development. Researchers may use one of several approaches for correcting faulty genes:
Gene therapy involves the manipulation of genes to fight or prevent diseases. Put most simply, it introduces a 'good' gene into a person who has a disease caused by a 'bad' gene.
There are two forms of gene therapy. The first, somatic gene therapy, involves introducing a 'good' gene into targeted cells with the end result of treating the patient - but not the patient's future children because these genes do not get passed along to offspring. In other words, even though some of the patient's genes may be altered to treat a disease, the likelihood remains that the same disease will affect the patient's children. This is the form of gene therapy that is being done at most genetics laboratories throughout the world.
Germ line gene therapy involves modifying the genes in sperm- or egg-producing cells, which will then pass any genetic changes to future generations as well. In experimenting with this type of therapy, scientists injected fragments of DNA into fertilized mouse eggs. The mice grew into adults and their offspring had the new gene. Scientists found that growth and fertility problems could be corrected with this form of therapy, which led them to hypothesize that the same could be true for humans. However, although it has potential for preventing inherited disease, this form of gene therapy is extremely controversial and currently very little research is being done in this area, both for technical and ethical reasons.
Transgenic animals have been used for simulating diseases and testing new therapies, egg. Cardiovascular and neurodegenerative diseases. Animal models provide an opportunity to test methods for the prevention or delay of disease in humans. Some examples:
|
Genetic alteration |
Method of alteration |
Human disease equivalent |
|
Introduction of mutant collagen gene into wild type mice |
Nuclear microinjection of inducible minigene |
Osteogenesis imperfecta |
|
Inactivation of mouse gene encoding hypoxanthine-guanine phosphoribosyl transferase (HPRT) |
Insertion of retrovirus into HPRT locus in embryonic stem cells |
HPRT deficiency |
|
Mutation at locus for X-linked muscular dystrophy |
Male mutagenesis followed by identification of female carriers |
X-linked muscular dystrophy |
|
Introduction of activated human ras and c-myc oncogenes |
Nuclear microinjection of inducible minigene |
Induction of malignancy |
|
Introduction of mutant (Z)allele of human alpha -1-antitrypsin gene |
Microinjection of DNA fragment bearing mutant allele |
Neonatal hepatitis |
|
Introduction of HIV tat gene |
Microinjection of DNA fragment |
Kaposi's sarcoma |
|
Introduction of beta-globin sickle gene |
Microinjection |
Sickle-cell anaemia |
|
Introduction of mouse renin gene |
Microinjection |
Hypertension |
|
Introduction of (beta)-amyloid protein precursor (APP gene) |
Microinjection |
Alzheimer's disease |
Candidate Diseases for Gene Therapy
Gene therapy is likely to have the greatest success with diseases that are cause by single gene defects. By the end of 1993, gene therapy had been approved for use on such diseases as severe combined immune deficiency, familial hypercholesterolemia, cystic fibrosis, and Gaucher's disease. Most protocols to date are aimed toward the treatment of cancer; a few are also targeted toward AIDS. Numerous disorders are discussed as candidates for gene therapy: Parkinson's and Alzheimer's diseases, arthritis, and heart disease.
Now all we have to do is deliver the gene into the proper cells and put it to work. This is not an easy job. Gene delivery is one of the biggest challenges in the field of gene therapy.
What are some of the hallmarks of successful gene delivery?
1. TARGETING the right cells. If you want to deliver a gene into cells of the liver, it shouldn't wind up in the big toe. How can you ensure that the gene gets into the correct cells?
2. ACTIVATING the gene. A gene's journey is not over when it enters the cell. It must go to the cell's nucleus and be 'turned on,' meaning that its transcription and translation are activated to produce the protein product encoded by the gene. For gene delivery to be successful, the protein that is produced must function properly.
3. INTEGRATING the gene in the cells. You might want the gene to stay put and continue working in the target cells. If so, you need to ensure that the gene integrates into, or becomes part of the host cell's genetic material, or that the gene finds another way to survive in the nucleus without being trashed.
Choosing the best vector
There is no 'perfect vector' that can treat every disorder. Like any type of medical treatment, a gene therapy vector must be customized to address the unique features of the disorder. Part of the challenge in gene therapy is choosing the most suitable vector for treating the disorder. Below, find out more about the most commonly used types of gene therapy vectors.
Viral vectors
Mother Nature is a brilliant scientist! Over the last three billion years or so, she's developed an incredibly efficient means of delivering foreign genes into cells: the virus. When faced with the problem of gene delivery, scientists looked to viruses. Why reinvent the wheel if there's a perfectly good one out there? If we can modify viruses to deliver genes without making people sick, we may have a good set of gene therapy tools.
General advantages of viral vectors:
General drawbacks of viral vectors:
However, modern viral vectors have been engineered without most of the proteins that would cause an immune response.
Some of the different types of viruses used as gene therapy vectors:
LIMITATIONS OF CURRENT RECOMBINANT ADENOVIRUS VECTORS
Several hurdles are associated with the use of Ad2 and Ad5 viruses as gene delivery vehicles. In particular, several tissues of major therapeutic interest have been found to be refractory to transduction by Ad2- and Ad5-derived vectors. High dosages of recombinant vectors are therefore necessary to genetically engineer the target cells in vivo, which may in turn compromise the safety of the treated patients. Overcoming these hurdles requires the development of vectors that are (1) able to transduce specifically and efficiently the desired human cells while bypassing nontarget tissues; (2) not prone to neutralization by human serum; and (3) characterized by an acceptable toxicity profile.
GENERATION OF IMPROVED ADENOVIRUS VECTORS
From the previously listed limitations, it is clear that the use of Ad2- or Ad5-derived vectors for human gene transfer poses a dilemma. On one hand, the therapeutic viral dose must be elevated to overcome the presence of neutralizing antibodies and to compensate for the low efficiency of transduction of the target cells. On the other hand, a high viral dose causes undesired local and systemic toxicity and possibly results in the eradication of the transduced cells by the host immune response. To overcome this dilemma, adenoviral vectors must be generated that are not neutralized by the antiviral antibodies pre-existing in the serum and that are able to specifically and efficiently transduce the tissue of interest and not undesired organs. Several strategies have been explored to develop vectors that specifically target the cell types of interest
Adeno-associated virus (AAV)-based vectors have recently gained popularity as potential vectors for gene therapy. The major advantages of using AAV are nonpathogenicity, long-term expression, and relatively low immunogenecity. Although AAV was believed to infect both dividing and nondividing cells, recent studies have clearly demonstrated variations in transduction among cell types. The identification of the host cell receptor, coreceptors, and other factors that facilitate AAV entry and intracellular processing, leading to transgene expression, indicates that deficiency in one or more of these molecules can limit a successful transduction. Thus, it is apparent that modifications that will overcome these limitations will not only maximize the application of AAV vectors in gene therapy, but also lead to the development of targeted vectors for transgene delivery to specific cell types.
The eventual development of truly effective methods of in vivo gene delivery for gene therapy will require methods for delivering therapeutic genes to the correct target tissue specifically and efficiently, whether the gene delivery vector is a viral or a nonviral agent. A variety of viral vectors are available for such applications, including retroviral and lentiviral vectors. Retroviral vectors have been effective tools for developing many of our current concepts and techniques for models of gene therapy. They have many advantageous features, especially their ease of manipulation for vector construction and the generally stable transgene expression that results from provirus integration. They also have disadvantages, including their relatively low titers compared with several other viral vector systems, their instability in vivo, and their inability to transduce nonreplicating or slowly replicating cells such as neurons. Several of these deficiencies have been corrected partly by the development of methods to increase titers and alter vector tropism through methods for pseudotyping vectors with surrogate envelope components such as VSV-G protein. The development of the lentiviral vector system has also provided an efficient approach to gene transfer into post mitotic cells that exhibits many of the useful features of other retroviral systems and promises to become important technique in future clinical studies. There are at least two principal difficulties in the use of these and all other viral vectors for therapeutic gene delivery. In the case of retroviruses, our present understanding of virus structure is insufficient to allow the design and production of truly targeted vectors through insertion of potentially cell-specific ligands into the viral capsid. In addition, there is a dearth of rigorous information on the pharmacokinetic and pharmacodynamic properties of these viruses; that is, the in vivo fate of vector particles, their interactions with the vascular endothelium as the first tissue barrier that they encounter, and the mechanisms that define their tissue uptake. In the case of systemic administration of retroviral vectors, as in the preclinical and clinical studies aimed at correction of the factor VIII deficiency of classical haemophilia, very little information is readily available on the distribution of systemically delivered virus particles and the exact identification of the tissue source of the factor VIII protein. In addition, even though the studies involving in vivo delivery of AAV vectors to muscle or liver for treatment of haemophilia B are extremely promising, variable patient response and treatment responses are likely to result from insufficient knowledge of the pharmacological properties of the vectors and variations in the mechanisms of tissue uptake and cell entry.
Non-Viral Vectors
Although viruses can effectively deliver genetic material into a patient's cells, they do have some limitations. It is sometimes more efficient to deliver a gene using a non-viral vector, which has fewer size constraints and which won't generate an immune response.
Besides virus-mediated gene-delivery systems, there are several nonviral options for gene delivery. The simplest method is the direct introduction of therapeutic DNA into target cells. This approach is limited in its application because it can be used only with certain tissues and requires large amounts of DNA.
Another nonviral approach involves the creation of an artificial lipid sphere with an aqueous core. This liposome, which carries the therapeutic DNA, is capable of passing the DNA through the target cell's membrane.
Delivery of nucleic acids using liposomes holds great promise as a safe and nonimmunogenic approach to gene therapy. Furthermore, gene therapies that use these artificial reagents can be standardized and regulated as drugs rather than as biologics. Much effort has been devoted to the development of nonviral delivery due to the disadvantages of viruses used for gene delivery. The disadvantages of viral delivery include:
The advantages in using liposomes for gene therapy are several and include:
The disadvantage of nonviral delivery systems had been the low levels of delivery and gene expression produced by first generation complexes. However, recent advances have been made that dramatically improve transfection efficiencies of nonviral vectors. Delivery of nucleic acid-based therapeutics to specific target tissues, organs, or cells is desirable or required for certain applications. For example, lower amounts of complexes could be administered intravenously if the bulk of the injected material is delivered to target cells that are solely responsible for producing the therapeutic gene product. Injection of lower amounts of complexes would be most cost-effective, would provide another level of safety to the patient, and may produce greater efficacy for the treatment of certain diseases. Furthermore, strategies such as suicide gene approaches that are designed to kill target cells, such as tumour cells, require targeted delivery or gene expression to avoid killing normal cells. Cell or tissue-specific gene expression can also be achieved by creating plasmids containing specific promoters to produce expression exclusively in the target cells. Nonviral delivery vehicles have no target specificity, and therefore, retargeting is not required. Basically, ligands are used to coat nucleic acidliposome complexes to achieve specific delivery to cell surface receptors. To efficiently coat the surface of complexes, nucleic acids must be encapsulated within the delivery vehicle so that the ligand does not interfere with nucleic acid condensation. In addition, the encapsulated nucleic acid does not prevent the attachment of the ligand on the surface of the complexes.
ENCAPSULATION OF NUCLEIC ACIDS
Methods used to date for targeted delivery of nonviral delivery vehicles have produced inefficient gene expression in the target cells. Although gene expression is apparently produced primarily in the target cell, the levels of expression are lower in these cells than those produced using the nontargeted vehicle for delivery. Novel approaches involve targeted delivery of liposomes that are optimized for charge on the surface of complexes, cell entry by fusion with the membrane, and penetration across tight barriers in vivo to reach and diffuse through the target tissue /organ efficiently.
Much effort has gone into developing ways to enhance the specificity of liposomal delivery systems, most commonly by conjugating ligands to the liposome surface that will produce a specific interaction with the target cell. Ligands include vitamins, glycoproteins, peptides and oligonucleotides octamers, in addition to the most commonly used ligands, antibodies or antibody fragments. The first report of Mabs conjugated to liposomes was by Torchilin et al. (1979), who demonstrated that antimyosin-immunoliposome retained its ability to specifically bind to the receptor on the target cells. Various attempts have since been made to improve the efficiency of delivery of the liposome payload including development of pH-sensitive immunoliposomes. However, despite promising in vitro results, the in vivo use of these Mab-immunoliposomes has been hampered by lack of stability in circulation and their rapid clearance by the mononuclear phagocyte system (MPS) (liver and spleen). An example of this is the report by Matzku et al. (1990), who found virtually no uptake of Mab-immunoliposomes in either a human melanoma xenograft model or a syngeneic murine lymphoma model that spontaneously metastasizes to the liver. These investigators speculated that lack of uptake was the result of limited availability of the complex due to only moderate stability in circulation as well as inability of the immunoliposome to extravasate.
In an effort to sidestep the safety limitations of viral vectors and the cytotoxicity of liposomal carriers, investigators have used receptor-targeted molecular conjugates to direct gene transfer into mammalian cells in vitro. Complexes consisting of DNA, noncovalently bound to a poly-cation polymer that is chemically conjugated to a ligand, can bind a cell surface receptor and be internalized.
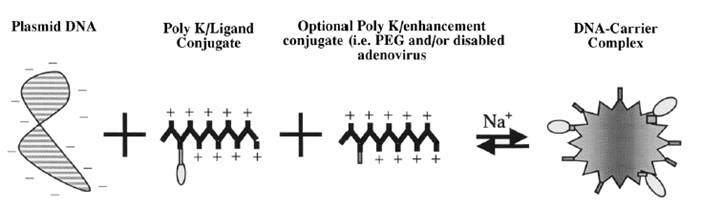
General scheme of receptor targeted DNA complex construction. DNA complexes are formed by mixing plasmid DNA with the molecular conjugate under the proper salt conditions. Molecular conjugates consist of a polycation coupled to a receptor ligand. Polycations modified with enhancers (e.g., PEG or adenovirus) may also be included in the DNA complex.
Therapeutic DNA also can get inside target cells by chemically linking the DNA to a molecule that will bind to special cell receptors. Once bound to these receptors, the therapeutic DNA constructs are engulfed by the cell membrane and passed into the interior of the target cell. This delivery system tends to be less effective than other options.
Researchers also are experimenting with introducing a 47th (artificial human) chromosome into target cells. This chromosome would exist autonomously alongside the standard 46 --not affecting their workings or causing any mutations. It would be a large vector capable of carrying substantial amounts of genetic code, and scientists anticipate that, because of its construction and autonomy, the body's immune systems would not attack it. A problem with this potential method is the difficulty in delivering such a large molecule to the nucleus of a target cell.
How do you know whether a disorder is a good candidate for gene therapy?
For any candidate disorder, you need to answer the following questions:
Eve Nichols describes the criteria for selection of disease candidates for human gene therapy: 1) the disease is an incurable, life-threatening disease; 2) organ, tissue and cell types affected by the disease have been identified; 3) the normal counterpart of the defective gene has been isolated and cloned; 4) the normal gene can be introduced into a substantial subfraction of the cells from the affected tissue; or that introduction of the gene into the available target tissue, such as bone marrow, will somehow alter the disease process in the tissue affected by the disease; 5) the gene can be expressed adequately (it will direct the production of enough normal protein to make a difference); and 6) techniques are available to verify the safety of the procedure.
Arguments in Favour of Gene Therapy
The central argument in favour of gene therapy is that it can be used to treat desperately ill patients, or to prevent the onset of horrible illnesses. Conventional treatment has failed for the candidate diseases for gene therapy, and for these patients, gene therapy is the only hope for a future. Many commentators liken somatic cell gene therapy to other new medical technologies, and argue that we have an obligation to treat patients if we can.
Eric Juengst summarized the arguments in favour of and against human germ-line gene therapy in 1991: 1) germ-line gene therapy offers a true cure, and not simply palliative or symptomatic treatment; 2) germ-line gene therapy may be the only effective way of addressing some genetic diseases; 3) by preventing the transmission of disease genes, the expense and risk of somatic cell therapy for multiple generations is avoided; 4) medicine should respond to the reproductive health needs of prospective parents at risk for transmitting serious genetic diseases; and 5) the scientific community has a right to free inquiry, within the bounds of acceptable human research.
While the development of germ-line gene therapy techniques will undoubtedly place some embryos at risk in the laboratory, once the successful techniques are developed, the therapy could help parents and researchers avoid the moral dilemma of disposing of 'defective' embryos in the lab if the embryos could be repaired.
Arguments Against Gene Therapy
Many persons who voice concerns about somatic cell gene therapy use a 'slippery slope' argument against it. They wonder whether it is possible to distinguish between 'good' and 'bad' uses of the gene modification techniques, and whether the potential for harmful abuse of the technology should keep us from developing more techniques. Other commentators have pointed to the difficulty of following up with patients in long-term clinical research. Gene therapy patients would need to be under surveillance for decades to monitor long-term effects of the therapy on future generations. Some are troubled that many gene therapy candidates are children too young to understand the ramifications of gene therapy treatment.
Others have pointed to potential conflict of interest problems pitting an individual's reproductive liberties and privacy interests against the interests of insurance companies, or society not to bear the financial burden of caring for a child with serious genetic defects. Issues of justice and resource allocation have also been raised: in a time of strain on our health care system, can we afford such expensive therapy? Who should receive gene therapy? If it is made available only to those who can afford it, 'the distribution of desirable biological traits among different socioeconomic and ethnic groups would become badly skewed'.
Arguments specifically against the development of germ-line gene therapy techniques include:
germ-line gene therapy experiments would involve too much scientific uncertainty and clinical risks, and the long term effects of such therapy are unknown;
such gene therapy would open the door to attempts at altering human traits not associated with disease, which could exacerbate problems of social discrimination;
as germ-line gene therapy involves research on early embryos and effects their offspring, such research essentially creates generations of unconsenting research subjects;
gene therapy is very expensive, and will never be cost effective enough to merit high social priority;
germ-line gene therapy would violate the rights of subsequent generations to inherit a genetic endowment that has not been intentionally modified.
The ethical issues posed by both somatic and germ-line gene therapies are international in scope. The documents listed below serve to demonstrate the variety of reactions to gene therapy, and to illuminate the complexity of this continuing public debate.
Theoretically, gene therapy can be targeted to somatic (body) or germ (egg and sperm) cells. In somatic gene therapy the recipient's genome is changed, but the change is not passed along to the next generation. This form of gene therapy is contrasted with germ line gene therapy, in which a goal is to pass the change on to offspring. Germ line gene therapy is not being actively investigated, at least in larger animals and humans, although a lot of discussion is being conducted about its value and desirability.
Gene therapy should not be
confused with cloning, which has been in the news so much in the past year,
Viruses have evolved a way of encapsulating and delivering their genes to human cells in a pathogenic manner. Scientists have tried to take advantage of the virus's biology and manipulate its genome to remove the disease-causing genes and insert therapeutic genes. These gene-delivery vehicles will make this field a reality.
In the mid-1980s, the focus of gene therapy was entirely on treating diseases caused by such single-gene defects as haemophilia, Duchenne's muscular dystrophy, and sickle cell anaemia. In the late 1980s and early 1990s, the concept of gene therapy expanded into a number of acquired diseases. When human testing of first-generation vectors began in 1990, scientists learned that the vectors didn't transfer genes efficiently and that they were not sufficiently weakened. Expression and use of the therapeutic genes did not last very long.
In 1995, a public debate led to the consensus that gene therapy has value although many unanswered questions require continued basic research. As the field has matured over the last decade, it has caught the attention of the biopharmaceutical industry, which has begun to sort out its own role in gene therapy. This is critical because ultimately this industry will bring gene therapies to large patient populations.
Recent gene therapy approaches
promise to avoid repeated injections, which can be painful, impractical, and
extremely expensive. One method uses a new vector called adeno-associated
virus, an organism that causes no known disease and doesn't trigger patient
immune response. The vector takes up residence in the cells, which then express
the corrected gene to manufacture the protein. In haemophilia treatments, for
example, a gene-carrying vector could be injected into a muscle, prompting the
muscle cells to produce Factor IX and thus prevent bleeding. This method would
end the need for injections of Factor IX --a derivative of pooled blood
products and a potential source of HIV and hepatitis infection. In studies by
Wilson and Kathy High (
Gene therapy is not a new field; it has been evolving for decades. Despite the best efforts of researchers around the world, however, gene therapy has seen only limited success. Why? The answer is that gene therapy poses one of the greatest technical challenges in modern medicine. It is very hard to introduce new genes into cells of the body. Let's look at some of the main technical issues in gene therapy.
Gene delivery and activation
Gene therapy will work only if we can deliver a normal gene to a large number of cells - say, several million - in a tissue. And they have to be the correct cells, in the correct tissue. Once the gene reaches its destination, it must be activated, or turned on to produce the protein encoded by the gene. Gene delivery and activation are the biggest obstacles facing gene therapy researchers
Introducing changes into the germ line
Targeting a gene to the correct cells is crucial to the success of any gene therapy treatment. Just as important, though, is making sure that the gene is not incorporated into the wrong cells. Delivering a gene to the wrong tissue would be inefficient and could cause health problems for the patient.
For example, improper targeting could incorporate the therapeutic gene into a patient's germ line, or reproductive cells, which ultimately produce sperm and eggs. Should this happen, the patient would pass the introduced gene on to his or her offspring. The consequences would vary, depending on the type of gene introduced.
Immune response
Our immune systems are very good at fighting off intruders such as bacteria, viruses and other biological substances. Gene delivery vectors must be able to escape the body's natural surveillance systems. Failure to do so can cause serious illness or even death.
The story of Jesse Gelsinger illustrates this challenge
well. Gelsinger, who had a rare liver disorder, participated in a 1999 gene
therapy trial at the
Disrupting important genes in target cells
The best gene therapy is the one that lasts. Ideally, we would want a gene that is introduced into a group of cells to remain there and continue working. For this to happen, the newly introduced gene must become a permanent part of each cell's genome, usually by integrating, or 'stitching' itself, into the cell's existing DNA. But what happens if the gene stitches itself into an inappropriate location, disrupting another gene?
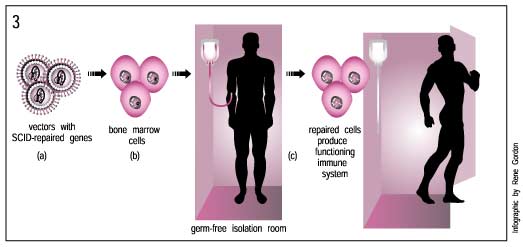
This happened recently in a gene therapy trial to treat several children with X-linked Severe Combined Immune Deficiency (SCID). People with this disorder have virtually no immune protection against bacteria and viruses. To escape infections and illnesses, they must live in a completely germ-free environment.
In the late 1990s, researchers tested a gene therapy treatment that would restore the function of a crucial gene, gamma c, to cells of the immune system. This treatment appeared very successful, restoring immune function to most of the children who received it. But later, two of these children developed leukemia. Researchers found that the leukemia occurred because the newly transferred gamma c gene had stitched itself into the wrong place, interrupting the function of a gene that normally helps regulate the rate at which cells divide. As a result, the cells began to divide out of control, causing the blood cancer leukemia. Although doctors have treated the children successfully with chemotherapy, the fact that they developed leukemia during treatment raises another important safety-related issue that gene therapy researchers must address.
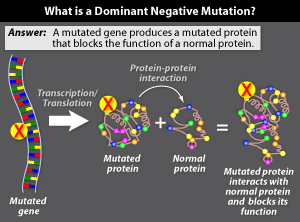 There are times, though, when adding a
'good' copy of the gene won't solve the problem. For example, if the
mutated gene encodes
a protein that prevents the
normal protein from doing its job, adding back the normal gene won't help. Mutated genes that
function this way are called dominant negative.
There are times, though, when adding a
'good' copy of the gene won't solve the problem. For example, if the
mutated gene encodes
a protein that prevents the
normal protein from doing its job, adding back the normal gene won't help. Mutated genes that
function this way are called dominant negative.
How to deal with a dominant negative?
To address this situation, you could either repair the mutated gene's product, or you could get rid of it altogether. Here are some of the newest methods that scientists are developing as potential approaches to gene therapy. Each of these techniques also requires a specific and efficient means of delivering the gene to the target cell.
A technique for repairing mutations:
SMaRT The term SMaRT stands for 'Spliceosome-Mediated RNA Trans-splicing.' This technique targets and repairs the messenger RNA (mRNA) transcripts copied from the mutated gene. Instead of attempting to replace the entire gene, this technique repairs just the section of the mRNA transcript that contains the mutation. After a gene is copied into mRNA, the cell uses RNA-based machinery called spliceosomes to cut out the non-coding introns and splice the exons together.
SMaRT involves three steps:
Delivery of an RNA strand that pairs specifically with the intron next to the mutated segment of mRNA. Once bound, this RNA strand prevents spliceosomes from including the mutated segment in the final, spliced RNA product.
Simultaneous delivery of a correct version of the segment to replace the mutated piece in the final mRNA product
Translation of the repaired mRNA to produce the normal, functional protein
SMaRT is a trademark of Intronn, Inc.
Techniques to prevent the production of a mutated protein:
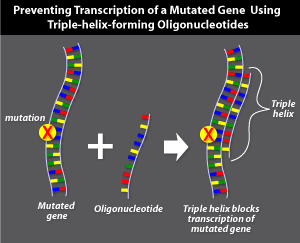 Triple-helix-forming
oligonucleotides
Triple-helix-forming
oligonucleotides
Triple-helix-forming oligonucleotide gene therapy targets the DNA sequence of a mutated gene to prevent its transcription. This technique involves the delivery of short, single-stranded pieces of DNA, called oligonucleotides that bind specifically in the groove between the double strands of the mutated gene's DNA. Binding produces a triple-helix structure that prevents that segment of DNA from being transcribed into mRNA.
Antisense
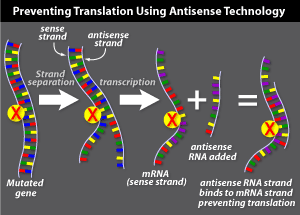 Antisense gene therapy aims to turn off a
mutated gene in a cell by targeting the mRNA transcripts copied from the gene.
Genes are made up of two paired DNA strands. During transcription, the sequence
of one strand is copied into a single strand of mRNA. This mRNA is called the
'sense' strand because it contains the code that will be read by the
cell as it makes a protein. The opposite strand is the 'antisense'
strand.
Antisense gene therapy aims to turn off a
mutated gene in a cell by targeting the mRNA transcripts copied from the gene.
Genes are made up of two paired DNA strands. During transcription, the sequence
of one strand is copied into a single strand of mRNA. This mRNA is called the
'sense' strand because it contains the code that will be read by the
cell as it makes a protein. The opposite strand is the 'antisense'
strand.
Antisense gene therapy involves the following steps:
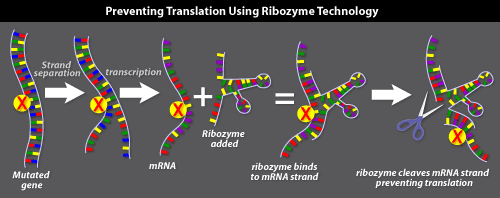
Ribozymes
Like antisense, ribozyme gene therapy aims to turn off a mutated gene in a cell by targeting the mRNA transcripts copied from the gene. This approach prevents the production of the mutated protein.
Ribozymes are RNA molecules that act as enzymes. Most often, they act as molecular scissors that cut RNA. For example, spliceosomes (described above) are believed to be a type of ribozyme
Ribozyme gene therapy involves the following steps:
RNA Interference
RNA interference (RNAi) is a phenomenon in which the introduction of double-stranded RNA (dsRNA) into a diverse range of organisms and cell types causes degradation of the complementary mRNA. Discovered in 1998, it has become clear now that RNA interference represents the major technological advance in Molecular Biology since the discovery of PCR and hence is called 'The Next Big Thing in Biotech'. RNAi is present in a wide variety of eukaryotic organisms including mammals. For the most part, it is believed that RNAi serves as an antiviral defence mechanism although there is preliminary evidence that it also plays a role in the formation and maintenance of heterochromatin during mitosis and meiosis.
SiRNAs have a well defined structure: a short (usually 21-nt) double-strand of RNA (dsRNA) with 2-nt 3' overhangs on either end:

Each strand has a 5' phosphate group and a 3' hydroxyl (-OH) group. This structure is the result of Dicer processing, an enzyme that converts either long dsRNAs or hairpin RNAs into siRNAs. SiRNAs can also be exogenously (artificially) introduced into cells by various transfection methods to bring about the specific knockdown of a gene of interest. Essentially any gene of which the sequence is known can thus be targeted based on sequence complementarity with an appropriately tailored siRNA. This has made siRNAs an important tool for gene function and drug target validation studies in the post-genomic era.
Upon introduction into the cell, long dsRNAs enter a cellular pathway that is commonly referred to as the RNA interference pathway. First the dsRNA's are cleaved into short 21-25 nucleotide small interfering RNAs, or siRNAs, by a ribonuclease known as Dicer. The siRNAs subsequently assemble with protein components into an RNA-induced silencing complex (RISC). The siRNA strands are then unwound to form activated RISCs. These activated RISCs then bind to complementary RNA molecules by base pairing interactions between the siRNA antisense strand and the mRNA. The bound mRNA is cleaved and sequence specific degradation of mRNA results in gene silencing.
Although there are different methods to generate siRNA for gene silencing, the easiest and most efficient way to achieve RNAi is to use synthetic small-interfering RNA (siRNA). siRNA are duplexes of short mixed oligonucleotides. Recent advances in molecular biology have shown that gene expression can be effectively silenced in a highly specific manner through the addition of double stranded RNA (dsRNA).
Preliminary studies in mammalian systems using long dsRNAs to initiate the RNAi response failed because they led to the induction of a non-specific Type I interferon response that produced extensive changes in protein expression and eventually resulted in cell death. Subsequent studies, however, using synthetic, short double-stranded RNAs that mimic the siRNAs produced by the enzyme dicer, sequence specific gene silencing could be achieved in mammalian cells without inducing the interferon response. siRNA technology is now extensively recognized as a powerful tool for the specific suppression of gene expression and is presently being used by researchers in a wide range of disciplines for the assessment of gene function.
RNAi intersects with a number of other pathways, so it is not surprising that on occasion non-specific effects are triggered by the experimental introduction of an siRNA. When a mammalian cell encounters a double-stranded RNA such as an siRNA, it may mistake it as a viral by-product and mount an immune response. Furthermore, since structurally related microRNAs modulate gene expression largely via incomplete complementarity with a target mRNA, unintended off-targeting may be effected by the introduction of an siRNA.
Introduction of too much siRNA can result in non-specific events due to activation of innate immune responses. Most papers suggest that this is probably due to activation of the dsRNA sensor PKR, although retinoic acid inducible Gene I (RIG-I) may also be involved. One promising method of reducing the non-specific effects is to convert the shRNA into a microRNA. MicroRNAs occur naturally, and by harnessing this endogenous pathway it should be possible to achieve similar gene knockdown at comparatively low concentrations of resulting siRNAs. This should minimise non-specific effects.
Opinion 1 -Given the ability to knockdown essentially any gene of interest, RNAi via siRNAs has generated a great deal of interest in both basic and applied biology. There is an increasing number of large-scale RNAi screens that are designed to identify the important genes in various biological pathways. As disease processes also depend on the activity of multiple genes, it is expected that by turning off their activity with siRNAs or their biosynthetic precursors, therapeutic benefit can be derived directly via RNAi. Indeed, phase I results of the first two therapeutic RNAi trials (indicated for age-related macular degeneration, aka AMD) reported at the end of 2005, demonstrate that siRNAs are well tolerated and have suitable pharmacokinetic properties. SiRNAs and related RNAi induction methods therefore stand to become an important new class of drugs in the foreseeable future.
Opinion 2 - Using siRNA's/shRNA's to knockdown specific genes is certainly a valuable tool in the laboratory. However, there are a great deal of challenges when it comes to taking a laboratory technique and applying it to living animals, especially humans. Firstly, siRNA's show different effectiveness in different cell types, apparently indiscriminately - some cells respond well to siRNA's and show a robust knockdown, others show no such knockdown (even despite efficient transfection). Secondly, and most importantly, the non-specific responses of si/shRNA's are still relatively poorly understood. Until these responses can be understood and overcome, the chances of using si/shRNA's outside of the lab, e.g. as an effective new class of drug, remain slim.
The term miRNA was first introduced in a set of three articles in Science (26 October ).
miRNA (micro-RNA) is a form of single-stranded RNA which is typically 20-25 nucleotides long, and is thought to regulate the expression of other genes. miRNAs are RNA genes which are transcribed from DNA, but are not translated into protein. The DNA sequence that codes for an miRNA gene is longer than the miRNA. This DNA sequence includes the miRNA sequence and an approximate reverse complement. When this DNA sequence is transcribed into a single-stranded RNA molecule, the miRNA sequence and its reverse-complement base pair to form a double stranded RNA hairpin loop; this forms a primary miRNA structure (pri-miRNA). In animals, the nuclear enzyme Drosha cleaves the base of the hairpin to form pre-miRNA. The pre-miRNA molecule is then actively transported out of the nucleus into the cytoplasm by Exportin 5, a carrier protein. The Dicer enzyme then cuts 20-25 nucleotides from the base of the hairpin to release the mature miRNA. In plants, which lack Drosha homologues, pri- and pre-miRNA processing by Dicer probably takes place in the nucleus, and mature miRNA duplexes are exported to the cytosol by Exportin 5.
The function of miRNAs appears to be in gene regulation. For that purpose, a miRNA is complementary to a part of one or more messenger RNAs (mRNAs). Animal miRNAs are usually complementary to a site in the 3' UTR whereas plant miRNAs are usually complementary to coding regions of mRNAs. The annealing of the miRNA to the mRNA then inhibits protein translation, but sometimes facilitates cleavage of the mRNA. This is thought to be the primary mode of action of plant miRNAs. In such cases, the formation of the double-stranded RNA through the binding of the miRNA triggers the degradation of the mRNA transcript through a process similar to RNA interference (RNAi), though in other cases it is believed that the miRNA complex blocks the protein translation machinery or otherwise prevents protein translation without causing the mRNA to be degraded. miRNAs may also target methylation of genomic sites which correspond to targeted mRNAs. miRNAs function in association with a complement of proteins collectively termed the miRNP.
This effect was first described for the worm Caenorhabditis elegans in by R. C. Lee of Harvard University. As of , miRNAs have been confirmed in various plants and animals, including C. elegans, human and the plant Arabidopsis thaliana. Genes have been found in bacteria that are similar in the sense that they control mRNA abundance or translation by binding an mRNA by base pairing, however they are not generally considered to be miRNAs because the Dicer enzyme is not involved.
miRNA has been found to have links with some types of cancer.
A study of mice altered to produce excess c-myc a protein implicated in several cancers shows that miRNA has an effect on the development of cancer. Mice that were engineered to produce a surplus of types of miRNA found in lymphoma cells developed the disease within 50 days and died two weeks later. In contrast, mice without the surplus miRNA lived over 100 days.
Another study found that two types of miRNA inhibit the E2F1 protein, which regulates cell proliferation. miRNA appears to bind to messenger RNA before it can be translated to proteins that switch genes on and off.
By measuring activity among 217 genes encoding miRNA, patterns of gene activity that can distinguish types of cancers can be discerned. miRNA signatures may enable classification of cancer. This will allow doctors to determine the original tissue type which spawned a cancer and to be able to target a treatment course based on the original tissue type. miRNA profiling has already been able to determine whether patients with chronic lymphocytic leukemia had slow growing or aggressive forms of the cancer.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 5067
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved