| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Drug Therapy for Hypercholesterolemia and Dyslipidemia
Overview
|
Hyperlipidemia is a major cause of atherosclerosis and atherosclerosis-associated conditions, such as coronary heart disease, ischemic cerebrovascular disease, and peripheral vascular disease. This chapter covers the normal metabolism of lipoproteins, the pathophysiology of dyslipidemia and atherosclerosis, and drugs used to treat dyslipidemia. Drugs covered include HMG-CoA reductase inhibitorsthe statinswhich are the most effective and best-tolerated drugs currently in use for treating dyslipidemia; bile acidbinding resins; nicotinic acid (niacin); and fibric acid derivatives. The chapter concludes with a brief discussion of potential new classes of antidyslipidemic drugs that are undergoing clinical or preclinical evaluation. |
Drug Therapy for Hypercholesterolemia and Dyslipidemia: Introduction
|
Although the incidence of
atherosclerosis-related vascular disease events is declining in the Recognition of hypercholesterolemia as a risk factor has led to the development of drugs that reduce cholesterol levels. These drugs have been used in well-controlled studies of patients with high cholesterol levels caused primarily by elevated levels of low-density lipoproteins (LDL). The results of these trials indicate that CHD mortality is reduced by as much as 30% to 40% and that nonfatal events are similarly reduced when hypercholesterolemic patients are treated with moderate doses of hypolipidemic drugs [Scandinavian Simvastatin Survival Study Group, 1994; Shepherd et al., 1995; The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group, 1998]. Clinical trial data support extending the benefit of lipid-lowering
therapy to high-risk patients whose major lipid risk factor is a reduced
plasma level of high-density-lipoprotein cholesterol (HDL-C) even if the LDL
cholesterol (LDL-C) levels of these patients do not meet the existing
threshold values for initiating hypolipidemic drug therapy (The Expert Panel,
1993). In patients with low HDL-C and average LDL-C levels, appropriate drug
therapy reduced CHD endpoint events by 20% to 35% (Downs et al., 1998;
Rubins et al., 1999). Since 40% of patients with CHD in the Hypertriglyceridemia (elevated levels of triglycerides), if severe (>1000 mg/dl), requires therapy to prevent pancreatitis. Moderately elevated triglyceride levels (150 to 400 mg/dl) also are of concern because they often occur as part of a syndrome distinguished by insulin resistance, obesity, hypertension, and substantially increased CHD risk. The atherogenic dyslipidemia in patients with this insulin resistance or metabolic syndrome is characterized by moderately elevated triglycerides, low HDL-C levels, and lipid-depleted LDL (sometimes referred to as 'small, dense LDL') (Reaven, 1995; Grundy, 1998a). The metabolic syndrome is common in CHD patients; hence, identification of moderate hypertriglyceridemia in a patient, even if the total cholesterol level is normal, should trigger an evaluation to identify this disorder (National Cholesterol Education Program Expert Panel, 2001). Hyperlipidemia (elevated levels of triglycerides or cholesterol) and reduced HDL-C levels occur as a consequence of several factors that affect the concentrations of the various plasma lipoproteins. These factors may be lifestyle or behavioral (e.g., diet or exercise), genetic (e.g., mutations in a gene regulating lipoprotein levels), or metabolic conditions (e.g., diabetes mellitus) that influence plasma lipoprotein metabolism. An understanding of these factors requires a brief description of lipoprotein metabolism. More detailed descriptions can be found elsewhere (Breslow, 1994; Ginsberg and Goldberg, 1998; Mahley et al., 1998). |
Plasma Lipoprotein Metabolism
|
Lipoproteins are macromolecules that contain lipids and proteins known as apolipoproteins or apoproteins. The lipid constituents include free and esterified cholesterol, triglycerides, and phospholipids. The apoproteins are very important since they provide structural stability to the lipoproteins, and a number of apoproteins function as ligands in lipoproteinreceptor interactions or are cofactors in enzymatic processes that regulate lipoprotein metabolism. In all spherical lipoproteins, the most water-insoluble lipids (cholesteryl esters and triglycerides) are core components, and the more polar, water-soluble components (apoproteins, phospholipids, and unesterified cholesterol) are located on the surface. Table 361 lists the major classes of lipoproteins and describes a number of their properties. Table 362 provides information about apoproteins that have well-defined roles in plasma lipoprotein metabolism. These apolipoproteins include apolipoprotein (apo) A-I, apoA-II, apoA-IV, apoB-100, apoB-48, apoC-I, apoC-II, apoC-III, apoE, and apo(a). Except for apo(a), the lipid-binding regions of all apoproteins contain structural features called amphipathic helices that interact with the polar, hydrophilic lipids (such as surface phospholipids) and with the aqueous plasma environment in which the lipoproteins circulate. Differences in the non-lipid-binding regions are responsible for the functional specificities of the apolipoproteins. Chylomicrons Chylomicrons are synthesized from the fatty acids of dietary triglycerides and cholesterol absorbed from the small intestine by epithelial cells. Triglyceride synthesis is regulated by diacylglycerol transferase, an enzyme that regulates triglyceride synthesis in many tissues (Farese et al., 2000). After they are synthesized in the endoplasmic reticulum, triglycerides are transferred by microsomal triglyceride transfer protein (MTP) to the site where newly synthesized apoB-48 is available to form chylomicrons. Dietary cholesterol is esterified by one of two forms of the enzyme acyl coenzyme A:cholesterol acyltransferase (ACAT). This enzyme, ACAT-2, is found in the intestine and in the liver, where cellular free cholesterol is esterified before triglyceride-rich lipoproteins [chylomicrons and very-low-density lipoproteins (VLDL)] are assembled. In the intestine, ACAT-2 regulates the absorption of dietary cholesterol, and it may be a potential pharmacological target for reducing blood cholesterol levels (Cases et al., 1998). [A second ACAT enzyme, ACAT-1, is expressed in macrophages, including foam cells, adrenocortical cells, and skin sebaceous glands. Although ACAT-1 esterifies cholesterol and promotes foam-cell development, ACAT-1 knockout mice do not have a reduced susceptibility for developing atherosclerosis (Accad et al., 2000).] Chylomicrons are the largest of the plasma lipoproteins and are the
only lipoproteins that float to the top of a tube of plasma that has been
allowed to stand undisturbed for 12 hours. The buoyancy of chylomicrons
reflects their 98% to 99% fat content, of which 85% is dietary triglyceride.
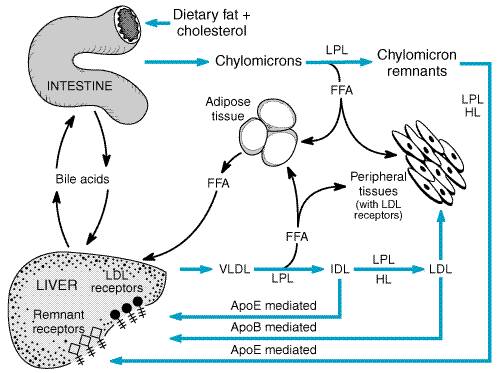
In chylomicrons, the ratio of triglycerides to cholesterol is The apolipoproteins of chylomicrons include some (apoB-48, apoA-I, and apoA-IV) that are synthesized by intestinal epithelial cells and others (apoE and apoC-I, C-II, and C-III) acquired from HDL after chylomicrons have been secreted into the lymph and enter the plasma (Table 362). The apoB-48 of chylomicrons is one of two forms of apoB present in lipoproteins. ApoB-48, synthesized only by intestinal epithelial cells, is unique to chylomicrons, whereas apoB-100 is synthesized by the liver and incorporated into VLDL and intermediate-density lipoproteins (IDL) and LDL, which are products of VLDL catabolism. The apparent molecular weight of apoB-48 is 48% that of apoB-100, which accounts for the name 'apoB-48.' This is because the amino acid sequence of apoB-48 is identical to the first 2152 of the 4536 residues of apoB-100. An RNA editing mechanism unique to the intestine accounts for the premature termination of the translation of the apoB-100 mRNA (Innerarity et al., 1996). ApoB-48 does not contain the portion of the sequence of apoB-100 that allows apoB-100 to bind to the LDL receptor, so apoB-48 appears to function primarily as a structural component of chylomicrons. After gaining entry to the circulation via the thoracic duct, chylomicrons are metabolized initially at the capillary luminal surface of tissues that synthesize lipoprotein lipase (LPL), a triglyceride hydrolase (Figure 361). These tissues include adipose tissue, skeletal and cardiac muscle, and breast tissue of lactating women. As the triglycerides are hydrolyzed by LPL, the resulting free fatty acids are taken up and utilized by the adjacent tissues. The interaction of chylomicrons and LPL requires apoC-II as an absolute cofactor that mediates the interaction of LPL and chylomicron triglycerides. The absence of functional LPL or functional apoC-II prevents the hydrolysis of triglycerides in chylomicrons and results in severe hypertriglyceridemia and pancreatitis during childhood or even infancy (chylomicronemia syndrome). Recently, a variety of new, potentially atherogenic roles for LPL have been identified that affect the metabolism and uptake of atherogenic lipoproteins by the liver, the arterial wall, and the dyslipidemia of insulin resistance (Mead et al., 1999).
The concentration of chylomicrons can be controlled only by reducing dietary fat consumption. There is no current therapeutic approach that will enhance chylomicron catabolism except for insulin replacement in patients with type I diabetes mellitus (insulin has a 'permissive effect' on LPL-mediated triglyceride hydrolysis). Chylomicron Remnants After LPL-mediated removal of much of the dietary triglycerides, the chylomicron remnants, which still contain all of the dietary cholesterol, detach from the capillary surface and within minutes are removed from the circulation by the liver in a multistep process mediated by apoE (Figure 361) (Mahley and Ji, 1999). First, the remnants are sequestered by the interaction of apoE with heparan sulfate proteoglycans on the surface of hepatocytes and are processed by hepatic lipase (HL), which further reduces the remnant triglyceride content. Next, apoE mediates remnant uptake by interacting with the hepatic LDL receptor or the LDL receptorrelated protein (LRP) (Krieger and Herz, 1994). The LRP is a receptor with multiple functions that recognizes a variety of ligandsincluding apoE, HL, and LPLand several ligands unrelated to lipid metabolism. In plasma lipid metabolism, the LRP is important because it is the backup receptor responsible for the uptake of apoE-enriched remnants of chylomicrons and VLDL. Cell-surface heparan sulfate proteoglycans facilitate the interaction of apoE-containing remnant lipoproteins with the LRP, which mediates uptake by hepatocytes (Mahley and Huang, 1999). Inherited absence of either functional HL (very rare) or functional apoE impedes remnant clearance by the LDL receptor and the LRP, resulting in a hyperlipidemia characterized by an increase of triglyceride- and cholesterol-rich remnant lipoproteins in the plasma (type III hyperlipoproteinemia) (Mahley and Rall, 2001). Chylomicron remnants are not precursors of LDL. However, during the initial hydrolysis of chylomicron triglycerides by LPL, apoA-I and phospholipids are shed from the surface of chylomicrons and remain in the plasma. This is one mechanism by which nascent (precursor) HDL are generated. Very-Low-Density Lipoproteins VLDL are produced in the liver and are synthesized when triglyceride production is stimulated by an increased flux of free fatty acids or by increased de novo synthesis of fatty acids by the liver. The VLDL are 400 to 1000 in diameter and are large enough to cause plasma turbidity, but VLDL particles, unlike chylomicrons, do not float spontaneously to the top of a tube of plasma that is allowed to stand undisturbed for 12 hours. ApoB-100, apoE, and apoC-I, C-II, and C-III are synthesized constitutively by the liver and incorporated into VLDL (Table 362). If triglycerides are not available to form VLDL, the newly synthesized apoB-100 is degraded by hepatocytes. Triglycerides are synthesized in the endoplasmic reticulum and, along with other lipid constituents, are transferred by MTP to the site in the endoplasmic reticulum where newly synthesized apoB-100 is available to form nascent (precursor) VLDL. The nascent VLDL incorporate small amounts of apoE and the C apoproteins within the liver before secretion, but most of these apoproteins are acquired from plasma HDL after the VLDL are secreted by the liver. Without MTP, hepatic triglycerides cannot be transferred to apoB-100. As a consequence, patients with dysfunctional MTP fail to make any of the apoB-containing lipoproteins (VLDL, IDL, or LDL). MTP also plays a key role in the synthesis of chylomicrons in the intestine, and mutations of MTP that result in the inability of triglycerides to be transferred to either apoB-100 in the liver or apoB-48 in the intestine prevent VLDL and chylomicron production and cause the genetic disorder abetalipoproteinemia (Gregg and Wetterau, 1994). Plasma VLDL are then catabolized by LPL in the capillary beds in a process similar to the lipolytic processing described for chylomicrons (Figure 361). When triglyceride hydrolysis is nearly complete, the VLDL remnants, usually termed IDL, are released from the capillary endothelium and reenter the circulation. ApoB-100containing small VLDL and IDL (VLDL remnants), which have a half-life of less than 30 minutes, have two potential fates. About 40% to 60% are bound by LDL receptors or the LRP, which recognizes ligands (apoB-100 and apoE) on the remnants, and are cleared from the plasma primarily by the liver. The remainder of the IDL are further acted upon by LPL and HLwhich remove additional triglycerides, C apoproteins, and apoEand are converted to plasma LDL. Virtually all LDL particles in the plasma are derived from VLDL. ApoE plays a major role in the metabolism of triglyceride-rich lipoproteins (chylomicrons, chylomicron remnants, VLDL, and IDL) and has a number of major functions related to the binding and uptake of plasma lipoproteins and to the redistribution of lipids locally among cells (Mahley and Rall, 2000; Mahley, 1988; Mahley and Huang, 1999). About half of the apoE in the plasma of fasting subjects is associated with triglyceride-rich lipoproteins, and the other half is a constituent of HDL. ApoE controls the catabolism of the apoE-containing lipoproteins by mediating their binding to cell-surface heparan sulfate proteoglycans (especially in the liver) and to LDL receptors and the LRP (Mahley and Ji, 1999). About three-fourths of the apoE in plasma is synthesized by the liver and the remainder by a variety of tissues. The brain is the second most abundant site of apoE mRNA synthesis, which occurs primarily in astrocytes. ApoE also is synthesized by macrophages, where it appears to play a role in modulating cholesterol accumulation. In transgenic mice, overexpression of apoE by macrophages inhibits hypercholesterolemia-induced atherogenesis (Bellosta et al., 1995; Hasty et al., 1999). There are three commonly occurring alleles of the apoE gene (designated
Single amino acid substitutions result from the genetic polymorphisms in the apoE gene (Mahley and Rall, 2000; Mahley, 1988). ApoE2, with a cysteine at residue 158, differs from apoE3, which has arginine at this site. ApoE3, with a cysteine at residue 112, differs from apoE4, which has arginine at this site. These single amino differences affect both receptor binding and lipid binding of the three apoE isoforms. Both apoE3 and apoE4 can bind to the LDL receptor, but apoE2 binds much less effectively and, as a consequence, causes the remnant lipoprotein dyslipidemia of type III hyperlipoproteinemia. ApoE2 and apoE3 bind preferentially to the phospholipids of HDL, whereas apoE4 binds preferentially to VLDL triglycerides. Low-Density Lipoproteins The LDL particles arising from the catabolism of IDL have a half-life of 1.5 to 2 days, which accounts for the higher plasma concentration of LDL than of VLDL and IDL. In subjects without hypertriglyceridemia, two-thirds of plasma cholesterol is found in the LDL. Plasma clearance of LDL particles is mediated primarily by LDL receptors; a small component is mediated by nonreceptor clearance mechanisms (Brown and Goldstein, 1986). Defective or absent LDL receptors cause high levels of plasma LDL and familial hypercholesterolemia (Brown and Goldstein, 1986; Hobbs et al., 1992). ApoB-100, the only apoprotein of LDL, is the ligand that binds LDL to its receptor. Residues 3000 to 3700 in the carboxyl-terminal sequence are critical for binding. Mutations in this region disrupt binding and are a cause of hypercholesterolemia (familial defective apoB-100) (Innerarity et al., 1990; Pullinger et al., 1995). The liver expresses a large complement of LDL receptors and removes The most effective dietary (decreased consumption of saturated fat and cholesterol) and pharmacological (treatment with statins) treatments of hypercholesterolemia act by enhancing hepatic LDL receptor expression (Bilheimer et al., 1983; Woollett and Dietschy, 1994). Regulation of LDL receptor expression is part of a complex process by which cells regulate their free cholesterol content. This regulatory process is mediated by transcription factors called sterol regulatory binding element proteins (SREBPs) (Brown and Goldstein, 1998). SREBPs enhance LDL receptor expression when cellular cholesterol content is reduced. LDL become atherogenic when they are modified by oxidation (Steinberg, 1997), a required step for LDL uptake by the scavenger receptors of macrophages. This process leads to foam-cell formation in arterial lesions. At least two scavenger receptors (SRs) are involved (SR-AI/II and CD36). Knocking out either receptor in transgenic mice retards the uptake of oxidized LDL by macrophages. Expression of the two receptors is differentially regulated; SR-AI/II appears to be more important in early atherogenesis, and CD36 more important as foam cells form during lesion progression (Nakata et al., 1999; Dhaliwal and Steinbrecher, 1999). High-Density Lipoproteins The metabolism of HDL is complex because of the multiple mechanisms by which HDL particles are modified in the plasma compartment and by which HDL particles are synthesized (Breslow, 1994; Segrest et al., 2000; Tall et al., 2000). ApoA-I is the major HDL apoprotein, and its plasma concentration is a more powerful inverse predictor of CHD risk than is the HDL-C level (Maciejko et al., 1983). ApoA-I synthesis is required for normal production of HDL. Mutations in the apoA-I gene that cause HDL deficiency are variable in their clinical expression and often are associated with accelerated atherogenesis (Assmann et al., 2001). Conversely, overexpression of apoA-I in transgenic mice protects against experimentally induced atherogenesis (Plump et al., 1994). Mature HDL can be separated by ultracentrifugation into HDL2
(d = 1.063 to 1.125 g/ml), which are larger, more cholesterol-rich
lipoproteins (70 to 100 in diameter), and HDL3 (d = 1.125 to
1.21 g/ml), which are smaller particles (50 to 70 in diameter). In
addition, two major subclasses of mature HDL particles in the plasma can be
differentiated by their content of the major HDL apoproteins, apoA-I and
apoA-II (Duriez and Fruchart, 1999). Mature HDL particles have The precursor of most of the plasma HDL is a discoidal particle
containing apoA-I and phospholipid, called pre- Discoidal pre- The membrane transporter ATP-binding cassette protein 1 (ABC-1) facilitates the transfer of free cholesterol from cells to HDL (Young and Fielding, 1999; Oram and Vaughan, 2000). When ABC-1 is defective, the acquisition of cholesterol by HDL is greatly diminished, and HDL levels are markedly reduced because poorly lipidated nascent HDL are metabolized rapidly. Dysfunctional mutants of ABC-1 cause the defect observed in Tangier disease, a genetic disorder characterized by extremely low levels of HDL and cholesterol accumulation in the liver, spleen, tonsils, and neurons of peripheral nerves. After free cholesterol is acquired by the pre- As the cholesteryl ester content of the HDL2 increases, the cholesteryl esters of these particles begin to be exchanged for triglycerides derived from any of the triglyceride-containing lipoproteins (chylomicrons, VLDL, remnant lipoproteins, and LDL). This exchange is mediated by the cholesteryl ester transfer protein and, in human beings, accounts for the removal of about two-thirds of the cholesterol associated with HDL. The transferred cholesterol subsequently is metabolized as part of the lipoprotein into which it was transferred. The triglyceride that is transferred into HDL2 is hydrolyzed in the liver by HL, a process that regenerates smaller, spherical HDL3 particles that recirculate and acquire additional free cholesterol from tissues containing excess free cholesterol. HL activity is regulated and modulates HDL-C levels. Both androgens and estrogens affect HL gene expression, but with opposite effects (Haffner et al., 1983; Brinton, 1996). Androgens increase HL activity, which accounts for the lower HDL-C values observed in men than in women. Estrogens reduce HL activity, but their impact on HDL-C levels in women is substantially less than that of androgens on HDL-C levels in men. HL appears to have a pivotal role in regulating HDL-C levels, as HL activity is increased in many patients with low HDL-C levels. HDL are protective lipoproteins that decrease the risk of CHD; thus, high levels of HDL are desirable. This protective effect may result from the participation of HDL in reverse cholesterol transport, the process by which excess cholesterol is acquired from cells and transferred to the liver for excretion. HDL also may inhibit oxidative modification of LDL through the action of paraoxonase, an HDL-associated antioxidant protein. Lipoprotein(a) Lipoprotein(a) [Lp(a)] is composed of an LDL particle that has a
second apoprotein in addition to apoB-100 (Berg, 1994). The second
apoprotein, apo(a), is attached to apoB-100 by at least one disulfide bond
and does not function as a lipid-binding apoprotein. |
Hyperlipidemia and Atherosclerosis
|
Despite a continuing decline in the incidence
of atherosclerosis-related deaths in the past 35 years, deaths from CHD,
cerebrovascular disease, and peripheral vascular disease accounted for 30% of
the 2.3 million deaths in the These statistics illustrate the importance of identifying and managing risk factors for CHD. The major known risk factors are elevated LDL-C, reduced HDL-C, cigarette smoking, hypertension, type 2 diabetes mellitus, advancing age, and a family history of premature (men < 55 years; women < 65 years) CHD events in a first-degree relative. Control of the modifiable risk factors is especially important in preventing premature CHD (events in men below 55 years or in women below 65 years). Observational studies suggest that modifiable risk factors account for 85% of excess risk (risk over and above that of individuals with optimal risk-factor profiles) for premature CHD (Stamler et al., 1986; Wilson et al., 1998). Furthermore, these studies indicate that, when total cholesterol levels are below 160 mg/dl, CHD risk is markedly attenuated, even in the presence of additional risk factors (Grundy et al., 1998). This pivotal role of hypercholesterolemia in atherogenesis gave rise to the almost universally accepted cholesterol-diet-CHD hypothesis (Thompson and Barter, 1999). The cholesterol-diet-CHD hypothesis states that elevated plasma cholesterol levels cause CHD, that diets rich in saturated fat (animal fat) and cholesterol raise cholesterol levels, and that the lowering of cholesterol levels reduces CHD risk. Although the relationship between cholesterol, diet, and CHD was recognized nearly 50 years ago, proof that cholesterol lowering was safe and prevented CHD death required extensive epidemiological studies and clinical trials. Epidemiological Studies Epidemiological studies have demonstrated the importance of the relationship between excess saturated fat consumption and elevated cholesterol levels. Reducing the consumption of dietary saturated fat and cholesterol is the cornerstone of population-based approaches to the management of hypercholesterolemia (National Cholesterol Education Program, 1990). In addition, it is clearly established that the higher the cholesterol level, the higher the CHD risk (Stamler et al., 1986). Clinical Trials Studies of the efficacy of cholesterol lowering began in the 1960s. However, it was not until the advent of powerful cholesterol-reducing drugs known as statins that clear-cut evidence of the benefit of cholesterol lowering became available (Illingworth and Durrington, 1999). Several important trials in the 1970s and 1980s showed that average cholesterol reductions of about 10% resulted in 20% reductions in nonfatal CHD events, but these trials were not large enough to detect an effect on mortality (Lipid Research Clinics Program, 1984a; Committee of Principal Investigators, 1984; Frick et al., 1987; Durrington and Illingworth, 1998). In fact, increases in noncardiac mortality in these trials raised concerns about the safety of cholesterol-lowering therapy (Wysowski and Gross, 1990). In 1994, the Scandinavian Simvastatin Survival Study (4S), a secondary prevention trial, proved for the first time that lowering cholesterol levels with simvastatin reduced total mortality among CHD patients with normal HDL levels and high mean baseline LDL-C levels (188 mg/dl). Simvastatin therapy reduced LDL-C by an average of 35%, CHD mortality by 42%, nonfatal CHD events by 40%, and total mortality by 30%. Simvastatin therapy did not increase noncardiac mortality from any cause (Scandinavian Simvastatin Survival Study Group, 1994). Subsequently, the efficacy and safety of statin therapy in patients with established CHD at baseline was evaluated in the Cholesterol and Recurrent Events (CARE) trial and the Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) study [Sacks et al., 1996; The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group, 1998]. In CARE and LIPID, the average baseline LDL-C and HDL-C levels (139 and 39 mg/dl in CARE and 150 and 36 mg/dl in LIPID) were lower than in 4S. Treatment with pravastatin reduced LDL-C levels by 25% (CARE) and 28% (LIPID) and was associated with a 24% reduction in CHD death in LIPID and 29% and 28% reductions in nonfatal myocardial infarctions in LIPID and CARE, respectively. The results of the 4S, CARE, and LIPID trials indicated that CHD
patients with baseline LDL-C values above 130 mg/dl benefit from
lipid-lowering therapy. Subgroup analyses of patients in these trials with
baseline LDL-C levels between 100 and 130 mg/dl did not consistently show a
benefit from cholesterol-lowering drug therapy (Grundy, 1998b). More
recently, however, the Veterans Affairs High Density Lipoprotein Intervention
Trial (VA HIT) showed that CHD patients with LDL-C There also have been clinical trials of lipid lowering in patients who had no evidence of vascular disease at baseline (primary prevention trials). The West of Scotland Coronary Prevention Study (WOSCOPS) (Shepherd et al., 1995) demonstrated a benefit of pravastatin therapy in male patients with baseline LDL-C >155 mg/dl. The average LDL-C in WOSCOPS was high (192 mg/dl), and the mean HDL-C level was 44 mg/dl. The average on-treatment LDL-C was 142 mg/dl, a 26% decrease from baseline, and this resulted in a 31% reduction in CHD death and nonfatal myocardial infarction. The second major statin trial in patients without vascular disease was the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS) (Downs et al., 1998). This trial included men and women who at baseline had only moderately elevated levels of LDL-C (average, 156 mg/dl) and who were primarily at risk because of age (men >45 years of age; women >55 years of age) and because of low HDL-C levels (average, 37 mg/dl). In this trial, lovastatin reduced LDL-C by 26% and primary endpoint events (fatal and nonfatal myocardial infarction and unstable angina pectoris) by 37%. The trials described above provided evidence that supported the 2001
revision of the National Cholesterol Education Program (NCEP) guidelines for
the management of dyslipidemic patients. These trials demonstrated efficacy
in the prevention of vascular disease events and provided convincing evidence
for the short-term ( National Cholesterol Education Program Guidelines for Treatment: Managing Patients with Dyslipidemia The current NCEP guidelines for management of patients with lipid disorders are of two types. One is a population-based approach, which is intended to lower blood cholesterol by dietary recommendations: Reduce total calories from fat to less than 30% and from saturated fat to less than 10%; consume less than 300 mg of cholesterol per day; and maintain desirable body weight (National Cholesterol Education Program, 1990). The second is the patient-based approach described in the 2001 report of the NCEP Adult Treatment Panel III, which continues to focus on lowering LDL-C levels as the primary goal of therapy (National Cholesterol Education Program Expert Panel, 2001). The 2001 Adult Treatment Panel III guidelines for the management of adults 20 years and older recommend a complete lipoprotein profile (total cholesterol, LDL-C, HDL-C, and triglycerides) rather than screening for total cholesterol and HDL-C alone. Fasting for 12 hours is required to accurately measure the triglyceride and LDL-C levels [LDL-C = total cholesterol (triglyceride 5) HDL-C]. The classification of lipid levels is shown in Table 363. If the values for total cholesterol, LDL-C, and triglycerides are in the lowest category and the HDL-C level is not low, lifestyle recommendations (diet and exercise) should be made to ensure maintenance of a normal lipid profile. Other vascular disease risk factors (Table 364), if present, should be assessed and treated individually. For patients with elevated levels of total cholesterol, LDL-C, or triglycerides, or reduced HDL-C values, further treatment is based on the patient's risk-factor status (Table 364) and LDL-C levels (Table 365). All patients should receive instruction about dietary restriction of saturated fat and cholesterol. Patients with CHD or a CHD equivalent (symptomatic peripheral or carotid vascular disease, abdominal aortic aneurysm, >20% ten-year CHD risk or diabetes mellitus) should immediately start appropriate lipid-lowering therapy. Patients without CHD or CHD equivalent should be managed with lifestyle advice (diet, exercise, weight management) for 3 to 6 months before drug therapy is implemented. Before drug therapy is initiated, however, secondary causes of hyperlipidemia should be excluded. Most secondary causes (Table 366) can be excluded by ascertaining the patient's medication history and by measuring serum creatinine, liver function tests, fasting glucose, and thyroid-stimulating hormone levels. Treatment of the disorder causing secondary dyslipidemia may preclude the necessity of treatment with hypolipidemic drugs. Risk Assessment Using The 2001 NCEP guidelines and those of the European Atherosclerosis Society (Wood et al., 1998) employ risk assessment tables devised from the Framingham Heart Study in an attempt to match the intensity of treatment to the severity of CHD risk in patients without a prior history of symptomatic atherosclerotic vascular disease. High risk or 'CHD equivalent' status is defined as >20% chance of sustaining a CHD event in the next ten years. The tables used to determine a patient's absolute risk do not take into account risk associated with a family history of premature CHD or obesity. As a consequence, the risk may be seriously underestimated, resulting in insufficiently aggressive management. After calculation of the risk score, more aggressive therapy should be considered for obese patients or for patients with a family history of premature CHD. Arterial Wall Biology and Plaque Stability More effective lipid-lowering agents and a better understanding of atherogenesis have helped to prove that aggressive lipid-lowering therapy has many beneficial effects over and above those obtained by simply decreasing lipid deposition in the arterial wall. Arteriographic trials have shown that, although aggressive lipid lowering results only in very small increases in lumen diameter, it promptly decreases acute coronary events (Brown et al., 1993). Lesions causing less than 60% occlusion are responsible for more than two-thirds of the acute events. Aggressive lipid-lowering therapy may prevent acute events through its positive effects on the arterial wall; it corrects endothelial dysfunction, corrects abnormal vascular reactivity (spasms), and improves plaque stability. Atherosclerotic lesions containing a large lipid core, large numbers of macrophages, and a poorly formed fibrous cap (Brown et al., 1993; Gutstein and Fuster, 1999) are prone to plaque rupture and acute thrombosis. Aggressive lipid lowering appears to alter plaque architecture, resulting in less lipid, fewer macrophages, and a larger collagen and smooth muscle cellrich fibrous cap. Stabilization of plaque susceptibility to thrombosis appears to be a direct result of LDL-C lowering or an indirect result of changes in cholesterol and lipoprotein metabolism or arterial wall biology (see below, 'Potential Cardioprotective Effects Other Than LDL Lowering?'). Who and When to Treat? Large-scale trials with statins have provided new insights into which patients with dyslipidemia should be treated and when treatment should be initiated. Gender Both men and women benefit from lipid-lowering therapy. In fact, CARE
and AFCAPS/TexCAPS showed greater benefit in women. Statins, rather than
hormone-replacement therapy, are now recommended by the American Heart
Association and the Age Age >45 years in men and >55 years in women is considered to be a CHD risk factor. The statin trials have shown that patients >65 years of age benefit from therapy as much as do younger patients. Old age per se is not a reason to withhold drug therapy in an otherwise healthy person. Cerebrovascular Disease Patients In most studies of patients with cerebrovascular disease, plasma cholesterol levels correlate positively with risk of ischemic stroke. In clinical trials, statins reduced stroke and/or transient ischemic attacks in patients with and without CHD (Hebert et al., 1997). Peripheral Vascular Disease Patients Statins have proved beneficial in patients with peripheral vascular disease. Hypertensive Patients and Smokers The risk reduction for coronary events in hypertensive patients and in smokers is similar to that in subjects without these risk factors. Type 2 Diabetes Mellitus Patients with type 2 diabetes benefit very significantly from aggressive lipid lowering (see'Treatment of Type 2 Diabetes,' below). PostMyocardial Infarction or Revascularization Patients As soon as CHD is diagnosed, it is essential to begin lipid-lowering therapy (NCEP guidelines: LDL-C < 100 mg/dl). Compliance with drug therapy is greatly enhanced if treatment is initiated in the hospital (Fonarow and Gawlinski, 2000). It remains to be determined if statin therapy alters restenosis after angioplasty; however, the NHLBI Post Coronary Artery Bypass Graft trial showed that statin therapy improved the long-term outcome after bypass surgery and that the lower the LDL-C, the better (The Post Coronary Artery Bypass Graft Trial Investigators, 1997). Can Cholesterol Levels Be Lowered Too Much? Are there total and LDL cholesterol levels below which adverse health consequences begin to increase? Observational studies initially were confusing. In the United States and western Europe, low cholesterol levels appeared to be associated with an increase in noncardiac mortality from chronic pulmonary disease, chronic liver disease, cancer (many primary sites), and hemorrhagic stroke. However, more recent data indicate that it is the noncardiac diseases that cause the low plasma cholesterol levels and not the low cholesterol levels that cause the noncardiac diseases (Law et al., 1994). One exception may be hemorrhagic stroke. In the Multiple Risk Factor Intervention Trial (MRFIT), hemorrhagic stroke occurred more frequently in hypertensive patients with total cholesterol levels below 160 mg/dl; however, the increased incidence of hemorrhagic stroke was more than offset by reduced CHD risk due to the low cholesterol levels (Neaton et al., 1992). In addition, in a study of the Chinese population, in which cholesterol levels rarely exceeded 160 mg/dl, lower levels of total cholesterol were not associated with increases in hemorrhagic stroke or any other cause of noncardiac mortality (Chen et al., 1991). Abetalipoproteinemia and hypobetalipoproteinemia, two rare disorders in human beings that are associated with extremely low total cholesterol levels, are instructive because affected individuals have reduced CHD risk and no increase in noncardiac mortality (Welty et al., 1998). Patients who are homozygous for the mutations that cause these disorders have total cholesterol levels below 50 mg/dl and triglyceride levels below 25 mg/dl. Individuals consuming very low levels of total fat (less than 5% of total calories) and vegetarians, who consume no animal fat, usually have total cholesterol levels below 150 mg/dl and have no increase in noncardiac mortality (Appleby et al., 1999). Based on the lack of harm associated with low total cholesterol levels in these various groups, reducing cholesterol levels to similarly low levels with drugs does not appear to be contraindicated. With the advent of more efficacious cholesterol-lowering agents, it soon may be possible to test the benefits and risks of lowering total cholesterol levels below 150 mg/dl. Will Greater Lipid Lowering Further Reduce CHD? Despite the remarkable results of the statin trials (a 25% to 40%
reduction in events), there are still 60% to 75% as many events in the
treatment groups of the statin trials as in the placebo groups. Do these
results suggest that even more aggressive lipid lowering is required (Grundy,
1998b)? The investigators who conducted the AFCAPS/TexCAPS trial suggested
that the 1993 NCEP guidelines are still too conservative for high-risk
subjects without CHD, like those enrolled in the AFCAPS/TexCAPS study (Downs et
al., 1998). They state that drug therapyalong with a prudent diet,
regular exercise, and risk factor modificationshould be used to lower the
risk of the first acute major coronary event in primary prevention candidates
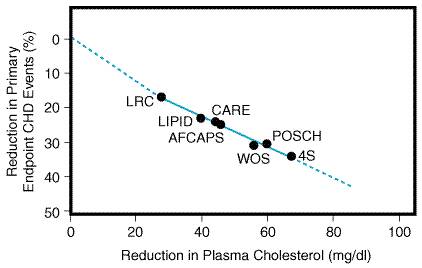
who are older (men As more effective lipid-lowering drugs and better combinations of therapies are developed, we will be able to lower lipid levels more effectively. But will lower cholesterol levels translate into a further reduction of clinical events (Figure 362)? Many researchers believe that the answer is yes. In addition, as statins become generic drugs, more aggressive treatment of wider segments of the population will become more cost-effective.
Treatment of Type 2 Diabetes Diabetes mellitus is an independent predictor of high risk for CHD. CHD morbidity is two to four times higher in patients with diabetes than in nondiabetics, and the mortality from CHD is up to 100% higher in diabetic patients than in nondiabetics over a 6-year period (Grundy et al., 1999). Glucose control is essential, but this provides only minimal benefit with respect to CHD prevention. Aggressive treatment of diabetic dyslipidemia through diet, weight control, and drugs (in most cases) is critical in reducing risk. Diabetic dyslipidemia is usually characterized by high triglycerides, low HDL-C, and moderate elevations of total cholesterol and LDL-C. Recent recommendations from the American Heart Association and the American Diabetes Association indicate that the treatment guidelines for diabetic patients should be the same as for patients with CHD (Grundy et al., 1999). The revised 2001 NCEP guidelines also will reflect this recommendation. Haffner et al. (1998) reported that diabetics without diagnosed CHD are at the same level of risk as nondiabetics with established CHD. This is consistent with the recommendation to reduce plasma LDL-C levels of all diabetics to <100 mg/dl, irrespective of whether they have had a prior ischemic vascular disease event. The American Diabetes Association further recommends that the first line of treatment for a diabetic dyslipidemia usually should be a statin (Grundy et al., 1999). Clinical trials with simvastatin, pravastatin, and lovastatin have clearly established in post hoc analyses that diabetics profit from cholesterol lowering as much as other subgroups or even more. For example, diabetics in the 4S, CARE, AFCAPS/TexCAPS, and LIPID trials had 55%, 25%, 43%, and 19% reductions in events, respectively. The Diabetes Atherosclerosis Intervention Study recently showed a significant benefit of treating type 2 diabetics with fenofibrate. This 3-year arteriographic study demonstrated a 40% decrease of focal coronary stenoses (p= 0.029) (Diabetes Atherosclerosis Intervention Study Investigators, 2001). Metabolic Syndrome The 2001 NCEP guidelines recognize the increased CHD risk associated with the insulin-resistant, prediabetic state described under the rubric of 'metabolic syndrome.' This syndrome consists of a constellation of five CHD risk factors (Table 367). The 2001 NCEP guidelines arbitrarily define the presence of three or more of these risk factors as indicating that a patient is affected. Treatment should focus on weight loss and increased physical activity since overweight and obesity usually preclude optimal risk factor reduction. Specific treatment of increased LDL-C and triglyceride levels and low HDL-C levels should be undertaken as well. Treatment of Hypertriglyceridemia The 2001 NCEP guidelines reflect the increased CHD risk associated with the presence of triglyceride levels above 150 mg/dl. Three categories of hypertriglyceridemia are recognized (Table 363), and treatment is recommended based on the degree of elevation. Weight loss, increased exercise, and alcohol restriction are important for all hypertriglyceridemic patients. The LDL-C goal should be ascertained based on each patient's risk factor or CHD status (Table 365). If triglycerides remain above 200 mg/dl after the LDL-C goal is reached, further reduction in triglycerides may be achieved by increasing the dose of a statin or of niacin. Combination therapy (statin plus niacin or statin plus fibrate) may be required, but caution is necessary with these combinations to avoid myopathy (see Statins in Combination with Other Lipid-lowering Drugs, below). Treatment of Low HDL-C The most frequent risk factor for premature CHD is low HDL-C. In a
study of 321 men with angiographically documented CHD, In low-HDL-C patients, the total cholesterol/HDL-C ratio is a
particularly useful predictor of CHD risk. The Results from AFCAPS/TexCAPS (a primary prevention trial) and VA HIT (a secondary prevention trial) are particularly relevant. Patients in these trials had average or low LDL-C, low HDL-C, and high total cholesterol/ HDL-C ratios and treatment greatly reduced clinical events in both trials. The 2001 NCEP guidelines extend treatment to include some, but not all, patients typical of those who benefited in AFCAPS/TexCAPS and VA HIT. See Table 368 for a summary of trial results (Downs et al., 1998; Rubins et al., 1999). The treatment of low HDL-C patients according to the 2001 NCEP
guidelines is focused on lowering LDL-C to the target level based on the
patient's risk factor or CHD status (Table 365) and a reduction of
VLDL cholesterol (estimated by dividing the plasma triglyceride level by 5)
below 30 mg/dl. If this strategy results in a ratio of total
cholesterol/HDL-C that is |
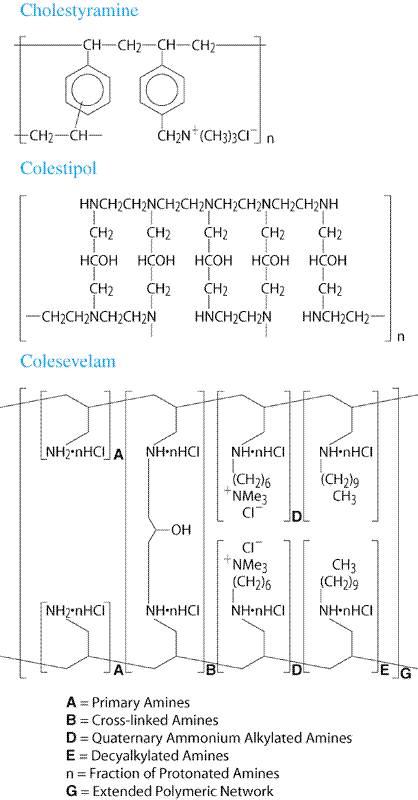
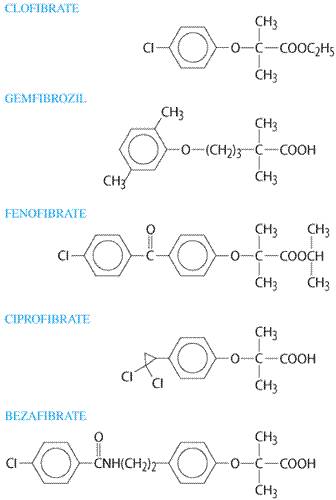
Drug Therapy of Dyslipidemia
|
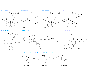
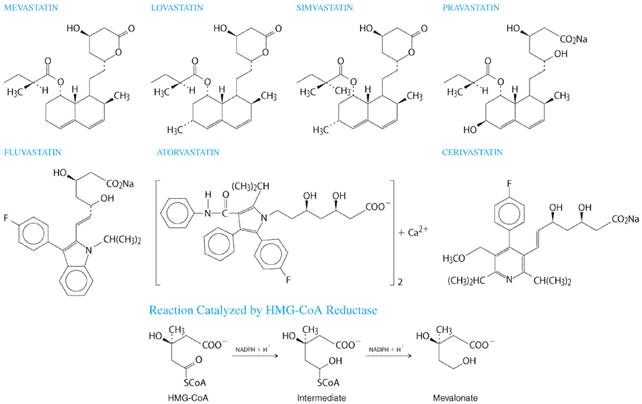
In addition to the present discussion, the topic of drug therapy for dyslipidemia has been extensively reviewed by Durrington and Illingworth (1998). Statins The statins are the most effective and best-tolerated agents for treating dyslipidemia. These drugs are competitive inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, which catalyzes an early, rate-limiting step in cholesterol biosynthesis. Higher doses of the more potent statins (e.g., atorvastatin and simvastatin) also can reduce triglyceride levels caused by elevated VLDL levels. Some statins also are indicated for raising HDL-C levels, although the clinical significance of these effects on HDL-C remains to be proven. Five large, well-controlled clinical trials have documented the efficacy and safety of simvastatin, pravastatin, and lovastatin in reducing fatal and nonfatal CHD events, strokes, and total mortality [Scandinavian Simvastatin Survival Study Group, 1994; Shepherd et al., 1995; Sacks et al., 1996; Downs et al., 1998; The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group, 1998]. Rates of adverse events in all five trials were the same in the placebo groups and in the groups receiving active drug. This was true with regard to noncardiac illness and the two laboratory tests, hepatic transaminases and creatine kinase (CK), that have been most frequently used to monitor patients taking statins. History Statins were isolated from a mold, Penicillium citrinium, and identified as inhibitors of cholesterol biosynthesis in 1976 by Endo and colleagues (Endo et al., 1976). Subsequently, Brown et al., (1978) established that statins act by inhibiting HMG-CoA reductase. The first statin studied in human beings was compactin, renamed mevastatin, which demonstrated the therapeutic potential of this class of drugs (Yamamoto et al., 1984). However, Alberts and colleagues at Merck developed the first statin (lovastatin; formerly known as mevinolin) that was approved for use in human beings, which was isolated from Aspergillus terreus (Alberts et al., 1980; Bilheimer et al., 1983). Since the approval of lovastatin by the United States Food and Drug Administration (FDA) in 1987, five other statins have been approved. Two of these, pravastatin and simvastatin, are chemically modified derivatives of lovastatin (seeFigure 363). The more recently approved statinsatorvastatin, fluvastatin, and cerivastatinare synthetic compounds. More statins are under development.
Chemistry The structural formulas of the original statin (mevastatin) and the six statins currently available in the United States are shown in Figure 363 along with the reaction (conversion of HMG-CoA to mevalonate) catalyzed by HMG-CoA reductase, the enzyme they competitively inhibit. The statins possess a side group that is structurally similar to HMG-CoA. Mevastatin, lovastatin, simvastatin, and pravastatin are fungal metabolites, and each contains a hexahydronapthalene ring. Lovastatin differs from mevastatin in having a methyl group at carbon 3. There are two major side chains. One is a methylbutyrate ester (lovastatin and pravastatin) or a dimethylbutyrate ester (simvastatin). The other contains a hydroxy acid that forms a six-membered analog of the intermediate compound in the HMG-CoA reductase reaction (Figure 363). Fluvastatin, atorvastatin, and cerivastatin are entirely synthetic compounds containing a heptanoic acid side chain that forms a structural analog of the HMG-CoA intermediate. As a result of their structural similarity to HMG-CoA, statins are reversible competitive inhibitors of the enzyme's natural substrate, HMG-CoA. The inhibition constant (Ki) of cerivastatin is 0.01 nM (Bischoff et al., 1997); all other statins have a Ki in the 1-nM range. The dissociation constant of HMG-CoA is three orders of magnitude higher than this value. Lovastatin and simvastatin are lactone prodrugs that are modified in the liver to active hydroxy acid forms. Since they are lactones, they are less soluble in water than are the other statins, a difference that appears to have little if any clinical significance. Pravastatin (an acid in the active form), fluvastatin and cerivastatin (sodium salts), and atorvastatin (a calcium salt), are all administered in the active, open-ring form. Mechanism of Action Statins exert their major effectreduction of LDL levelsthrough a mevalonic acidlike moiety that competitively inhibits HMG-CoA reductase by product inhibition (Alberts et al., 1980). Statins affect blood cholesterol levels by inhibiting cholesterogenesis in the liver, which results in increased expression of the LDL receptor gene. In response to the reduced free cholesterol content within hepatocytes, membrane-bound SREBPs are cleaved by a protease and translocated to the nucleus. The transcription factors are then bound by the sterol-responsive element of the LDL receptor gene, enhancing transcription and ultimately increasing the synthesis of LDL receptors (Brown and Goldstein, 1998). Degradation of LDL receptors also is reduced (Brown et al., 1978). The greater number of LDL receptors on the surface of hepatocytes results in increased removal of LDL from the blood (Bilheimer et al., 1983), thereby lowering LDL-C levels. Some studies suggest that statins also can reduce LDL levels by enhancing the removal of LDL precursors (VLDL and IDL) and by decreasing hepatic VLDL production (Grundy and Vega, 1985; Arad et al., 1990; Aguilar-Salinas et al., 1998). Since VLDL remnants and IDL are enriched in apoE, a statin-induced increase in the number of LDL receptors, which recognize both apoB-100 and apoE, enhances the clearance of these LDL precursors (Gaw et al., 1993). The reduction in hepatic VLDL production induced by statins is thought to be mediated by reduced synthesis of cholesterol, a required component of VLDL (Thompson et al., 1996). This mechanism also likely accounts for the triglyceride-lowering effect of statins (Ginsberg, 1998) and may account for the approximately 25% reduction of LDL-C levels in patients with homozygous familial hypercholesterolemia treated with 80 mg of either atorvastatin or simvastatin (Raal et al., 1997, 2000). Triglyceride Reduction by Statins Triglyceride levels greater than 250 mg/dl are reduced substantially by statins, and the percent reduction achieved is similar to the percent reduction in LDL-C (Stein et al., 1998). Accordingly, hypertriglyceridemic patients taking the highest doses (80 mg/day) of two of the most potent statins (simvastatin, atorvastatin) experience a 35% to 45% reduction in LDL-C and a similar reduction in fasting triglyceride levels (Bakker-Arkema et al., 1996; Ose et al., 2000). If baseline triglyceride levels are below 250 mg/dl, reductions in triglycerides do not exceed 25% irrespective of the dose or statin used (Stein et al., 1998). Similar reductions (35% to 45%) in triglycerides can be accomplished with usual doses of fibrates or niacin (see below), although these drugs do not reduce LDL-C to the same extent as 80-mg doses of atorvastatin or simvastatin. Effect of Statins on HDL-C Levels Most studies of patients treated with statins have systematically excluded patients with low HDL-C levels. In studies of patients with elevated LDL-C levels and gender-appropriate HDL-C levels (40 to 50 mg/dl for men; 50 to 60 mg/dl for women), an increase in HDL-C of 5% to 10% was observed, irrespective of the dose or statin employed. However, in patients with reduced HDL-C levels (<35 mg/dl), preliminary studies suggest that statins differ in their effects on HDL-C levels. Simvastatin, at its highest dose of 80 mg, increases HDL-C and apoA-I levels more than a comparable dose of atorvastatin (Crouse et al., 1999; Crouse et al., 2000). However, more studies are needed to ascertain the effects of statins on HDL-C in patients with low HDL-C levels and to determine if statin effects on HDL-C are clinically meaningful. Effects of Statins on LDL-C Levels Statins lower LDL-C by 20% to 55%, depending on the dose and statin
used. In large trials comparing the effects of the various statins,
equivalent doses appear to be 5 mg of simvastatin = Table 369 provides information regarding the doses of the various statins that are required to reduce LDL-C by 20% to 55%. The percent reductions achieved with the various doses are the same regardless of the absolute value of the baseline LDL-C level. The statins are effective in virtually all patients with high LDL-C levels. The exception is patients with homozygous familial hypercholesterolemia, who have very attenuated responses to the usual doses of statins, because both alleles of the LDL receptor gene code for dysfunctional LDL receptors; the partial response in these patients is due to a reduction in hepatic VLDL synthesis associated with the inhibition of HMG-CoA reductasemediated cholesterol synthesis (Raal et al., 1997, 2000). Statin therapy does not reduce Lp(a) levels (Kostner et al., 1989). Potential Cardioprotective Effects Other Than LDL Lowering? Although the statins clearly exert their major effects on CHD by lowering LDL-C and improving the lipid profile as reflected in plasma cholesterol levels (Thompson and Barter, 1999) (Figure 362), a multitude of potentially cardioprotective effects are being ascribed to these drugs (Davignon and Laaksonen, 1999), largely on the basis of in vitro and ex vivo data. However, the mechanisms of action for nonlipid roles of statins have not been established, and it is not known whether these potential pleiotropic effects represent a class-action effect, differ among statins, or are biologically relevant. Until these questions are resolved, selection of a specific statin should not be based on any one of these effects. Nevertheless, the potential importance of the nonlipid roles of statins requires some discussion. Statins and Endothelial Function A variety of studies have established that the vascular endothelium plays a dynamic role in vasoconstriction/relaxation and that hypercholesterolemia modulates these processes directly. Acetylcholine-induced vasodilation of coronary arteries is depressed in patients with hypercholesterolemia and in patients with vascular disease (Treasure et al., 1995). Statin therapy improves coronary vasodilation in response to acetylcholine. The vasomotor response is controlled by nitric oxide, which is synthesized by endothelial cell nitric oxide synthase. Statins stabilize endothelial cell nitric oxide synthase mRNA, thereby enhancing synthesis of endothelial cell nitric oxide (Laufs et al., 1998). Statin therapy reverses endothelial dysfunction as monitored by vasoactivity within as short a period as one month (O'Driscoll et al., 1997), but similar results have been observed after a single acute reduction of LDL levels by apheresis (Tamai et al., 1997). In nonhuman primates fed a high-cholesterol diet, statin therapy improved endothelial function independently of significant changes in plasma cholesterol levels (Williams et al., 1998). Statins and Plaque Stability As discussed earlier, the vulnerability of plaques to rupture and thrombosis is of greater clinical relevance than the degree of stenosis they cause (Gutstein and Fuster, 1999). Statins may affect plaque stability in a variety of ways. There are reports that statins inhibit monocyte infiltration into the artery wall in a rabbit model (Bustos et al., 1998) and inhibit macrophage secretion of matrix metalloproteinases in vitro (Bellosta et al., 1998). The metalloproteinases degrade all extracellular matrix components and thus weaken the fibrous cap of atherosclerotic plaques. Statins also appear to modulate the cellularity of the artery wall by inhibiting proliferation of smooth muscle cells and enhancing apoptotic cell death (Corsini et al., 1998). It is debatable whether these effects would be beneficial or harmful if they occurred in vivo. Reduced proliferation of smooth muscle cells and enhanced apoptosis could retard initial hyperplasia and restenosis but also could weaken the fibrous cap and destabilize the lesion. Interestingly, statin-induced suppression of cell proliferation and the induction of apoptosis have been extended to tumor biology. The effects of statins on isoprenoid biosynthesis and protein prenylation associated with reduced availability of mevalonate may alter the development of malignancies (Davignon and Laaksonen, 1999). Statins and Inflammation Appreciation of the importance of inflammatory processes in atherogenesis is growing (Ross, 1999), and statins have been suggested to have an antiinflammatory role (Rossen, 1997). In a retrospective analysis of blood samples from the CARE trial, Ridker et al. (1998) demonstrated that the C-reactive protein concentration was a marker for high CHD risk and that statin therapy decreased baseline C-reactive protein levels and risk of CHD independently of cholesterol lowering. It remains to be determined whether the C-reactive protein is simply a marker of inflammation or it contributes to the pathogenesis of atherosclerosis. Statins and Lipoprotein Oxidation Oxidative modification of LDL appears to play a key role in mediating the uptake of lipoprotein cholesterol by macrophages and in other processes, including cytotoxicity within lesions (Steinberg, 1997). Statins reduce the susceptibility of lipoproteins to oxidation both in vitro and ex vivo (Kleinveld et al., 1993; Hussein et al., 1997b). Furthermore, atorvastatin has been reported to stabilize or increase the plasma level of paraoxonase, the antioxidation enzyme associated with plasma HDL (Aviram et al., 1998). Statins and Coagulation Statins reduce platelet aggregation (Hussein et al., 1997a), and in vitro model systems indicate that statins reduce the deposition of platelet thrombi on porcine aorta (Lacoste et al., 1995). In addition, the different statins have variable effects on fibrinogen levels, the significance of which remains to be determined (Rosenson and Tangney, 1998). Elevated plasma fibrinogen levels are associated with an increase in the incidence of CHD (Ernst and Resch, 1993). However, it remains to be determined whether fibrinogen is involved in the pathogenesis or is a marker of disease. Statins in Combination with Other Lipid-Lowering Drugs Statins plus the bile acidbinding resins cholestyramine and colestipol (see below) produce 20% to 30% greater reductions in LDL-C than can be achieved with statins alone (Tikkanen, 1996). Preliminary data indicate that colesevelam hydrochloride plus a statin lowers LDL-C by 8% to 16% more than do statins alone. Niacin also can enhance the effect of statins, but the occurrence of myopathy increases when statin doses greater than 25% of maximum (e.g., 20 mg of simvastatin or atorvastatin) are used with niacin (Guyton and Capuzzi, 1998). A fibrate (clofibrate, gemfibrozil, or fenofibrate; see below) plus a statin is particularly useful in patients with hypertriglyceridemia and a high LDL-C level. This combination does increase the risk of myopathy, but it usually is safe to use a fibrate at its usual maximal dose plus no more than 25% of each statin's maximal dose (Tikkanen, 1996; Athyros et al., 1997). Triple therapy with resins, niacin, and statins can reduce LDL-C by up to 70% (Malloy et al., 1987). Absorption, Fate, and Excretion All the statins are administered as the active Lovastatin and simvastatin are administered as prodrugs and are converted to their active hydroxy acid forms in the liver. About 30% of lovastatin and up to 85% of simvastatin are absorbed. About 50% of the active metabolites are protein bound. The liver is the primary route of excretion. Pravastatin is administered in its active Fluvastatin also is administered in its active form as a sodium salt and is almost completely absorbed. The drug is metabolized (50% to 80%) to inactive metabolites, and >90% of its excretion is hepatic (Corsini et al., 1999). Fluvastatin is the only statin with saturable first-pass hepatic metabolism; consequently, it is the only statin to achieve peak plasma concentrations in the micromolar range. However, the clinical relevance of this is unclear, as fluvastatin and its metabolites have the shortest elimination half-life of available statins (Dain et al., 1993; Corsini et al., 1999). Atorvastatin is administered as a calcium salt. It is extensively transformed in the liver to ortho- and parahydroxylated derivatives, which account for about 70% of the circulating inhibitory activity of HMG-CoA reductase. Atorvastatin and its active metabolites are metabolized principally in the liver, where they are excreted into the bile. Atorvastatin has a half-life of about 20 hours, but the half-life of plasma HMG-CoA reductase inhibitory activity is up to 30 hours. All of the other statins have half-lives of only 1 to 4 hours. The clinical significance, if any, of the prolonged half-life of atorvastatin is unclear, but it is thought to play a role in the greater efficacy of atorvastatin compared with the other statins (Christians et al., 1998; Corsini et al., 1999). Cerivastatin is given in its active form as a sodium salt. Absorption is greater than 98%. In the liver, cerivastatin is transformed into two major and two minor active metabolites. About 70% of these metabolites are excreted in the feces, and the remainder is excreted in the urine (Corsini et al., 1999). Adverse Effects and Drug Interactions Hepatotoxicity The initial postmarketing surveillance studies of the various statins revealed up to 1% incidence of elevations in hepatic transaminase to values greater than three times the upper limit of normal. The incidence appears to be dose related. However, in the placebo-controlled outcome trials, in which 20- to 40-mg doses of simvastatin, lovastatin, or pravastatin were used, the incidence of threefold elevations in hepatic transaminases was not significantly increased in the active drug treatment groups [Scandinavian Simvastatin Survival Study Group, 1994; Shepherd et al., 1995; Sacks et al., 1996; Downs et al., 1998; The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group, 1998]. Serious hepatopathy must be rare, although it appears to occur (Nakad et al., 1999). Primarily because of the safety data from the clinical trials, it is reasonable to measure alanine aminotransferase (ALT) at baseline and 3 to 6 months after therapy is initiated or after increasing the dose. If the ALT values are normal, it is not necessary to repeat the ALT test more than every 6 to 12 months. Myopathy The only major adverse effect of clinical significance associated with statin use is myopathy. All statins have been associated with myopathy and rhabdomyolysis (Pogson et al., 1999; Physicians' Desk Reference, 2001). The incidence of myopathy is low (<0.1%) in patients taking statins without concomitant administration of drugs that enhance the risk of myopathy. Two classes of drugs, fibrates (gemfibrozil, clofibrate, and fenofibrate) and niacin, also are lipid-lowering drugs and can cause myopathy by themselves (see below). When statins are administered with fibrates or niacin, the myopathy is probably caused by an enhanced inhibition of skeletal muscle sterol synthesis (a pharmacodynamic interaction) (Christians et al., 1998). The other drugs are those that, like most statins, are metabolized by the 3A4 isoform of cytochrome P450 (CYP3A4) and include certain macrolide antibiotics (e.g., erythromycin), azole antifungals (e.g., itraconazole), cyclosporine, a phenylpiperazine antidepressant, nefazodone, and protease inhibitors (Christians et al., 1998; Fichtenbaum et al., 2000; Dresser et al., 2000). This interaction is pharmacokinetic and is associated with increased plasma concentrations of statins and their active metabolites (Christians et al., 1998). Atorvastatin, cerivastatin, lovastatin, and simvastatin are primarily metabolized by CYP3A4, but cerivastatin also can be metabolized by CYP2C8. Recently, cerivastatin plus gemfibrozil therapy has been contraindicated because of a number of case reports of myopathy (Physicians' Desk Reference, 2001). Fluvastatin is mostly (50% to 80%) metabolized by CYP2C9 to inactive metabolites, but CYP3A4 and CYP2C8 also contribute to the metabolism of fluvastatin. Pravastatin, however, is not metabolized to any appreciable extent by the CYP system (Corsini et al., 1999) but is excreted unchanged in the urine. Pravastatin and fluvastatin, which are not extensively metabolized by the CYP3A4 system, may be less likely to cause myopathy when used with one of the predisposing drugs. However, because cases of myopathy have been reported with both drugs, the benefits of combined therapy with any statin should be carefully weighed against the risk of myopathy. The myopathy syndrome is characterized by intense myalgia, first in the arms and thighs and then in the entire body (similar to flu-related myalgia) along with fatigue. The symptoms progress as long as patients continue to take the drug or drugs that induce the myopathy. Myoglobinuria and renal failure have been reported (Pogson et al., 1999). Serum creatine kinase (CK) levels in affected patients typically are 10-fold higher than the upper limit of normal. As soon as myopathy is suspected, a blood sample should be drawn to document the presence of a 10-fold elevation of CK, as many patients complain of muscle pain that is not due to true statin-induced myopathy. The statin, and any other drug suspected of contributing to myopathy, should be discontinued if true myopathy is suspected, even if it is not possible to measure CK activity to document the presence of myopathy. Rhabdomyolysis should be excluded and renal function monitored. Since myopathy rarely occurs in the absence of combination therapy, routine CK monitoring is not recommended unless the statins are used with one of the predisposing drugs. Such monitoring is not sufficient to protect patients, as myopathy can occur months to years after combined therapy is initiated. As a rule, statins may be used in combination with one of these predisposing drugs with reduced risk of myopathy if the statin is administered at no more than 25% of its maximal dose (Christians et al., 1998). Although cataracts were a concern initially, careful monitoring of patients in the early days of statin use failed to document any statin-induced eye pathology (Bradford et al., 1991). Differences in the solubility of the statins (hydrophilic versus hydrophobic) prompted speculation that the more lipid-soluble drugs might be more likely to penetrate the central nervous system. However, cerebrospinal fluid concentrations of the two lipid-soluble statins, lovastatin and simvastatin, are very low, which is probably due to extensive plasma protein binding of these drugs. Differences in lipid solubility among statins do not appear to be clinically relevant. Pregnancy The safety of statins during pregnancy has not been established. Women wishing to conceive should not take statins. During their childbearing years, women taking statins should use highly effective contraceptive procedures. Nursing mothers also are advised to avoid taking statins. Therapeutic Uses Each statin has a low recommended starting dose that reduces LDL-C by 20% to 30%. Since studies have documented that a majority of dyslipidemic patients remain on their initial dose and are not titrated to achieve their target LDL-C level, this approach leads to undertreatment. For this reason, it is advisable to start patients on a dose that will achieve the patient's target goal for LDL-C lowering. For example, a patient with a baseline LDL-C of 150 mg/dl and a goal of 100 mg/dl requires a 33% reduction in LDL-C and should be started on a dose that will provide it (seeTable 369). The manufacturer's initial recommended dose of lovastatin ( MEVACOR ) is 20 mg and is slightly more effective if taken with the evening meal than if it is taken at bedtime. If it is more convenient, bedtime dosing is preferable to missing doses. The statins that have half-lives of 4 hours or less should be given in the evening because hepatic cholesterol synthesis is maximal between midnight and 2:00 a.m. The dose may be increased every 3 to 6 weeks up to a maximum of 80 mg per day. The 80-mg dose is slightly (2% to 3%) more effective if given as 40 mg twice daily. The FDA-approved starting dose of simvastatin ( ZOCOR ) for most patients is 20 mg at bedtime unless the required LDL-C reduction exceeds 45%, in which case a 40-mg starting dose is indicated. The maximal dose is 80 mg, and the drug should be taken at bedtime. In patients taking cyclosporine, fibrates, or niacin, the daily dose should not exceed 20 mg. Pravastatin ( PRAVACHOL ) therapy is initiated with a 10- or 20-mg dose that may be increased to 40 mg. This drug should be taken at bedtime. Since pravastatin is a hydroxy acid, it is bound by bile-acid sequestrants, which reduces its absorption. Practically, this is rarely a problem since the resins should be taken before meals and pravastatin should be taken at bedtime. The starting dose of fluvastatin ( LESCOL ) is 20 or 40 mg, and the maximum is 80 mg per day. Like pravastatin, it is administered as a hydroxy acid and should be taken at bedtime, several hours after ingesting a bile-acid sequestrant. Atorvastatin ( LIPITOR ) has a long half-life, which allows administration of this statin at any time of the day. The starting dose is 10 mg, and the maximum is 80 mg per day. Cerivastatin (BAYCOL) is available in doses ranging between 0.2 and 0.4 mg. It should be taken at bedtime and several hours after a dose of a bile-acid sequestrant. The maximal FDA-approved dose is 0.8 mg. The choice of statins should be based on efficacy and cost. Can the dose of a particular statin reduce the patient's LDL-C to the target level? Cost should be the next discriminating factor. However, three drugs (lovastatin, simvastatin, and pravastatin) have been used safely in clinical trials involving thousands of subjects for 5 or more years. The documented safety records of these statins should be considered, especially when initiating therapy in younger patients. Once drug treatment is initiated, it is almost always lifelong. Baseline determinations of ALT and repeat testing at 3 to 6 months are recommended. If ALT is normal after the initial 3 to 6 months, then it need not be repeated more than once every 6 to 12 months. CK measurements are not routinely necessary unless the patient also is taking a drug that enhances the risk of myopathy. However, even if CK levels are monitored every 3 to 4 months, patients receiving combined therapy may develop myopathy months to years after starting the therapy. Bile-Acid Sequestrants The two established bile-acid sequestrants or resins (cholestyramine and colestipol) are among the oldest of the hypolipidemic drugs, and they are probably the safest, since they are not absorbed from the intestine (West et al., 1980; Groot et al., 1983). These resins are the only hypocholesterolemic drugs currently recommended for children 11 to 20 years of age, although data now are emerging that document the safety of statin therapy of children in this age range (National Cholesterol Education Program, 1991; Stein et al., 1999). Because statins are so effective as monotherapy, the resins are most often used as second agents if statin therapy does not lower LDL-C levels sufficiently. When used with a statin, cholestyramine and colestipol usually are prescribed at submaximal doses. Maximal doses can reduce LDL-C by up to 25% but are associated with unacceptable gastrointestinal side effects (bloating and constipation) that limit compliance. Colesevelam is a new bile-acid sequestrant that is prepared as an anhydrous gel and taken as a tablet. It lowers LDL-C by 18% at its maximum dose. The safety and efficacy of colesevelam have not been studied in pediatric patients or pregnant women. Cholestyramine was used in the Coronary Primary Prevention Trial, one of the first studies to document that lowering LDL-C prevents heart disease events (Lipid Research Clinics Program, 1984a; Lipid Research Clinics Program, 1984b). Cholestyramine therapy reduced total cholesterol and LDL-C by 13% and 20%, respectively, compared with diet-induced reductions of 5% in total cholesterol and 8% in LDL-C. CHD events (fatal and nonfatal) were reduced by 19%, suggesting that a 1% reduction in total cholesterol is associated with at least a 2% reduction in CHD events. Chemistry Cholestyramine and colestipol are anion-exchange resins. Cholestyramine, a polymer of styrene and divinylbenzene with active sites formed from trimethylbenzylammonium groups, is a quaternary amine (Figure 364). Colestipol, a co-polymer of diethylenetriamine and 1-chloro-2,3-epoxypropane, is a mixture of tertiary and quaternary diamines (Figure 364). Cholestyramine and colestipol are hygroscopic powders administered as chloride salts and are insoluble in water. Colesevelam is a polymer, poly(allylamine hydrochloride), cross-linked with epichlorohydrin and alkylated with 1-bromodecane and (6-bromohexyl)-trimethylammonium bromide (Figure 364). It is a hydrophilic gel and insoluble in water.
Mechanism of Action The bile-acid sequestrants are highly positively charged and bind negatively charged bile acids. Because of their large size, resins are not absorbed, and the bound bile acids are excreted in the stool. Since over 95% of bile acids are normally reabsorbed, interruption of this process depletes the liver's pool of bile acids, and hepatic bile-acid synthesis increases. As a result, hepatic cholesterol content declines, stimulating the production of LDL receptors, an effect similar to that of statins (Bilheimer et al., 1983). The increase in hepatic LDL receptors increases LDL clearance and lowers LDL-C levels, but this effect is partially offset by the enhanced cholesterol synthesis caused by upregulation of HMG-CoA reductase (Shepherd et al., 1980; Reihnr et al., 1990). Inhibition of reductase activity by a statin substantially increases the effectiveness of the resins. The resin-induced increase in bile-acid production is accompanied by an increase in hepatic triglyceride synthesis, which is of consequence in patients with significant hypertriglyceridemia (baseline triglyceride level >250 mg/dl). In such patients, bile-acid sequestrant therapy may cause striking increases in triglyceride levels. An initial report suggests that colesevelam may not raise triglyceride levels significantly; however, baseline triglyceride levels in the patients studied were not elevated (Davidson et al., 1999). Consequently, use of colesevelam to lower LDL-C levels in hypertriglyceridemic patients should be accompanied by frequent (every 1 to 2 weeks) monitoring of fasting triglyceride levels until the triglyceride level is stable, or its use should be avoided until data are published supporting the use of colesevelam in these patients. Effects on Lipoprotein Levels The reduction in LDL-C is dose-dependent. Doses of 8 to 12 g of cholestyramine or 10 to 15 g of colestipol are associated with 12% to 18% reductions in LDL-C (Casdorph, 1975; Hunninghake et al., 1981). Maximal doses (24 g of cholestyramine or 30 g of colestipol) may reduce LDL-C by as much as 25%, but cause gastrointestinal side effects and are poorly tolerated by most patients. One to two weeks is sufficient to attain maximal LDL-C reduction by a given resin dose. In patients with normal triglyceride levels, triglycerides may increase transiently and then return to baseline. HDL-C levels increase 4% to 5% (Witztum et al., 1979). Statins plus resins or niacin plus resins can reduce LDL-C by as much as 40% to 60% (Kane et al., 1981; Illingworth et al., 1981; Davignon and Pearson, 1998). Colesevelam in doses of 3.0 to 3.75 g reduces LDL-C levels significantly in a dose-dependent manner by 9% to 19%. Adverse Effects and Drug Interactions The resins are quite safe, as they are not systemically absorbed (West et al., 1980). Since they are administered as a chloride salt, rare instances of hyperchloremic acidosis have been reported. Severe hypertriglyceridemia is a contraindication to the use of cholestyramine and colestipol since these resins increase triglyceride levels (Crouse, 1987). At present, there are insufficient data on the effect of colesevelam on triglyceride levels. Cholestyramine and colestipol both are available as a powder that must be mixed with water and drunk as a slurry. The gritty sensation is objectionable to patients initially but can be tolerated easily. Colestipol is available in a tablet form that reduces the complaint of grittiness but not the gastrointestinal symptoms. Colesevelam is available as a hard capsule that absorbs water and creates a soft, gelatinous material that allegedly minimizes the potential for gastrointestinal irritation. The main objections of patients taking cholestyramine and colestipol are bloating and dyspepsia. However, these symptoms can be substantially reduced if the drug is completely suspended in liquid several hours before ingestion. For example, evening doses can be mixed in the morning and refrigerated; morning doses can be mixed the previous evening and refrigerated. Constipation still may occur but sometimes can be prevented by adequate daily water intake and psyllium, if necessary. Fecal impaction has been reported. Limited data from 106 patients treated with colesevelam for six weeks suggest that it may not cause the dyspepsia, bloating, and constipation observed in patients treated with cholestyramine or colestipol (Davidson et al., 1999). Cholestyramine and colestipol bind many other drugs and interfere with their absorption. Drugs affected include, but are not limited to, some thiazides, furosemide, propranolol, l-thyroxine, some cardiac glycosides, coumarin anticoagulants, and some of the statins (Farmer and Gotto, 1994). The effect of cholestyramine and colestipol on the absorption of most drugs has not been studied. For this reason, it is wise to administer all drugs either 1 hour before or 3 to 4 hours after a dose of cholestyramine or colestipol. Colesevelam does not appear to interfere with the absorption of fat-soluble vitamins or of drugs such as digoxin, lovastatin, warfarin, metoprolol, quinidine, and valproic acid. However, the maximum concentration and the area under the curve of sustained-release verapamil were reduced by 31% and 11%, respectively, when coadministered with colesevelam. Since the effect of colesevelam on the absorption of other drugs has not been tested, it seems prudent to recommend that patients take other medications 1 hour before or 3 or 4 hours after a dose of colesevelam. Therapeutic Uses Cholestyramine resin QUESTRAN, QUESTRAN LIGHT, LOCHOLEST LIGHT, PREVALITE) is available in bulk (with scoops that measure a 4-g dose) or in individual packets of 4 g. Additional flavorings are added to increase palatability. The 'light' preparations contain artificial sweeteners rather than sucrose. Colestipol hydrochloride ( COLESTID, FLAVORED COLESTID) is available in bulk, in individual packets containing 5 g of colestipol, or as 1-g tablets. The powdered forms of cholestyramine (4 g per dose) and colestipol (5 g per dose) are either mixed with a fluid (water or juice) and drunk as a slurry or mixed with crushed ice in a blender. Each patient should be instructed to determine the most palatable slurry. Resins should never be taken in the dry form. Ideally, patients should take the resins before breakfast and before supper, starting with one scoop or packet twice daily, and then increase the number of scoops or packets after several weeks or longer as needed and as tolerated. Most studies that have carefully monitored patients using resins have found that patients generally will not take more than two doses (scoops or packets) twice a day. Colesevelam hydrochloride WELCHOL) is available as a solid tablet containing 0.625 g of colesevelam. The starting dose is either 3 tablets taken twice daily with meals or all 6 tablets taken with a meal. The tablets should be taken with a liquid. The maximum daily dose is 7 tablets (4.375 g). Nicotinic Acid (Niacin) Nicotinic acid (niacin, pyridine-3-carboxylic acid) is one of the oldest drugs used to treat dyslipidemia and is the most versatile in that it favorably affects virtually all lipid parameters (Altschul et al., 1955; Knopp, 1998). Niacin is a water-soluble B-complex vitamin that functions as a vitamin only after its conversion to nicotinamide adenine dinucleotide (seeChapter 63: Water-Soluble Vitamins: The Vitamin B Complex and Ascorbic Acid). Oral nicotinamide may be used as a source of niacin for its vitamin functions, but nicotinamide does not affect lipid levels. The hypolipidemic effects of niacin require larger doses than are required for its vitamin effects. Niacin is the best agent available for increasing HDL-C (increments of 30% to 40%); it also lowers triglycerides by 35% to 45% (as effectively as fibrates and the more potent statins) and reduces LDL-C levels by 20% to 30% (Knopp et al., 1985; Vega and Grundy, 1994; Martin-Jadraque et al., 1996). Niacin also is the only lipid-lowering drug that reduces Lp(a) levels significantly, by about 40% (Carlson et al., 1989). However, adequate control of other lipid abnormalities renders an elevation of Lp(a) harmless (Maher et al., 1995). The only other drugs that significantly lower Lp(a) levels are estrogen and neomycin (Gurakar et al., 1985; Espeland et al., 1998; Shlipak et al., 2000). Despite its salutary effect on lipids, niacin has side effects that limit its use (see'Adverse Effects,' below). Mechanism of Action Niacin has multiple effects on lipoprotein metabolism. In adipose tissue, niacin inhibits the lipolysis of triglycerides by hormone-sensitive lipase, which reduces transport of free fatty acids to the liver and decreases hepatic triglyceride synthesis (Grundy et al., 1981). In the liver, niacin reduces triglyceride synthesis by inhibiting both the synthesis and esterification of fatty acids, effects that increase apoB degradation (Jin et al., 1999). Reduction of triglyceride synthesis reduces hepatic VLDL production, which accounts for the reduced LDL levels. Niacin also enhances LPL activity, which promotes the clearance of chylomicrons and VLDL triglycerides. Niacin raises HDL-C levels by decreasing the fractional clearance of apoA-I in HDL rather than by enhancing HDL synthesis (Blum et al., 1977). This effect is due to a reduction in the hepatic clearance of HDL-apoA-I, but not of cholesteryl esters, thereby increasing the apoA-I content of plasma and augmenting reverse cholesterol transport (Jin et al., 1997). Effects on Plasma Lipoprotein Levels Niacin in doses of 2 to 6 g per day reduces triglycerides by 35% to 50%, and the maximal effect occurs within 4 to 7 days (Figge et al., 1988). Reductions of 25% in LDL-C levels are possible with doses of 4.5 to 6 g per day, but 3 to 6 weeks are required for maximal LDL reductions to be observed. HDL-C increases less in patients with low HDL-C levels (<35 mg/dl) than in those with higher levels. Average increases of 15% to 30% occur in patients with low HDL-C levels; greater increases may occur in patients with normal HDL-C levels at baseline (Vega and Grundy, 1994). Combination therapy with resins can reduce LDL-C levels by as much as 40% to 60% (Malloy et al., 1987). Absorption, Fate, and Excretion The pharmacological doses of regular (crystalline) niacin (>1 g per day) used to treat dyslipidemia are almost completely absorbed, and peak plasma concentrations (up to 0.24 mM) are achieved within 30 to 60 minutes. The half-life is about 60 minutes, which accounts for the necessity of twice or thrice daily dosing. At lower doses, most niacin is taken up by the liver; only the major metabolite, nicotinuric acid, is found in the urine. At higher doses, a greater proportion of the drug is excreted in the urine as unchanged nicotinic acid (Iwaki et al., 1996). Adverse Effects Two of niacin's side effects, flushing and dyspepsia, limit patient
compliance. The cutaneous effects include flushing and pruritus of the face
and upper trunk, skin rashes, and acanthosis nigricans. The most common, medically serious side effects are hepatotoxicity, which causes elevated serum transaminases, and hyperglycemia. Both regular (crystalline) niacin and sustained-release niacin, which was developed to reduce flushing and itching, have been reported to cause severe liver toxicity, and sustained-released niacin can cause fulminant hepatic failure (Christensen et al., 1961; Mullin et al., 1989; McKenney et al., 1994; Tatet al., 1998). A newer preparation of sustained-release niacin ( NIASPAN ) appears to be less likely to cause severe hepatotoxicity (Capuzzi et al., 1998), perhaps simply because it is administered once daily instead of more frequently (Guyton et al., 1998). The incidence of flushing and pruritus with this preparation is not substantially different from that with regular niacin. Patients using sustained-release niacin may develop hepatic toxicity at any time, but severe toxicity appears to occur when patients take more than 2 g of sustained-release, over-the-counter preparations. Affected patients experience flu-like fatigue and weakness. Usually, aspartate transaminase and ALT are elevated, serum albumin levels decline, and all serum lipid levels decline substantially. In fact, concurrent reductions in LDL-C and HDL-C in a patient taking niacin should prompt concern about niacin toxicity. In patients with diabetes mellitus, niacin-induced insulin resistance can cause severe hyperglycemia, and the drug usually is not recommended for use in these patients (Knopp et al., 1985; Henkin et al., 1991; Schwartz, 1993). In fact, niacin use in patients with diabetes mellitus often mandates a change to insulin therapy. If niacin is prescribed for patients with known or suspected diabetes, blood glucose levels should be monitored at least once a week until proven to be stable. Niacin also elevates uric acid levels and may reactivate gout. A history of gout is a relative contraindication for niacin use. More rare side effects include toxic amblyopia and toxic maculopathy, which are reversible. Atrial tachyarrhythmias and atrial fibrillation have been reported, more commonly in elderly patients. Niacin, at doses used in human beings, has been associated with birth defects in experimental animals and should not be taken by pregnant women. Therapeutic Uses Niacin is indicated for hypertriglyceridemia and elevated LDL-C; it is especially useful in patients with both hypertriglyceridemia and low HDL-C levels. There are two commonly available forms of niacin. Crystalline niacin (immediate-release or regular) refers to niacin tablets that dissolve quickly after ingestion. Sustained-release niacin refers to preparations that continuously release niacin for 6 to 8 hours after ingestion. Crystalline niacin tablets do not require a prescription and are available in a variety of strengths from 50-mg to 500-mg tablets. To minimize the flushing and pruritus, it is best to start with a low dose (e.g., 100 mg twice daily taken after breakfast and supper). The dose may be increased stepwise every 7 days by 100 to 200 mg to a total daily dose of 1.5 to 2.0 g. After 2 to 4 weeks at this dose, transaminases, serum albumin, fasting glucose, and uric acid levels should be measured. Lipid levels should be checked and the dose increased further until the desired effect on plasma lipids is achieved. After a stable dose is attained, blood should be drawn every 3 to 6 months to monitor for the various toxicities. Since concurrent use of niacin and statins can cause myopathy, the dose of the statin should be maintained at no more than 25% of each particular statin's maximal dose. Patients also should be instructed to expect flu-like muscle aches throughout the body if myopathy occurs. Routine measurement of CK in patients taking niacin and statins does not assure that severe myopathy will be prevented or detected, as patients have developed myopathy after several years of concomitant use of niacin with a statin. Over-the-counter, sustained-release niacin preparations are effective up to a total daily dose of 2.0 g per day. All doses of sustained-release niacin, but particularly doses above 2 g per day, have been reported to cause hepatotoxicity, which may occur soon after beginning therapy or after several years of use (Knopp et al., 1985). Although the exact incidence of hepatotoxicity from use of sustained-release niacin is unknown because it can be purchased without a prescription, the potential for severe liver damage should preclude its use in most patients, including those who have taken an equivalent dose of crystalline niacin safely for many years and are considering switching to a sustained-release preparation (Mullin et al., 1989). Fibric Acid Derivatives History Thorp and Waring (1962) reported that ethyl chlorophenoxyisobutyrate
lowered lipid levels in rats. In 1967, the ester form (clofibrate) was
approved for use in the Two subsequent trials, the Helsinki Heart Study and the Veterans Affairs HDL Intervention Trial, have reported favorable effects of gemfibrozil therapy on fatal and nonfatal cardiac events without an increase in morbidity or mortality (Frick et al., 1987; Rubins et al., 1999). Chemistry Clofibrate, the prototype of the fibric acid derivatives, is the ethyl
ester of p-chlorophenoxyisobutyrate. Gemfibrozil is a nonhalogenated
phenoxypentanoic acid and thus is distinct from the halogenated fibrates. A
number of fibric acid analogs (e.g., fenofibrate, bezafibrate, and ciprofibrate)
have been developed and are used in
Mechanism of Action Despite extensive studies in human beings, the mechanisms by which
fibrates lower lipoprotein levels, or raise HDL levels, remain unclear (Grundy
and Vega, 1987; Illingworth, 1991). Recent studies suggest that many of the
effects of these compounds on blood lipids are mediated by their interaction
with peroxisome proliferatoractivated receptors (PPARs) (Kersten et al.,
2000), which regulate gene transcription. Three PPAR isotypes ( LDL levels rise in many patients, especially hypertriglyceridemic
patients, treated with gemfibrozil. However, LDL levels are unchanged or fall
in others, especially those whose triglyceride levels are not elevated or who
are taking a second-generation agent, such as fenofibrate, bezafibrate, or
ciprofibrate. The fall of LDL levels may be due in part to changes in the
cholesterol and triglyceride contents of LDL that are mediated by cholesteryl
ester transfer protein activity; such changes can alter the affinity of LDL
for the LDL receptor (Eisenberg et al., 1984). There also is evidence
that a PPAR Most of the fibric acid agents have potential antiatherothrombotic effects, including inhibition of coagulation and enhancement of fibrinolysis. These salutary effects also could alter cardiovascular outcomes by mechanisms unrelated to any hypolipidemic activity (Watts and Dimmitt, 1999). Effects on Lipoprotein Levels The effects of the fibric acid agents on lipoprotein levels differ widely depending on the starting lipoprotein profile, the presence or absence of a genetic hyperlipoproteinemia, the associated environmental influences, and the drug used. Patients with type III hyperlipoproteinemia (dysbetalipoproteinemia) are among the most sensitive responders to fibrates (Mahley and Rall, 2001). Elevated triglyceride and cholesterol levels are dramatically lowered, and tuberoeruptive and palmar xanthomas may regress completely. Angina and intermittent claudication also improve (Kuo et al., 1988). In patients with mild hypertriglyceridemia (e.g., triglycerides <400 mg/dl), fibrate treatment decreases triglyceride levels by up to 50% and increases HDL-C concentrations about 15%; LDL-C levels may be unchanged or increase. The second-generation agents, such as fenofibrate, bezafibrate, and ciprofibrate, lower VLDL levels to a degree similar to that produced by gemfibrozil, but they also are more likely to decrease LDL levels by 15% to 20%. In patients with more marked hypertriglyceridemia (e.g., 400 to 1000 mg/dl), a similar fall in triglycerides occurs, but LDL increases of 10% to 30% are seen frequently. Normotriglyceridemic patients with heterozygous familial hypercholesterolemia usually experience little change in LDL levels with gemfibrozil; with the other fibric acid agents, reductions as great as 20% may occur in some patients. Fibrates usually are the drugs of choice for treating severe hypertriglyceridemia and the chylomicronemia syndrome. While the primary therapy is to remove alcohol and as much fat from the diet as possible, fibrates help both by increasing triglyceride clearance and by decreasing hepatic triglyceride synthesis. In patients with chylomicronemia syndrome, fibrate maintenance therapy and a low-fat diet keep triglyceride levels well below 1000 mg/dl and thus prevent episodes of pancreatitis. Gemfibrozil was used in the Helsinki Heart Study, a primary prevention trial of 4081 hyperlipidemic men who received either placebo or gemfibrozil for 5 years (Frick et al., 1987). Gemfibrozil reduced total cholesterol by 10% and LDL-C by 11%, raised HDL-C levels by 11%, and decreased triglycerides by 35%. Overall, there was a 34% decrease in fatal and nonfatal cardiovascular events without any effect on total mortality. No increased incidence of gallstones or cancers was observed. Subgroup analysis suggested that the greatest benefit occurred in the subjects with the highest levels of VLDL or combined VLDL and LDL and in those with the lowest HDL-C levels (<35 mg/dl). It also is possible that gemfibrozil affected the outcome by influencing platelet function, coagulation factor synthesis, or LDL size. In a recent secondary prevention trial, gemfibrozil reduced fatal and nonfatal CHD events by 22% despite a lack of effect on LDL-C levels. HDL-C levels increased by 6%, which may have contributed to the favorable outcome (Rubins et al., 1999). Absorption, Fate, and Excretion All of the fibrate drugs are absorbed rapidly and efficiently (>90%) when given with a meal but less efficiently when taken on an empty stomach. The ester bond is hydrolyzed rapidly, and peak plasma concentrations are attained within 1 to 4 hours. More than 95% of these drugs in plasma are bound to protein, nearly exclusively to albumin. The half-lives of fibrates differ significantly (Miller and Spence, 1998), ranging from 1.1 hours (gemfibrozil) to 20 hours (fenofibrate). The drugs are widely distributed throughout the body, and concentrations in liver, kidney, and intestine exceed the plasma level. Gemfibrozil is transferred across the placenta. The fibrate drugs are excreted predominantly as glucuronide conjugates; 60% to 90% of an oral dose is excreted in the urine, with smaller amounts appearing in the feces. Excretion of these drugs is impaired in renal failure, though excretion of gemfibrozil was reported to be less severely compromised in renal insufficiency than was excretion of other fibrates (Evans et al., 1987). Nevertheless, the use of fibrates is contraindicated in patients with renal failure. Adverse Effects and Drug Interactions Fibric acid compounds usually are well tolerated (Miller and Spence, 1998). Side effects may occur in 5% to 10% of patients but most often are not sufficient to cause discontinuation of the drug. Gastrointestinal side effects occur in up to 5% of patients. Other side effects are reported infrequently and include rash, urticaria, hair loss, myalgias, fatigue, headache, impotence, and anemia. Minor increases in liver transaminases and decreases in alkaline phosphatase have been reported. Clofibrate, bezafibrate, and fenofibrate have been reported to potentiate the action of oral anticoagulants, in part by displacing them from their binding sites on albumin. Careful monitoring of the prothrombin time and reduction in dosage of the anticoagulant may be appropriate when treatment with a fibrate is begun. A myositis flu-like syndrome occasionally occurs in subjects taking clofibrate, gemfibrozil, and fenofibrate, and may occur in up to 5% of patients treated with a combination of an HMG-CoA reductase inhibitor and gemfibrozil, if higher doses of the reductase inhibitor are used. Patients on this combination should be instructed to be aware of the potential symptoms and should be followed at 3-month intervals with careful history and determination of CK values until a stable pattern is established. Use of fibrates with cerivastatin should be avoided because a number of cases of rhabdomyolysis have resulted from combined gemfibrozil-cerivastatin therapy (Guyton et al., 1999; Alexandridis et al., 2000). All of the fibrates increase the lithogenicity of bile. In the Coronary Drug Project and the WHO trial, clofibrate use was associated with increased risk of gallstone formation. However, no significant increase was seen in the Helsinki Heart Study with the use of gemfibrozil or in the VA HIT. Placebo-controlled clinical trial data for fenofibrate are not available. Renal failure is a relative contraindication to the use of fibric acid agents, as is hepatic dysfunction. Combined statin-fibrate therapy should be avoided in patients with compromised renal function. Gemfibrozil should be used with caution and at a reduced dosage to treat the hyperlipidemia of renal failure. Fibrates should not be used by children or pregnant women. Therapeutic Uses Clofibrate (ATROMID-S) is available for oral administration. The usual dose is 2 g per day in divided doses. This compound is little used, but it still may be useful in patients who do not tolerate gemfibrozil or fenofibrate. Gemfibrozil ( LOPID ) is usually administered as a 600-mg dose taken twice a day 30 minutes before the morning and evening meals. Fenofibrate (TRICOR) is available as single-dose capsules of 67, 134, and 200 mg. Fibrates are the drugs of choice for treating hyperlipidemic subjects with type III hyperlipoproteinemia as well as subjects with severe hypertriglyceridemia (triglycerides >1000 mg/dl), who are at risk for pancreatitis. Based on the VA HIT results, fibrates appear to have an important role in subjects with familial combined hyperlipidemia, who predominantly have elevated VLDL levels and low HDL-C levels. When fibrates are used in such patients, the LDL levels need to be monitored; if LDL levels rise, the addition of a low dose of a statin may be needed. Alternatively, many experts now treat such patients first with a statin, and only subsequently add a 600-mg dose of gemfibrozil once or twice a day to further lower VLDL levels. If this combination is used, there should be careful monitoring for myositis. |
Prospectus
|
New Lipid-Lowering Agents Statins More potent statins capable of lowering LDL-C by >65% are being developed. Some of these agents also may have greater efficacy in reducing triglycerides and raising HDL-C. A new statin, ZD4522, in developmental clinical trials has been reported to reduce LDL-C by 65% (Olsson et al., 2000). MTP Inhibitor MTP transfers triglycerides and other nonpolar lipids to the apolipoproteins of nascent lipoproteins as they form in the intestine and liver and is required for the synthesis and secretion of chylomicrons and VLDL. For example, an MTP inhibitor targeted to the liver would decrease VLDL production, thereby decreasing plasma triglyceride levels and ultimately reducing LDL production from VLDL. One such MTP inhibitor, BMS-201038, is in clinical trials (Wetterau et al., 1998). Dietary and Biliary Cholesterol Absorption Inhibitor Ezetimibe is an azetidione-based cholesterol absorption inhibitor that blocks the intestinal absorption of cholesterol, resulting in lowered plasma total cholesterol and LDL-C levels (van Heek et al., 2000). The drug undergoes glucuronidation in the intestine, and the absorbed glucuronide, an active metabolite, is excreted into the bile by the liver. Due to its enterohepatic circulation, the half-life of the active metabolite is 22 hours in human beings, which indicates that once daily dosing is sufficient (Zhu et al., 2000). The maximum effective dose is 10 mg daily, which provides a 19% reduction in LDL-C (Lipka et al., 2000). A 19% reduction of LDL-C by ezetimibe is equivalent to three doublings of a statin from its baseline dose, since each doubling of a statin dose after the starting dose provides an additional 6% decrease in LDL-C (Pedersen and Tobert, 1996). In human beings, the combination of simvastatin (20 mg; the starting dose of simvastatin) plus ezetimibe (10 mg) provides a 52% reduction in LDL-C (Kosoglou et al., 2000). This 50% reduction achieved with low-dose simvastatin plus ezetimibe (10 mg) is equivalent to that of 80 mg of simvastatin alone. Since there is virtually no myopathy when statins are used at their starting doses, the combination of ezetimibe plus low-dose statin promises an additional margin of safety. Early studies suggest that there are no dangerous interactions between ezetimibe and statins, but further investigations are required to adequately document this initial impression. ACAT Inhibitors To date, ACAT inhibitors have failed to reach the marketplace. However, the recognition that there are at least two forms of this enzyme with specific tissue sites of expression suggests that it may be possible to develop an inhibitor that specifically inhibits the assimilation of dietary cholesterol. ACAT-1 is expressed in several tissues, including macrophages (Chang et al., 1993). Avasimibe, an inhibitor of this enzyme, appears to reduce macrophage and cholesteryl ester contents of lesions in cholesterol-fed rabbits and could affect atherosclerotic lesion development, an effect that could stabilize lesions (Bocan et al., 2000). Avasimibe reduced plasma triglycerides and LDL-C levels by up to 50% in miniature swine; however, hepatictriglyceride content increased two- to sevenfold in a dose-dependent manner (Burnett et al., 1999). Interestingly, ACAT-1 knockout mice did not have reduced susceptibility to developing atherosclerosis (Accad et al., 2000). ACAT-2 is expressed in the liver and intestine and appears to play a role in cholesteryl ester formation for VLDL and chylomicron production (Cases et al., 1998). An inhibitor of this form of ACAT could reduce plasma lipids. For further discussion of hyperlipoproteinemias and other disorders of
lipid metabolism, seeChapter 335 in Harrison's Principles of
Internal Medicine, 16th ed., |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3373
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved